
UNITED NATIONS ENVIRONMENT PROGRAMME
INTERNATIONAL LABOUR ORGANISATION
WORLD HEALTH ORGANIZATION
INTERNATIONAL PROGRAMME ON CHEMICAL SAFETY
ENVIRONMENTAL HEALTH CRITERIA 215
Vinyl Chloride
This report contains the collective views of an international group of
experts and does not necessarily represent the decisions or the stated
policy of the United Nations Environment Programme, the International
Labour Organisation, or the World Health Organization.
Published under the joint sponsorship of the United Nations
Environment Programme, the International Labour Organisation, and the
World Health Organization, and produced within the framework of the
Inter-Organization Programme for the Sound Management of Chemicals.
World Health Organization
Geneva, 1999
The International Programme on Chemical Safety (IPCS),
established in 1980, is a joint venture of the United Nations
Environment Programme (UNEP), the International Labour Organisation
(ILO), and the World Health Organization (WHO). The overall objectives
of the IPCS are to establish the scientific basis for assessment of
the risk to human health and the environment from exposure to
chemicals, through international peer-review processes, as a
prerequisite for the promotion of chemical safety, and to provide
technical assistance in strengthening national capacities for the
sound management of chemicals.
The Inter-Organization Programme for the Sound Management of
Chemicals (IOMC) was established in 1995 by UNEP, ILO, the Food and
Agriculture Organization of the United Nations, WHO, the United
Nations Industrial Development Organization and the Organisation for
Economic Co-operation and Development (Participating Organizations),
following recommendations made by the 1992 UN Conference on
Environment and Development to strengthen cooperation and increase
coordination in the field of chemical safety. The purpose of the IOMC
is to promote coordination of the policies and activities pursued by
the Participating Organizations, jointly or separately, to achieve the
sound management of chemicals in relation to human health and the
environment.
WHO Library Cataloguing in Publication Data
Vinyl chloride.
(Environmental health criteria ; 215)
1.Vinyl chloride - analysis 2.Vinyl chloride - toxicity
3.Vinyl chloride - adverse effects 4.Environmental exposure
5.Occupational exposure I.International Programme on
Chemical Safety II.Series
ISBN 92 4 157215 9 (NLM Classification: QV 633)
ISSN 0250-863X
The World Health Organization welcomes requests for permission to
reproduce or translate its publications, in part or in full.
Applications and enquiries should be addressed to the Office of
Publications, World Health Organization, Geneva, Switzerland, which
will be glad to provide the latest information on any changes made to
the text, plans for new editions, and reprints and translations
already available.
(c) World Health Organization 1999
Publications of the World Health Organization enjoy copyright
protection in accordance with the provisions of Protocol 2 of the
Universal Copyright Convention. All rights reserved.
The designations employed and the presentation of the material in
this publication do not imply the expression of any opinion whatsoever
on the part of the Secretariat of the World Health Organization
concerning the legal status of any country, territory, city or area or
of its authorities, or concerning the delimitation of its frontiers or
boundaries.
The mention of specific companies or of certain manufacturers'
products does not imply that they are endorsed or recommended by the
World Health Organization in preference to others of a similar nature
that are not mentioned. Errors and omissions excepted, the names of
proprietary products are distinguished by initial capital letters.
CONTENTS
ENVIRONMENTAL HEALTH CRITERIA FOR VINYL CHLORIDE
1. SUMMARY
1.1. Identity, physical and chemical properties, and
analytical methods
1.2. Sources of human and environmental exposure
1.3. Environmental transport, distribution and transformation
1.4. Environmental levels and human exposure
1.5. Kinetics and metabolism in laboratory animals and
humans
1.6. Effects on laboratory mammals and in vitro test
systems
1.7. Effects on humans
1.8. Effects on other organisms in the laboratory and
field
2. IDENTITY, PHYSICAL AND CHEMICAL PROPERTIES, AND ANALYTICAL
METHODS
2.1. Identity
2.2. Physical and chemical properties
2.3. Conversion factors
2.4. Analytical methods
2.4.1. General analytical methods and
detection
2.4.2. Sample preparation, extraction and analysis
for different matrices
2.4.2.1 Air
2.4.2.2 Water
2.4.2.3 PVC resins and PVC products
2.4.2.4 Food, liquid drug and cosmetic
products
2.4.2.5 Biological samples
2.4.2.6 Human monitoring
2.4.2.7 Workplace air monitoring
3. SOURCES OF HUMAN AND ENVIRONMENTAL EXPOSURE
3.1. Natural occurrence
3.2. Anthropogenic sources
3.2.1. Production levels and processes
3.2.1.1 Production of VC
3.2.1.2 Production of PVC from VC
3.2.1.3 PVC products
3.2.2. Emissions from VC/PVC plants
3.2.2.1 Sources of emission during the
production of VC
3.2.2.2 Emission of VC and dioxins from VC/PVC
plants during production
3.2.3. Accidental releases of VC
3.2.3.1 PVC plant and transport accidents
3.2.3.2 Leakage and discharge from VC/PVC
plants
3.2.4. VC residues in virgin PVC resin and products
3.2.4.1 VC residues in different PVC samples
3.2.4.2 VC residues in PVC products
3.2.4.3 VC formation as a result of heating PVC
3.2.5. Other sources of VC
3.2.5.1 VC as a degradation product of
chlorinated hydrocarbons
3.2.5.2 VC formation from tobacco
3.3. Uses
4. ENVIRONMENTAL TRANSPORT, DISTRIBUTION AND TRANSFORMATION
4.1. Transport and distribution between media
4.1.1. Air
4.1.2. Water and sediments
4.1.3. Soil and sewage sludge
4.1.4. Biota
4.2. Transformation
4.2.1. Microbial degradation
4.2.2. Abiotic degradation
4.2.2.1 Photodegradation
4.2.2.2 Hydrolysis
4.2.3. Other interactions
4.3. Bioaccumulation
4.4. Ultimate fate following use
4.4.1. Waste disposal
4.4.2. Fate of VC processed to PVC
5. ENVIRONMENTAL LEVELS AND HUMAN EXPOSURE
5.1. Environmental levels
5.1.1. Air
5.1.1.1 Outdoor air
5.1.1.2 Indoor air
5.1.2. Water and sediment
5.1.3. Soil and sewage sludge
5.1.3.1 Soil
5.1.3.2 Sewage sludge
5.1.4. Food, feed and other products
5.1.5. Terrestrial and aquatic organisms
5.2. General population exposure
5.2.1. Estimations
5.2.2. Monitoring data of human tissues
or fluids
5.3. Occupational exposure
6. KINETICS AND METABOLISM IN LABORATORY ANIMALS AND HUMANS
6.1. Absorption
6.1.1. Oral exposure
6.1.2. Inhalation exposure
6.1.3. Dermal exposure
6.2. Distribution and retention
6.2.1. Oral exposure
6.2.2. Inhalation exposure
6.2.3. Partition coefficients in vitro
6.3. Metabolic transformation
6.4. Elimination and excretion
6.4.1. Oral exposure
6.4.2. Inhalation exposure
6.5. Reaction with body components
6.5.1. Formation of DNA adducts
6.5.2. Alkylation of proteins
6.6. Modelling of pharmacokinetic data for
vinyl chloride
7. EFFECTS ON LABORATORY MAMMALS AND IN VITRO TEST SYSTEMS
7.1. Acute toxicity
7.2. Short-term toxicity
7.2.1. Oral exposure
7.2.2. Inhalation exposure
7.2.3. Dermal exposure
7.3. Long-term toxicity - effects other than tumours
7.3.1. Oral exposure
7.3.2. Inhalation exposure
7.4. Skin and eye irritation; sensitization
7.5. Reproductive toxicity, embryotoxicity and
teratogenicity
7.5.1. Male reproductive toxicity
7.5.2. Embryotoxicity and teratogenicity
7.6. Special studies
7.6.1. Neurotoxicity
7.6.2. Immunotoxicity
7.6.3. Cardiovascular effects
7.6.4. Hepatotoxicity
7.7. Carcinogenicity
7.7.1. Oral exposure
7.7.2. Inhalation exposure
7.7.2.1 Short-term exposure
7.7.2.2 Long-term exposure
7.7.3. The effect of age on susceptibility to
tumour induction
7.7.4. The effect of gender on susceptibility to
tumour induction
7.7.5. Carcinogenicity of metabolites
7.8. Genotoxicity
7.8.1. In vitro studies
7.8.2. In vivo studies
7.8.3. Genotoxicity of VC metabolites
7.8.4. Other toxic effects of VC metabolites
7.8.5. Mutagenic and promutagenic properties of DNA
adducts formed by VC metabolites
7.8.6. Mutations in VC-induced tumours
7.9. Factors modifying toxicity
7.10. Mechanisms of toxicity - mode of action
7.10.1. Mechanisms of VC disease
7.10.2. Mechanism of carcinogenesis
8. EFFECTS ON HUMANS
8.1. General population
8.2. Controlled human studies
8.3. Occupational exposure
8.3.1. Overview
8.3.2. Non-neoplastic effects
8.3.2.1 Acute toxicity
8.3.2.2 Effects of short- and long-term
exposure
8.3.2.3 Organ effects
8.3.3. Neoplastic effects
8.3.3.1 Liver and biliary tract cancers
8.3.3.2 Brain and central nervous
system (CNS)
8.3.3.3 Respiratory tract
8.3.3.4 Lymphatic and haematopoietic
cancers
8.3.3.5 Malignant melanoma
8.3.3.6 Breast cancer
8.3.3.7 Other cancer sites
8.4. Genotoxicity studies
8.4.1. Cytogenetic studies of VC-exposed
workers
8.4.2. Mutations at the hypoxanthine guanine
phosphoribosyltransferase (hprt) locus
8.4.3. Mutations in ASL from VC-exposed
workers
8.4.3.1 p53 gene
8.4.3.2 ras genes
8.5. Studies on biological markers
8.5.1. Excretion of metabolites
8.5.2. Genetic assays
8.5.3. Enzyme studies
8.5.4. von Willebrand factor
8.5.5. p53 and ras proteins
8.6. Susceptible subpopulations
8.6.1. Age susceptibility
8.6.2. Immunological susceptibility
8.6.3. Polymorphic genes in VC metabolism
9. EFFECTS ON OTHER ORGANISMS IN THE LABORATORY AND FIELD
9.1. Laboratory experiments
9.1.1. Microorganisms
9.1.1.1 Water
9.1.1.2 Soil
9.1.2. Aquatic organisms
9.1.2.1 Invertebrates
9.1.2.2 Vertebrates
9.2. Field observations
9.2.1. Aquatic organisms
10. EVALUATION OF HUMAN HEALTH RISKS AND EFFECTS ON THE
ENVIRONMENT
10.1. Evaluation of human health effects
10.1.1. Hazard identification
10.1.1.1 Non-neoplastic effects
10.1.1.2 Neoplastic effects
10.1.2. Dose-response analysis
10.1.2.1 Non-neoplastic effects
10.1.2.2 Neoplastic effects
10.1.3. Human exposure
10.1.3.1 General population
10.1.3.2 Occupational exposure
10.1.4. Risk characterization
10.2. Evaluation of effects on the environment
11. RECOMMENDATIONS FOR PROTECTION OF HUMAN HEALTH
11.1. Public health
11.2. Occupational health
12. FURTHER RESEARCH
13. PREVIOUS EVALUATIONS BY INTERNATIONAL BODIES
REFERENCES
ANNEX 1. REGULATIONS CONCERNING VINYL CHLORIDE
ANNEX 2. PHYSIOLOGICAL MODELLING AND RECENT RISK ASSESSMENTS
ANNEX 3. EXECUTIVE SUMMARY OF VINYL CHLORIDE PANEL REPORT
RESUME
RESUMEN
NOTE TO READERS OF THE CRITERIA MONOGRAPHS
Every effort has been made to present information in the criteria
monographs as accurately as possible without unduly delaying their
publication. In the interest of all users of the Environmental Health
Criteria monographs, readers are requested to communicate any errors
that may have occurred to the Director of the International Programme
on Chemical Safety, World Health Organization, Geneva, Switzerland, in
order that they may be included in corrigenda.
* * *
A detailed data profile and a legal file can be obtained from the
International Register of Potentially Toxic Chemicals, Case postale
356, 1219 Châtelaine, Geneva, Switzerland (telephone no. + 41 22 -
9799111, fax no. + 41 22 - 7973460, E-mail irptc@unep.ch).
Environmental Health Criteria
PREAMBLE
Objectives
In 1973 the WHO Environmental Health Criteria Programme was
initiated with the following objectives:
(i) to assess information on the relationship between exposure to
environmental pollutants and human health, and to provide
guidelines for setting exposure limits;
(ii) to identify new or potential pollutants;
(iii) to identify gaps in knowledge concerning the health effects of
pollutants;
(iv) to promote the harmonization of toxicological and
epidemiological methods in order to have internationally
comparable results.
The first Environmental Health Criteria (EHC) monograph, on
mercury, was published in 1976 and since that time an ever-increasing
number of assessments of chemicals and of physical effects have been
produced. In addition, many EHC monographs have been devoted to
evaluating toxicological methodology, e.g. for genetic, neurotoxic,
teratogenic and nephrotoxic effects. Other publications have been
concerned with epidemiological guidelines, evaluation of short-term
tests for carcinogens, biomarkers, effects on the elderly and so
forth.
Since its inauguration the EHC Programme has widened its scope,
and the importance of environmental effects, in addition to health
effects, has been increasingly emphasized in the total evaluation of
chemicals.
The original impetus for the Programme came from World Health
Assembly resolutions and the recommendations of the 1972 UN Conference
on the Human Environment. Subsequently the work became an integral
part of the International Programme on Chemical Safety (IPCS), a
cooperative programme of UNEP, ILO and WHO. In this manner, with the
strong support of the new partners, the importance of occupational
health and environmental effects was fully recognized. The EHC
monographs have become widely established, used and recognized
throughout the world.
The recommendations of the 1992 UN Conference on Environment and
Development and the subsequent establishment of the Intergovernmental
Forum on Chemical Safety with the priorities for action in the six
programme areas of Chapter 19, Agenda 21, all lend further weight to
the need for EHC assessments of the risks of chemicals.
Scope
The criteria monographs are intended to provide critical reviews
on the effect on human health and the environment of chemicals and of
combinations of chemicals and physical and biological agents. As such,
they include and review studies that are of direct relevance for the
evaluation. However, they do not describe every study carried out.
Worldwide data are used and are quoted from original studies, not from
abstracts or reviews. Both published and unpublished reports are
considered and it is incumbent on the authors to assess all the
articles cited in the references. Preference is always given to
published data. Unpublished data are used only when relevant published
data are absent or when they are pivotal to the risk assessment. A
detailed policy statement is available that describes the procedures
used for unpublished proprietary data so that this information can be
used in the evaluation without compromising its confidential nature
(WHO (1990) Revised Guidelines for the Preparation of Environmental
Health Criteria Monographs. PCS/90.69, Geneva, World Health
Organization).
In the evaluation of human health risks, sound human data,
whenever available, are preferred to animal data. Animal and in vitro
studies provide support and are used mainly to supply evidence
missing from human studies. It is mandatory that research on human
subjects is conducted in full accord with ethical principles,
including the provisions of the Helsinki Declaration.
The EHC monographs are intended to assist national and
international authorities in making risk assessments and subsequent
risk management decisions. They represent a thorough evaluation of
risks and are not, in any sense, recommendations for regulation or
standard setting. These latter are the exclusive purview of national
and regional governments.
Content
The layout of EHC monographs for chemicals is outlined below.
* Summary - a review of the salient facts and the risk evaluation
of the chemical
* Identity - physical and chemical properties, analytical methods
* Sources of exposure
* Environmental transport, distribution and transformation
* Environmental levels and human exposure
* Kinetics and metabolism in laboratory animals and humans
* Effects on laboratory mammals and in vitro test systems
* Effects on humans
* Effects on other organisms in the laboratory and field
* Evaluation of human health risks and effects on the environment
* Conclusions and recommendations for protection of human health
and the environment
* Further research
* Previous evaluations by international bodies, e.g. IARC, JECFA,
JMPR
Selection of chemicals
Since the inception of the EHC Programme, the IPCS has organized
meetings of scientists to establish lists of priority chemicals for
subsequent evaluation. Such meetings have been held in Ispra, Italy,
1980; Oxford, United Kingdom, 1984; Berlin, Germany, 1987; and North
Carolina, USA, 1995. The selection of chemicals has been based on the
following criteria: the existence of scientific evidence that the
substance presents a hazard to human health and/or the environment;
the possible use, persistence, accumulation or degradation of the
substance shows that there may be significant human or environmental
exposure; the size and nature of populations at risk (both human and
other species) and risks for environment; international concern, i.e.
the substance is of major interest to several countries; adequate data
on the hazards are available.
If an EHC monograph is proposed for a chemical not on the
priority list, the IPCS Secretariat consults with the Cooperating
Organizations and all the Participating Institutions before embarking
on the preparation of the monograph.
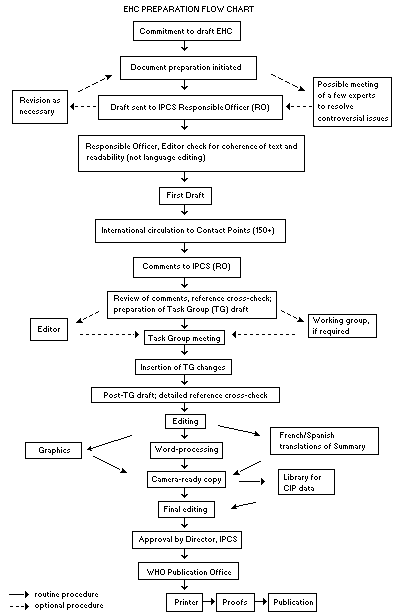
Procedures
The order of procedures that result in the publication of an EHC
monograph is shown in the flow chart on p. xv. A designated staff
member of IPCS, responsible for the scientific quality of the
document, serves as Responsible Officer (RO). The IPCS Editor is
responsible for layout and language. The first draft, prepared by
consultants or, more usually, staff from an IPCS Participating
Institution, is based initially on data provided from the
International Register of Potentially Toxic Chemicals, and reference
data bases such as Medline and Toxline.
The draft document, when received by the RO, may require an
initial review by a small panel of experts to determine its scientific
quality and objectivity. Once the RO finds the document acceptable as
a first draft, it is distributed, in its unedited form, to well over
150 EHC contact points throughout the world who are asked to comment
on its completeness and accuracy and, where necessary, provide
additional material. The contact points, usually designated by
governments, may be Participating Institutions, IPCS Focal Points, or
individual scientists known for their particular expertise. Generally
some four months are allowed before the comments are considered by the
RO and author(s). A second draft incorporating comments received and
approved by the Director, IPCS, is then distributed to Task Group
members, who carry out the peer review, at least six weeks before
their meeting.
The Task Group members serve as individual scientists, not as
representatives of any organization, government or industry. Their
function is to evaluate the accuracy, significance and relevance of
the information in the document and to assess the health and
environmental risks from exposure to the chemical. A summary and
recommendations for further research and improved safety aspects are
also required. The composition of the Task Group is dictated by the
range of expertise required for the subject of the meeting and by the
need for a balanced geographical distribution.
 The three cooperating organizations of the IPCS recognize the
important role played by nongovernmental organizations.
Representatives from relevant national and international associations
may be invited to join the Task Group as observers. Although observers
may provide a valuable contribution to the process, they can only
speak at the invitation of the Chairperson. Observers do not
participate in the final evaluation of the chemical; this is the sole
responsibility of the Task Group members. When the Task Group
considers it to be appropriate, it may meet in camera.
All individuals who as authors, consultants or advisers
participate in the preparation of the EHC monograph must, in addition
to serving in their personal capacity as scientists, inform the RO if
at any time a conflict of interest, whether actual or potential, could
be perceived in their work. They are required to sign a conflict of
interest statement. Such a procedure ensures the transparency and
probity of the process.
When the Task Group has completed its review and the RO is
satisfied as to the scientific correctness and completeness of the
document, it then goes for language editing, reference checking and
preparation of camera-ready copy. After approval by the Director,
IPCS, the monograph is submitted to the WHO Office of Publications for
printing. At this time a copy of the final draft is sent to the
Chairperson and Rapporteur of the Task Group to check for any errors.
It is accepted that the following criteria should initiate the
updating of an EHC monograph: new data are available that would
substantially change the evaluation; there is public concern for
health or environmental effects of the agent because of greater
exposure; an appreciable time period has elapsed since the last
evaluation.
All Participating Institutions are informed, through the EHC
progress report, of the authors and institutions proposed for the
drafting of the documents. A comprehensive file of all comments
received on drafts of each EHC monograph is maintained and is
available on request. The Chairpersons of Task Groups are briefed
before each meeting on their role and responsibility in ensuring that
these rules are followed.
WHO TASK GROUP ON ENVIRONMENTAL HEALTH CRITERIA FOR VINYL CHLORIDE
Members
Dr A. Barbin, International Agency for Research on Cancer, Lyon,
France
Professor V.J. Feron, TNO Nutrition and Food Research Institute, HE
Zeist, Netherlands
Ms P. Heikkilä, Uusimaa Regional Institute of Occupational Health,
Helsinki, Finland
Dr J. Kielhorn, Chemical Risk Assessment, Fraunhofer Institute for
Toxicology and Aerosol Research, Hanover, Germany (Co-Rapporteur)
Professor M. Kogevinas, Respiratory and Environmental Health Research
Unit, Municipal Institute of Medical Investigation (IMIM), Barcelona,
Spain
Mr H. Malcolm, Institute of Terrestrial Ecology, Monks Wood, Abbots
Ripton, Huntingdon, Cambridgeshire, United Kingdom (Co-Rapporteur)
Dr W. Pepelko, National Center for Environmental Assessment, Office of
Research and Development, US EPA, Washington DC, USA
Dr A. Pintér, National Institute of Environmental Health, Budapest,
Hungary (Vice-Chairman)
Dr L. Simonato, Department of Oncology, University of Padua, Venetian
Tumours Registry, Padua, Italy
Professor H. Vainio, Division of Health Risk Assessment, National
Institute of Environmental Medicine, Karolinska Institute, Stockholm,
Sweden (Chairman)
Dr E.M. Ward, Division of Surveillance Hazard, Evaluation and Field
Studies, National Institute for Occupational Safety and Health
(NIOSH), Robert Taft Laboratory, Cincinnati, Ohio, USA (Contact
address: Environmental Cancer Epidemiology, International Agency for
Research on Cancer, Lyon, France
Dr J.M. Zielinski, Biostatistics and Research Coordination Division,
Ottawa, Ontario, Canada
Secretariat
Dr A. Aitio, International Programme on Chemical Safety, World Health
Organization, Geneva, Switzerland (Secretary)
Dr I. Mangelsdorf, Chemical Risk Assessment, Fraunhofer Institute for
Toxicology and Aerosol Research, Hanover, Germany
Dr C. Melber, Chemical Risk Assessment, Fraunhofer Institute for
Toxicology and Aerosol Research, Hanover, Germany
Dr U. Wahnschaffe, Chemical Risk Assessment, Fraunhofer Institute for
Toxicology and Aerosol Research, Hanover, Germany
WHO TASK GROUP ON ENVIRONMENTAL HEALTH CRITERIA FOR VINYL CHLORIDE
A WHO Task Group on Environmental Health Criteria for Vinyl
Chloride met at the Fraunhofer Institute for Toxicology and Aerosol
Research, Hanover, Germany, from 25 to 29 January 1999. Professor H.
Muhle welcomed the participants on behalf of the Institute and its
Director, Professor U. Heinrich. Dr A. Aitio, IPCS, welcomed the
participants on behalf of the Director, IPCS, and the three IPCS
co-operating organisations (UNEP, ILO, and WHO). The Group reviewed
and revised the draft and made an evaluation of the risks for human
health and the environment from exposure to vinyl chloride.
The first and second drafts of this monograph were prepared,
under the co-ordination of Dr I. Mangelsdorf, by the authors
Dr J. Kielhorn, Dr C. Melber and Dr U. Wahnschaffe. In the preparation
of the second draft, the comments received from the IPCS contact
points were carefully considered.
Dr A. Aitio of the IPCS Central Unit was responsible for the
scientific aspects of the monograph, and Dr P.G. Jenkins for the
technical editing.
The efforts of all who helped in the preparation and finalisation
of the monograph are gratefully acknowledged.
* * *
The Federal Ministry for the Environment, Nature Conservation and
Nuclear Safety, Germany, contributed financially to the preparation of
this Environmental Health Criteria monograph, and the meeting was
organised by the Fraunhofer Institute for Toxicology and Aerosol
Research.
ABBREVIATIONS
Epsilon A 1, N 6-ethenoadenine
Epsilon C 3, N 4-ethenocytosine
Epsilon dA 1, N 6-etheno-2'-deoxyadenosine
Epsilon dC 3, N 4-etheno-2'-deoxycytidine
Epsilon G ethenoguanine
7-OEG 7-(2'-oxoethyl)guanine
ALAT alanine aminotransferase
ASAT aspartate aminotransferase
ASL angiosarcoma of the liver
BCF bioconcentration factor
CA chromosomal aberration
CAA chloroacetaldehyde
CEO chloroethylene oxide
CI confidence interval (95% unless otherwise stated)
CNS central nervous system
CYP2E1 cytochrome P-450 isozyme 2e1
ECD electron capture detection
EDC 1,2-dichloroethane
FDA Food and Drug Administration (USA)
FID flame ionization detector
GC gas chromatography
GST glutathione S-transferase
HCC hepatocellular carcinoma
HLA human-leukocyte-associated antigen
HPLC high performance liquid chromatography
HWD hazardous waste dump
IR infrared
LOAEL lowest-observed-adverse-effect level
MN micronuclei
MOR morbidity odds ratio
MS mass spectrometry
MSW municipal solid waste
NER non-extractable residue
NOAEC no-observed-adverse-effect concentration
NOAEL no-observed-adverse-effect level
NOEL no-observed-effect level
PCDD polychlorinated dibenzodioxin
PCDF polychlorinated dibenzofuran
PCE tetrachloroethene (perchloroethene)
PID photoionization detector
PVC polyvinyl chloride
SCE sister chromatid exchange
SIR standardized incidence ratio
SLRL sex-linked recessive lethal
SMR standardized mortality ratio
TCE trichloroethene
TEQ toxic equivalent quantity
UV ultraviolet
VC vinyl chloride
VOC volatile organic compound
1. SUMMARY
This monograph deals with vinyl chloride (VC) monomer itself and
is not an evaluation of polyvinyl chloride (PVC), the polymer of VC.
Exposures to VC in mixtures are not addressed.
1.1 Identity, physical and chemical properties, and analytical
methods
Under ambient conditions, VC is a colourless, flammable gas with
a slightly sweet odour. It has a high vapour pressure, a high value
for Henry's Law constant and a relatively low water solubility. It is
heavier than air and is soluble in almost all organic solvents. It is
transported in liquid form under pressure.
At ambient temperatures in the absence of air, dry purified VC is
highly stable and non-corrosive but above 450°C, or in the presence of
sodium or potassium hydroxide, partial decomposition can occur.
Combustion of VC in air produces carbon dioxide and hydrogen chloride.
With air and oxygen, very explosive peroxides can be formed,
necessitating a continuous monitoring and limitation of the oxygen
content, particularly in VC recovery plants. In the presence of water,
hydrochloric acid is formed.
Polymerization reactions to PVC are technically the most
important reactions from an industrial view, but addition reactions
with other halogens at the double bond, e.g., to yield
1,1,2-trichloroethane or 1,1-dichloroethane, are also important.
The concentration of VC in air can be monitored by trapping it on
adsorbents and, after liquid or thermal desorption, analysis by gas
chromatography. In ambient air measurements, several adsorbents in
series or refrigerated traps may be needed to increase the efficiency
of trapping. Peak concentrations at workplaces can be measured with
direct-reading instruments based on, for instance, FID or PID. In
continuous monitoring, IR and GC/FID analysers combined with data
logging and processing have been used. In analysis of VC in liquids
and solids, direct injection, extraction and more increasingly head
space or purge-and-trap techniques are applied. Also in these samples,
VC is analysed by GC fitted to, for instance, FID or MS detectors.
1.2 Sources of human and environmental exposure
VC is not known to occur naturally although it has been found in
landfill gas and groundwater as a degradation product of chlorinated
hydrocarbons deposited as solvent wastes in landfills or in the
environment of workplaces using such solvents. VC is also present in
cigarette smoke.
VC is produced industrially by two main reactions: a) the
hydrochlorination of acetylene; and b) thermal cracking (at about
500°C) of 1,2-dichloroethane (EDC) produced by direct chlorination
(ethylene and chlorine) or oxychlorination (ethylene, HCl and air/O2)
of ethylene in the "balanced process". The latter process is the most
usual nowadays.
The world production of PVC (and therefore VC) in 1998 was about
27 million tonnes. PVC accounts for 20% of plastics material usage and
is used in most industrial sectors. About 95% of the world production
of VC is used for the production of PVC. The remainder goes into the
production of chlorinated solvents, primarily 1,1,1-trichloroethane
(10 000 tonnes/year).
Three main processes are used for the commercial production of
PVC: suspension (providing 80% of world production), emulsion (12%)
and mass or bulk (8%). Most of the case studies describing adverse
effects of VC concern plants using the suspension (also called
dispersion) process.
There have been reports of VC release through accidents in PVC
plants or during transportation. VC recovery has been introduced in
many countries to recover residual non-converted VC from
polymerization and other sources of the process such as in off-gas and
water effluents. Where special precautions are not taken, VC can be
detected in PVC resins and products.
The level of residual VC in PVC has been regulated since the late
1970s in many countries. Since then, release of VC from the thermal
degradation of PVC is either not detectable or is at very low levels.
Dioxins can be formed as contaminants in VC production. The
levels of dioxins emitted into the environment are controversial.
1.3 Environmental transport, distribution and transformation
Owing to its high vapour pressure, VC released to the atmosphere
is expected to exist almost entirely in the vapour phase. There are
indications for wet deposition.
VC has a relatively low solubility in water and has a low
adsorption capacity to particulate matter and sediment. Volatilization
of VC is the most rapid process for removal of VC introduced into
surface waters. Half-lives reported for volatilization from surface
waters range from about 1 to 40 h.
Volatilization half-lives from soil were calculated to be 0.2-0.5
days. Estimated losses of VC (after one year under a 1 m soil cover)
ranged from 0.1-45%, depending on soil type. Soil sorption
coefficients estimated from physicochemical data indicate a low
sorption potential and therefore a high mobility in soil. Another
important distribution route is leaching through the soil into
groundwater where VC may persist for years.
Laboratory experiments with aquatic organisms showed some
bioaccumulation, but no biomagnification within the foodchain.
With few exceptions, VC is not easily degraded by unadapted
microbial consortia under environmental conditions. Maximum
unacclimated biodegradation half-lives of VC were estimated to be in
the order of several months or years. However, special enrichment or
pure (e.g., Mycobacterium sp.) cultures are capable of degrading VC
under optimal culture conditions. The main degradation products were
glycolic acid or carbon dioxide after aerobic conversion and ethane,
ethene, methane or chloromethane after anaerobic transformation.
Frequently, the degradation reaction of VC proceeded faster with
aerobes than with anaerobes.
Reaction with photochemically produced OH radicals is the
dominant atmospheric transformation process, resulting in calculated
tropospheric half-lives of 1 to 4 days. Several critical compounds,
such as chloroacetaldehyde, formaldehyde and formyl chloride, are
generated during experimental photolysis reactions.
Photolytic reactions as well as chemical hydrolysis are thought
to be of minor importance in aqueous media. However, the presence of
photosensitizers may enhance the transformation of VC.
There are indications for reactions of VC with chlorine or
chloride used for water disinfection, thus leading to
chloroacetaldehyde and other undesirable compounds. Another
possibility for interaction is with salts, many of which have the
ability to form complexes with VC, perhaps resulting in increased
solubility.
Methods employed (with differing success) for removal of VC from
contaminated waters include stripping, extraction, adsorption and
oxidation. Some in situ bioremediation techniques (for groundwater
or soil) couple evaporative and other methods with microbial
treatment. VC in waste gases can be recycled, incinerated or
microbially degraded. Most of the VC produced industrially is bound in
PVC articles. Their incineration involves a risk of formation of
PCDDs/PCDFs and other unwanted chlorinated organic compounds.
1.4 Environmental levels and human exposure
There is very little exposure of the general population to VC.
Atmospheric concentrations of VC in ambient air are low, usually
less than 3 µg/m3. Exposure of the general population may be higher
in situations where large amounts of VC are accidentally released to
the environment, such as in a spill during transportation. However,
such exposure is likely to be transient. Near VC/PVC industry and
waste disposal sites, much higher concentrations (up to 8000 µg/m3
and 100 µg/m3, respectively) have been recorded.
Indoor air concentrations in houses adjacent to land fills
reached maximal concentrations of 1000 µg/m3.
The main route of occupational exposure is via inhalation and
occurs primarily in VC/PVC plants. Occupational exposures to VC
amounted to several thousands of mg/m3 in the 1940s and 1950s, and
were several hundreds of mg/m3 in the 1960s and early 1970s. After
the recognition of the carcinogenic hazards of VC, occupational
exposure standards were set at approximately 13-26 mg/m3 (5-10 ppm)
in most countries in the 1970s. Compliance with these guidelines has
considerably lowered workplace VC concentrations, but even in the
1990s higher concentrations have been reported and may still be
encountered in some countries.
VC has occasionally been detected in surface waters, sediment and
sewage sludges, with maxima of 570 µg/litre, 580 µg/kg, and
62 000 µg/litre, respectively. Soil samples near an abandoned chemical
cleaning shop contained very high VC concentrations (up to 900 mg/kg).
Maximal VC concentrations in groundwater or leachate from areas
contaminated with chlorinated hydrocarbons amounted to 60 000 µg/litre
(or more). High concentrations (up to 200 mg/litre) were detected in
well water in the vicinity of a PVC plant 10 years after leakages.
The few data available show that VC can be present in tissues of
small aquatic invertebrates and fish.
In the majority of drinking-water samples analysed, VC was not
present at detectable concentrations. The maximum VC concentration
reported in finished drinking-water was 10 µg/litre. There is a lack
of recent data on VC concentrations in drinking-water, but these
levels are expected to be below 10 µg/litre. If contaminated water is
used as the source of drinking-water, higher exposures may occur. Some
recent studies have identified VC in PVC-bottled drinking-water at
levels below 1 µg/litre. The frequency of occurrence of VC in such
water is expected to be higher than in tap water.
Packaging with certain PVC materials can result in VC
contamination of foodstuff, pharmaceutical or cosmetic products,
including liquors (up to 20 mg/kg), vegetable oils (up to 18 mg/kg),
vinegars (up to 9.8 mg/kg) and mouthwashes (up to 7.9 mg/kg). Owing to
the legislative action of many countries, a significant reduction in
VC levels and/or in the number of positive samples has been achieved
since the early 1970s.
Dietary exposure to VC from PVC packages used for food has been
calculated by several agencies and, based upon estimated average
intakes in the United Kingdom and USA, an exposure of < 0.0004 µg/kg
per day was estimated for the late 1970s and early 1980s. An early
study identified VC in tobacco smoke at the ng/cigarette range.
1.5 Kinetics and metabolism in laboratory animals and humans
VC is rapidly and well absorbed after inhalation or oral
exposure. The primary route of exposure to VC is inhalation. In animal
and human studies, under steady-state conditions, approximately 40% of
inspired VC is absorbed after exposure by inhalation. Animal studies
showed an absorption of more than 95% after oral exposure. Dermal
absorption of VC in the gaseous state is not significant.
Data from oral and inhalation studies on rats indicate rapid and
widespread distribution of VC. Rapid metabolism and excretion limits
accumulation of VC in the body. Placental transfer of VC occurs
rapidly in rats. No studies on distribution after dermal exposure have
been reported.
The main route of metabolism of VC after inhalation or oral
uptake involves oxidation by cytochrome P-450 (CYP2E1) to form
chloroethylene oxide (CEO), a highly reactive, short-lived epoxide
which rapidly rearranges to form chloroacetaldehyde (CAA). The primary
detoxification reaction of these two reactive metabolites as well as
chloroacetic acid, the dehydrogenation product of CAA, is conjugation
with glutathione catalysed by glutathione S-transferase. The
conjugation products are further modified to substituted cysteine
derivatives (S-(2-hydroxyethyl)-cysteine, N-acetyl- S-
(2-hydroxyethyl)cysteine, S-carboxymethyl cysteine and
thiodiglycolic acid) and are excreted via urine. The metabolite
carbon dioxide is exhaled in air.
CYP2E1 and glutathione S-transferase isoenzymes are known to
have large inter-species and inter-individual variation in activity.
After inhalative or oral exposure to low doses, VC is
metabolically eliminated and non-volatile metabolites are excreted
mainly in the urine. Comparative investigations of VC uptake via
inhalation revealed a lower velocity of metabolic elimination in
humans than in laboratory animals, on a body weight basis. However,
when corrected on a body surface area basis, the metabolic clearance
of VC in humans becomes comparable to that of other mammalian species.
With increasing oral or inhalative exposure, the major route of
excretion in animals is exhalation of unchanged VC, indicating
saturation of metabolic pathways. Independently of applied dose, the
excretion of metabolites via faeces is only a minor route. No studies
were located that specifically investigated excretion via the bile.
CEO is thought to be the most important metabolite in vivo,
concerning the mutagenic and carcinogenic effects of VC. CEO reacts
with DNA to produce the major adduct 7-(2'-oxoethyl)guanine (7-OEG),
and, at lower levels, the exocyclic etheno adducts,
1, N6-ethenoadenine (Epsilon A), 3, N4-ethenocytosine (Epsilon C)
and N2,3-ethenoguanine (Epsilon G). The etheno DNA adducts exhibit
pro-mutagenic properties, in contrast to the major adduct 7-OEG.
7-OEG, Epsilon A, Epsilon C and Epsilon G have been measured in
various tissues from rodents exposed to VC. Physiologically based
toxicokinetic (PBTK) models have been developed to describe the
relationship between target tissue dose and toxic end-points for VC.
1.6 Effects on laboratory mammals and in vitro test systems
VC appears to be of low acute toxicity when administered to
various species by inhalation. The 2-h LC50 for rat, mouse,
guinea-pig and rabbit were reported to be 390 000, 293 000, 595 000
and 295 000 mg/m3, respectively. No data are available on acute
toxicity after oral or dermal application. VC has a narcotic effect
after acute inhalation administration. In rats, mice and hamsters,
death was preceded by increased motor activity, ataxia and
convulsions, followed by respiratory failure. In dogs, severe cardiac
arrythmias occurred under narcosis after inhalative exposure to
260 000 mg/m3. After acute inhalation exposure to VC in rats,
pathological findings included congestion of the internal organs,
particularly lung, liver and kidney, as well as pulmonary oedema.
No studies or relevant data are available for assessing effects
of dermal exposure, skin irritation or sensitizing property of VC.
Short-term oral exposure to VC for 13 weeks in rats resulted in a
no-observed-effect level (NOEL), based on increase in liver weight, of
30 mg/kg.
In various species, the main target organ for short-term (up to
6 months) inhalation exposure to VC was the liver. Increases in
relative liver weights and hepatocellular changes were noted in rats
at 26 mg/m3 (the lowest dose level tested); at higher levels
(> 260 mg/m3) more pronounced liver changes occurred in a
dose-related manner. Other target organs were the kidney, lung and
testis. Rats, mice and rabbits seem to be more sensitive than
guinea-pigs and dogs.
Long-term exposure to VC by inhalation resulted in statistically
significant increases in mortality in some strains of rats at a dose
of as low as 260 mg/m3, in mice at 130 mg/m3 and in hamsters at
520mg/m3 for various lengths of exposure. Rats exposed to
130 mg/m3 showed reduced body weight and increased relative spleen
weight, hepatocellular degeneration and proliferation of cells lining
the liver sinusoids. Exposure to higher levels produced degenerative
alteration in the testis, tubular nephrosis and focal degeneration of
the myocardium in rats. For rats and mice exposed via inhalation, the
no-observed-adverse-effect level (NOAEL) concerning non-neoplastic
effects is below 130 mg/m3.
Chronic feeding studies showed increased mortality, increased
liver weights and morphological alteration of the liver.
After oral exposure, liver cell polymorphism (variation in size
and shape of hepatocytes and their nuclei) could be seen in rats at
levels as low as 1.3 mg/kg body weight. The NOAEL was 0.13 mg/kg body
weight.
Long-term feeding studies in rats with VC in PVC granules yielded
significantly increased tumour incidences of liver angiosarcoma (ASL)
at 5.0 mg/kg body weight per day and neoplastic liver nodules
(females) and hepatocellular carcinoma (HCC) (males) at 1.3 mg/kg body
weight per day.
In inhalation studies with VC in Sprague-Dawley rats, a clear
dose-response relationship was observed for ASL and, at high
concentrations, Zymbal gland carcinomas. No clear dose-dependency for
hepatoma or extrahepatic angiosarcoma, nephroblastomas,
neuroblastomas, or mammary malignant tumours was observed. In mice,
the spectrum of tumours induced by long-term inhalation exposure is
similar to that observed in rats but an increase in lung tumours was
only observed in mice. In hamsters, an increased tumour incidence of
ASL, mammary gland and acoustic duct tumours, melanomas, stomach and
skin epithelial tumours was reported.
The mutagenic and genotoxic effects of VC have been detected in a
number of in vitro test systems, predominantly after metabolic
activation. VC is mutagenic in the Ames test in S. typhimurium
strains TA100, TA1530 and TA1535 but not in TA98, TA1537 and TA1538,
indicating mutations as a result of base-pair substitutions
(transversion and transition) rather than frameshift mutations. This
is in agreement with the finding that etheno-DNA adducts formed by the
reactive metabolites CEO and CAA convert to actual mutations by
base-pair substitutions.
Other gene mutation assays in bacteria, yeast cells and mammalian
cells have revealed positive results exclusively in the presence of
metabolic activation. Mutagenic effects were also reported in a human
cell line containing cloned cytochrome P-450IIE1, which is capable of
metabolizing VC. Gene mutation was also detected in plant
( Tradescantia ) cuttings exposed to VC. In gene conversion assays,
positive results were reported with Saccharomyces cerevisiae in the
presence of a metabolic activation system. VC exposure induced
unscheduled DNA synthesis in rat hepatocytes and increased
sister-chromatid exchange (SCE) in human lymphocytes after addition of
exogenic activation system. No growth inhibition was detected in DNA
repair-deficient bacteria without metabolic activation. Cell
transformation assays revealed positive results both with and without
metabolic activation.
VC exposure induced gene mutation and mitotic recombination in
Drosophila melanogaster but not gene mutation in mammalian germ
cells. VC showed clastogenic effects in rodents, increased SCE in
hamsters and induced DNA breaks in mice. In host-mediated (rat)
assays, VC induced gene conversion and forward mutations in yeast.
CEO and CAA were found to be mutagenic in different test systems.
CEO is a potent mutagen, whereas CAA is highly toxic. CEO and CAA were
found to be carcinogenic in mice, CEO being much more active than CAA.
Mutations of the ras and p53 genes were analysed in liver
tumours induced by VC in Sprague-Dawley rats: base-pair substitutions
were found in the Ha- ras gene in hepatocellular carcinoma (HCC) and
in the p53 gene in ASL. These mutations are in agreement with the
observed formation and persistence of etheno adducts in liver DNA,
following exposure of rats to VC, and with the known pro-mutagenic
properties of etheno adducts.
Studies into the mechanism of carcinogenicity of VC suggest that
the reactive epoxide intermediate CEO interacts with DNA to form
etheno adducts, which result in a base-pair substitution leading to
neoplastic transformation.
1.7 Effects on humans
Concentrations of VC in the region of 2590 mg/m3 (1000 ppm),
which were not unusual prior to 1974, over periods ranging from 1
month to several years, have been reported to cause a specific
pathological syndrome found in VC workers called the "vinyl chloride
illness". Symptoms described were earache and headache, dizziness,
unclear vision, fatigue and lack of appetite, nausea, sleeplessness,
breathlessness, stomachache, pain in the liver/spleen area, pain and
tingling sensation in the arms/legs, cold sensation at the
extremities, loss of libido and weight loss. Clinical findings
included scleroderma-like changes in the fingers with subsequent bony
changes in the tips of the fingers described as acroosteolysis,
peripheral circulatory changes identical with the classical picture of
Raynaud's disease and enlargement of the liver and spleen with a
specific histological appearance, and respiratory manifestations.
Studies in humans have not been adequate to confirm effects on
the reproductive system. A few morbidity studies have reported
elevated incidence of circulatory diseases among vinyl chloride
workers. However, large cohort studies have found lower cardiovascular
disease mortality.
There is strong and consistent evidence from epidemiological
studies that VC exposure causes the rare tumour, angiosarcoma of the
liver. Brain tumours and hepatocellular carcinoma of the liver may
also be associated with VC, although the evidence cannot be considered
definitive. Other cancer sites reported to be in excess, but less
consistently, include lung, lymphatic and haematopoietic tissue, and
skin.
VC is mutagenic and clastogenic in humans. Frequencies of
chromosomal aberrations (CA), micronuclei (MN) and SCE in the
peripheral blood lymphocytes of workers exposed to high levels of VC
have been shown to be raised compared to controls. Although in many
studies the exposure concentrations and duration of exposure were only
estimated, a dose-response relationship and a "normalization" of
genotoxic effects with time after reduction of exposure can be seen.
Point mutations have been detected in p53 and ras genes in
tumours from highly exposed (before 1974) autoclave workers with liver
angiosarcoma (ASL) and another VC worker with hepatocellular
carcinoma.
Biological markers that have been investigated as indicators for
VC exposure or VC-induced effects include a) excretion of VC
metabolites (e.g., thiodiglycolic acid), b) genetic assays (e.g.,
chromosomal abnormalities or micronucleus assay), c) levels of enzymes
(e.g., in liver function tests), d) serum oncoproteins (p21 and p53)
and/or their antibodies as biomarkers of VC-induced effects.
Children living near landfill sites and other point sources may
be at increased risk based on suggested evidence of early life
sensitivity in animal studies. However, there is no direct evidence in
humans.
In epidemiological studies, a clear dose-response is only evident
for ASL alone or in combination with other liver tumours. Only one
epidemiological study has sufficient data for quantitative
dose-response estimation.
1.8 Effects on other organisms in the laboratory and field
There is a lack of standard toxicity data on the survival and
reproduction of aquatic organisms exposed to VC. Care must be taken
when interpreting the data that are available, as most of it was
generated from tests where the exposure concentration was not measured
and therefore losses due to volatilization were not taken into
account.
The lowest concentration of VC that caused an effect in
microorganisms was 40 mg/litre. This was an EC50 value based upon
inhibition of respiration in anaerobic microorganisms in a batch assay
over 3.5 days.
The lowest concentration that caused an effect in higher
organisms was 210 mg/litre (48-h LC50 for a freshwater fish); with a
corresponding no-observed-adverse-effect concentration (NOAEC) of 128
mg/litre. Effects due to VC have been reported at lower concentrations
in other species, but the ecological significance of these effects was
not verified.
VC concentrations predicted to be non-hazardous to freshwater
fish were calculated to range from 0.088 to 29 mg/litre.
There is a paucity of data concerning the effects of VC on
terrestrial organisms.
2. IDENTITY, PHYSICAL AND CHEMICAL PROPERTIES, AND ANALYTICAL METHODS
This monograph deals with vinyl chloride (VC) monomer itself and
is not an evaluation of polyvinyl chloride (PVC), the polymer of VC.
2.1 Identity
Chemical formula: C2H3Cl
Chemical structure: H2C=CHCl
Relative molecular mass: 62.5
Common names: Vinyl chloride
CAS chemical name: Ethene, chloro-
IUPAC name: Chloroethene
CAS Registry 75-01-4
number:
EC Number: 602-023-007
EINECS Number: 2008310
Synonyms: vinyl chloride monomer,
monochloroethene;
monochloroethylene;
1-chloroethylene, chloroethene,
chloroethylene
Purity 99.9% (by weight); water: max.
120 mg/kg; HCl: max. 1 mg/kg
(BUA, 1989)
Up to the 1960s the purity was not
so high (Lester et al., 1963)
Typical trace 10-100 mg/kg range: chloromethane,
components chloroethane; 1-10 mg/kg range:
ethyne
(acetylene), 1,3-butadiene, butene,
1,2-dichloroethane, ethene,
propadiene
(allene), propene, 1-butyne-3-ene
(vinyl acetylene) (BUA, 1989)
2.2 Physical and chemical properties
Some physical properties of VC are given in Table 1. Under
ambient conditions, vinyl chloride is a colourless, flammable gas with
a slightly sweet odour. It is heavier than air and has relatively low
solubility. There are discrepancies in the literature with regard to
Henry's Law constant (air-water partition coefficient, Hc). Whereas
some authors give a value between 1 and 3 kPa.m3/mol, other sources
quote a value two orders higher. Large uncertainties in the absolute
aqueous solubility in older studies probably contribute most to these
discrepancies (Ashworth et al., 1988). It is an azeotrope with water:
0.1 parts water/100 parts vinyl chloride (Bönnighausen, 1986; Rossberg
et al., 1986). VC is soluble in almost all organic solvents.
Since it is a gas that is heavier than air, VC can spread over
the ground creating an exposure long distances away from the original
source and can form explosive mixtures. The odour threshold value is
very subjective (see Table 1) and is far above the present accepted
occupational safety threshold values (see Annex 1).
VC is transported as a compressed liquid. As it does not tend to
polymerize easily, liquid VC (in the absence of oxygen and water) can
be stored and transported without polymerization inhibitors
(Bönnighausen, 1986).
At ambient temperatures in the absence of air, dry purified VC is
highly stable and non-corrosive. Above 450°C, partial decomposition
occurs yielding acetylene, hydrogen chloride and trace amounts of
2-chloro-1,3-butadiene (chloroprene) (Rossberg et al., 1986). This
reaction also occurs by lower temperatures (at 30°C and under) in the
presence of sodium or potassium hydroxide (Bönnighausen, 1986).
Combustion of VC in air produces carbon dioxide and hydrogen
chloride. Under oxygen deficient conditions, traces of phosgene may be
formed (Rossberg et al., 1986). In chlorine-atom-initiated oxidation
of VC, the vinyl chloride peroxide formed decomposes to formaldehyde,
hydrogen chloride and carbon monoxide (Bauer & Sabel, 1975; Sanhueza
et al., 1976).
With air and oxygen, very explosive peroxides can be formed
(Rossberg et al., 1986). There are reports of explosions in vinyl
chloride plants (Terwiesch, 1982). In VC recovery plants there is a
higher chance of explosion, which necessitates continuous monitoring
and limitation of the oxygen content.
Table 1. Some physical and chemical properties of vinyl chloride
Melting point -153.8 °C Bönnighausen (1986);
Dreher (1986)
Boiling point -13.4 °C Bönnighausen (1986);
(at 101.3 kPa) Dreher (1986)
Flash point -78 °C Bönnighausen (1986);
(open cup) Dreher (1986)
Autoignition 472 °C Bönnighausen (1986);
temperature Dreher (1986)
Critical temperature 156 °C Bönnighausen (1986);
Dreher (1986)
Critical pressure 5600 kPa Bönnighausen (1986);
Dreher (1986)
Explosion limits in air 3.8-29.3 vol% Bönnighausen (1986)
in air (20 °C);
4-22 vol% Dreher (1986)
Decomposition 450 °C Bönnighausen (1986)
temperature
Density (20 °C) 0.910 g/cm3 Bönnighausen (1986)
Vapour pressure at -20 °C 78 kPa Dreher (1986)
0 °C 165 kPa
20 °C 333 kPa
Solubility of VC in 0.95 wt% (9.5 g/litre) DeLassus & Schmidt
water; extrapolated over temperature (1981)
from low pressure range
experiments 1.1 g/litre Euro Chlor (1999)
over range 15-85 °C
at 20 °C
Solubility of water 0.02 ml (-20 °C) Bönnighausen (1986)
in 100 g VC 0.08 ml (+20 °C)
Henry's Law Constant 1.96 at 17.5 °C Gossett (1987)
(Hc) (kPa.m3/mol) 2.0-2.8 at 25 °C Ashworth et al. (1988)
18.8 at 20 °C Euro Chlor (1999)
Table 1. (cont'd)
Solubility in organic soluble in most Dreher (1986)
solvents organic liquids and
solvents;
insoluble in lower Bönnighausen (1986)
polyalcohols
log n-octanol/water 1.58 (measured; BUA (1989)
partition coefficient 22 °C)
(log Kow) 1.36 (calculated) BUA (1989)
1.52 Gossett et al. (1983)
Odour threshold value 26-52 mg/m3 by Hori et al. (1972)
some, but by all at
2600 mg/m3
650 mg/m3 Baretta et al. (1969)
10 700 mg/m3 Patty (1963)
Polymerization reactions to form PVC are the most important
reactions from an industrial view (see section 3.2.1.2).
nH2C = CHCl -> (- H2C - CHCl -)n; Delta HR = -71.2 kJ/mol
The reaction is exothermic. Addition reactions with other halogens at
the double bond, for instance, to yield 1,1,2-trichloroethane or
1,1-dichloroethane, are also important. Catalytic halogen exchange by
hydrogen fluoride gives vinyl fluoride (Rossberg et al., 1986). In the
presence of water, hydrochloric acid is formed which attacks most
metals and alloys. This hydrolysis probably proceeds via a peroxide
intermediate (Lederer, 1959).
Vinyl chlorine reacts with chlorine to form trichloroethane.
1,1-Dichloroethane is formed from the exothermal reaction of VC with
hydrogen chloride in the presence of iron compounds.
2.3 Conversion factors
1 ppm = 2.59 mg/m3 at 20°C and 101.3 kPa
1 mg/m3 = 0.386 ppm
2.4 Analytical methods
2.4.1 General analytical methods and detection
Stringent regulations for the production, use and handling of
carcinogenic VC have been made in several countries (see Annex 1)
necessitating the usage of reliable methods to detect trace amounts of
this compound in air, water and in PVC articles in such human contact
applications as food packing, medical equipment and potable water
transport.
VC in air has been monitored by trapping it on different
adsorbents, e.g., activated charcoal, molecular sieve and carbotrap.
VC can be removed from adsorbents by liquid or thermal desorption
and analysed by GC fitted with FID, PID or MS detection. In ambient
air measurements, several adsorbents in sieves or refrigerated traps
have been used to increase the efficiency of trapping. In continuous
monitoring of workplace and ambient concentration, IR and GC/FID
analysers can be used.
Direct injection, extraction and more increasingly head space or
purge and trap techniques have been applied for analysis of liquids
and solids. VC can be detected by GC fitted to, for instance, FID,
PID, MS or Hall detectors.
A pre-concentration step and chemical derivatization may increase
sensitivity.
An overview of analytical methods for detecting VC in various
matrices is given in Table 2.
2.4.2 Sample preparation, extraction and analysis for different
matrices
2.4.2.1 Air
Most methods are based on that of Hill et al. (1976a), using
adsorption on activated charcoal, desorption with carbon disulfide and
analysis by GC/FID. Kruschel et al. (1994) used a three-stage carbon
molecular sieve adsorbent cartridge to collect a wide range of
selected polar and non-polar VOCs. After purging with helium prior to
analysis, levels of water and other interfering compounds were reduced
sufficiently to allow cryogenic preconcentration and focusing of the
sample onto the head of the analytical column. VC was detected at
levels below the detection limit of former methods.
Landfill gas monitoring has been carried out by trapping VC on a
molecular sieve, and samples have been analysed using, for instance,
GC/MS (Bruckmann & Mülder, 1982) or GC/ECD with prior conversion to
the 1,2-dibromo derivative (Wittsiepe et al., 1996).
Table 2. Analytical methodsa
Matrix Sampling/preparation Separation Detector Detection Comments References
limitb
Air
Expired air collected in 50 ml GC FID 50 ppb Baretta et al. (1969)
pipettes; direct (packed (130 µg/m3)
injection column)
Expired air multistage cryogenic GC FID; MS low ppb low reproducibility; Conkle et al. (1975)
trapping; thermal (packed & cap.) long sampling time
desorption
Expired air 500 ml charcoal tubes GC 0.3 mg/m3 Krajewski & Dobecki
(1978, 1980)
Expired air 1 litre canister; capGC MS n.g. for collecting Pleil & Lindstrom
pressurized with alveolar samples; e.g., (1997)
neutral gas; cryogenic 16 and 25 µg/m3
concentration
Air in car charcoal tube, CS2 GC FID 10 ppb Going (1976); Hedley
interior desorption (26 µg/m3) et al. (1976)
Ambient air activated charcoal/CS2 GC FID 2.6 mg/m3 Hill et al. (1976a)
Ambient air silica gel at -78°C, GC FID 2.6 mg/m3 IARC (1978)
thermal desorption
Ambient air activated charcoal GC n.g. 0.5 ppb Dimmick (1981)
column; 24-h sampling (1.3 µg/m3)
Ambient air sampling (1 to 10 HRGC MS 1 ng VOCs Kruschel et al.
litre) on carbon trap; FID (0.3 µg/m3) (1994)
thermal desorption
Table 2. (cont'd)
Matrix Sampling/preparation Separation Detector Detection Comments References
limitb
Ambient air solid phase sample capGC IMS 2 mg/m3 new method for Simpson et al.
trap preconcentration field monitoring (1996)
Landfill gas (20 litre) carbon capGC ECD 82 ng/m3 Wittsiepe et al.
molecular sieve; CS2 (1996)
desorption;
conversion to
1,2,-dibromo
derivative
Tobacco charcoal tube, CS2 GLC ECD 15 pg per Hoffmann et al.
smoke extraction; injection (1976)
conversion to
1,2,-dibromo
derivative
Workplace CS2 desorption GC (packed FID 0.04 µg working range NIOSH (1994)
air column) (5 litre 0.4 to 40 mg/m3 (based on Hill
sample) et al., 1976a)
Workplace charcoal sorbent GC (packed FID 5 mg/m3 Kollar et al.
air tube; extraction column) (3 dm3 sample) (1988)
with
nitro-methane
Workplace carbon trap, GC FID 2.6 mg/m3 Hung et al. (1996)
air thermal desorption
Workplace activated charcoal, GC FID 0.1 mg/m3 working range HSE (1987);
air CS2 desorption 0.07-25 mg/m3 for ASTM (1993)
30-litre samples
Workplace continuous process GC FID n.g. Pau et al. (1988)
air sampling
Table 2. (cont'd)
Matrix Sampling/preparation Separation Detector Detection Comments References
limitb
Workplace continuous pyrolysis detection 1 mg/m3 Nakano et al.
air sampling of HCl (1996)
Water
Water purge & trap GC MC n.g. in PVC pipes Dressman &
McFarren (1978)
Water purge & trap GC MS 0.05 µg/litre Schlett &
Pfeifer (1993)
Water headspace capGC MS 1 µg/litre Gryder-Boutet &
Kennish (1988)
Water purge & trap capGC PID-ELCD n.g. modification of US Driscoll et al.
EPA Methods 601 (1987)
& 602 for VOC
Water purge & trap capGC PID-ELCD 0.1 µg/litre VOC Ho (1989)
Water purge & trap; capGC ECD 1.6 ng/litre Wittsiepe et al.
CS2 desorption; (1990, 1993)
1,2-dibromo
derivatization
Water GC PID and n.g. VCM loss during Soule et al.
Hall laboratory holding (1996)
detector time
Water purge & trap GC FID n.g. VOC Lopez-Avila et
al. (1987a)
Table 2. (cont'd)
Matrix Sampling/preparation Separation Detector Detection Comments References
limitb
Water CS2 desorption; capGC ECD 1.6 ng/litre Wittsiepe et al.
conversion to (1990, 1996)
1,2,-dibromo
derivative
Bottled headspace with GC MS 10 ng/litre Benfenati et al.
drinking- thermal desorption (1991)
water cold-trap
injector (TCT)
Water solid phase capGC FID n.g. Shirey (1995)
micro-extraction MS
Food, liquids, biological fluids and tissues
Liquid headspace GC FID 0.1 ppb Watson et al.
drugs; (1979)
cosmetics
Food; headspace GLC confirmation 10 ppb Williams (1976a)
liquids with MS
Liquids derivatization to GLC ECD 15 µg/litre Williams (1976b)
1-chloro-1,2- (vinegar);
dibromoethane 50 µg/litre oil
Food direct injection GC FID 2-5 µg/kg detection limit UK MAFF (1978)
depends on medium
Food headspace GC FID 1 µg/kg IARC (1978)
Table 2. (cont'd)
Matrix Sampling/preparation Separation Detector Detection Comments References
limitb
Oil GC FID 5 µg/litre Rösli et al.
(1975) based on
Williams & Miles
(1975)
Food headspace GC n.g. 2-5 µg/kg UK MAFF (1978)
Intravenous headspace capGC FID 1 µg/litre Arbin et al.
solutions (1983)
Blood (rat) headspace GC FID 5 µg/litre Zuccato et al.
ethanol-water (1979)
extraction
Tissues freezing, GC FID 30 µg/kg Zuccato et al.
(rat) homogenization (1979)
then as above
Urine dry; dissolution GC MS 50 µg/litre TDGA Müller et al.
in methanol; (1979)
methylation with
diazomethane
Urine extraction and GC FID 10 mg/litre TDGA; standard: Draminski &
silylation o-phthalic acid Trojanowska
(1981)
Urine conversion to GC MS < 0.5 TDGA; standard: Pettit (1986)
dibutyl ester µmol/litre pimelic acid
PVC
PVC products charcoal tube, GC FID 10 ppb Going (1976)
CS2 desorption (26 µg/m3)
Table 2. (cont'd)
Matrix Sampling/preparation Separation Detector Detection Comments References
limitb
PVC headspace packed column 5 ppb ASTM (1985)
GC
PVC headspace capGC FID update suggestion for Wright et al.
FID-PID ASTM (1985) (1992)
PVC extraction/headspace GC FID 0.1 mg/kg Puschmann (1975);
IARC (1978)
PVC HPLC < 1 ppm for temperatures Kontominas et al.
packaging simulating storage (1985)
of foods conditions (8 to 27 °C)
PVC dynamic headspace GC FID low ppb Poy et al. (1987)
with a sparging and
focusing step
before thermal
desorption
PVC film or GC FID 2.2 ng Gilbert et al.
resin (5 ppb (w/w)) (1975)
Packaging MS 8.7 pg
materials
PVC bags purge/trap GC FID/ECD 0.3 ppb Thomas & Ramstad
(Tenax/charcoal) (1992)
Table 2. (cont'd)
a Abbreviations: capGC = capillary gas chromatography; ECD = electron capture; ELCD = electrolytic conductivity detector;
FID = flame ionization detector; GC = gas chromatography; GLC = gas-liquid chromatography; HRGL = high-resolution gas
chromatography; IMS = ion mobility spectrometry; MC = microcoulometric titration detector; MS = mass spectrometry;
PID = photoionization detector; SPME = solid-phase microextraction; TDGA = determination of the metabolite, thiodiglycolic
acid; VOC = volatile organic chemical (a general method); n.g. = not given
b The % recovery was not given in most cases
2.4.2.2 Water
VC is first purged from the water and then collected for GC
analysis by headspace/purge and trap. VC is highly volatile and has a
low specific retention volume on Tenax-GC, the most commonly used
trapping medium in purge/trap analysis. Combination traps such as
Tenax/silica gel/charcoal (Ho, 1989) or Tenax/OV-1/silica (Lopez-Avila
et al., 1987b) have been used. Another approach is to bypass the trap
altogether by purging directly onto a cryocooled capillary column
(Gryder-Boutet & Kennish, 1988; Pankow & Rosen, 1988; Cochran, 1988;
Cochran & Henson, 1988), but here there are complications due to the
need to remove water when stripping from an aqueous solution. A more
recent adaptation of the headspace method uses solid-phase
microextraction (SPME) in which a stationary phase, usually
poly(dimethylsiloxane), coated on a fused-silica fibre is used to
extract aqueous samples in completely filled sealed vials (Shirey,
1995).
It should be noted when measuring VC content in water or
groundwater that samples should be analysed as soon as possible, as
the VC content decreases with holding time (Soule et al., 1996).
2.4.2.3 PVC resins and PVC products
For the quantification of residual VC in PVC, a solid and a
solution approach have been used. The former involves the
equilibration of a solid polymer sample at 90°C in a sealed system,
followed by headspace analysis with single or multiple extraction
(Berens et al., 1975; Kolb, 1982). The solution approach involves the
equilibration of a 10% solution of PVC in dimethylacetamide in a
sealed system, followed by analysis of the headspace gas (Puschmann,
1975). A automatic dynamic headspace method involving a sparging and a
focusing step before desorption into the GC column has been developed
to increase the sensitivity of the solution approach method (Poy et
al., 1987).
2.4.2.4 Food, liquid drug and cosmetic products
For monitoring VC in foods in contact with PVC packaging,
headspace GC is the usual method. Before the levels of VC allowed in
PVC was regulated in the 1970s (see Annex I), many of the VC levels
observed were high enough to be determined by direct injection
methods. Limits of detection are given as 2, 5 and 5 µg/kg for
aqueous, ethanolic, and oleaginous medium respectively using
headspace-GC-FID (UK MAFF, 1978). Williams (1976a,b) reported a
gas-liquid chromatographic method using subsequent GC-MS confirmation,
and a further GC/ECD method requiring derivatization to
1-chloro-1,2-dibromoethane for determination of VC content in liquid
foods.
Methods for VC levels in liquid drug and cosmetic preparations
were described by Watson et al. (1979). A weighed aliquot of the
commercial product in a tightly septum-sealed vial with accurately
known headspace volume is heated to 50°C for 30 min. A portion of the
warm headspace gas is then injected into a GC equipped with FID and a
styrene-divinylbenzene porous polymer column.
2.4.2.5 Biological samples
There are few data on VC analysis in biological tissues. The only
report available was on rat blood and tissues (Zuccato et al., 1979).
2.4.2.6 Human monitoring
Methods for measuring VC concentrations in exhaled air (breath)
have been described (see Table 2) but, although useful for studying
metabolism, they are not well suited for biological monitoring due to
the short half times in the body and the saturable metabolism of VC.
Metabolites of vinyl chloride have been identified in the urine
of rats (Müller et al., 1976; Green & Hathway, 1977) and humans
(Müller et al., 1979) using GC-MS. As there is a strong correlation
between VC exposure in humans and increased excretion of
thiodiglycolic acid (Müller et al. 1978), this metabolite has been
using for monitoring purposes. It is, however, not specific for VC as
certain drugs and other C2 compounds also have thiodiglycolic acid as
a urinary metabolite (Müller et al., 1979). Since thiodiglycolic acid
is also detected in unexposed subjects (Müller et al., 1979; van
Sittert & de Jong, 1985) and even premature babies (Pettit, 1986),
this approach can only be used to demonstrate high levels of exposure.
A discussion of biological markers for VC exposures and VC-induced
liver cancer is presented in section 8.5.
Thiodiglycolic acid has been determined by dissolving the dried
urine residue in methanol, methylating with diazomethane and analysing
with GC-MS (Müller et al., 1979), analysing the metabolite as its
dibutyl ester by GC-MS using selected ion monitoring (Pettit, 1986),
and by using GC/FID (Draminski & Trojanowska, 1981). Care must be
taken with methods which analyse for VC metabolites, as these
metabolites are not specific to VC.
A specific and sensitive new method has been reported for the
quantification of the VC metabolite N-acetyl- S-(2-hydroxyethyl)
cysteine by exchange solid-phase extraction and isotope dilution
HPLC-tandem mass spectrometry (Barr & Ashley, 1998). This method may
prove useful for monitoring occupational VC exposure, as the detection
limit of 0.68 µg/litre is low enough to detect this metabolite even in
people with no overt exposure to VC, ethylene oxide or ethylene
dibromide.
2.4.2.7 Workplace air monitoring
Before the 1960s, when it was established that VC was a
carcinogenic substance, halogen detectors and explosimeters were used,
non-specific for VC, with detection limits of 518-1295 mg/m3 (200-500
ppm). Gradually more sophisticated techniques became available for
detection of low ppm levels of VC, such as IR analysers, FID, PID
(ECETOC, 1988) and more recently mass spectrometry. In order to check
for leaks or for control measurements during cleaning and repair work,
detector tubes or direct-reading instruments with FID or PID can be
used, although they are not specific for VC and regular calibration is
necessary (Depret & Bindelle, 1998).
Continuous analyses based on, for instance, IR, GC/FID or HCl
detection have been developed (IARC, 1978; Pau et al., 1988; Nakano et
al., 1996). Analysers can be equipped for computerized data logging
and processing. The detection limit of an IR analyser depends on, for
instance, path length and is about 1.3 mg/m3 (IARC, 1978). The
analyser detecting HCl from VC pyrolyzed in a quartz tube was reported
to have a detection limit of 1 mg/m3 when the sampling time was 40
seconds (Nakano et al., 1996).
Breathing zone concentrations can be measured by sampling VC with
portable pumps or diffusion on activated charcoal (Nelms et al., 1977;
Heger et al., 1981; ASTM, 1993; NIOSH, 1994; Du et al., 1996). By
using thermosorption tubes (carbotrap 110-400), detection limits can
be decreased and the use of carbon disulfide in desorption avoided
(Hung et al., 1996).
Passive monitors for occupational personal monitoring of exposure
to VC are commercially available.
3. SOURCES OF HUMAN AND ENVIRONMENTAL EXPOSURE
3.1 Natural occurrence
VC is not known to occur naturally.
3.2 Anthropogenic sources
Anthropogenic sources of VC include the intentional manufacture
of the compound for further processing, primarily to PVC, and
unintentional formation of VC in, for instance, sanitary landfills, as
a degradation product of chlorinated hydrocarbons such as those used
as solvents, and the subsequent presence of VC in emitted gases and
groundwater. VC is also found in tobacco smoke.
3.2.1 Production levels and processes
VC was first synthesized by Regnault in 1835. It was not until
the 1930s that techniques were devised to polymerize VC into stable
forms of PVC.
VC production methods were altered in 1974 in many countries
after the confirmation that VC was a human carcinogen. Manufacturers
developed closed production methods to reduce exposure of the
workforce.
Annual total world production of VC, which is approximately equal
to PVC production, was about 17 million tonnes in 1985 and over 26
million tonnes in 1995 (see Table 3). More than half the world's
capacity (64%) in 1985 was concentrated in Western Europe and the USA.
Since that time many new VC/PVC plants have been opened or are under
construction in SE Asia, Eastern Europe, the Indian subcontinent and
developing and oil-producing countries. Thus there has been a
geographical shift of VC/PVC production.
The leading producers of PVC and therefore also of VC are the
USA, Japan, Germany, France and SE Asian countries such as Taiwan and
China (CHEM-FACTS, 1992). The capacity has only increased moderately
in the USA and Western Europe in recent years. Significant increases
in production have been reported for Japan and Taiwan. All the
countries of Eastern Europe have PVC plants and have exported PVC to
Western European countries. The PVC capacity and imports and exports
for each country are given in CHEM-FACTS (1992).
VC is produced in Western Europe by 14 companies. The plants are
in Belgium (3 plants), France (3 plants), Germany (8 plants), Italy (4
plants), the Netherlands (1 plant), Spain (2 plants), Sweden (2
plants), United Kingdom (1 plant) (Euro Chlor, 1999).
Table 3. World PVC (and therefore VC) production/capacity 1980-1998
Region Production/capacity in 1000 tonnes/year
1980a 1985a 1990a 1995b 1998c
World capacity 16 000 17 000 20 700 26 400 approx. 27 000
World production 11 750 14 200 18 300
North America total 3200 3390 4700 6070
Suspension and mass 2810 2990
Vinyl acetate copolymer 200 210
Emulsion 190 190
Western Europe total 3900 4330 4800 5750 approx. 5600
Suspension and mass 33 350 3700
Vinyl acetate copolymer 130 130
Emulsion 420 500
Eastern Europe 925 1100 1200 2700d
Former Soviet Union 370 700 760
China 150 400 790
Japan 1400 1550 2070 8200e
SE Asia 330 600 900
South America 400 540 780
Rest of the world 1075 1590 2300 3680
a Allsopp & Vianello (1992)
b Rehm & Werner (1996)
c Although exact figures are not available, an increase in world production
was seen in 1998 but no great increase in Western Europe (personal
communication, European Council of Vinyl Manufacturers, 1999)
d Including former Soviet Republics
e Total Asia
The manufacture of VC/PVC is one of the largest consumers of
chlorine.
3.2.1.1 Production of VC
VC is produced industrially by two main reactions, the first is
hydrochlorination of acetylene, which proceeds via the following
reaction:
C2H2 + HCl -> CH2=CHCl
This route was used in the past when acetylene, produced via calcium
carbide from coal, was one of the important basic feedstocks for the
chemical industry (Rossberg et al., 1986). Today all USA and most
Western European manufacturers use the "balanced process" described
below. However, many Eastern European countries such as Poland and the
countries of the former Soviet Union still use acetylene to
manufacture VC because of relatively cheap raw materials such as
calcium carbide and natural gas. Mercury has been used as a catalyst,
although a new catalyst has now been developed in Russia, based on
platinum metal salts instead of mercury, which has increased yields
with acetylene conversion from 95 to 99% (Randall, 1994).
The second major production process involves thermal cracking
[reaction iii] (at about 500°C) of 1,2-dichloroethane (EDC), produced
by direct chlorination [reaction i] of ethylene or oxychlorination
[reaction ii] of ethylene in the "balanced process".
[i] CH2=CH2 + Cl2 -> Cl CH2-CH2 Cl
[ii] CH2=CH2 + 2 HCl + 5 O2 -> Cl CH2-CH2 Cl + H2O
[iii] Cl CH2-CH2 Cl -> CH2=CHCl + HCl
After the cracking (pyrolysis), HCl and unconverted EDC are separated
from VC by two steps of distillation and recycled. The VC is stored
either under pressure at ambient temperature or refrigerated at
approximately atmospheric pressure (European Council of Vinyl
Manufacturers, 1994). More than 90% of the VC produced today is based
on this route (Rossberg et al., 1986; Allsopp & Vianello, 1992).
Other methods of industrial production include:
a) VC from crack gases, where unpurified acetylene and ethylene are
chlorinated together, acetylene being first chlorinated to
1,2-dichloroethane.
b) VC from ethane, which is readily available in some countries. The
major drawback is that ethane must first be functionalized by
substitution reactions giving rise to a variety of side chain
reactions and therefore the reaction must be kinetically
controlled to obtain maximal VC yield.
3.2.1.2 Production of PVC from VC
Many PVC plants are fully integrated beginning with ethylene and
chlorine (or sodium chloride).
VC is a gas at ambient temperatures but is handled as a
compressed volatile liquid in all polymerization operations. PVC
polymerization reactors are thick-walled jacketed steel vessels with a
pressure rating of 1725 kPa. The polymerization of VC is strongly
exothermic. The explosive limits of VC in air are 4-22 vol%, and the
plant must be designed and operated with this in mind, particularly
when handling unreacted VC in the recovery system (Allsopp & Vianello,
1992).
Three main processes are used for the commercial production of
PVC: suspension (providing 80% of world production), emulsion (12%)
and mass or bulk (8%). In Western Europe, the proportion of PVC
produced by the different processes is: 80% suspension; 13% mass; 5%
emulsion and 2% copolymers (Wrede, 1995).
In the suspension (also called dispersion) process,
polymerization takes place at 40-70°C (depending on the type of PVC
being produced) in a reactor (autoclave) of 25-150 m3 capacity fitted
with a jacket and/or condenser for heat removal, as the reaction is
strongly exothermic. Precautions have to be taken in order to avoid
explosive mixtures with air. Liquid VC under its autogenous vapour
pressure is dispersed in water by vigorous stirring to form droplets
of average diameter 30-40 µm. The polymerization takes place within
these droplets and is started by addition of initiators dissolved in
the monomer. Stabilizers are added to prevent the drops rejoining and
to prevent the already polymerized PVC particles from agglomerating.
The reaction conditions can be exactly controlled and the properties
of the product, such as relative molecular mass, can be controlled
exactly. Once polymerization has ended, the autoclave charge is
emptied into degassing tanks, and the non-polymerized VC is degassed,
compressed and stored for reuse (ECETOC, 1988; Allsopp & Vianello,
1992).
During the polymerization process, the PVC is dispersed in the
aqueous phase and it cannot be prevented that a film of PVC forms on
the inside wall of the reactor. This film interferes with the transfer
of heat between the reactor and contents, and the process has to be
interrupted periodically to allow the reactor to be cleaned. The
autoclave, after being emptied, is opened, rinsed and washed either
with solvents or more usually by means of automatic high-pressure jets
(ECETOC, 1988). The latest development in this area is the use of
proprietary build-up suppressants, which are applied before every PVC
batch. After each batch, low pressure rinse with water can remove
loose polymers and the batch cycle is ready to restart. The reactor
needs then to be opened for a thorough cleaning only after 500 or more
batches (Randall, 1994).
Before awareness of the toxicity of VC, it was the autoclave
cleaning personnel who were primarily highly exposed to the compound.
In the past autoclaves were cleaned manually; the inside had to be
scraped with a spatula, or sometimes hammer and chisel to remove the
encrusted polymer adhering to the walls of the vessel and mixing
devices. Lumps of polymer often released monomer when broken,
resulting in high concentrations of VC in the autoclave. Before about
1970, it was usual to check that the level was below 1036 mg/m3 (400
ppm), i.e. two orders of magnitude below the lower explosion limit of
VC. Occupational exposure limits are now 18 mg/m3 (7 ppm) or less
(see Annex I). Further details of workplaces with a former high
exposure to VC are given in section 5.3 and Jones (1981).
Once polymerization has ended, the polymerization batch is
transferred to the stripping unit and then to the slurry tank. The
slurry is a suspension of PVC in water that has to be permanently
stirred; it is then dewatered in a centrifuge decanter and dried. The
resulting dried powder is either stored in silos or bagged. PVC is
then further processed into ready-to-use resins (Depret & Bindelle,
1998).
3.2.1.3 PVC products
PVC is a polymer of VC with 700-1500 monomeric units. It is
relatively inexpensive and is used in a wide range of applications.
PVC is a generic name. Each producer makes a range of PVC polymers,
which vary in morphology and in molecular mass according to the
intended use. PVC resins are rarely used alone but can be mixed with
heat stabilizers, e.g., lead, zinc and tin compounds (Allsopp &
Vianello, 1992), lubricants, plasticizers (e.g., diethylhexyl
phthalate) fillers and other additives, all of which can
influence its physical and mechanical properties (Williamson &
Kavanagh, 1987; Allsopp & Vianello, 1992). Such additives may
constitute up to 60% of the total weight in some finished PVC plastics
(Froneberg et al., 1982). These plastics are formed into a multitude
of consumer products by extrusion, thermoprocessing and rotational
moulding, and into rigid or flexible film by extrusion or calendering.
PVC accounts for 20% of plastic material usage and is used in
most industrial sectors (ECETOC, 1988; European Council of Vinyl
Manufacturers, 1994).
* Packaging
* bottles (produced by blow-moulding) for containing liquid
foods, beverages, cooking oils, vinegar, etc.
* rigid film (calendered or extruded) which is converted into
tubes and shaped containers by subsequent vacuum or
pressure-forming for packaging of various foodstuffs
* flexible film (made by blowing or calendering) for wrapping
solid foods such as cheese, meat, vegetables, fresh fruit,
etc.
* coatings in metal cans
* Building - floor coverings, wall coverings, windows, roller
shutters, piping; 58% of the water supply network and 80% of the
waste water disposal systems in Europe use pipes and fittings
made from PVC.
* Electrical appliances - wires and cables insulation
* Medical care - equipment such as blood bags and gloves;
pharmaceutical and cosmetic packaging. Worldwide, more than 25%
of all plastic-based medical devices used in hospitals are made
from PVC (Hansen, 1991)
* Agriculture - piping; drainage; tubing in dairy industry
* Automobiles - car dash boards and lateral trimming
* Toys
At present, the largest use of PVC is in the building sector
(Rehm & Werner, 1996).
3.2.2 Emissions from VC/PVC plants
3.2.2.1 Sources of emission during the production of VC
Process waste, by-products and unreacted material from a balanced
process and from a PVC plant include (Randall, 1994): a) light/heavy
ends from EDC purification (from direct chlorination and
oxychlorination reactors); b) heavy ends from VC purification (from
pyrolysis reactor); c) pyrolysis coke/tars (from thermal reactions in
the pyrolysis reactor); d) spent catalyst (from direct chlorination,
oxychlorination); e) recovery of unreacted VC (from PVC reactor); f)
offspec batches (from PVC reactor); g) aqueous streams (from EDC
washing, vent scrubbing, oxychlorination water; from centrifuge, VC
stripping, slurry tanks); h) vent gases (from distillation columns,
flash drums, reactor vents, storage tanks, vessel openings; from VC
recovery systems, dryer stacks, centrifuge vent, blending, storage
facilities); i) spills and leaks (sampling, pumps, flanges, pipes,
loading/unloading, valves; bag filling, agitator seals); and j)
equipment cleaning (tanks, towers, heat exchangers, piping; PVC
reactor, product dryer, recovery system).
a) Strategies for minimizing emissions
During the sixties, some control of exposure levels was
introduced resulting in an important decrease of estimated ambient
levels of VC.
In the 1970s, efforts were focused on controlling emissions at
the most significant emission points: reactors, filters and storage
tanks. Elementary modifications of equipment, room and local
ventilation by fans, provisional operation procedures, etc., enabled
the reduction of exposure levels in working areas.
Technical developments have achieved further reductions:
* removal of residual VC from the PVC suspension by stripping
between polymerization and drying with a flow of steam or in
closed-loop systems;
* appropriate collection of residual vents to thermal oxidizers or
other abatement systems;
* reduction of all sources of fugitive emission by maintenance and
upgraded equipment;
* high pressure internal cleaning of the autoclaves to remove PVC
crusts;
* intensive removal of VC before opening or a closed process
design.
Appropriate working procedures, personnel awareness and high
standard equipment, associated with good maintenance practices, are
the recommended ways to reduce fugitive emission to very low levels
(Depret & Bindelle, 1998).
b) Disposal of by-products
The main waste streams of EDC/VC production process in Europe are
light ends (gases: VC, EDC, HCl, ethylene, dioxins; aqueous effluents:
EDC, copper, dioxins) and heavy ends (viscous tars). According to the
PVC Information Council the total amount produced is approximately
0.03 tonnes of by-products per tonne of VC produced (European Council
of Vinyl Manufacturers, 1994). The fractions are either used as
feedstocks for other processes or combusted under controlled
conditions (> 900°C) or by catalytic oxidation to produce CO2, CO,
HCl and water which can be recycled (see above). Exit gases should be
treated using HCl absorbers and gas scrubbers. Spent catalyst, metal
sludges and coke from EDC cracking should be disposed of in controlled
hazardous waste dumps (HWD) or incinerated under controlled
conditions. Sludges from effluent purification should be either
combusted under controlled conditions or deposited in HWD (European
Council of Vinyl Manufacturers, 1994).
3.2.2.2 Emission of VC and dioxins from VC/PVC plants during
production
a) VC
An estimated 550 tonnes of VC was released into air, 451 kg to
water and 1554 kg to soil from manufacturing and processing facilities
in the USA in 1992, although this was not an exhaustive list (ATSDR,
1997). From a VC capacity of 6 200 000 tonnes, an average emission of
80 g/tonne can be calculated (Depret & Bindelle, 1998).
Euro Chlor (1999) reported emissions of VC from suspension PVC
plants in Europe to be 448 tonnes/year, 22 tonnes/year being released
in waste water. An estimated emission value of 300 tonnes VC/year was
provided by German producers (BUA, 1989), i.e., about 55 g VC/tonne
PVC. Total VC emissions in England and Wales were reported to be
3800 tonnes in 1993 and 18 990 tonnes in 1994 (HMIP, 1996).
An estimated 200 000 tonnes of VC was released into the worldwide
atmosphere during 1982 (based on a worldwide PVC production of
17 million tonnes), i.e., 12 kg/tonne of PVC produced (Hartmans et
al., 1985). In 1974, a VC emission of 0.5 kg/tonne PVC was estimated
(Kopetz et al., 1986).
b) Dioxins
PCDDs/PCDFs, some of which are classified as human carcinogens
(IARC, 1997), are formed during EDC/VC production. Concentrations in
VC distillation residues (PU4043) (probably "heavy ends") for two
production factories were 3192 and 5602 ng ITEQ (international toxic
equivalent)/kg, which was estimated to be 12 to 30 g of dioxin/year at
a production level of 200 000 tonnes VC/year in heavy ends (Stringer
et al., 1995). However, the VC/PVC industry argues that, although
dioxins may be found in production wastes, these are incinerated and
are not ultimately emitted to the environment (Fairley, 1997), at
least in those countries where waste streams are regulated (not all
countries possess such high standards of waste incineration). Some
companies do not incinerate their waste products but dispose of them
in other ways, e.g., deep-well injection (Stringer et al., 1995).
Wastewater from VC production can also be contaminated with
PCDDs/PCDFs, in particular if they contain suspended solids.
Installation of filtration devices should lower solid levels and
subsequently PCDD/PCDF emissions (Stringer et al., 1995). Using such
filtration devices, PCDD/PCDF concentrations in wastewater from
EDC/VC/PVC facilities in the USA ranged from not detectable to 6.7
pg/litre TEQ (Carroll et al., 1996).
Annual global emission of dioxins into the environment from
EDC/VC manufacture has been estimated to be 0.002-0.09 kg TEQ by the
European PVC industry but 1.8 kg by Greenpeace (Miller, 1993).
Virgin suspension PVC resin from 11 major production sites in
Europe was found not to contain any process-generated PCDDs/PCDFs at
concentrations above the limits of quantification (2 ppt) (Wagenaar et
al., 1998).
3.2.3 Accidental releases of VC
3.2.3.1 PVC plant and transport accidents
An explosion occurred at a VC recovery plant in 1978 in Germany,
which was set off by vinyl chloride peroxides (Terwiesch, 1982; see
also section 2.2).
A freight train with 12 tank cars of VC was derailed in McGregor,
Manitoba, Canada in March, 1980 under near-blizzard conditions and
-20°C and two of the tanks released VC (Charlton et al., 1983). One
car lost 47 500 litres in the first hour and the other car lost
23 100 litres at an initial rate of 1400 litres/hour, decreasing to
45 litres/hour after 31 h. In the first 15 min, 5680 litres of VC
vapourized by free surface evaporation; thereafter only 2.5% of the
discharging liquid evaporated rapidly. The remaining liquid in the
snow bank was assumed to have evaporated at a rate of 15% per hour,
leaving about 900 litres of VC in the snow bank after 36 h. Although
there was an explosion hazard, no fire occurred.
Two incidences, in 1988 and 1996, occurred in Germany involving
accidental release of VC due to derailment of trains transporting the
liquid substance (Neuhoff, 1988; Anon, 1996). Both accidents were
followed by explosion and fire. Derailment of a goods train occurred
1 km from Schönebeck (near Magdeburg in Germany) and the subsequent
explosion and fire produced a 600- to 800-m black column of smoke. In
all, 1044 tonnes of VC were involved of which 261 tonnes burnt and
350 tonnes could be reclaimed after the fire; 153 tonnes of HCl were
released. Median measurements of the numerous air samples taken at the
place of accident or surroundings did not exceed the German technical
guidance level of 5 vol ppm, although these were not taken until 14 h
after the fire. Maximum concentrations of VC measured were 78 mg/m3
(30 ppm) near the train and 26 mg/m3 (10 ppm) at a distance of 200 m
from the centre of the fire (Hahn et al., 1998). Levels in nearby
industrial sewage pipes were up to 1250 ppm (Anon, 1996). A cytogenic
analysis was carried out on some of the general population exposed to
VC from this accident (Hüttner & Nikolova, 1998; see section 8.1 and
Table 42).
3.2.3.2 Leakage and discharge from VC/PVC plants
Leakage from a waste liquor basin of a VC/PVC plant in Finland
caused high concentrations of VC and dichloroethene in groundwater in
1974. Concentrations of up to 484 mg/litre of the chlorocarbons were
measured in groundwater in the mid-1980s (Nystén, 1988).
It should be noted that in the 1960s and before, when the
toxicity of VC was not known, large amounts of PVC production sludges
containing VC were dumped onto landfills (and possibly still are in
countries where there are no adequate restrictions).
3.2.4 VC residues in virgin PVC resin and products
3.2.4.1 VC residues in different PVC samples
VC is not soluble in PVC nor is it absorbed or adsorbed in the
resin particle. It is entrapped and can escape to the ambient air
(Wheeler, 1981). PVC in a bulk-container loses its residual monomer at
a rate of 25 to 50% per month. Heating tends to accelerate this step,
but when the residual monomer has disappeared PVC is not a significant
source of VC.
Since the 1970s when VC was confirmed as a human carcinogen, it
has been mandatory in many countries to "degas" PVC after
polymerization (see section 3.3.1) and before further processing.
There are limit values for VC content in PVC (see Annex I). For
example, in 1974 raw PVC usually contained more than 1000 ppm of
residual VC. This was subsequently reduced to 10 ppm by regulation
(German Environmental Office, 1978). In a survey of 45 samples of raw
PVC from various countries carried out in 1976-1977, over a third had
VC residues of > 1 ppm, but a third had residues of over 50 ppm, with
4 samples over 200 ppm. The samples with the highest residue level
came from Hungary, Rumania, Italy and the USA (German Environmental
Office, 1978).
3.2.4.2 VC residues in PVC products
In a survey of PVC products carried out in 1976-1977, the
following indoor articles had a VC content of > 0.05 ppm: bathroom
tiles, piping, plastic bottle for table oil, and kitchen film. The
highest concentrations were found in music records, those bought
recently having a VC content of up to 210 ppm and one record 10 years
old having a content of 970 ppm. In record shops and other rooms
containing many records, this could have been an important source of
VC. In contrast, the VC content of toys, kitchen utensils, food
wrappings, wallpaper and car interiors was < 0.05 ppm (German
Environmental Office, 1978).
Whereas in 1974 the typical level of residual VC in PVC bottles
was 50 mg/kg, introduction of improved manufacturing practices at the
polymer resin processing stage reduced this to 3 mg/kg by 1975. In a
1978 survey, 22 out of 24 PVC bottles contained VC at less than
0.4 mg/kg (UK MAFF, 1978).
In a more recent survey, VC residues in various PVC samples were
given as follows: rigid water bottle (850 ppb); thin plasticized food
film (3 ppb); monopolymer powder (10 ppb); copolymer film (15 ppb)
(Poy et al., 1987). PVC film is still used widely for food packaging.
For example, in Denmark, in 1990, 129/239 samples of cling-film used
for cheese wrapping were of PVC (Svensson, 1994).
Residual VC could not be detected (< 0.1 ppm) in two PVC
products from Thailand (Smerasta et al., 1991) or in PVC and products
from it in Poland (Stareczek, 1988). PVC medical devices are regulated
in the USA and have to meet certain requirements (a maximum of 5 ppb
residual monomer for flexible compounds and a 10 ppb ceiling for rigid
compounds (Rakus et al., 1991)).
Levels of VC found in food and pharmaceutical articles are given
in section 5.1.4. Annex I gives current regulations for VC content in
various PVC products.
3.2.4.3 VC formation as a result of heating PVC
a) Thermal degradation of PVC
PVC is thermally stable below 225°C. Between 225°C and its
ignition temperature of 475°C, thermal degradation results in the
release of about 50 compounds (Boettner et al. 1969). PVC does not
degrade back to VC. Thermal degradation of two types of bulk PVC
samples at 148-232°C resulted in the release of long-chain aliphatic
alcohols, toluene, benzene, various chlorinated species, and a major
peak of HCl. The main components released at 260°C-315°C were aromatic
hydrocarbons such as benzene, phenol and adipates, along with various
aliphatic alcohols, alkenes, anhydrides, some of them chlorinated, and
carbon monoxide (Froneberg et al., 1982). When 1 kg of PVC is heated
to 300°C, it releases about 13 g HCl and 5 g CO.
b) Release of VC from heating PVC
Various PVC resins from different producers were tested for VC
evolution over the 130 to 500°C range using a heating rate of
10°C/min. A consistently low level of VC, amounting to 15-30 ppm
(based on resin), was found in the volatile decomposition products
from all of the samples tested, regardless of resin type or
manufacturing source (USA) (Wakeman & Johnson, 1978). A 100-mg sample
was programmed for heating from 200 to 450°C at 3°C/min; this resulted
in the formation of a total of 23.2 ppm VC, the major portion being
generated in the 275-350°C region. Dehydrochlorination occurred most
rapidly between 250 and 275°C. During this period only 2.3 ppm of VC
was formed. The VC evolved by heating PVC is the VC monomer entrapped
in the PVC resin.
At temperatures required for thermoforming PVC for food packaging
applications (90-120°C for a few seconds), no detectable VC was formed
in up to 1 h of exposure at 130°C (detection limit of analysis in air
- 1 ppb). Temperatures for calendering and extrusion operations are
175-210°C. Maximum VC levels determined at 210°C were 0.5 ppm (resin
basis) after 5 min and 1.2 ppm after 30 min (Wakeman & Johnson, 1978).
In more recent studies into VC formation during the thermal
welding of plasticized PVC sheeting (about 225°C), in normal field
situations such as piping in sewers, VC concentrations were usually
not above the detection limit of 0.05 ppm. Only where there was poor
ventilation were higher levels detected (0.2 ppm VC; 1.0-3.5 ppm HCl)
(Williamson & Kavanagh, 1987).
3.2.5 Other sources of VC
3.2.5.1 VC as a degradation product of chlorinated hydrocarbons
VC as a gas, in leachate and groundwater (see Table 4), has been
found in landfills and surroundings where there were no VC/PVC
production facilities in the vicinity. It was found that VC can be
formed, under anaerobic conditions, from the reductive halogenation of
the more highly chlorinated chloroethenes: tetrachloroethylene (PCE),
trichloroethene (TCE), and the dichloroethene isomers ( cis-1,2-DCE,
trans-1,2-DCE, and 1,1-DCE) (Parsons et al., 1984; Vogel & McCarty,
1985; McCarty, 1996, 1997, see Fig. 1). PCE and TCE are widely used as
industrial solvents in particular for degreasing and cleaning metal
parts and electronic components, and in dry cleaning. Production
levels for 1984 were 260 and 200 thousand tonnes for PCE and TCE,
respectively (Wolf et al., 1987). Careless handling, storage and
disposal, as well as the high chemical stability of these compounds,
have made them, and consequently VC, some of the most frequently
encountered groundwater contaminants (Arneth et al., 1988). Although
VC may be further degraded to less chlorinated and non-chlorinated
ethenes, and possibly finally to carbon dioxide and ethane, this
proceeds only at a slow rate under highly reducing conditions
(Freedman & Gossett, 1989; DiStefano et al., 1991; De Bruin et al.,
1992; see also section 4.2). As a consequence, VC can be detected in
landfill sites in and surrounding areas through spreading. Reports
from several countries show high levels of VC contamination of soil
and groundwater, aquifers and wells (see Table 4 and section 5.1).
There have recently been several field studies in
PCE/TCE-contaminated landfill sites and aquifers (Major et al., 1991,
1995; Fiorenza et al., 1994; Lee et al., 1995, see Table 5). These
have shown that under anaerobic conditions, PCE and TCE can be
intrinsically biodegraded to ethene by indigenous methanogenic,
acetogenic and sulfate-reducing bacteria. Furthermore, under aerobic
conditions there is a potential for direct or co-metabolic oxidation
of DCE and VC. Therefore, an efficient bioremediation of chlorinated
ethene-contaminated aquifers may occur in contaminant plumes
characterized by upgradient anaerobic and downgradient aerobic zones,
such as where anaerobic, chlorinated ethene plumes discharge to
aerobic surface water bodies. However, this depends on the ability of
the stream-bed microbial community to degrade efficiently and
completely DCE and VC over a range of contaminant concentrations (Cox
et al., 1995; Bradley & Chapelle, 1998a). It should be noted that this
bioremediation occurs under specific conditions. The biodegradation
studies listed in chapter 4 give conflicting results.
The three cooperating organizations of the IPCS recognize the
important role played by nongovernmental organizations.
Representatives from relevant national and international associations
may be invited to join the Task Group as observers. Although observers
may provide a valuable contribution to the process, they can only
speak at the invitation of the Chairperson. Observers do not
participate in the final evaluation of the chemical; this is the sole
responsibility of the Task Group members. When the Task Group
considers it to be appropriate, it may meet in camera.
All individuals who as authors, consultants or advisers
participate in the preparation of the EHC monograph must, in addition
to serving in their personal capacity as scientists, inform the RO if
at any time a conflict of interest, whether actual or potential, could
be perceived in their work. They are required to sign a conflict of
interest statement. Such a procedure ensures the transparency and
probity of the process.
When the Task Group has completed its review and the RO is
satisfied as to the scientific correctness and completeness of the
document, it then goes for language editing, reference checking and
preparation of camera-ready copy. After approval by the Director,
IPCS, the monograph is submitted to the WHO Office of Publications for
printing. At this time a copy of the final draft is sent to the
Chairperson and Rapporteur of the Task Group to check for any errors.
It is accepted that the following criteria should initiate the
updating of an EHC monograph: new data are available that would
substantially change the evaluation; there is public concern for
health or environmental effects of the agent because of greater
exposure; an appreciable time period has elapsed since the last
evaluation.
All Participating Institutions are informed, through the EHC
progress report, of the authors and institutions proposed for the
drafting of the documents. A comprehensive file of all comments
received on drafts of each EHC monograph is maintained and is
available on request. The Chairpersons of Task Groups are briefed
before each meeting on their role and responsibility in ensuring that
these rules are followed.
WHO TASK GROUP ON ENVIRONMENTAL HEALTH CRITERIA FOR VINYL CHLORIDE
Members
Dr A. Barbin, International Agency for Research on Cancer, Lyon,
France
Professor V.J. Feron, TNO Nutrition and Food Research Institute, HE
Zeist, Netherlands
Ms P. Heikkilä, Uusimaa Regional Institute of Occupational Health,
Helsinki, Finland
Dr J. Kielhorn, Chemical Risk Assessment, Fraunhofer Institute for
Toxicology and Aerosol Research, Hanover, Germany (Co-Rapporteur)
Professor M. Kogevinas, Respiratory and Environmental Health Research
Unit, Municipal Institute of Medical Investigation (IMIM), Barcelona,
Spain
Mr H. Malcolm, Institute of Terrestrial Ecology, Monks Wood, Abbots
Ripton, Huntingdon, Cambridgeshire, United Kingdom (Co-Rapporteur)
Dr W. Pepelko, National Center for Environmental Assessment, Office of
Research and Development, US EPA, Washington DC, USA
Dr A. Pintér, National Institute of Environmental Health, Budapest,
Hungary (Vice-Chairman)
Dr L. Simonato, Department of Oncology, University of Padua, Venetian
Tumours Registry, Padua, Italy
Professor H. Vainio, Division of Health Risk Assessment, National
Institute of Environmental Medicine, Karolinska Institute, Stockholm,
Sweden (Chairman)
Dr E.M. Ward, Division of Surveillance Hazard, Evaluation and Field
Studies, National Institute for Occupational Safety and Health
(NIOSH), Robert Taft Laboratory, Cincinnati, Ohio, USA (Contact
address: Environmental Cancer Epidemiology, International Agency for
Research on Cancer, Lyon, France
Dr J.M. Zielinski, Biostatistics and Research Coordination Division,
Ottawa, Ontario, Canada
Secretariat
Dr A. Aitio, International Programme on Chemical Safety, World Health
Organization, Geneva, Switzerland (Secretary)
Dr I. Mangelsdorf, Chemical Risk Assessment, Fraunhofer Institute for
Toxicology and Aerosol Research, Hanover, Germany
Dr C. Melber, Chemical Risk Assessment, Fraunhofer Institute for
Toxicology and Aerosol Research, Hanover, Germany
Dr U. Wahnschaffe, Chemical Risk Assessment, Fraunhofer Institute for
Toxicology and Aerosol Research, Hanover, Germany
WHO TASK GROUP ON ENVIRONMENTAL HEALTH CRITERIA FOR VINYL CHLORIDE
A WHO Task Group on Environmental Health Criteria for Vinyl
Chloride met at the Fraunhofer Institute for Toxicology and Aerosol
Research, Hanover, Germany, from 25 to 29 January 1999. Professor H.
Muhle welcomed the participants on behalf of the Institute and its
Director, Professor U. Heinrich. Dr A. Aitio, IPCS, welcomed the
participants on behalf of the Director, IPCS, and the three IPCS
co-operating organisations (UNEP, ILO, and WHO). The Group reviewed
and revised the draft and made an evaluation of the risks for human
health and the environment from exposure to vinyl chloride.
The first and second drafts of this monograph were prepared,
under the co-ordination of Dr I. Mangelsdorf, by the authors
Dr J. Kielhorn, Dr C. Melber and Dr U. Wahnschaffe. In the preparation
of the second draft, the comments received from the IPCS contact
points were carefully considered.
Dr A. Aitio of the IPCS Central Unit was responsible for the
scientific aspects of the monograph, and Dr P.G. Jenkins for the
technical editing.
The efforts of all who helped in the preparation and finalisation
of the monograph are gratefully acknowledged.
* * *
The Federal Ministry for the Environment, Nature Conservation and
Nuclear Safety, Germany, contributed financially to the preparation of
this Environmental Health Criteria monograph, and the meeting was
organised by the Fraunhofer Institute for Toxicology and Aerosol
Research.
ABBREVIATIONS
Epsilon A 1, N 6-ethenoadenine
Epsilon C 3, N 4-ethenocytosine
Epsilon dA 1, N 6-etheno-2'-deoxyadenosine
Epsilon dC 3, N 4-etheno-2'-deoxycytidine
Epsilon G ethenoguanine
7-OEG 7-(2'-oxoethyl)guanine
ALAT alanine aminotransferase
ASAT aspartate aminotransferase
ASL angiosarcoma of the liver
BCF bioconcentration factor
CA chromosomal aberration
CAA chloroacetaldehyde
CEO chloroethylene oxide
CI confidence interval (95% unless otherwise stated)
CNS central nervous system
CYP2E1 cytochrome P-450 isozyme 2e1
ECD electron capture detection
EDC 1,2-dichloroethane
FDA Food and Drug Administration (USA)
FID flame ionization detector
GC gas chromatography
GST glutathione S-transferase
HCC hepatocellular carcinoma
HLA human-leukocyte-associated antigen
HPLC high performance liquid chromatography
HWD hazardous waste dump
IR infrared
LOAEL lowest-observed-adverse-effect level
MN micronuclei
MOR morbidity odds ratio
MS mass spectrometry
MSW municipal solid waste
NER non-extractable residue
NOAEC no-observed-adverse-effect concentration
NOAEL no-observed-adverse-effect level
NOEL no-observed-effect level
PCDD polychlorinated dibenzodioxin
PCDF polychlorinated dibenzofuran
PCE tetrachloroethene (perchloroethene)
PID photoionization detector
PVC polyvinyl chloride
SCE sister chromatid exchange
SIR standardized incidence ratio
SLRL sex-linked recessive lethal
SMR standardized mortality ratio
TCE trichloroethene
TEQ toxic equivalent quantity
UV ultraviolet
VC vinyl chloride
VOC volatile organic compound
1. SUMMARY
This monograph deals with vinyl chloride (VC) monomer itself and
is not an evaluation of polyvinyl chloride (PVC), the polymer of VC.
Exposures to VC in mixtures are not addressed.
1.1 Identity, physical and chemical properties, and analytical
methods
Under ambient conditions, VC is a colourless, flammable gas with
a slightly sweet odour. It has a high vapour pressure, a high value
for Henry's Law constant and a relatively low water solubility. It is
heavier than air and is soluble in almost all organic solvents. It is
transported in liquid form under pressure.
At ambient temperatures in the absence of air, dry purified VC is
highly stable and non-corrosive but above 450°C, or in the presence of
sodium or potassium hydroxide, partial decomposition can occur.
Combustion of VC in air produces carbon dioxide and hydrogen chloride.
With air and oxygen, very explosive peroxides can be formed,
necessitating a continuous monitoring and limitation of the oxygen
content, particularly in VC recovery plants. In the presence of water,
hydrochloric acid is formed.
Polymerization reactions to PVC are technically the most
important reactions from an industrial view, but addition reactions
with other halogens at the double bond, e.g., to yield
1,1,2-trichloroethane or 1,1-dichloroethane, are also important.
The concentration of VC in air can be monitored by trapping it on
adsorbents and, after liquid or thermal desorption, analysis by gas
chromatography. In ambient air measurements, several adsorbents in
series or refrigerated traps may be needed to increase the efficiency
of trapping. Peak concentrations at workplaces can be measured with
direct-reading instruments based on, for instance, FID or PID. In
continuous monitoring, IR and GC/FID analysers combined with data
logging and processing have been used. In analysis of VC in liquids
and solids, direct injection, extraction and more increasingly head
space or purge-and-trap techniques are applied. Also in these samples,
VC is analysed by GC fitted to, for instance, FID or MS detectors.
1.2 Sources of human and environmental exposure
VC is not known to occur naturally although it has been found in
landfill gas and groundwater as a degradation product of chlorinated
hydrocarbons deposited as solvent wastes in landfills or in the
environment of workplaces using such solvents. VC is also present in
cigarette smoke.
VC is produced industrially by two main reactions: a) the
hydrochlorination of acetylene; and b) thermal cracking (at about
500°C) of 1,2-dichloroethane (EDC) produced by direct chlorination
(ethylene and chlorine) or oxychlorination (ethylene, HCl and air/O2)
of ethylene in the "balanced process". The latter process is the most
usual nowadays.
The world production of PVC (and therefore VC) in 1998 was about
27 million tonnes. PVC accounts for 20% of plastics material usage and
is used in most industrial sectors. About 95% of the world production
of VC is used for the production of PVC. The remainder goes into the
production of chlorinated solvents, primarily 1,1,1-trichloroethane
(10 000 tonnes/year).
Three main processes are used for the commercial production of
PVC: suspension (providing 80% of world production), emulsion (12%)
and mass or bulk (8%). Most of the case studies describing adverse
effects of VC concern plants using the suspension (also called
dispersion) process.
There have been reports of VC release through accidents in PVC
plants or during transportation. VC recovery has been introduced in
many countries to recover residual non-converted VC from
polymerization and other sources of the process such as in off-gas and
water effluents. Where special precautions are not taken, VC can be
detected in PVC resins and products.
The level of residual VC in PVC has been regulated since the late
1970s in many countries. Since then, release of VC from the thermal
degradation of PVC is either not detectable or is at very low levels.
Dioxins can be formed as contaminants in VC production. The
levels of dioxins emitted into the environment are controversial.
1.3 Environmental transport, distribution and transformation
Owing to its high vapour pressure, VC released to the atmosphere
is expected to exist almost entirely in the vapour phase. There are
indications for wet deposition.
VC has a relatively low solubility in water and has a low
adsorption capacity to particulate matter and sediment. Volatilization
of VC is the most rapid process for removal of VC introduced into
surface waters. Half-lives reported for volatilization from surface
waters range from about 1 to 40 h.
Volatilization half-lives from soil were calculated to be 0.2-0.5
days. Estimated losses of VC (after one year under a 1 m soil cover)
ranged from 0.1-45%, depending on soil type. Soil sorption
coefficients estimated from physicochemical data indicate a low
sorption potential and therefore a high mobility in soil. Another
important distribution route is leaching through the soil into
groundwater where VC may persist for years.
Laboratory experiments with aquatic organisms showed some
bioaccumulation, but no biomagnification within the foodchain.
With few exceptions, VC is not easily degraded by unadapted
microbial consortia under environmental conditions. Maximum
unacclimated biodegradation half-lives of VC were estimated to be in
the order of several months or years. However, special enrichment or
pure (e.g., Mycobacterium sp.) cultures are capable of degrading VC
under optimal culture conditions. The main degradation products were
glycolic acid or carbon dioxide after aerobic conversion and ethane,
ethene, methane or chloromethane after anaerobic transformation.
Frequently, the degradation reaction of VC proceeded faster with
aerobes than with anaerobes.
Reaction with photochemically produced OH radicals is the
dominant atmospheric transformation process, resulting in calculated
tropospheric half-lives of 1 to 4 days. Several critical compounds,
such as chloroacetaldehyde, formaldehyde and formyl chloride, are
generated during experimental photolysis reactions.
Photolytic reactions as well as chemical hydrolysis are thought
to be of minor importance in aqueous media. However, the presence of
photosensitizers may enhance the transformation of VC.
There are indications for reactions of VC with chlorine or
chloride used for water disinfection, thus leading to
chloroacetaldehyde and other undesirable compounds. Another
possibility for interaction is with salts, many of which have the
ability to form complexes with VC, perhaps resulting in increased
solubility.
Methods employed (with differing success) for removal of VC from
contaminated waters include stripping, extraction, adsorption and
oxidation. Some in situ bioremediation techniques (for groundwater
or soil) couple evaporative and other methods with microbial
treatment. VC in waste gases can be recycled, incinerated or
microbially degraded. Most of the VC produced industrially is bound in
PVC articles. Their incineration involves a risk of formation of
PCDDs/PCDFs and other unwanted chlorinated organic compounds.
1.4 Environmental levels and human exposure
There is very little exposure of the general population to VC.
Atmospheric concentrations of VC in ambient air are low, usually
less than 3 µg/m3. Exposure of the general population may be higher
in situations where large amounts of VC are accidentally released to
the environment, such as in a spill during transportation. However,
such exposure is likely to be transient. Near VC/PVC industry and
waste disposal sites, much higher concentrations (up to 8000 µg/m3
and 100 µg/m3, respectively) have been recorded.
Indoor air concentrations in houses adjacent to land fills
reached maximal concentrations of 1000 µg/m3.
The main route of occupational exposure is via inhalation and
occurs primarily in VC/PVC plants. Occupational exposures to VC
amounted to several thousands of mg/m3 in the 1940s and 1950s, and
were several hundreds of mg/m3 in the 1960s and early 1970s. After
the recognition of the carcinogenic hazards of VC, occupational
exposure standards were set at approximately 13-26 mg/m3 (5-10 ppm)
in most countries in the 1970s. Compliance with these guidelines has
considerably lowered workplace VC concentrations, but even in the
1990s higher concentrations have been reported and may still be
encountered in some countries.
VC has occasionally been detected in surface waters, sediment and
sewage sludges, with maxima of 570 µg/litre, 580 µg/kg, and
62 000 µg/litre, respectively. Soil samples near an abandoned chemical
cleaning shop contained very high VC concentrations (up to 900 mg/kg).
Maximal VC concentrations in groundwater or leachate from areas
contaminated with chlorinated hydrocarbons amounted to 60 000 µg/litre
(or more). High concentrations (up to 200 mg/litre) were detected in
well water in the vicinity of a PVC plant 10 years after leakages.
The few data available show that VC can be present in tissues of
small aquatic invertebrates and fish.
In the majority of drinking-water samples analysed, VC was not
present at detectable concentrations. The maximum VC concentration
reported in finished drinking-water was 10 µg/litre. There is a lack
of recent data on VC concentrations in drinking-water, but these
levels are expected to be below 10 µg/litre. If contaminated water is
used as the source of drinking-water, higher exposures may occur. Some
recent studies have identified VC in PVC-bottled drinking-water at
levels below 1 µg/litre. The frequency of occurrence of VC in such
water is expected to be higher than in tap water.
Packaging with certain PVC materials can result in VC
contamination of foodstuff, pharmaceutical or cosmetic products,
including liquors (up to 20 mg/kg), vegetable oils (up to 18 mg/kg),
vinegars (up to 9.8 mg/kg) and mouthwashes (up to 7.9 mg/kg). Owing to
the legislative action of many countries, a significant reduction in
VC levels and/or in the number of positive samples has been achieved
since the early 1970s.
Dietary exposure to VC from PVC packages used for food has been
calculated by several agencies and, based upon estimated average
intakes in the United Kingdom and USA, an exposure of < 0.0004 µg/kg
per day was estimated for the late 1970s and early 1980s. An early
study identified VC in tobacco smoke at the ng/cigarette range.
1.5 Kinetics and metabolism in laboratory animals and humans
VC is rapidly and well absorbed after inhalation or oral
exposure. The primary route of exposure to VC is inhalation. In animal
and human studies, under steady-state conditions, approximately 40% of
inspired VC is absorbed after exposure by inhalation. Animal studies
showed an absorption of more than 95% after oral exposure. Dermal
absorption of VC in the gaseous state is not significant.
Data from oral and inhalation studies on rats indicate rapid and
widespread distribution of VC. Rapid metabolism and excretion limits
accumulation of VC in the body. Placental transfer of VC occurs
rapidly in rats. No studies on distribution after dermal exposure have
been reported.
The main route of metabolism of VC after inhalation or oral
uptake involves oxidation by cytochrome P-450 (CYP2E1) to form
chloroethylene oxide (CEO), a highly reactive, short-lived epoxide
which rapidly rearranges to form chloroacetaldehyde (CAA). The primary
detoxification reaction of these two reactive metabolites as well as
chloroacetic acid, the dehydrogenation product of CAA, is conjugation
with glutathione catalysed by glutathione S-transferase. The
conjugation products are further modified to substituted cysteine
derivatives (S-(2-hydroxyethyl)-cysteine, N-acetyl- S-
(2-hydroxyethyl)cysteine, S-carboxymethyl cysteine and
thiodiglycolic acid) and are excreted via urine. The metabolite
carbon dioxide is exhaled in air.
CYP2E1 and glutathione S-transferase isoenzymes are known to
have large inter-species and inter-individual variation in activity.
After inhalative or oral exposure to low doses, VC is
metabolically eliminated and non-volatile metabolites are excreted
mainly in the urine. Comparative investigations of VC uptake via
inhalation revealed a lower velocity of metabolic elimination in
humans than in laboratory animals, on a body weight basis. However,
when corrected on a body surface area basis, the metabolic clearance
of VC in humans becomes comparable to that of other mammalian species.
With increasing oral or inhalative exposure, the major route of
excretion in animals is exhalation of unchanged VC, indicating
saturation of metabolic pathways. Independently of applied dose, the
excretion of metabolites via faeces is only a minor route. No studies
were located that specifically investigated excretion via the bile.
CEO is thought to be the most important metabolite in vivo,
concerning the mutagenic and carcinogenic effects of VC. CEO reacts
with DNA to produce the major adduct 7-(2'-oxoethyl)guanine (7-OEG),
and, at lower levels, the exocyclic etheno adducts,
1, N6-ethenoadenine (Epsilon A), 3, N4-ethenocytosine (Epsilon C)
and N2,3-ethenoguanine (Epsilon G). The etheno DNA adducts exhibit
pro-mutagenic properties, in contrast to the major adduct 7-OEG.
7-OEG, Epsilon A, Epsilon C and Epsilon G have been measured in
various tissues from rodents exposed to VC. Physiologically based
toxicokinetic (PBTK) models have been developed to describe the
relationship between target tissue dose and toxic end-points for VC.
1.6 Effects on laboratory mammals and in vitro test systems
VC appears to be of low acute toxicity when administered to
various species by inhalation. The 2-h LC50 for rat, mouse,
guinea-pig and rabbit were reported to be 390 000, 293 000, 595 000
and 295 000 mg/m3, respectively. No data are available on acute
toxicity after oral or dermal application. VC has a narcotic effect
after acute inhalation administration. In rats, mice and hamsters,
death was preceded by increased motor activity, ataxia and
convulsions, followed by respiratory failure. In dogs, severe cardiac
arrythmias occurred under narcosis after inhalative exposure to
260 000 mg/m3. After acute inhalation exposure to VC in rats,
pathological findings included congestion of the internal organs,
particularly lung, liver and kidney, as well as pulmonary oedema.
No studies or relevant data are available for assessing effects
of dermal exposure, skin irritation or sensitizing property of VC.
Short-term oral exposure to VC for 13 weeks in rats resulted in a
no-observed-effect level (NOEL), based on increase in liver weight, of
30 mg/kg.
In various species, the main target organ for short-term (up to
6 months) inhalation exposure to VC was the liver. Increases in
relative liver weights and hepatocellular changes were noted in rats
at 26 mg/m3 (the lowest dose level tested); at higher levels
(> 260 mg/m3) more pronounced liver changes occurred in a
dose-related manner. Other target organs were the kidney, lung and
testis. Rats, mice and rabbits seem to be more sensitive than
guinea-pigs and dogs.
Long-term exposure to VC by inhalation resulted in statistically
significant increases in mortality in some strains of rats at a dose
of as low as 260 mg/m3, in mice at 130 mg/m3 and in hamsters at
520mg/m3 for various lengths of exposure. Rats exposed to
130 mg/m3 showed reduced body weight and increased relative spleen
weight, hepatocellular degeneration and proliferation of cells lining
the liver sinusoids. Exposure to higher levels produced degenerative
alteration in the testis, tubular nephrosis and focal degeneration of
the myocardium in rats. For rats and mice exposed via inhalation, the
no-observed-adverse-effect level (NOAEL) concerning non-neoplastic
effects is below 130 mg/m3.
Chronic feeding studies showed increased mortality, increased
liver weights and morphological alteration of the liver.
After oral exposure, liver cell polymorphism (variation in size
and shape of hepatocytes and their nuclei) could be seen in rats at
levels as low as 1.3 mg/kg body weight. The NOAEL was 0.13 mg/kg body
weight.
Long-term feeding studies in rats with VC in PVC granules yielded
significantly increased tumour incidences of liver angiosarcoma (ASL)
at 5.0 mg/kg body weight per day and neoplastic liver nodules
(females) and hepatocellular carcinoma (HCC) (males) at 1.3 mg/kg body
weight per day.
In inhalation studies with VC in Sprague-Dawley rats, a clear
dose-response relationship was observed for ASL and, at high
concentrations, Zymbal gland carcinomas. No clear dose-dependency for
hepatoma or extrahepatic angiosarcoma, nephroblastomas,
neuroblastomas, or mammary malignant tumours was observed. In mice,
the spectrum of tumours induced by long-term inhalation exposure is
similar to that observed in rats but an increase in lung tumours was
only observed in mice. In hamsters, an increased tumour incidence of
ASL, mammary gland and acoustic duct tumours, melanomas, stomach and
skin epithelial tumours was reported.
The mutagenic and genotoxic effects of VC have been detected in a
number of in vitro test systems, predominantly after metabolic
activation. VC is mutagenic in the Ames test in S. typhimurium
strains TA100, TA1530 and TA1535 but not in TA98, TA1537 and TA1538,
indicating mutations as a result of base-pair substitutions
(transversion and transition) rather than frameshift mutations. This
is in agreement with the finding that etheno-DNA adducts formed by the
reactive metabolites CEO and CAA convert to actual mutations by
base-pair substitutions.
Other gene mutation assays in bacteria, yeast cells and mammalian
cells have revealed positive results exclusively in the presence of
metabolic activation. Mutagenic effects were also reported in a human
cell line containing cloned cytochrome P-450IIE1, which is capable of
metabolizing VC. Gene mutation was also detected in plant
( Tradescantia ) cuttings exposed to VC. In gene conversion assays,
positive results were reported with Saccharomyces cerevisiae in the
presence of a metabolic activation system. VC exposure induced
unscheduled DNA synthesis in rat hepatocytes and increased
sister-chromatid exchange (SCE) in human lymphocytes after addition of
exogenic activation system. No growth inhibition was detected in DNA
repair-deficient bacteria without metabolic activation. Cell
transformation assays revealed positive results both with and without
metabolic activation.
VC exposure induced gene mutation and mitotic recombination in
Drosophila melanogaster but not gene mutation in mammalian germ
cells. VC showed clastogenic effects in rodents, increased SCE in
hamsters and induced DNA breaks in mice. In host-mediated (rat)
assays, VC induced gene conversion and forward mutations in yeast.
CEO and CAA were found to be mutagenic in different test systems.
CEO is a potent mutagen, whereas CAA is highly toxic. CEO and CAA were
found to be carcinogenic in mice, CEO being much more active than CAA.
Mutations of the ras and p53 genes were analysed in liver
tumours induced by VC in Sprague-Dawley rats: base-pair substitutions
were found in the Ha- ras gene in hepatocellular carcinoma (HCC) and
in the p53 gene in ASL. These mutations are in agreement with the
observed formation and persistence of etheno adducts in liver DNA,
following exposure of rats to VC, and with the known pro-mutagenic
properties of etheno adducts.
Studies into the mechanism of carcinogenicity of VC suggest that
the reactive epoxide intermediate CEO interacts with DNA to form
etheno adducts, which result in a base-pair substitution leading to
neoplastic transformation.
1.7 Effects on humans
Concentrations of VC in the region of 2590 mg/m3 (1000 ppm),
which were not unusual prior to 1974, over periods ranging from 1
month to several years, have been reported to cause a specific
pathological syndrome found in VC workers called the "vinyl chloride
illness". Symptoms described were earache and headache, dizziness,
unclear vision, fatigue and lack of appetite, nausea, sleeplessness,
breathlessness, stomachache, pain in the liver/spleen area, pain and
tingling sensation in the arms/legs, cold sensation at the
extremities, loss of libido and weight loss. Clinical findings
included scleroderma-like changes in the fingers with subsequent bony
changes in the tips of the fingers described as acroosteolysis,
peripheral circulatory changes identical with the classical picture of
Raynaud's disease and enlargement of the liver and spleen with a
specific histological appearance, and respiratory manifestations.
Studies in humans have not been adequate to confirm effects on
the reproductive system. A few morbidity studies have reported
elevated incidence of circulatory diseases among vinyl chloride
workers. However, large cohort studies have found lower cardiovascular
disease mortality.
There is strong and consistent evidence from epidemiological
studies that VC exposure causes the rare tumour, angiosarcoma of the
liver. Brain tumours and hepatocellular carcinoma of the liver may
also be associated with VC, although the evidence cannot be considered
definitive. Other cancer sites reported to be in excess, but less
consistently, include lung, lymphatic and haematopoietic tissue, and
skin.
VC is mutagenic and clastogenic in humans. Frequencies of
chromosomal aberrations (CA), micronuclei (MN) and SCE in the
peripheral blood lymphocytes of workers exposed to high levels of VC
have been shown to be raised compared to controls. Although in many
studies the exposure concentrations and duration of exposure were only
estimated, a dose-response relationship and a "normalization" of
genotoxic effects with time after reduction of exposure can be seen.
Point mutations have been detected in p53 and ras genes in
tumours from highly exposed (before 1974) autoclave workers with liver
angiosarcoma (ASL) and another VC worker with hepatocellular
carcinoma.
Biological markers that have been investigated as indicators for
VC exposure or VC-induced effects include a) excretion of VC
metabolites (e.g., thiodiglycolic acid), b) genetic assays (e.g.,
chromosomal abnormalities or micronucleus assay), c) levels of enzymes
(e.g., in liver function tests), d) serum oncoproteins (p21 and p53)
and/or their antibodies as biomarkers of VC-induced effects.
Children living near landfill sites and other point sources may
be at increased risk based on suggested evidence of early life
sensitivity in animal studies. However, there is no direct evidence in
humans.
In epidemiological studies, a clear dose-response is only evident
for ASL alone or in combination with other liver tumours. Only one
epidemiological study has sufficient data for quantitative
dose-response estimation.
1.8 Effects on other organisms in the laboratory and field
There is a lack of standard toxicity data on the survival and
reproduction of aquatic organisms exposed to VC. Care must be taken
when interpreting the data that are available, as most of it was
generated from tests where the exposure concentration was not measured
and therefore losses due to volatilization were not taken into
account.
The lowest concentration of VC that caused an effect in
microorganisms was 40 mg/litre. This was an EC50 value based upon
inhibition of respiration in anaerobic microorganisms in a batch assay
over 3.5 days.
The lowest concentration that caused an effect in higher
organisms was 210 mg/litre (48-h LC50 for a freshwater fish); with a
corresponding no-observed-adverse-effect concentration (NOAEC) of 128
mg/litre. Effects due to VC have been reported at lower concentrations
in other species, but the ecological significance of these effects was
not verified.
VC concentrations predicted to be non-hazardous to freshwater
fish were calculated to range from 0.088 to 29 mg/litre.
There is a paucity of data concerning the effects of VC on
terrestrial organisms.
2. IDENTITY, PHYSICAL AND CHEMICAL PROPERTIES, AND ANALYTICAL METHODS
This monograph deals with vinyl chloride (VC) monomer itself and
is not an evaluation of polyvinyl chloride (PVC), the polymer of VC.
2.1 Identity
Chemical formula: C2H3Cl
Chemical structure: H2C=CHCl
Relative molecular mass: 62.5
Common names: Vinyl chloride
CAS chemical name: Ethene, chloro-
IUPAC name: Chloroethene
CAS Registry 75-01-4
number:
EC Number: 602-023-007
EINECS Number: 2008310
Synonyms: vinyl chloride monomer,
monochloroethene;
monochloroethylene;
1-chloroethylene, chloroethene,
chloroethylene
Purity 99.9% (by weight); water: max.
120 mg/kg; HCl: max. 1 mg/kg
(BUA, 1989)
Up to the 1960s the purity was not
so high (Lester et al., 1963)
Typical trace 10-100 mg/kg range: chloromethane,
components chloroethane; 1-10 mg/kg range:
ethyne
(acetylene), 1,3-butadiene, butene,
1,2-dichloroethane, ethene,
propadiene
(allene), propene, 1-butyne-3-ene
(vinyl acetylene) (BUA, 1989)
2.2 Physical and chemical properties
Some physical properties of VC are given in Table 1. Under
ambient conditions, vinyl chloride is a colourless, flammable gas with
a slightly sweet odour. It is heavier than air and has relatively low
solubility. There are discrepancies in the literature with regard to
Henry's Law constant (air-water partition coefficient, Hc). Whereas
some authors give a value between 1 and 3 kPa.m3/mol, other sources
quote a value two orders higher. Large uncertainties in the absolute
aqueous solubility in older studies probably contribute most to these
discrepancies (Ashworth et al., 1988). It is an azeotrope with water:
0.1 parts water/100 parts vinyl chloride (Bönnighausen, 1986; Rossberg
et al., 1986). VC is soluble in almost all organic solvents.
Since it is a gas that is heavier than air, VC can spread over
the ground creating an exposure long distances away from the original
source and can form explosive mixtures. The odour threshold value is
very subjective (see Table 1) and is far above the present accepted
occupational safety threshold values (see Annex 1).
VC is transported as a compressed liquid. As it does not tend to
polymerize easily, liquid VC (in the absence of oxygen and water) can
be stored and transported without polymerization inhibitors
(Bönnighausen, 1986).
At ambient temperatures in the absence of air, dry purified VC is
highly stable and non-corrosive. Above 450°C, partial decomposition
occurs yielding acetylene, hydrogen chloride and trace amounts of
2-chloro-1,3-butadiene (chloroprene) (Rossberg et al., 1986). This
reaction also occurs by lower temperatures (at 30°C and under) in the
presence of sodium or potassium hydroxide (Bönnighausen, 1986).
Combustion of VC in air produces carbon dioxide and hydrogen
chloride. Under oxygen deficient conditions, traces of phosgene may be
formed (Rossberg et al., 1986). In chlorine-atom-initiated oxidation
of VC, the vinyl chloride peroxide formed decomposes to formaldehyde,
hydrogen chloride and carbon monoxide (Bauer & Sabel, 1975; Sanhueza
et al., 1976).
With air and oxygen, very explosive peroxides can be formed
(Rossberg et al., 1986). There are reports of explosions in vinyl
chloride plants (Terwiesch, 1982). In VC recovery plants there is a
higher chance of explosion, which necessitates continuous monitoring
and limitation of the oxygen content.
Table 1. Some physical and chemical properties of vinyl chloride
Melting point -153.8 °C Bönnighausen (1986);
Dreher (1986)
Boiling point -13.4 °C Bönnighausen (1986);
(at 101.3 kPa) Dreher (1986)
Flash point -78 °C Bönnighausen (1986);
(open cup) Dreher (1986)
Autoignition 472 °C Bönnighausen (1986);
temperature Dreher (1986)
Critical temperature 156 °C Bönnighausen (1986);
Dreher (1986)
Critical pressure 5600 kPa Bönnighausen (1986);
Dreher (1986)
Explosion limits in air 3.8-29.3 vol% Bönnighausen (1986)
in air (20 °C);
4-22 vol% Dreher (1986)
Decomposition 450 °C Bönnighausen (1986)
temperature
Density (20 °C) 0.910 g/cm3 Bönnighausen (1986)
Vapour pressure at -20 °C 78 kPa Dreher (1986)
0 °C 165 kPa
20 °C 333 kPa
Solubility of VC in 0.95 wt% (9.5 g/litre) DeLassus & Schmidt
water; extrapolated over temperature (1981)
from low pressure range
experiments 1.1 g/litre Euro Chlor (1999)
over range 15-85 °C
at 20 °C
Solubility of water 0.02 ml (-20 °C) Bönnighausen (1986)
in 100 g VC 0.08 ml (+20 °C)
Henry's Law Constant 1.96 at 17.5 °C Gossett (1987)
(Hc) (kPa.m3/mol) 2.0-2.8 at 25 °C Ashworth et al. (1988)
18.8 at 20 °C Euro Chlor (1999)
Table 1. (cont'd)
Solubility in organic soluble in most Dreher (1986)
solvents organic liquids and
solvents;
insoluble in lower Bönnighausen (1986)
polyalcohols
log n-octanol/water 1.58 (measured; BUA (1989)
partition coefficient 22 °C)
(log Kow) 1.36 (calculated) BUA (1989)
1.52 Gossett et al. (1983)
Odour threshold value 26-52 mg/m3 by Hori et al. (1972)
some, but by all at
2600 mg/m3
650 mg/m3 Baretta et al. (1969)
10 700 mg/m3 Patty (1963)
Polymerization reactions to form PVC are the most important
reactions from an industrial view (see section 3.2.1.2).
nH2C = CHCl -> (- H2C - CHCl -)n; Delta HR = -71.2 kJ/mol
The reaction is exothermic. Addition reactions with other halogens at
the double bond, for instance, to yield 1,1,2-trichloroethane or
1,1-dichloroethane, are also important. Catalytic halogen exchange by
hydrogen fluoride gives vinyl fluoride (Rossberg et al., 1986). In the
presence of water, hydrochloric acid is formed which attacks most
metals and alloys. This hydrolysis probably proceeds via a peroxide
intermediate (Lederer, 1959).
Vinyl chlorine reacts with chlorine to form trichloroethane.
1,1-Dichloroethane is formed from the exothermal reaction of VC with
hydrogen chloride in the presence of iron compounds.
2.3 Conversion factors
1 ppm = 2.59 mg/m3 at 20°C and 101.3 kPa
1 mg/m3 = 0.386 ppm
2.4 Analytical methods
2.4.1 General analytical methods and detection
Stringent regulations for the production, use and handling of
carcinogenic VC have been made in several countries (see Annex 1)
necessitating the usage of reliable methods to detect trace amounts of
this compound in air, water and in PVC articles in such human contact
applications as food packing, medical equipment and potable water
transport.
VC in air has been monitored by trapping it on different
adsorbents, e.g., activated charcoal, molecular sieve and carbotrap.
VC can be removed from adsorbents by liquid or thermal desorption
and analysed by GC fitted with FID, PID or MS detection. In ambient
air measurements, several adsorbents in sieves or refrigerated traps
have been used to increase the efficiency of trapping. In continuous
monitoring of workplace and ambient concentration, IR and GC/FID
analysers can be used.
Direct injection, extraction and more increasingly head space or
purge and trap techniques have been applied for analysis of liquids
and solids. VC can be detected by GC fitted to, for instance, FID,
PID, MS or Hall detectors.
A pre-concentration step and chemical derivatization may increase
sensitivity.
An overview of analytical methods for detecting VC in various
matrices is given in Table 2.
2.4.2 Sample preparation, extraction and analysis for different
matrices
2.4.2.1 Air
Most methods are based on that of Hill et al. (1976a), using
adsorption on activated charcoal, desorption with carbon disulfide and
analysis by GC/FID. Kruschel et al. (1994) used a three-stage carbon
molecular sieve adsorbent cartridge to collect a wide range of
selected polar and non-polar VOCs. After purging with helium prior to
analysis, levels of water and other interfering compounds were reduced
sufficiently to allow cryogenic preconcentration and focusing of the
sample onto the head of the analytical column. VC was detected at
levels below the detection limit of former methods.
Landfill gas monitoring has been carried out by trapping VC on a
molecular sieve, and samples have been analysed using, for instance,
GC/MS (Bruckmann & Mülder, 1982) or GC/ECD with prior conversion to
the 1,2-dibromo derivative (Wittsiepe et al., 1996).
Table 2. Analytical methodsa
Matrix Sampling/preparation Separation Detector Detection Comments References
limitb
Air
Expired air collected in 50 ml GC FID 50 ppb Baretta et al. (1969)
pipettes; direct (packed (130 µg/m3)
injection column)
Expired air multistage cryogenic GC FID; MS low ppb low reproducibility; Conkle et al. (1975)
trapping; thermal (packed & cap.) long sampling time
desorption
Expired air 500 ml charcoal tubes GC 0.3 mg/m3 Krajewski & Dobecki
(1978, 1980)
Expired air 1 litre canister; capGC MS n.g. for collecting Pleil & Lindstrom
pressurized with alveolar samples; e.g., (1997)
neutral gas; cryogenic 16 and 25 µg/m3
concentration
Air in car charcoal tube, CS2 GC FID 10 ppb Going (1976); Hedley
interior desorption (26 µg/m3) et al. (1976)
Ambient air activated charcoal/CS2 GC FID 2.6 mg/m3 Hill et al. (1976a)
Ambient air silica gel at -78°C, GC FID 2.6 mg/m3 IARC (1978)
thermal desorption
Ambient air activated charcoal GC n.g. 0.5 ppb Dimmick (1981)
column; 24-h sampling (1.3 µg/m3)
Ambient air sampling (1 to 10 HRGC MS 1 ng VOCs Kruschel et al.
litre) on carbon trap; FID (0.3 µg/m3) (1994)
thermal desorption
Table 2. (cont'd)
Matrix Sampling/preparation Separation Detector Detection Comments References
limitb
Ambient air solid phase sample capGC IMS 2 mg/m3 new method for Simpson et al.
trap preconcentration field monitoring (1996)
Landfill gas (20 litre) carbon capGC ECD 82 ng/m3 Wittsiepe et al.
molecular sieve; CS2 (1996)
desorption;
conversion to
1,2,-dibromo
derivative
Tobacco charcoal tube, CS2 GLC ECD 15 pg per Hoffmann et al.
smoke extraction; injection (1976)
conversion to
1,2,-dibromo
derivative
Workplace CS2 desorption GC (packed FID 0.04 µg working range NIOSH (1994)
air column) (5 litre 0.4 to 40 mg/m3 (based on Hill
sample) et al., 1976a)
Workplace charcoal sorbent GC (packed FID 5 mg/m3 Kollar et al.
air tube; extraction column) (3 dm3 sample) (1988)
with
nitro-methane
Workplace carbon trap, GC FID 2.6 mg/m3 Hung et al. (1996)
air thermal desorption
Workplace activated charcoal, GC FID 0.1 mg/m3 working range HSE (1987);
air CS2 desorption 0.07-25 mg/m3 for ASTM (1993)
30-litre samples
Workplace continuous process GC FID n.g. Pau et al. (1988)
air sampling
Table 2. (cont'd)
Matrix Sampling/preparation Separation Detector Detection Comments References
limitb
Workplace continuous pyrolysis detection 1 mg/m3 Nakano et al.
air sampling of HCl (1996)
Water
Water purge & trap GC MC n.g. in PVC pipes Dressman &
McFarren (1978)
Water purge & trap GC MS 0.05 µg/litre Schlett &
Pfeifer (1993)
Water headspace capGC MS 1 µg/litre Gryder-Boutet &
Kennish (1988)
Water purge & trap capGC PID-ELCD n.g. modification of US Driscoll et al.
EPA Methods 601 (1987)
& 602 for VOC
Water purge & trap capGC PID-ELCD 0.1 µg/litre VOC Ho (1989)
Water purge & trap; capGC ECD 1.6 ng/litre Wittsiepe et al.
CS2 desorption; (1990, 1993)
1,2-dibromo
derivatization
Water GC PID and n.g. VCM loss during Soule et al.
Hall laboratory holding (1996)
detector time
Water purge & trap GC FID n.g. VOC Lopez-Avila et
al. (1987a)
Table 2. (cont'd)
Matrix Sampling/preparation Separation Detector Detection Comments References
limitb
Water CS2 desorption; capGC ECD 1.6 ng/litre Wittsiepe et al.
conversion to (1990, 1996)
1,2,-dibromo
derivative
Bottled headspace with GC MS 10 ng/litre Benfenati et al.
drinking- thermal desorption (1991)
water cold-trap
injector (TCT)
Water solid phase capGC FID n.g. Shirey (1995)
micro-extraction MS
Food, liquids, biological fluids and tissues
Liquid headspace GC FID 0.1 ppb Watson et al.
drugs; (1979)
cosmetics
Food; headspace GLC confirmation 10 ppb Williams (1976a)
liquids with MS
Liquids derivatization to GLC ECD 15 µg/litre Williams (1976b)
1-chloro-1,2- (vinegar);
dibromoethane 50 µg/litre oil
Food direct injection GC FID 2-5 µg/kg detection limit UK MAFF (1978)
depends on medium
Food headspace GC FID 1 µg/kg IARC (1978)
Table 2. (cont'd)
Matrix Sampling/preparation Separation Detector Detection Comments References
limitb
Oil GC FID 5 µg/litre Rösli et al.
(1975) based on
Williams & Miles
(1975)
Food headspace GC n.g. 2-5 µg/kg UK MAFF (1978)
Intravenous headspace capGC FID 1 µg/litre Arbin et al.
solutions (1983)
Blood (rat) headspace GC FID 5 µg/litre Zuccato et al.
ethanol-water (1979)
extraction
Tissues freezing, GC FID 30 µg/kg Zuccato et al.
(rat) homogenization (1979)
then as above
Urine dry; dissolution GC MS 50 µg/litre TDGA Müller et al.
in methanol; (1979)
methylation with
diazomethane
Urine extraction and GC FID 10 mg/litre TDGA; standard: Draminski &
silylation o-phthalic acid Trojanowska
(1981)
Urine conversion to GC MS < 0.5 TDGA; standard: Pettit (1986)
dibutyl ester µmol/litre pimelic acid
PVC
PVC products charcoal tube, GC FID 10 ppb Going (1976)
CS2 desorption (26 µg/m3)
Table 2. (cont'd)
Matrix Sampling/preparation Separation Detector Detection Comments References
limitb
PVC headspace packed column 5 ppb ASTM (1985)
GC
PVC headspace capGC FID update suggestion for Wright et al.
FID-PID ASTM (1985) (1992)
PVC extraction/headspace GC FID 0.1 mg/kg Puschmann (1975);
IARC (1978)
PVC HPLC < 1 ppm for temperatures Kontominas et al.
packaging simulating storage (1985)
of foods conditions (8 to 27 °C)
PVC dynamic headspace GC FID low ppb Poy et al. (1987)
with a sparging and
focusing step
before thermal
desorption
PVC film or GC FID 2.2 ng Gilbert et al.
resin (5 ppb (w/w)) (1975)
Packaging MS 8.7 pg
materials
PVC bags purge/trap GC FID/ECD 0.3 ppb Thomas & Ramstad
(Tenax/charcoal) (1992)
Table 2. (cont'd)
a Abbreviations: capGC = capillary gas chromatography; ECD = electron capture; ELCD = electrolytic conductivity detector;
FID = flame ionization detector; GC = gas chromatography; GLC = gas-liquid chromatography; HRGL = high-resolution gas
chromatography; IMS = ion mobility spectrometry; MC = microcoulometric titration detector; MS = mass spectrometry;
PID = photoionization detector; SPME = solid-phase microextraction; TDGA = determination of the metabolite, thiodiglycolic
acid; VOC = volatile organic chemical (a general method); n.g. = not given
b The % recovery was not given in most cases
2.4.2.2 Water
VC is first purged from the water and then collected for GC
analysis by headspace/purge and trap. VC is highly volatile and has a
low specific retention volume on Tenax-GC, the most commonly used
trapping medium in purge/trap analysis. Combination traps such as
Tenax/silica gel/charcoal (Ho, 1989) or Tenax/OV-1/silica (Lopez-Avila
et al., 1987b) have been used. Another approach is to bypass the trap
altogether by purging directly onto a cryocooled capillary column
(Gryder-Boutet & Kennish, 1988; Pankow & Rosen, 1988; Cochran, 1988;
Cochran & Henson, 1988), but here there are complications due to the
need to remove water when stripping from an aqueous solution. A more
recent adaptation of the headspace method uses solid-phase
microextraction (SPME) in which a stationary phase, usually
poly(dimethylsiloxane), coated on a fused-silica fibre is used to
extract aqueous samples in completely filled sealed vials (Shirey,
1995).
It should be noted when measuring VC content in water or
groundwater that samples should be analysed as soon as possible, as
the VC content decreases with holding time (Soule et al., 1996).
2.4.2.3 PVC resins and PVC products
For the quantification of residual VC in PVC, a solid and a
solution approach have been used. The former involves the
equilibration of a solid polymer sample at 90°C in a sealed system,
followed by headspace analysis with single or multiple extraction
(Berens et al., 1975; Kolb, 1982). The solution approach involves the
equilibration of a 10% solution of PVC in dimethylacetamide in a
sealed system, followed by analysis of the headspace gas (Puschmann,
1975). A automatic dynamic headspace method involving a sparging and a
focusing step before desorption into the GC column has been developed
to increase the sensitivity of the solution approach method (Poy et
al., 1987).
2.4.2.4 Food, liquid drug and cosmetic products
For monitoring VC in foods in contact with PVC packaging,
headspace GC is the usual method. Before the levels of VC allowed in
PVC was regulated in the 1970s (see Annex I), many of the VC levels
observed were high enough to be determined by direct injection
methods. Limits of detection are given as 2, 5 and 5 µg/kg for
aqueous, ethanolic, and oleaginous medium respectively using
headspace-GC-FID (UK MAFF, 1978). Williams (1976a,b) reported a
gas-liquid chromatographic method using subsequent GC-MS confirmation,
and a further GC/ECD method requiring derivatization to
1-chloro-1,2-dibromoethane for determination of VC content in liquid
foods.
Methods for VC levels in liquid drug and cosmetic preparations
were described by Watson et al. (1979). A weighed aliquot of the
commercial product in a tightly septum-sealed vial with accurately
known headspace volume is heated to 50°C for 30 min. A portion of the
warm headspace gas is then injected into a GC equipped with FID and a
styrene-divinylbenzene porous polymer column.
2.4.2.5 Biological samples
There are few data on VC analysis in biological tissues. The only
report available was on rat blood and tissues (Zuccato et al., 1979).
2.4.2.6 Human monitoring
Methods for measuring VC concentrations in exhaled air (breath)
have been described (see Table 2) but, although useful for studying
metabolism, they are not well suited for biological monitoring due to
the short half times in the body and the saturable metabolism of VC.
Metabolites of vinyl chloride have been identified in the urine
of rats (Müller et al., 1976; Green & Hathway, 1977) and humans
(Müller et al., 1979) using GC-MS. As there is a strong correlation
between VC exposure in humans and increased excretion of
thiodiglycolic acid (Müller et al. 1978), this metabolite has been
using for monitoring purposes. It is, however, not specific for VC as
certain drugs and other C2 compounds also have thiodiglycolic acid as
a urinary metabolite (Müller et al., 1979). Since thiodiglycolic acid
is also detected in unexposed subjects (Müller et al., 1979; van
Sittert & de Jong, 1985) and even premature babies (Pettit, 1986),
this approach can only be used to demonstrate high levels of exposure.
A discussion of biological markers for VC exposures and VC-induced
liver cancer is presented in section 8.5.
Thiodiglycolic acid has been determined by dissolving the dried
urine residue in methanol, methylating with diazomethane and analysing
with GC-MS (Müller et al., 1979), analysing the metabolite as its
dibutyl ester by GC-MS using selected ion monitoring (Pettit, 1986),
and by using GC/FID (Draminski & Trojanowska, 1981). Care must be
taken with methods which analyse for VC metabolites, as these
metabolites are not specific to VC.
A specific and sensitive new method has been reported for the
quantification of the VC metabolite N-acetyl- S-(2-hydroxyethyl)
cysteine by exchange solid-phase extraction and isotope dilution
HPLC-tandem mass spectrometry (Barr & Ashley, 1998). This method may
prove useful for monitoring occupational VC exposure, as the detection
limit of 0.68 µg/litre is low enough to detect this metabolite even in
people with no overt exposure to VC, ethylene oxide or ethylene
dibromide.
2.4.2.7 Workplace air monitoring
Before the 1960s, when it was established that VC was a
carcinogenic substance, halogen detectors and explosimeters were used,
non-specific for VC, with detection limits of 518-1295 mg/m3 (200-500
ppm). Gradually more sophisticated techniques became available for
detection of low ppm levels of VC, such as IR analysers, FID, PID
(ECETOC, 1988) and more recently mass spectrometry. In order to check
for leaks or for control measurements during cleaning and repair work,
detector tubes or direct-reading instruments with FID or PID can be
used, although they are not specific for VC and regular calibration is
necessary (Depret & Bindelle, 1998).
Continuous analyses based on, for instance, IR, GC/FID or HCl
detection have been developed (IARC, 1978; Pau et al., 1988; Nakano et
al., 1996). Analysers can be equipped for computerized data logging
and processing. The detection limit of an IR analyser depends on, for
instance, path length and is about 1.3 mg/m3 (IARC, 1978). The
analyser detecting HCl from VC pyrolyzed in a quartz tube was reported
to have a detection limit of 1 mg/m3 when the sampling time was 40
seconds (Nakano et al., 1996).
Breathing zone concentrations can be measured by sampling VC with
portable pumps or diffusion on activated charcoal (Nelms et al., 1977;
Heger et al., 1981; ASTM, 1993; NIOSH, 1994; Du et al., 1996). By
using thermosorption tubes (carbotrap 110-400), detection limits can
be decreased and the use of carbon disulfide in desorption avoided
(Hung et al., 1996).
Passive monitors for occupational personal monitoring of exposure
to VC are commercially available.
3. SOURCES OF HUMAN AND ENVIRONMENTAL EXPOSURE
3.1 Natural occurrence
VC is not known to occur naturally.
3.2 Anthropogenic sources
Anthropogenic sources of VC include the intentional manufacture
of the compound for further processing, primarily to PVC, and
unintentional formation of VC in, for instance, sanitary landfills, as
a degradation product of chlorinated hydrocarbons such as those used
as solvents, and the subsequent presence of VC in emitted gases and
groundwater. VC is also found in tobacco smoke.
3.2.1 Production levels and processes
VC was first synthesized by Regnault in 1835. It was not until
the 1930s that techniques were devised to polymerize VC into stable
forms of PVC.
VC production methods were altered in 1974 in many countries
after the confirmation that VC was a human carcinogen. Manufacturers
developed closed production methods to reduce exposure of the
workforce.
Annual total world production of VC, which is approximately equal
to PVC production, was about 17 million tonnes in 1985 and over 26
million tonnes in 1995 (see Table 3). More than half the world's
capacity (64%) in 1985 was concentrated in Western Europe and the USA.
Since that time many new VC/PVC plants have been opened or are under
construction in SE Asia, Eastern Europe, the Indian subcontinent and
developing and oil-producing countries. Thus there has been a
geographical shift of VC/PVC production.
The leading producers of PVC and therefore also of VC are the
USA, Japan, Germany, France and SE Asian countries such as Taiwan and
China (CHEM-FACTS, 1992). The capacity has only increased moderately
in the USA and Western Europe in recent years. Significant increases
in production have been reported for Japan and Taiwan. All the
countries of Eastern Europe have PVC plants and have exported PVC to
Western European countries. The PVC capacity and imports and exports
for each country are given in CHEM-FACTS (1992).
VC is produced in Western Europe by 14 companies. The plants are
in Belgium (3 plants), France (3 plants), Germany (8 plants), Italy (4
plants), the Netherlands (1 plant), Spain (2 plants), Sweden (2
plants), United Kingdom (1 plant) (Euro Chlor, 1999).
Table 3. World PVC (and therefore VC) production/capacity 1980-1998
Region Production/capacity in 1000 tonnes/year
1980a 1985a 1990a 1995b 1998c
World capacity 16 000 17 000 20 700 26 400 approx. 27 000
World production 11 750 14 200 18 300
North America total 3200 3390 4700 6070
Suspension and mass 2810 2990
Vinyl acetate copolymer 200 210
Emulsion 190 190
Western Europe total 3900 4330 4800 5750 approx. 5600
Suspension and mass 33 350 3700
Vinyl acetate copolymer 130 130
Emulsion 420 500
Eastern Europe 925 1100 1200 2700d
Former Soviet Union 370 700 760
China 150 400 790
Japan 1400 1550 2070 8200e
SE Asia 330 600 900
South America 400 540 780
Rest of the world 1075 1590 2300 3680
a Allsopp & Vianello (1992)
b Rehm & Werner (1996)
c Although exact figures are not available, an increase in world production
was seen in 1998 but no great increase in Western Europe (personal
communication, European Council of Vinyl Manufacturers, 1999)
d Including former Soviet Republics
e Total Asia
The manufacture of VC/PVC is one of the largest consumers of
chlorine.
3.2.1.1 Production of VC
VC is produced industrially by two main reactions, the first is
hydrochlorination of acetylene, which proceeds via the following
reaction:
C2H2 + HCl -> CH2=CHCl
This route was used in the past when acetylene, produced via calcium
carbide from coal, was one of the important basic feedstocks for the
chemical industry (Rossberg et al., 1986). Today all USA and most
Western European manufacturers use the "balanced process" described
below. However, many Eastern European countries such as Poland and the
countries of the former Soviet Union still use acetylene to
manufacture VC because of relatively cheap raw materials such as
calcium carbide and natural gas. Mercury has been used as a catalyst,
although a new catalyst has now been developed in Russia, based on
platinum metal salts instead of mercury, which has increased yields
with acetylene conversion from 95 to 99% (Randall, 1994).
The second major production process involves thermal cracking
[reaction iii] (at about 500°C) of 1,2-dichloroethane (EDC), produced
by direct chlorination [reaction i] of ethylene or oxychlorination
[reaction ii] of ethylene in the "balanced process".
[i] CH2=CH2 + Cl2 -> Cl CH2-CH2 Cl
[ii] CH2=CH2 + 2 HCl + 5 O2 -> Cl CH2-CH2 Cl + H2O
[iii] Cl CH2-CH2 Cl -> CH2=CHCl + HCl
After the cracking (pyrolysis), HCl and unconverted EDC are separated
from VC by two steps of distillation and recycled. The VC is stored
either under pressure at ambient temperature or refrigerated at
approximately atmospheric pressure (European Council of Vinyl
Manufacturers, 1994). More than 90% of the VC produced today is based
on this route (Rossberg et al., 1986; Allsopp & Vianello, 1992).
Other methods of industrial production include:
a) VC from crack gases, where unpurified acetylene and ethylene are
chlorinated together, acetylene being first chlorinated to
1,2-dichloroethane.
b) VC from ethane, which is readily available in some countries. The
major drawback is that ethane must first be functionalized by
substitution reactions giving rise to a variety of side chain
reactions and therefore the reaction must be kinetically
controlled to obtain maximal VC yield.
3.2.1.2 Production of PVC from VC
Many PVC plants are fully integrated beginning with ethylene and
chlorine (or sodium chloride).
VC is a gas at ambient temperatures but is handled as a
compressed volatile liquid in all polymerization operations. PVC
polymerization reactors are thick-walled jacketed steel vessels with a
pressure rating of 1725 kPa. The polymerization of VC is strongly
exothermic. The explosive limits of VC in air are 4-22 vol%, and the
plant must be designed and operated with this in mind, particularly
when handling unreacted VC in the recovery system (Allsopp & Vianello,
1992).
Three main processes are used for the commercial production of
PVC: suspension (providing 80% of world production), emulsion (12%)
and mass or bulk (8%). In Western Europe, the proportion of PVC
produced by the different processes is: 80% suspension; 13% mass; 5%
emulsion and 2% copolymers (Wrede, 1995).
In the suspension (also called dispersion) process,
polymerization takes place at 40-70°C (depending on the type of PVC
being produced) in a reactor (autoclave) of 25-150 m3 capacity fitted
with a jacket and/or condenser for heat removal, as the reaction is
strongly exothermic. Precautions have to be taken in order to avoid
explosive mixtures with air. Liquid VC under its autogenous vapour
pressure is dispersed in water by vigorous stirring to form droplets
of average diameter 30-40 µm. The polymerization takes place within
these droplets and is started by addition of initiators dissolved in
the monomer. Stabilizers are added to prevent the drops rejoining and
to prevent the already polymerized PVC particles from agglomerating.
The reaction conditions can be exactly controlled and the properties
of the product, such as relative molecular mass, can be controlled
exactly. Once polymerization has ended, the autoclave charge is
emptied into degassing tanks, and the non-polymerized VC is degassed,
compressed and stored for reuse (ECETOC, 1988; Allsopp & Vianello,
1992).
During the polymerization process, the PVC is dispersed in the
aqueous phase and it cannot be prevented that a film of PVC forms on
the inside wall of the reactor. This film interferes with the transfer
of heat between the reactor and contents, and the process has to be
interrupted periodically to allow the reactor to be cleaned. The
autoclave, after being emptied, is opened, rinsed and washed either
with solvents or more usually by means of automatic high-pressure jets
(ECETOC, 1988). The latest development in this area is the use of
proprietary build-up suppressants, which are applied before every PVC
batch. After each batch, low pressure rinse with water can remove
loose polymers and the batch cycle is ready to restart. The reactor
needs then to be opened for a thorough cleaning only after 500 or more
batches (Randall, 1994).
Before awareness of the toxicity of VC, it was the autoclave
cleaning personnel who were primarily highly exposed to the compound.
In the past autoclaves were cleaned manually; the inside had to be
scraped with a spatula, or sometimes hammer and chisel to remove the
encrusted polymer adhering to the walls of the vessel and mixing
devices. Lumps of polymer often released monomer when broken,
resulting in high concentrations of VC in the autoclave. Before about
1970, it was usual to check that the level was below 1036 mg/m3 (400
ppm), i.e. two orders of magnitude below the lower explosion limit of
VC. Occupational exposure limits are now 18 mg/m3 (7 ppm) or less
(see Annex I). Further details of workplaces with a former high
exposure to VC are given in section 5.3 and Jones (1981).
Once polymerization has ended, the polymerization batch is
transferred to the stripping unit and then to the slurry tank. The
slurry is a suspension of PVC in water that has to be permanently
stirred; it is then dewatered in a centrifuge decanter and dried. The
resulting dried powder is either stored in silos or bagged. PVC is
then further processed into ready-to-use resins (Depret & Bindelle,
1998).
3.2.1.3 PVC products
PVC is a polymer of VC with 700-1500 monomeric units. It is
relatively inexpensive and is used in a wide range of applications.
PVC is a generic name. Each producer makes a range of PVC polymers,
which vary in morphology and in molecular mass according to the
intended use. PVC resins are rarely used alone but can be mixed with
heat stabilizers, e.g., lead, zinc and tin compounds (Allsopp &
Vianello, 1992), lubricants, plasticizers (e.g., diethylhexyl
phthalate) fillers and other additives, all of which can
influence its physical and mechanical properties (Williamson &
Kavanagh, 1987; Allsopp & Vianello, 1992). Such additives may
constitute up to 60% of the total weight in some finished PVC plastics
(Froneberg et al., 1982). These plastics are formed into a multitude
of consumer products by extrusion, thermoprocessing and rotational
moulding, and into rigid or flexible film by extrusion or calendering.
PVC accounts for 20% of plastic material usage and is used in
most industrial sectors (ECETOC, 1988; European Council of Vinyl
Manufacturers, 1994).
* Packaging
* bottles (produced by blow-moulding) for containing liquid
foods, beverages, cooking oils, vinegar, etc.
* rigid film (calendered or extruded) which is converted into
tubes and shaped containers by subsequent vacuum or
pressure-forming for packaging of various foodstuffs
* flexible film (made by blowing or calendering) for wrapping
solid foods such as cheese, meat, vegetables, fresh fruit,
etc.
* coatings in metal cans
* Building - floor coverings, wall coverings, windows, roller
shutters, piping; 58% of the water supply network and 80% of the
waste water disposal systems in Europe use pipes and fittings
made from PVC.
* Electrical appliances - wires and cables insulation
* Medical care - equipment such as blood bags and gloves;
pharmaceutical and cosmetic packaging. Worldwide, more than 25%
of all plastic-based medical devices used in hospitals are made
from PVC (Hansen, 1991)
* Agriculture - piping; drainage; tubing in dairy industry
* Automobiles - car dash boards and lateral trimming
* Toys
At present, the largest use of PVC is in the building sector
(Rehm & Werner, 1996).
3.2.2 Emissions from VC/PVC plants
3.2.2.1 Sources of emission during the production of VC
Process waste, by-products and unreacted material from a balanced
process and from a PVC plant include (Randall, 1994): a) light/heavy
ends from EDC purification (from direct chlorination and
oxychlorination reactors); b) heavy ends from VC purification (from
pyrolysis reactor); c) pyrolysis coke/tars (from thermal reactions in
the pyrolysis reactor); d) spent catalyst (from direct chlorination,
oxychlorination); e) recovery of unreacted VC (from PVC reactor); f)
offspec batches (from PVC reactor); g) aqueous streams (from EDC
washing, vent scrubbing, oxychlorination water; from centrifuge, VC
stripping, slurry tanks); h) vent gases (from distillation columns,
flash drums, reactor vents, storage tanks, vessel openings; from VC
recovery systems, dryer stacks, centrifuge vent, blending, storage
facilities); i) spills and leaks (sampling, pumps, flanges, pipes,
loading/unloading, valves; bag filling, agitator seals); and j)
equipment cleaning (tanks, towers, heat exchangers, piping; PVC
reactor, product dryer, recovery system).
a) Strategies for minimizing emissions
During the sixties, some control of exposure levels was
introduced resulting in an important decrease of estimated ambient
levels of VC.
In the 1970s, efforts were focused on controlling emissions at
the most significant emission points: reactors, filters and storage
tanks. Elementary modifications of equipment, room and local
ventilation by fans, provisional operation procedures, etc., enabled
the reduction of exposure levels in working areas.
Technical developments have achieved further reductions:
* removal of residual VC from the PVC suspension by stripping
between polymerization and drying with a flow of steam or in
closed-loop systems;
* appropriate collection of residual vents to thermal oxidizers or
other abatement systems;
* reduction of all sources of fugitive emission by maintenance and
upgraded equipment;
* high pressure internal cleaning of the autoclaves to remove PVC
crusts;
* intensive removal of VC before opening or a closed process
design.
Appropriate working procedures, personnel awareness and high
standard equipment, associated with good maintenance practices, are
the recommended ways to reduce fugitive emission to very low levels
(Depret & Bindelle, 1998).
b) Disposal of by-products
The main waste streams of EDC/VC production process in Europe are
light ends (gases: VC, EDC, HCl, ethylene, dioxins; aqueous effluents:
EDC, copper, dioxins) and heavy ends (viscous tars). According to the
PVC Information Council the total amount produced is approximately
0.03 tonnes of by-products per tonne of VC produced (European Council
of Vinyl Manufacturers, 1994). The fractions are either used as
feedstocks for other processes or combusted under controlled
conditions (> 900°C) or by catalytic oxidation to produce CO2, CO,
HCl and water which can be recycled (see above). Exit gases should be
treated using HCl absorbers and gas scrubbers. Spent catalyst, metal
sludges and coke from EDC cracking should be disposed of in controlled
hazardous waste dumps (HWD) or incinerated under controlled
conditions. Sludges from effluent purification should be either
combusted under controlled conditions or deposited in HWD (European
Council of Vinyl Manufacturers, 1994).
3.2.2.2 Emission of VC and dioxins from VC/PVC plants during
production
a) VC
An estimated 550 tonnes of VC was released into air, 451 kg to
water and 1554 kg to soil from manufacturing and processing facilities
in the USA in 1992, although this was not an exhaustive list (ATSDR,
1997). From a VC capacity of 6 200 000 tonnes, an average emission of
80 g/tonne can be calculated (Depret & Bindelle, 1998).
Euro Chlor (1999) reported emissions of VC from suspension PVC
plants in Europe to be 448 tonnes/year, 22 tonnes/year being released
in waste water. An estimated emission value of 300 tonnes VC/year was
provided by German producers (BUA, 1989), i.e., about 55 g VC/tonne
PVC. Total VC emissions in England and Wales were reported to be
3800 tonnes in 1993 and 18 990 tonnes in 1994 (HMIP, 1996).
An estimated 200 000 tonnes of VC was released into the worldwide
atmosphere during 1982 (based on a worldwide PVC production of
17 million tonnes), i.e., 12 kg/tonne of PVC produced (Hartmans et
al., 1985). In 1974, a VC emission of 0.5 kg/tonne PVC was estimated
(Kopetz et al., 1986).
b) Dioxins
PCDDs/PCDFs, some of which are classified as human carcinogens
(IARC, 1997), are formed during EDC/VC production. Concentrations in
VC distillation residues (PU4043) (probably "heavy ends") for two
production factories were 3192 and 5602 ng ITEQ (international toxic
equivalent)/kg, which was estimated to be 12 to 30 g of dioxin/year at
a production level of 200 000 tonnes VC/year in heavy ends (Stringer
et al., 1995). However, the VC/PVC industry argues that, although
dioxins may be found in production wastes, these are incinerated and
are not ultimately emitted to the environment (Fairley, 1997), at
least in those countries where waste streams are regulated (not all
countries possess such high standards of waste incineration). Some
companies do not incinerate their waste products but dispose of them
in other ways, e.g., deep-well injection (Stringer et al., 1995).
Wastewater from VC production can also be contaminated with
PCDDs/PCDFs, in particular if they contain suspended solids.
Installation of filtration devices should lower solid levels and
subsequently PCDD/PCDF emissions (Stringer et al., 1995). Using such
filtration devices, PCDD/PCDF concentrations in wastewater from
EDC/VC/PVC facilities in the USA ranged from not detectable to 6.7
pg/litre TEQ (Carroll et al., 1996).
Annual global emission of dioxins into the environment from
EDC/VC manufacture has been estimated to be 0.002-0.09 kg TEQ by the
European PVC industry but 1.8 kg by Greenpeace (Miller, 1993).
Virgin suspension PVC resin from 11 major production sites in
Europe was found not to contain any process-generated PCDDs/PCDFs at
concentrations above the limits of quantification (2 ppt) (Wagenaar et
al., 1998).
3.2.3 Accidental releases of VC
3.2.3.1 PVC plant and transport accidents
An explosion occurred at a VC recovery plant in 1978 in Germany,
which was set off by vinyl chloride peroxides (Terwiesch, 1982; see
also section 2.2).
A freight train with 12 tank cars of VC was derailed in McGregor,
Manitoba, Canada in March, 1980 under near-blizzard conditions and
-20°C and two of the tanks released VC (Charlton et al., 1983). One
car lost 47 500 litres in the first hour and the other car lost
23 100 litres at an initial rate of 1400 litres/hour, decreasing to
45 litres/hour after 31 h. In the first 15 min, 5680 litres of VC
vapourized by free surface evaporation; thereafter only 2.5% of the
discharging liquid evaporated rapidly. The remaining liquid in the
snow bank was assumed to have evaporated at a rate of 15% per hour,
leaving about 900 litres of VC in the snow bank after 36 h. Although
there was an explosion hazard, no fire occurred.
Two incidences, in 1988 and 1996, occurred in Germany involving
accidental release of VC due to derailment of trains transporting the
liquid substance (Neuhoff, 1988; Anon, 1996). Both accidents were
followed by explosion and fire. Derailment of a goods train occurred
1 km from Schönebeck (near Magdeburg in Germany) and the subsequent
explosion and fire produced a 600- to 800-m black column of smoke. In
all, 1044 tonnes of VC were involved of which 261 tonnes burnt and
350 tonnes could be reclaimed after the fire; 153 tonnes of HCl were
released. Median measurements of the numerous air samples taken at the
place of accident or surroundings did not exceed the German technical
guidance level of 5 vol ppm, although these were not taken until 14 h
after the fire. Maximum concentrations of VC measured were 78 mg/m3
(30 ppm) near the train and 26 mg/m3 (10 ppm) at a distance of 200 m
from the centre of the fire (Hahn et al., 1998). Levels in nearby
industrial sewage pipes were up to 1250 ppm (Anon, 1996). A cytogenic
analysis was carried out on some of the general population exposed to
VC from this accident (Hüttner & Nikolova, 1998; see section 8.1 and
Table 42).
3.2.3.2 Leakage and discharge from VC/PVC plants
Leakage from a waste liquor basin of a VC/PVC plant in Finland
caused high concentrations of VC and dichloroethene in groundwater in
1974. Concentrations of up to 484 mg/litre of the chlorocarbons were
measured in groundwater in the mid-1980s (Nystén, 1988).
It should be noted that in the 1960s and before, when the
toxicity of VC was not known, large amounts of PVC production sludges
containing VC were dumped onto landfills (and possibly still are in
countries where there are no adequate restrictions).
3.2.4 VC residues in virgin PVC resin and products
3.2.4.1 VC residues in different PVC samples
VC is not soluble in PVC nor is it absorbed or adsorbed in the
resin particle. It is entrapped and can escape to the ambient air
(Wheeler, 1981). PVC in a bulk-container loses its residual monomer at
a rate of 25 to 50% per month. Heating tends to accelerate this step,
but when the residual monomer has disappeared PVC is not a significant
source of VC.
Since the 1970s when VC was confirmed as a human carcinogen, it
has been mandatory in many countries to "degas" PVC after
polymerization (see section 3.3.1) and before further processing.
There are limit values for VC content in PVC (see Annex I). For
example, in 1974 raw PVC usually contained more than 1000 ppm of
residual VC. This was subsequently reduced to 10 ppm by regulation
(German Environmental Office, 1978). In a survey of 45 samples of raw
PVC from various countries carried out in 1976-1977, over a third had
VC residues of > 1 ppm, but a third had residues of over 50 ppm, with
4 samples over 200 ppm. The samples with the highest residue level
came from Hungary, Rumania, Italy and the USA (German Environmental
Office, 1978).
3.2.4.2 VC residues in PVC products
In a survey of PVC products carried out in 1976-1977, the
following indoor articles had a VC content of > 0.05 ppm: bathroom
tiles, piping, plastic bottle for table oil, and kitchen film. The
highest concentrations were found in music records, those bought
recently having a VC content of up to 210 ppm and one record 10 years
old having a content of 970 ppm. In record shops and other rooms
containing many records, this could have been an important source of
VC. In contrast, the VC content of toys, kitchen utensils, food
wrappings, wallpaper and car interiors was < 0.05 ppm (German
Environmental Office, 1978).
Whereas in 1974 the typical level of residual VC in PVC bottles
was 50 mg/kg, introduction of improved manufacturing practices at the
polymer resin processing stage reduced this to 3 mg/kg by 1975. In a
1978 survey, 22 out of 24 PVC bottles contained VC at less than
0.4 mg/kg (UK MAFF, 1978).
In a more recent survey, VC residues in various PVC samples were
given as follows: rigid water bottle (850 ppb); thin plasticized food
film (3 ppb); monopolymer powder (10 ppb); copolymer film (15 ppb)
(Poy et al., 1987). PVC film is still used widely for food packaging.
For example, in Denmark, in 1990, 129/239 samples of cling-film used
for cheese wrapping were of PVC (Svensson, 1994).
Residual VC could not be detected (< 0.1 ppm) in two PVC
products from Thailand (Smerasta et al., 1991) or in PVC and products
from it in Poland (Stareczek, 1988). PVC medical devices are regulated
in the USA and have to meet certain requirements (a maximum of 5 ppb
residual monomer for flexible compounds and a 10 ppb ceiling for rigid
compounds (Rakus et al., 1991)).
Levels of VC found in food and pharmaceutical articles are given
in section 5.1.4. Annex I gives current regulations for VC content in
various PVC products.
3.2.4.3 VC formation as a result of heating PVC
a) Thermal degradation of PVC
PVC is thermally stable below 225°C. Between 225°C and its
ignition temperature of 475°C, thermal degradation results in the
release of about 50 compounds (Boettner et al. 1969). PVC does not
degrade back to VC. Thermal degradation of two types of bulk PVC
samples at 148-232°C resulted in the release of long-chain aliphatic
alcohols, toluene, benzene, various chlorinated species, and a major
peak of HCl. The main components released at 260°C-315°C were aromatic
hydrocarbons such as benzene, phenol and adipates, along with various
aliphatic alcohols, alkenes, anhydrides, some of them chlorinated, and
carbon monoxide (Froneberg et al., 1982). When 1 kg of PVC is heated
to 300°C, it releases about 13 g HCl and 5 g CO.
b) Release of VC from heating PVC
Various PVC resins from different producers were tested for VC
evolution over the 130 to 500°C range using a heating rate of
10°C/min. A consistently low level of VC, amounting to 15-30 ppm
(based on resin), was found in the volatile decomposition products
from all of the samples tested, regardless of resin type or
manufacturing source (USA) (Wakeman & Johnson, 1978). A 100-mg sample
was programmed for heating from 200 to 450°C at 3°C/min; this resulted
in the formation of a total of 23.2 ppm VC, the major portion being
generated in the 275-350°C region. Dehydrochlorination occurred most
rapidly between 250 and 275°C. During this period only 2.3 ppm of VC
was formed. The VC evolved by heating PVC is the VC monomer entrapped
in the PVC resin.
At temperatures required for thermoforming PVC for food packaging
applications (90-120°C for a few seconds), no detectable VC was formed
in up to 1 h of exposure at 130°C (detection limit of analysis in air
- 1 ppb). Temperatures for calendering and extrusion operations are
175-210°C. Maximum VC levels determined at 210°C were 0.5 ppm (resin
basis) after 5 min and 1.2 ppm after 30 min (Wakeman & Johnson, 1978).
In more recent studies into VC formation during the thermal
welding of plasticized PVC sheeting (about 225°C), in normal field
situations such as piping in sewers, VC concentrations were usually
not above the detection limit of 0.05 ppm. Only where there was poor
ventilation were higher levels detected (0.2 ppm VC; 1.0-3.5 ppm HCl)
(Williamson & Kavanagh, 1987).
3.2.5 Other sources of VC
3.2.5.1 VC as a degradation product of chlorinated hydrocarbons
VC as a gas, in leachate and groundwater (see Table 4), has been
found in landfills and surroundings where there were no VC/PVC
production facilities in the vicinity. It was found that VC can be
formed, under anaerobic conditions, from the reductive halogenation of
the more highly chlorinated chloroethenes: tetrachloroethylene (PCE),
trichloroethene (TCE), and the dichloroethene isomers ( cis-1,2-DCE,
trans-1,2-DCE, and 1,1-DCE) (Parsons et al., 1984; Vogel & McCarty,
1985; McCarty, 1996, 1997, see Fig. 1). PCE and TCE are widely used as
industrial solvents in particular for degreasing and cleaning metal
parts and electronic components, and in dry cleaning. Production
levels for 1984 were 260 and 200 thousand tonnes for PCE and TCE,
respectively (Wolf et al., 1987). Careless handling, storage and
disposal, as well as the high chemical stability of these compounds,
have made them, and consequently VC, some of the most frequently
encountered groundwater contaminants (Arneth et al., 1988). Although
VC may be further degraded to less chlorinated and non-chlorinated
ethenes, and possibly finally to carbon dioxide and ethane, this
proceeds only at a slow rate under highly reducing conditions
(Freedman & Gossett, 1989; DiStefano et al., 1991; De Bruin et al.,
1992; see also section 4.2). As a consequence, VC can be detected in
landfill sites in and surrounding areas through spreading. Reports
from several countries show high levels of VC contamination of soil
and groundwater, aquifers and wells (see Table 4 and section 5.1).
There have recently been several field studies in
PCE/TCE-contaminated landfill sites and aquifers (Major et al., 1991,
1995; Fiorenza et al., 1994; Lee et al., 1995, see Table 5). These
have shown that under anaerobic conditions, PCE and TCE can be
intrinsically biodegraded to ethene by indigenous methanogenic,
acetogenic and sulfate-reducing bacteria. Furthermore, under aerobic
conditions there is a potential for direct or co-metabolic oxidation
of DCE and VC. Therefore, an efficient bioremediation of chlorinated
ethene-contaminated aquifers may occur in contaminant plumes
characterized by upgradient anaerobic and downgradient aerobic zones,
such as where anaerobic, chlorinated ethene plumes discharge to
aerobic surface water bodies. However, this depends on the ability of
the stream-bed microbial community to degrade efficiently and
completely DCE and VC over a range of contaminant concentrations (Cox
et al., 1995; Bradley & Chapelle, 1998a). It should be noted that this
bioremediation occurs under specific conditions. The biodegradation
studies listed in chapter 4 give conflicting results.
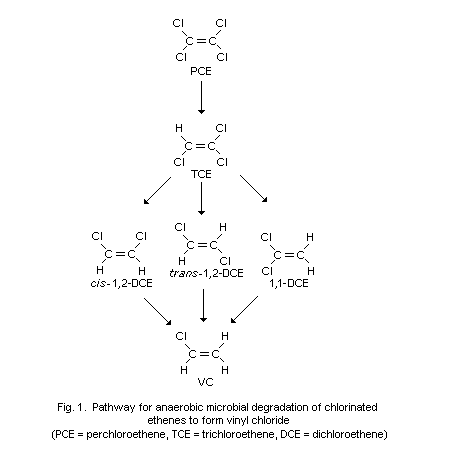 Table 4. Vinyl chloride found in landfill/waste disposal sites as a gas, in leachate and in groundwater
formed probably from degradation of higher chloroethenes
Sample Place (year) of sampling Valuea Concentrations Reference
Landfill gas 2 landfills USA max 230 mg/m3 Lipsky & Jacot (1985)
average 34 mg/m3
Landfill gas landfill, UK max 11 mg/m3 Ward et al. (1996)
plume, 100 m from boundary 40 mg/m3
due to subsurface migration (1991)
Landfill gas landfill, Braunschweig, Germany mean 9 mg/m3 Henning & Richter (1985)
Gas effluents garbage dump, Berlin, Germany 0.27 mg/m3 Höfler et al. (1986)
Gas Germany, average Janson (1989)
industrial waste disposal site 41 mg/m3
municipal waste disposal site 10 mg/m3
Gas Germany, waste disposal site range 0.03-0.3 mg/m3 Bruckmann & Mülder (1982)
Landfill gas UK, 7 waste disposal sites range < 0.1-87 mg/m3 Allen et al. (1997)
Soil air Germany, solvent waste sites 3 max out 128 mg/m3, Köster (1989)
of 200 47 mg/m3,
5 mg/m3
Leachate MSW, Wisconsin, USA (1982) range 61 µg/litre Sabel & Clark (1984)
Leachate USA sites established before 1980 range 8-61 µg/litre Chilton & Chilton (1992)
(6 chosen sites)
Leachate or industrial landfill range 140-32 500, Brown & Donnelly (1988)
groundwater municipal landfill 20-61 000
plume µg/litre
Groundwater, Germany, contaminated water range < 5-460 µg/litre Brauch et al. (1987)
Wells range 15-1000 µg/litre
Groundwater Germany, solvent waste site 3 max 1000 µg/litre, Köster (1989)
samples/200 500 µg/litre,
200 µg/litre
Table 4. (cont'd)
Sample Place (year) of sampling Valuea Concentrations Reference
Groundwater Germany max 120 µg/litre Milde et al. (1988)
Groundwater Santa Clara Valley, USA (near range 50-500 µg/litre Wolf et al. (1987)
plants manufacturing electronic
equipment which use
significant amounts of
chlorinated solvents)
Groundwater Germany: 136 samples from max 12 000 µg/litre Dieter & Kerndorff (1993)
down-gradient wells of 100 waste mean 1694 µg/litre
disposal sites
Groundwater sand aquifer near industrial max > 5 µg/litre Semprini et al. (1995)
site, Michigan, USA. Concentration at 10 m;
increased with depth consistent 56 400 µg/litre
with methane at 23 m
Outwash aquifer Gloucester landfill, Canada (1988) range < 1-40 µg/litre Lesage et al. (1990)
a This column indicates whether the concentration is a maximum (max), average or range value
Each landfill site has individual conditions (e.g., presence of
other solvents such as acetone and methanol), so that the degradation
rates cannot be directly compared. The most extensively studied site
of intrinsic chlorinated solvent biodegradation is the St Joseph
(Michigan, USA) Superfund site where groundwater concentrations of TCE
as high as 100 mg/litre have been found, with extensive transformation
to cis-DCE, VC and ethene. Conversion of TCE to ethene was most
complete where methane production was highest and where removal of
nitrate and sulfate by reduction was most complete (McCarty, 1996;
Weaver et al., 1996). At another site in the USA (Dover Air Force
Base), half-lives of 1 to 2 years have been estimated for each stage
in the reaction chain (e.g., DCE to VC; VC to ethene) (Ellis et al.,
1996). The degradability of chlorinated aliphatic compounds was
studied under methanogenic conditions in batch reactors with leachate
from eight landfill sites in Denmark. PCE and TCE were found to be
degraded in only three of the eight leachates, with significantly
different conversion rates. In one leachate, complete conversion of
chlorinated ethenes, including conversion of VC, was observed within
40 days, while another leachate showed only 50% conversion of PCE
(Kromann et al., 1998).
No known microorganism can aerobically destroy PCE. Laboratory
studies have shown that some anaerobic bacteria (e.g., Dehalobacter
restrictus ) use chlorinated solvents for respiration
(halorespiration), breaking them down in the process to form
cis-dichloroethene, although restricted diet conditions are
necessary (Sharma & McCarty, 1996). Recently, a coccoid bacterium has
been isolated (provisionally named Dehalococcoides ethenogenes
strain 195) which, together with extracts from mixed microbial
cultures, can dechlorinate PCE, removing further chlorine atoms to
form vinyl chloride and finally ethene (Maymó-Gatell et al., 1997).
Escape of landfill gas from the disposal site can take place via
the surface (emission) or into the ambient soil (migration). VC is
emitted from the landfill surface into the ambient air (Wittsiepe et
al., 1996). Awareness of this problem has encouraged the development
of in situ bioremediation of chlorinated solvents and VC using
anaerobic or aerobic co-metabolic processes (Dolan & McCarty, 1995b;
Jain & Criddle, 1995; Semprini, 1995; see section 4.2).
The estimated emission of vinyl chloride from landfill sites in
the USA is 60-33 000 tonnes/annum (Lahl et al., 1991).
3.2.5.2 VC formation from tobacco
VC was identified in the smoke of all 13 cigarettes tested
(1.3-16 ng/cigarette) and in both small cigars tested (14-27
ng/cigar). The level correlates directly with the chloride content of
the tobacco. Filter tips with charcoal reduce selectively the VC
content of cigarette smoke (Hoffmann et al., 1976).
Table 5. Some examples of formation of VC through biodegradation of tetrachloroethene
in landfill sites (concentrations in mg/litre unless stated otherwise)a
Site Sample PCE TCE cis-DCE VC Ethene Reference
Chemical transfer groundwater 4.4 1.7 5.8 0.22 0.01 Major et al.
factory facility, downgradient well n.d. none 76 9.7 0.42 (1991)
North Toronto,
Canada
Carpet backing groundwater n.d. 56 4.2 0.076 Fiorenza et al.
manufacturing downgradient from n.d. 4.5 5.2 low (1994)
plant, Ontario, lagoon
Canada
Refuse landfills 7.15 5.09 not 5.6 McCarty &
(average of 8) (ppmv) (ppmv) measured (ppmv) Reinhard (1993)
Landfill groundwater 0.54 2.6 2.2 2.7 33 Lee et al. (1995)
well 3.4 14 44 54 43
15 270 140 48 14
Heavily polluted groundwater 20 70 20 2 Middeldorp et
site (solvent al. (1998)
distributor) in
Netherlands
a PCE = tetrachloroethene; TCE = trichloroethene; DCE = dichloroethene; n.d. = not detected
3.3 Uses
About 95% of the world production of VC is used for the
production of PVC. The remainder is used for the production of
chlorinated solvents, primarily 1,1,1-trichloroethane (10 000 tonnes
per year; European Council of Vinyl Manufacturers, 1994), via the more
toxic 1,1,2-trichloroethane and 1,1-dichloroethane.
VC was previously used as a refrigerant (Danziger, 1960) and as a
propellant in aerosol sprays for a variety of products, such as
pesticides, drugs and cosmetics (Wolf et al., 1987). These uses have
been banned since 1974 in the USA and in other countries.
4. ENVIRONMENTAL TRANSPORT, DISTRIBUTION AND TRANSFORMATION
4.1 Transport and distribution between media
Depending on the sources, VC can enter the environment via air,
water or soil. The most critical matrices are probably air and
groundwater. Euro Chlor (1999) calculated the partitioning of VC into
environmental compartments, based upon the Mackay Level 1 model, to be
99.99% air, 0.01% water, < 0.01% soil and < 0.01% sediment.
4.1.1 Air
Owing to its high vapour pressure (saturation vapour pressure
P0 > 10-4 mmHg; see also section 2), VC released to the
atmosphere is expected, based on calculations of Eisenreich et al.
(1981), to exist almost entirely in the vapour phase. The atmospheric
life-time of VC is limited by its reaction with photochemically
produced OH radicals (see section 4.2.2).
VC is volatilized to the atmosphere from area sources such as
landfill sites. Models have been developed to predict such short-range
dispersion. These models were validated by VC concentrations ranging
from < 5 to 31 µg/m3 (< 2 to 12 ppb) measured in the vicinity of a
landfill in Los Angeles 3 years after it last received any waste
containing VC (Chitgopekar et al., 1990).
No data are available on wet deposition.
In order to model distribution processes, liquid : air partition
coefficients have been determined (Gargas et al., 1989). Coefficients
of 0.43 and of 24.4 were obtained for 0.9% saline : air and for olive
oil : air, respectively.
4.1.2 Water and sediments
VC has a relatively low solubility in water (see section 2), and
the solubility can be increased by the presence of salts (see section
4.2.3).
Experimental data on adsorption to particulate matter in the
water column or to sediment are not available. A partition constant
(unitless) of 8.2 for a sediment-water system was calculated (from a
Kow value of 17) by Mabey et al. (1982), indicating a low adsorption
capacity. A high input of VC into water may lead to low-level
long-term storage in the associated sediment (Hill et al., 1976b).
Volatilization of VC acts as a significant transport mechanism.
It is considered to be the most rapid route for removal of VC from
surface water, but to be an unlikely pathway for disappearance from
groundwater that is not directly exposed to air (Smith & Dragun,
1984). Volatilization parameters such as vapour pressure and Henry's
Law constants indicate that VC is highly volatile. Another factor, the
reaeration rate ratio (rate constant for loss of VC from aqueous
solution divided by the rate constant for oxygen uptake by the same
solution), was reported to be 0.675 at 25°C (theoretical calculation
by Mabey et al., 1982) and approximately 2 (experimental measurement
by Hill et al., 1976b). The results of measurements or calculations of
volatilization half-lives of VC from water bodies are given in Table
6; they range from < 1 h to 5 h (measured in models for disturbed or
quiescent water) and from 2.5 to 43 h (calculated for natural water
bodies). In groundwater, however, VC may remain for months or years
(ATSDR, 1990).
More or less complex models have been developed to describe the
stability of VC in aquatic ecosystems (Hill et al., 1976b; Miller,
1992).
4.1.3 Soil and sewage sludge
Owing to its high vapour pressure, VC can be expected to
volatilize rapidly, especially from dry soil surfaces. No experimental
data are available. However, volatilization of VC from soil can be
predicted based upon its physicochemical properties. The amount of VC
volatilized from a soil depth of 1 m in 1 year was reported to range
from 16 to 45% in a sandy soil and 0.1 to 0.7% in a clay soil. These
calculations were based upon a Henry's Law constant of 0.44 and a
degradation half-life of between 30 and 180 days (Jury et al., 1992).
It has been observed at landfill sites that subsurface migration
of VC is a significant transport mechanism (Hodgson et al., 1992;
Little et al., 1992; Ward et al., 1996). Quantitative experimental
data on the potential of VC for gaseous subsurface migration (see
section 5.1.1) were not found.
Because of its solubility in water, VC can be leached through the
soil to groundwater. Additionally, the high solubility of VC in many
organic solvents may increase its mobility at special locations, e.g.,
landfills or waste disposal sites. Standard experimental studies on
soil sorption of VC are lacking. The soil adsorption coefficients of
VC were estimated from its water solubility, octanol/water partition
coefficient and from the molecular topology and quantitative
structure-activity relationship analysis method according to equations
given by Chiou et al. (1979), Kenaga & Goring (1980), Lyman et al.
(1982, 1990) and Sabljic (1984). The Koc values obtained ranged from
14 to 240, indicating a low adsorption tendency and therefore a high
mobility of VC introduced into soil (US EPA , 1985a; Stephens et al.,
1986; ATSDR, 1997).
A field study performed to determine non-extractable (bound)
residues (NER) of highly volatile chlorinated hydrocarbons gave a low
value of 2.4% for VC, as measured in a lysimeter after one growth
period in the upper (10 cm) soil layer (Klein et al., 1989).
VC was assumed not to appear frequently in sewage sludge due to
its low adsorption potential (log Kow < 2.0) and its high
volatilization tendency (Wild & Jones, 1992).
Table 6. Half-lives reported for volatilization of VC from water
Source Method Conditions Half-life Reference
Dilute aqueous experimental 22-25°C, Dilling et al. (1975);
solution (200 ml) (laboratory) rapid continuous 25.8 min Dilling (1977);
stored in an stirring
open container continuous stirring (200 rpm) 26-27.6 min Callahan et al. (1979)
discontinuous stirring approx. 80-90
(5% of time) min
quiescent, no stirring 290 min
Flowing channel experimental water input: 35 litre/second 0.9 h Scherb (1978)
(field) flow velocity: < 0.50 m/second
depth: 30 cm
Stream calculation based on Hc = 243 kPa.m3/mol 2.5 h Lyman et al. (1990)
depth: 1 m
flow velocity: 1 m/second
wind current: 3 m/second
calculation based on reaeration rate ratio of US EPA (1985a)
2; assumed oxygen reaeration ratesa:
Pond 0.008 h-1 43.3 h
River 0.04 h-1 8.7 h
Lake 0.01 h-1 34.7 h
a According to Tsivoglou (1967); Lyman et al. (1990)
4.1.4 Biota
VC has been identified in environmental samples of fish tissue
(see section 5.1.5) and in several species of aquatic laboratory
animals (molluscs, crustaceans, insects and vertebrates) and algae
experimentally exposed to VC-containing water (see section 4.3).
Reports on the presence of VC in terrestrial plants and animals have
not been found in the literature.
Further experimental data, e.g., in what way and to what extent
VC is capable of entering the biota, are lacking. However, a few
estimations have been made on the basis of the physicochemical
properties of VC. They refer to uptake of VC by terrestrial plants and
animals. Plant uptake was considered to be unlikely (Ryan et al.,
1988; Shimp et al., 1993) because, at an assumed half-life of
< 10 days (Ryan et al., 1988), VC should be lost from the system
rather than be taken up by the plant. Another study (Wild & Jones,
1992) screened organic contaminants for possible transfers into plants
and animals by summarizing approach data. Within the three categories
used (high, moderate, low potential) VC was classified in the
following way:
* retention by root surface: low;
* uptake and translocation: moderate;
* foliar uptake: high;
* transfer to animal tissues by soil ingestion: low;
* transfer to animal tissues by foliage ingestion: moderate.
4.2 Transformation
4.2.1 Microbial degradation
There have been many studies on the biodegradation of VC under
various simulated environmental conditions and these are listed in
Tables 8 to 10. The biodegradation studies have given contradictory
results, with no evidence of degradation under some aerobic conditions
such as surface water (Hill et al., 1976b) and sewage (Helfgott et
al., 1977) and under anaerobic conditions such as groundwater
(Barrio-Lage et al., 1990). Unacclimated biodegradation half-lives of
VC were generally estimated to be of the order of several months or
years (Howard et al., 1991). However, other studies reported complete
degradation in 3 months in simulated aerobic groundwater (Davis &
Carpenter, 1990). Where degradation is reported care must be taken to
ensure that the loss of VC is due to degradation and not from other
losses such as volatilization from the test system. Phelps et al.
(1991b) reported that > 99% of VC was lost from a bioactive reactor
compared to 60% from the control reactor.
Significant microbial degradation of VC under aerobic and
anaerobic conditions has been detected in studies using enrichment or
pure cultures isolated mostly from sites contaminated with different
organic chemicals (Tables 8 to 10). Vinyl chloride cannot be used by
most microorganisms as sole carbon source, but it can be
degraded/metabolized in the presence of propane, methanol,
3-chloropropanol, propylene, isopropene and glucose. However, in some
cases VC can even serve as sole substrate, as is seen with
Mycobacterium sp. (Table 9). The main degradation products include
glycolic acid or CO2 after aerobic conversion (Tables 7 to 9) and
ethane, ethene, methane or chloromethane after anaerobic
transformation (Table 10). Anaerobic mineralization of VC to CO2 has
been demonstrated (Table 10) and may occur under special conditions
(Bradley & Chapelle, 1998b).
A complete mass balance was given by degradation studies with
radiolabelled VC, for example, in aerobic resting cell suspensions of
Rhodococcus sp. A starting concentration of 1 mg [1,2-14C] VC
produced more than 66% 14CO2 and 20% 14C aqueous phase products, and
10% was incorporated into the biomass (Malachowsky et al., 1994).
Aerobic cultures of Mycobacterium aurum growing on a special filter
material were reported to mineralize VC quantitatively according to
the following equation:
VC + microorganisms -> biomass + HCl + CO2 + H2O (Meier, 1994).
The underlying reaction mechanisms for the aerobic and anaerobic
degradation of VC have been postulated to be oxidative and reductive
dehalogenations, respectively, involving a variety of pathways (Vogel
et al., 1987; Barrio-Lage et al., 1990; Ensley, 1991; Castro et al.,
1992a; Leisinger, 1992; Castro, 1993; Meier, 1994; Hartmans, 1995;
Jain & Criddle, 1995).
Frequently, the degradation reaction of VC proceeds more readily
with aerobes than with anaerobes (Tables 7 to 10). The reverse occurs
(Freedman & Gossett, 1989; Semprini et al., 1995) with PCE, an
important precursor of VC in the environment (see chapter 3). Thus,
two-stage treatment systems consisting of anaerobic (first stage) and
aerobic (second stage) cultures have been proposed to achieve the
complete degradation of a range of alkenes having different degrees of
chlorination (Leisinger, 1992; Murray & Richardson, 1993; Nelson &
Jewell, 1993). Recently, a chlorobenzoate-enriched biofilm reactor
using Desulfomonile tiedjei DCB-1 was developed which degraded PCE
under anaerobic conditions without any detectable VC remaining
(Fathepure & Tiedje, 1994).
There have been many efforts to use the VC-degrading capacities
of microorganisms in practical applications such as the purification
of waste gases (Meier, 1994) or of municipal waste waters (Narayanan
et al., 1995) and the remediation of landfill leachates (Lesage et
al., 1993), groundwaters (McCarty, 1993; Holliger, 1995) and
contaminated soils (Schulz-Berendt, 1993). Possible limitations arise
from physical (temperature, accessibility of substrate), chemical (pH,
redox state, concentration of VC and other contaminants, presence of
additional secondary substrates, salinity) and biological (presence of
predators, competition phenomena, adsorption of microbes to surfaces)
factors (Van der Meer et al., 1992).
Table 7. Aerobic degradation of vinyl chloride by mixed microbial consortia from different sites
Inoculum Test design/ Measured Initial Duration Efficiency of Reference
conditionsa parameter concentration degradation
Surface water room VC 20 ml/2.9 ml 41 h no degradation Hill et al. (1976b)
samples temperature
Mixed consortium 21°C; VC 20-120 mg/litre several no degradaion Hill et al. (1976b)
from natural + / - nutrients weeks
aquatic systems
Mixed consortium 20°C oxygen 25 days no degradation Helfgott et al.
from domestic + nutrients demand (1977)
sewage
Mixed consortium 25°C; 14CO2 0.05 mg/litre 5 days 21.5% degradation Freitag et al.
from activated + nutrients (1982, 1985)
municipal sewage
sludge
Naturally simulated 14CO2, 1 mg/kg 108 days > 99% degradation Davis &
occurring aquifers: VC soil-water 65% mineralization Carpenter
consortium from soil-water (CO2) (1990)
groundwater microcosms 0.1 mg/kg 109 days 50% mineralization
(prepared with soil-water (CO2)
sub-surface soil
and groundwater
(20°C)
Consortium aquifer sediment 14CO2, 17 µmol/litre 84 h 22-39% Bradley &
indigenous to microcosms VC mineralization Chapelle (1996)
anaerobic aquifer (CO2)
systems
(contaminated
with CHs)
Table 7. (cont'd)
Inoculum Test design/ Measured Initial Duration Efficiency of Reference
conditionsa parameter concentration degradation
Consortium creek bed sediment 14CO2, 0.2-57 µmol/litre 24 h 6.2-58% Bradley &
indigenous to microcosms VC mineralization Chapelle (1998a)
creek sediment (CO2)
(contaminated
with DCE)
Consortium from soil microcosms VC 5.3 mg/litre 95 h little change Dolan & McCarty
aquifer material in VC (1995a)
(from a concentration
VC-contaminated
site)
a CH = chlorinated hydrocarbons; DCE = dichloroethene
Table 8. Elimination of vinyl chloride in aerobic tests with mixed microbial consortia utilizing special substratesa
Inoculum Additional Test Efficiency of Remarks Reference
substrate degradation
Mixed methane laboratory removal of up inhibition by Fogel et al. (1986,
methanotrophs studies to 100% within methane and 1,1-DCE 1987); Strandberg et al.
4 h-30 days possible; toxic (1989); Uchiyama et al.
effects of VC and VC (1989); Nelson & Jewell
products possible (1993); Dolan & McCarty
(1995a); Chang & Alvarez-Cohen
(1996)
methane field study about 95% inhibition by Semprini et al. (1990, 1991)
(groundwater) in-situ methane possible
transformation
i.c. =
0.03 mg/litre
Mixed propane laboratory > 99% loss after > 60% loss in Phelps et al. (1991b);
microbial study 30 days control Lackey et al. (1994)
consortia i.c. =
4-20 mg/litre
methane laboratory 82 to > 99% Phelps et al. (1991b);
plus study loss after 10-21 Lackey et al. (1994)
propane days; i.c. =
1-20 mg/litre
Mixed methane laboratory 2.3 µmol VC per relative TCs from Dolan & McCarty (1995a)
methanotrophs plus study mg of cells during highest to lowest:
formate 26 h i.c. = trans-DCE;
14.8 mg/litre cis-DCE; VC;
TCE; 1.1-DCE
Table 8. (cont'd)
Inoculum Additional Test Efficiency of Remarks Reference
substrate degradation
Mixed methane laboratory removal of up to Deipser (1998)
consortia study > 99% within 15
days i.c. =
0.55 mg/litre
a i.c.= Initial concentration; DCE = dichloroethene; TCE = trichloroethene;
TC = transformation capacity
Table 9. Survey on isolated bacterial cultures capable of degrading vinyl chloride under aerobic conditions
Inoculum Additional Major degradation Remarks Reference
substrate producta
Mixed culture n. sp. CO2 (> 67%) Malachowsky et al. (1991)
consisting of
Rhodococcus rhodochrous
and 2 bacteria of the
order Actinomycetales
Bacterium of the order propane, CO2 (> 67%) Phelps et al. (1991a)
Actinomycetales glucose or
acetate
Alcaligenes denitrificans isoprene n. sp. Ewers et al. (1990)
ssp. Xylosoxidans
Methylosinus trichosporium methane n. sp. inactivation Tsien et al. (1989); Chang
OB3b possible & Alvarez-Cohen (1996)
methane glycolic Castro et al. (1992a)
acid (44%;
determined at
68% conversion)
Mycobacterium sp. ; no CO2 ; initially: VC as primary Hartmans et al. (1985, 1992);
M. aurum chlorooxirane substrate; Hartmans & DeBont (1992);
(epoxide) inhibition Meier (1994)
possible
Mycobacterium no CO2, HCl maximum growth Hauschild et al. (1994)
aurum L1 rates at
1 mmol
VC/litre
Mycobacterium vaccae propane n. sp. Wackett et al. (1989)
(JOB 5)
Table 9. (cont'd)
Inoculum Additional Major degradation Remarks Reference
substrate producta
Nitrosomonas europaea ammonia n. sp. Vannelli et al. (1990)
Pseudomonas sp. 3-chloro-propanol glycolic acid Castro et al. (1992b)
(71%; determined
at 25%
conversion)
Rhodococcus sp. propane CO2 (> 66%) Malachowsky et al. (1994)
Rhodococcus isoprene n. sp. Ewers et al. (1990)
erythropolis
Xanthobacter propylene n. sp. Ensign et al. (1992)
(strain Py 2)
a The degradation efficiency is given in parentheses; n. sp. = not specified
Table 10. Survey on anaerobic microbial degradation of vinyl chloridea
Inoculum Test design/ Conditions Major degradation Efficiency of Remarks Reference
products degradation
Mixed methanogenic liquid cultures n. sp. i.c. = 400 µg/litre Brauch et al.
consortium (from (20°C) groundwater (1987)
PCE-, TCE-, with sterile sand approx. 50% (100%)
VC-contaminated after 4 (11) weeks
groundwater)
without sterile sand approx. 20% (55%)
after 4 (11) weeks
Mixed consortium incubation of n.sp. 94% within 16 days Nerger &
(from groundwater plus i.c. = 18 µg/litre Mergler-Völkl
TCE-contaminated waste water (1988)
water) (9 : 1)( 21°C)
Mixed consortium digesters filled n.sp. low degradation Deipser (1998)
(from compost) with mature sieved (0.2 mg/m3 compost/h)
compost from private
households
Mixed consortium soil-groundwater (< 1% CO2) no degradation in 5 Barrio-Lage et
(from natural microcosms (25°C) months al. (1990)
sites) i.c. = 2 mg/litre
Mixed consortium flow-through column methane plus 89% degradation traces of Barrio-Lage et
(from natural packed with soil, ethene (82%), (in 9-15 days) chloromethane al. (1990)
sites) a. s.: mixture of CO2 (7%)
phenol, citrate,
ammonium
dihydrogenphosphate,
methanol and methane
Table 10. (cont'd)
Inoculum Test design/ Conditions Major degradation Efficiency of Remarks Reference
products degradation
Consortium aquifer sediment CO2 15-34% mineralization Bradley &
indigenous microcosms, anaerobic in 84 h (versus 3-5% Chapelle (1996)
to anaerobic conditions plus without Fe-EDTA
aquifer systems Fe(III) as Fe-EDTA amendment)
(contaminated i.c. = 17 µmol/litre
with CHs)
Mixed varying conditions chloroethane slow degradation Baek et al.
methanogenic or 15-30°C or ethene (1990); Carter &
methanol-enriched (plus ethane) Jewell (1993);
consortia Skeen et al.
(1995)
Enriched PCE- and batch cultures ethene partial to nearly inhibition by Freedman &
TCE-degrading (35°C) complete degradation PCE possible Gossett (1989);
consortia DiStefano et al.
(1991); Tandoi
et al. (1994)
Mixed anaerobic fixed-bed columnb ethene plus almost complete De Bruin et al.
consortia from 24°C a.s.: ethane conversion of (1992)
river sediment lactate PCE (95-98%)
and wastewater via VC
sludge
Methanobacterium resting cell no degradation Castro et al.
thermoautotrophicum suspensions (60°C) i.c. = 10-3 (1994)
mol/litre
Table 10. (cont'd)
Inoculum Test design/ Conditions Major degradation Efficiency of Remarks Reference
products degradation
"Dehalococcoides anaerobic H2-PCEb ethene 90% conversion decay in rate Maymó-Gatell
ethenogenes strain enrichment culture of PCE via VC of VC et al. (1995,
195"c + mixed conversion 1997)
microbial consortia
a a.s. = additional substrate; i.c. = initial concentration; n.sp. = not specified; CH = chlorinated hydrocarbons;
PCE = tetrachloroethene; TCE = trichloroethene
b starting material PCE
c preliminary name
4.2.2 Abiotic degradation
4.2.2.1 Photodegradation
Studies on the photodegradation of VC are summarized in Table 11.
They include direct and indirect photolysis.
VC in the vapour phase or in water does not absorb wavelengths
above 220 nm or 218 nm, respectively (Hill et al., 1976b). However,
solar radiation reaching the troposphere lacks wavelengths below about
290 nm due to the stratospheric ozone shield. So, direct photolysis of
VC is expected to be insignificant under environmental conditions,
because there is no overlap between the absorption spectrum of VC and
the sunlight radiation spectrum (Callahan et al., 1979). Consistently,
no photodegradation was observed with pure VC in the gas phase or in
water at wavelengths above 220 nm. After irradiation at 185 nm, VC was
photolysed (Table 11).
In the environment, indirect photolysis occurs and includes
reactions of VC in the presence of photosensitizers and those
(Table 12) with photochemically produced reactive particles.
A variety of photolytic products was formed after irradiation of
VC under several experimental conditions (Table 11). Some
intermediates, e.g., chloroacetaldehyde, were of considerable
photochemical stability. Therefore, the photooxidation is unsuitable
as a means of removing VC from waste gases (Gürtler et al., 1994). On
the other hand, treatment of water contaminated with VC and other
halogenated organic compounds by means of UV-enhanced oxidation
(UV/ozone or UV/hydrogen peroxide) was reported to be successful (Zeff
& Barich, 1992).
The atmospheric fate of VC depends on its reaction with reactive
particles such as free OH and NO3 radicals, Cl atoms, ozone and
singlet oxygen. As can be seen from Table 12, the reaction with OH
radicals is the dominant transformation process, showing calculated
tropospheric half-lives of 1-2 days or more. Factors influencing
indirectly (via OH radical concentration) the lifetime of VC are the
degree of air pollution and solar radiation, leading to spatial,
diurnal and seasonal variations (e.g., Hesstvedt et al., 1976).
Reaction products include formaldehyde (HCHO) and formyl chloride
(HCOCl), the latter being a stable potential toxicant (Tuazon et al.,
1988; Pitts, 1993).
Rate constants for the photodegradation of VC in aqueous
solutions have been reported to range from 6.99 × 109 mol-1 second-1
(Grosjean & Williams, 1992) to 7.1 × 109 mol-1 second-1 (Klöpffer et
al., 1985). Mabey et al. (1982) reported that photolysis of VC was not
an environmentally relevant process. They reported oxidation rate
constants of < 108 mol -1h -1 and 3 mol -1h -1 for reactions with
singlet oxygen (1O2) and peroxy radicals (RO2), respectively. On
the basis of an average OH radical concentration of 10-17 mol in
Table 11. Survey on vinyl chloride photolysis studies
Medium Irradiation Photolytic degradation Photolytic productsa Reference
(Duration)
VC in a high-vacuum medium pressure yes primary products: radicals Fujimoto et al. (1970)
system arc (C2H3, Cl); C2H2, HCl
VC in air sunlight (outdoors) yes n.sp. Pearson & McConnell
(half-life = 11 weeks ± 50%) (1975)
xenon arc (> 290 nm) yes CO (90%); HCl
VC in air high pressure yes (> 99% within 15 min) chloroacetaldehyde, Kagiya et al. (1975)
mercury lamp HCl, CO2
outdoors yes (55% within 2 days)
VC in air (dry) > 230 nm (4 h) yes chloroacetaldehyde (primary Müller & Korte (1977)
product), HCl, CO, formyl
chloride
VC in air < 400 nm (45 min) yes CO2, CO, H2O, HCl, HCOOH, Woldbaek & Klaboe
C2H2 (1978)
sunshine (3-45 h) very slow
VC (adsorbed on > 290 nm (n.sp.) yes n.sp. Freitag et al. (1985)
silica gel) (15.3% of applied amount)
VC in an oxygen 185 nm (up to 50 min) yes formyl chloride, Gürtler et al. (1994)
atmosphereb (quantum yield: 2-3) monochloroacetaldehyde,
acetylene, CO, CO2,
monochloroacetyl chloride,
HClc, formic acidc
254 nm (up to 6 h) no
VC in air xenon lamp yes n.sp. Haag et al. (1996)
(initial k: 0.09 second-1)
Table 11. (cont'd)
Medium Irradiation Photolytic degradation Photolytic productsa Reference
(Duration)
VC in air plus UV (> 290 nm) yes formic acid, HCl, CO, Cox et al. (1974);
nitrogen oxides (up to 22 h) (half-life = 1-7 h, + NO formaldehyde, ozone, Dilling et al. (1976);
half-life = 18 h, - NO) (other minor products) Gay et al. (1976);
Carassiti et al.
xenon lamp yes formaldehyde, Hcl (1977) Kanno et al.
(0-120 min) (1977)
< 400 nm (25 min) yes at low NO2 conc.: the same Woldbaek & Klaboe
(increase in reaction rate products as observed in air (1978)
as compared to NO2/NO (see above);
being absent) at high NO2 conc.:
additionally nitrosyl
chloride, N2O
VC in air plus xenon lamp yes n.sp. Haag et al. (1996)
1,1-DCE (initial k: 0.15 second-1)
VC in pure water > 300 nm no Hill et al. (1976b)
(10 mg/litre) (90 h)
VC in natural water > 300 nm no
samples (20 h)
(10 mg/litre)
VC in water plus > 300 nm yes (rapid) various products
photosensitizers
VC in PVC plant > 300 nm yes (half-life = 40 h) n.sp.
effluent sunlight (25 h) very little
a n.sp. = not specified; DCE = dichloroethene
b direct photolysis
c in the presence of water vapour
Table 12. Rate constants and half-lives for gas-phase reactions of vinyl chloride with OH radicals and other reactive particles
VC reaction Rate constant Temperature Assumed atmospheric Calculated Reference
with a (in units of (°C) c concentration of the half-life c,d
cm3/molecule-sec) b reactive particle c
* OH 5.6 × 10-12 27 1 × 106 molecules/cm3 1.4 days Cox et al. (1974);
(measured) US EPA (1985a)
* OH 4.5 × 10-12 23 (296 K) 1 × 106 molecules/cm3 1.8 days Howard (1976); US
(measured) EPA (1985a)
* OH 6.60 × 10-12 26 (299 K) n.sp. n.sp. Perry et al. (1977)
5.01 × 10-12 85 (358 K)
3.95 × 10-12 149 (423K)
(measured)
* OH 6.60 × 10-12 26 1 × 106 molecules/cm3 1.2 days US EPA (1985a)
(Perry et al., 1977)
* OH 6.60 × 10-12 room temperature 1 × 106 molecules/cm3 (3.5 days) Atkinson et al.
(Perry et al., 1977) (298 ± 2 K) (12-h daytime average, (1979, 1987);
Crutzen, 1982) Atkinson (1985)
* OH 6.60 × 10-12 room temperature n.sp. n.sp. Atkinson (1987)
(measured)
5.3 × 10-12
(calculated)
* OH 6.60 × 10-12 25 ± 2 5 × 105 molecules/cm3 (approx 3 days) Tuazon et al. (1988)
(Perry et al., 1977) (298 ± 2 K)
* OH 6.60 × 10-12 26 5 × 105 molecules/cm3 2.2-2.7 days BUA (1989)
(Perry et al., 1977) (Crutzen, 1982)
* OH 6.8 × 10-12 27 (300 K) 5 × 105 molecules/cm3 approx 2.4 days BUA (1989)
(Becker et al., 1984) (Crutzen, 1982)
Table 12. (cont'd)
VC reaction Rate constant Temperature Assumed atmospheric Calculated Reference
with a (in units of (°C) c concentration of the half-life c,d
cm3/molecule-sec) b reactive particle c
* OH 6.60 × 10-12 26 8 × 105 molecules/cm3 1.5 days Howard (1989)
(Perry et al., 1977)
* OH 4.0 × 10-12 26 (299 K) n.sp. n.sp. Kirchner et al.
cm3/mol-sec (1990)
(measured)
* OH 10.6 × 10-12 n.sp. n.sp. n.sp. Klamt (1993)
(calculated)
* OH n.sp. n.sp. 1 × 106 molecules/cm3 (42 h) Pitts (1993)
(24-h average)
* OH 6.60 × 10-12 n.sp. 6.5 × 105 molecules/cm3 (2.7 days) Helmig et al.
(Perry et al., 1977) (estimated global mean; (1996)
Tie et al., 1992)
* OH 6.60 × 10-12 26 7.5 × 105 molecules/cm3 1.6 days Palm (1997)
(Perry et al., 1977) (24-h average, BUA, 1993) personal
communication
* NO3 2.3 × 10-16 room temperature 2.4 × 109 molecules/cm3 (42 days) Atkinson et al.
(measured) (298 ± 2 K) (12-h nighttime average, (1987)
Platt et al., 1984)
* NO3 1.4 × 10-16 23 (296 ± 1 K) n.sp. n.sp. Andersson &
(measured) Ljungström (1989)
Cl 12.7 × 10-11 25 (298 ± 2 K) n.sp. n.sp. Atkinson & Aschmann
(measured) (1987); Grosjean &
Williams (1992)
Table 12. (cont'd)
VC reaction Rate constant Temperature Assumed atmospheric Calculated Reference
with a (in units of (°C) c concentration of the half-life c,d
cm3/molecule-sec) b reactive particle c
O3 1.9 × 10-19 25 n.sp. n.sp. Gay et al. (1976);
Singh et al. (1984)
O3 2.0 × 10-18 n.sp. n.sp. 4 days Hendry & Kenley
(1979);
ECETOC (1983)
O3 2.45 (± 0.45) × 10-19 room temperature n.sp. n.sp. Zhang et al. (1983)
(approx. 25)
O3 2.5 × 10-19 25 7 × 1011 molecules/cm3 (66 days) Atkinson & Carter
(Zhang et al., 1983) (Singh et al., 1978) (1984); Atkinson et
al. (1987)
O3 2.45 × 10-19 25 1 × 1012 molecules/cm3 33 days US EPA (1985a)
(Zhang et al., 1983)
O3 1.2 × 106 27 1.6 × 1012 molecules/cm3 4.2 days Lyman et al. (1982,
cm3/mole-sec 1990); US EPA (1985a)
O3 1.7 × 10-19 22 7 × 1011 molecules/cm3 67 days Klöpffer et al.
(1988)
O3 2.5 × 10-19 (calculated n.sp. n.sp. n.sp. Meylan & Howard
according to AOP) (1993)
19 × 10-19 (calculated n.sp. n.sp. n.sp.
according to FAP)
O (3P) 8.6 × 10 -13 25 2.5 × 104 molecules/cm3 373 days Sanhueza & Heicklen
(1975); US EPA
(1985a)
Table 12. (cont'd)
VC reaction Rate constant Temperature Assumed atmospheric Calculated Reference
with a (in units of (°C) c concentration of the half-life c,d
cm3/molecule-sec) b reactive particle c
O (3P) 5.98 × 10 -13 25 2.5 × 104 molecules/cm3 532 days Atkinson & Pitts
(1977); US EPA
(1985a)
a O(3P) = oxygen atom
b AOP = Atmospheric Oxidation Program (currently used by US EPA); FAP = Fate of Atmospheric Pollutants (part of US EPA's Graphical
Exposure Modeling system, GEMS)
c n.sp. = not specified
d Values in parentheses reported as 'lifetime'
natural water, the kOH rate constant resulted in a half-life of
approximately 110 days (US EPA, 1985a). Both of the other reactions
appeared to be negligible.
4.2.2.2 Hydrolysis
Observations on chemical hydrolysis of VC derive from experiments
with effluent water from a VC plant (pH = 4.3-9.4; 50°C; 57 h;
Callahan et al., 1979), water of different pH values (85°C; 27 h; Hill
et al., 1976b; Mabey et al., 1982), water saturated with O2 (85°C;
12 h; Hill et al., 1976b), water plus ethanol (120°C; Rappoport & Gal,
1969) and two natural water samples (pH = 6.1/4.2 for river/swamp
water, both at room temperature and at 85°C; 41 h; Hill et al.,
1976b). In all cases, no or only slow hydrolysis occurred. The
hydrolytic half-life was estimated to be < 10 years at 25°C (Hill et
al., 1976b). Hydrolysis experiments under strongly alkaline, high
temperature (and therefore environmentally irrelevant) conditions
resulted in a polymerization of VC (Jeffers & Wolfe, 1996).
4.2.3 Other interactions
Under experimental conditions, VC and chlorine in water form
chloroacetaldehyde, chloroacetic acid and other unidentified compounds
(Ando & Sayato, 1984).
Many salts have the ability to form complexes with VC, thus
possibly leading to an increase in its solubility (Callahan et al.,
1979).
4.3 Bioaccumulation
Owing to its high vapour pressure and low octanol/water partition
coefficient, VC is expected to have little tendency for
bioaccumulation (BUA, 1989). Theoretical calculations resulted in
bioconcentration factors (BCFs) of 2.8 (based on a log Kow of
approximately 0.9) and around 7 (based on a water solubility of
2763 mg/litre) in aquatic organisms (US EPA, 1985a). A BCF of 5.7
(based on log Kow = 1.23) was calculated by Mabey et al. (1982) for
aquatic microorganisms.
Experiments performed with 14C-VC (initial concentration:
250 µg/litre) in a closed laboratory model aquatic ecosystem gave the
following results: after 3 days at 26.7°C 34% of the 14C was found in
the water and 65% in the air. The organisms from different trophic
levels contained 14C residues (in VC equivalents, µg/kg) of 1307
(alga, Oedogonium cardiacum), 621 (waterflea, Daphnia magna), 123
(snail, Physa sp.), 1196 (mosquito larva, Culex pipiens
quinquefasciatus) and 312 (fish, Gambusia affinis) (Lu et al.,
1977). These values led to BCFs ranging from 3 to 31 when compared to
the VC water concentration of 42 µg/litre, indicating some
bioaccumulation, but no biomagnification within the food chain.
Another study determined BCFs for green algae (Chlorella fusca)
after a 24-h exposure to 0.05 mg VC/litre and for fish (golden ide,
Leuciscus idus melanotus) exposed to a constant average
concentration of 0.05 mg VC/litre over 3 days. BCFs of 40 and < 10,
respectively, were found (Freitag et al., 1985).
After 5 days of incubation of 0.05 mg VC/litre in activated
sewage sludge, an accumulation factor of 1100 (based on the
distribution of VC between sludge, dry weight and water) was observed
(Freitag et al., 1985).
4.4 Ultimate fate following use
4.4.1 Waste disposal
Several methods have been employed for removal of VC from waste
water: stripping with air, steam or inert gas (Nathan, 1978;
Cocciarini & Campaña, 1992; Hwang et al., 1992), extraction (Nathan,
1978) or adsorption onto activated charcoal or adsorbent resin
(Nathan, 1978; Dummer & Schmidhammer, 1983, 1984).
Like VC waste gases produced during other processes (see
chapter 3), the recovered VC can be recycled or incinerated (US EPA,
1982; BUA, 1989). Special biological filters have been developed for
degrading VC in waste emissions (Meier, 1994, 1996; see also section
4.2.1).
Incineration leading to the total destruction of VC requires
temperatures ranging from 450°C to 1600°C and residence times of
seconds for gases and liquids, or hours for solids (HSDB, 1995).
Photochemical oxidations (Topudurti, 1992; Zeff & Barich, 1992;
Berman & Dong, 1994; see also section 4.2.2) are further methods of VC
elimination. UV-enhanced oxidation (oxidants used: ozone, hydrogen
peroxide) was applied for purification of polluted waters, (e.g.,
waste, leachate, groundwater) (Zeff & Barich, 1992). Treatment with
sodium dichromate in concentrated sulfuric acid was recommended for
the destruction of small quantities of VC, for instance, from
experimental laboratories (HSDB, 1995).
Chlorinated volatile organic compounds (VOCs) can be removed from
drinking-water/groundwater by treatment with activated charcoal
(Schippert, 1987), air stripping (after water is pumped to the
surface) (Boyden et al., 1992) or by air sparging (applied in situ)
(Pankow et al., 1993). Recently, on-site and in situ bioremediation
techniques, which couple evaporative or other methods with microbial
treatment, have been developed for restoration of groundwater systems
(Roberts et al., 1989; Portier et al., 1992, 1993; Fredrickson et al.,
1993; McCarty, 1993; Lackey et al., 1994) or soils (Schulz-Berendt,
1993) contaminated with VC and other VOCs.
4.4.2 Fate of VC processed to PVC
Most of the VC produced is used for the manufacture of PVC (see
section 3) and will therefore be connected with the fate of PVC. PVC
and articles made from it can be disposed of in landfills,
incineration or feedstock recycling. While rigid PVC is an extremely
persistent material, flexible PVC may be less recalcitrant to
disintegration (Harris & Sarvadi, 1994). At incineration, PVC plastics
do not depolymerize to form VC (Harris & Sarvadi, 1994), but produce
volatile aliphatic hydrocarbons and volatile chlorinated organic
compounds (Nishikawa et al., 1992; see also section 3.2.4). There is
evidence for formation of PCDFs/PCDDs (Theisen et al., 1989, 1991;
IPCS, 1989).
5. ENVIRONMENTAL LEVELS AND HUMAN EXPOSURE
5.1 Environmental levels
There is very little exposure of the general population to VC.
Concentrations of VC in ambient air are low, usually less than
3 µg/m3. Exposure of the general population may be higher in
situations where large amounts of VC are accidentally released to the
environment, such as a spill during transportation. However, such
exposure is likely to be transient. Near VC/PVC industry and waste
disposal sites, relatively much higher concentrations, up to
8000 µg/m3 and 100 µg/m3, respectively, have been observed. VC has
only rarely been detected in surface waters, sediment or sewage
sludges. Maximal VC concentrations in groundwater or leachate from
areas contaminated with chlorinated hydrocarbons amount to 60 000
µg/litre.
5.1.1 Air
5.1.1.1 Outdoor air
Atmospheric air levels of VC in rural/remote and suburban/urban
areas range from not detectable to 24 µg/m3 (Table 13). Higher values
were recorded in industrial areas, with maxima in the vicinity of
VC/PVC producing or processing plants, even at distances of 5 km. Peak
concentrations were as high as 86 mg/m3 (33 ppm) and 17 mg/m3 (7
ppm), measured, respectively, in 1974 in the USA and 1983 in China
(Table 13). In many countries, VC concentrations near plants have
decreased with time due to regulatory measures (Table 13).
In more recent years attention has been paid to the occurrence of
VC near waste disposal sites, where levels of up to 0.18 mg/m3
(70 ppb) have been detected (Table 13). This is much less than the
significant amounts of VC found in undiluted landfill gas (see
section 3). Bruckmann & Mülder (1982) assumed that gas discharges of
landfills are diluted by a factor of 104 when entering the
atmosphere.
VC emissions of < 0.5 µg/m3 (remote from VC plants) or of a few
µg/m3 (near VC plants) in Germany and the Netherlands were derived
from a computer model (Besemer et al., 1984; WHO, 1987; LAI, 1992).
Ambient air concentrations in three geographical areas of the USA were
computed to range from trace to about 3 µg/m3 (Pellizzari et al.,
1979).
Table 13. Vinyl chloride measured in ambient (atmospheric) aira
Country; site and Valueb Concentrations Reference
year of sampling (µg/m3)
Rural/remote areas single 0.1 Kruschel et
Canada: pine forest al. (1994)
near Barrie, Ontario
year n. sp.
Germany: Westerland, mean values 6.6-24 Bauer
Schwarzwald, (1981)
Lüneburger Heide,
Bayer Wald; prior to
1977
Germany: Taunus; mean value 0.1 Dulson
1975 (n = 4) (1978)
USA: rural Northwest (n = n. sp.) n.d. (< 0.013) Grimsrud &
USA (Pullman, Rasmussen
Washington); (1975)
1974-1975
USA: over ocean (n = n. sp.) n.d. (< 26) Lillian et
Sandy Hook, NJ al. (1975)
(3 miles offshore);
1974
Suburban/urban areas
Canada: urban (22) meanc 0.06 Dann &
and rural (1) sites; (n = 1370; 4% Wang (1992)
1989-1990 > detection)
Germany: Berlin range (n=78) n.d. (n.sp.) - 3.5 Dulson
(3 sites); 1977 means 0.3-0.4 (1978);
Lahmann
(1980)
Germany: Frankfurt/M. n = 1 2.6 Bergert &
(city); 1974 Betz (1976)
Germany: Frankfurt/M. mean (n = 16) 21.1 Arendt et
(suburban); 1975 al. (1977)
Germany: Merkenich annual mean 3.6 German Expert
(North-Rhine Group on the
Westfalia); 1980 Environment
(1988)
Table 13. (cont'd)
Country; site and Valueb Concentrations Reference
year of sampling (µg/m3)
Germany: Cologne means 0.5-15.3 Anon (1981);
(3 stations); (n = n.sp.) BUA (1989)
1979-1986
Germany: several mean values 0.2-11 Bouscaren
sites; year n. sp. et al. (1987)
Germany: Hamburg annual means 2.4-9.0 (including Bruckmann
(12 sites in the city); (n = 300) 1-butene) et al. (1988)
1986-1987
USA: New Jersey geom. mean 1981: 0 Harkov et
(3 urban sites: (3/113)d and 1982: 0 al. (1983,
Newark, Elizabeth, (0/105)d (detection limit: 1984)
Camden); 1981-1982 0.013)
USA (eastern): urban daily mean 0.62 Dann &
sites; 1989 (n=397; 2% Wang (1992)
> detection)
Industrial sites
Canada: Shawinigan range n.d. - 117 Thériault
(vicinity of VC et al. (1983)
polymerization
plant); year n. sp.
China: dormitories for n = 16×3 Zhao et al.
workers (50, 100 and (4 h/day for (1994)
1000 m from a VC 4 days at
polymerization plant) 3 stations)
1983 maxima 17 400 / 5900 / 1500
daily means 4810 / 1080 / 580
1984 maxima 7800 / 2500 / 1400
daily means 4080 / 720 / 500
1986 maxima 6600 / 2000 / 1100
daily means 3120 / 820 / 520
1988 maxima 12 700 / 1000 / 300
daily means 4430 / 320 / 200
1989 maxima 3100 / 700 / 300
daily means 760 / 220 / 170
Table 13. (cont'd)
Country; site and Valueb Concentrations Reference
year of sampling (µg/m3)
Finland: 1.2 km from Kinnunen
a PVC plant (1996)
1993 range of 0.1-1.3
monthly means
1994 range of 0.1-0.8
monthly means
1995 range of < 0.1-0.1
monthly means
range of single 0.1-11
measurements
(n approx. 4000)
1996 range of 0.1-0.4 Kinnunen
monthly means (1997)
range of single 0.1-13
measurements
(n approx. 7000)
Germany: n.sp.; maximum 252 Bouscaren
prior to 1980 et al. (1987)
Germany: Ruhrgebiet 99th percentile 69 BUA (1989)
West; year n. sp.
Germany: 1978-1982 max. (n = n.sp.) 113 Bauer (1981)
Germany: Marl (80 m mean (n = 24) 213 Rohrschneider
and 500 m above mean (n = 9) 108 et al. (1971)
chemical plant);1970
Germany: Frankfurt/M. n.sp. 4.5 Atri (1985)
(industrial area);
year n.sp.
Netherlands: > 600 m maximum ca. 600 Besemer
from VC plant; mean 210 et al.
1976-1977 (1984)
0-500 m distant range (n = 200) < 21-504
500 m distant; 1978 range (n = 100) 13-55
mean 18
Netherlands: range 7.8-181 Guicherit &
VC/PVC plants; (n = n.sp.) Schulting
1980 (1985)
Table 13. (cont'd)
Country; site and Valueb Concentrations Reference
year of sampling (µg/m3)
UK: 5 VC plants; overall means < 13-228 Turner et al.
1984 (April-July) daily means 23-218 (1984)
(ranges related
to the 5 plants,
total n = 440)
Plant VI (7 stations, max. (n = 28); 20 202 (0 km),
0-4.8 km distant) 880 (0.8 km)
Plant IX (15 stations, means (n = 180); 311-1373
0.8-5 km distant) maximum 8806 (5 km)
USA: 1974-1975; VC (n = 708) US EPA
plant (Narco) means 3.4-262 (1975);
(4 stations, geom. means 0-28 Dimmick
< 250 - > 1000 m maximum 27 045 (< 250 m) (1981)
distant)
PVC plant (Aberdeen) (n = 438)
(4 stations, means 11.6-958
< 250 - > 1000 m geom. means 3.7-372
distant) maximum 23 430 (300 m)
PVC plant (Louisville) (n = 712)
(4 stations, means 11.4-101
< 250 - > 1000 m geom. means 5-30
distant) maximum 814 (250-400 m)
USA: residential areas range (n = 30) n.d. - 104 US EPA
near chemical plants (1975)
(n = 15); 1974
USA: Houston, Texas; range (n = 18.) 8-3238 Gordon &
1974 Meeks
(1977)
USA: residential max. (n = n.sp.) > 2560 Fishbein
areas in the vicinity mean (n = n.sp.) 44 (1979)
of VC/PVC plants;
prior to 1975
USA: vicinity of max. 34 McMurry &
VC/PVC plants (Texas, Tarr (1978)
7 stations); 1977
Table 13. (cont'd)
Country; site and Valueb Concentrations Reference
year of sampling (µg/m3)
Areas in the vicinity
of waste sites
Belgium: Mellery; prior no details 19 Lakhanisky
to 1990 et al. (1993)
Germany: surroundings no details < 3 Pudill (1993)
of a hazardous waste
site; about 1988
Germany: 2 landfills range < 0.082-0.65 Wittsiepe
near Bochum; 1990-1991 (n = 16) et al. (1996)
USA: West Covina, range (24-h av; 26-104 Camarena &
California; 1981-1984 n = 32) max 130 Coy (1984)
USA: West Covina, 24-h averages 13-31 (n = 5 days) Baker &
California; 1984 5-day average 23 MacKay
site A (200 m distant 24-h averages 5.7-18 (1985)
from landfill) 5-day average 10.4 (n = 5 days)
site B (20 m distant
from landfill)
USA: southern max. 18 Stephens
California, 1981 range 5-18 et al. (1986)
after 1981 (6 stations)
USA: California; max. 30 Little et
prior to 1990 al. (1992)
USA: New York range n.d. (5.4) - 16 Lipsky &
(2 sites) 1982 Jacot
(1985)
Miscellaneous
USA: all outdoor site mean 8.5 Shah &
types; prior to 1988 (n = 574) Singh
(1988)
a n.d. = not detected (detection limit in parentheses, if specified);
n.sp. = not specified
b This column indicates whether the concentration is a maximum (max),
average or range value
c Values below detection set to 0.5 method detection limit (0.1 µg/m3)
d Values in parentheses = no. detected / no. sampled
5.1.1.2 Indoor air
Indoor air concentrations of VC in houses near landfills in the
USA reached concentrations of up to 1 mg/m3 air (Little et al., 1992:
2 landfills, maxima of 0.13 and 0.3 mg/m3; Stephens et al., 1986:
1 landfill, maximum of 1 mg/m3), thus exceeding the maximum outdoor
levels reported in Table 13 for areas adjacent to landfills. Moreover,
the Californian monitoring programme, collecting a total of 500 air
samples at two outdoor and four indoor sites downwind of a landfill,
revealed that the 120 samples containing the most VC
(> 0.025 mg/m3; 10 ppb) were taken inside homes (Little et al.,
1992). It is assumed that in addition to atmospheric transport,
subsurface migration of VC accounts for the elevated indoor air levels
of VC (Wood & Porter, 1987; Hodgson et al., 1992; Little et al.,
1992).
A room being painted with a red latex paint based on a terpolymer
of vinyl chloride, vinyl acetate and ethylene showed VC levels of
75 and 10 µg/m3 (29 and 4 ppb), respectively, during and some time
(less than one day) after painting (Going, 1976).
In the early 1970s it was investigated whether VC was present in
car interiors as a result of volatization from PVC. Measurements of VC
concentrations in the interior of seven different new 1975 automobiles
gave positive results for two of them. Levels of 1036 to 3108 µg/m3
(0.4 to 1.2 ppm) were detected (detection limit: 130 µg/m3; 0.05 ppm)
(Hedley et al., 1976). Another study (Going, 1976) did not find VC in
the interior ambient air of 16 new and used cars and 4 mobile homes
(detection limit: 26 µg/m3; 10 ppb). It should be noted that since
this time the levels of VC in PVC resins have been drastically reduced
(see section 3.2.4).
5.1.2 Water and sediment
Owing to its high volatility, VC has rarely been detected in
surface waters. The concentrations measured generally do not exceed
10 µg/litre, with a maximum of 570 µg/litre from contaminated sites
(Table 14).
Much higher levels of up to 56 000 µg/litre have been found in
groundwater samples from contaminated sites (Table 15).
The levels in drinking-water supplies ranged from not detected to
2 µg/litre in samples collected in 100 German cities in 1977. In a
state-wide USA study performed in 1981-1982, random samples (taken
from randomly selected water systems) had concentration ranges of n.d.
to 1.1 µg/litre, and non-random samples (taken from systems that were
likely to be contaminated with VOCs) varied from n.d. to 8 µg/litre.
Prior to 1980 single maximum values of up to 380 µg/litre were
reported from the USA (Table 16).
Table 14. Vinyl chloride concentrations measured in surface watera
Country, source Year of sampling Valueb Concentrations Remarks Reference
(µg/litre)
Germany
River Rhine (prior to) 1978 typical conc. 1 Anna & Alberti (1978)
(n = many)
River Rhine 1982 < 0.2 Malle (1984)
River Rhine 1990 range (n = 78) < 0.01-0.031 Wittsiepe (1990)
Tributaries of Rhine (prior to) 1978 typical conc. < 1-5 Anna & Alberti (1978)
(Northrhine-Westfalia) (n = many)
Surface water from n.sp. range < 0.0004-0.4 > 150 samples Wittsiepe et al. (1990)
unspecified sites (n = n.sp.)
in former FRG
River Main 1990 range (n = 22) < 0.004-0.008 Wittsiepe (1990)
River Lippe 1989 range (n = 54) 0.12-0.4 receiving Wittsiepe (1990)
wastewater from
VC/PVC plants
River Ruhr (plus 1990 range (n = 60) < 0.0004-0.005 Wittsiepe (1990)
artificial lake) (lake: up to
0.06)
River Wupper 1989 range (n = 36) up to 0.069 Wittsiepe (1990)
River Saale 1990 range (n = 4) up to 69 receiving Wittsiepe (1990)
wastewater from
an industrial
area of the
former GDR
Table 14. (cont'd)
Country, source Year of sampling Valueb Concentrations Remarks Reference
(µg/litre)
Japan
Rivers in Osaka 1995 range (n = 28) up to 1.2 (3/28)c Yamamoto et
al. (1997)
USA
Delaware River 1976-1977 (n = 11) n. d. Sheldon & Hites
(1978)
Surface water from 1977-1979 maximum 566 (21/606)c Page (1981)
different sites (n = 606)
in New Jersey median 0
Surface water from n.sp. maximum 9.8 Burmaster (1982);
9 states (n = n.sp.) Dyksen & Hess (1982)
Final effluent from a 1980-1981 mean 6.2 Gossett et al. (1983)
waste-water treatment (n = 5)
plant in Los Angeles
Surface waters prior to 1984 median < 5 (63/1048)c Staples et al. (1985)
(n = 1048)
Indian River Lagoon n.sp. (n = n.sp.) n.d. (< 1.0) Wang et al. (1985)
(near water discharge
of VC), several
stations
Surface water near 1985-1990 (n = n.sp.) n.d. Hallbourg et al. (1992)
3 landfills in
Florida
Table 14. (cont'd)
Country, source Year of sampling Valueb Concentrations Remarks Reference
(µg/litre)
Surface water at a 1989-1990 range 0.23 Chen & Zoltek (1995)
landfill in Florida 1992-1993 (n = 5) n.d.
(Orange County)
a n.d = not detected (detection limit in parentheses, if specified); n.sp. = not specified;
GDR = German Democratic Republic
b This column indicates whether the concentration is a maximum (max), average or range
value
c Values in parentheses = no. detected / no. sampled
One reason for the occurrence of VC in drinking-water may be that
residual VC can migrate from PVC pipes used in some water distribution
systems into the water flowing through them. This has been found out
by field (Dressman & McFarren, 1978) and experimental (Banzer, 1979;
Nakamura & Mimura, 1979; Ando & Sayato, 1984) studies. The extent of
leaching depended on the VC concentration in the pipe material. In the
field study the highest VC concentrations (1.4 µg/litre) consistently
occurred in water from new pipes, whereas the lowest level (0.03
µg/litre) was found in the oldest (9 years of age) distribution
system. VC concentrations in landfill leachate samples amounted to up
to 61 mg/litre (Table 17). A gross analysis of water (no
specification) available for the USA and based on 5553 observations
reported maximum and median concentrations of VC as high as
202.6 mg/litre and 107 µg/litre, respectively (US EPA, 1985a).
No VC was detected in urban stormwater run-off from 15 cities in
the USA (n = 86) during a monitoring project concerning priority
pollutants (Cole et al., 1984).
Generally, a time trend cannot be derived from the water analysis
data available.
Most sediment samples contain very low VC concentrations, even at
rather contaminated sites. Sediment was monitored for VC during
1981-1982 in Florida (USA) at several stations (n = 8) of the Indian
River Lagoon and a conveying canal. Although the latter received
discharged water having VC concentrations of 34-135 µg/litre, no VC
was detected in the sediment samples (3 from each station, collected
monthly over a year), the detection limit being 2 ng/g (Wang et al.,
1985). The same was true for surface water (Table 14) and oyster
(section 5.1.5) samples from this site. Sediment samples (n = 2) taken
near the discharge zone of a wastewater treatment plant in Los Angeles
County (California, USA) contained < 0.5 µg VC/kg dry weight. The
corresponding water concentration of VC was 6.2 µg/litre (Gossett et
al., 1983). A survey of 343 sediment samples from the USA gave a
median VC concentration of < 0.5 µg/kg dry weight (Staples et al.,
1985). However, higher VC concentrations were also reported. According
to US EPA (1985a), VC was detected in sediment samples (no further
details given) in the USA at levels ranging from 0-580 µg/kg (n = 649;
median = 23 µg/kg).
5.1.3 Soil and sewage sludge
5.1.3.1 Soil
Subsurface soil samples near the waste pit of an abandoned
chemical cleaning shop in southern Finland showed VC concentrations as
high as 900 mg/kg (Salkinoja-Salonen et al., 1995). After an
accidental spillage of VC into snow in 1980, VC levels as high as
500 mg/kg were measured in the soil at up to 2 m depth (Charlton
et al., 1983).
5.1.3.2 Sewage sludge
VC has been detected in municipal sewage sludges in the USA. The
concentrations ranged from 3 to 110 mg/kg dry weight (corresponding to
145 to 3292 µg/litre), being detected in 3 of 13 samples, with a
median concentration of 5.7 mg/kg dry weight (corresponding to
250 µg/litre) (Naylor & Loehr, 1982). Another study reported a mean
concentration of 35.4 mg/kg dry weight for 6 of 44 samples (Fricke
et al., 1985). A range of 8-62 000 µg/litre was found in 35 of 435 raw
sludge samples (Burns & Roe, 1982).
5.1.4 Food, feed and other products
VC is not a general contaminant of foodstuff and pharmaceutical
or cosmetic products, but it can be detected after contact of these
products with PVC packaging materials. The use of PVC as packaging
material for food, drink and drugs began in the early sixties (chapter
3), whereas legislative action for safeguarding consumers from
exposure to VC did not begin until the early seventies (starting with
a ban on the use of PVC containers for packaging alcoholic beverages
in the USA; Anon, 1973). Current EC and US FDA regulations on the
level of VC in PVC materials intended to come into contact with
foodstuffs are listed in Annex 1.
VC concentrations measured in PVC-packed food and drink of
several countries are compiled in Table 18. A maximum value of
20 mg/kg was found in liquors. Other positive samples included
vegetable oils (up to 18 mg/kg), vinegars (up to 9.8 mg/kg),
margarines (up to 0.25 mg/kg), fruit drinks (> 0.2 mg/kg) and bottled
water (< 0.6 µg/litre).
Retail surveys of foods showed a significant reduction in VC
levels and/or in the number of positive samples since 1974 (UK MAFF,
1978, 1984; van Lierop, 1979; Codex Committee, 1984).
The latest data were from the 1990s (Table 18) and refer to
bottled drinking-water. In addition to small amounts of VC, there were
also indications for the presence of possible reaction products of VC
with chlorine (Fayad et al., 1997).
Pharmaceutical and cosmetic products were less frequently
monitored. The highest concentration, amounting to 7.9 mg/kg, was
detected in mouthwashes (Table 19).
Reports on analyses of animal feed were not available.
The potential for leaching of residual VC from PVC packaging into
the contents has been demonstrated by a variety of product analyses
(Tables 18 and 19) and by experimental studies using food or food
simulants (Daniels & Proctor, 1975; Hocking, 1975; Tester, 1976;
Diachenko et al., 1977; Pfab & Mücke, 1977; UK MAFF, 1978; Chan et
al., 1978; vom Bruck et al., 1979; van Lierop, 1979; Benfenati et al.,
1991; Thomas & Ramstad, 1992). Altogether, the results indicated that
Table 15. Vinyl chloride concentrations measured in groundwatera
Country; source Year of Valueb Concentrations Remarks Reference
of groundwater sampling (µg/litre)
Canada
from a VC spill site 1980 maximum; 10 weeks 10 000 Charlton et al.
after the spill < 20 (1983)
beneath a landfill 1988 range (n = 37) < 1-40 (5/37)c Lesage et al.
near Ottawa (1990)
Finland
from village wells mid-1980 range 5-200 mg/litre Nystén (1988);
(contaminated by leakage of (including DCE) Salkinoja-Salonen
a waste liquor basin of et al. (1995)
VC/PVC industry - detected
in 1974)
Germany
from different sites mean values n.d. (< 5) - 460 Brauch et al.
(4 sites) (1987)
from different wells mean values 15-1040
of a large surface
contamination (5 wells)
from a catchment area of range (n = 30) < 1-120 Milde et al.
a water-works (1988); Nerger &
(TCE / PCE-contaminated) Mergler-Völkl
(1988)
contaminated by waste until 1988 range (n = 113) < 1-12 000 (14/113)c Schleyer et al.
disposal sites (92 mean 2700 (1988)
sites) median 475
Table 15. (cont'd)
Country; source Year of Valueb Concentrations Remarks Reference
of groundwater sampling (µg/litre)
from a site contaminated n.sp. (n = 3) corresponding air Köster (1989)
with chlorinated pockets in soil
hydrocarbons 1000 128 000 µg/m3
500 47 000 µg/m3
200 5000 µg/m3
from a site contaminated ca. 1989 max. (n = 5) 110 Leschber et
with chlorinated al. (1990)
hydrocarbons (in Berlin)
from a contaminated site n.sp. range (n = 3) 710-1670 Kästner (1991)
(in Braunschweig)
of a catchment area of a 1989 (n = n.sp.) up to 0.130 Wittsiepe et al.
waterworks (1990)
Contaminated by accidental 1989 (n = n.sp.) up to 3
spillage of TCE and PCE
Contaminated by waste n.sp. mean (n = 136) 1693 18% positive Dieter &
disposal sites (approx. max. 12 000 Kerndorff (1993)
100 sites)
USA
from different sites in 1977-1979 max. (n = 1060) 9.5 (4/1060)c Page (1981)
New Jersey
from 9 states max. 380 7% positive Dyksen & Hess
(1982)
monitoring wells near n.sp. max. 635 present in Stuart (1983)
industrial waste sites in 3 out of 9 wells
Connecticut
Table 15. (cont'd)
Country; source Year of Valueb Concentrations Remarks Reference
of groundwater sampling (µg/litre)
from Nassau County, 1980 range (n = >100) 1.6-2.5 Connor (1984)
Long Island
from Miami, Florida n.sp. mean (n = 3) 6.8 Parsons et al.
(1984)
from a TCE spill site in n.sp. mean (n = 3) 82 Parsons et al.
Vero Beach, Florida (1984)
monitoring wells near MSW n.sp. present (in Sabel & Clark
landfills in Minnesota 5/20 sites), (1984)
but not quantified
near 3 plants manufacturing n.sp. range (n = 3) 50-500 chlorinated White Paper
electronics equipment solvents stored in (1984) cited in
(Santa Clara Valley) underground tanks Wolf et al.
(1987)
near solvent recovery range (n = 4) Cline & Viste
facilities (1985)
Connecticut 1980 n.d. (< 10) - 2700
Wisconsin 1983 n.d. (< 10) - 210
from a residential area 1983 maximum 2800 contamination Andreoli
in Long Island, New York from dry-cleaning (1985)
(several wells) shop
near a landfill in (about max. 692 Shechter (1985)
New Jersey 1981) (n = approx. 100)
monitoring wells around 1985 max. 2600 seasonal, spatial Stephens et al.
hazardous waste landfill and analytical (1986)
in S. California variations
Table 15. (cont'd)
Country; source Year of Valueb Concentrations Remarks Reference
of groundwater sampling (µg/litre)
monitoring wells near 1990 range (n = 64) n.d. (1-2) - 12 (5/64)c EMO (1992)
an airforce base in Ohio
near 3 landfills in Florida 1985-1990 mean values < 0.22-26.5 Hallbourg et al.
(10 locations) (1992)
monitoring wells at a 1989-1990 range (n = 3) n.d. - 0.23 increase of VC Chen & Zoltek
landfill in central 1992-1993 4.8-48.4 with time, decrease (1995)
Florida (Orange County) in total VOC
from an industrial site; (n = n.sp.) 51-146 Topudurti (1992)
recycling operations
(1940-1987); California
near a former waste disposal (n = n.sp.) US EPA (1992)
facility in Wisconsin:
on-property max. 77
off-property max. 5
Contaminated by 1991 maxima < 5-56 400 Semprini et al.
TCE > 10 years before minima < 5-321 (1995)
(17 sites)
near Plattburg, New York n.sp. (n = 2) 8 and 384 Bradley & Chapelle
(2 locations) (1996)
a n.d. = not detected (detection limit in parentheses, if specified); n.sp. = not specified; TCE = trichloroethene;
PCE = tetrachloroethene; VOC = volatile organic compounds; max. = maximum; MSW = municipal solid waste
b This column indicates whether the concentration is a maximum (max), average or range value
c Values in parentheses = no. detected / no. sampled
Table 16. Vinyl chloride measured in drinking-water suppliesa
Country; source Year of sampling Valueb Concentration Remarks Reference
(µg/litre)
Germany
drinking-water supplies 1977 range n.d. - 1.7 Bauer (1981)
of 100 cities
drinking-water n.sp. (n = n.sp.) ca. 1.6 ng/litre Wittsiepe et al.
(no details) (1993)
USA
finished drinking-water (prior to) 1975 max. (n = n.sp.) 10 Fishbein (1979)
water supplies from (prior to) 1975 max. (n = n.sp.) 5.6 & 0.27
Florida and Philadelphia
raw drinking-water 1975-1979 2 positives 2.2 & 9.4 (2/13)c CEQ (1981)
(13 cities)
finished drinking-water 1 positive 9.4 (1/25)c
(25 cities)
drinking-water wells (prior to) 1980 max. (n = n.sp.) 50 CEQ (1981);
(state New York) Burmaster (1982);
Craun (1984)
drinking-water of (prior to) 1981 range 0.05-0.18 Kraybill (1983)
113 cities mean 0.052
drinking-water 1977-1981 range of trace - 380 (82/1288)c Cotruvo et al.
supplies (166) positives (1986)
Table 16. (cont'd)
Country; source Year of sampling Valueb Concentration Remarks Reference
(µg/litre)
finished water supplies 1981-1982 max.: Westrick et al.
(using groundwater random samples: 1.1 (1/466)c (1984)
sources) from 51 states (n = 466)
in the USA non-random 8.4 (6/479)c
samples:
(n = 479)
private wells in 1982 max. 4.5 (1/63)c Goodenkauf &
Nebraska (n = 63) Atkinson (1986)
a n.d. = not detected (detection limit in parentheses, if specified); n.sp. = not specified
b This column indicates whether the concentration is a maximum (max), average or range value
c Values in parentheses = no. detected / no. sampled
Table 17. Vinyl chloride concentrations measured in leachatea
Country; source Year of sampling Valueb Concentrations Remarks Reference
(µg/litre)
Canada
MSW landfill leachate 1988 n.sp. 14 Lesage et al. (1993)
from Guelph (Ontario) 1989 n.sp. 23
USA
landfill leachates n.sp. (n = 6) not quantified present in 1/6 Sabel & Clark (1984)
from Minnesota samples
landfill leachates prior to 1982 max. (n = 4) 61 (1/4)c
from Wisconsin
landfill leachates prior to 1985 range (n = 5) n.d. Cline & Viste (1985)
(municipal and (< 10) - 120
industrial)
landfill leachates: prior to 1988 range total number of Brown & Donnelly
(n = n.sp.) landfills: 58 (1988)
municipal 20-61 000
industrial 140-32 500
MSW leachates (several (prior to) 1988 range 8-61
states in the USA) (n = n.sp.)
median 40 (6/?)c Chilton & Chilton
(1992)
a n.d. = not detected (detection limit in parentheses, if specified); n.sp. = not specified;
MSW = municipal solid waste
b This column indicates whether the concentration is a maximum (max), average or range value
c Values in parentheses = no. detected / no. sampled
the amount of VC migrating into food or solvent was proportional to
the VC concentration in the PVC packaging, to storage time and to
increasing temperature.
Until 1974 the use of VC as a propellant in aerosol sprays was
allowed in the USA (chapter 3). Realistic application of such aerosol
products (hairspray, deodorant, insecticide, disinfectant, furniture
polish or window cleaner) resulted in high (ppm range) indoor air
concentrations of VC (Gay et al., 1975).
There have been no reports on VC levels found in food,
pharmaceutical or cosmetic products in recent years. This may be due
to regulatory measures for PVC packaging in several countries (see
Annex 1).
5.1.5 Terrestrial and aquatic organisms
Oysters ( Crassostrea virginica) from the Indian River Lagoon in
Florida (USA) contained no detectable levels of VC (detection limit:
0.4 ng/g). The samples were collected weekly (1981) and monthly (1982)
at three sites (Wang et al., 1985). VC was also not detected in the
corresponding water and sediment samples (section 5.1.2).
Another study (Gossett et al., 1983) did find low concentrations
of VC (< 0.3 µg/kg wet weight) in a sample of small invertebrates
from just above the bottom sediments (n = 1), in a muscle sample of
shrimp (n = 1) and in liver samples of several fish species (n = 4).
The animals were collected in 1981 in final effluent waters from a
wastewater treatment plant in Los Angeles County (Palos Verdes,
California, USA) that had VC concentrations of 6.2 µg/litre (section
5.1.2).
Data on fish tissue (no further details given) available from the
US EPA STORET database were reported by US EPA (1985a). VC levels
ranged from 0-250 mg/kg (n = 530; median: 6 mg/kg).
5.2 General population exposure
Exposure of the general population to VC is possible by several
routes. They include inhalation of air polluted with VC (section
5.1.1), mainly in the vicinity of VC/PVC plants or waste disposal
sites, intake of contaminated drinking-water (sections 5.1.2 and
5.1.4), ingestion of food, beverages and medicines packed in PVC
(section 5.1.4), and absorption through skin from PVC-wrapped
cosmetics (section 5.1.4).
Normally, the general population is exposed to only small amounts
of VC, if at all. However, the exposure varies according to the
countries' regulatory measures, the occurrence of accidents or the
spread of precursor substances.
Table 18. Levels of vinyl chloride in food and drink packaged, stored or transported in PVC articles
Country Yeara Product No.a,b Concentrationsc Reference
(µg/kg)
Canada n.sp. alcoholic beverages 22 < 25-1600 Williams & Miles
vinegars 28 n.d. (10) - 8400 (1975)
peanut oil 10 300-3300
Canada n.sp. alcoholic beverages 10 n.d. (10) - 2100 Williams (1976a)
vinegars 10 300-7800
vinegars 9 14-9800 Williams (1976b)
sherry 3 500-2400
peanut oil 3 3800-18 000
Canada n.sp. oil 5 80-2100 Page & O'Grady (1977)
vinegars 5 10-5700
Canada 1981-1982 vinegars n.sp. 27-43 Codex Committee (1984)
other foods n.sp. < 10
Italy n.sp. drinking-water 10 0.013-0.083 Benfenati et al. (1991)
(PVC-bottled)
Netherlands 1975 oil samples 8 > 50 Van Lierop (1979)
salad dressing 1 250
margarine 4 60-250
ready-made salads n.sp. > 50
(whole
batch)
wine n.sp. 760
Netherlands 1976 margarine 1 60 Van Lierop (1979)
fish 1 90
biscuit 3 20-130
Table 18. (cont'd)
Country Yeara Product No.a,b Concentrationsc Reference
(µg/kg)
1977 peanut butter 1 4100 Van Lierop (1979)
(very small
individually wrapped
portions)
1978 various foods: 67 (3 +) n.d. (0.1) - 2 Van Lierop (1979)
soya oil 1 0.3
vinegar 1 0.6
salmon salad 1 2
Norway 1975 butter/margarine 16 n.d. (2) Ehtesham-Ud Din
salad 14 2-15 et al. (1977)
juices 8 6-25
vinegar 27 6-2790
mustard 5 9-14
Saudi Arabia n.sp. drinking-water 9 × 48d < 0.6 Fayad et al. (1997)
(PVC-bottled)
Sweden 1974 edible fats 127 n.d. (2) - 127 Fuchs et al. (1975)
Sweden 1975-1976 various foods 104 n.d. (2) - 600 Albanus et al. (1979)
(edible fats and oils, (mostly: < 10)
ketchup, vinegar, lime
juice, fruit syrup)
Switzerland 1973-1975 edible oils 41 n.d. (5) - 1750 Rösli et al. (1975)
United Kingdom n.sp. spirits n.sp. 0-250 Davies & Perry (1975)
Table 18. (cont'd)
Country Yeara Product No.a,b Concentrationsc Reference
(µg/kg)
United Kingdom 1974 fruit drinks 25 10 - > 200 UK MAFF (1978)
1977 13 < 10
1974 cooking oil 23 10 - > 200
1977 7 < 2
1975 butter, soft margarine 51 < 2 - 200
1977 9 < 2
USA 1971 vegetable oil 1 7000 Breder et al. (1975)
1974 3 700
USA 1973 spirits n.sp. up to 20 000 Anon (1973);
US FDA (1973)
USA 1973-1975 alcoholic beverages n.sp. 11 000-25 000 Codex Committee (1984)
a n.sp. = not specified
b + = number of samples positive
c n.d. = not detected (detection limit in parentheses, if specified)
d 9 brands (locally produced and imported)
Table 19. Vinyl chloride detected in pharmaceutical and cosmetic
products packaged in PVC materials
Product No.a Concentrationb Reference
(µg/kg or µg/litre)
Blood coagulant 30 n.d. (15) Breder et al.
solutions (1975)
Mouthwashes 11 n.d. (20-30) - 7900
Several capsules, tablets 11 n.d. (10-30) Watson et al.
and mouthwashes (1977)
Large volume 5 n.d. (0.1) - < 1 Watson et al.
parenterals (1979)
Mouthwashes 5 7-120
Shampoos 4 9-17
Body oils 4 n.d. (0.1) - 41
Intravenous solutions n.sp. n.d. (1) Arbin et al.
(many) (1983)
Cefmetazole sodium 4 n.d. (0.3) - <1 Thomas &
(Zefazone(R) sterile Ramstad
powder) (1992)
a n.sp. = not specified
b n.d. = not detected (detection limit in parentheses, if specified)
5.2.1 Estimations
Estimations of the respiratory intake of VC reported for the USA
ranged from 0 to 48.3 mg per person per day, based on exposure values
of 0 to 2.1 mg/m3 and assuming that 23 m3 of air are inhaled per day
(US EPA, 1985b). According to Seiber (1996) over 100 000 Californians,
particularly those living near landfills, may be exposed to VC levels
of 2.59 µg/m3 (1 ppb) or more.
A European study (Besemer et al., 1984) evaluating the exposure
of the Dutch population to VC in ambient air assumed an average
exposure of about 0.2 µg/m3, which resulted in a calculated daily
intake of 4 µg VC per person. Small fractions of the population were
concluded to be exposed to higher average levels: 0.01% (> 8.5
millions) to > 5 µg/m3, 0.04% to 4-5 µg/m3, and 0.05% to
3-4 µg/m3. The corresponding estimated daily intakes of VC were
> 100 µg, 80 µg and 60 µg, respectively.
Intake via drinking-water from public water supplies in the USA
was estimated to exceed 1 µg/litre for 0.9% of the population,
5 µg/litre for 0.3% and 10 µg/litre for 0.1% of the population. These
would result in daily VC intakes (assuming a 70-kg man and 2 litres of
water/day) of > 2, 10, and 20 µg/day. Maximal values were estimated
to be approximately 120 µg/day (US EPA, 1985b).
Another study (Benfenati et al., 1991) estimated the daily intake
of VC from PVC-bottled drinking-water bought in Italian supermarkets.
Based on the analytical results (Table 18) and assuming a consumption
of 2 litres of water per person per day and a storage time of 2 months
for the PVC bottles, the authors calculated that the oral intake could
exceed 100 ng VC per person per day.
Evaluation of the results of food surveillance programmes from
the United Kingdom led to a calculated maximum likely VC intake of
1.3 µg/day per person in 1974, of 0.1 µg/day per person in 1976, and
less than 0.02 µg/day per person by 1978 (UK MAFF, 1978, 1984). These
calculations were based on the typical daily consumption of fruit
drink, cooking oil and soft margarine.
Evaluating and summing up possible maximum user intakes of VC
from PVC-bottled liquor, wine and oil, and food packaged in PVC
materials, the US FDA calculated a maximum lifetime-averaged exposure
of 25 ng/person per day (US FDA, 1986). An earlier review reported an
estimated dietary intake of VC of 40 ng/day per person in the USA
(Codex Committee, 1984).
The intake by food and drinking-water in the Netherlands was
estimated (without specifying details) to be about 0.1 µg/day per
person or less (Besemer et al., 1984). Estimates (no details given) of
the average human intake for Switzerland were reported to be 3 ng/kg
body weight per day (Lutz & Schlatter, 1993).
5.2.2 Monitoring data of human tissues or fluids
Monitoring human tissues or fluids as an indirect measure of
exposure to VC has not frequently been applied. As with workers in the
plastics industry (section 5.3), the urine of premature babies was
found to contain large amounts of thiodiglycolic acid (thiodiacetic
acid), a metabolite of VC (chapter 6), but the relationship to
possible VC exposure was questionable (Pettit, 1986).
5.3 Occupational exposure
The main route of occupational exposure to VC is via inhalation
(Sittig, 1985), while dermal absorption is considered to be negligible
(ECETOC, 1988).
Industrial environments associated with VC exposure include VC
production plants, VC polymerization (PVC production) plants and PVC
processing factories. Estimates of numbers of workers exposed to VC
were, for example, in the USA (1981-1983) in the range of 80 000
(ATSDR, 1997) or in Sweden (1975-1980) more than 5000 (Holm et al.,
1982). Since, at the onset of VC/PVC production in the USA and Western
Europe, VC was not recognized as a toxic compound, no precautions
against contact were provided for nor was regular workplace monitoring
performed. Therefore, only sporadic measurements or retrospective
estimates (Table 20) of exposures are available for the period prior
to 1975. Published data from various countries on VC contamination of
workplace air throughout the early and later periods are compiled in
Table 21 and 22. Highest exposures occurred in the VC/PVC production
plants, with peak concentrations of several thousand ppm whereas much
lower exposure levels were measured in processing plants. Owing to
standard-setting and legislative regulations by national authorities
and technical improvements, levels dropped markedly to values of a few
ppm in many countries. Official exposure limits can serve as an
additional indication of approximate VC concentrations occurring in
plants in many countries. These limits have declined gradually (IARC,
1979). Generally, standards require that exposures do not exceed 13 to
26 mg/m3 (5 to 10 ppm) (Torkelson, 1994; Rippen, 1995; ACGIH, 1999).
However, even in the 1990s the standards were not always realized
in all countries (Table 21).
Whereas in industrialized countries factories that could not
satisfy the rigorous regulations of the early 1970s to reduce VC
emissions were forced to close down, in the countries of eastern
Europe and developing countries this was not possible for
socioeconomic reasons and large plants with old-fashioned technologies
continued to function (Hozo et al., 1996).
Table 20. Retrospective estimates of daily occupational
exposures to vinyl chloride prior to 1975a
Country Period VC exposure Reference
(mg/m3)
Germany "first years" > 2600 Szadkowski & Lehnert
prior to 1971 1300 (1982)
1971 260
1974 5.2-7.8
Norway 1950-1954 5200 Hansteen et al. (1978);
1955-1959 2600 Heldaas et al. (1984)
1960-1967 1300
1968-1972 260
1973-1974 207
Sweden 1945-1954 1300 Holm et al. (1982)
1955-1964 780
1965-1969 520
1970-1974 130
United Kingdom 1945-1955 2600 Barnes (1976);
1955-1960 1040-1300 Anderson et al. (1980);
1960-1970 780-1040 Purchase et al. (1987)
mid 1973 390
1975 13
1940-1955 1300-2070 Jones et al. (1988)
1956-1974 390-1300
USA 1945-1955 2600 Wu et al. (1989)
1955-1970 780-1300
1970-1974 260-520
1975 < 2.6-13
a Exposure levels during autoclave cleaning may have been as high as
7800 mg/m3 (Barnes, 1976)
Table 21. Levels of vinyl chloride reported for workplace air samples in VC/PVC production plants
Country Workplace Yearc Concentrations reported Reference
(mg/m3)c
China PVC production plant n.sp. 30-210 Bao et al. (1982)
Croatia plastics industry n.sp. mean = 13 Fucic et al. (1990a)
5200 (occasional peak)
Croatia VC/PVC plant 1949-1987 mean = 543 Hozo et al. (1996,
up to 1300 (occasional peak) 1997)
Former n.sp. n.sp. 2-41 Hrivnak et al. (1990)
Czechoslovakia
Egypt VC/PVC plant n.sp. 0.05-18 (8-h TWA) Rashad et al. (1994)
Finland PVC production plant, n.d. range
breathing zone 1981-1985 1.6 < 0.3-57 Viinanen (1993)
concentrations (TWA) 1986-1989 1.6 < 0.3-46
1993 0.3 < 0.3-26
France PVC production plant 1977-1978 2.3-7.3 (range of monthly Haguenoer et al.
means) (1979)
Germany PVC production 1974 < 65-181 Fleig & Thiess (1974)
department
PVC production plant 1977 1.3-91 German Environmental Office (1978)
PVC production plant 1979 12 (12-h TWA; stationary); Heger et al. (1981)
15.5 (12-h TWA; personal)
Germany 24 plants 1981-1984 3% of 33 samples: > 5 Coenen (1986);
(90 percentile: < 1); BIA (1996)
(shift means)
Table 21. (cont'd)
Country Workplace Yearc Concentrations reported Reference
(mg/m3)c
46 plants 1989-1992 all of 117 samples: < 5
(90 percentile: < 0.1);
(shift means)
Italy VC/PVC plants 1950-1985 < 13- > 1300 Pirastu et al. (1991)
The Netherlands PVC plant 1976-1977 2.6-26 (8-h TWA) De Jong et al. (1988)
Norway PVC plant 1974 65 Hansteen et al.
(1978)
Poland VC/PVC plant 1974 (30-600)a Studniarek et al.
(several departments) 1975 (30-270)a (1989)
1976 (15-60)a
1977 (6-150)a
1978 (1-30)a
1979 (1-15)a
1981 (0.1-36)a
1982 (0.1-12)a
(autoclave cleaners) 1974 (990)a
1982 (9-180)a
breathing zone of VC 1986 21.3 Dobecki &
synthesis mechanic 1987 66.9 Romaniwicz (1993)
1988 43.7
1989 0.7
1990 0.2
Romania PVC production plant 1965-1967 112-554 Anghelescu et al.
(1969)
Russia VC/PVC plant Gáliková et al. (1994)
16 probes (whole plant) 1990-1993 1-9 (range of annual means)
under the reactor 1990-1993 up to 200 (range of annual
means)
Table 21. (cont'd)
Country Workplace Yearc Concentrations reported Reference
(mg/m3)c
in compressor room 1990-1993 up to 400 (range of annual
mean)
Singapore PVC production plant 1976
after 1983 2.6-54 (15.3)a Ho et al. (1991)
up to 26 (short-term) (3.9)a
Sweden PVC production plant 1974-1981 0.26-114 (8-h TWA) Holm et al. (1982)
PVC production plant 1974-1980 0.26-5.7 (6-h TWA)
Taiwan PVC plants (n = 5): n.sp. Du et al. (1996)
15 different operation range (n=114): n.d.
units (e.g. outside (0.13) - 1009 range (n=4):
reaction tank)b 6-1009 (mean: 296; median: 86)
15 different job titles range of TWA (n=85):
(e.g. tank supplier)b n.d. - 3680 range (n=9):
5.7-3680 (mean: 660; median:
23.7)
United Kingdom PVC production plant "early days" 7800 Barnes (1976)
(full-time autoclave
cleaner)
USA PVC plant 1950-1959 up to 10 400; 13-2140 Ott et al. (1975)
(8-h TWA)
1960-1963 up to 1300; 13-620 (8-h TWA)
PVC plant n.sp. up to 650 (weekly TWA) Baretta et al. (1969)
USA VC/PVC plants 1973 up to 390 (TWA); peaks Rowe (1975)
2600-10 400
Table 21. (cont'd)
Country Workplace Yearc Concentrations reported Reference
(mg/m3)c
Former USSR VC/PVC plants early 1950s 100-800 Smulevich et al.
(1988)
PVC producing plant 50-800 Filatova &
(occasionally 87 300) Gronsberg (1957)
Former PVC production plant 1974 > 195 Orusev et al. (1976)
Yugoslavia
a Concentrations in parentheses designate geometric means
b Showing highest mean VC concentration
c n.sp.= not specified; TWA = time-weighted average; n.d. = not detected
Table 22. Levels of vinyl chloride reported for workplace air samples in PVC processing plants
Country Workplace Year Concentrations reported Reference
(mg/m3)
China PVC processing plant n.sp. > 30 Bao et al. (1982)
Germany PVC processing department 1974 < 2.6-67 Fleig & Thiess (1974)
Germany polymer extrusion (17 plants) 1989-1992 all of 33 samples: BIA (1996)
< 8 (90 percentile:
< 0.15) (shift means)
Sweden PVC processing plant 1974 < 0.26-0.8 Holm et al. (1982)
Sweden PVC processing plant prior to 1975 >13 - > 26 (8-h TWA) Lundberg et al.
(1993)
Russia PVC processing plant prior to 1990 0.007-1.26 Solionova et al.
(rubber footwear plant) (1992)
Russia PVC processing plant prior to 1966 < 113.6 Bol'shakov (1969)
(synthetic leather plant)
United Kingdom PVC processing plants n.sp. 0.4-0.9 Murdoch & Hammond
(cable factories) (1977)
USA automotive assembly plant(s) 1970s 0.13-7.8 Nelson et al. (1993)
(2 personal samples)
Autoclave cleaners in Croatia were exposed to extremely high
concentrations of VC (between 1295 and 3885 mg/m3; 500 and 1500 ppm).
A retrospective investigation of exposure to VC has been conducted
with 37 autoclave workers (emptying and cleaning) in Split, Croatia,
who were exposed to VC in a suspension polymerization plant. The
investigation covered the period from 1969 to 1987, when the factory
was closed because of its VC emissions. At the beginning, measurements
were done by simple means (Draeger's tubes) and later, from 1980 on,
by infra-red spectroscopy. The 37 workers were exposed to average VC
concentrations of 1412 mg/m3 (543 ppm) (Hozo et al., 1996; Hozo,
1998).
6. KINETICS AND METABOLISM IN LABORATORY ANIMALS AND HUMANS
6.1 Absorption
Animal and human studies have shown that VC is readily and
rapidly absorbed. The primary route of exposure to VC is inhalation.
However, the net uptake after exposure by inhalation is only 30-40% of
inspired VC. This is due to the fact that VC is taken up rapidly until
it reaches a blood concentration in equilibrium with that based upon
inspired concentration and the blood-to-air partition coefficient.
Uptake then decreases to an amount sufficient to replace that
metabolized. The importance of metabolism was shown by Bolt et al.
(1977) who showed that uptake of VC ceased once equilibrium was
reached.
While uptake by the oral route is near 100%, any VC not
metabolized during first pass through the liver will be expired. Thus,
the net dose may be less than the uptake, especially at high doses
resulting in saturation of metabolizing enzymes.
6.1.1 Oral exposure
In a study reported by Watanabe & Gehring (1976) and Watanabe et
al. (1976a), male rats received a single dose by gavage of 0.05, 1.0,
20 or 100 mg/kg body weight and 14C-labelled VC and the excreted
radioactivity was determined for 72 h. As only 2.4, 2.2, 1.0 and 0.5%,
respectively, of the administered radioactivity (Table 23) was
recovered in the faeces (total recovery 91, 89, 81 and 82%,
respectively), it can be assumed that there is nearly complete
absorption of VC in the gastrointestinal tract. Likewise, oral
administration of 0.25 or 450 mg/kg body weight 14C-VC to male rats
resulted in excretion of only 4.6 or 0.7%, respectively (Table 23), of
the applied radioactivity via faeces 0-72 h after application (Green &
Hathway, 1975).
Similar results were obtained by Feron et al. (1981), who fed
rats with different amounts of VC monomer in PVC powder via the diet.
The average amount of VC detected in faeces was 8, 10 and 17% for oral
intakes of 2.3, 7.0 and 21.2 mg/kg body weight per day, respectively.
Since the VC excreted in the faeces was considered by the authors to
be still enclosed in PVC granules and was not bioavailable, it was
concluded that the available VC was nearly completely absorbed in the
gastrointestinal tract.
Studies conducted by Withey (1976) have shown the rapid
absorption of VC from the gastrointestinal tract in male rats after a
single gavage application of 44 to 92 mg/kg body weight in aqueous
solution. Highest blood levels were always measured within 10 min
after administration. After an oral dose of 10 mg VC/kg body weight,
peak levels in brain, liver, kidney and lung were measured 5 min after
dosing, indicating rapid absorption from gastrointestinal tract
(Zuccato et al., 1979).
Table 23. Excretion of radioactivity in rats (in % of applied dose) 72 h after a single oral dose of VCa
Dose in mg/kg VC in CO2 in Urine Faeces Carcass and Total recovery Reference
body weight exhaled air exhaled air tissues
0.05 1.4 9.0 68.3 2.4 10.1 91.2 Watanabe et al. (1976a)
0.25 3.7 13.5 75.1 4.6 n.g. n.g. Green & Hathway (1975)
1.0 2.1 13.3 59.3 2.2 11.1 88.8 Watanabe et al. (1976a)
20 41.6 4.8 22.6 1.0 11.0 81.0 Watanabe & Gehring (1976)
100 66.6 2.5 10.8 0.5 1.8 82.3 Watanabe et al. (1976a)
450 91.9 0.7 5.4 0.7 n.g. n.g. Green & Hathway (1975)
a n.g. = not given
6.1.2 Inhalation exposure
Bolt et al. (1977) blocked the metabolism of VC in male rats by
i.p. injection of 6-nitro-1,2,3-benzothiadiazole (50 mg/kg body
weight), a compound that inhibits most microsomal monooxygenases. The
rats were exposed to approximately 1.2 mg/m3 14C-labelled VC in a
closed system 30 min after the injection. VC was taken up by the
animals until equilibrium was reached between VC in the atmosphere and
in the organism after 15 min, suggesting a rapid uptake of VC.
Similarly, equilibrium blood levels were observed by Withey (1976) in
male rats 30 min after the start of exposure to 18 200 mg VC/m3 (head
only).
Bolt et al. (1976) measured the decline of 14C-VC in a closed
system due to uptake by 3 male rats using an initial concentration of
130 mg/m3. The radioactivity in the air of the exposure system
decreased with a half-life of 1.13 h. Calculation of the clearance of
VC revealed an absorption of inspired VC of about 40%. These results
are in accordance with the data of Hefner et al. (1975a), who reported
in kinetic studies on male rats a similar decline at the same exposure
concentration.
Krajewski et al. (1980) measured the retention of VC in the lungs
of five human male volunteers exposed for 6 h to 7.5, 15, 30 or
60 mg/m3 through a gas mask. The percentage of retention (mean 42%)
was independent of the VC concentration and reached the highest level
of 46% in the first 15 min of exposure.
Buchter et al. (1978) reported toxicokinetic experiments on
themselves. In an open system, about 3-5 min after the start of
inhalation of 6.5 mg VC/m3 in the exposure atmosphere, an equilibrium
was reached between VC inspired and VC expired; 26% (person A) and 28%
(person B) of the VC inspired were removed by the body, probably by
absorption and subsequent metabolism. Metabolism started within the
first minutes of exposure, suggesting fast absorption of VC.
Breath samples were measured of volunteers who had showered in
VC-contaminated well water (Pleil & Lindstrom, 1997). For a brief
10-min shower exposure of 25 µg/m3 (inhalation) and 4 µg/litre
(dermal contact in water), 0.9 µg absorbed dose of VC and a blood
concentration of 0.01 µg/litre was calculated.
6.1.3 Dermal exposure
Hefner et al. (1975b) exposed the whole body (excluding the head)
of male rhesus monkeys to 2080 (2.5 h) or 18 200 mg/m3 (2.0 h)
14C-labelled VC. Very little VC was absorbed through the skin.
Although minuscule, the quantity absorbed appeared
concentration-dependent. The major portion of the VC absorbed was
eliminated by the lungs. The authors argued that no significant
percutaneous absorption would be expected under occupational exposure
conditions.
6.2 Distribution and retention
Data from inhalation and oral studies on rats indicate rapid and
widespread distribution of VC. Rapid distribution of VC was also
reported in humans after inhalation exposure. Rapid metabolism and
excretion limit accumulation of VC in the body. Data on distribution
after oral exposure showed similar results. No studies are available
concerning distribution after dermal exposure. Under conditions of
blocked VC metabolism, VC has been found to accumulate in adipose
tissue.
6.2.1 Oral exposure
Green & Hathway (1975) investigated the distribution in the
tissues of young rats 0.25, 2 and 4 h after oral administration of
30 mg/kg body weight [14C]-VC using whole-animal autoradiography. At
15 min after gavage, most radioactivity was detected in the liver,
followed by the gut (no data about the stomach), and there were small
amounts in the lung and kidney. After 2 h, label was observed in the
liver, kidney, small intestine, stomach, skin, para-auricular region
(probably Zymbal glands), and there were small amounts in lung and
heart. A similar distribution was determined after 4 h, with
additional label in the thymus, salivary gland and Harders gland, but
no radioactivity in the stomach lumen.
The distribution of VC and VC metabolites remaining in the body
(11.1% of recovered radioactivity) 72 h after a single oral
administration via gavage of 1 mg/kg body weight 14C-VC was described
as follows (expressed as % of administered radioactivity per gram
tissue): liver 0.182, skin 0.076, carcass 0.046, plasma 0.053, muscle
0.031, lung 0.061, fat 0.045 (no data about amount in the kidney). A
similar distribution pattern was observed using doses of 0.05 or
100 mg/kg body weight (Watanabe et al., 1976a).
6.2.2 Inhalation exposure
Whole body autoradiograms of male rats showed radioactivity in
the liver, bile duct, digestive lumen and kidneys when animals were
exposed for 5 min to 52 g/m3 14C-labelled VC and sacrificed 10 min
after the end of the exposure period. Animals sacrificed 2-3 h after
the exposure period showed a wider distribution of radioactivity, with
most of the labelled substances being observed in the liver, urinary
system, digestive lumen, lacrimal glands, skin and thymus (Duprat et
al., 1977).
Buchter et al. (1977) pretreated male rats i.p. with 50 mg/kg
body weight 6-nitro-1,2,3-benzothiadiazole (to block VC metabolism)
and exposed the rats in a closed system for 5 h to VC concentrations
of between 65 and 26 000 mg/m3. Authors observed an equilibrium
between 14C-labelled VC in the gas phase and the exposed organism.
The distribution of VC was independent of the exposure concentration.
The following distribution of VC (labelled and unlabelled) in
different organs (expressed as mol VC in 1 g tissue per mol VC in 1 ml
air) was reported immediately after the exposure period: blood 0.65,
liver 0.62, spleen 0.59, kidney 0.59, muscle 0.68 and adipose tissue
8.3. These results indicated that unmetabolized VC is accumulated in
adipose tissue due to its lipophilicity. In contrast, without blockage
of VC metabolism, most radioactivity was detected in kidney and liver
(5 h, 260 mg/m3).
In similar studies Bolt et al. (1976) measured radioactivity
(representing mostly VC metabolites) in different organs immediately
after exposure of male rats to 14C-VC (initial concentration 130
mg/m3) for 5 h. Highest levels were detected in the kidney (2.13%;
expressed as % of incorporated VC per g tissue) and liver (1.86%),
followed by spleen (0.73%), muscle (0.32%), adipose tissue (0.22%) and
brain (0.17%).
Distribution pattern changed with longer post-exposure
observation periods. Watanabe et al. (1976b) reported the following
percentages of 14C activity (mostly VC metabolites) in rats per gram
tissue 72 h after inhalation exposure to 26 or 2600 mg/m3
14C-labelled VC, respectively, for 6 h: liver (0.139; 0.145), kidney
(0.079; 0.057), skin (0.072; 0.115); carcass (0.048; 0.049), plasma
(0.051; not detected), muscle (0.052; 0.038), lung (0.065; 0.046), fat
(0.026; not detected). There was no significant difference between low
and high dose. 72 h after the exposure period most of radioactivity
had been excreted; 13.8% (low dose), 14.5% (high dose) of total
recovered radioactivity remained in carcass and tissues (Table 25). A
similar distribution pattern were presented by Watanabe et al. (1978b)
using the same experimental design but rats exposed once or repeatedly
to 13 000 mg/m3. Furthermore, the authors detected no significant
difference between single and repeated exposure.
Ungváry et al. (1978) presented evidence for the permeability of
the placenta to VC. After exposure of pregnant rats to 5500, 18 000 or
33 000 mg VC/m3 for 2.5 h on day 18 of gestation, VC was detected in
fetal (13, 23 and 31 µg/ml, respectively) and maternal blood (19, 32
and 49 µg/ml), as well as in amniotic fluid (4, 5 and 14 µg/ml).
Toxicokinetic experiments on humans (self-experiments) were
reported by Buchter et al. (1978). Using an open system with a
concentration of 6.5 mg VC/m3 inspired air, the concentration in the
expired air reached a constant concentration after about 5 min
exposure in subject A and 7 min in subject B, indicating the end of
the distribution phase.
6.2.3 Partition coefficients in vitro
In vitro studies using the vial equilibration method (3 h
incubation, blood and tissue homogenates from male Sprague-Dawley
rats) revealed the following VC partition coefficients for male rats:
blood/air 2.4; fat/blood 10.0; muscle/blood 0.4; liver/blood 0.7; and
kidney/blood 0.7 (Barton et al., 1995). In similar experiments
tissue/air partition coefficients were obtained for different rodent
species (Gargas et al., 1989; Clement International Corporation, 1990;
Table 24). These data suggested that the concentration of VC in
adipose tissue is higher than in other tissues. Furthermore, in all
species in which both sexes were tested, partition coefficients for
fat/air were greater in females than in males (Table 24).
6.3 Metabolic transformation
The main route of metabolism of VC in the liver into non-volatile
compounds after inhalative or oral uptake involves 3 steps: a) the
oxidation by cytochrome P-450 to form chloroethylene oxide (CEO, also
known as 2-chlorooxirane), a highly reactive, short-lived epoxide that
rapidly rearranges to form chloroacetaldehyde (CAA); b) the
detoxification of these two reactive metabolites as well as
chloroacetic acid, the dehydrogenation product of CAA, through
conjugation with glutathione catalysed by glutathione S-transferase;
c) the modification of the conjugation products to substituted
cysteine derivatives, which are excreted via urine. The main metabolic
pathways are shown in Fig. 2. At high dose levels the metabolism of VC
is saturable.
The first step in VC metabolism requires microsomal
mixed-function oxidases (cytochrome P-450 enzymes) together with
oxygen and NADPH as cofactors. This was confirmed by studies in vitro
(Barbin et al., 1975; Guengerich et al., 1979) and in vivo
(Reynolds et al., 1975a,b; Guengerich & Watanabe, 1979; Bartsch et
al., 1979; Guengerich et al., 1981). The major catalyst of the
oxidation is CYP2E1 in humans. This has been demonstrated by in vitro
systems using purified human CYP2E1 or by inhibition of catalytic
activity in human liver microsomes with rabbit anti-human CYP2E1. In
liver microsomes from uninduced rats, VC is activated solely by
CYP2E1, at concentrations ranging from 1 to 106 ppm in the gas phase,
according to Michaelis-Menten kinetics (El Ghissassi et al., 1998).
The following kinetic constants were determined:
Km = 7.42 ± 0.37 µmol/litre;
Vmax = 4674 ± 46 nmol.mg protein -1 min-1.
Comparison of the Vmax obtained in this study to the Vmax determined
in vivo in rats (Gehring et al., 1978; Filser & Bolt, 1979) shows
that virtually all the metabolic activation of VC in vivo occurs in
the liver.
Table 24. Tissue/air VC partition coefficients for rodent tissuesa
Species; strain Sex Blood/ Liver/ Muscle/ Fat/
air air air air
Rat; F344 male 1.60 1.99 2.06 11.8
female 1.55 2.05 2.39 21.1
Rat; CDBR male 1.79 3.0 2.18 14.6
female 2.12 1.66 1.28 19.2
Rat; Wistar male 2.10 2.69 2.72 10.2
female 1.62 1.48 1.06 22.3
Mouse; B6C3F1 male 2.83 n.g. n.g. n.g.
female 2.56 n.g. n.g. n.g.
Mouse; CD-1 male 2.27 n.g. n.g. n.g.
female 2.37 n.g. n.g. n.g.
Hamster; male 2.74 3.38 2.56 14.3
Syrian golden female 2.21 1.31 1.96 21.1
a Data from Clement International Corporation (1990); vial
equilibration method (Gargas et al., 1989), blood or tissue
homogenates incubated for 1-4 h until equilibrium was achieved,
as indicated by two consecutive time points without significant
difference; n.g. = not given
Table 25. Percentage of 14C activity eliminated during 72 h following
inhalation exposure to [14C]-vinyl chloride for 6 h in male ratsa
Exposure groups 26 mg/m3 2600 mg/m3 13 000 mg/m3
(number of animals) (4) (4) (2)
Expired as unchanged VC 1.6 12.3 54.5
Expired as CO2 12.1 12.3 8.0
Urine 68.0 56.3 27.1
Faeces 4.4 4.2 3.2
Carcass and tissues 13.8 14.5 7.3
a Expressed as percentage of the total 14C activity recovered
(similar experimental design; Watanabe & Gehring, 1976;
Watanabe et al., 1976b, 1978b)
Table 4. Vinyl chloride found in landfill/waste disposal sites as a gas, in leachate and in groundwater
formed probably from degradation of higher chloroethenes
Sample Place (year) of sampling Valuea Concentrations Reference
Landfill gas 2 landfills USA max 230 mg/m3 Lipsky & Jacot (1985)
average 34 mg/m3
Landfill gas landfill, UK max 11 mg/m3 Ward et al. (1996)
plume, 100 m from boundary 40 mg/m3
due to subsurface migration (1991)
Landfill gas landfill, Braunschweig, Germany mean 9 mg/m3 Henning & Richter (1985)
Gas effluents garbage dump, Berlin, Germany 0.27 mg/m3 Höfler et al. (1986)
Gas Germany, average Janson (1989)
industrial waste disposal site 41 mg/m3
municipal waste disposal site 10 mg/m3
Gas Germany, waste disposal site range 0.03-0.3 mg/m3 Bruckmann & Mülder (1982)
Landfill gas UK, 7 waste disposal sites range < 0.1-87 mg/m3 Allen et al. (1997)
Soil air Germany, solvent waste sites 3 max out 128 mg/m3, Köster (1989)
of 200 47 mg/m3,
5 mg/m3
Leachate MSW, Wisconsin, USA (1982) range 61 µg/litre Sabel & Clark (1984)
Leachate USA sites established before 1980 range 8-61 µg/litre Chilton & Chilton (1992)
(6 chosen sites)
Leachate or industrial landfill range 140-32 500, Brown & Donnelly (1988)
groundwater municipal landfill 20-61 000
plume µg/litre
Groundwater, Germany, contaminated water range < 5-460 µg/litre Brauch et al. (1987)
Wells range 15-1000 µg/litre
Groundwater Germany, solvent waste site 3 max 1000 µg/litre, Köster (1989)
samples/200 500 µg/litre,
200 µg/litre
Table 4. (cont'd)
Sample Place (year) of sampling Valuea Concentrations Reference
Groundwater Germany max 120 µg/litre Milde et al. (1988)
Groundwater Santa Clara Valley, USA (near range 50-500 µg/litre Wolf et al. (1987)
plants manufacturing electronic
equipment which use
significant amounts of
chlorinated solvents)
Groundwater Germany: 136 samples from max 12 000 µg/litre Dieter & Kerndorff (1993)
down-gradient wells of 100 waste mean 1694 µg/litre
disposal sites
Groundwater sand aquifer near industrial max > 5 µg/litre Semprini et al. (1995)
site, Michigan, USA. Concentration at 10 m;
increased with depth consistent 56 400 µg/litre
with methane at 23 m
Outwash aquifer Gloucester landfill, Canada (1988) range < 1-40 µg/litre Lesage et al. (1990)
a This column indicates whether the concentration is a maximum (max), average or range value
Each landfill site has individual conditions (e.g., presence of
other solvents such as acetone and methanol), so that the degradation
rates cannot be directly compared. The most extensively studied site
of intrinsic chlorinated solvent biodegradation is the St Joseph
(Michigan, USA) Superfund site where groundwater concentrations of TCE
as high as 100 mg/litre have been found, with extensive transformation
to cis-DCE, VC and ethene. Conversion of TCE to ethene was most
complete where methane production was highest and where removal of
nitrate and sulfate by reduction was most complete (McCarty, 1996;
Weaver et al., 1996). At another site in the USA (Dover Air Force
Base), half-lives of 1 to 2 years have been estimated for each stage
in the reaction chain (e.g., DCE to VC; VC to ethene) (Ellis et al.,
1996). The degradability of chlorinated aliphatic compounds was
studied under methanogenic conditions in batch reactors with leachate
from eight landfill sites in Denmark. PCE and TCE were found to be
degraded in only three of the eight leachates, with significantly
different conversion rates. In one leachate, complete conversion of
chlorinated ethenes, including conversion of VC, was observed within
40 days, while another leachate showed only 50% conversion of PCE
(Kromann et al., 1998).
No known microorganism can aerobically destroy PCE. Laboratory
studies have shown that some anaerobic bacteria (e.g., Dehalobacter
restrictus ) use chlorinated solvents for respiration
(halorespiration), breaking them down in the process to form
cis-dichloroethene, although restricted diet conditions are
necessary (Sharma & McCarty, 1996). Recently, a coccoid bacterium has
been isolated (provisionally named Dehalococcoides ethenogenes
strain 195) which, together with extracts from mixed microbial
cultures, can dechlorinate PCE, removing further chlorine atoms to
form vinyl chloride and finally ethene (Maymó-Gatell et al., 1997).
Escape of landfill gas from the disposal site can take place via
the surface (emission) or into the ambient soil (migration). VC is
emitted from the landfill surface into the ambient air (Wittsiepe et
al., 1996). Awareness of this problem has encouraged the development
of in situ bioremediation of chlorinated solvents and VC using
anaerobic or aerobic co-metabolic processes (Dolan & McCarty, 1995b;
Jain & Criddle, 1995; Semprini, 1995; see section 4.2).
The estimated emission of vinyl chloride from landfill sites in
the USA is 60-33 000 tonnes/annum (Lahl et al., 1991).
3.2.5.2 VC formation from tobacco
VC was identified in the smoke of all 13 cigarettes tested
(1.3-16 ng/cigarette) and in both small cigars tested (14-27
ng/cigar). The level correlates directly with the chloride content of
the tobacco. Filter tips with charcoal reduce selectively the VC
content of cigarette smoke (Hoffmann et al., 1976).
Table 5. Some examples of formation of VC through biodegradation of tetrachloroethene
in landfill sites (concentrations in mg/litre unless stated otherwise)a
Site Sample PCE TCE cis-DCE VC Ethene Reference
Chemical transfer groundwater 4.4 1.7 5.8 0.22 0.01 Major et al.
factory facility, downgradient well n.d. none 76 9.7 0.42 (1991)
North Toronto,
Canada
Carpet backing groundwater n.d. 56 4.2 0.076 Fiorenza et al.
manufacturing downgradient from n.d. 4.5 5.2 low (1994)
plant, Ontario, lagoon
Canada
Refuse landfills 7.15 5.09 not 5.6 McCarty &
(average of 8) (ppmv) (ppmv) measured (ppmv) Reinhard (1993)
Landfill groundwater 0.54 2.6 2.2 2.7 33 Lee et al. (1995)
well 3.4 14 44 54 43
15 270 140 48 14
Heavily polluted groundwater 20 70 20 2 Middeldorp et
site (solvent al. (1998)
distributor) in
Netherlands
a PCE = tetrachloroethene; TCE = trichloroethene; DCE = dichloroethene; n.d. = not detected
3.3 Uses
About 95% of the world production of VC is used for the
production of PVC. The remainder is used for the production of
chlorinated solvents, primarily 1,1,1-trichloroethane (10 000 tonnes
per year; European Council of Vinyl Manufacturers, 1994), via the more
toxic 1,1,2-trichloroethane and 1,1-dichloroethane.
VC was previously used as a refrigerant (Danziger, 1960) and as a
propellant in aerosol sprays for a variety of products, such as
pesticides, drugs and cosmetics (Wolf et al., 1987). These uses have
been banned since 1974 in the USA and in other countries.
4. ENVIRONMENTAL TRANSPORT, DISTRIBUTION AND TRANSFORMATION
4.1 Transport and distribution between media
Depending on the sources, VC can enter the environment via air,
water or soil. The most critical matrices are probably air and
groundwater. Euro Chlor (1999) calculated the partitioning of VC into
environmental compartments, based upon the Mackay Level 1 model, to be
99.99% air, 0.01% water, < 0.01% soil and < 0.01% sediment.
4.1.1 Air
Owing to its high vapour pressure (saturation vapour pressure
P0 > 10-4 mmHg; see also section 2), VC released to the
atmosphere is expected, based on calculations of Eisenreich et al.
(1981), to exist almost entirely in the vapour phase. The atmospheric
life-time of VC is limited by its reaction with photochemically
produced OH radicals (see section 4.2.2).
VC is volatilized to the atmosphere from area sources such as
landfill sites. Models have been developed to predict such short-range
dispersion. These models were validated by VC concentrations ranging
from < 5 to 31 µg/m3 (< 2 to 12 ppb) measured in the vicinity of a
landfill in Los Angeles 3 years after it last received any waste
containing VC (Chitgopekar et al., 1990).
No data are available on wet deposition.
In order to model distribution processes, liquid : air partition
coefficients have been determined (Gargas et al., 1989). Coefficients
of 0.43 and of 24.4 were obtained for 0.9% saline : air and for olive
oil : air, respectively.
4.1.2 Water and sediments
VC has a relatively low solubility in water (see section 2), and
the solubility can be increased by the presence of salts (see section
4.2.3).
Experimental data on adsorption to particulate matter in the
water column or to sediment are not available. A partition constant
(unitless) of 8.2 for a sediment-water system was calculated (from a
Kow value of 17) by Mabey et al. (1982), indicating a low adsorption
capacity. A high input of VC into water may lead to low-level
long-term storage in the associated sediment (Hill et al., 1976b).
Volatilization of VC acts as a significant transport mechanism.
It is considered to be the most rapid route for removal of VC from
surface water, but to be an unlikely pathway for disappearance from
groundwater that is not directly exposed to air (Smith & Dragun,
1984). Volatilization parameters such as vapour pressure and Henry's
Law constants indicate that VC is highly volatile. Another factor, the
reaeration rate ratio (rate constant for loss of VC from aqueous
solution divided by the rate constant for oxygen uptake by the same
solution), was reported to be 0.675 at 25°C (theoretical calculation
by Mabey et al., 1982) and approximately 2 (experimental measurement
by Hill et al., 1976b). The results of measurements or calculations of
volatilization half-lives of VC from water bodies are given in Table
6; they range from < 1 h to 5 h (measured in models for disturbed or
quiescent water) and from 2.5 to 43 h (calculated for natural water
bodies). In groundwater, however, VC may remain for months or years
(ATSDR, 1990).
More or less complex models have been developed to describe the
stability of VC in aquatic ecosystems (Hill et al., 1976b; Miller,
1992).
4.1.3 Soil and sewage sludge
Owing to its high vapour pressure, VC can be expected to
volatilize rapidly, especially from dry soil surfaces. No experimental
data are available. However, volatilization of VC from soil can be
predicted based upon its physicochemical properties. The amount of VC
volatilized from a soil depth of 1 m in 1 year was reported to range
from 16 to 45% in a sandy soil and 0.1 to 0.7% in a clay soil. These
calculations were based upon a Henry's Law constant of 0.44 and a
degradation half-life of between 30 and 180 days (Jury et al., 1992).
It has been observed at landfill sites that subsurface migration
of VC is a significant transport mechanism (Hodgson et al., 1992;
Little et al., 1992; Ward et al., 1996). Quantitative experimental
data on the potential of VC for gaseous subsurface migration (see
section 5.1.1) were not found.
Because of its solubility in water, VC can be leached through the
soil to groundwater. Additionally, the high solubility of VC in many
organic solvents may increase its mobility at special locations, e.g.,
landfills or waste disposal sites. Standard experimental studies on
soil sorption of VC are lacking. The soil adsorption coefficients of
VC were estimated from its water solubility, octanol/water partition
coefficient and from the molecular topology and quantitative
structure-activity relationship analysis method according to equations
given by Chiou et al. (1979), Kenaga & Goring (1980), Lyman et al.
(1982, 1990) and Sabljic (1984). The Koc values obtained ranged from
14 to 240, indicating a low adsorption tendency and therefore a high
mobility of VC introduced into soil (US EPA , 1985a; Stephens et al.,
1986; ATSDR, 1997).
A field study performed to determine non-extractable (bound)
residues (NER) of highly volatile chlorinated hydrocarbons gave a low
value of 2.4% for VC, as measured in a lysimeter after one growth
period in the upper (10 cm) soil layer (Klein et al., 1989).
VC was assumed not to appear frequently in sewage sludge due to
its low adsorption potential (log Kow < 2.0) and its high
volatilization tendency (Wild & Jones, 1992).
Table 6. Half-lives reported for volatilization of VC from water
Source Method Conditions Half-life Reference
Dilute aqueous experimental 22-25°C, Dilling et al. (1975);
solution (200 ml) (laboratory) rapid continuous 25.8 min Dilling (1977);
stored in an stirring
open container continuous stirring (200 rpm) 26-27.6 min Callahan et al. (1979)
discontinuous stirring approx. 80-90
(5% of time) min
quiescent, no stirring 290 min
Flowing channel experimental water input: 35 litre/second 0.9 h Scherb (1978)
(field) flow velocity: < 0.50 m/second
depth: 30 cm
Stream calculation based on Hc = 243 kPa.m3/mol 2.5 h Lyman et al. (1990)
depth: 1 m
flow velocity: 1 m/second
wind current: 3 m/second
calculation based on reaeration rate ratio of US EPA (1985a)
2; assumed oxygen reaeration ratesa:
Pond 0.008 h-1 43.3 h
River 0.04 h-1 8.7 h
Lake 0.01 h-1 34.7 h
a According to Tsivoglou (1967); Lyman et al. (1990)
4.1.4 Biota
VC has been identified in environmental samples of fish tissue
(see section 5.1.5) and in several species of aquatic laboratory
animals (molluscs, crustaceans, insects and vertebrates) and algae
experimentally exposed to VC-containing water (see section 4.3).
Reports on the presence of VC in terrestrial plants and animals have
not been found in the literature.
Further experimental data, e.g., in what way and to what extent
VC is capable of entering the biota, are lacking. However, a few
estimations have been made on the basis of the physicochemical
properties of VC. They refer to uptake of VC by terrestrial plants and
animals. Plant uptake was considered to be unlikely (Ryan et al.,
1988; Shimp et al., 1993) because, at an assumed half-life of
< 10 days (Ryan et al., 1988), VC should be lost from the system
rather than be taken up by the plant. Another study (Wild & Jones,
1992) screened organic contaminants for possible transfers into plants
and animals by summarizing approach data. Within the three categories
used (high, moderate, low potential) VC was classified in the
following way:
* retention by root surface: low;
* uptake and translocation: moderate;
* foliar uptake: high;
* transfer to animal tissues by soil ingestion: low;
* transfer to animal tissues by foliage ingestion: moderate.
4.2 Transformation
4.2.1 Microbial degradation
There have been many studies on the biodegradation of VC under
various simulated environmental conditions and these are listed in
Tables 8 to 10. The biodegradation studies have given contradictory
results, with no evidence of degradation under some aerobic conditions
such as surface water (Hill et al., 1976b) and sewage (Helfgott et
al., 1977) and under anaerobic conditions such as groundwater
(Barrio-Lage et al., 1990). Unacclimated biodegradation half-lives of
VC were generally estimated to be of the order of several months or
years (Howard et al., 1991). However, other studies reported complete
degradation in 3 months in simulated aerobic groundwater (Davis &
Carpenter, 1990). Where degradation is reported care must be taken to
ensure that the loss of VC is due to degradation and not from other
losses such as volatilization from the test system. Phelps et al.
(1991b) reported that > 99% of VC was lost from a bioactive reactor
compared to 60% from the control reactor.
Significant microbial degradation of VC under aerobic and
anaerobic conditions has been detected in studies using enrichment or
pure cultures isolated mostly from sites contaminated with different
organic chemicals (Tables 8 to 10). Vinyl chloride cannot be used by
most microorganisms as sole carbon source, but it can be
degraded/metabolized in the presence of propane, methanol,
3-chloropropanol, propylene, isopropene and glucose. However, in some
cases VC can even serve as sole substrate, as is seen with
Mycobacterium sp. (Table 9). The main degradation products include
glycolic acid or CO2 after aerobic conversion (Tables 7 to 9) and
ethane, ethene, methane or chloromethane after anaerobic
transformation (Table 10). Anaerobic mineralization of VC to CO2 has
been demonstrated (Table 10) and may occur under special conditions
(Bradley & Chapelle, 1998b).
A complete mass balance was given by degradation studies with
radiolabelled VC, for example, in aerobic resting cell suspensions of
Rhodococcus sp. A starting concentration of 1 mg [1,2-14C] VC
produced more than 66% 14CO2 and 20% 14C aqueous phase products, and
10% was incorporated into the biomass (Malachowsky et al., 1994).
Aerobic cultures of Mycobacterium aurum growing on a special filter
material were reported to mineralize VC quantitatively according to
the following equation:
VC + microorganisms -> biomass + HCl + CO2 + H2O (Meier, 1994).
The underlying reaction mechanisms for the aerobic and anaerobic
degradation of VC have been postulated to be oxidative and reductive
dehalogenations, respectively, involving a variety of pathways (Vogel
et al., 1987; Barrio-Lage et al., 1990; Ensley, 1991; Castro et al.,
1992a; Leisinger, 1992; Castro, 1993; Meier, 1994; Hartmans, 1995;
Jain & Criddle, 1995).
Frequently, the degradation reaction of VC proceeds more readily
with aerobes than with anaerobes (Tables 7 to 10). The reverse occurs
(Freedman & Gossett, 1989; Semprini et al., 1995) with PCE, an
important precursor of VC in the environment (see chapter 3). Thus,
two-stage treatment systems consisting of anaerobic (first stage) and
aerobic (second stage) cultures have been proposed to achieve the
complete degradation of a range of alkenes having different degrees of
chlorination (Leisinger, 1992; Murray & Richardson, 1993; Nelson &
Jewell, 1993). Recently, a chlorobenzoate-enriched biofilm reactor
using Desulfomonile tiedjei DCB-1 was developed which degraded PCE
under anaerobic conditions without any detectable VC remaining
(Fathepure & Tiedje, 1994).
There have been many efforts to use the VC-degrading capacities
of microorganisms in practical applications such as the purification
of waste gases (Meier, 1994) or of municipal waste waters (Narayanan
et al., 1995) and the remediation of landfill leachates (Lesage et
al., 1993), groundwaters (McCarty, 1993; Holliger, 1995) and
contaminated soils (Schulz-Berendt, 1993). Possible limitations arise
from physical (temperature, accessibility of substrate), chemical (pH,
redox state, concentration of VC and other contaminants, presence of
additional secondary substrates, salinity) and biological (presence of
predators, competition phenomena, adsorption of microbes to surfaces)
factors (Van der Meer et al., 1992).
Table 7. Aerobic degradation of vinyl chloride by mixed microbial consortia from different sites
Inoculum Test design/ Measured Initial Duration Efficiency of Reference
conditionsa parameter concentration degradation
Surface water room VC 20 ml/2.9 ml 41 h no degradation Hill et al. (1976b)
samples temperature
Mixed consortium 21°C; VC 20-120 mg/litre several no degradaion Hill et al. (1976b)
from natural + / - nutrients weeks
aquatic systems
Mixed consortium 20°C oxygen 25 days no degradation Helfgott et al.
from domestic + nutrients demand (1977)
sewage
Mixed consortium 25°C; 14CO2 0.05 mg/litre 5 days 21.5% degradation Freitag et al.
from activated + nutrients (1982, 1985)
municipal sewage
sludge
Naturally simulated 14CO2, 1 mg/kg 108 days > 99% degradation Davis &
occurring aquifers: VC soil-water 65% mineralization Carpenter
consortium from soil-water (CO2) (1990)
groundwater microcosms 0.1 mg/kg 109 days 50% mineralization
(prepared with soil-water (CO2)
sub-surface soil
and groundwater
(20°C)
Consortium aquifer sediment 14CO2, 17 µmol/litre 84 h 22-39% Bradley &
indigenous to microcosms VC mineralization Chapelle (1996)
anaerobic aquifer (CO2)
systems
(contaminated
with CHs)
Table 7. (cont'd)
Inoculum Test design/ Measured Initial Duration Efficiency of Reference
conditionsa parameter concentration degradation
Consortium creek bed sediment 14CO2, 0.2-57 µmol/litre 24 h 6.2-58% Bradley &
indigenous to microcosms VC mineralization Chapelle (1998a)
creek sediment (CO2)
(contaminated
with DCE)
Consortium from soil microcosms VC 5.3 mg/litre 95 h little change Dolan & McCarty
aquifer material in VC (1995a)
(from a concentration
VC-contaminated
site)
a CH = chlorinated hydrocarbons; DCE = dichloroethene
Table 8. Elimination of vinyl chloride in aerobic tests with mixed microbial consortia utilizing special substratesa
Inoculum Additional Test Efficiency of Remarks Reference
substrate degradation
Mixed methane laboratory removal of up inhibition by Fogel et al. (1986,
methanotrophs studies to 100% within methane and 1,1-DCE 1987); Strandberg et al.
4 h-30 days possible; toxic (1989); Uchiyama et al.
effects of VC and VC (1989); Nelson & Jewell
products possible (1993); Dolan & McCarty
(1995a); Chang & Alvarez-Cohen
(1996)
methane field study about 95% inhibition by Semprini et al. (1990, 1991)
(groundwater) in-situ methane possible
transformation
i.c. =
0.03 mg/litre
Mixed propane laboratory > 99% loss after > 60% loss in Phelps et al. (1991b);
microbial study 30 days control Lackey et al. (1994)
consortia i.c. =
4-20 mg/litre
methane laboratory 82 to > 99% Phelps et al. (1991b);
plus study loss after 10-21 Lackey et al. (1994)
propane days; i.c. =
1-20 mg/litre
Mixed methane laboratory 2.3 µmol VC per relative TCs from Dolan & McCarty (1995a)
methanotrophs plus study mg of cells during highest to lowest:
formate 26 h i.c. = trans-DCE;
14.8 mg/litre cis-DCE; VC;
TCE; 1.1-DCE
Table 8. (cont'd)
Inoculum Additional Test Efficiency of Remarks Reference
substrate degradation
Mixed methane laboratory removal of up to Deipser (1998)
consortia study > 99% within 15
days i.c. =
0.55 mg/litre
a i.c.= Initial concentration; DCE = dichloroethene; TCE = trichloroethene;
TC = transformation capacity
Table 9. Survey on isolated bacterial cultures capable of degrading vinyl chloride under aerobic conditions
Inoculum Additional Major degradation Remarks Reference
substrate producta
Mixed culture n. sp. CO2 (> 67%) Malachowsky et al. (1991)
consisting of
Rhodococcus rhodochrous
and 2 bacteria of the
order Actinomycetales
Bacterium of the order propane, CO2 (> 67%) Phelps et al. (1991a)
Actinomycetales glucose or
acetate
Alcaligenes denitrificans isoprene n. sp. Ewers et al. (1990)
ssp. Xylosoxidans
Methylosinus trichosporium methane n. sp. inactivation Tsien et al. (1989); Chang
OB3b possible & Alvarez-Cohen (1996)
methane glycolic Castro et al. (1992a)
acid (44%;
determined at
68% conversion)
Mycobacterium sp. ; no CO2 ; initially: VC as primary Hartmans et al. (1985, 1992);
M. aurum chlorooxirane substrate; Hartmans & DeBont (1992);
(epoxide) inhibition Meier (1994)
possible
Mycobacterium no CO2, HCl maximum growth Hauschild et al. (1994)
aurum L1 rates at
1 mmol
VC/litre
Mycobacterium vaccae propane n. sp. Wackett et al. (1989)
(JOB 5)
Table 9. (cont'd)
Inoculum Additional Major degradation Remarks Reference
substrate producta
Nitrosomonas europaea ammonia n. sp. Vannelli et al. (1990)
Pseudomonas sp. 3-chloro-propanol glycolic acid Castro et al. (1992b)
(71%; determined
at 25%
conversion)
Rhodococcus sp. propane CO2 (> 66%) Malachowsky et al. (1994)
Rhodococcus isoprene n. sp. Ewers et al. (1990)
erythropolis
Xanthobacter propylene n. sp. Ensign et al. (1992)
(strain Py 2)
a The degradation efficiency is given in parentheses; n. sp. = not specified
Table 10. Survey on anaerobic microbial degradation of vinyl chloridea
Inoculum Test design/ Conditions Major degradation Efficiency of Remarks Reference
products degradation
Mixed methanogenic liquid cultures n. sp. i.c. = 400 µg/litre Brauch et al.
consortium (from (20°C) groundwater (1987)
PCE-, TCE-, with sterile sand approx. 50% (100%)
VC-contaminated after 4 (11) weeks
groundwater)
without sterile sand approx. 20% (55%)
after 4 (11) weeks
Mixed consortium incubation of n.sp. 94% within 16 days Nerger &
(from groundwater plus i.c. = 18 µg/litre Mergler-Völkl
TCE-contaminated waste water (1988)
water) (9 : 1)( 21°C)
Mixed consortium digesters filled n.sp. low degradation Deipser (1998)
(from compost) with mature sieved (0.2 mg/m3 compost/h)
compost from private
households
Mixed consortium soil-groundwater (< 1% CO2) no degradation in 5 Barrio-Lage et
(from natural microcosms (25°C) months al. (1990)
sites) i.c. = 2 mg/litre
Mixed consortium flow-through column methane plus 89% degradation traces of Barrio-Lage et
(from natural packed with soil, ethene (82%), (in 9-15 days) chloromethane al. (1990)
sites) a. s.: mixture of CO2 (7%)
phenol, citrate,
ammonium
dihydrogenphosphate,
methanol and methane
Table 10. (cont'd)
Inoculum Test design/ Conditions Major degradation Efficiency of Remarks Reference
products degradation
Consortium aquifer sediment CO2 15-34% mineralization Bradley &
indigenous microcosms, anaerobic in 84 h (versus 3-5% Chapelle (1996)
to anaerobic conditions plus without Fe-EDTA
aquifer systems Fe(III) as Fe-EDTA amendment)
(contaminated i.c. = 17 µmol/litre
with CHs)
Mixed varying conditions chloroethane slow degradation Baek et al.
methanogenic or 15-30°C or ethene (1990); Carter &
methanol-enriched (plus ethane) Jewell (1993);
consortia Skeen et al.
(1995)
Enriched PCE- and batch cultures ethene partial to nearly inhibition by Freedman &
TCE-degrading (35°C) complete degradation PCE possible Gossett (1989);
consortia DiStefano et al.
(1991); Tandoi
et al. (1994)
Mixed anaerobic fixed-bed columnb ethene plus almost complete De Bruin et al.
consortia from 24°C a.s.: ethane conversion of (1992)
river sediment lactate PCE (95-98%)
and wastewater via VC
sludge
Methanobacterium resting cell no degradation Castro et al.
thermoautotrophicum suspensions (60°C) i.c. = 10-3 (1994)
mol/litre
Table 10. (cont'd)
Inoculum Test design/ Conditions Major degradation Efficiency of Remarks Reference
products degradation
"Dehalococcoides anaerobic H2-PCEb ethene 90% conversion decay in rate Maymó-Gatell
ethenogenes strain enrichment culture of PCE via VC of VC et al. (1995,
195"c + mixed conversion 1997)
microbial consortia
a a.s. = additional substrate; i.c. = initial concentration; n.sp. = not specified; CH = chlorinated hydrocarbons;
PCE = tetrachloroethene; TCE = trichloroethene
b starting material PCE
c preliminary name
4.2.2 Abiotic degradation
4.2.2.1 Photodegradation
Studies on the photodegradation of VC are summarized in Table 11.
They include direct and indirect photolysis.
VC in the vapour phase or in water does not absorb wavelengths
above 220 nm or 218 nm, respectively (Hill et al., 1976b). However,
solar radiation reaching the troposphere lacks wavelengths below about
290 nm due to the stratospheric ozone shield. So, direct photolysis of
VC is expected to be insignificant under environmental conditions,
because there is no overlap between the absorption spectrum of VC and
the sunlight radiation spectrum (Callahan et al., 1979). Consistently,
no photodegradation was observed with pure VC in the gas phase or in
water at wavelengths above 220 nm. After irradiation at 185 nm, VC was
photolysed (Table 11).
In the environment, indirect photolysis occurs and includes
reactions of VC in the presence of photosensitizers and those
(Table 12) with photochemically produced reactive particles.
A variety of photolytic products was formed after irradiation of
VC under several experimental conditions (Table 11). Some
intermediates, e.g., chloroacetaldehyde, were of considerable
photochemical stability. Therefore, the photooxidation is unsuitable
as a means of removing VC from waste gases (Gürtler et al., 1994). On
the other hand, treatment of water contaminated with VC and other
halogenated organic compounds by means of UV-enhanced oxidation
(UV/ozone or UV/hydrogen peroxide) was reported to be successful (Zeff
& Barich, 1992).
The atmospheric fate of VC depends on its reaction with reactive
particles such as free OH and NO3 radicals, Cl atoms, ozone and
singlet oxygen. As can be seen from Table 12, the reaction with OH
radicals is the dominant transformation process, showing calculated
tropospheric half-lives of 1-2 days or more. Factors influencing
indirectly (via OH radical concentration) the lifetime of VC are the
degree of air pollution and solar radiation, leading to spatial,
diurnal and seasonal variations (e.g., Hesstvedt et al., 1976).
Reaction products include formaldehyde (HCHO) and formyl chloride
(HCOCl), the latter being a stable potential toxicant (Tuazon et al.,
1988; Pitts, 1993).
Rate constants for the photodegradation of VC in aqueous
solutions have been reported to range from 6.99 × 109 mol-1 second-1
(Grosjean & Williams, 1992) to 7.1 × 109 mol-1 second-1 (Klöpffer et
al., 1985). Mabey et al. (1982) reported that photolysis of VC was not
an environmentally relevant process. They reported oxidation rate
constants of < 108 mol -1h -1 and 3 mol -1h -1 for reactions with
singlet oxygen (1O2) and peroxy radicals (RO2), respectively. On
the basis of an average OH radical concentration of 10-17 mol in
Table 11. Survey on vinyl chloride photolysis studies
Medium Irradiation Photolytic degradation Photolytic productsa Reference
(Duration)
VC in a high-vacuum medium pressure yes primary products: radicals Fujimoto et al. (1970)
system arc (C2H3, Cl); C2H2, HCl
VC in air sunlight (outdoors) yes n.sp. Pearson & McConnell
(half-life = 11 weeks ± 50%) (1975)
xenon arc (> 290 nm) yes CO (90%); HCl
VC in air high pressure yes (> 99% within 15 min) chloroacetaldehyde, Kagiya et al. (1975)
mercury lamp HCl, CO2
outdoors yes (55% within 2 days)
VC in air (dry) > 230 nm (4 h) yes chloroacetaldehyde (primary Müller & Korte (1977)
product), HCl, CO, formyl
chloride
VC in air < 400 nm (45 min) yes CO2, CO, H2O, HCl, HCOOH, Woldbaek & Klaboe
C2H2 (1978)
sunshine (3-45 h) very slow
VC (adsorbed on > 290 nm (n.sp.) yes n.sp. Freitag et al. (1985)
silica gel) (15.3% of applied amount)
VC in an oxygen 185 nm (up to 50 min) yes formyl chloride, Gürtler et al. (1994)
atmosphereb (quantum yield: 2-3) monochloroacetaldehyde,
acetylene, CO, CO2,
monochloroacetyl chloride,
HClc, formic acidc
254 nm (up to 6 h) no
VC in air xenon lamp yes n.sp. Haag et al. (1996)
(initial k: 0.09 second-1)
Table 11. (cont'd)
Medium Irradiation Photolytic degradation Photolytic productsa Reference
(Duration)
VC in air plus UV (> 290 nm) yes formic acid, HCl, CO, Cox et al. (1974);
nitrogen oxides (up to 22 h) (half-life = 1-7 h, + NO formaldehyde, ozone, Dilling et al. (1976);
half-life = 18 h, - NO) (other minor products) Gay et al. (1976);
Carassiti et al.
xenon lamp yes formaldehyde, Hcl (1977) Kanno et al.
(0-120 min) (1977)
< 400 nm (25 min) yes at low NO2 conc.: the same Woldbaek & Klaboe
(increase in reaction rate products as observed in air (1978)
as compared to NO2/NO (see above);
being absent) at high NO2 conc.:
additionally nitrosyl
chloride, N2O
VC in air plus xenon lamp yes n.sp. Haag et al. (1996)
1,1-DCE (initial k: 0.15 second-1)
VC in pure water > 300 nm no Hill et al. (1976b)
(10 mg/litre) (90 h)
VC in natural water > 300 nm no
samples (20 h)
(10 mg/litre)
VC in water plus > 300 nm yes (rapid) various products
photosensitizers
VC in PVC plant > 300 nm yes (half-life = 40 h) n.sp.
effluent sunlight (25 h) very little
a n.sp. = not specified; DCE = dichloroethene
b direct photolysis
c in the presence of water vapour
Table 12. Rate constants and half-lives for gas-phase reactions of vinyl chloride with OH radicals and other reactive particles
VC reaction Rate constant Temperature Assumed atmospheric Calculated Reference
with a (in units of (°C) c concentration of the half-life c,d
cm3/molecule-sec) b reactive particle c
* OH 5.6 × 10-12 27 1 × 106 molecules/cm3 1.4 days Cox et al. (1974);
(measured) US EPA (1985a)
* OH 4.5 × 10-12 23 (296 K) 1 × 106 molecules/cm3 1.8 days Howard (1976); US
(measured) EPA (1985a)
* OH 6.60 × 10-12 26 (299 K) n.sp. n.sp. Perry et al. (1977)
5.01 × 10-12 85 (358 K)
3.95 × 10-12 149 (423K)
(measured)
* OH 6.60 × 10-12 26 1 × 106 molecules/cm3 1.2 days US EPA (1985a)
(Perry et al., 1977)
* OH 6.60 × 10-12 room temperature 1 × 106 molecules/cm3 (3.5 days) Atkinson et al.
(Perry et al., 1977) (298 ± 2 K) (12-h daytime average, (1979, 1987);
Crutzen, 1982) Atkinson (1985)
* OH 6.60 × 10-12 room temperature n.sp. n.sp. Atkinson (1987)
(measured)
5.3 × 10-12
(calculated)
* OH 6.60 × 10-12 25 ± 2 5 × 105 molecules/cm3 (approx 3 days) Tuazon et al. (1988)
(Perry et al., 1977) (298 ± 2 K)
* OH 6.60 × 10-12 26 5 × 105 molecules/cm3 2.2-2.7 days BUA (1989)
(Perry et al., 1977) (Crutzen, 1982)
* OH 6.8 × 10-12 27 (300 K) 5 × 105 molecules/cm3 approx 2.4 days BUA (1989)
(Becker et al., 1984) (Crutzen, 1982)
Table 12. (cont'd)
VC reaction Rate constant Temperature Assumed atmospheric Calculated Reference
with a (in units of (°C) c concentration of the half-life c,d
cm3/molecule-sec) b reactive particle c
* OH 6.60 × 10-12 26 8 × 105 molecules/cm3 1.5 days Howard (1989)
(Perry et al., 1977)
* OH 4.0 × 10-12 26 (299 K) n.sp. n.sp. Kirchner et al.
cm3/mol-sec (1990)
(measured)
* OH 10.6 × 10-12 n.sp. n.sp. n.sp. Klamt (1993)
(calculated)
* OH n.sp. n.sp. 1 × 106 molecules/cm3 (42 h) Pitts (1993)
(24-h average)
* OH 6.60 × 10-12 n.sp. 6.5 × 105 molecules/cm3 (2.7 days) Helmig et al.
(Perry et al., 1977) (estimated global mean; (1996)
Tie et al., 1992)
* OH 6.60 × 10-12 26 7.5 × 105 molecules/cm3 1.6 days Palm (1997)
(Perry et al., 1977) (24-h average, BUA, 1993) personal
communication
* NO3 2.3 × 10-16 room temperature 2.4 × 109 molecules/cm3 (42 days) Atkinson et al.
(measured) (298 ± 2 K) (12-h nighttime average, (1987)
Platt et al., 1984)
* NO3 1.4 × 10-16 23 (296 ± 1 K) n.sp. n.sp. Andersson &
(measured) Ljungström (1989)
Cl 12.7 × 10-11 25 (298 ± 2 K) n.sp. n.sp. Atkinson & Aschmann
(measured) (1987); Grosjean &
Williams (1992)
Table 12. (cont'd)
VC reaction Rate constant Temperature Assumed atmospheric Calculated Reference
with a (in units of (°C) c concentration of the half-life c,d
cm3/molecule-sec) b reactive particle c
O3 1.9 × 10-19 25 n.sp. n.sp. Gay et al. (1976);
Singh et al. (1984)
O3 2.0 × 10-18 n.sp. n.sp. 4 days Hendry & Kenley
(1979);
ECETOC (1983)
O3 2.45 (± 0.45) × 10-19 room temperature n.sp. n.sp. Zhang et al. (1983)
(approx. 25)
O3 2.5 × 10-19 25 7 × 1011 molecules/cm3 (66 days) Atkinson & Carter
(Zhang et al., 1983) (Singh et al., 1978) (1984); Atkinson et
al. (1987)
O3 2.45 × 10-19 25 1 × 1012 molecules/cm3 33 days US EPA (1985a)
(Zhang et al., 1983)
O3 1.2 × 106 27 1.6 × 1012 molecules/cm3 4.2 days Lyman et al. (1982,
cm3/mole-sec 1990); US EPA (1985a)
O3 1.7 × 10-19 22 7 × 1011 molecules/cm3 67 days Klöpffer et al.
(1988)
O3 2.5 × 10-19 (calculated n.sp. n.sp. n.sp. Meylan & Howard
according to AOP) (1993)
19 × 10-19 (calculated n.sp. n.sp. n.sp.
according to FAP)
O (3P) 8.6 × 10 -13 25 2.5 × 104 molecules/cm3 373 days Sanhueza & Heicklen
(1975); US EPA
(1985a)
Table 12. (cont'd)
VC reaction Rate constant Temperature Assumed atmospheric Calculated Reference
with a (in units of (°C) c concentration of the half-life c,d
cm3/molecule-sec) b reactive particle c
O (3P) 5.98 × 10 -13 25 2.5 × 104 molecules/cm3 532 days Atkinson & Pitts
(1977); US EPA
(1985a)
a O(3P) = oxygen atom
b AOP = Atmospheric Oxidation Program (currently used by US EPA); FAP = Fate of Atmospheric Pollutants (part of US EPA's Graphical
Exposure Modeling system, GEMS)
c n.sp. = not specified
d Values in parentheses reported as 'lifetime'
natural water, the kOH rate constant resulted in a half-life of
approximately 110 days (US EPA, 1985a). Both of the other reactions
appeared to be negligible.
4.2.2.2 Hydrolysis
Observations on chemical hydrolysis of VC derive from experiments
with effluent water from a VC plant (pH = 4.3-9.4; 50°C; 57 h;
Callahan et al., 1979), water of different pH values (85°C; 27 h; Hill
et al., 1976b; Mabey et al., 1982), water saturated with O2 (85°C;
12 h; Hill et al., 1976b), water plus ethanol (120°C; Rappoport & Gal,
1969) and two natural water samples (pH = 6.1/4.2 for river/swamp
water, both at room temperature and at 85°C; 41 h; Hill et al.,
1976b). In all cases, no or only slow hydrolysis occurred. The
hydrolytic half-life was estimated to be < 10 years at 25°C (Hill et
al., 1976b). Hydrolysis experiments under strongly alkaline, high
temperature (and therefore environmentally irrelevant) conditions
resulted in a polymerization of VC (Jeffers & Wolfe, 1996).
4.2.3 Other interactions
Under experimental conditions, VC and chlorine in water form
chloroacetaldehyde, chloroacetic acid and other unidentified compounds
(Ando & Sayato, 1984).
Many salts have the ability to form complexes with VC, thus
possibly leading to an increase in its solubility (Callahan et al.,
1979).
4.3 Bioaccumulation
Owing to its high vapour pressure and low octanol/water partition
coefficient, VC is expected to have little tendency for
bioaccumulation (BUA, 1989). Theoretical calculations resulted in
bioconcentration factors (BCFs) of 2.8 (based on a log Kow of
approximately 0.9) and around 7 (based on a water solubility of
2763 mg/litre) in aquatic organisms (US EPA, 1985a). A BCF of 5.7
(based on log Kow = 1.23) was calculated by Mabey et al. (1982) for
aquatic microorganisms.
Experiments performed with 14C-VC (initial concentration:
250 µg/litre) in a closed laboratory model aquatic ecosystem gave the
following results: after 3 days at 26.7°C 34% of the 14C was found in
the water and 65% in the air. The organisms from different trophic
levels contained 14C residues (in VC equivalents, µg/kg) of 1307
(alga, Oedogonium cardiacum), 621 (waterflea, Daphnia magna), 123
(snail, Physa sp.), 1196 (mosquito larva, Culex pipiens
quinquefasciatus) and 312 (fish, Gambusia affinis) (Lu et al.,
1977). These values led to BCFs ranging from 3 to 31 when compared to
the VC water concentration of 42 µg/litre, indicating some
bioaccumulation, but no biomagnification within the food chain.
Another study determined BCFs for green algae (Chlorella fusca)
after a 24-h exposure to 0.05 mg VC/litre and for fish (golden ide,
Leuciscus idus melanotus) exposed to a constant average
concentration of 0.05 mg VC/litre over 3 days. BCFs of 40 and < 10,
respectively, were found (Freitag et al., 1985).
After 5 days of incubation of 0.05 mg VC/litre in activated
sewage sludge, an accumulation factor of 1100 (based on the
distribution of VC between sludge, dry weight and water) was observed
(Freitag et al., 1985).
4.4 Ultimate fate following use
4.4.1 Waste disposal
Several methods have been employed for removal of VC from waste
water: stripping with air, steam or inert gas (Nathan, 1978;
Cocciarini & Campaña, 1992; Hwang et al., 1992), extraction (Nathan,
1978) or adsorption onto activated charcoal or adsorbent resin
(Nathan, 1978; Dummer & Schmidhammer, 1983, 1984).
Like VC waste gases produced during other processes (see
chapter 3), the recovered VC can be recycled or incinerated (US EPA,
1982; BUA, 1989). Special biological filters have been developed for
degrading VC in waste emissions (Meier, 1994, 1996; see also section
4.2.1).
Incineration leading to the total destruction of VC requires
temperatures ranging from 450°C to 1600°C and residence times of
seconds for gases and liquids, or hours for solids (HSDB, 1995).
Photochemical oxidations (Topudurti, 1992; Zeff & Barich, 1992;
Berman & Dong, 1994; see also section 4.2.2) are further methods of VC
elimination. UV-enhanced oxidation (oxidants used: ozone, hydrogen
peroxide) was applied for purification of polluted waters, (e.g.,
waste, leachate, groundwater) (Zeff & Barich, 1992). Treatment with
sodium dichromate in concentrated sulfuric acid was recommended for
the destruction of small quantities of VC, for instance, from
experimental laboratories (HSDB, 1995).
Chlorinated volatile organic compounds (VOCs) can be removed from
drinking-water/groundwater by treatment with activated charcoal
(Schippert, 1987), air stripping (after water is pumped to the
surface) (Boyden et al., 1992) or by air sparging (applied in situ)
(Pankow et al., 1993). Recently, on-site and in situ bioremediation
techniques, which couple evaporative or other methods with microbial
treatment, have been developed for restoration of groundwater systems
(Roberts et al., 1989; Portier et al., 1992, 1993; Fredrickson et al.,
1993; McCarty, 1993; Lackey et al., 1994) or soils (Schulz-Berendt,
1993) contaminated with VC and other VOCs.
4.4.2 Fate of VC processed to PVC
Most of the VC produced is used for the manufacture of PVC (see
section 3) and will therefore be connected with the fate of PVC. PVC
and articles made from it can be disposed of in landfills,
incineration or feedstock recycling. While rigid PVC is an extremely
persistent material, flexible PVC may be less recalcitrant to
disintegration (Harris & Sarvadi, 1994). At incineration, PVC plastics
do not depolymerize to form VC (Harris & Sarvadi, 1994), but produce
volatile aliphatic hydrocarbons and volatile chlorinated organic
compounds (Nishikawa et al., 1992; see also section 3.2.4). There is
evidence for formation of PCDFs/PCDDs (Theisen et al., 1989, 1991;
IPCS, 1989).
5. ENVIRONMENTAL LEVELS AND HUMAN EXPOSURE
5.1 Environmental levels
There is very little exposure of the general population to VC.
Concentrations of VC in ambient air are low, usually less than
3 µg/m3. Exposure of the general population may be higher in
situations where large amounts of VC are accidentally released to the
environment, such as a spill during transportation. However, such
exposure is likely to be transient. Near VC/PVC industry and waste
disposal sites, relatively much higher concentrations, up to
8000 µg/m3 and 100 µg/m3, respectively, have been observed. VC has
only rarely been detected in surface waters, sediment or sewage
sludges. Maximal VC concentrations in groundwater or leachate from
areas contaminated with chlorinated hydrocarbons amount to 60 000
µg/litre.
5.1.1 Air
5.1.1.1 Outdoor air
Atmospheric air levels of VC in rural/remote and suburban/urban
areas range from not detectable to 24 µg/m3 (Table 13). Higher values
were recorded in industrial areas, with maxima in the vicinity of
VC/PVC producing or processing plants, even at distances of 5 km. Peak
concentrations were as high as 86 mg/m3 (33 ppm) and 17 mg/m3 (7
ppm), measured, respectively, in 1974 in the USA and 1983 in China
(Table 13). In many countries, VC concentrations near plants have
decreased with time due to regulatory measures (Table 13).
In more recent years attention has been paid to the occurrence of
VC near waste disposal sites, where levels of up to 0.18 mg/m3
(70 ppb) have been detected (Table 13). This is much less than the
significant amounts of VC found in undiluted landfill gas (see
section 3). Bruckmann & Mülder (1982) assumed that gas discharges of
landfills are diluted by a factor of 104 when entering the
atmosphere.
VC emissions of < 0.5 µg/m3 (remote from VC plants) or of a few
µg/m3 (near VC plants) in Germany and the Netherlands were derived
from a computer model (Besemer et al., 1984; WHO, 1987; LAI, 1992).
Ambient air concentrations in three geographical areas of the USA were
computed to range from trace to about 3 µg/m3 (Pellizzari et al.,
1979).
Table 13. Vinyl chloride measured in ambient (atmospheric) aira
Country; site and Valueb Concentrations Reference
year of sampling (µg/m3)
Rural/remote areas single 0.1 Kruschel et
Canada: pine forest al. (1994)
near Barrie, Ontario
year n. sp.
Germany: Westerland, mean values 6.6-24 Bauer
Schwarzwald, (1981)
Lüneburger Heide,
Bayer Wald; prior to
1977
Germany: Taunus; mean value 0.1 Dulson
1975 (n = 4) (1978)
USA: rural Northwest (n = n. sp.) n.d. (< 0.013) Grimsrud &
USA (Pullman, Rasmussen
Washington); (1975)
1974-1975
USA: over ocean (n = n. sp.) n.d. (< 26) Lillian et
Sandy Hook, NJ al. (1975)
(3 miles offshore);
1974
Suburban/urban areas
Canada: urban (22) meanc 0.06 Dann &
and rural (1) sites; (n = 1370; 4% Wang (1992)
1989-1990 > detection)
Germany: Berlin range (n=78) n.d. (n.sp.) - 3.5 Dulson
(3 sites); 1977 means 0.3-0.4 (1978);
Lahmann
(1980)
Germany: Frankfurt/M. n = 1 2.6 Bergert &
(city); 1974 Betz (1976)
Germany: Frankfurt/M. mean (n = 16) 21.1 Arendt et
(suburban); 1975 al. (1977)
Germany: Merkenich annual mean 3.6 German Expert
(North-Rhine Group on the
Westfalia); 1980 Environment
(1988)
Table 13. (cont'd)
Country; site and Valueb Concentrations Reference
year of sampling (µg/m3)
Germany: Cologne means 0.5-15.3 Anon (1981);
(3 stations); (n = n.sp.) BUA (1989)
1979-1986
Germany: several mean values 0.2-11 Bouscaren
sites; year n. sp. et al. (1987)
Germany: Hamburg annual means 2.4-9.0 (including Bruckmann
(12 sites in the city); (n = 300) 1-butene) et al. (1988)
1986-1987
USA: New Jersey geom. mean 1981: 0 Harkov et
(3 urban sites: (3/113)d and 1982: 0 al. (1983,
Newark, Elizabeth, (0/105)d (detection limit: 1984)
Camden); 1981-1982 0.013)
USA (eastern): urban daily mean 0.62 Dann &
sites; 1989 (n=397; 2% Wang (1992)
> detection)
Industrial sites
Canada: Shawinigan range n.d. - 117 Thériault
(vicinity of VC et al. (1983)
polymerization
plant); year n. sp.
China: dormitories for n = 16×3 Zhao et al.
workers (50, 100 and (4 h/day for (1994)
1000 m from a VC 4 days at
polymerization plant) 3 stations)
1983 maxima 17 400 / 5900 / 1500
daily means 4810 / 1080 / 580
1984 maxima 7800 / 2500 / 1400
daily means 4080 / 720 / 500
1986 maxima 6600 / 2000 / 1100
daily means 3120 / 820 / 520
1988 maxima 12 700 / 1000 / 300
daily means 4430 / 320 / 200
1989 maxima 3100 / 700 / 300
daily means 760 / 220 / 170
Table 13. (cont'd)
Country; site and Valueb Concentrations Reference
year of sampling (µg/m3)
Finland: 1.2 km from Kinnunen
a PVC plant (1996)
1993 range of 0.1-1.3
monthly means
1994 range of 0.1-0.8
monthly means
1995 range of < 0.1-0.1
monthly means
range of single 0.1-11
measurements
(n approx. 4000)
1996 range of 0.1-0.4 Kinnunen
monthly means (1997)
range of single 0.1-13
measurements
(n approx. 7000)
Germany: n.sp.; maximum 252 Bouscaren
prior to 1980 et al. (1987)
Germany: Ruhrgebiet 99th percentile 69 BUA (1989)
West; year n. sp.
Germany: 1978-1982 max. (n = n.sp.) 113 Bauer (1981)
Germany: Marl (80 m mean (n = 24) 213 Rohrschneider
and 500 m above mean (n = 9) 108 et al. (1971)
chemical plant);1970
Germany: Frankfurt/M. n.sp. 4.5 Atri (1985)
(industrial area);
year n.sp.
Netherlands: > 600 m maximum ca. 600 Besemer
from VC plant; mean 210 et al.
1976-1977 (1984)
0-500 m distant range (n = 200) < 21-504
500 m distant; 1978 range (n = 100) 13-55
mean 18
Netherlands: range 7.8-181 Guicherit &
VC/PVC plants; (n = n.sp.) Schulting
1980 (1985)
Table 13. (cont'd)
Country; site and Valueb Concentrations Reference
year of sampling (µg/m3)
UK: 5 VC plants; overall means < 13-228 Turner et al.
1984 (April-July) daily means 23-218 (1984)
(ranges related
to the 5 plants,
total n = 440)
Plant VI (7 stations, max. (n = 28); 20 202 (0 km),
0-4.8 km distant) 880 (0.8 km)
Plant IX (15 stations, means (n = 180); 311-1373
0.8-5 km distant) maximum 8806 (5 km)
USA: 1974-1975; VC (n = 708) US EPA
plant (Narco) means 3.4-262 (1975);
(4 stations, geom. means 0-28 Dimmick
< 250 - > 1000 m maximum 27 045 (< 250 m) (1981)
distant)
PVC plant (Aberdeen) (n = 438)
(4 stations, means 11.6-958
< 250 - > 1000 m geom. means 3.7-372
distant) maximum 23 430 (300 m)
PVC plant (Louisville) (n = 712)
(4 stations, means 11.4-101
< 250 - > 1000 m geom. means 5-30
distant) maximum 814 (250-400 m)
USA: residential areas range (n = 30) n.d. - 104 US EPA
near chemical plants (1975)
(n = 15); 1974
USA: Houston, Texas; range (n = 18.) 8-3238 Gordon &
1974 Meeks
(1977)
USA: residential max. (n = n.sp.) > 2560 Fishbein
areas in the vicinity mean (n = n.sp.) 44 (1979)
of VC/PVC plants;
prior to 1975
USA: vicinity of max. 34 McMurry &
VC/PVC plants (Texas, Tarr (1978)
7 stations); 1977
Table 13. (cont'd)
Country; site and Valueb Concentrations Reference
year of sampling (µg/m3)
Areas in the vicinity
of waste sites
Belgium: Mellery; prior no details 19 Lakhanisky
to 1990 et al. (1993)
Germany: surroundings no details < 3 Pudill (1993)
of a hazardous waste
site; about 1988
Germany: 2 landfills range < 0.082-0.65 Wittsiepe
near Bochum; 1990-1991 (n = 16) et al. (1996)
USA: West Covina, range (24-h av; 26-104 Camarena &
California; 1981-1984 n = 32) max 130 Coy (1984)
USA: West Covina, 24-h averages 13-31 (n = 5 days) Baker &
California; 1984 5-day average 23 MacKay
site A (200 m distant 24-h averages 5.7-18 (1985)
from landfill) 5-day average 10.4 (n = 5 days)
site B (20 m distant
from landfill)
USA: southern max. 18 Stephens
California, 1981 range 5-18 et al. (1986)
after 1981 (6 stations)
USA: California; max. 30 Little et
prior to 1990 al. (1992)
USA: New York range n.d. (5.4) - 16 Lipsky &
(2 sites) 1982 Jacot
(1985)
Miscellaneous
USA: all outdoor site mean 8.5 Shah &
types; prior to 1988 (n = 574) Singh
(1988)
a n.d. = not detected (detection limit in parentheses, if specified);
n.sp. = not specified
b This column indicates whether the concentration is a maximum (max),
average or range value
c Values below detection set to 0.5 method detection limit (0.1 µg/m3)
d Values in parentheses = no. detected / no. sampled
5.1.1.2 Indoor air
Indoor air concentrations of VC in houses near landfills in the
USA reached concentrations of up to 1 mg/m3 air (Little et al., 1992:
2 landfills, maxima of 0.13 and 0.3 mg/m3; Stephens et al., 1986:
1 landfill, maximum of 1 mg/m3), thus exceeding the maximum outdoor
levels reported in Table 13 for areas adjacent to landfills. Moreover,
the Californian monitoring programme, collecting a total of 500 air
samples at two outdoor and four indoor sites downwind of a landfill,
revealed that the 120 samples containing the most VC
(> 0.025 mg/m3; 10 ppb) were taken inside homes (Little et al.,
1992). It is assumed that in addition to atmospheric transport,
subsurface migration of VC accounts for the elevated indoor air levels
of VC (Wood & Porter, 1987; Hodgson et al., 1992; Little et al.,
1992).
A room being painted with a red latex paint based on a terpolymer
of vinyl chloride, vinyl acetate and ethylene showed VC levels of
75 and 10 µg/m3 (29 and 4 ppb), respectively, during and some time
(less than one day) after painting (Going, 1976).
In the early 1970s it was investigated whether VC was present in
car interiors as a result of volatization from PVC. Measurements of VC
concentrations in the interior of seven different new 1975 automobiles
gave positive results for two of them. Levels of 1036 to 3108 µg/m3
(0.4 to 1.2 ppm) were detected (detection limit: 130 µg/m3; 0.05 ppm)
(Hedley et al., 1976). Another study (Going, 1976) did not find VC in
the interior ambient air of 16 new and used cars and 4 mobile homes
(detection limit: 26 µg/m3; 10 ppb). It should be noted that since
this time the levels of VC in PVC resins have been drastically reduced
(see section 3.2.4).
5.1.2 Water and sediment
Owing to its high volatility, VC has rarely been detected in
surface waters. The concentrations measured generally do not exceed
10 µg/litre, with a maximum of 570 µg/litre from contaminated sites
(Table 14).
Much higher levels of up to 56 000 µg/litre have been found in
groundwater samples from contaminated sites (Table 15).
The levels in drinking-water supplies ranged from not detected to
2 µg/litre in samples collected in 100 German cities in 1977. In a
state-wide USA study performed in 1981-1982, random samples (taken
from randomly selected water systems) had concentration ranges of n.d.
to 1.1 µg/litre, and non-random samples (taken from systems that were
likely to be contaminated with VOCs) varied from n.d. to 8 µg/litre.
Prior to 1980 single maximum values of up to 380 µg/litre were
reported from the USA (Table 16).
Table 14. Vinyl chloride concentrations measured in surface watera
Country, source Year of sampling Valueb Concentrations Remarks Reference
(µg/litre)
Germany
River Rhine (prior to) 1978 typical conc. 1 Anna & Alberti (1978)
(n = many)
River Rhine 1982 < 0.2 Malle (1984)
River Rhine 1990 range (n = 78) < 0.01-0.031 Wittsiepe (1990)
Tributaries of Rhine (prior to) 1978 typical conc. < 1-5 Anna & Alberti (1978)
(Northrhine-Westfalia) (n = many)
Surface water from n.sp. range < 0.0004-0.4 > 150 samples Wittsiepe et al. (1990)
unspecified sites (n = n.sp.)
in former FRG
River Main 1990 range (n = 22) < 0.004-0.008 Wittsiepe (1990)
River Lippe 1989 range (n = 54) 0.12-0.4 receiving Wittsiepe (1990)
wastewater from
VC/PVC plants
River Ruhr (plus 1990 range (n = 60) < 0.0004-0.005 Wittsiepe (1990)
artificial lake) (lake: up to
0.06)
River Wupper 1989 range (n = 36) up to 0.069 Wittsiepe (1990)
River Saale 1990 range (n = 4) up to 69 receiving Wittsiepe (1990)
wastewater from
an industrial
area of the
former GDR
Table 14. (cont'd)
Country, source Year of sampling Valueb Concentrations Remarks Reference
(µg/litre)
Japan
Rivers in Osaka 1995 range (n = 28) up to 1.2 (3/28)c Yamamoto et
al. (1997)
USA
Delaware River 1976-1977 (n = 11) n. d. Sheldon & Hites
(1978)
Surface water from 1977-1979 maximum 566 (21/606)c Page (1981)
different sites (n = 606)
in New Jersey median 0
Surface water from n.sp. maximum 9.8 Burmaster (1982);
9 states (n = n.sp.) Dyksen & Hess (1982)
Final effluent from a 1980-1981 mean 6.2 Gossett et al. (1983)
waste-water treatment (n = 5)
plant in Los Angeles
Surface waters prior to 1984 median < 5 (63/1048)c Staples et al. (1985)
(n = 1048)
Indian River Lagoon n.sp. (n = n.sp.) n.d. (< 1.0) Wang et al. (1985)
(near water discharge
of VC), several
stations
Surface water near 1985-1990 (n = n.sp.) n.d. Hallbourg et al. (1992)
3 landfills in
Florida
Table 14. (cont'd)
Country, source Year of sampling Valueb Concentrations Remarks Reference
(µg/litre)
Surface water at a 1989-1990 range 0.23 Chen & Zoltek (1995)
landfill in Florida 1992-1993 (n = 5) n.d.
(Orange County)
a n.d = not detected (detection limit in parentheses, if specified); n.sp. = not specified;
GDR = German Democratic Republic
b This column indicates whether the concentration is a maximum (max), average or range
value
c Values in parentheses = no. detected / no. sampled
One reason for the occurrence of VC in drinking-water may be that
residual VC can migrate from PVC pipes used in some water distribution
systems into the water flowing through them. This has been found out
by field (Dressman & McFarren, 1978) and experimental (Banzer, 1979;
Nakamura & Mimura, 1979; Ando & Sayato, 1984) studies. The extent of
leaching depended on the VC concentration in the pipe material. In the
field study the highest VC concentrations (1.4 µg/litre) consistently
occurred in water from new pipes, whereas the lowest level (0.03
µg/litre) was found in the oldest (9 years of age) distribution
system. VC concentrations in landfill leachate samples amounted to up
to 61 mg/litre (Table 17). A gross analysis of water (no
specification) available for the USA and based on 5553 observations
reported maximum and median concentrations of VC as high as
202.6 mg/litre and 107 µg/litre, respectively (US EPA, 1985a).
No VC was detected in urban stormwater run-off from 15 cities in
the USA (n = 86) during a monitoring project concerning priority
pollutants (Cole et al., 1984).
Generally, a time trend cannot be derived from the water analysis
data available.
Most sediment samples contain very low VC concentrations, even at
rather contaminated sites. Sediment was monitored for VC during
1981-1982 in Florida (USA) at several stations (n = 8) of the Indian
River Lagoon and a conveying canal. Although the latter received
discharged water having VC concentrations of 34-135 µg/litre, no VC
was detected in the sediment samples (3 from each station, collected
monthly over a year), the detection limit being 2 ng/g (Wang et al.,
1985). The same was true for surface water (Table 14) and oyster
(section 5.1.5) samples from this site. Sediment samples (n = 2) taken
near the discharge zone of a wastewater treatment plant in Los Angeles
County (California, USA) contained < 0.5 µg VC/kg dry weight. The
corresponding water concentration of VC was 6.2 µg/litre (Gossett et
al., 1983). A survey of 343 sediment samples from the USA gave a
median VC concentration of < 0.5 µg/kg dry weight (Staples et al.,
1985). However, higher VC concentrations were also reported. According
to US EPA (1985a), VC was detected in sediment samples (no further
details given) in the USA at levels ranging from 0-580 µg/kg (n = 649;
median = 23 µg/kg).
5.1.3 Soil and sewage sludge
5.1.3.1 Soil
Subsurface soil samples near the waste pit of an abandoned
chemical cleaning shop in southern Finland showed VC concentrations as
high as 900 mg/kg (Salkinoja-Salonen et al., 1995). After an
accidental spillage of VC into snow in 1980, VC levels as high as
500 mg/kg were measured in the soil at up to 2 m depth (Charlton
et al., 1983).
5.1.3.2 Sewage sludge
VC has been detected in municipal sewage sludges in the USA. The
concentrations ranged from 3 to 110 mg/kg dry weight (corresponding to
145 to 3292 µg/litre), being detected in 3 of 13 samples, with a
median concentration of 5.7 mg/kg dry weight (corresponding to
250 µg/litre) (Naylor & Loehr, 1982). Another study reported a mean
concentration of 35.4 mg/kg dry weight for 6 of 44 samples (Fricke
et al., 1985). A range of 8-62 000 µg/litre was found in 35 of 435 raw
sludge samples (Burns & Roe, 1982).
5.1.4 Food, feed and other products
VC is not a general contaminant of foodstuff and pharmaceutical
or cosmetic products, but it can be detected after contact of these
products with PVC packaging materials. The use of PVC as packaging
material for food, drink and drugs began in the early sixties (chapter
3), whereas legislative action for safeguarding consumers from
exposure to VC did not begin until the early seventies (starting with
a ban on the use of PVC containers for packaging alcoholic beverages
in the USA; Anon, 1973). Current EC and US FDA regulations on the
level of VC in PVC materials intended to come into contact with
foodstuffs are listed in Annex 1.
VC concentrations measured in PVC-packed food and drink of
several countries are compiled in Table 18. A maximum value of
20 mg/kg was found in liquors. Other positive samples included
vegetable oils (up to 18 mg/kg), vinegars (up to 9.8 mg/kg),
margarines (up to 0.25 mg/kg), fruit drinks (> 0.2 mg/kg) and bottled
water (< 0.6 µg/litre).
Retail surveys of foods showed a significant reduction in VC
levels and/or in the number of positive samples since 1974 (UK MAFF,
1978, 1984; van Lierop, 1979; Codex Committee, 1984).
The latest data were from the 1990s (Table 18) and refer to
bottled drinking-water. In addition to small amounts of VC, there were
also indications for the presence of possible reaction products of VC
with chlorine (Fayad et al., 1997).
Pharmaceutical and cosmetic products were less frequently
monitored. The highest concentration, amounting to 7.9 mg/kg, was
detected in mouthwashes (Table 19).
Reports on analyses of animal feed were not available.
The potential for leaching of residual VC from PVC packaging into
the contents has been demonstrated by a variety of product analyses
(Tables 18 and 19) and by experimental studies using food or food
simulants (Daniels & Proctor, 1975; Hocking, 1975; Tester, 1976;
Diachenko et al., 1977; Pfab & Mücke, 1977; UK MAFF, 1978; Chan et
al., 1978; vom Bruck et al., 1979; van Lierop, 1979; Benfenati et al.,
1991; Thomas & Ramstad, 1992). Altogether, the results indicated that
Table 15. Vinyl chloride concentrations measured in groundwatera
Country; source Year of Valueb Concentrations Remarks Reference
of groundwater sampling (µg/litre)
Canada
from a VC spill site 1980 maximum; 10 weeks 10 000 Charlton et al.
after the spill < 20 (1983)
beneath a landfill 1988 range (n = 37) < 1-40 (5/37)c Lesage et al.
near Ottawa (1990)
Finland
from village wells mid-1980 range 5-200 mg/litre Nystén (1988);
(contaminated by leakage of (including DCE) Salkinoja-Salonen
a waste liquor basin of et al. (1995)
VC/PVC industry - detected
in 1974)
Germany
from different sites mean values n.d. (< 5) - 460 Brauch et al.
(4 sites) (1987)
from different wells mean values 15-1040
of a large surface
contamination (5 wells)
from a catchment area of range (n = 30) < 1-120 Milde et al.
a water-works (1988); Nerger &
(TCE / PCE-contaminated) Mergler-Völkl
(1988)
contaminated by waste until 1988 range (n = 113) < 1-12 000 (14/113)c Schleyer et al.
disposal sites (92 mean 2700 (1988)
sites) median 475
Table 15. (cont'd)
Country; source Year of Valueb Concentrations Remarks Reference
of groundwater sampling (µg/litre)
from a site contaminated n.sp. (n = 3) corresponding air Köster (1989)
with chlorinated pockets in soil
hydrocarbons 1000 128 000 µg/m3
500 47 000 µg/m3
200 5000 µg/m3
from a site contaminated ca. 1989 max. (n = 5) 110 Leschber et
with chlorinated al. (1990)
hydrocarbons (in Berlin)
from a contaminated site n.sp. range (n = 3) 710-1670 Kästner (1991)
(in Braunschweig)
of a catchment area of a 1989 (n = n.sp.) up to 0.130 Wittsiepe et al.
waterworks (1990)
Contaminated by accidental 1989 (n = n.sp.) up to 3
spillage of TCE and PCE
Contaminated by waste n.sp. mean (n = 136) 1693 18% positive Dieter &
disposal sites (approx. max. 12 000 Kerndorff (1993)
100 sites)
USA
from different sites in 1977-1979 max. (n = 1060) 9.5 (4/1060)c Page (1981)
New Jersey
from 9 states max. 380 7% positive Dyksen & Hess
(1982)
monitoring wells near n.sp. max. 635 present in Stuart (1983)
industrial waste sites in 3 out of 9 wells
Connecticut
Table 15. (cont'd)
Country; source Year of Valueb Concentrations Remarks Reference
of groundwater sampling (µg/litre)
from Nassau County, 1980 range (n = >100) 1.6-2.5 Connor (1984)
Long Island
from Miami, Florida n.sp. mean (n = 3) 6.8 Parsons et al.
(1984)
from a TCE spill site in n.sp. mean (n = 3) 82 Parsons et al.
Vero Beach, Florida (1984)
monitoring wells near MSW n.sp. present (in Sabel & Clark
landfills in Minnesota 5/20 sites), (1984)
but not quantified
near 3 plants manufacturing n.sp. range (n = 3) 50-500 chlorinated White Paper
electronics equipment solvents stored in (1984) cited in
(Santa Clara Valley) underground tanks Wolf et al.
(1987)
near solvent recovery range (n = 4) Cline & Viste
facilities (1985)
Connecticut 1980 n.d. (< 10) - 2700
Wisconsin 1983 n.d. (< 10) - 210
from a residential area 1983 maximum 2800 contamination Andreoli
in Long Island, New York from dry-cleaning (1985)
(several wells) shop
near a landfill in (about max. 692 Shechter (1985)
New Jersey 1981) (n = approx. 100)
monitoring wells around 1985 max. 2600 seasonal, spatial Stephens et al.
hazardous waste landfill and analytical (1986)
in S. California variations
Table 15. (cont'd)
Country; source Year of Valueb Concentrations Remarks Reference
of groundwater sampling (µg/litre)
monitoring wells near 1990 range (n = 64) n.d. (1-2) - 12 (5/64)c EMO (1992)
an airforce base in Ohio
near 3 landfills in Florida 1985-1990 mean values < 0.22-26.5 Hallbourg et al.
(10 locations) (1992)
monitoring wells at a 1989-1990 range (n = 3) n.d. - 0.23 increase of VC Chen & Zoltek
landfill in central 1992-1993 4.8-48.4 with time, decrease (1995)
Florida (Orange County) in total VOC
from an industrial site; (n = n.sp.) 51-146 Topudurti (1992)
recycling operations
(1940-1987); California
near a former waste disposal (n = n.sp.) US EPA (1992)
facility in Wisconsin:
on-property max. 77
off-property max. 5
Contaminated by 1991 maxima < 5-56 400 Semprini et al.
TCE > 10 years before minima < 5-321 (1995)
(17 sites)
near Plattburg, New York n.sp. (n = 2) 8 and 384 Bradley & Chapelle
(2 locations) (1996)
a n.d. = not detected (detection limit in parentheses, if specified); n.sp. = not specified; TCE = trichloroethene;
PCE = tetrachloroethene; VOC = volatile organic compounds; max. = maximum; MSW = municipal solid waste
b This column indicates whether the concentration is a maximum (max), average or range value
c Values in parentheses = no. detected / no. sampled
Table 16. Vinyl chloride measured in drinking-water suppliesa
Country; source Year of sampling Valueb Concentration Remarks Reference
(µg/litre)
Germany
drinking-water supplies 1977 range n.d. - 1.7 Bauer (1981)
of 100 cities
drinking-water n.sp. (n = n.sp.) ca. 1.6 ng/litre Wittsiepe et al.
(no details) (1993)
USA
finished drinking-water (prior to) 1975 max. (n = n.sp.) 10 Fishbein (1979)
water supplies from (prior to) 1975 max. (n = n.sp.) 5.6 & 0.27
Florida and Philadelphia
raw drinking-water 1975-1979 2 positives 2.2 & 9.4 (2/13)c CEQ (1981)
(13 cities)
finished drinking-water 1 positive 9.4 (1/25)c
(25 cities)
drinking-water wells (prior to) 1980 max. (n = n.sp.) 50 CEQ (1981);
(state New York) Burmaster (1982);
Craun (1984)
drinking-water of (prior to) 1981 range 0.05-0.18 Kraybill (1983)
113 cities mean 0.052
drinking-water 1977-1981 range of trace - 380 (82/1288)c Cotruvo et al.
supplies (166) positives (1986)
Table 16. (cont'd)
Country; source Year of sampling Valueb Concentration Remarks Reference
(µg/litre)
finished water supplies 1981-1982 max.: Westrick et al.
(using groundwater random samples: 1.1 (1/466)c (1984)
sources) from 51 states (n = 466)
in the USA non-random 8.4 (6/479)c
samples:
(n = 479)
private wells in 1982 max. 4.5 (1/63)c Goodenkauf &
Nebraska (n = 63) Atkinson (1986)
a n.d. = not detected (detection limit in parentheses, if specified); n.sp. = not specified
b This column indicates whether the concentration is a maximum (max), average or range value
c Values in parentheses = no. detected / no. sampled
Table 17. Vinyl chloride concentrations measured in leachatea
Country; source Year of sampling Valueb Concentrations Remarks Reference
(µg/litre)
Canada
MSW landfill leachate 1988 n.sp. 14 Lesage et al. (1993)
from Guelph (Ontario) 1989 n.sp. 23
USA
landfill leachates n.sp. (n = 6) not quantified present in 1/6 Sabel & Clark (1984)
from Minnesota samples
landfill leachates prior to 1982 max. (n = 4) 61 (1/4)c
from Wisconsin
landfill leachates prior to 1985 range (n = 5) n.d. Cline & Viste (1985)
(municipal and (< 10) - 120
industrial)
landfill leachates: prior to 1988 range total number of Brown & Donnelly
(n = n.sp.) landfills: 58 (1988)
municipal 20-61 000
industrial 140-32 500
MSW leachates (several (prior to) 1988 range 8-61
states in the USA) (n = n.sp.)
median 40 (6/?)c Chilton & Chilton
(1992)
a n.d. = not detected (detection limit in parentheses, if specified); n.sp. = not specified;
MSW = municipal solid waste
b This column indicates whether the concentration is a maximum (max), average or range value
c Values in parentheses = no. detected / no. sampled
the amount of VC migrating into food or solvent was proportional to
the VC concentration in the PVC packaging, to storage time and to
increasing temperature.
Until 1974 the use of VC as a propellant in aerosol sprays was
allowed in the USA (chapter 3). Realistic application of such aerosol
products (hairspray, deodorant, insecticide, disinfectant, furniture
polish or window cleaner) resulted in high (ppm range) indoor air
concentrations of VC (Gay et al., 1975).
There have been no reports on VC levels found in food,
pharmaceutical or cosmetic products in recent years. This may be due
to regulatory measures for PVC packaging in several countries (see
Annex 1).
5.1.5 Terrestrial and aquatic organisms
Oysters ( Crassostrea virginica) from the Indian River Lagoon in
Florida (USA) contained no detectable levels of VC (detection limit:
0.4 ng/g). The samples were collected weekly (1981) and monthly (1982)
at three sites (Wang et al., 1985). VC was also not detected in the
corresponding water and sediment samples (section 5.1.2).
Another study (Gossett et al., 1983) did find low concentrations
of VC (< 0.3 µg/kg wet weight) in a sample of small invertebrates
from just above the bottom sediments (n = 1), in a muscle sample of
shrimp (n = 1) and in liver samples of several fish species (n = 4).
The animals were collected in 1981 in final effluent waters from a
wastewater treatment plant in Los Angeles County (Palos Verdes,
California, USA) that had VC concentrations of 6.2 µg/litre (section
5.1.2).
Data on fish tissue (no further details given) available from the
US EPA STORET database were reported by US EPA (1985a). VC levels
ranged from 0-250 mg/kg (n = 530; median: 6 mg/kg).
5.2 General population exposure
Exposure of the general population to VC is possible by several
routes. They include inhalation of air polluted with VC (section
5.1.1), mainly in the vicinity of VC/PVC plants or waste disposal
sites, intake of contaminated drinking-water (sections 5.1.2 and
5.1.4), ingestion of food, beverages and medicines packed in PVC
(section 5.1.4), and absorption through skin from PVC-wrapped
cosmetics (section 5.1.4).
Normally, the general population is exposed to only small amounts
of VC, if at all. However, the exposure varies according to the
countries' regulatory measures, the occurrence of accidents or the
spread of precursor substances.
Table 18. Levels of vinyl chloride in food and drink packaged, stored or transported in PVC articles
Country Yeara Product No.a,b Concentrationsc Reference
(µg/kg)
Canada n.sp. alcoholic beverages 22 < 25-1600 Williams & Miles
vinegars 28 n.d. (10) - 8400 (1975)
peanut oil 10 300-3300
Canada n.sp. alcoholic beverages 10 n.d. (10) - 2100 Williams (1976a)
vinegars 10 300-7800
vinegars 9 14-9800 Williams (1976b)
sherry 3 500-2400
peanut oil 3 3800-18 000
Canada n.sp. oil 5 80-2100 Page & O'Grady (1977)
vinegars 5 10-5700
Canada 1981-1982 vinegars n.sp. 27-43 Codex Committee (1984)
other foods n.sp. < 10
Italy n.sp. drinking-water 10 0.013-0.083 Benfenati et al. (1991)
(PVC-bottled)
Netherlands 1975 oil samples 8 > 50 Van Lierop (1979)
salad dressing 1 250
margarine 4 60-250
ready-made salads n.sp. > 50
(whole
batch)
wine n.sp. 760
Netherlands 1976 margarine 1 60 Van Lierop (1979)
fish 1 90
biscuit 3 20-130
Table 18. (cont'd)
Country Yeara Product No.a,b Concentrationsc Reference
(µg/kg)
1977 peanut butter 1 4100 Van Lierop (1979)
(very small
individually wrapped
portions)
1978 various foods: 67 (3 +) n.d. (0.1) - 2 Van Lierop (1979)
soya oil 1 0.3
vinegar 1 0.6
salmon salad 1 2
Norway 1975 butter/margarine 16 n.d. (2) Ehtesham-Ud Din
salad 14 2-15 et al. (1977)
juices 8 6-25
vinegar 27 6-2790
mustard 5 9-14
Saudi Arabia n.sp. drinking-water 9 × 48d < 0.6 Fayad et al. (1997)
(PVC-bottled)
Sweden 1974 edible fats 127 n.d. (2) - 127 Fuchs et al. (1975)
Sweden 1975-1976 various foods 104 n.d. (2) - 600 Albanus et al. (1979)
(edible fats and oils, (mostly: < 10)
ketchup, vinegar, lime
juice, fruit syrup)
Switzerland 1973-1975 edible oils 41 n.d. (5) - 1750 Rösli et al. (1975)
United Kingdom n.sp. spirits n.sp. 0-250 Davies & Perry (1975)
Table 18. (cont'd)
Country Yeara Product No.a,b Concentrationsc Reference
(µg/kg)
United Kingdom 1974 fruit drinks 25 10 - > 200 UK MAFF (1978)
1977 13 < 10
1974 cooking oil 23 10 - > 200
1977 7 < 2
1975 butter, soft margarine 51 < 2 - 200
1977 9 < 2
USA 1971 vegetable oil 1 7000 Breder et al. (1975)
1974 3 700
USA 1973 spirits n.sp. up to 20 000 Anon (1973);
US FDA (1973)
USA 1973-1975 alcoholic beverages n.sp. 11 000-25 000 Codex Committee (1984)
a n.sp. = not specified
b + = number of samples positive
c n.d. = not detected (detection limit in parentheses, if specified)
d 9 brands (locally produced and imported)
Table 19. Vinyl chloride detected in pharmaceutical and cosmetic
products packaged in PVC materials
Product No.a Concentrationb Reference
(µg/kg or µg/litre)
Blood coagulant 30 n.d. (15) Breder et al.
solutions (1975)
Mouthwashes 11 n.d. (20-30) - 7900
Several capsules, tablets 11 n.d. (10-30) Watson et al.
and mouthwashes (1977)
Large volume 5 n.d. (0.1) - < 1 Watson et al.
parenterals (1979)
Mouthwashes 5 7-120
Shampoos 4 9-17
Body oils 4 n.d. (0.1) - 41
Intravenous solutions n.sp. n.d. (1) Arbin et al.
(many) (1983)
Cefmetazole sodium 4 n.d. (0.3) - <1 Thomas &
(Zefazone(R) sterile Ramstad
powder) (1992)
a n.sp. = not specified
b n.d. = not detected (detection limit in parentheses, if specified)
5.2.1 Estimations
Estimations of the respiratory intake of VC reported for the USA
ranged from 0 to 48.3 mg per person per day, based on exposure values
of 0 to 2.1 mg/m3 and assuming that 23 m3 of air are inhaled per day
(US EPA, 1985b). According to Seiber (1996) over 100 000 Californians,
particularly those living near landfills, may be exposed to VC levels
of 2.59 µg/m3 (1 ppb) or more.
A European study (Besemer et al., 1984) evaluating the exposure
of the Dutch population to VC in ambient air assumed an average
exposure of about 0.2 µg/m3, which resulted in a calculated daily
intake of 4 µg VC per person. Small fractions of the population were
concluded to be exposed to higher average levels: 0.01% (> 8.5
millions) to > 5 µg/m3, 0.04% to 4-5 µg/m3, and 0.05% to
3-4 µg/m3. The corresponding estimated daily intakes of VC were
> 100 µg, 80 µg and 60 µg, respectively.
Intake via drinking-water from public water supplies in the USA
was estimated to exceed 1 µg/litre for 0.9% of the population,
5 µg/litre for 0.3% and 10 µg/litre for 0.1% of the population. These
would result in daily VC intakes (assuming a 70-kg man and 2 litres of
water/day) of > 2, 10, and 20 µg/day. Maximal values were estimated
to be approximately 120 µg/day (US EPA, 1985b).
Another study (Benfenati et al., 1991) estimated the daily intake
of VC from PVC-bottled drinking-water bought in Italian supermarkets.
Based on the analytical results (Table 18) and assuming a consumption
of 2 litres of water per person per day and a storage time of 2 months
for the PVC bottles, the authors calculated that the oral intake could
exceed 100 ng VC per person per day.
Evaluation of the results of food surveillance programmes from
the United Kingdom led to a calculated maximum likely VC intake of
1.3 µg/day per person in 1974, of 0.1 µg/day per person in 1976, and
less than 0.02 µg/day per person by 1978 (UK MAFF, 1978, 1984). These
calculations were based on the typical daily consumption of fruit
drink, cooking oil and soft margarine.
Evaluating and summing up possible maximum user intakes of VC
from PVC-bottled liquor, wine and oil, and food packaged in PVC
materials, the US FDA calculated a maximum lifetime-averaged exposure
of 25 ng/person per day (US FDA, 1986). An earlier review reported an
estimated dietary intake of VC of 40 ng/day per person in the USA
(Codex Committee, 1984).
The intake by food and drinking-water in the Netherlands was
estimated (without specifying details) to be about 0.1 µg/day per
person or less (Besemer et al., 1984). Estimates (no details given) of
the average human intake for Switzerland were reported to be 3 ng/kg
body weight per day (Lutz & Schlatter, 1993).
5.2.2 Monitoring data of human tissues or fluids
Monitoring human tissues or fluids as an indirect measure of
exposure to VC has not frequently been applied. As with workers in the
plastics industry (section 5.3), the urine of premature babies was
found to contain large amounts of thiodiglycolic acid (thiodiacetic
acid), a metabolite of VC (chapter 6), but the relationship to
possible VC exposure was questionable (Pettit, 1986).
5.3 Occupational exposure
The main route of occupational exposure to VC is via inhalation
(Sittig, 1985), while dermal absorption is considered to be negligible
(ECETOC, 1988).
Industrial environments associated with VC exposure include VC
production plants, VC polymerization (PVC production) plants and PVC
processing factories. Estimates of numbers of workers exposed to VC
were, for example, in the USA (1981-1983) in the range of 80 000
(ATSDR, 1997) or in Sweden (1975-1980) more than 5000 (Holm et al.,
1982). Since, at the onset of VC/PVC production in the USA and Western
Europe, VC was not recognized as a toxic compound, no precautions
against contact were provided for nor was regular workplace monitoring
performed. Therefore, only sporadic measurements or retrospective
estimates (Table 20) of exposures are available for the period prior
to 1975. Published data from various countries on VC contamination of
workplace air throughout the early and later periods are compiled in
Table 21 and 22. Highest exposures occurred in the VC/PVC production
plants, with peak concentrations of several thousand ppm whereas much
lower exposure levels were measured in processing plants. Owing to
standard-setting and legislative regulations by national authorities
and technical improvements, levels dropped markedly to values of a few
ppm in many countries. Official exposure limits can serve as an
additional indication of approximate VC concentrations occurring in
plants in many countries. These limits have declined gradually (IARC,
1979). Generally, standards require that exposures do not exceed 13 to
26 mg/m3 (5 to 10 ppm) (Torkelson, 1994; Rippen, 1995; ACGIH, 1999).
However, even in the 1990s the standards were not always realized
in all countries (Table 21).
Whereas in industrialized countries factories that could not
satisfy the rigorous regulations of the early 1970s to reduce VC
emissions were forced to close down, in the countries of eastern
Europe and developing countries this was not possible for
socioeconomic reasons and large plants with old-fashioned technologies
continued to function (Hozo et al., 1996).
Table 20. Retrospective estimates of daily occupational
exposures to vinyl chloride prior to 1975a
Country Period VC exposure Reference
(mg/m3)
Germany "first years" > 2600 Szadkowski & Lehnert
prior to 1971 1300 (1982)
1971 260
1974 5.2-7.8
Norway 1950-1954 5200 Hansteen et al. (1978);
1955-1959 2600 Heldaas et al. (1984)
1960-1967 1300
1968-1972 260
1973-1974 207
Sweden 1945-1954 1300 Holm et al. (1982)
1955-1964 780
1965-1969 520
1970-1974 130
United Kingdom 1945-1955 2600 Barnes (1976);
1955-1960 1040-1300 Anderson et al. (1980);
1960-1970 780-1040 Purchase et al. (1987)
mid 1973 390
1975 13
1940-1955 1300-2070 Jones et al. (1988)
1956-1974 390-1300
USA 1945-1955 2600 Wu et al. (1989)
1955-1970 780-1300
1970-1974 260-520
1975 < 2.6-13
a Exposure levels during autoclave cleaning may have been as high as
7800 mg/m3 (Barnes, 1976)
Table 21. Levels of vinyl chloride reported for workplace air samples in VC/PVC production plants
Country Workplace Yearc Concentrations reported Reference
(mg/m3)c
China PVC production plant n.sp. 30-210 Bao et al. (1982)
Croatia plastics industry n.sp. mean = 13 Fucic et al. (1990a)
5200 (occasional peak)
Croatia VC/PVC plant 1949-1987 mean = 543 Hozo et al. (1996,
up to 1300 (occasional peak) 1997)
Former n.sp. n.sp. 2-41 Hrivnak et al. (1990)
Czechoslovakia
Egypt VC/PVC plant n.sp. 0.05-18 (8-h TWA) Rashad et al. (1994)
Finland PVC production plant, n.d. range
breathing zone 1981-1985 1.6 < 0.3-57 Viinanen (1993)
concentrations (TWA) 1986-1989 1.6 < 0.3-46
1993 0.3 < 0.3-26
France PVC production plant 1977-1978 2.3-7.3 (range of monthly Haguenoer et al.
means) (1979)
Germany PVC production 1974 < 65-181 Fleig & Thiess (1974)
department
PVC production plant 1977 1.3-91 German Environmental Office (1978)
PVC production plant 1979 12 (12-h TWA; stationary); Heger et al. (1981)
15.5 (12-h TWA; personal)
Germany 24 plants 1981-1984 3% of 33 samples: > 5 Coenen (1986);
(90 percentile: < 1); BIA (1996)
(shift means)
Table 21. (cont'd)
Country Workplace Yearc Concentrations reported Reference
(mg/m3)c
46 plants 1989-1992 all of 117 samples: < 5
(90 percentile: < 0.1);
(shift means)
Italy VC/PVC plants 1950-1985 < 13- > 1300 Pirastu et al. (1991)
The Netherlands PVC plant 1976-1977 2.6-26 (8-h TWA) De Jong et al. (1988)
Norway PVC plant 1974 65 Hansteen et al.
(1978)
Poland VC/PVC plant 1974 (30-600)a Studniarek et al.
(several departments) 1975 (30-270)a (1989)
1976 (15-60)a
1977 (6-150)a
1978 (1-30)a
1979 (1-15)a
1981 (0.1-36)a
1982 (0.1-12)a
(autoclave cleaners) 1974 (990)a
1982 (9-180)a
breathing zone of VC 1986 21.3 Dobecki &
synthesis mechanic 1987 66.9 Romaniwicz (1993)
1988 43.7
1989 0.7
1990 0.2
Romania PVC production plant 1965-1967 112-554 Anghelescu et al.
(1969)
Russia VC/PVC plant Gáliková et al. (1994)
16 probes (whole plant) 1990-1993 1-9 (range of annual means)
under the reactor 1990-1993 up to 200 (range of annual
means)
Table 21. (cont'd)
Country Workplace Yearc Concentrations reported Reference
(mg/m3)c
in compressor room 1990-1993 up to 400 (range of annual
mean)
Singapore PVC production plant 1976
after 1983 2.6-54 (15.3)a Ho et al. (1991)
up to 26 (short-term) (3.9)a
Sweden PVC production plant 1974-1981 0.26-114 (8-h TWA) Holm et al. (1982)
PVC production plant 1974-1980 0.26-5.7 (6-h TWA)
Taiwan PVC plants (n = 5): n.sp. Du et al. (1996)
15 different operation range (n=114): n.d.
units (e.g. outside (0.13) - 1009 range (n=4):
reaction tank)b 6-1009 (mean: 296; median: 86)
15 different job titles range of TWA (n=85):
(e.g. tank supplier)b n.d. - 3680 range (n=9):
5.7-3680 (mean: 660; median:
23.7)
United Kingdom PVC production plant "early days" 7800 Barnes (1976)
(full-time autoclave
cleaner)
USA PVC plant 1950-1959 up to 10 400; 13-2140 Ott et al. (1975)
(8-h TWA)
1960-1963 up to 1300; 13-620 (8-h TWA)
PVC plant n.sp. up to 650 (weekly TWA) Baretta et al. (1969)
USA VC/PVC plants 1973 up to 390 (TWA); peaks Rowe (1975)
2600-10 400
Table 21. (cont'd)
Country Workplace Yearc Concentrations reported Reference
(mg/m3)c
Former USSR VC/PVC plants early 1950s 100-800 Smulevich et al.
(1988)
PVC producing plant 50-800 Filatova &
(occasionally 87 300) Gronsberg (1957)
Former PVC production plant 1974 > 195 Orusev et al. (1976)
Yugoslavia
a Concentrations in parentheses designate geometric means
b Showing highest mean VC concentration
c n.sp.= not specified; TWA = time-weighted average; n.d. = not detected
Table 22. Levels of vinyl chloride reported for workplace air samples in PVC processing plants
Country Workplace Year Concentrations reported Reference
(mg/m3)
China PVC processing plant n.sp. > 30 Bao et al. (1982)
Germany PVC processing department 1974 < 2.6-67 Fleig & Thiess (1974)
Germany polymer extrusion (17 plants) 1989-1992 all of 33 samples: BIA (1996)
< 8 (90 percentile:
< 0.15) (shift means)
Sweden PVC processing plant 1974 < 0.26-0.8 Holm et al. (1982)
Sweden PVC processing plant prior to 1975 >13 - > 26 (8-h TWA) Lundberg et al.
(1993)
Russia PVC processing plant prior to 1990 0.007-1.26 Solionova et al.
(rubber footwear plant) (1992)
Russia PVC processing plant prior to 1966 < 113.6 Bol'shakov (1969)
(synthetic leather plant)
United Kingdom PVC processing plants n.sp. 0.4-0.9 Murdoch & Hammond
(cable factories) (1977)
USA automotive assembly plant(s) 1970s 0.13-7.8 Nelson et al. (1993)
(2 personal samples)
Autoclave cleaners in Croatia were exposed to extremely high
concentrations of VC (between 1295 and 3885 mg/m3; 500 and 1500 ppm).
A retrospective investigation of exposure to VC has been conducted
with 37 autoclave workers (emptying and cleaning) in Split, Croatia,
who were exposed to VC in a suspension polymerization plant. The
investigation covered the period from 1969 to 1987, when the factory
was closed because of its VC emissions. At the beginning, measurements
were done by simple means (Draeger's tubes) and later, from 1980 on,
by infra-red spectroscopy. The 37 workers were exposed to average VC
concentrations of 1412 mg/m3 (543 ppm) (Hozo et al., 1996; Hozo,
1998).
6. KINETICS AND METABOLISM IN LABORATORY ANIMALS AND HUMANS
6.1 Absorption
Animal and human studies have shown that VC is readily and
rapidly absorbed. The primary route of exposure to VC is inhalation.
However, the net uptake after exposure by inhalation is only 30-40% of
inspired VC. This is due to the fact that VC is taken up rapidly until
it reaches a blood concentration in equilibrium with that based upon
inspired concentration and the blood-to-air partition coefficient.
Uptake then decreases to an amount sufficient to replace that
metabolized. The importance of metabolism was shown by Bolt et al.
(1977) who showed that uptake of VC ceased once equilibrium was
reached.
While uptake by the oral route is near 100%, any VC not
metabolized during first pass through the liver will be expired. Thus,
the net dose may be less than the uptake, especially at high doses
resulting in saturation of metabolizing enzymes.
6.1.1 Oral exposure
In a study reported by Watanabe & Gehring (1976) and Watanabe et
al. (1976a), male rats received a single dose by gavage of 0.05, 1.0,
20 or 100 mg/kg body weight and 14C-labelled VC and the excreted
radioactivity was determined for 72 h. As only 2.4, 2.2, 1.0 and 0.5%,
respectively, of the administered radioactivity (Table 23) was
recovered in the faeces (total recovery 91, 89, 81 and 82%,
respectively), it can be assumed that there is nearly complete
absorption of VC in the gastrointestinal tract. Likewise, oral
administration of 0.25 or 450 mg/kg body weight 14C-VC to male rats
resulted in excretion of only 4.6 or 0.7%, respectively (Table 23), of
the applied radioactivity via faeces 0-72 h after application (Green &
Hathway, 1975).
Similar results were obtained by Feron et al. (1981), who fed
rats with different amounts of VC monomer in PVC powder via the diet.
The average amount of VC detected in faeces was 8, 10 and 17% for oral
intakes of 2.3, 7.0 and 21.2 mg/kg body weight per day, respectively.
Since the VC excreted in the faeces was considered by the authors to
be still enclosed in PVC granules and was not bioavailable, it was
concluded that the available VC was nearly completely absorbed in the
gastrointestinal tract.
Studies conducted by Withey (1976) have shown the rapid
absorption of VC from the gastrointestinal tract in male rats after a
single gavage application of 44 to 92 mg/kg body weight in aqueous
solution. Highest blood levels were always measured within 10 min
after administration. After an oral dose of 10 mg VC/kg body weight,
peak levels in brain, liver, kidney and lung were measured 5 min after
dosing, indicating rapid absorption from gastrointestinal tract
(Zuccato et al., 1979).
Table 23. Excretion of radioactivity in rats (in % of applied dose) 72 h after a single oral dose of VCa
Dose in mg/kg VC in CO2 in Urine Faeces Carcass and Total recovery Reference
body weight exhaled air exhaled air tissues
0.05 1.4 9.0 68.3 2.4 10.1 91.2 Watanabe et al. (1976a)
0.25 3.7 13.5 75.1 4.6 n.g. n.g. Green & Hathway (1975)
1.0 2.1 13.3 59.3 2.2 11.1 88.8 Watanabe et al. (1976a)
20 41.6 4.8 22.6 1.0 11.0 81.0 Watanabe & Gehring (1976)
100 66.6 2.5 10.8 0.5 1.8 82.3 Watanabe et al. (1976a)
450 91.9 0.7 5.4 0.7 n.g. n.g. Green & Hathway (1975)
a n.g. = not given
6.1.2 Inhalation exposure
Bolt et al. (1977) blocked the metabolism of VC in male rats by
i.p. injection of 6-nitro-1,2,3-benzothiadiazole (50 mg/kg body
weight), a compound that inhibits most microsomal monooxygenases. The
rats were exposed to approximately 1.2 mg/m3 14C-labelled VC in a
closed system 30 min after the injection. VC was taken up by the
animals until equilibrium was reached between VC in the atmosphere and
in the organism after 15 min, suggesting a rapid uptake of VC.
Similarly, equilibrium blood levels were observed by Withey (1976) in
male rats 30 min after the start of exposure to 18 200 mg VC/m3 (head
only).
Bolt et al. (1976) measured the decline of 14C-VC in a closed
system due to uptake by 3 male rats using an initial concentration of
130 mg/m3. The radioactivity in the air of the exposure system
decreased with a half-life of 1.13 h. Calculation of the clearance of
VC revealed an absorption of inspired VC of about 40%. These results
are in accordance with the data of Hefner et al. (1975a), who reported
in kinetic studies on male rats a similar decline at the same exposure
concentration.
Krajewski et al. (1980) measured the retention of VC in the lungs
of five human male volunteers exposed for 6 h to 7.5, 15, 30 or
60 mg/m3 through a gas mask. The percentage of retention (mean 42%)
was independent of the VC concentration and reached the highest level
of 46% in the first 15 min of exposure.
Buchter et al. (1978) reported toxicokinetic experiments on
themselves. In an open system, about 3-5 min after the start of
inhalation of 6.5 mg VC/m3 in the exposure atmosphere, an equilibrium
was reached between VC inspired and VC expired; 26% (person A) and 28%
(person B) of the VC inspired were removed by the body, probably by
absorption and subsequent metabolism. Metabolism started within the
first minutes of exposure, suggesting fast absorption of VC.
Breath samples were measured of volunteers who had showered in
VC-contaminated well water (Pleil & Lindstrom, 1997). For a brief
10-min shower exposure of 25 µg/m3 (inhalation) and 4 µg/litre
(dermal contact in water), 0.9 µg absorbed dose of VC and a blood
concentration of 0.01 µg/litre was calculated.
6.1.3 Dermal exposure
Hefner et al. (1975b) exposed the whole body (excluding the head)
of male rhesus monkeys to 2080 (2.5 h) or 18 200 mg/m3 (2.0 h)
14C-labelled VC. Very little VC was absorbed through the skin.
Although minuscule, the quantity absorbed appeared
concentration-dependent. The major portion of the VC absorbed was
eliminated by the lungs. The authors argued that no significant
percutaneous absorption would be expected under occupational exposure
conditions.
6.2 Distribution and retention
Data from inhalation and oral studies on rats indicate rapid and
widespread distribution of VC. Rapid distribution of VC was also
reported in humans after inhalation exposure. Rapid metabolism and
excretion limit accumulation of VC in the body. Data on distribution
after oral exposure showed similar results. No studies are available
concerning distribution after dermal exposure. Under conditions of
blocked VC metabolism, VC has been found to accumulate in adipose
tissue.
6.2.1 Oral exposure
Green & Hathway (1975) investigated the distribution in the
tissues of young rats 0.25, 2 and 4 h after oral administration of
30 mg/kg body weight [14C]-VC using whole-animal autoradiography. At
15 min after gavage, most radioactivity was detected in the liver,
followed by the gut (no data about the stomach), and there were small
amounts in the lung and kidney. After 2 h, label was observed in the
liver, kidney, small intestine, stomach, skin, para-auricular region
(probably Zymbal glands), and there were small amounts in lung and
heart. A similar distribution was determined after 4 h, with
additional label in the thymus, salivary gland and Harders gland, but
no radioactivity in the stomach lumen.
The distribution of VC and VC metabolites remaining in the body
(11.1% of recovered radioactivity) 72 h after a single oral
administration via gavage of 1 mg/kg body weight 14C-VC was described
as follows (expressed as % of administered radioactivity per gram
tissue): liver 0.182, skin 0.076, carcass 0.046, plasma 0.053, muscle
0.031, lung 0.061, fat 0.045 (no data about amount in the kidney). A
similar distribution pattern was observed using doses of 0.05 or
100 mg/kg body weight (Watanabe et al., 1976a).
6.2.2 Inhalation exposure
Whole body autoradiograms of male rats showed radioactivity in
the liver, bile duct, digestive lumen and kidneys when animals were
exposed for 5 min to 52 g/m3 14C-labelled VC and sacrificed 10 min
after the end of the exposure period. Animals sacrificed 2-3 h after
the exposure period showed a wider distribution of radioactivity, with
most of the labelled substances being observed in the liver, urinary
system, digestive lumen, lacrimal glands, skin and thymus (Duprat et
al., 1977).
Buchter et al. (1977) pretreated male rats i.p. with 50 mg/kg
body weight 6-nitro-1,2,3-benzothiadiazole (to block VC metabolism)
and exposed the rats in a closed system for 5 h to VC concentrations
of between 65 and 26 000 mg/m3. Authors observed an equilibrium
between 14C-labelled VC in the gas phase and the exposed organism.
The distribution of VC was independent of the exposure concentration.
The following distribution of VC (labelled and unlabelled) in
different organs (expressed as mol VC in 1 g tissue per mol VC in 1 ml
air) was reported immediately after the exposure period: blood 0.65,
liver 0.62, spleen 0.59, kidney 0.59, muscle 0.68 and adipose tissue
8.3. These results indicated that unmetabolized VC is accumulated in
adipose tissue due to its lipophilicity. In contrast, without blockage
of VC metabolism, most radioactivity was detected in kidney and liver
(5 h, 260 mg/m3).
In similar studies Bolt et al. (1976) measured radioactivity
(representing mostly VC metabolites) in different organs immediately
after exposure of male rats to 14C-VC (initial concentration 130
mg/m3) for 5 h. Highest levels were detected in the kidney (2.13%;
expressed as % of incorporated VC per g tissue) and liver (1.86%),
followed by spleen (0.73%), muscle (0.32%), adipose tissue (0.22%) and
brain (0.17%).
Distribution pattern changed with longer post-exposure
observation periods. Watanabe et al. (1976b) reported the following
percentages of 14C activity (mostly VC metabolites) in rats per gram
tissue 72 h after inhalation exposure to 26 or 2600 mg/m3
14C-labelled VC, respectively, for 6 h: liver (0.139; 0.145), kidney
(0.079; 0.057), skin (0.072; 0.115); carcass (0.048; 0.049), plasma
(0.051; not detected), muscle (0.052; 0.038), lung (0.065; 0.046), fat
(0.026; not detected). There was no significant difference between low
and high dose. 72 h after the exposure period most of radioactivity
had been excreted; 13.8% (low dose), 14.5% (high dose) of total
recovered radioactivity remained in carcass and tissues (Table 25). A
similar distribution pattern were presented by Watanabe et al. (1978b)
using the same experimental design but rats exposed once or repeatedly
to 13 000 mg/m3. Furthermore, the authors detected no significant
difference between single and repeated exposure.
Ungváry et al. (1978) presented evidence for the permeability of
the placenta to VC. After exposure of pregnant rats to 5500, 18 000 or
33 000 mg VC/m3 for 2.5 h on day 18 of gestation, VC was detected in
fetal (13, 23 and 31 µg/ml, respectively) and maternal blood (19, 32
and 49 µg/ml), as well as in amniotic fluid (4, 5 and 14 µg/ml).
Toxicokinetic experiments on humans (self-experiments) were
reported by Buchter et al. (1978). Using an open system with a
concentration of 6.5 mg VC/m3 inspired air, the concentration in the
expired air reached a constant concentration after about 5 min
exposure in subject A and 7 min in subject B, indicating the end of
the distribution phase.
6.2.3 Partition coefficients in vitro
In vitro studies using the vial equilibration method (3 h
incubation, blood and tissue homogenates from male Sprague-Dawley
rats) revealed the following VC partition coefficients for male rats:
blood/air 2.4; fat/blood 10.0; muscle/blood 0.4; liver/blood 0.7; and
kidney/blood 0.7 (Barton et al., 1995). In similar experiments
tissue/air partition coefficients were obtained for different rodent
species (Gargas et al., 1989; Clement International Corporation, 1990;
Table 24). These data suggested that the concentration of VC in
adipose tissue is higher than in other tissues. Furthermore, in all
species in which both sexes were tested, partition coefficients for
fat/air were greater in females than in males (Table 24).
6.3 Metabolic transformation
The main route of metabolism of VC in the liver into non-volatile
compounds after inhalative or oral uptake involves 3 steps: a) the
oxidation by cytochrome P-450 to form chloroethylene oxide (CEO, also
known as 2-chlorooxirane), a highly reactive, short-lived epoxide that
rapidly rearranges to form chloroacetaldehyde (CAA); b) the
detoxification of these two reactive metabolites as well as
chloroacetic acid, the dehydrogenation product of CAA, through
conjugation with glutathione catalysed by glutathione S-transferase;
c) the modification of the conjugation products to substituted
cysteine derivatives, which are excreted via urine. The main metabolic
pathways are shown in Fig. 2. At high dose levels the metabolism of VC
is saturable.
The first step in VC metabolism requires microsomal
mixed-function oxidases (cytochrome P-450 enzymes) together with
oxygen and NADPH as cofactors. This was confirmed by studies in vitro
(Barbin et al., 1975; Guengerich et al., 1979) and in vivo
(Reynolds et al., 1975a,b; Guengerich & Watanabe, 1979; Bartsch et
al., 1979; Guengerich et al., 1981). The major catalyst of the
oxidation is CYP2E1 in humans. This has been demonstrated by in vitro
systems using purified human CYP2E1 or by inhibition of catalytic
activity in human liver microsomes with rabbit anti-human CYP2E1. In
liver microsomes from uninduced rats, VC is activated solely by
CYP2E1, at concentrations ranging from 1 to 106 ppm in the gas phase,
according to Michaelis-Menten kinetics (El Ghissassi et al., 1998).
The following kinetic constants were determined:
Km = 7.42 ± 0.37 µmol/litre;
Vmax = 4674 ± 46 nmol.mg protein -1 min-1.
Comparison of the Vmax obtained in this study to the Vmax determined
in vivo in rats (Gehring et al., 1978; Filser & Bolt, 1979) shows
that virtually all the metabolic activation of VC in vivo occurs in
the liver.
Table 24. Tissue/air VC partition coefficients for rodent tissuesa
Species; strain Sex Blood/ Liver/ Muscle/ Fat/
air air air air
Rat; F344 male 1.60 1.99 2.06 11.8
female 1.55 2.05 2.39 21.1
Rat; CDBR male 1.79 3.0 2.18 14.6
female 2.12 1.66 1.28 19.2
Rat; Wistar male 2.10 2.69 2.72 10.2
female 1.62 1.48 1.06 22.3
Mouse; B6C3F1 male 2.83 n.g. n.g. n.g.
female 2.56 n.g. n.g. n.g.
Mouse; CD-1 male 2.27 n.g. n.g. n.g.
female 2.37 n.g. n.g. n.g.
Hamster; male 2.74 3.38 2.56 14.3
Syrian golden female 2.21 1.31 1.96 21.1
a Data from Clement International Corporation (1990); vial
equilibration method (Gargas et al., 1989), blood or tissue
homogenates incubated for 1-4 h until equilibrium was achieved,
as indicated by two consecutive time points without significant
difference; n.g. = not given
Table 25. Percentage of 14C activity eliminated during 72 h following
inhalation exposure to [14C]-vinyl chloride for 6 h in male ratsa
Exposure groups 26 mg/m3 2600 mg/m3 13 000 mg/m3
(number of animals) (4) (4) (2)
Expired as unchanged VC 1.6 12.3 54.5
Expired as CO2 12.1 12.3 8.0
Urine 68.0 56.3 27.1
Faeces 4.4 4.2 3.2
Carcass and tissues 13.8 14.5 7.3
a Expressed as percentage of the total 14C activity recovered
(similar experimental design; Watanabe & Gehring, 1976;
Watanabe et al., 1976b, 1978b)
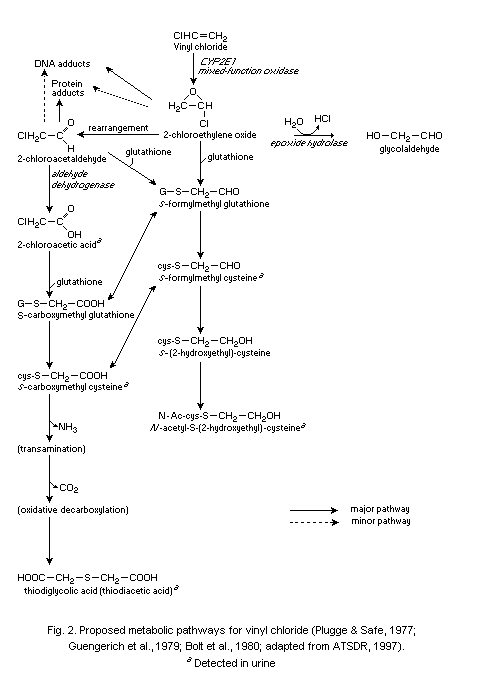 Applying the pharmacokinetic model developed by Andersen et al.
(1987) to describe the metabolism of inhaled gases and vapours, the
uptake of VC by rats in vivo, as determined by Gehring et al. (1978)
and by Filser & Bolt (1979), could be accurately predicted.
Using S9 extracts from human liver samples, Sabadie et al. (1980)
observed a great interindividual variability in the capacity to
activate VC into mutagenic metabolites. This is in agreement with the
observation of Guengerich et al. (1991) who found that levels of
CYP2E1 varied considerably among individual humans. Sabadie et al.
(1980) noted that the average activity of human samples is similar to
that of rat samples.
Chloroethylene oxide (CEO) has a half-life of only 1.6 min at pH
7.4 and 37°C (Malaveille et al., 1975). It can spontaneously rearrange
to CAA (Barbin et al., 1975) or hydrolyse to glycolaldehyde
(Guengerich et al., 1979; Guengerich, 1992). The latter reaction can
also be catalysed by epoxide hydrolase (Fig. 2).
Evidence for the detoxification of reactive VC metabolites
through conjugation with hepatic glutathione catalysed by glutathione
S-transferase (GST) has been shown by measuring the decrease of the
hepatic non-protein sulfhydryl content (primarily glutathione) and the
excretion of thiodiglycolic acid via urine in rats after inhalative
exposure to high levels (> 260 mg/m3) of VC (Watanabe & Gehring,
1976; Watanabe et al., 1976c, 1978a; Jedrychowski et al., 1984).
The conjugation products S-carboxymethyl glutathione and
S-formylmethyl glutathione are excreted in the urine of animals as
substituted cysteine derivatives [ N-acetyl- S-(2-
hydroxyethyl)cysteine, S-carboxymethyl cysteine and thiodiglycolic
acid] and the metabolite CO2 in exhaled air (Green & Hathway, 1975,
1977; Watanabe et al., 1976a,b; Müller et al., 1976; Bolt et al.,
1980). Thiodiglycolic acid has been detected in the urine of workers
occupationally exposed to 0.36-18.2 mg/m3 (Müller et al., 1978).
An alternative pathway has been suggested from inhibition studies
of VC metabolism with ethanol (Hefner et al., 1975c). Pretreatment of
rats with ethanol (5 ml/kg body weight) significantly reduced the
depression of the concentration of non-protein sulfhydryl in the liver
caused by exposure to 2780 mg VC/m3 for 105 min. This inhibition was
less pronounced in rats exposed to < 260 mg/m3. It is postulated
that at low concentrations, a sequential oxidation to 2-chloroethanol,
2-chloroacetaldehyde and 2-chloroacetic acid, involving alcohol
dehydrogenase, takes place. It is speculated that ethanol inhibits
specific P-450 enzymes. However, this hypothesis has not been
substantiated by further experimental data and this pathway has not
been recognized as a direct pathway in recent physiologically based
pharmacokinetic (PBPK) models and risk assessments based on them
(sections 6.6 and 10).
VC exposure does not result in enzyme induction but, on the
contrary, causes destruction of the cytochrome P-450 protein
responsible for its biotransformation (Pessayre et al., 1979; Du et
al., 1982). The impaired rate of oxidative metabolism associated with
P-450 destruction may partly explain the phenomenon of saturation of
the VC metabolism already at relatively low dosage levels and on the
other hand explain the tolerance to liver damage in experimental
animals subjected to continuous intermittent exposure to high VC
concentrations (73 000 mg/m3, 7 h/day, 5 days/week for 6 weeks).
Enzymes metabolizing VC were shown to be saturated in rats at a
concentration of 650 mg/m3 (rats exposed in a closed system; Bolt et
al., 1977; Filser & Bolt, 1979). In rhesus monkeys saturation of
metabolic elimination of VC was observed at atmospheric concentrations
greater than 520 mg/m3 (closed system; Buchter et al., 1980).
Saturation of metabolism occurred in rats at a single oral dose of
20 mg/kg body weight by gavage (Watanabe & Gehring, 1976; Table 23).
Saturation conditions were not reached in humans at an inhalation
exposure to 60 mg/m3 for 6 h (Krajewski et al., 1980).
In closed systems, after the initial absorption of VC until
equilibrium between atmosphere and organism, the continued absorption
is attributed to metabolism (Bolt et al., 1977). Rats exposed to VC
concentrations that did not exceed the above-mentioned threshold of
saturation metabolized VC in accordance with first-order kinetics with
a half-time of 86 min. At concentrations above saturation, the
elimination followed zero-order kinetics (Hefner et al., 1975a,c;
Filser & Bolt, 1979).
Although VC is primarily metabolized in the hepatocyte
(Ottenwälder & Bolt, 1980), the primary target cell for
carcinogenicity in the liver is the sinusoidal cell, as can be seen
from the incidence of ASL in both animals and humans. Non-parenchymal
cells have only 12% of the activity of hepatocytes in transforming VC
into reactive, alkylating metabolites (Ottenwälder & Bolt, 1980). VC
does not induce DNA damage in isolated non-parenchymal liver cells, as
measured by an alkaline comet assay (Kuchenmeister et al., 1996),
whereas it does in isolated hepatocytes. However, the majority of the
DNA adduct studies have been conducted or related to the hepatocyte.
It can be postulated that the majority of reactive metabolites can
leave the intact hepatocyte to produce tumours in the sinusoidal cells
(Laib & Bolt, 1980). The greater susceptibility of the sinusoidal
cells to the carcinogenic effects of VC may also result from the
inability of the sinusoidal cells to repair one or more of the DNA
adducts produced by VC as efficiently as the hepatocyte.
6.4 Elimination and excretion
After low doses, VC is metabolically eliminated and non-volatile
metabolites excreted mainly in the urine. At doses that saturate the
metabolism, the major route of excretion is exhalation of unchanged
VC. Independent of applied dose, the excretion of metabolites via
faeces is only a minor route. The metabolic clearance of VC is slower
in humans than in experimental animals, on a body weight basis.
However, it is comparable in several mammalian species, including
humans, when calculated on a body surface area basis.
6.4.1 Oral exposure
Male rats were gavaged with different doses of 14C-labelled VC,
and the radioactivity excreted was determined during the following
72 h (Green & Hathway, 1975; Watanabe & Gehring, 1976; Watanabe et
al., 1976a). Results are presented in Table 23. With low doses
radioactivity was mainly excreted as conjugated metabolites via the
urine or exhaled as 14C-labelled CO2 (section 6.3), but with doses
of 20 mg/kg body weight or more the main elimination route was
exhalation of unchanged VC (Table 23), suggesting saturated
metabolism. A minor route of excretion at all doses tested is via the
faeces.
Measuring the elimination of radioactivity in the urine as a
function of time revealed biphasic elimination at dose levels up to
100 mg/kg body weight with a half-life of approximately 4.6 h in the
initial rapid phase (first-order kinetics) (Watanabe et al., 1976a).
At low doses (1 mg/kg body weight) pulmonary elimination (as
CO2) during the first 4 h was monophasic with a half-life of 58 min,
but with a dose of 100 mg/kg body weight elimination of mainly
unchanged VC was biphasic with an initial rapid phase (half-life
14.4 min) followed by a slow phase (half-life 41 min) (Watanabe et
al., 1976a).
Most of the radioactivity excreted via urine or exhaled as the
metabolite CO2 was eliminated during the first 24 h after gavaging of
0.25 or 450 mg/kg body weight, whereas elimination of unchanged VC via
the lung was complete within 3-4 h (Green & Hathway, 1975).
Pretreatment of rats with unlabelled VC (up to 300 mg/kg body
weight per day orally for 60 days) had no effect on the rate of
elimination of a single oral dose of 14C-VC (oral application on
day 1 and 60) (Green & Hathway, 1975), suggesting that VC did not
induce its metabolism.
6.4.2 Inhalation exposure
Metabolic elimination of VC has been investigated in different
species, measuring the decline of VC in the gas phase of a closed
system into which VC was initially injected (Buchter et al., 1978;
Filser & Bolt, 1979; Buchter et al., 1980). Using VC concentrations
that did not exceed the saturation threshold (section 6.3), the
following first-order metabolic clearance rates for VC (expressed in
litre/h per kg body weight; initial concentration in mg/m3 in
parentheses) were determined: rat 11.0 (< 650), mouse 25.6 (130),
gerbil 12.48 (130), rabbit 2.74 (130), rhesus monkey 3.55 (< 520),
humans 2.08 (26). Because the metabolism of VC is perfusion-limited
(Filser & Bolt, 1979), comparison of clearance rates should be made on
a body surface area basis rather than a body weight basis. In this
case, these six mammalian species exhibit similar clearance rates.
With exposure concentrations above the "saturation point"
(> 650 mg/m3), the maximum velocity of metabolic elimination in rats
was 110 µmol/h per kg body weight (Filser & Bolt, 1979) or 3.6 mg/h
per kg body weight (Barton et al., 1995).
Elimination and excretion of 14C in rats within 72 h after a 6-h
exposure to 26, 2600 or 13 000 mg/m3 14C-labelled VC is shown in
Table 25 (similar experimental design; Watanabe et al., 1976b, 1978b;
Watanabe & Gehring, 1976). The amount of expired VC increased with the
exposure concentration, whereas the relative urinary excretion of
metabolites decreased, indicating a saturation of metabolism. Minor
decreases were seen in the proportion excreted via the faeces or
expired as CO2.
Measuring the time course of expiration in these experiments, the
pattern of pulmonary elimination of unchanged VC was similar at all
exposure concentrations, following first-order kinetics with
half-lives of 20.4, 22.4 (Watanabe et al., 1976b) and 30 min (Watanabe
et al., 1978b), respectively. After a 6-h exposure to 26 and
2600 mg/m3, elimination of the 14C-label via urine as a function of
time revealed a biphasic excretion of radioactivity with estimated
half-lives for the first (rapid) phase of 4.6 and 4.1 h (Watanabe et
al., 1976b). Because of extremely variable excretion curves in the
second phase, no attempts were made to estimate the half-lives of the
slow phase, which accounted for less than 3% of the radioactivity
excreted in the urine. Similar results were presented by Bolt et al.
(1976). Rats exposed for 5 h to 130 mg/m3 14C-VC excreted 70% of
incorporated radioactivity during the first 24 h after exposure in
urine and less than 3% in the following 3 days.
The rate of elimination of a single inhalative exposure to
14C-VC is not influenced by prior repeated exposure to the same
concentration (13 000 mg/m3) of unlabelled VC 6 h/day, 5 days/week
for 7 weeks (Watanabe et al., 1978b).
In human volunteers, the mean concentration of VC in the expired
air up to 30 min after a 6-h exposure to 7.5-60 mg/m3 reached no more
than 5% of the inhaled concentration (Krajewski et al., 1980).
When male volunteers were exposed to 130 (n=6), 650 (n=4) or
1300 mg/m3 (n=4) for 7.5 h, the VC concentration in expired air was
2.6, 23 or 52 mg/m3, respectively, 1 h after exposure (Baretta et
al., 1969).
6.5 Reaction with body components
6.5.1 Formation of DNA adducts
In vitro, both CEO and CAA can form etheno adducts with nucleic
acid bases (Fig. 3; Bolt, 1986; Bartsch et al., 1994; Barbin, 1998),
but the former exhibits greater reactivity (Guengerich, 1992). In
addition, 7-OEG has been characterized as a major reaction product of
CEO with guanine (Scherer et al., 1981), whereas CAA does not yield
this adduct (Oesch & Doerjer, 1982). 1, N6-Ethenoadenosine and
3, N4-ethenocytidine were characterized as reaction products of VC
with ribonucleosides in the presence of a microsomal activation system
(Barbin et al., 1975; Laib & Bolt, 1978). Analysis of DNA incubated
in vitro with rat liver microsomes, an NADPH-regenerating system
and [14C]-VC revealed the formation of 7-OEG, the major DNA adduct,
and of 1, N6-etheno-2'-deoxyadenosine (Epsilon dA) and
3, N4-etheno-2'-deoxycytidine (Epsilon dC) (Laib et al., 1981).
More recently, Müller et al. (1997) quantified six adducts in DNA
treated with CEO, including 7-OEG, the four ethenobases and
5,6,7,9-tetrahydro-7-hydroxy-9-oxoimidazo[1,2-alpha]purine. The
reactivity of CAA towards double-stranded B-DNA is very low
(Guengerich, 1992). CAA reacts with unpaired A and C bases to yield
Epsilon A and Epsilon C, respectively. Treatment of DNA with CAA has
also been reported to result in the formation of N2,3-Epsilon G and
1, N2-Epsilon G moieties (Oesch & Doerjer, 1982; Kusmierek & Singer,
1992; Guengerich, 1992).
The formation of Epsilon dC and tentatively of Epsilon dA in
liver DNA from rats exposed to VC in their drinking-water for 2 years
was reported by Green & Hathway (1978). In subsequent studies, Epsilon
A and Epsilon C were found in the nucleotides in hydrolysates of rat
liver RNA and 7-OEG but not Epsilon A or Epsilon C in DNA after
exposure to [14C]-VC (Laib & Bolt, 1977, 1978; Laib et al., 1981).
Similar results were found in mice (Osterman-Golkar et al., 1977).
More recent studies, using analytical methods (HPLC and fluorescence
spectrophotometry, monoclonal antibodies and negative-ion chemical
ionization with mass spectrometry using electrophore labelling; Fedtke
et al., 1989, 1990a; Eberle et al., 1989) or experimental designs with
greater sensitivity (young rats, short delay between exposure and
analysis), have led to conclusive demonstration of Epsilon dA and
Epsilon dC as DNA adducts in different rat organs after inhalation
exposure to VC (Table 26). The concentration of DNA adducts was 3- to
8-fold higher in the liver than in the lung and kidney, reflecting the
higher capacity of the liver for metabolic activation of VC (Fedtke et
al., 1990b; Swenberg et al., 1992). Both etheno bases (Epsilon C and
Epsilon A) accumulated in rat liver DNA during intermittent exposure
to VC (1300 mg/m3). Only Epsilon C accumulated in rat lung and
kidney, Epsilon A appearing to accumulate principally in the target
organ, the liver (Guichard et al., 1996). Subsequently, analysis of
further tissues showed increased levels of Epsilon A in the testis,
but not in the brain and spleen of rats exposed intermittently for 8
weeks; levels of Epsilon C increased in the testis and spleen but not
in the brain (Barbin, in press).
Applying the pharmacokinetic model developed by Andersen et al.
(1987) to describe the metabolism of inhaled gases and vapours, the
uptake of VC by rats in vivo, as determined by Gehring et al. (1978)
and by Filser & Bolt (1979), could be accurately predicted.
Using S9 extracts from human liver samples, Sabadie et al. (1980)
observed a great interindividual variability in the capacity to
activate VC into mutagenic metabolites. This is in agreement with the
observation of Guengerich et al. (1991) who found that levels of
CYP2E1 varied considerably among individual humans. Sabadie et al.
(1980) noted that the average activity of human samples is similar to
that of rat samples.
Chloroethylene oxide (CEO) has a half-life of only 1.6 min at pH
7.4 and 37°C (Malaveille et al., 1975). It can spontaneously rearrange
to CAA (Barbin et al., 1975) or hydrolyse to glycolaldehyde
(Guengerich et al., 1979; Guengerich, 1992). The latter reaction can
also be catalysed by epoxide hydrolase (Fig. 2).
Evidence for the detoxification of reactive VC metabolites
through conjugation with hepatic glutathione catalysed by glutathione
S-transferase (GST) has been shown by measuring the decrease of the
hepatic non-protein sulfhydryl content (primarily glutathione) and the
excretion of thiodiglycolic acid via urine in rats after inhalative
exposure to high levels (> 260 mg/m3) of VC (Watanabe & Gehring,
1976; Watanabe et al., 1976c, 1978a; Jedrychowski et al., 1984).
The conjugation products S-carboxymethyl glutathione and
S-formylmethyl glutathione are excreted in the urine of animals as
substituted cysteine derivatives [ N-acetyl- S-(2-
hydroxyethyl)cysteine, S-carboxymethyl cysteine and thiodiglycolic
acid] and the metabolite CO2 in exhaled air (Green & Hathway, 1975,
1977; Watanabe et al., 1976a,b; Müller et al., 1976; Bolt et al.,
1980). Thiodiglycolic acid has been detected in the urine of workers
occupationally exposed to 0.36-18.2 mg/m3 (Müller et al., 1978).
An alternative pathway has been suggested from inhibition studies
of VC metabolism with ethanol (Hefner et al., 1975c). Pretreatment of
rats with ethanol (5 ml/kg body weight) significantly reduced the
depression of the concentration of non-protein sulfhydryl in the liver
caused by exposure to 2780 mg VC/m3 for 105 min. This inhibition was
less pronounced in rats exposed to < 260 mg/m3. It is postulated
that at low concentrations, a sequential oxidation to 2-chloroethanol,
2-chloroacetaldehyde and 2-chloroacetic acid, involving alcohol
dehydrogenase, takes place. It is speculated that ethanol inhibits
specific P-450 enzymes. However, this hypothesis has not been
substantiated by further experimental data and this pathway has not
been recognized as a direct pathway in recent physiologically based
pharmacokinetic (PBPK) models and risk assessments based on them
(sections 6.6 and 10).
VC exposure does not result in enzyme induction but, on the
contrary, causes destruction of the cytochrome P-450 protein
responsible for its biotransformation (Pessayre et al., 1979; Du et
al., 1982). The impaired rate of oxidative metabolism associated with
P-450 destruction may partly explain the phenomenon of saturation of
the VC metabolism already at relatively low dosage levels and on the
other hand explain the tolerance to liver damage in experimental
animals subjected to continuous intermittent exposure to high VC
concentrations (73 000 mg/m3, 7 h/day, 5 days/week for 6 weeks).
Enzymes metabolizing VC were shown to be saturated in rats at a
concentration of 650 mg/m3 (rats exposed in a closed system; Bolt et
al., 1977; Filser & Bolt, 1979). In rhesus monkeys saturation of
metabolic elimination of VC was observed at atmospheric concentrations
greater than 520 mg/m3 (closed system; Buchter et al., 1980).
Saturation of metabolism occurred in rats at a single oral dose of
20 mg/kg body weight by gavage (Watanabe & Gehring, 1976; Table 23).
Saturation conditions were not reached in humans at an inhalation
exposure to 60 mg/m3 for 6 h (Krajewski et al., 1980).
In closed systems, after the initial absorption of VC until
equilibrium between atmosphere and organism, the continued absorption
is attributed to metabolism (Bolt et al., 1977). Rats exposed to VC
concentrations that did not exceed the above-mentioned threshold of
saturation metabolized VC in accordance with first-order kinetics with
a half-time of 86 min. At concentrations above saturation, the
elimination followed zero-order kinetics (Hefner et al., 1975a,c;
Filser & Bolt, 1979).
Although VC is primarily metabolized in the hepatocyte
(Ottenwälder & Bolt, 1980), the primary target cell for
carcinogenicity in the liver is the sinusoidal cell, as can be seen
from the incidence of ASL in both animals and humans. Non-parenchymal
cells have only 12% of the activity of hepatocytes in transforming VC
into reactive, alkylating metabolites (Ottenwälder & Bolt, 1980). VC
does not induce DNA damage in isolated non-parenchymal liver cells, as
measured by an alkaline comet assay (Kuchenmeister et al., 1996),
whereas it does in isolated hepatocytes. However, the majority of the
DNA adduct studies have been conducted or related to the hepatocyte.
It can be postulated that the majority of reactive metabolites can
leave the intact hepatocyte to produce tumours in the sinusoidal cells
(Laib & Bolt, 1980). The greater susceptibility of the sinusoidal
cells to the carcinogenic effects of VC may also result from the
inability of the sinusoidal cells to repair one or more of the DNA
adducts produced by VC as efficiently as the hepatocyte.
6.4 Elimination and excretion
After low doses, VC is metabolically eliminated and non-volatile
metabolites excreted mainly in the urine. At doses that saturate the
metabolism, the major route of excretion is exhalation of unchanged
VC. Independent of applied dose, the excretion of metabolites via
faeces is only a minor route. The metabolic clearance of VC is slower
in humans than in experimental animals, on a body weight basis.
However, it is comparable in several mammalian species, including
humans, when calculated on a body surface area basis.
6.4.1 Oral exposure
Male rats were gavaged with different doses of 14C-labelled VC,
and the radioactivity excreted was determined during the following
72 h (Green & Hathway, 1975; Watanabe & Gehring, 1976; Watanabe et
al., 1976a). Results are presented in Table 23. With low doses
radioactivity was mainly excreted as conjugated metabolites via the
urine or exhaled as 14C-labelled CO2 (section 6.3), but with doses
of 20 mg/kg body weight or more the main elimination route was
exhalation of unchanged VC (Table 23), suggesting saturated
metabolism. A minor route of excretion at all doses tested is via the
faeces.
Measuring the elimination of radioactivity in the urine as a
function of time revealed biphasic elimination at dose levels up to
100 mg/kg body weight with a half-life of approximately 4.6 h in the
initial rapid phase (first-order kinetics) (Watanabe et al., 1976a).
At low doses (1 mg/kg body weight) pulmonary elimination (as
CO2) during the first 4 h was monophasic with a half-life of 58 min,
but with a dose of 100 mg/kg body weight elimination of mainly
unchanged VC was biphasic with an initial rapid phase (half-life
14.4 min) followed by a slow phase (half-life 41 min) (Watanabe et
al., 1976a).
Most of the radioactivity excreted via urine or exhaled as the
metabolite CO2 was eliminated during the first 24 h after gavaging of
0.25 or 450 mg/kg body weight, whereas elimination of unchanged VC via
the lung was complete within 3-4 h (Green & Hathway, 1975).
Pretreatment of rats with unlabelled VC (up to 300 mg/kg body
weight per day orally for 60 days) had no effect on the rate of
elimination of a single oral dose of 14C-VC (oral application on
day 1 and 60) (Green & Hathway, 1975), suggesting that VC did not
induce its metabolism.
6.4.2 Inhalation exposure
Metabolic elimination of VC has been investigated in different
species, measuring the decline of VC in the gas phase of a closed
system into which VC was initially injected (Buchter et al., 1978;
Filser & Bolt, 1979; Buchter et al., 1980). Using VC concentrations
that did not exceed the saturation threshold (section 6.3), the
following first-order metabolic clearance rates for VC (expressed in
litre/h per kg body weight; initial concentration in mg/m3 in
parentheses) were determined: rat 11.0 (< 650), mouse 25.6 (130),
gerbil 12.48 (130), rabbit 2.74 (130), rhesus monkey 3.55 (< 520),
humans 2.08 (26). Because the metabolism of VC is perfusion-limited
(Filser & Bolt, 1979), comparison of clearance rates should be made on
a body surface area basis rather than a body weight basis. In this
case, these six mammalian species exhibit similar clearance rates.
With exposure concentrations above the "saturation point"
(> 650 mg/m3), the maximum velocity of metabolic elimination in rats
was 110 µmol/h per kg body weight (Filser & Bolt, 1979) or 3.6 mg/h
per kg body weight (Barton et al., 1995).
Elimination and excretion of 14C in rats within 72 h after a 6-h
exposure to 26, 2600 or 13 000 mg/m3 14C-labelled VC is shown in
Table 25 (similar experimental design; Watanabe et al., 1976b, 1978b;
Watanabe & Gehring, 1976). The amount of expired VC increased with the
exposure concentration, whereas the relative urinary excretion of
metabolites decreased, indicating a saturation of metabolism. Minor
decreases were seen in the proportion excreted via the faeces or
expired as CO2.
Measuring the time course of expiration in these experiments, the
pattern of pulmonary elimination of unchanged VC was similar at all
exposure concentrations, following first-order kinetics with
half-lives of 20.4, 22.4 (Watanabe et al., 1976b) and 30 min (Watanabe
et al., 1978b), respectively. After a 6-h exposure to 26 and
2600 mg/m3, elimination of the 14C-label via urine as a function of
time revealed a biphasic excretion of radioactivity with estimated
half-lives for the first (rapid) phase of 4.6 and 4.1 h (Watanabe et
al., 1976b). Because of extremely variable excretion curves in the
second phase, no attempts were made to estimate the half-lives of the
slow phase, which accounted for less than 3% of the radioactivity
excreted in the urine. Similar results were presented by Bolt et al.
(1976). Rats exposed for 5 h to 130 mg/m3 14C-VC excreted 70% of
incorporated radioactivity during the first 24 h after exposure in
urine and less than 3% in the following 3 days.
The rate of elimination of a single inhalative exposure to
14C-VC is not influenced by prior repeated exposure to the same
concentration (13 000 mg/m3) of unlabelled VC 6 h/day, 5 days/week
for 7 weeks (Watanabe et al., 1978b).
In human volunteers, the mean concentration of VC in the expired
air up to 30 min after a 6-h exposure to 7.5-60 mg/m3 reached no more
than 5% of the inhaled concentration (Krajewski et al., 1980).
When male volunteers were exposed to 130 (n=6), 650 (n=4) or
1300 mg/m3 (n=4) for 7.5 h, the VC concentration in expired air was
2.6, 23 or 52 mg/m3, respectively, 1 h after exposure (Baretta et
al., 1969).
6.5 Reaction with body components
6.5.1 Formation of DNA adducts
In vitro, both CEO and CAA can form etheno adducts with nucleic
acid bases (Fig. 3; Bolt, 1986; Bartsch et al., 1994; Barbin, 1998),
but the former exhibits greater reactivity (Guengerich, 1992). In
addition, 7-OEG has been characterized as a major reaction product of
CEO with guanine (Scherer et al., 1981), whereas CAA does not yield
this adduct (Oesch & Doerjer, 1982). 1, N6-Ethenoadenosine and
3, N4-ethenocytidine were characterized as reaction products of VC
with ribonucleosides in the presence of a microsomal activation system
(Barbin et al., 1975; Laib & Bolt, 1978). Analysis of DNA incubated
in vitro with rat liver microsomes, an NADPH-regenerating system
and [14C]-VC revealed the formation of 7-OEG, the major DNA adduct,
and of 1, N6-etheno-2'-deoxyadenosine (Epsilon dA) and
3, N4-etheno-2'-deoxycytidine (Epsilon dC) (Laib et al., 1981).
More recently, Müller et al. (1997) quantified six adducts in DNA
treated with CEO, including 7-OEG, the four ethenobases and
5,6,7,9-tetrahydro-7-hydroxy-9-oxoimidazo[1,2-alpha]purine. The
reactivity of CAA towards double-stranded B-DNA is very low
(Guengerich, 1992). CAA reacts with unpaired A and C bases to yield
Epsilon A and Epsilon C, respectively. Treatment of DNA with CAA has
also been reported to result in the formation of N2,3-Epsilon G and
1, N2-Epsilon G moieties (Oesch & Doerjer, 1982; Kusmierek & Singer,
1992; Guengerich, 1992).
The formation of Epsilon dC and tentatively of Epsilon dA in
liver DNA from rats exposed to VC in their drinking-water for 2 years
was reported by Green & Hathway (1978). In subsequent studies, Epsilon
A and Epsilon C were found in the nucleotides in hydrolysates of rat
liver RNA and 7-OEG but not Epsilon A or Epsilon C in DNA after
exposure to [14C]-VC (Laib & Bolt, 1977, 1978; Laib et al., 1981).
Similar results were found in mice (Osterman-Golkar et al., 1977).
More recent studies, using analytical methods (HPLC and fluorescence
spectrophotometry, monoclonal antibodies and negative-ion chemical
ionization with mass spectrometry using electrophore labelling; Fedtke
et al., 1989, 1990a; Eberle et al., 1989) or experimental designs with
greater sensitivity (young rats, short delay between exposure and
analysis), have led to conclusive demonstration of Epsilon dA and
Epsilon dC as DNA adducts in different rat organs after inhalation
exposure to VC (Table 26). The concentration of DNA adducts was 3- to
8-fold higher in the liver than in the lung and kidney, reflecting the
higher capacity of the liver for metabolic activation of VC (Fedtke et
al., 1990b; Swenberg et al., 1992). Both etheno bases (Epsilon C and
Epsilon A) accumulated in rat liver DNA during intermittent exposure
to VC (1300 mg/m3). Only Epsilon C accumulated in rat lung and
kidney, Epsilon A appearing to accumulate principally in the target
organ, the liver (Guichard et al., 1996). Subsequently, analysis of
further tissues showed increased levels of Epsilon A in the testis,
but not in the brain and spleen of rats exposed intermittently for 8
weeks; levels of Epsilon C increased in the testis and spleen but not
in the brain (Barbin, in press).
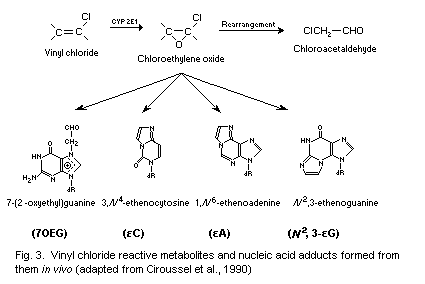 The rate of reaction of CAA with nucleic bases is slower than
with CEO (Zajdela et al., 1980). In in vitro studies, CEO but not
CAA was shown to be the main entity giving rise to etheno adducts
(Guengerich, 1992). Furthermore, as discussed in section 7.8, CEO but
not CAA show similar toxicity/mutagenic profiles to VC in a
metabolically competent human B-lymphoblastoid line (Chiang et al.,
1997). These findings seem to corroborate the original suggestion that
it is CEO rather than CAA that is the main source of etheno adducts
(Van Duuren, 1975; Guengerich et al., 1981; Gwinner et al., 1983).
In pre-weanling rats exposed to VC, 7-OEG had a half-life of
approx. 62 h, while the etheno adducts are highly persistent with a
half-life for Epsilon G of approx. 30 days (Swenberg et al., 1992).
Studies on the persistence of Epsilon A and Epsilon C in the liver of
adult rats have shown that there is no significant decrease in adduct
levels 2 months after termination of VC exposure (Guichard et al.,
1996). This is in contrast with the known repair of etheno adducts in
vitro (Dosanjh et al., 1994). The rat and human 3-methyladenine DNA
glycosylases can excise Epsilon A from DNA (Saparbaev et al., 1995).
Epsilon C can be released by the human mismatch-specific thymine-DNA
glycosylase (Saparbaev & Laval, 1998).
DNA adduct formation seems to be age-dependent; about 5- to
6-fold more DNA adducts were determined in young animals compared to
adults (Laib et al., 1989). Similar results were presented by Fedtke
et al. (1990b) who exposed adult and 10-day-old Sprague-Dawley rats
(Table 26). Ciroussel et al. (1990) exposed 7-day-old and 13-week-old
rats (strain BD IV) for 2 weeks to VC and detected 6 times more DNA
adducts in the liver of young rats compared to adults (Table 26).
It should be noted that background levels of etheno bases have
been found in various tissues in unexposed rodents (Guichard et al.,
1996; Fernando et al., 1996; Barbin, in press) and humans (Nair et
al., 1995, 1997). Lipid peroxidation products have been shown to react
with nucleic acid bases yielding etheno adducts (El Ghissassi et al.,
1995a; Chung et al., 1996; Bartsch et al., 1997). The role of lipid
peroxidation products in the endogenous formation of background levels
of etheno bases is further supported by the finding of elevated levels
of Epsilon dA and Epsilon dC in the liver from Long Evans Cinnamon
rats (Nair et al., 1996) and from patients with Wilson's disease or
with primary haemochromatosis (Nair et al., 1998).
Etheno adducts are also formed via substituted oxiranes formed
from other vinyl monomers, e.g., vinyl bromide (Bolt, 1994) and vinyl
and ethyl carbamate (urethane) (Park et al., 1993).
6.5.2 Alkylation of proteins
Bolt et al. (1980) studied covalent binding of radiolabel to
proteins in rats exposed to 14C-labelled VC. The target of alkylation
is the free sulfhydryl group of proteins. The liver always showed the
highest binding rate. The fraction of VC that was irreversibly bound
to proteins was independent of the VC dose applied, indicating no
threshold effect even at low doses. In vivo studies on rats
(Guengerich & Watanabe, 1979) have shown that the amount of total VC
metabolites bound in the liver to proteins is twice that bound to DNA,
RNA and lipids. Osterman-Golkar et al. (1977) reported the alkylation
of cysteine (S-(2-hydroxyethyl)cysteine) and histidine ( (N-1-and
N-3) hydroxyethylhistidine) of the globin precipitate of haemoglobin
and small amounts of the alkylated histidines in proteins from testis
in mice exposed to 1,2-14C-vinyl chloride.
In vitro studies have shown that incubation of rat liver
microsomes with 14C-labelled VC results in NADPH-dependent microsomal
uptake and covalent binding to microsomal proteins (Kappus et al.,
1975, 1976; Baker & Ronnenberg, 1993). It has been suggested that VC
metabolites might be involved in the destruction of the haem moiety of
cytochrome P-450 in the liver (Guengerich & Strickland, 1977).
6.6 Modelling of pharmacokinetic data for vinyl chloride
There has been progress in recent years in the development of
physiologically based toxicokinetic (PBTK) models describing the
toxicokinetics of chemicals. These theoretical models permit
predictions of the dose of active metabolites reaching target tissues
in different species, including humans, and, therefore, have improved
the toxicokinetic extrapolation in cancer risk assessments. PBTK
models have also been used as a tool to examine the behaviour of VC in
mammalian systems. The description of PBTK models for VC is presented
in Annex 2.
Table 26. Detection of DNA adducts in vivo after VC inhalation in ratsa
Strain; sex; Treatment; Investigated Alkylated bases in Comments References
age post-exposure organs DNA (max. concentration
survival time in pmol/µmol unmodified
base in specified organs)
BD IV; male & 1300 mg/m3, 7 h/day, liver, lung, epsilon-dA (0.131, 0.105, higher sensitivity of Ciroussel
female; 7 days 7 days/week for brain, kidney 0.06, b.d.l.); epsilon-dC young rats compared et al.(1990)
2 weeks; none (0.492, 0.246, 0.216, with adults
b.d.l.) (see section 7.7.4)
BD IV; male; 1300 mg/m3, 7 h/day, liver epsilon-dA (0.019); Ciroussel
13 weeks 7 days/week for epsilon-dC (0.080) et al. (1990)
2 weeks; none
S.-D.; male & 1560 mg/m3, 4 h/day liver, lung, 7-OEG (162, 20, 29, <10, higher sensitivity of Fedtke et
female; 10 days for 5 days; kidney, <10); N2,3-epsilon-G (1.81, young rats compared al. (1990b)
0, 3, 7, 14 days brain, spleen 0.21, 0.31, <0.12, <0.12); with adults (see
measured immediately a.e. section 7.7.4)
concerning liver
adducts
S.-D.; female; 1560 mg/m3, 4 h/day liver, lung, 7-OEG (43, 20, n.g.); DNA adduct formation Fedtke et
adult for 5 days; kidney N2,3-epsilon-G (0.47, max. at the end of al. (1990b)
0, 3, 7, 14 days 0.27, <0.12); measured exposure; most DNA
immediately a.e. adducts in the liver
S.-D.; male & 5200 mg/m3, 7 h/day liver, lung epsilon-dA (0.05, 0.13) more DNA adducts in the Eberle et
female; 11 days on days 1-9 and 24 h epsilon-dC (0.16, 0.33) lung than in the liver al. (1989)
on day 10; none
S.-D.; male & 1560 mg/m3, 4 h/day liver, lung, 7-OEG (162, 20, 29); DNA adduct Swenberg
female; 10 days for 5 days; kidney N2,3-epsilon-G (1.81, 0.21, max. at the end et al.
0, 3, 7, 14 days 0.31); epsilon-dC (0.98, 0.30, exposure; most DNA (1992)
0.29); epsilon-dA (0.21, 0.065, adducts in the liver
0.04); measured immediately a.e.
Table 26. (cont'd)
Strain; sex; Treatment; Investigated Alkylated bases in Comments References
age post-exposure organs DNA (max. concentration
survival time in pmol/µmol unmodified
base in specified organs)
S.-D.; male; 1300 mg/m3, 4 h/day, liver, lung, epsilon-dA (liver: background exposure-time-dependent Guichard
6 weeks 5 days/week for 1, kidney, 0.0004, a.e. up to 0.045; increase of adducts in et al. (1996)
2, 4, 8 weeks; none lymphocytes background lung and kidney up to liver (ca. 100-fold);
0.033, no increase a.e.); no (lymphocytes) or
epsilon-dC (liver: background slight increase (ca.
0.0007, a.e. up to 0.08; kidney: 2-fold in kidney, ca.
background 0.086, a.e. up to 5-fold in lung) of
0.16; lung: background 0.072, adducts in other
a.e. up to 0.38) organs but higher
background levels
8 weeks brain, testis, epsilon-dA (increase in testis, Barbin
spleen not in brain or spleen) (in press)
epsilon-dC (increase in testis
and spleen, but not in brain)
a a.e. = after exposure; b.d.l. = below detection limit; n.g. = not given; S.-D. = Sprague-Dawley; epsilon-dA = 1,N6-ethenodeoxyadenosine;
epsilon-dC = 3,N4-ethenodeoxycytidine; 7-OEG = 7-(2-oxoethyl)guanine; epsilon-G = ethenoguanine
7. EFFECTS ON LABORATORY MAMMALS AND IN VITRO TEST SYSTEMS
7.1 Acute toxicity
VC appears to be of low acute toxicity when administered to
various species by inhalation. A summary of acute toxicity is given in
Table 27. No data are available on acute toxicity after dermal
application.
VC has a narcotic effect (see also section 7.6) after inhalative
administration of high doses. In rats, mice and hamsters, death was
preceded by increased motor activity, twitching of extremities,
tremor, ataxia, tonic-clonic convulsions and accelerated respiration
(Patty et al., 1930; Mastromatteo et al., 1960; Prodan et al., 1975).
In dogs, severe cardiac arrhythmias occurred under narcosis after
inhalative exposure to 260 000 mg VC/m3 (Oster et al., 1947). Similar
results were reported by Carr et al. (1949). Pneumonitis was more
frequent in experimental mice than in controls 8 and 18 months after a
single 1-h exposure to 1000, 3000, 13 000 or 130 000 mg/m3 (Hehir et
al., 1981).
After acute inhalative exposure to VC, pathological findings in
the rat included congestion of the internal organs, particularly lung,
liver and kidney as well as pulmonary oedema (Patty et al., 1930;
Mastromatteo et al., 1960; Lester et al., 1963; Prodan et al., 1975).
Exposure to VC (3900 mg/m3) for 24 h did not cause pathological
changes in male and female rats or female New Zealand rabbits. In
mice, exposure to the same concentration for 4 and 8 h resulted in
circulatory changes, while longer exposure (12 and 24 h) caused
vasomotor paralysis followed by characteristic shock with subsequent
alterations in the liver and lungs (Tátrai & Ungváry, 1981).
7.2 Short-term toxicity
7.2.1 Oral exposure
Groups of 15 male and 15 female weanling Wistar rats were gavaged
with VC dissolved in soya-bean oil (0, 30, 100 and 300 mg/kg body
weight, once daily, 6 days/week for 13 weeks). The treatment caused no
noticeable changes in appearance or behaviour, body weight gain or
food intake. The total number of white blood cells and the sugar
content of the blood were slightly decreased by the intermediate and
high dose levels. The activities of serum ASAT and ALAT and of urinary
ASAT were decreased in males given the top dose. There were no other
significant changes in the haematological or biochemical indices and
no treatment-related alterations were observed in the microscopic
constituents of the urine. The relative weight of the liver in males
and females showed a tendency to increase with increasing doses of VC
but the difference from the controls was statistically significant
only at the highest dose level. The NOEL was given as 30 mg/kg body
weight (Feron et al., 1975). In contrast, in another study, all male
and female Wistar rats (presumably a total of 15 rats) receiving once
daily 300 mg VC/kg body weight in peroxide-free corn oil by gavage
died within 60 days of treatment (Knight & Gibbons, 1987).
7.2.2 Inhalation exposure
A summary of the non-neoplastic effects of VC after short-term
inhalation is presented in Table 28. Information on carcinogenic
effects after short-term inhalation are presented in section 7.7 and
Table 30.
In various species, the main target of VC toxicity is the liver.
A dose-related significant increase in relative liver weight was found
in male rats at exposure levels of 26, 260 and 7800 mg/m3 (Bi et al.,
1985). Degenerative effects on liver parenchyma were reported in
rabbits at a dose level of 520 mg/m3 (Torkelson et al., 1961), in
rats at 1300 mg/m3 (Torkelson et al., 1961; Wisniewska-Knypl et al.,
1980), and in mice at 2600 mg/m3 (Lee et al., 1977). Bi et al. (1985)
reported a decreased relative testis weight in rats at exposure levels
of 260 and 7800 mg/m3; however, the effect was not dose-related.
These authors also reported a higher incidence and severity of damage
to the testicular seminiferous tubules at all dose levels tested (26,
260 and 7000 mg/m3), the differences with the controls being
statistically significant only at the two highest dose levels.
However, the severity of the testicular damage was clearly
dose-related (correlation coefficient 0.993; P < 0.01) suggesting
an adverse effect of VC on the testes already at 26 mg/m3. Effects on
relative liver weight were detected in rats at 26 mg/m3 (Bi et al.,
1985) (see also Table 28 and section 7.5.1). Effects on the kidney
(Lee et al., 1977; Feron et al., 1979a,b; Himeno et al., 1983) and the
lung (Suzuki, 1980) were observed in rats and/or mice at higher doses
(Table 28). For mice a LOEL of 130 mg/m3 was given (decreased
survival; Hong et al., 1981). Rats, mice and rabbits seem to be more
sensitive than guinea-pigs and dogs (Torkelson et al., 1961; Hong et
al., 1981).
7.2.3 Dermal exposure
No studies were available on short-term dermal exposure.
7.3 Long-term toxicity - effects other than tumours
7.3.1 Oral exposure
Studies on effects induced by long-term oral application of VC in
rats are presented in detail in Table 29. Studies on other species are
not available.
Long-term feeding studies in male and female rats showed
increased mortality in males at doses > 5.0 mg/kg body weight per
day (Feron et al., 1981) and in females at doses of > 1.3 mg/kg
Table 27. Toxicity of VC after acute inhalation exposurea
Species Duration of Parameter Value References
exposure in g/m3
Rat 2 h LC50 390 Prodan et
LC100 525 al. (1975)
Rat 30 min LC100 780 Mastromatteo
et al. (1960)
Rat 1 h ataxia, 130 Hehir et al.
hyperventilation (1981)
Mouse 2 h LC50 293 Prodan et al.
LC100 375 (1975)
Mouse 30 min LC100 780 Mastromatteo
et al. (1960)
Guinea-pig 2 h LC50 595 Prodan et al.
LC100 700 (1975)
Guinea-pig 30 min death, 780 Mastromatteo
threshold dose et al. (1960)
Guinea-pig 2-6 h deep narcosis, 260 Patty et al.
no death (1930)
Guinea-pig 18-55 min death, 390-650 Patty et al.
threshold dose (1930)
Guinea-pig 90 min narcosis, 65-130 Patty et al.
threshold dose (1930)
Rabbit 2 h LC50 295 Prodan et al.
LC100 700 (1975)
a Cited studies not conducted according to present-day standards
Table 28. Toxicity of vinyl chloride in animals after short-term inhalation exposures - non-neoplastic effectsa
Species, strain; Doses in Exposure duration; Significant effects References
number of animals mg/m3 frequency;
per dose per post-exposure
exposure period observation period
Rat, Wistar; 8 0, 26, 260, 3 or 6 mo; 26 mg/m3: relative spleen and heart weight Bi et al.
(3 mo) or 30 7800 6 d/week, 6 h/d; * (6 mo); incidence of testicular seminiferous (1985)
(6 mo) m rats none tubule damage * ! (exposure period not specified)
> 26 mg/m3: relative liver weight * (6 mo) 260 mg/m3:
relative heart weight * (3 mo) > 260 mg/m3: relative
testis weight ** (6 mo); incidence of testicular
tubule damage * 7800 mg/m3: relative kidney and
spleen weight * (3 mo)
Rat; n.g.; at high 0, 130, 260, 4.5 (high dose) 130 mg/m3: NOEL (body and organ weight, Torkelson
dose 10 f & 520, 1300 or 6 mo; survival, haematology, clinical chemistry, urine et al.
10 m (control 5 d/week, analysis, histopathology) 260 mg/m3: relative liver (1961)
5 f & 5 m); other 7 h/d; none weight * (m+f) 1300 mg/m3: granular degeneration
groups 20-24 m in centrilobular liver parenchyma #;
& 24 f liver weight * (m)
Rat, Wistar; 0, 130, 1, 3, 6 mo; 130 mg/m3: slight changes such as proliferation Wisniewska-Knypl
8-10 m 1300, 5 d/week, 5 h/d; of hepatocellular SER (3-6 mo)# et al.
52 000 none > 1300 mg/m3: liver weight * (1-6 mo), ultrastructural (1980)
hepatocellular changes (swollen mitochondria, lipid
droplets *) after 3 and 6 mo #
Rat, 0, 2465 24.5 weeks; 2465 mg/m3: mortality *# (m+f), haematology and Groth et
Sprague-Dawley; 7 h/d, 5 d/week; clinical chemistry <-> al. (1981)
110-128 rats up to 19 weeks
per sex
Rat, Wistar; 0, 13 000 4, 13, 26 weeks; 13 000 mg/m3, > 4 weeks: body weight **, Feron et al.
10 f & 10 m 5 d/week, 7 h/d; blood clotting time ** > 13 weeks: liver function (1979a,b)
none (BSB-retention test) **; liver and kidney
weight * (m+f)
Table 28. (cont'd)
Species, strain; Doses in Exposure duration; Significant effects References
number of animals mg/m3 frequency;
per dose per post-exposure
exposure period observation period
26 weeks: spleen weight * (m+f); clear cell foci Feron &
and basophilic foci in the liver * (m+f)# Kroes (1979)
Rat, Sherman; 0, 52 000 92 d; 5 d/week, 52 000 mg/m3: relative liver weight * and Lester et al.
12-15 rats/sex 8 h/d; none spleen weight ** (m+f); white blood cell counts **; (1963)
swelling of hepatocytes with vacuolization,
compression of sinusoids #
Mouse, CD-1; 0, 130, 650, 1, 3, 6 mo; > 130 mg/m3: survival after 6 mo exposure ** # (low Hong et al.
8-28 mice/sex 2600 5 d/week, 6 h/d; dose: m, 1/8 versus 22/28 in control; f, 0/8 versus (1981)
12 mo 23/28; tumour incidences no differences)
Mouse, CD-1; 0, 2600 3-9 exposures; 2600 mg/m3: early deaths (2 m + 1 f)!; pathological Lee et al.
36 mice/sex 5 d/week, 6 h/d; changes in dead animals: acute toxic hepatitis (1977)
none (congestion, diffused necrosis), tubular necrosis
in renal cortex
Mouse, n.g.; 6500 1 or 6 mo; 6500 mg/m3: hyperplastic nodules and dilatated Schaffner
5 m (1 mo) or (no control) 5 d/week, 5 h/d; sinusoids in liver parenchyma after 6 mo (1979)
14 m (6 mo) none exposure #
Mouse, CD-1; 0, 6500, 5-6 mo; > 6500 mg/m3: proliferation and hypertrophy of Suzuki
3-16 m 15 600 5 d/week, 5 h/d; bronchiolar cells, hypersecretion of bronchial (1980)
2-37 d and bronchiolar epithelium, hyperplasia of
alveolar epithelium, bronchiolar inflammation #
(only pulmonary effects recorded; effects not
dose related)
Mouse, CD-1; 13 000 10 weeks; 13 000 mg/m3: focal lung hyperplasia, proliferation Himeno et
10 m (no control) 5 d/week, 4 h/d; of sinusoidal cells of the liver, proliferative al. (1983)
none effects in renal glomeruli, giant cells in testis #
Table 28. (cont'd)
Species, strain; Doses in Exposure duration; Significant effects References
number of animals mg/m3 frequency;
per dose per post-exposure
exposure period observation period
Mouse, ICR; a) 0, 13 000, a) 5 to 6 d, basophilic stippled erythrocytes * (#) in a) and b), Kudo et
n.g. 26 000; b) 62 d; a) 4 h/d, related effect not dose al. (1990)
b) 78 to 104 b) continuously;
none
Hamster, 0, 520 6 mo; 5 d/week, 520 mg/m3: survival ** Drew et
golden Syrian; 6 h/d; life span al. (1983)
56 f
Guinea-pig, 0, 130, 6 mo; 5 d/week, 520 mg/m3: NOEL (body and organ weight, Torkelson
n.g.; 10-12 m 260, 520 7 h/d; none survival, clinical chemistry, histopathology) et al. (1961)
& 8-12 f
Rabbit, n.g.; 0, 130, 6 mo; 5 d/week, 260 mg/m3: NOEL (body and organ weight, Torkelson
3 rabbits/sex 260, 520 7 h/d; none survival, clinical chemistry, histopathology) et al. (1961)
520 mg/m3: degeneration of centrilobular
liver parenchyma (m+f) with periportal cellular
infiltration (f) #
Dog; n.g.; 0, 130, 6 mo; 5 d/week, 130-520 mg/m3: no effects recorded (body and Torkelson
1 dog/sex 260, 520 7 h/d; none organ weight, survival, haematology, clinical et al. (1961)
chemistry, urine analysis, histopathology) #
a d = day; mo = month, m = male; f = female; n.g. = not given; ! = increase not significant; # = no data about significance;
* = increased; ** = decreased; <-> = no change
body weight per day (Feron et al., 1981; Til et al., 1983, 1991). At
14.1 mg/kg body weight per day, blood clotting time was decreased and
alpha-fetoprotein levels in blood serum were increased (Feron et al.,
1981). Skin fibrosis was observed at 30 mg/kg body weight per day
administered by gavage (Knight & Gibbons, 1987).
As for short-term exposure, the primary target organ of VC in
rats after long-term oral exposure is the liver. Female rats appeared
to be more sensitive than males to the hepatotoxicity of VC
(section 7.7.4). Increased relative liver weights were found at
14.1 mg/kg body weight per day after feeding periods of 6 or 12 months
(Feron et al., 1981). Morphological alterations of the liver included
extensive hepatocellular necrosis at doses > 5 mg/kg body weight
per day, foci of haematopoiesis at 14.1 mg/kg body weight per day, and
cysts and liver cell polymorphism (variation in size and shape of
hepatocytes and their nuclei) at doses > 1.3 mg/kg body weight per
day (Feron et al., 1981; Til et al., 1983, 1991). Foci of
hepatocellular alteration (clear cell, mixed cell, eosinophilic and
basophilic foci) were common findings. Clear cell, mixed cell and
eosinophilic foci occurred at doses > 1.3 mg/kg body weight per day
and basophilic foci at doses > 0.014 mg/kg body weight per day (Til
et al., 1983, 1991).
7.3.2 Inhalation exposure
A summary of non-neoplastic and neoplastic effects after
long-term intermittent inhalation of VC is presented in Table 32 with
details of exposure regime (see also section 7.7). Long-term exposure
to VC by inhalation resulted in increased mortality in rats exposed to
a dose of 260 mg/m3 for 12, 18 and 24 months, in mice exposed to
130 mg/m3 for 6, 12 and 18 months and in hamsters exposed to
520 mg/m3 for 6, 12 and 18 months (Drew et al., 1983). Maltoni et al.
(1984) reported increased mortality in rats (BT15) and hamsters (BT8)
at lower dose levels (2.6 and 130 mg/m3, respectively), but no
statistical evaluation was performed. Rats exposed to 130 mg/m3
showed reduced body weight and increased relative spleen weight (Sokal
et al., 1980; see below). At this dose morphological alterations were
reported in rat liver, such as hepatocellular lipid accumulation and
mitochondrial swellings (Wisniewska-Knypl et al., 1980) as well as
proliferation of cells lining the liver sinusoids (Sokal et al.,
1980). Exposure to higher doses revealed degenerative alteration in
the testis (1300 mg/m3; Sokal et al., 1980) and tubular nephrosis and
focal degeneration of the myocardium (13 000 mg/m3; Feron & Kroes,
1979) in rats.
Male Wistar rats (42-80 per group) exposed to 0, 130, 1300 and
52 000 mg VC/m3, 5 days/week, 5 h daily, for 10 months, showed
significantly reduced body weight in all treatment groups; general
condition and behaviour were not altered. Relative organ weights of
spleen and heart (except at the mid dose) were significantly elevated
at > 130 mg/m3, as well as liver and kidney weights at mid- and
high-dose levels and testis weights at the high-dose level. X-ray
analysis did not show any skeletal alterations. Histopathology
revealed statistically significantly increased incidences of nuclear
polymorphism (nuclei of variable size and irregular shape) of
hepatocytes and proliferation of reticuloendothelial cells lining
liver sinusoids at the two highest dose levels. Fatty degeneration of
hepatocytes was found at all exposure levels. Ultrastructural changes
in hepatocytes seen at all exposure levels, but not in controls,
included swollen and giant mitochondria with broken cristae,
proliferation and dilatation of smooth endoplasmic reticulum, nuclear
membrane invaginations, areas of cytoplasmic degradation and increased
numbers of small lipid droplets (Sokal et al., 1980; Wisniewska-Knypl
et al., 1980). In addition, Sokal et al. (1980) reported necrotic foci
of the spermatogenic epithelium and disorders of spermatogenesis
accompanied by large multinuclear syncytial cells in the testis
predominantly at 1300 mg/m3. Haematology, urine analysis and clinical
chemistry did not reveal differences of toxicological significance.
Thus NOEL for rats or mice concerning non-neoplastic effects
could not be derived, since effects were observed at the lowest levels
studied (130 mg/m3).
7.4 Skin and eye irritation; sensitization
No relevant data on skin irritation were identified. No studies
were available on sensitizing effects of VC in animals. Erythema and
second-degree burns were reported in a worker after accidental
exposure to liquid VC (see section 8.3.2.1). Dryness of eyes and nose
was reported by volunteers exposed to 1300 mg/m3 (see section 8.2).
7.5 Reproductive toxicity, embryotoxicity and teratogenicity
7.5.1 Male reproductive toxicity
Inhalation studies on rats showed some evidence of reduced
fertility and morphological alterations of the testis. It should be
noted that none of the studies cited were conducted according to
current guidelines (OECD, 1983a,b).
In dominant lethal studies on mice no reduction in fertility was
observed (Anderson et al., 1976; Table 35). However, reduced fertility
was noted in male CD rats (12 per group) mated once on week 11 of
exposure (0, 130, 650 or 2600 mg/m3; 6 h/day, 5 days per week). VC
treatment decreased dose-dependently the ratio of pregnant to mated
females; this was significant at mid- and high-dose level (Short et
al., 1977; see also section 7.8.2).
Bi et al. (1985; for details see Table 28 and section 7.2.2)
observed decreased relative testis weight at 260 mg/m3 and
morphological alterations in the testis of rats even at the lowest
dose of 26 mg/m3. Morphological alterations in the testis of rats
were also reported by Sokal et al. (1980, see section 7.3.2).
VC was administered to adult female CD rats at 0, 24, 260 and
2860 mg/m3, 6 h/day, 5 days/week for at least 10 weeks prior to
mating until day 4 of lactation (94 + days) in a two-generation study.
Alterations in reproductive performance and fertility were not
detected at any dose level tested. Centrilobular hypertrophy in the
liver and increased relative liver weights, however, were noted at all
dose levels tested in a dose-related manner (Shah, 1998).
7.5.2 Embryotoxicity and teratogenicity
Although the available studies did not follow guideline
standards, the information leads to the conclusion that there is
embryotoxicity or fetal toxicity, including increased numbers of
resorptions, decreased numbers of live fetuses and delayed development
at dose levels producing maternal toxicity. VC treatment did not
induce gross malformations. There is evidence for the permeability of
the placenta to VC (Ungváry et al., 1978; see section 6.2.2).
John et al. (1977, 1981) investigated mice, rats and rabbits for
teratogenic effects of inhaled VC. In all three species a similar
experimental design was used. Pregnant CF-1 mice were exposed 7 h/day
to 0, 130 or 1300 mg VC/m3 on day 6-15 of gestation. For both
concentrations tested, concurrent control groups were sham-exposed.
Animals were observed daily and maternal body weight recorded at
several intervals (no further data). The mice were sacrificed on day
18 of gestation. After determination of external anomalies, one-third
of each litter (19-26 litters per group) was examined for soft tissue
anomalies and the other mice for skeletal anomalies. Exposure to
1300 mg/m3 led to deaths (5 of 29 bred females, P < 0.05), reduced
maternal body weight gain (P < 0.05) and food consumption
(P< 0.05). No maternal toxicity was apparent in females exposed to
130 mg/m3. The number of live fetuses per litter and fetal body
weight were significantly decreased and the number of resorptions
significantly increased at 1300 mg/m3, but these values were within
the range observed for the second concurrent control group or for
historical controls. No soft tissue or external anomalies were
detected. Significantly increased incidences of three skeletal
variants (delayed skull and sternebrae ossification, unfused
sternebrae) in the high-dose group were indicative of delayed skeletal
development. No developmental toxicity was observed at 130 mg/m3.
Sprague-Dawley rats were exposed to 0, 1300 or 6500 mg/m3 on
gestation day 6-15 and dams were sacrificed on day 21 of gestation.
Low-dose exposure resulted in reduced maternal weight gain
(P< 0.05). At the high-dose level further maternal effects like
reduced food consumption and increased liver weight were observed
(P< 0.05), and one out of 17 pregnant rats died (no further
information). Examination of 16-31 litters per group revealed
significantly decreased fetal weight in the low-dose but not in the
high-dose group. Significantly increased incidences in dilated ureter
were observed at 6500 mg/m3.
Rabbits were exposed on gestation day 6-18 to 0, 1300 or
6500 mg/m3 and sacrificed on gestation day 29. Except for reduced
food consumption in the low-dose group and 1 death in 7 bred females
of the high-dose group, no further evidence of maternal toxicity was
observed. In the high-dose group only 5 litters were examined, but in
other groups 11-19 litters. Compared to the concurrent control, litter
size was significantly reduced in the low-dose group (but not at
6500 mg/m3). However, there was an increase in litter size in this
treatment group compared to controls concurrent to the high-dose
group. The incidence of delayed ossification of the sternebrae was
increased at 1300 mg/m3, but not in the high-dose group (John et al.,
1977, 1981).
Ungváry et al. (1978) exposed pregnant CFY rats continuously to 0
or 4000 mg VC/m3 during the first, second or last third of pregnancy
(gestation day 0-8, 7-13 or 13-20). Dams were sacrificed on gestation
day 20 and living, dead or resorbed fetuses were recorded. Placenta
and fetuses were weighed and fetuses macroscopically investigated. One
half of each litter was examined for soft tissue anomalies including
histopathology of organs with abnormalities and the other half was
processed for investigation of the skeletal system. Maternal weight
gain was significantly reduced in pregnant rats exposed during the
last third of pregnancy. Increased relative liver weight was observed
in dams exposed during the first or second third, but histopathology
revealed no pathological changes in the liver of any VC-treated rat.
No further signs of maternal toxicity were reported. Examination of 13
to 28 litters per group revealed increased fetal loss in per cent of
total implantation sites after exposure during gestation day 0-8
compared with the concurrent control. However, this value was not
significant compared with other control groups (e.g., control exposed
gestation day 13-20) of the same study. None of the soft tissue or
skeletal anomalies were attributed to VC treatment.
Exposure of 40 pregnant white Wistar rats to VC (mean level of
6.15 mg/m3 during whole gestation) resulted in elevated embryonic
mortality, lowered fetal weight, and induction of external and
internal anomalies in the development of the fetus (Mirkova et al.,
1978).
7.6 Special studies
7.6.1 Neurotoxicity
Profound narcosis was reported in guinea-pigs exposed to
65 000 mg VC/m3 for 90 min (Patty et al., 1930). Ataxia was observed
at this dose level after 5 min of exposure. The anaesthetic action of
VC was also observed in dogs (Oster et al., 1947) and mice (Peoples &
Leake, 1933). Mastromatteo et al. (1960) reported deep narcosis in
rats and mice exposed to 260 000 mg/m3 for 30 min. The narcotic
effect was preceded by increased motor activity after 5 min of
exposure, twitching of extremities (after 10 min), ataxia (after
15 min) and tremor (after 15 min). Rats exposed to 130 000 mg/m3 for
60 min showed ataxia preceded by hyperactivity but no narcotic effect
(Hehir et al., 1981).
Neuropathological alterations were observed in rats exposed to
78 000 mg/m3 (4 h/day, 5 days/week) for 12 months (Viola, 1970; Viola
et al., 1971). During the exposure period, the rats were slightly
soporific. Histopathology revealed diffuse degeneration in the gray
and white matter of the brain and at the level of the white matter
zones of reactive gliosis. In the cerebellum, atrophy of the granular
layer and degeneration of Purkinje cells were most prominent. In
addition, peripheral nerve bundles were often surrounded and invaded
by fibrotic processes.
Reports on neurotoxicity in humans occupationally exposed to VC
are given in section 8.3.2.3.
7.6.2 Immunotoxicity
Sharma & Gehring (1979) investigated mitogen-stimulated
transformation in splenic lymphocytes isolated from mice exposed to
26, 260 or 2600 mg/m3 for 2, 4 or 8 weeks (6 h/day, 5 days/week). The
treatment produced no effects on body or organ weight, except
increased spleen weight in high-dose groups, no effects on
haematological parameters and no pathological alterations at necropsy.
VC exposure caused stimulation of spontaneous lymphocyte
transformation in lymphocyte cultures prepared from mice exposed for 2
weeks to the high dose and from mice exposed for 4 weeks at all dose
levels, but this was not dose-dependent. The response of lymphocytes
to phytomitogens was increased at 2600 mg/m3 after exposure for
2 weeks and at all dose levels after exposure for 4 or 8 weeks, with
more pronounced effects at the mid-dose level. Stimulation of
lymphocyte transformation was not observed in lymphocytes from
unexposed mice cultured in the presence of VC, indicating that
metabolites of VC formed in vivo may be responsible for this effect.
Exposure of mice to 26 mg/m3 for 6 months (Bi et al., 1985;
Table 28) or rats to 130 mg/m3 for 10 months (Sokal et al., 1980;
section 7.3.2) induced increased relative spleen weight, whereas much
higher doses (52 000 mg/m3 for 92 days) produced decreased spleen
weight and reduced white blood cell counts (Lester et al., 1963; see
Table 28).
Reports on immunological and lymphoreticular effects in humans
occupationally exposed to VC are given in section 8.3.2.
7.6.3 Cardiovascular effects
Viola (1970) demonstrated thickening of the walls of small
arterial vessels (in some vessels blockage of lumen) due to
endothelial fibrosis and proliferation of endothelial cells in rats
exposed to 78 000 mg/m3 (4 h/day, 5 days/week) for 12 months.
Exposure of rats to 13 000 mg/m3 for 12 months (Feron & Kroes, 1979;
see Table 32) resulted in thickened walls of arteries and focal
degenerations of the myocardia.
Oster et al. (1947) observed cardiac arrhythmias (e.g.,
ventricular extrasystoles and fibrillation, auriculoventricular block)
in dogs at a dose level of 260 000 mg/m3.
Bi et al. (1985) reported increased relative heart weight in rats
exposed for 6 months to 26 mg/m3 or for 3 months to 260 mg/m3 (Table
28).
Impaired peripheral circulation as well as other cardiovascular
effects in occupational exposed humans are described in section 8.3.2.
7.6.4 Hepatotoxicity
Data on hepatotoxicity are presented in sections 7.2 and 7.3.
7.7 Carcinogenicity
VC causes a wide spectrum of tumours in animals and this spectrum
is similar in a number of different species (see Table 33). For
example in rats, the following tumours have been described after VC
inhalation exposure: liver and other angiosarcomas, other liver
tumours, mammary gland carcinoma, nephroblastoma, neuroblastoma,
stomach tumours and Zymbal gland tumours. The lowest dose at which an
increase in tumour incidences was observed when rats were exposed by
inhalation was 130 mg/m3 for liver angiosarcoma (ASL) and 13 mg/m3
for mammary tumours. There is evidence that animals are more
susceptible to tumour induction early in life. There is also evidence
that liver tumours are induced in female rats at lower doses than in
males.
VC is also carcinogenic in animals after oral application. The
spectrum of tumours is similar to that observed after inhalation
exposure. The lowest observed dose producing a carcinogenic effect
(ASL) in rats was 1.3 mg/kg body weight per day.
7.7.1 Oral exposure
Details of studies on the carcinogenicity of VC in rats after
oral administration are tabulated in Table 29. There are three gavage
studies: two studies with 52 or 59 weeks gavage and a 32- or 18-week
follow-up and a 2-year lifetime gavage study with small numbers of
animals and high mortality at high doses (Maltoni et al., 1981 [BT11
and BT27] and Knight & Gibbons, 1987). The two feeding studies used
PVC powder containing VC incorporated into the diet; the numbers of
animals were large, and the administration comprised the whole
lifetime of the animals (Feron et al., 1981; Til et al., 1983, 1991).
Table 29. Long-term toxicity/carcinogenicity of vinyl chloride in experimental animals after oral administrationa
Species; strain; Dose; 1) Effects other than tumours (dose in mg/kg bw/d); Reference
initial number of route of exposure; 2) number of animals for histopathological evaluation of
animals per dose; exposure period; neoplastic effects;
vehicle frequency of treatment; 3) type of tumour: number of animals with this tumour/dose
post-exposure group (unless otherwise given)
observation period
Rat; Wistar; 0, 1.7, 5.0, 14.1 1) > 1.7 mg/kg: haematology, biochemistry, urine analyis, Feron
60-80 rats/sex; mg/kg bw./d, organ function, body weight and food consumption <-> et al.
PVC powder bioavailable d (oral mortality !* (f); liver clear cell foci (f+m), basophilic (1981) e
(vehicle) intake 0, 1.8, 5.6, foci (f), eosinophilic foci (m+f) !*; liver-cell
containing VC 17.0 mg/kg bw/day); polymorphism !* (m); liver cysts !* (f);> 5.0 mg/kg: general
incorporated oral feed; lifespan condition ¡ (m+f, at mo 18); mortality !* (f+m); liver
into the diet study, terminated basophilic foci !* (m); extensive liver necrosis !* (f);
at week 135 (m) 14.1 mg/kg: extensive liver necrosis and liver cysts !* (m); focal
and 144 (f); diet haematopoiesis in liver !* (m); liver weight !* (f+m at mo
provided 4 h/d; 6, f at mo 12, interim sacrifice); blood clotting time at mo
none 6 and alpha-fetoprotein at mo 12 ¡* (f+m);
2) 55, 58, 56, 59 m and 57, 58, 59, 57 f;
3) ASL in m: 0, 0, 6*, 27* and in f: 0, 0, 2, 9*;neoplastic liver
nodules in m: 0, 1, 7*, 23* and in f : 2, 26*, 39*, 44*;
hepatocellular carcinoma in m: 0, 1, 2, 8* and in f: 0, 4, 19*,
29*; lung angiosarcoma in m: 0, 0, 4*, 19* and in f: 0, 0, 1, 5*
Rat; Wistar; 100 0, 0.014, 0.13, 1.3 1) > 0.014 mg/kg: bw. and food consumption <-> (m+f); liver Til et al.
rats/sex except mg/kg bw./d, basophilic foci !* (f); 1.3 mg/kg: mortality !* (f; at (1983,
high dose (50 bioavailable d (oral week 149); liver glutathione level at week 40 or 80 <-> 1991) f
per sex); PVC intake 0, 0.018, (satellite groups); liver clear cell (m+f), basophilic (m),
powder (vehicle) 0.17, 1.7 mg/kg eosinophilic (f), and mixed cell foci (f) !*; liver cysts !*
containing VC bw./day); oral feed; (f ); moderate to severe liver-cell polymorphism !* (m+f );
incorporated into lifespan study, 2) 99, 99, 99, 49 m and 98, 100, 96, 49 f;3) ASL in m: 0, 0,
the diet terminated at week 0, 1 and 0, 0, 0, 2 in f; neoplastic liver nodules in m 0,
149 (m) and 150 (f); 0, 0, 3 and 0, 1, 1, 10* in f; hepatocellular carcinoma in
diet provided 4 h/d; m 0, 0, 0, 3* and 1, 0, 1, 3 in f
none
Table 29. (cont'd)
Species; strain; Dose; 1) Effects other than tumours (dose in mg/kg bw/d); Reference
initial number of route of exposure; 2) number of animals for histopathological evaluation of
animals per dose; exposure period; neoplastic effects;
vehicle frequency of treatment; 3) type of tumour: number of animals with this tumour/dose
post-exposure group (unless otherwise given)
observation period
Rat; 0, 3.33, 16.6, 50 1) > 16.6 mg/kg: survival ¡# (m); 3.33 & 50: bw ¡# (m); Maltoni
Sprague-Dawley; mg/kg bw./d; 2) 40 f and 40 m per group; et al.
40 rats/sex; pure gavage; 52 weeks; 3) ASL in m: 0, 0, 4, 8* and in f: 0, 0, 6*, 9*; (1981,
virgin olive oil once daily, Zymbal gland carcinoma in m: 0, 0, 1, 1 and in f: 1, 0, 1984)
4-5 d/week; up to 1, 0; extrahepatic angiosarcoma in m: 0 in all groups and [BT 11] b
84 weeks c in f: 0, 2, 0, 2; nephroblastoma in m: 0, 0, 2, 1 and in
f: 0, 0, 1, 1;
Rat; 0, 0.03, 0.3, 1.0 1) > 0.03 mg/kg: survival and bw <->#; Maltoni
Sprague-Dawley; 75 mg/kg bw./d; 2) 75, 75, 73, 75 m and 75, 75, 73, 75 f; et al.
rats/sex; pure gavage; 59 weeks; 3) ASL in m: 0, 0, 0, 1 and in f: 0, 0, 1, 2; (1981,
virgin olive oil once daily, Zymbal gland carcinoma in m: 0, 0, 0, 2 and in f: 1, 0, 1984)
4-5 d/week; up to 0, 3; extrahepatic angiosarcoma in m: 0 in all groups and [BT 27] b
77 weeks c in f: 0, 0, 0, 1; nephroblastoma 0 in all groups
Rat; Wistar; 0, 3, 30, 300 mg/kg 1) 3 mg/kg: bw <-> , rat skin composition <->, mortality Knight &
10-20 rats/sex; bw./d; gavage; 1/16 (control n.g.) 30 mg/kg: bw <->, mortality 5/15; Gibbons
peroxide-free 95-125 weeks; biochemical parameters for skin fibrosis !* 300 mg/kg: (1987)
corn oil once daily; none mortality 15/15 within first 60 days of treatment;
2) 20, 16, 15, 10;
3) Liver tumors (predominantly angiosarcomas):
0, 1, 11, 10 #
a In all studies vehicle-treated controls; analysis of VC concentration in vehicle in all studies except Knight & Gibbons (1987);
bw. = body weight; d = day; f = females; m = males; mo = months; n.g. = not given; * = effect significant at P < 0.05;
# = no statistical evaluation; <-> = unchanged; ¡ = decreased; ! = increased
b Study number in experiments done by Maltoni and coworkers
c Animals were kept until spontaneous death or sacrificed at the end of given post exposure observation period
d Bioavailability studied in an ancillary study
e Study comparable to OECD guidelines 451 with acceptable restrictions (OECD, 1981a,b)
f Study comparable to OECD guidelines 453 (OECD, 1981a,b)
A statistically significant increase in the incidence of ASL was
seen in Sprague-Dawley rats at a dose level of 16.6 mg VC/kg body
weight per day (gavage; study BT 11), and some similar tumours were
also observed at dose levels of 0.3 and 1.0 mg/kg body weight per day
(Maltoni et al., 1981). Hepatic angiosarcomas were also observed in
gavage studies in Wistar rats (Knight & Gibbons, 1987). This tumour
type is very rare in untreated rats (4 in several thousand rats of the
colony used by Maltoni et al., 1981). Because of short dosage and
follow-up in the first study, and small number of animals due to early
mortality in the third, these studies probably did not reflect the
total carcinogenic potential of VC.
The results of the two feeding studies carried out by another
working group (Feron et al., 1981; Til et al., 1983, 1991) confirmed
the findings presented by Maltoni et al. (1981) concerning ASL
(induction at 1.3 mg/kg body weight per day, significant at 5.0 mg
VC/kg body weight per day). Furthermore these feeding studies
presented evidence for significantly increased tumour incidences of
neoplastic liver nodules (females) and of hepatocellular carcinoma
(HCC) (males) at 1.3 mg/kg body weight per day (Til et al., 1991).
7.7.2 Inhalation exposure
After the first reports of the carcinogenicity of VC in rats
(Viola, 1970; Viola et al., 1971), Maltoni and coworkers intensively
investigated the effects of inhalation exposure to VC in different
laboratory animals.a Study results were published in Maltoni et al.
(1974), Maltoni & Lefemine (1975) and Maltoni et al. (1979), and the
final report in Maltoni et al. (1981, 1984). Detailed information on
these inhalation studies as well as pertinent studies from other
working groups is tabulated in Table 30 (short-term studies) and Table
32 (long-term studies). A summary of tumour types induced by long-term
inhalation exposure to vinyl chloride in different species is given in
Table 33. Further studies on inhalative carcinogenicity of VC not
discussed in this section, but leading to similar results to the
studies tabulated below, were performed on rats (short-term: Hong et
al., 1981; long-term: Viola et al., 1971; Maltoni et al., 1974; Lee et
al., 1977; Maltoni et al., 1981, 1983, 1984; Bi et al., 1985; Maltoni
& Cotti, 1988; Froment et al., 1994) and mice (short-term: Suzuki,
1978, 1981; Schaffner, 1979; Hehir et al., 1981; Himeno et al., 1983;
Adkins et al., 1986; long-term: Keplinger et al., 1975; Lee et al.,
1977; Holmberg et al., 1979; Drew et al., 1983).
7.7.2.1 Short-term exposure
Several carcinogenicity studies have been performed where animals
were exposed by inhalation for rather short periods (up to 6 months)
and kept for different post-exposure periods up to lifetime
(Table 30). The tumour spectrum in rats and mice is very similar to
a Studies performed at Beautivoglio (BT) Laboratories; studies
numerated BT1-BT27
Table 30. Short-term inhalation studies on carcinogenicity of vinyl chloride in experimental animalsa
Species; strain; Doses in mg/m3; 1) Number of animals for histopathological References
age at start of exposure period; evaluation;
experiment; initial frequency of 2) type of tumour: number of animals with
number of animals per treatment; this tumour/dose group (unless otherwise
dose per exposure post-exposure given); other observations
period; type of observation period
control;
Rat; 0, 15 600, 26 000; 1) 227, 120, 118; Maltoni
Sprague-Dawley; 5 weeks; 4 h/d, 2) (#) ASL: 0, 0, 1; extrahepatic et al.
11 weeks; 120 5 d/week; angiosarcoma: 0, 0, 0; Zymbal gland (1981)
(control 240) 149 weeksc carcinoma: 0, 9, 9; hepatoma: 0, 0, 1; [BT 10]b
f & m; untreated nephroblastoma: 0, 1, 0;
control neuroblastoma: 0, 1, 0
Rat; 0, 15 600, 26 000; 1) 227, 118, 119; Maltoni
Sprague-Dawley; 25 weeks; 1 h/d, 2) (#) ASL: 0, 3, 1; extrahepatic angiosarcoma: et al.
11 weeks; 120 4 d/week; 0, 2, 0; Zymbal gland carcinoma: 0, 5, 9 (1981)
(control 240) 129 weeksc [BT 10]b
f & m; untreated
control
Rat; 15 600, 26 000; 1) 18 m and 24 f at low dose, 24 m and Maltoni
Sprague-Dawley; 5 weeks; 4 h/d, 20 f at high dose; et al.
1-day-old; 5 d/week; 119c 2) ASL in m: 5, 6 and in f:12, 9; (1981)
see number of extrahepatic angiosarcoma in m: 5, 6 [BT 14]b
animals for and inf: 12, 9; hepatoma in m: 9, 13
evaluation; no control; and in f: 11, 7
Rat; 0 or 2465; 1) 84-128 rats per sex per age group; Groth
Sprague-Dawley; 24.5 weeks; 7 h/d, 2) angiosarcomas (mostly in liver) in the 4 et al.
6, 18, 32, or 52 weeks; 5 d/week; scheduled age groups in exposed m: 1.2, 2.2, 7.4, (1981)
110-128 rats/sex sacrifice at 3, 6 and 18% and in exposed f: 2.3, 7.2, 28, 13%;
per age group; 9 months, only 1 subcutaneous angiosarcoma (m) in
treated control termination at all control groups (739 mice) #; older
week 43 adults more susceptible to
angiosarcoma-inducing effect
Table 30. (cont'd)
Species; strain; Doses in mg/m3; 1) Number of animals for histopathological References
age at start of exposure period; evaluation;
experiment; initial frequency of 2) type of tumour: number of animals with
number of animals per treatment; this tumour/dose group (unless otherwise
dose per exposure post-exposure given); other observations
period; type of observation period
control;
Rat; Wistar or 0, 6.5, 13, 26, 52, 1) 2-27 rats of each sex per dose per strain; Laib
Sprague-Dawley; 104, 208; 3 weeks; 2) dose dependent increase in ATPase-deficient et al.
3 days; 2-27 rats 8 h/d, 5 d/week; liver foci (preneoplastic lesion), (1985a)
per sex per strain; 10 weeks presumably significant at > 52 mg/m3
treated control ( m Sprague-Dawley rats at > 104 mg/m3);
f more susceptable than m in both strains
Rat; Wistar; 0 or 5200; 1) 2-10 rats of each sex per dose per exposure Laib
age-dependency 5-83 days, see 2); period; et al.
studied early in life, 8 h/d, 7 d/week; rats 2) age dependent increase in ATPase-deficient (1985b)
see 2); 4-10 rats sacrificed at the age liver foci lesion) (preneoplastic studied,
per sex; of 4 months rats exposed a) during gestation or at
treated control postnatal day 1-5 (b), 1-11 (c), 1-17 (d),
1-47 (e), 1-83 (f), 7-28 (g), or 21-49 (h);
no effect in a & b, foci area steeply
increased in c and d but not further
enhanced in e and f, foci area not lowered
in g but only a few foci in h
Mouse; CD1; 0, 2.6, 26, 260, 780, 1) see 2); Suzuki
5-6 weeks; 1560; 4 weeks; 2) pulmonary tumour incidences (#):week 0 (1983)
30-60 m; treated 6 h/d, 5 d/week; post-exposure: no tumour week 12: 0/18,
control 0, 12, 40 weeks 0/10, 0/9, 0/6, 6/9, 8/9 week 40: 0/17, 1/9,
3/9, 6/9, 5/7, 6/7; study focused on lung
tumours (no details on other tumours);
tumours derived from type II alveolar cells
Table 30. (cont'd)
Species; strain; Doses in mg/m3; 1) Number of animals for histopathological References
age at start of exposure period; evaluation;
experiment; initial frequency of 2) type of tumour: number of animals with
number of animals per treatment; this tumour/dose group (unless otherwise
dose per exposure post-exposure given); other observations
period; type of observation period
control;
Mouse; CD-1; 0, 130, 650, 2600; 1) 60, 40, 44, 38 m and 60, 40, 40, 38 f Hong et al.
2 months; 1, 3, 6 months; for cumulative effects; (1981)
8-28 mice/sex; 6 h/d, 5 d/week; 2) cumulative (f+m); ASL: 1, 2, 13*, 18*;
treated control 12 months extrahepatic angiosarcoma 0, 2, 6*, 2;
bronchioloalveolar tumor 16, 18, 52*, 50*;
cumulative (f): metastatic lung
adenocarcinoma 0, 4*, 2, 6*; mammary
carcinoma 4, 10*, 13*, 6; tumours at
different organ sites in f and m
increased in proportion to dose or duration
of exposure at higher dose levels
a d = day; f = females; m = males; n.g. = not given; # = no statistical evaluation; * = significant at P < 0.05
b Study number in experiments done by Maltoni and coworkers
c Animals were kept until spontaneous death or sacrificed at the end of given post-exposure observation period
that observed in long-term inhalation studies (compare with Table 32).
Hehir et al. (1981) presented evidence that even one single high dose
(> 13 000 mg/m3) resulted in a dose-related increase of pulmonary
tumours in mice. This effect was also demonstrated by repeated
administration to mice at lower dose levels with exposure periods
varying between 1 and 6 months (Hong et al., 1981; Suzuki, 1983).
Incidences of ASL, extrahepatic angiosarcoma and mammary gland
carcinoma were elevated in mice with increasing exposure periods at
doses of 130 or 650 mg/m3 (Hong et al., 1981). Also in rats,
angiosarcomas, predominantly in the liver, were induced by VC
inhalation for 25 weeks (15 600 mg/m3, 1 h/day, Maltoni et al., 1981;
2465 mg/m3, 7 h/day, Groth et al., 1981). Short-term exposure led to
increased incidences of carcinoma of the Zymbal gland, a sebaceous
gland of the ear canal in rats, at high doses (> 15 600 mg/m3;
Maltoni et al., 1981, BT10).
Short-term exposure studies on the effect of age on
susceptibility to tumour induction are discussed in section 7.7.3.
7.7.2.2 Long-term exposure
a) Rats
Although differences in experimental design, notably in the
duration of follow-up, and the low overall frequency of tumours tend
to confuse the picture, a dose-response relationship could be observed
in studies BT1, BT2, BT9 and BT15 on Sprague-Dawley rats for ASL
(Table 31, Fig. 4 and chapter 10) and Zymbal gland carcinomas, while
it was less clear for nephroblastomas, neuroblastomas and mammary
malignant tumours.
Lee et al. (1978) and Drew et al. (1983) reported similar
findings on the incidence of ASL. Female rats seem to be more
susceptible to ASL tumours than males (Lee et al., 1978; Maltoni et
al., 1981). ASL was detected at 26 mg/m3 and the incidence was
statistically significant at 130 mg/m3, which was at a lower level
than other tumour types with the exception of mammary gland tumours
(13 mg/m3; see Table 33) (Maltoni et al., 1981). VC exposure also
caused angiosarcomas at extrahepatic sites (Maltoni et al., 1981, BT14
see Table 30; Lee et al., 1978; Drew et al., 1983).
Increased incidences of other tumour types than those reported by
Maltoni et al. (1981) in Sprague-Dawley rats were shown by Drew et al.
(1983) in Fischer-344 rats (neoplastic liver nodules and
hepatocellular carcinoma) and by Feron & Kroes (1979) in Wistar rats
(tumour of the nasal cavity). These were single dose experiments.
Drew et al. (1983; see Table 32) studied the effect of exposure
duration (6, 12, 18 or 24 months) on tumour incidences and
demonstrated that longer exposure periods, and thus greater cumulative
exposures, led to an increase in tumorigenic responses concerning
liver angiosarcoma, angiosarcoma at all sites, and hepatocellular
Table 31. Incidence of angiosarcomas of the liver (ASL) and mammary
tumours observed in Sprague-Dawley ratsa
Exposure Concentration ASL ASL ASL Mammary
(ppm) (mg/m3) (males) (females) (males + adenocarcinomas
females)
0 0 0/173 0/239 0/412 12/239
1 2.6 0/58 0/60 0/118 12/60
5 13 0/59 0/60 0/119 22/60
10 26 0/59 1/60 1/119 11/60
25 65 1/60 4/60 5/120 15/60
50 130 2/174 13/180 15/354 61/180
100 260 0/60 1/60 1/120 3/60
150 390 1/60 5/60 6/120 6/60
200 520 7/60 5/60 12/120 5/60
250 650 1/29 2/30 3/59 2/30
500 1300 0/30 6/30 6/60 1/30
2500 6500 6/30 7/30 13/60 2/30
6000 15 600 3/29 10/30 13/59 0/30
10 000 3/30
a The data are combined from the experiments BT1, BT2, BT9 and BT15 conducted by Maltoni et al. (1984).
Control animals from several experiments are combined in the 0 ppm group. Animals were exposed
4 h/day, 5 days/week for 52 weeks; the follow-up was until the death of the animals or until week
83 (BT1), 90 (BT2), 90 (BT9) or 95 (BT15). Tumours were scored at the time of death. Adapted from
Reitz et al. (1996)
The rate of reaction of CAA with nucleic bases is slower than
with CEO (Zajdela et al., 1980). In in vitro studies, CEO but not
CAA was shown to be the main entity giving rise to etheno adducts
(Guengerich, 1992). Furthermore, as discussed in section 7.8, CEO but
not CAA show similar toxicity/mutagenic profiles to VC in a
metabolically competent human B-lymphoblastoid line (Chiang et al.,
1997). These findings seem to corroborate the original suggestion that
it is CEO rather than CAA that is the main source of etheno adducts
(Van Duuren, 1975; Guengerich et al., 1981; Gwinner et al., 1983).
In pre-weanling rats exposed to VC, 7-OEG had a half-life of
approx. 62 h, while the etheno adducts are highly persistent with a
half-life for Epsilon G of approx. 30 days (Swenberg et al., 1992).
Studies on the persistence of Epsilon A and Epsilon C in the liver of
adult rats have shown that there is no significant decrease in adduct
levels 2 months after termination of VC exposure (Guichard et al.,
1996). This is in contrast with the known repair of etheno adducts in
vitro (Dosanjh et al., 1994). The rat and human 3-methyladenine DNA
glycosylases can excise Epsilon A from DNA (Saparbaev et al., 1995).
Epsilon C can be released by the human mismatch-specific thymine-DNA
glycosylase (Saparbaev & Laval, 1998).
DNA adduct formation seems to be age-dependent; about 5- to
6-fold more DNA adducts were determined in young animals compared to
adults (Laib et al., 1989). Similar results were presented by Fedtke
et al. (1990b) who exposed adult and 10-day-old Sprague-Dawley rats
(Table 26). Ciroussel et al. (1990) exposed 7-day-old and 13-week-old
rats (strain BD IV) for 2 weeks to VC and detected 6 times more DNA
adducts in the liver of young rats compared to adults (Table 26).
It should be noted that background levels of etheno bases have
been found in various tissues in unexposed rodents (Guichard et al.,
1996; Fernando et al., 1996; Barbin, in press) and humans (Nair et
al., 1995, 1997). Lipid peroxidation products have been shown to react
with nucleic acid bases yielding etheno adducts (El Ghissassi et al.,
1995a; Chung et al., 1996; Bartsch et al., 1997). The role of lipid
peroxidation products in the endogenous formation of background levels
of etheno bases is further supported by the finding of elevated levels
of Epsilon dA and Epsilon dC in the liver from Long Evans Cinnamon
rats (Nair et al., 1996) and from patients with Wilson's disease or
with primary haemochromatosis (Nair et al., 1998).
Etheno adducts are also formed via substituted oxiranes formed
from other vinyl monomers, e.g., vinyl bromide (Bolt, 1994) and vinyl
and ethyl carbamate (urethane) (Park et al., 1993).
6.5.2 Alkylation of proteins
Bolt et al. (1980) studied covalent binding of radiolabel to
proteins in rats exposed to 14C-labelled VC. The target of alkylation
is the free sulfhydryl group of proteins. The liver always showed the
highest binding rate. The fraction of VC that was irreversibly bound
to proteins was independent of the VC dose applied, indicating no
threshold effect even at low doses. In vivo studies on rats
(Guengerich & Watanabe, 1979) have shown that the amount of total VC
metabolites bound in the liver to proteins is twice that bound to DNA,
RNA and lipids. Osterman-Golkar et al. (1977) reported the alkylation
of cysteine (S-(2-hydroxyethyl)cysteine) and histidine ( (N-1-and
N-3) hydroxyethylhistidine) of the globin precipitate of haemoglobin
and small amounts of the alkylated histidines in proteins from testis
in mice exposed to 1,2-14C-vinyl chloride.
In vitro studies have shown that incubation of rat liver
microsomes with 14C-labelled VC results in NADPH-dependent microsomal
uptake and covalent binding to microsomal proteins (Kappus et al.,
1975, 1976; Baker & Ronnenberg, 1993). It has been suggested that VC
metabolites might be involved in the destruction of the haem moiety of
cytochrome P-450 in the liver (Guengerich & Strickland, 1977).
6.6 Modelling of pharmacokinetic data for vinyl chloride
There has been progress in recent years in the development of
physiologically based toxicokinetic (PBTK) models describing the
toxicokinetics of chemicals. These theoretical models permit
predictions of the dose of active metabolites reaching target tissues
in different species, including humans, and, therefore, have improved
the toxicokinetic extrapolation in cancer risk assessments. PBTK
models have also been used as a tool to examine the behaviour of VC in
mammalian systems. The description of PBTK models for VC is presented
in Annex 2.
Table 26. Detection of DNA adducts in vivo after VC inhalation in ratsa
Strain; sex; Treatment; Investigated Alkylated bases in Comments References
age post-exposure organs DNA (max. concentration
survival time in pmol/µmol unmodified
base in specified organs)
BD IV; male & 1300 mg/m3, 7 h/day, liver, lung, epsilon-dA (0.131, 0.105, higher sensitivity of Ciroussel
female; 7 days 7 days/week for brain, kidney 0.06, b.d.l.); epsilon-dC young rats compared et al.(1990)
2 weeks; none (0.492, 0.246, 0.216, with adults
b.d.l.) (see section 7.7.4)
BD IV; male; 1300 mg/m3, 7 h/day, liver epsilon-dA (0.019); Ciroussel
13 weeks 7 days/week for epsilon-dC (0.080) et al. (1990)
2 weeks; none
S.-D.; male & 1560 mg/m3, 4 h/day liver, lung, 7-OEG (162, 20, 29, <10, higher sensitivity of Fedtke et
female; 10 days for 5 days; kidney, <10); N2,3-epsilon-G (1.81, young rats compared al. (1990b)
0, 3, 7, 14 days brain, spleen 0.21, 0.31, <0.12, <0.12); with adults (see
measured immediately a.e. section 7.7.4)
concerning liver
adducts
S.-D.; female; 1560 mg/m3, 4 h/day liver, lung, 7-OEG (43, 20, n.g.); DNA adduct formation Fedtke et
adult for 5 days; kidney N2,3-epsilon-G (0.47, max. at the end of al. (1990b)
0, 3, 7, 14 days 0.27, <0.12); measured exposure; most DNA
immediately a.e. adducts in the liver
S.-D.; male & 5200 mg/m3, 7 h/day liver, lung epsilon-dA (0.05, 0.13) more DNA adducts in the Eberle et
female; 11 days on days 1-9 and 24 h epsilon-dC (0.16, 0.33) lung than in the liver al. (1989)
on day 10; none
S.-D.; male & 1560 mg/m3, 4 h/day liver, lung, 7-OEG (162, 20, 29); DNA adduct Swenberg
female; 10 days for 5 days; kidney N2,3-epsilon-G (1.81, 0.21, max. at the end et al.
0, 3, 7, 14 days 0.31); epsilon-dC (0.98, 0.30, exposure; most DNA (1992)
0.29); epsilon-dA (0.21, 0.065, adducts in the liver
0.04); measured immediately a.e.
Table 26. (cont'd)
Strain; sex; Treatment; Investigated Alkylated bases in Comments References
age post-exposure organs DNA (max. concentration
survival time in pmol/µmol unmodified
base in specified organs)
S.-D.; male; 1300 mg/m3, 4 h/day, liver, lung, epsilon-dA (liver: background exposure-time-dependent Guichard
6 weeks 5 days/week for 1, kidney, 0.0004, a.e. up to 0.045; increase of adducts in et al. (1996)
2, 4, 8 weeks; none lymphocytes background lung and kidney up to liver (ca. 100-fold);
0.033, no increase a.e.); no (lymphocytes) or
epsilon-dC (liver: background slight increase (ca.
0.0007, a.e. up to 0.08; kidney: 2-fold in kidney, ca.
background 0.086, a.e. up to 5-fold in lung) of
0.16; lung: background 0.072, adducts in other
a.e. up to 0.38) organs but higher
background levels
8 weeks brain, testis, epsilon-dA (increase in testis, Barbin
spleen not in brain or spleen) (in press)
epsilon-dC (increase in testis
and spleen, but not in brain)
a a.e. = after exposure; b.d.l. = below detection limit; n.g. = not given; S.-D. = Sprague-Dawley; epsilon-dA = 1,N6-ethenodeoxyadenosine;
epsilon-dC = 3,N4-ethenodeoxycytidine; 7-OEG = 7-(2-oxoethyl)guanine; epsilon-G = ethenoguanine
7. EFFECTS ON LABORATORY MAMMALS AND IN VITRO TEST SYSTEMS
7.1 Acute toxicity
VC appears to be of low acute toxicity when administered to
various species by inhalation. A summary of acute toxicity is given in
Table 27. No data are available on acute toxicity after dermal
application.
VC has a narcotic effect (see also section 7.6) after inhalative
administration of high doses. In rats, mice and hamsters, death was
preceded by increased motor activity, twitching of extremities,
tremor, ataxia, tonic-clonic convulsions and accelerated respiration
(Patty et al., 1930; Mastromatteo et al., 1960; Prodan et al., 1975).
In dogs, severe cardiac arrhythmias occurred under narcosis after
inhalative exposure to 260 000 mg VC/m3 (Oster et al., 1947). Similar
results were reported by Carr et al. (1949). Pneumonitis was more
frequent in experimental mice than in controls 8 and 18 months after a
single 1-h exposure to 1000, 3000, 13 000 or 130 000 mg/m3 (Hehir et
al., 1981).
After acute inhalative exposure to VC, pathological findings in
the rat included congestion of the internal organs, particularly lung,
liver and kidney as well as pulmonary oedema (Patty et al., 1930;
Mastromatteo et al., 1960; Lester et al., 1963; Prodan et al., 1975).
Exposure to VC (3900 mg/m3) for 24 h did not cause pathological
changes in male and female rats or female New Zealand rabbits. In
mice, exposure to the same concentration for 4 and 8 h resulted in
circulatory changes, while longer exposure (12 and 24 h) caused
vasomotor paralysis followed by characteristic shock with subsequent
alterations in the liver and lungs (Tátrai & Ungváry, 1981).
7.2 Short-term toxicity
7.2.1 Oral exposure
Groups of 15 male and 15 female weanling Wistar rats were gavaged
with VC dissolved in soya-bean oil (0, 30, 100 and 300 mg/kg body
weight, once daily, 6 days/week for 13 weeks). The treatment caused no
noticeable changes in appearance or behaviour, body weight gain or
food intake. The total number of white blood cells and the sugar
content of the blood were slightly decreased by the intermediate and
high dose levels. The activities of serum ASAT and ALAT and of urinary
ASAT were decreased in males given the top dose. There were no other
significant changes in the haematological or biochemical indices and
no treatment-related alterations were observed in the microscopic
constituents of the urine. The relative weight of the liver in males
and females showed a tendency to increase with increasing doses of VC
but the difference from the controls was statistically significant
only at the highest dose level. The NOEL was given as 30 mg/kg body
weight (Feron et al., 1975). In contrast, in another study, all male
and female Wistar rats (presumably a total of 15 rats) receiving once
daily 300 mg VC/kg body weight in peroxide-free corn oil by gavage
died within 60 days of treatment (Knight & Gibbons, 1987).
7.2.2 Inhalation exposure
A summary of the non-neoplastic effects of VC after short-term
inhalation is presented in Table 28. Information on carcinogenic
effects after short-term inhalation are presented in section 7.7 and
Table 30.
In various species, the main target of VC toxicity is the liver.
A dose-related significant increase in relative liver weight was found
in male rats at exposure levels of 26, 260 and 7800 mg/m3 (Bi et al.,
1985). Degenerative effects on liver parenchyma were reported in
rabbits at a dose level of 520 mg/m3 (Torkelson et al., 1961), in
rats at 1300 mg/m3 (Torkelson et al., 1961; Wisniewska-Knypl et al.,
1980), and in mice at 2600 mg/m3 (Lee et al., 1977). Bi et al. (1985)
reported a decreased relative testis weight in rats at exposure levels
of 260 and 7800 mg/m3; however, the effect was not dose-related.
These authors also reported a higher incidence and severity of damage
to the testicular seminiferous tubules at all dose levels tested (26,
260 and 7000 mg/m3), the differences with the controls being
statistically significant only at the two highest dose levels.
However, the severity of the testicular damage was clearly
dose-related (correlation coefficient 0.993; P < 0.01) suggesting
an adverse effect of VC on the testes already at 26 mg/m3. Effects on
relative liver weight were detected in rats at 26 mg/m3 (Bi et al.,
1985) (see also Table 28 and section 7.5.1). Effects on the kidney
(Lee et al., 1977; Feron et al., 1979a,b; Himeno et al., 1983) and the
lung (Suzuki, 1980) were observed in rats and/or mice at higher doses
(Table 28). For mice a LOEL of 130 mg/m3 was given (decreased
survival; Hong et al., 1981). Rats, mice and rabbits seem to be more
sensitive than guinea-pigs and dogs (Torkelson et al., 1961; Hong et
al., 1981).
7.2.3 Dermal exposure
No studies were available on short-term dermal exposure.
7.3 Long-term toxicity - effects other than tumours
7.3.1 Oral exposure
Studies on effects induced by long-term oral application of VC in
rats are presented in detail in Table 29. Studies on other species are
not available.
Long-term feeding studies in male and female rats showed
increased mortality in males at doses > 5.0 mg/kg body weight per
day (Feron et al., 1981) and in females at doses of > 1.3 mg/kg
Table 27. Toxicity of VC after acute inhalation exposurea
Species Duration of Parameter Value References
exposure in g/m3
Rat 2 h LC50 390 Prodan et
LC100 525 al. (1975)
Rat 30 min LC100 780 Mastromatteo
et al. (1960)
Rat 1 h ataxia, 130 Hehir et al.
hyperventilation (1981)
Mouse 2 h LC50 293 Prodan et al.
LC100 375 (1975)
Mouse 30 min LC100 780 Mastromatteo
et al. (1960)
Guinea-pig 2 h LC50 595 Prodan et al.
LC100 700 (1975)
Guinea-pig 30 min death, 780 Mastromatteo
threshold dose et al. (1960)
Guinea-pig 2-6 h deep narcosis, 260 Patty et al.
no death (1930)
Guinea-pig 18-55 min death, 390-650 Patty et al.
threshold dose (1930)
Guinea-pig 90 min narcosis, 65-130 Patty et al.
threshold dose (1930)
Rabbit 2 h LC50 295 Prodan et al.
LC100 700 (1975)
a Cited studies not conducted according to present-day standards
Table 28. Toxicity of vinyl chloride in animals after short-term inhalation exposures - non-neoplastic effectsa
Species, strain; Doses in Exposure duration; Significant effects References
number of animals mg/m3 frequency;
per dose per post-exposure
exposure period observation period
Rat, Wistar; 8 0, 26, 260, 3 or 6 mo; 26 mg/m3: relative spleen and heart weight Bi et al.
(3 mo) or 30 7800 6 d/week, 6 h/d; * (6 mo); incidence of testicular seminiferous (1985)
(6 mo) m rats none tubule damage * ! (exposure period not specified)
> 26 mg/m3: relative liver weight * (6 mo) 260 mg/m3:
relative heart weight * (3 mo) > 260 mg/m3: relative
testis weight ** (6 mo); incidence of testicular
tubule damage * 7800 mg/m3: relative kidney and
spleen weight * (3 mo)
Rat; n.g.; at high 0, 130, 260, 4.5 (high dose) 130 mg/m3: NOEL (body and organ weight, Torkelson
dose 10 f & 520, 1300 or 6 mo; survival, haematology, clinical chemistry, urine et al.
10 m (control 5 d/week, analysis, histopathology) 260 mg/m3: relative liver (1961)
5 f & 5 m); other 7 h/d; none weight * (m+f) 1300 mg/m3: granular degeneration
groups 20-24 m in centrilobular liver parenchyma #;
& 24 f liver weight * (m)
Rat, Wistar; 0, 130, 1, 3, 6 mo; 130 mg/m3: slight changes such as proliferation Wisniewska-Knypl
8-10 m 1300, 5 d/week, 5 h/d; of hepatocellular SER (3-6 mo)# et al.
52 000 none > 1300 mg/m3: liver weight * (1-6 mo), ultrastructural (1980)
hepatocellular changes (swollen mitochondria, lipid
droplets *) after 3 and 6 mo #
Rat, 0, 2465 24.5 weeks; 2465 mg/m3: mortality *# (m+f), haematology and Groth et
Sprague-Dawley; 7 h/d, 5 d/week; clinical chemistry <-> al. (1981)
110-128 rats up to 19 weeks
per sex
Rat, Wistar; 0, 13 000 4, 13, 26 weeks; 13 000 mg/m3, > 4 weeks: body weight **, Feron et al.
10 f & 10 m 5 d/week, 7 h/d; blood clotting time ** > 13 weeks: liver function (1979a,b)
none (BSB-retention test) **; liver and kidney
weight * (m+f)
Table 28. (cont'd)
Species, strain; Doses in Exposure duration; Significant effects References
number of animals mg/m3 frequency;
per dose per post-exposure
exposure period observation period
26 weeks: spleen weight * (m+f); clear cell foci Feron &
and basophilic foci in the liver * (m+f)# Kroes (1979)
Rat, Sherman; 0, 52 000 92 d; 5 d/week, 52 000 mg/m3: relative liver weight * and Lester et al.
12-15 rats/sex 8 h/d; none spleen weight ** (m+f); white blood cell counts **; (1963)
swelling of hepatocytes with vacuolization,
compression of sinusoids #
Mouse, CD-1; 0, 130, 650, 1, 3, 6 mo; > 130 mg/m3: survival after 6 mo exposure ** # (low Hong et al.
8-28 mice/sex 2600 5 d/week, 6 h/d; dose: m, 1/8 versus 22/28 in control; f, 0/8 versus (1981)
12 mo 23/28; tumour incidences no differences)
Mouse, CD-1; 0, 2600 3-9 exposures; 2600 mg/m3: early deaths (2 m + 1 f)!; pathological Lee et al.
36 mice/sex 5 d/week, 6 h/d; changes in dead animals: acute toxic hepatitis (1977)
none (congestion, diffused necrosis), tubular necrosis
in renal cortex
Mouse, n.g.; 6500 1 or 6 mo; 6500 mg/m3: hyperplastic nodules and dilatated Schaffner
5 m (1 mo) or (no control) 5 d/week, 5 h/d; sinusoids in liver parenchyma after 6 mo (1979)
14 m (6 mo) none exposure #
Mouse, CD-1; 0, 6500, 5-6 mo; > 6500 mg/m3: proliferation and hypertrophy of Suzuki
3-16 m 15 600 5 d/week, 5 h/d; bronchiolar cells, hypersecretion of bronchial (1980)
2-37 d and bronchiolar epithelium, hyperplasia of
alveolar epithelium, bronchiolar inflammation #
(only pulmonary effects recorded; effects not
dose related)
Mouse, CD-1; 13 000 10 weeks; 13 000 mg/m3: focal lung hyperplasia, proliferation Himeno et
10 m (no control) 5 d/week, 4 h/d; of sinusoidal cells of the liver, proliferative al. (1983)
none effects in renal glomeruli, giant cells in testis #
Table 28. (cont'd)
Species, strain; Doses in Exposure duration; Significant effects References
number of animals mg/m3 frequency;
per dose per post-exposure
exposure period observation period
Mouse, ICR; a) 0, 13 000, a) 5 to 6 d, basophilic stippled erythrocytes * (#) in a) and b), Kudo et
n.g. 26 000; b) 62 d; a) 4 h/d, related effect not dose al. (1990)
b) 78 to 104 b) continuously;
none
Hamster, 0, 520 6 mo; 5 d/week, 520 mg/m3: survival ** Drew et
golden Syrian; 6 h/d; life span al. (1983)
56 f
Guinea-pig, 0, 130, 6 mo; 5 d/week, 520 mg/m3: NOEL (body and organ weight, Torkelson
n.g.; 10-12 m 260, 520 7 h/d; none survival, clinical chemistry, histopathology) et al. (1961)
& 8-12 f
Rabbit, n.g.; 0, 130, 6 mo; 5 d/week, 260 mg/m3: NOEL (body and organ weight, Torkelson
3 rabbits/sex 260, 520 7 h/d; none survival, clinical chemistry, histopathology) et al. (1961)
520 mg/m3: degeneration of centrilobular
liver parenchyma (m+f) with periportal cellular
infiltration (f) #
Dog; n.g.; 0, 130, 6 mo; 5 d/week, 130-520 mg/m3: no effects recorded (body and Torkelson
1 dog/sex 260, 520 7 h/d; none organ weight, survival, haematology, clinical et al. (1961)
chemistry, urine analysis, histopathology) #
a d = day; mo = month, m = male; f = female; n.g. = not given; ! = increase not significant; # = no data about significance;
* = increased; ** = decreased; <-> = no change
body weight per day (Feron et al., 1981; Til et al., 1983, 1991). At
14.1 mg/kg body weight per day, blood clotting time was decreased and
alpha-fetoprotein levels in blood serum were increased (Feron et al.,
1981). Skin fibrosis was observed at 30 mg/kg body weight per day
administered by gavage (Knight & Gibbons, 1987).
As for short-term exposure, the primary target organ of VC in
rats after long-term oral exposure is the liver. Female rats appeared
to be more sensitive than males to the hepatotoxicity of VC
(section 7.7.4). Increased relative liver weights were found at
14.1 mg/kg body weight per day after feeding periods of 6 or 12 months
(Feron et al., 1981). Morphological alterations of the liver included
extensive hepatocellular necrosis at doses > 5 mg/kg body weight
per day, foci of haematopoiesis at 14.1 mg/kg body weight per day, and
cysts and liver cell polymorphism (variation in size and shape of
hepatocytes and their nuclei) at doses > 1.3 mg/kg body weight per
day (Feron et al., 1981; Til et al., 1983, 1991). Foci of
hepatocellular alteration (clear cell, mixed cell, eosinophilic and
basophilic foci) were common findings. Clear cell, mixed cell and
eosinophilic foci occurred at doses > 1.3 mg/kg body weight per day
and basophilic foci at doses > 0.014 mg/kg body weight per day (Til
et al., 1983, 1991).
7.3.2 Inhalation exposure
A summary of non-neoplastic and neoplastic effects after
long-term intermittent inhalation of VC is presented in Table 32 with
details of exposure regime (see also section 7.7). Long-term exposure
to VC by inhalation resulted in increased mortality in rats exposed to
a dose of 260 mg/m3 for 12, 18 and 24 months, in mice exposed to
130 mg/m3 for 6, 12 and 18 months and in hamsters exposed to
520 mg/m3 for 6, 12 and 18 months (Drew et al., 1983). Maltoni et al.
(1984) reported increased mortality in rats (BT15) and hamsters (BT8)
at lower dose levels (2.6 and 130 mg/m3, respectively), but no
statistical evaluation was performed. Rats exposed to 130 mg/m3
showed reduced body weight and increased relative spleen weight (Sokal
et al., 1980; see below). At this dose morphological alterations were
reported in rat liver, such as hepatocellular lipid accumulation and
mitochondrial swellings (Wisniewska-Knypl et al., 1980) as well as
proliferation of cells lining the liver sinusoids (Sokal et al.,
1980). Exposure to higher doses revealed degenerative alteration in
the testis (1300 mg/m3; Sokal et al., 1980) and tubular nephrosis and
focal degeneration of the myocardium (13 000 mg/m3; Feron & Kroes,
1979) in rats.
Male Wistar rats (42-80 per group) exposed to 0, 130, 1300 and
52 000 mg VC/m3, 5 days/week, 5 h daily, for 10 months, showed
significantly reduced body weight in all treatment groups; general
condition and behaviour were not altered. Relative organ weights of
spleen and heart (except at the mid dose) were significantly elevated
at > 130 mg/m3, as well as liver and kidney weights at mid- and
high-dose levels and testis weights at the high-dose level. X-ray
analysis did not show any skeletal alterations. Histopathology
revealed statistically significantly increased incidences of nuclear
polymorphism (nuclei of variable size and irregular shape) of
hepatocytes and proliferation of reticuloendothelial cells lining
liver sinusoids at the two highest dose levels. Fatty degeneration of
hepatocytes was found at all exposure levels. Ultrastructural changes
in hepatocytes seen at all exposure levels, but not in controls,
included swollen and giant mitochondria with broken cristae,
proliferation and dilatation of smooth endoplasmic reticulum, nuclear
membrane invaginations, areas of cytoplasmic degradation and increased
numbers of small lipid droplets (Sokal et al., 1980; Wisniewska-Knypl
et al., 1980). In addition, Sokal et al. (1980) reported necrotic foci
of the spermatogenic epithelium and disorders of spermatogenesis
accompanied by large multinuclear syncytial cells in the testis
predominantly at 1300 mg/m3. Haematology, urine analysis and clinical
chemistry did not reveal differences of toxicological significance.
Thus NOEL for rats or mice concerning non-neoplastic effects
could not be derived, since effects were observed at the lowest levels
studied (130 mg/m3).
7.4 Skin and eye irritation; sensitization
No relevant data on skin irritation were identified. No studies
were available on sensitizing effects of VC in animals. Erythema and
second-degree burns were reported in a worker after accidental
exposure to liquid VC (see section 8.3.2.1). Dryness of eyes and nose
was reported by volunteers exposed to 1300 mg/m3 (see section 8.2).
7.5 Reproductive toxicity, embryotoxicity and teratogenicity
7.5.1 Male reproductive toxicity
Inhalation studies on rats showed some evidence of reduced
fertility and morphological alterations of the testis. It should be
noted that none of the studies cited were conducted according to
current guidelines (OECD, 1983a,b).
In dominant lethal studies on mice no reduction in fertility was
observed (Anderson et al., 1976; Table 35). However, reduced fertility
was noted in male CD rats (12 per group) mated once on week 11 of
exposure (0, 130, 650 or 2600 mg/m3; 6 h/day, 5 days per week). VC
treatment decreased dose-dependently the ratio of pregnant to mated
females; this was significant at mid- and high-dose level (Short et
al., 1977; see also section 7.8.2).
Bi et al. (1985; for details see Table 28 and section 7.2.2)
observed decreased relative testis weight at 260 mg/m3 and
morphological alterations in the testis of rats even at the lowest
dose of 26 mg/m3. Morphological alterations in the testis of rats
were also reported by Sokal et al. (1980, see section 7.3.2).
VC was administered to adult female CD rats at 0, 24, 260 and
2860 mg/m3, 6 h/day, 5 days/week for at least 10 weeks prior to
mating until day 4 of lactation (94 + days) in a two-generation study.
Alterations in reproductive performance and fertility were not
detected at any dose level tested. Centrilobular hypertrophy in the
liver and increased relative liver weights, however, were noted at all
dose levels tested in a dose-related manner (Shah, 1998).
7.5.2 Embryotoxicity and teratogenicity
Although the available studies did not follow guideline
standards, the information leads to the conclusion that there is
embryotoxicity or fetal toxicity, including increased numbers of
resorptions, decreased numbers of live fetuses and delayed development
at dose levels producing maternal toxicity. VC treatment did not
induce gross malformations. There is evidence for the permeability of
the placenta to VC (Ungváry et al., 1978; see section 6.2.2).
John et al. (1977, 1981) investigated mice, rats and rabbits for
teratogenic effects of inhaled VC. In all three species a similar
experimental design was used. Pregnant CF-1 mice were exposed 7 h/day
to 0, 130 or 1300 mg VC/m3 on day 6-15 of gestation. For both
concentrations tested, concurrent control groups were sham-exposed.
Animals were observed daily and maternal body weight recorded at
several intervals (no further data). The mice were sacrificed on day
18 of gestation. After determination of external anomalies, one-third
of each litter (19-26 litters per group) was examined for soft tissue
anomalies and the other mice for skeletal anomalies. Exposure to
1300 mg/m3 led to deaths (5 of 29 bred females, P < 0.05), reduced
maternal body weight gain (P < 0.05) and food consumption
(P< 0.05). No maternal toxicity was apparent in females exposed to
130 mg/m3. The number of live fetuses per litter and fetal body
weight were significantly decreased and the number of resorptions
significantly increased at 1300 mg/m3, but these values were within
the range observed for the second concurrent control group or for
historical controls. No soft tissue or external anomalies were
detected. Significantly increased incidences of three skeletal
variants (delayed skull and sternebrae ossification, unfused
sternebrae) in the high-dose group were indicative of delayed skeletal
development. No developmental toxicity was observed at 130 mg/m3.
Sprague-Dawley rats were exposed to 0, 1300 or 6500 mg/m3 on
gestation day 6-15 and dams were sacrificed on day 21 of gestation.
Low-dose exposure resulted in reduced maternal weight gain
(P< 0.05). At the high-dose level further maternal effects like
reduced food consumption and increased liver weight were observed
(P< 0.05), and one out of 17 pregnant rats died (no further
information). Examination of 16-31 litters per group revealed
significantly decreased fetal weight in the low-dose but not in the
high-dose group. Significantly increased incidences in dilated ureter
were observed at 6500 mg/m3.
Rabbits were exposed on gestation day 6-18 to 0, 1300 or
6500 mg/m3 and sacrificed on gestation day 29. Except for reduced
food consumption in the low-dose group and 1 death in 7 bred females
of the high-dose group, no further evidence of maternal toxicity was
observed. In the high-dose group only 5 litters were examined, but in
other groups 11-19 litters. Compared to the concurrent control, litter
size was significantly reduced in the low-dose group (but not at
6500 mg/m3). However, there was an increase in litter size in this
treatment group compared to controls concurrent to the high-dose
group. The incidence of delayed ossification of the sternebrae was
increased at 1300 mg/m3, but not in the high-dose group (John et al.,
1977, 1981).
Ungváry et al. (1978) exposed pregnant CFY rats continuously to 0
or 4000 mg VC/m3 during the first, second or last third of pregnancy
(gestation day 0-8, 7-13 or 13-20). Dams were sacrificed on gestation
day 20 and living, dead or resorbed fetuses were recorded. Placenta
and fetuses were weighed and fetuses macroscopically investigated. One
half of each litter was examined for soft tissue anomalies including
histopathology of organs with abnormalities and the other half was
processed for investigation of the skeletal system. Maternal weight
gain was significantly reduced in pregnant rats exposed during the
last third of pregnancy. Increased relative liver weight was observed
in dams exposed during the first or second third, but histopathology
revealed no pathological changes in the liver of any VC-treated rat.
No further signs of maternal toxicity were reported. Examination of 13
to 28 litters per group revealed increased fetal loss in per cent of
total implantation sites after exposure during gestation day 0-8
compared with the concurrent control. However, this value was not
significant compared with other control groups (e.g., control exposed
gestation day 13-20) of the same study. None of the soft tissue or
skeletal anomalies were attributed to VC treatment.
Exposure of 40 pregnant white Wistar rats to VC (mean level of
6.15 mg/m3 during whole gestation) resulted in elevated embryonic
mortality, lowered fetal weight, and induction of external and
internal anomalies in the development of the fetus (Mirkova et al.,
1978).
7.6 Special studies
7.6.1 Neurotoxicity
Profound narcosis was reported in guinea-pigs exposed to
65 000 mg VC/m3 for 90 min (Patty et al., 1930). Ataxia was observed
at this dose level after 5 min of exposure. The anaesthetic action of
VC was also observed in dogs (Oster et al., 1947) and mice (Peoples &
Leake, 1933). Mastromatteo et al. (1960) reported deep narcosis in
rats and mice exposed to 260 000 mg/m3 for 30 min. The narcotic
effect was preceded by increased motor activity after 5 min of
exposure, twitching of extremities (after 10 min), ataxia (after
15 min) and tremor (after 15 min). Rats exposed to 130 000 mg/m3 for
60 min showed ataxia preceded by hyperactivity but no narcotic effect
(Hehir et al., 1981).
Neuropathological alterations were observed in rats exposed to
78 000 mg/m3 (4 h/day, 5 days/week) for 12 months (Viola, 1970; Viola
et al., 1971). During the exposure period, the rats were slightly
soporific. Histopathology revealed diffuse degeneration in the gray
and white matter of the brain and at the level of the white matter
zones of reactive gliosis. In the cerebellum, atrophy of the granular
layer and degeneration of Purkinje cells were most prominent. In
addition, peripheral nerve bundles were often surrounded and invaded
by fibrotic processes.
Reports on neurotoxicity in humans occupationally exposed to VC
are given in section 8.3.2.3.
7.6.2 Immunotoxicity
Sharma & Gehring (1979) investigated mitogen-stimulated
transformation in splenic lymphocytes isolated from mice exposed to
26, 260 or 2600 mg/m3 for 2, 4 or 8 weeks (6 h/day, 5 days/week). The
treatment produced no effects on body or organ weight, except
increased spleen weight in high-dose groups, no effects on
haematological parameters and no pathological alterations at necropsy.
VC exposure caused stimulation of spontaneous lymphocyte
transformation in lymphocyte cultures prepared from mice exposed for 2
weeks to the high dose and from mice exposed for 4 weeks at all dose
levels, but this was not dose-dependent. The response of lymphocytes
to phytomitogens was increased at 2600 mg/m3 after exposure for
2 weeks and at all dose levels after exposure for 4 or 8 weeks, with
more pronounced effects at the mid-dose level. Stimulation of
lymphocyte transformation was not observed in lymphocytes from
unexposed mice cultured in the presence of VC, indicating that
metabolites of VC formed in vivo may be responsible for this effect.
Exposure of mice to 26 mg/m3 for 6 months (Bi et al., 1985;
Table 28) or rats to 130 mg/m3 for 10 months (Sokal et al., 1980;
section 7.3.2) induced increased relative spleen weight, whereas much
higher doses (52 000 mg/m3 for 92 days) produced decreased spleen
weight and reduced white blood cell counts (Lester et al., 1963; see
Table 28).
Reports on immunological and lymphoreticular effects in humans
occupationally exposed to VC are given in section 8.3.2.
7.6.3 Cardiovascular effects
Viola (1970) demonstrated thickening of the walls of small
arterial vessels (in some vessels blockage of lumen) due to
endothelial fibrosis and proliferation of endothelial cells in rats
exposed to 78 000 mg/m3 (4 h/day, 5 days/week) for 12 months.
Exposure of rats to 13 000 mg/m3 for 12 months (Feron & Kroes, 1979;
see Table 32) resulted in thickened walls of arteries and focal
degenerations of the myocardia.
Oster et al. (1947) observed cardiac arrhythmias (e.g.,
ventricular extrasystoles and fibrillation, auriculoventricular block)
in dogs at a dose level of 260 000 mg/m3.
Bi et al. (1985) reported increased relative heart weight in rats
exposed for 6 months to 26 mg/m3 or for 3 months to 260 mg/m3 (Table
28).
Impaired peripheral circulation as well as other cardiovascular
effects in occupational exposed humans are described in section 8.3.2.
7.6.4 Hepatotoxicity
Data on hepatotoxicity are presented in sections 7.2 and 7.3.
7.7 Carcinogenicity
VC causes a wide spectrum of tumours in animals and this spectrum
is similar in a number of different species (see Table 33). For
example in rats, the following tumours have been described after VC
inhalation exposure: liver and other angiosarcomas, other liver
tumours, mammary gland carcinoma, nephroblastoma, neuroblastoma,
stomach tumours and Zymbal gland tumours. The lowest dose at which an
increase in tumour incidences was observed when rats were exposed by
inhalation was 130 mg/m3 for liver angiosarcoma (ASL) and 13 mg/m3
for mammary tumours. There is evidence that animals are more
susceptible to tumour induction early in life. There is also evidence
that liver tumours are induced in female rats at lower doses than in
males.
VC is also carcinogenic in animals after oral application. The
spectrum of tumours is similar to that observed after inhalation
exposure. The lowest observed dose producing a carcinogenic effect
(ASL) in rats was 1.3 mg/kg body weight per day.
7.7.1 Oral exposure
Details of studies on the carcinogenicity of VC in rats after
oral administration are tabulated in Table 29. There are three gavage
studies: two studies with 52 or 59 weeks gavage and a 32- or 18-week
follow-up and a 2-year lifetime gavage study with small numbers of
animals and high mortality at high doses (Maltoni et al., 1981 [BT11
and BT27] and Knight & Gibbons, 1987). The two feeding studies used
PVC powder containing VC incorporated into the diet; the numbers of
animals were large, and the administration comprised the whole
lifetime of the animals (Feron et al., 1981; Til et al., 1983, 1991).
Table 29. Long-term toxicity/carcinogenicity of vinyl chloride in experimental animals after oral administrationa
Species; strain; Dose; 1) Effects other than tumours (dose in mg/kg bw/d); Reference
initial number of route of exposure; 2) number of animals for histopathological evaluation of
animals per dose; exposure period; neoplastic effects;
vehicle frequency of treatment; 3) type of tumour: number of animals with this tumour/dose
post-exposure group (unless otherwise given)
observation period
Rat; Wistar; 0, 1.7, 5.0, 14.1 1) > 1.7 mg/kg: haematology, biochemistry, urine analyis, Feron
60-80 rats/sex; mg/kg bw./d, organ function, body weight and food consumption <-> et al.
PVC powder bioavailable d (oral mortality !* (f); liver clear cell foci (f+m), basophilic (1981) e
(vehicle) intake 0, 1.8, 5.6, foci (f), eosinophilic foci (m+f) !*; liver-cell
containing VC 17.0 mg/kg bw/day); polymorphism !* (m); liver cysts !* (f);> 5.0 mg/kg: general
incorporated oral feed; lifespan condition ¡ (m+f, at mo 18); mortality !* (f+m); liver
into the diet study, terminated basophilic foci !* (m); extensive liver necrosis !* (f);
at week 135 (m) 14.1 mg/kg: extensive liver necrosis and liver cysts !* (m); focal
and 144 (f); diet haematopoiesis in liver !* (m); liver weight !* (f+m at mo
provided 4 h/d; 6, f at mo 12, interim sacrifice); blood clotting time at mo
none 6 and alpha-fetoprotein at mo 12 ¡* (f+m);
2) 55, 58, 56, 59 m and 57, 58, 59, 57 f;
3) ASL in m: 0, 0, 6*, 27* and in f: 0, 0, 2, 9*;neoplastic liver
nodules in m: 0, 1, 7*, 23* and in f : 2, 26*, 39*, 44*;
hepatocellular carcinoma in m: 0, 1, 2, 8* and in f: 0, 4, 19*,
29*; lung angiosarcoma in m: 0, 0, 4*, 19* and in f: 0, 0, 1, 5*
Rat; Wistar; 100 0, 0.014, 0.13, 1.3 1) > 0.014 mg/kg: bw. and food consumption <-> (m+f); liver Til et al.
rats/sex except mg/kg bw./d, basophilic foci !* (f); 1.3 mg/kg: mortality !* (f; at (1983,
high dose (50 bioavailable d (oral week 149); liver glutathione level at week 40 or 80 <-> 1991) f
per sex); PVC intake 0, 0.018, (satellite groups); liver clear cell (m+f), basophilic (m),
powder (vehicle) 0.17, 1.7 mg/kg eosinophilic (f), and mixed cell foci (f) !*; liver cysts !*
containing VC bw./day); oral feed; (f ); moderate to severe liver-cell polymorphism !* (m+f );
incorporated into lifespan study, 2) 99, 99, 99, 49 m and 98, 100, 96, 49 f;3) ASL in m: 0, 0,
the diet terminated at week 0, 1 and 0, 0, 0, 2 in f; neoplastic liver nodules in m 0,
149 (m) and 150 (f); 0, 0, 3 and 0, 1, 1, 10* in f; hepatocellular carcinoma in
diet provided 4 h/d; m 0, 0, 0, 3* and 1, 0, 1, 3 in f
none
Table 29. (cont'd)
Species; strain; Dose; 1) Effects other than tumours (dose in mg/kg bw/d); Reference
initial number of route of exposure; 2) number of animals for histopathological evaluation of
animals per dose; exposure period; neoplastic effects;
vehicle frequency of treatment; 3) type of tumour: number of animals with this tumour/dose
post-exposure group (unless otherwise given)
observation period
Rat; 0, 3.33, 16.6, 50 1) > 16.6 mg/kg: survival ¡# (m); 3.33 & 50: bw ¡# (m); Maltoni
Sprague-Dawley; mg/kg bw./d; 2) 40 f and 40 m per group; et al.
40 rats/sex; pure gavage; 52 weeks; 3) ASL in m: 0, 0, 4, 8* and in f: 0, 0, 6*, 9*; (1981,
virgin olive oil once daily, Zymbal gland carcinoma in m: 0, 0, 1, 1 and in f: 1, 0, 1984)
4-5 d/week; up to 1, 0; extrahepatic angiosarcoma in m: 0 in all groups and [BT 11] b
84 weeks c in f: 0, 2, 0, 2; nephroblastoma in m: 0, 0, 2, 1 and in
f: 0, 0, 1, 1;
Rat; 0, 0.03, 0.3, 1.0 1) > 0.03 mg/kg: survival and bw <->#; Maltoni
Sprague-Dawley; 75 mg/kg bw./d; 2) 75, 75, 73, 75 m and 75, 75, 73, 75 f; et al.
rats/sex; pure gavage; 59 weeks; 3) ASL in m: 0, 0, 0, 1 and in f: 0, 0, 1, 2; (1981,
virgin olive oil once daily, Zymbal gland carcinoma in m: 0, 0, 0, 2 and in f: 1, 0, 1984)
4-5 d/week; up to 0, 3; extrahepatic angiosarcoma in m: 0 in all groups and [BT 27] b
77 weeks c in f: 0, 0, 0, 1; nephroblastoma 0 in all groups
Rat; Wistar; 0, 3, 30, 300 mg/kg 1) 3 mg/kg: bw <-> , rat skin composition <->, mortality Knight &
10-20 rats/sex; bw./d; gavage; 1/16 (control n.g.) 30 mg/kg: bw <->, mortality 5/15; Gibbons
peroxide-free 95-125 weeks; biochemical parameters for skin fibrosis !* 300 mg/kg: (1987)
corn oil once daily; none mortality 15/15 within first 60 days of treatment;
2) 20, 16, 15, 10;
3) Liver tumors (predominantly angiosarcomas):
0, 1, 11, 10 #
a In all studies vehicle-treated controls; analysis of VC concentration in vehicle in all studies except Knight & Gibbons (1987);
bw. = body weight; d = day; f = females; m = males; mo = months; n.g. = not given; * = effect significant at P < 0.05;
# = no statistical evaluation; <-> = unchanged; ¡ = decreased; ! = increased
b Study number in experiments done by Maltoni and coworkers
c Animals were kept until spontaneous death or sacrificed at the end of given post exposure observation period
d Bioavailability studied in an ancillary study
e Study comparable to OECD guidelines 451 with acceptable restrictions (OECD, 1981a,b)
f Study comparable to OECD guidelines 453 (OECD, 1981a,b)
A statistically significant increase in the incidence of ASL was
seen in Sprague-Dawley rats at a dose level of 16.6 mg VC/kg body
weight per day (gavage; study BT 11), and some similar tumours were
also observed at dose levels of 0.3 and 1.0 mg/kg body weight per day
(Maltoni et al., 1981). Hepatic angiosarcomas were also observed in
gavage studies in Wistar rats (Knight & Gibbons, 1987). This tumour
type is very rare in untreated rats (4 in several thousand rats of the
colony used by Maltoni et al., 1981). Because of short dosage and
follow-up in the first study, and small number of animals due to early
mortality in the third, these studies probably did not reflect the
total carcinogenic potential of VC.
The results of the two feeding studies carried out by another
working group (Feron et al., 1981; Til et al., 1983, 1991) confirmed
the findings presented by Maltoni et al. (1981) concerning ASL
(induction at 1.3 mg/kg body weight per day, significant at 5.0 mg
VC/kg body weight per day). Furthermore these feeding studies
presented evidence for significantly increased tumour incidences of
neoplastic liver nodules (females) and of hepatocellular carcinoma
(HCC) (males) at 1.3 mg/kg body weight per day (Til et al., 1991).
7.7.2 Inhalation exposure
After the first reports of the carcinogenicity of VC in rats
(Viola, 1970; Viola et al., 1971), Maltoni and coworkers intensively
investigated the effects of inhalation exposure to VC in different
laboratory animals.a Study results were published in Maltoni et al.
(1974), Maltoni & Lefemine (1975) and Maltoni et al. (1979), and the
final report in Maltoni et al. (1981, 1984). Detailed information on
these inhalation studies as well as pertinent studies from other
working groups is tabulated in Table 30 (short-term studies) and Table
32 (long-term studies). A summary of tumour types induced by long-term
inhalation exposure to vinyl chloride in different species is given in
Table 33. Further studies on inhalative carcinogenicity of VC not
discussed in this section, but leading to similar results to the
studies tabulated below, were performed on rats (short-term: Hong et
al., 1981; long-term: Viola et al., 1971; Maltoni et al., 1974; Lee et
al., 1977; Maltoni et al., 1981, 1983, 1984; Bi et al., 1985; Maltoni
& Cotti, 1988; Froment et al., 1994) and mice (short-term: Suzuki,
1978, 1981; Schaffner, 1979; Hehir et al., 1981; Himeno et al., 1983;
Adkins et al., 1986; long-term: Keplinger et al., 1975; Lee et al.,
1977; Holmberg et al., 1979; Drew et al., 1983).
7.7.2.1 Short-term exposure
Several carcinogenicity studies have been performed where animals
were exposed by inhalation for rather short periods (up to 6 months)
and kept for different post-exposure periods up to lifetime
(Table 30). The tumour spectrum in rats and mice is very similar to
a Studies performed at Beautivoglio (BT) Laboratories; studies
numerated BT1-BT27
Table 30. Short-term inhalation studies on carcinogenicity of vinyl chloride in experimental animalsa
Species; strain; Doses in mg/m3; 1) Number of animals for histopathological References
age at start of exposure period; evaluation;
experiment; initial frequency of 2) type of tumour: number of animals with
number of animals per treatment; this tumour/dose group (unless otherwise
dose per exposure post-exposure given); other observations
period; type of observation period
control;
Rat; 0, 15 600, 26 000; 1) 227, 120, 118; Maltoni
Sprague-Dawley; 5 weeks; 4 h/d, 2) (#) ASL: 0, 0, 1; extrahepatic et al.
11 weeks; 120 5 d/week; angiosarcoma: 0, 0, 0; Zymbal gland (1981)
(control 240) 149 weeksc carcinoma: 0, 9, 9; hepatoma: 0, 0, 1; [BT 10]b
f & m; untreated nephroblastoma: 0, 1, 0;
control neuroblastoma: 0, 1, 0
Rat; 0, 15 600, 26 000; 1) 227, 118, 119; Maltoni
Sprague-Dawley; 25 weeks; 1 h/d, 2) (#) ASL: 0, 3, 1; extrahepatic angiosarcoma: et al.
11 weeks; 120 4 d/week; 0, 2, 0; Zymbal gland carcinoma: 0, 5, 9 (1981)
(control 240) 129 weeksc [BT 10]b
f & m; untreated
control
Rat; 15 600, 26 000; 1) 18 m and 24 f at low dose, 24 m and Maltoni
Sprague-Dawley; 5 weeks; 4 h/d, 20 f at high dose; et al.
1-day-old; 5 d/week; 119c 2) ASL in m: 5, 6 and in f:12, 9; (1981)
see number of extrahepatic angiosarcoma in m: 5, 6 [BT 14]b
animals for and inf: 12, 9; hepatoma in m: 9, 13
evaluation; no control; and in f: 11, 7
Rat; 0 or 2465; 1) 84-128 rats per sex per age group; Groth
Sprague-Dawley; 24.5 weeks; 7 h/d, 2) angiosarcomas (mostly in liver) in the 4 et al.
6, 18, 32, or 52 weeks; 5 d/week; scheduled age groups in exposed m: 1.2, 2.2, 7.4, (1981)
110-128 rats/sex sacrifice at 3, 6 and 18% and in exposed f: 2.3, 7.2, 28, 13%;
per age group; 9 months, only 1 subcutaneous angiosarcoma (m) in
treated control termination at all control groups (739 mice) #; older
week 43 adults more susceptible to
angiosarcoma-inducing effect
Table 30. (cont'd)
Species; strain; Doses in mg/m3; 1) Number of animals for histopathological References
age at start of exposure period; evaluation;
experiment; initial frequency of 2) type of tumour: number of animals with
number of animals per treatment; this tumour/dose group (unless otherwise
dose per exposure post-exposure given); other observations
period; type of observation period
control;
Rat; Wistar or 0, 6.5, 13, 26, 52, 1) 2-27 rats of each sex per dose per strain; Laib
Sprague-Dawley; 104, 208; 3 weeks; 2) dose dependent increase in ATPase-deficient et al.
3 days; 2-27 rats 8 h/d, 5 d/week; liver foci (preneoplastic lesion), (1985a)
per sex per strain; 10 weeks presumably significant at > 52 mg/m3
treated control ( m Sprague-Dawley rats at > 104 mg/m3);
f more susceptable than m in both strains
Rat; Wistar; 0 or 5200; 1) 2-10 rats of each sex per dose per exposure Laib
age-dependency 5-83 days, see 2); period; et al.
studied early in life, 8 h/d, 7 d/week; rats 2) age dependent increase in ATPase-deficient (1985b)
see 2); 4-10 rats sacrificed at the age liver foci lesion) (preneoplastic studied,
per sex; of 4 months rats exposed a) during gestation or at
treated control postnatal day 1-5 (b), 1-11 (c), 1-17 (d),
1-47 (e), 1-83 (f), 7-28 (g), or 21-49 (h);
no effect in a & b, foci area steeply
increased in c and d but not further
enhanced in e and f, foci area not lowered
in g but only a few foci in h
Mouse; CD1; 0, 2.6, 26, 260, 780, 1) see 2); Suzuki
5-6 weeks; 1560; 4 weeks; 2) pulmonary tumour incidences (#):week 0 (1983)
30-60 m; treated 6 h/d, 5 d/week; post-exposure: no tumour week 12: 0/18,
control 0, 12, 40 weeks 0/10, 0/9, 0/6, 6/9, 8/9 week 40: 0/17, 1/9,
3/9, 6/9, 5/7, 6/7; study focused on lung
tumours (no details on other tumours);
tumours derived from type II alveolar cells
Table 30. (cont'd)
Species; strain; Doses in mg/m3; 1) Number of animals for histopathological References
age at start of exposure period; evaluation;
experiment; initial frequency of 2) type of tumour: number of animals with
number of animals per treatment; this tumour/dose group (unless otherwise
dose per exposure post-exposure given); other observations
period; type of observation period
control;
Mouse; CD-1; 0, 130, 650, 2600; 1) 60, 40, 44, 38 m and 60, 40, 40, 38 f Hong et al.
2 months; 1, 3, 6 months; for cumulative effects; (1981)
8-28 mice/sex; 6 h/d, 5 d/week; 2) cumulative (f+m); ASL: 1, 2, 13*, 18*;
treated control 12 months extrahepatic angiosarcoma 0, 2, 6*, 2;
bronchioloalveolar tumor 16, 18, 52*, 50*;
cumulative (f): metastatic lung
adenocarcinoma 0, 4*, 2, 6*; mammary
carcinoma 4, 10*, 13*, 6; tumours at
different organ sites in f and m
increased in proportion to dose or duration
of exposure at higher dose levels
a d = day; f = females; m = males; n.g. = not given; # = no statistical evaluation; * = significant at P < 0.05
b Study number in experiments done by Maltoni and coworkers
c Animals were kept until spontaneous death or sacrificed at the end of given post-exposure observation period
that observed in long-term inhalation studies (compare with Table 32).
Hehir et al. (1981) presented evidence that even one single high dose
(> 13 000 mg/m3) resulted in a dose-related increase of pulmonary
tumours in mice. This effect was also demonstrated by repeated
administration to mice at lower dose levels with exposure periods
varying between 1 and 6 months (Hong et al., 1981; Suzuki, 1983).
Incidences of ASL, extrahepatic angiosarcoma and mammary gland
carcinoma were elevated in mice with increasing exposure periods at
doses of 130 or 650 mg/m3 (Hong et al., 1981). Also in rats,
angiosarcomas, predominantly in the liver, were induced by VC
inhalation for 25 weeks (15 600 mg/m3, 1 h/day, Maltoni et al., 1981;
2465 mg/m3, 7 h/day, Groth et al., 1981). Short-term exposure led to
increased incidences of carcinoma of the Zymbal gland, a sebaceous
gland of the ear canal in rats, at high doses (> 15 600 mg/m3;
Maltoni et al., 1981, BT10).
Short-term exposure studies on the effect of age on
susceptibility to tumour induction are discussed in section 7.7.3.
7.7.2.2 Long-term exposure
a) Rats
Although differences in experimental design, notably in the
duration of follow-up, and the low overall frequency of tumours tend
to confuse the picture, a dose-response relationship could be observed
in studies BT1, BT2, BT9 and BT15 on Sprague-Dawley rats for ASL
(Table 31, Fig. 4 and chapter 10) and Zymbal gland carcinomas, while
it was less clear for nephroblastomas, neuroblastomas and mammary
malignant tumours.
Lee et al. (1978) and Drew et al. (1983) reported similar
findings on the incidence of ASL. Female rats seem to be more
susceptible to ASL tumours than males (Lee et al., 1978; Maltoni et
al., 1981). ASL was detected at 26 mg/m3 and the incidence was
statistically significant at 130 mg/m3, which was at a lower level
than other tumour types with the exception of mammary gland tumours
(13 mg/m3; see Table 33) (Maltoni et al., 1981). VC exposure also
caused angiosarcomas at extrahepatic sites (Maltoni et al., 1981, BT14
see Table 30; Lee et al., 1978; Drew et al., 1983).
Increased incidences of other tumour types than those reported by
Maltoni et al. (1981) in Sprague-Dawley rats were shown by Drew et al.
(1983) in Fischer-344 rats (neoplastic liver nodules and
hepatocellular carcinoma) and by Feron & Kroes (1979) in Wistar rats
(tumour of the nasal cavity). These were single dose experiments.
Drew et al. (1983; see Table 32) studied the effect of exposure
duration (6, 12, 18 or 24 months) on tumour incidences and
demonstrated that longer exposure periods, and thus greater cumulative
exposures, led to an increase in tumorigenic responses concerning
liver angiosarcoma, angiosarcoma at all sites, and hepatocellular
Table 31. Incidence of angiosarcomas of the liver (ASL) and mammary
tumours observed in Sprague-Dawley ratsa
Exposure Concentration ASL ASL ASL Mammary
(ppm) (mg/m3) (males) (females) (males + adenocarcinomas
females)
0 0 0/173 0/239 0/412 12/239
1 2.6 0/58 0/60 0/118 12/60
5 13 0/59 0/60 0/119 22/60
10 26 0/59 1/60 1/119 11/60
25 65 1/60 4/60 5/120 15/60
50 130 2/174 13/180 15/354 61/180
100 260 0/60 1/60 1/120 3/60
150 390 1/60 5/60 6/120 6/60
200 520 7/60 5/60 12/120 5/60
250 650 1/29 2/30 3/59 2/30
500 1300 0/30 6/30 6/60 1/30
2500 6500 6/30 7/30 13/60 2/30
6000 15 600 3/29 10/30 13/59 0/30
10 000 3/30
a The data are combined from the experiments BT1, BT2, BT9 and BT15 conducted by Maltoni et al. (1984).
Control animals from several experiments are combined in the 0 ppm group. Animals were exposed
4 h/day, 5 days/week for 52 weeks; the follow-up was until the death of the animals or until week
83 (BT1), 90 (BT2), 90 (BT9) or 95 (BT15). Tumours were scored at the time of death. Adapted from
Reitz et al. (1996)
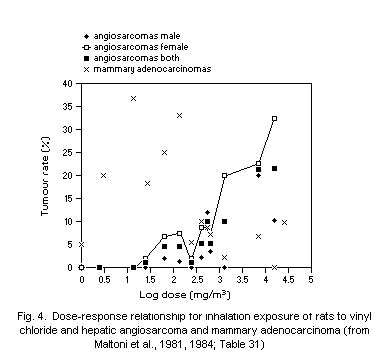 carcinoma in female F-344 rats. This exposure duration-related effect
was not observed in mice and hamsters, except angiosarcoma at all
sites in hamsters.
The lowest reported dose (13 mg/m3) to cause a statistically
significant increase in tumour incidences was demonstrated for mammary
adenocarcinoma in female rats (Maltoni et al. 1981; BT15). Although
this tumour type is common in untreated rats and no clear
dose-response relationship was shown (probably due to reduced survival
in the high-dose groups, Maltoni et al., 1984; BT1, BT2, BT9, BT15),
the increase in tumour rate was considered by the Task Group to be of
toxicological relevance, since the incidence was increased compared to
concurrent and historical controls of the same colony (Maltoni et al.,
1981, 1984) and similar results on mammary gland tumours were
presented in further studies on rats (Drew et al., 1983) and other
species (Lee et al., 1978; Maltoni et al., 1981, 1984; Drew et al.,
1983) (see also Table 33; Fig. 4).
b) Mice
In mice, the spectrum of tumours induced by long-term inhalation
exposure is similar to that observed in rats, but an increase in lung
tumours was only observed in mice (Lee et al., 1978; Maltoni et al.,
1981; Drew et al., 1983, see also Table 32). At a dose level of
130 mg/m3, incidences in angiosarcoma (liver, extrahepatic or all
sites combined), lung carcinoma, and mammary gland carcinoma showed a
statistically significant increase (Lee et al., 1978; Drew et al.,
1983). Doses lower than 130 mg/m3 were not investigated, but at the
dose levels investigated, the angiosarcoma frequencies were higher in
mice than in rats (Lee et al., 1978). No clear-cut relationship with
time of exposure (6-18 months) and incidence of ASL was observed; this
could, however, have been due to decreased survival and shorter
follow-up after longer exposure time (Drew et al., 1983).
c) Other species
Limited data are available on other species. In experiment BT8
with male Syrian golden hamsters, low frequencies of ASL, acoustic
duct tumours and melanomas were observed. In addition, an increased
incidence in forestomach and skin epithelial tumours was reported, but
these tumours were also observed in controls, there was no
dose-response relationship and a statistical evaluation was not
presented (Maltoni et al., 1981).
Female hamsters were exposed to a single dose for different
exposure periods (Drew et al., 1983). Angiosarcomas (all sites), skin
tumours and increased incidences in mammary gland carcinoma and
stomach adenomas (glandular portion of the stomach) were reported
(Drew et al., 1983).
In a study reported only as an abstract, Caputo et al. (1974)
reported increased incidences in skin acanthoma and lung
adenocarcinoma in rabbits exposed to VC.
Table 32. Inhalation studies on the long-term toxicity/carcinogenicity of vinyl chloride in experimental animalsa
Species; strain; age Doses in mg/m3; 1) Effects other than tumours Reference
at start of exposure period; (dose in mg/m3);
experiment; initial frequency of treatment; 2) number of animals for
number of animals per post-exposure histopathological evaluation of
dose per exposure observation period neoplastic effects;
period; type of 3) type of tumour: number of animals
control; with this tumour/dose group (unless
otherwise given)
Rat; 0, 130, 650, 1300, 1) > 650: survival ¡# (f, in m at > 1300); Maltoni et
Sprague-Dawley; 13 6500, 15 600, > 15 600: body weight ¡# (m, in f at 26 000); al. (1981,
weeks; 30 rats/sex; 26 000; 52 weeks; 2) 29-30 m and 29-30 f per group; 1984)
untreated control 4 h/d, 5 d/week; 3) ASL in m: 0, 0, 1, 0, 6*, 3, 3 and in [BT1]b
83 weeks c f : 0, 1, 2, 6*, 7*, 10*, 4; Zymbal gland
carcinoma in m: 0, 0, 0, 3, 1, 3, 10* and in f:
0, 0, 0, 1, 1, 4, 6*; hepatoma in f:
0, 0, 0, 5, 2, 1, 0;
nephroblastoma in m: 0, 0, 1, 2, 5*, 4, 3
and in f: 0, 1, 4*, 4*, 1, 1, 2;
neuroblastoma in m: 0, 0, 0, 0, 2, 2, 2 and in f:
0, 0, 0, 0, 2, 1, 5*;
mammary adenocarcinoma in f: 0, 2, 2, 1, 2, 0, 3
Rat; 0, 260, 390, 520; 1) > 130: survival ¡# (m, in f at 520); Maltoni et
Sprague-Dawley; 52 weeks; 4 h/d, 2) 59-60 rats per treatment group, control al. (1981,
13 weeks; 60 5 d/week; 91 weeks c 65 m and 100 f; 1984)
rats/sex, control 3) ASL in m: 0, 0, 1, 7* and in f: 0, 1, 5*, 5*; [BT2]b
85 m and 100 f; mammary adenocarcinoma in f: 1, 3, 6*, 5;
untreated control nephroblastoma in m: 0, 8*, 8*, 5* and in f:
0, 2, 3, 2
Table 32. (cont'd)
Species; strain; age Doses in mg/m3; 1) Effects other than tumours Reference
at start of exposure period; (dose in mg/m3);
experiment; initial frequency of treatment; 2) number of animals for
number of animals per post-exposure histopathological evaluation of
dose per exposure observation period neoplastic effects;
period; type of 3) type of tumour: number of animals
control; with this tumour/dose group (unless
otherwise given)
Rat; Sprague-Dawley; 0, 130; 52 weeks; 1) survival ¡# (f); Maltoni et
13 weeks; 150 rats 4 h/d, 5 d/week; 2) 144 m and 150 f, control 48 m and 50 f; al. (1981,
per sex, control 50 m 90 weeks c 3) ASL in m: 0, 2 and in f: 0, 12*; 1984)
and 50 f; mammary adenocarcinoma in f: 5, 59*; [BT9]b
untreated control no significant effects on incidences of other
tumour types
Rat; 0, 2.6, 13, 26, 65; 1) > 2.6 survival ¡# (m+f); Maltoni et
Sprague-Dawley; 52 weeks; 4 h/d, 2) 58-60 m and 60 f per group; al. (1981,
13 weeks; 60 rats/sex; 5 d/week; 3) ASL in m: 0, 0, 0, 0, 1 and in f: 0, 0, 0, 1, 1984)
untreated control 95 weeks c 4; mammary adenocarcinoma in f: 6, 12, 22*, [BT15]b
21*, 15*; nephroblastoma in m: 0, 0, 0, 0, 1;
hepatoma in f: 0, 0, 0, 0, 1
Rat; Spague-Dawley; 0, 6500; life time 1) survival & body weight ¡# Maltoni &
13 weeks (pregnant (up to 69 weeks); 2) 60, 54 dams, progeny 158, 63 m and 149, 64 f Cotti (1988)
rats) or gestation day 4 h/d, 5 d/week 3) tumour incidences in % (#):
12 (embryos); see for 7 weeks, than ASLd : 0, 50.0 in dams and 0, 56.2 in m and
number of animals for 7 h/d, 5 d/week; 0, 73.0 in f progeny; hepatocellular carcinoma:
for evaluation; none 0, 9.2 in dams, 0.6, 42.2 in m and 0, 60.3 in f
untreated control progeny mammary carcinoma: 7.4, 7.4 in dams, 5.4,
4.7 in f progeny; neuroblastomad: 0, 59.2 in dams,
0, 48.4 in m and 0, 42.8 in f progeny
Table 32. (cont'd)
Species; strain; age Doses in mg/m3; 1) Effects other than tumours Reference
at start of exposure period; (dose in mg/m3);
experiment; initial frequency of treatment; 2) number of animals for
number of animals per post-exposure histopathological evaluation of
dose per exposure observation period neoplastic effects;
period; type of 3) type of tumour: number of animals
control; with this tumour/dose group (unless
otherwise given)
Rat; Fischer-344; 0 or 260; 6, 12, 1) survival ¡* (exposure periods > 6 mo); Drew et al.
8-9 weeks, 2nd 18, 24 mo, 2nd 2) 112 (control), 76, 55, 55, 55 per exposure (1983)
experiment: 2, 8, experiment 6 or period; 2nd experiment 6 mo exposure: 112
14, 20 mo (6 mo 12 mo exposure (control), 76, 52, 51, 53 per age group; 2nd
exposure) or 2, 8, at different age; experiment 12 mo exposure: 112 (control), 55,
14 mo (12 mo 6 h/d, 5 d/week; 54, 49 per age group;
exposure); n.g. (only life span 3) ASL: 1, 4*, 11*,13*, 19*; angiosarcoma all
f, see number of sites: 2, 4, 12*, 15*, 24*; mammary fibroadenoma:
rats for evaluation); 24, 28*, 28*, 24*, 26*; mammary adenocarcinoma:
n.g. 5, 6, 11*, 9*, 5*; neoplastic liver nodules: 4,
15*, 20*, 7*, 6*; hepatocellular carcinoma: 1, 3,
4*, 8*, 9*; 2nd experiment 6 mo exposure: ASL: 1,
4*, 2, 0, 0; angiosarcoma all sites: 2, 4, 2, 0,
0; mammary fibroadenoma: 24, 28*, 23*, 17, 20;
mammary adenocarcinoma: 5, 6, 2, 3, 2; neoplastic
liver nodules: 4, 15*, 10*, 2, 4; hepatocellular
carcinoma: 1, 3, 6*, 0, 1;
2nd experiment 12 mo exposure: ASL: 1, 11*, 5*,
2; angiosarcoma all sites: 2, 12*, 5*, 2; mammary
fibroadenoma: 24, 28*, 16*, 15; mammary
adenocarcinoma: 5, 11*, 4, 0; neoplastic liver
nodules: 4, 20*, 4, 4; hepatocellular carcinoma:
1, 4*, 1, 0;
Table 32. (cont'd)
Species; strain; age Doses in mg/m3; 1) Effects other than tumours Reference
at start of exposure period; (dose in mg/m3);
experiment; initial frequency of treatment; 2) number of animals for
number of animals per post-exposure histopathological evaluation of
dose per exposure observation period neoplastic effects;
period; type of 3) type of tumour: number of animals
control; with this tumour/dose group (unless
otherwise given)
Rat; Wistar; 11 0, 130, 650, 1300, 1) > 130: survival & body weight ¡# Maltoni et
weeks; control 40 m, 6500, 15 600, 2) 38, 28, 28, 27, 25, 26, 27; al. (1981,
other groups 30 m; 26 000; 52 weeks; 3) (#):ASL: 0, 0, 1, 3, 3, 3, 8; hepatoma: 0, 0, 1984)
untreated control 4 h/d, 5 d/week; 0, 0, 1, 2, 0; nephroblastoma: 0, 1, 0, 2, 0, [BT17]b
113 weeks c 2, 1; neuroblastoma: 0, 0, 0, 0, 1, 1, 3;
Zymbal gland carcinoma: 0, 0, 0, 0, 0, 2, 2
Rat; Wistar; newly 0 or 13 000; 1) mortality !# (9 m and 10 f still alive at week Feron et al.
weaned; 62 rats/sex; 52 weeks, 10 rats 52); body weights ¡* (f+m); blood clotting time (1979a,b)
treated control per dose per sex ¡#; liver function ¡# (BSB-retention test); Feron &
sacrificed at week relative liver, kidney and spleen weight !*; Kroes
4, 13, 26 (see degree of tubular nephrosis !# (f+m); focal (1979)
section 7.2); 7 h/d, degeneration of myocardium and thickened walls
5 d/week; none of arteries # (f+m); haematopoietic activity
in the spleen !# (f+m); distended liver
sinusoids # (f+m);
2) 62 m and 62 f per group;
3) cumulative tumours (#):ASL: 0, 3 in m and 0, 6
in f ; Zymbal gland tumour: 0, 7 in m and 0, 4
in f; tumour of nasal cavity: 0, 10 in m and
0, 10 in f;
Table 32. (cont'd)
Species; strain; age Doses in mg/m3; 1) Effects other than tumours Reference
at start of exposure period; (dose in mg/m3);
experiment; initial frequency of treatment; 2) number of animals for
number of animals per post-exposure histopathological evaluation of
dose per exposure observation period neoplastic effects;
period; type of 3) type of tumour: number of animals
control; with this tumour/dose group (unless
otherwise given)
Rat; CD; 2 mo; 0, 130, 650, 2600; 1) > 650: survival ¡# (m+f); Lee et al.
36 rats/sex; 12 mo, 4 rats/dose 2) 35, 36, 36, 34 m and 35, 36, 34, 36 f; (1978)
treated control per sex terminated 3) tumours combined for all exposure periods:
at month 1, 2, 3, 6, ASL: 0, 0, 2, 6 in m and 0, 0, 10*, 15* in f;
9; 6 h/d, 5 d/week; lung angiosarcoma: 0, 0, 0, 4 in m and 0, 0,
none 3, 9* in f; other tumours not related to VC
treatment
Rat [no further data 0, 14, 25, 266, 1) no data Kurlyandski
provided] 3690; 52 weeks; 2) 70, 50, 39, 43, 51 m et al.
4.5 h/d, 5 d/week 3) ASL: 0, 0, 7.7, 9.3, 11.8%; (1981)
other angiosarcomas: 0, 1.0, 2.5, 0, 3.9%
other liver tumours: 2.8, 2.0, 2.6, 11.6, 13.7%
haemoblastoma: 4.3, 14.0, 15.4, 34.9, 2.0%
other tumours: 21.5, 28.1, 15.4, 9.3, 23.5%
Mouse; Swiss; 0, 130, 650, 1300, 1) > 130: survival ¡# (m+f); > 1300: body weight ¡# Maltoni et
11 weeks; 30 mice 6500, 15 600, (m, in f at > 650); al. (1981,
per sex, control 26 000; 30 weeks; 2) 80, 30, 30, 30, 29, 30, 26 m and 70, 30, 30, 1984)
80 m and 70 f; 4 h/d, 5 d/week; 30, 30, 30, 30 f; [BT4]b
untreated control 51 weeks c 3) combined (f+m) tumours (#): ASL: 0, 1, 18, 14,
16, 13, 10; liver angioma: 0,1, 11, 5, 5, 7, 6;
extrahepatic angiosarcoma: 1, 1, 3, 7, 8, 1, 1;
lung tumour: 15, 6, 41, 50, 40, 47, 46;
mammary carcinoma in f : 1, 13, 12, 9, 10, 9, 14
Table 32. (cont'd)
Species; strain; age Doses in mg/m3; 1) Effects other than tumours Reference
at start of exposure period; (dose in mg/m3);
experiment; initial frequency of treatment; 2) number of animals for
number of animals per post-exposure histopathological evaluation of
dose per exposure observation period neoplastic effects;
period; type of 3) type of tumour: number of animals
control; with this tumour/dose group (unless
otherwise given)
Mouse; Swiss CD-1; 0 or 130; 6, 12, 1) survival ¡* (all exposure periods); Drew et
8-9 weeks, 2nd 18 mo, 2nd 2) 71 (control), 67, 47, 45 per exposure period; al. (1983)
experiment: 2, 8, experiment 6 or 12 mo 2nd experiment 6 mo exposure: 71 (control), 67,
14 mo (6 or 12 mo exposure at 49, 53 per age group; 2nd experiment 12 mo
exposure); n.g. (only different age; exposure: 71 (control), 47, 46, 50 per age group;
f, see number of 6 h/d, 5 d/week; 3) angiosarcoma (all sites): 1, 29*, 30*, 20*;
mice for evaluation); lifespan mammary gland carcinoma: 2, 33*, 22*, 22*; lung
n.g. carcinoma: 9, 18*, 15*, 11*; 2nd experiment 6 mo
exposure: angiosarcoma (all sites): 1, 29*, 11*,
5; mammary gland carcinoma: 2, 33*, 13*, 2; lung
carcinoma: 9, 18*, 13*, 7;
2nd experiment 12 mo exposure: angiosarcoma
(all sites): 1, 30*, 17*, 3;
mammary gland carcinoma: 2, 22*, 8*, 0; lung
carcinoma: 9, 15*, 9*, 3;
Mouse; CD-1; 2 mo; 0, 130, 650, 2600; 1) > 650: survival ¡# (tumour development), all Lee et al.
total 36 mice/sex; 1, 2, 3, 6, 9, and mice in high-dose group and females in mid-dose (1978)
treated control 12 mo (4 mice per group died or were terminated at mo 10-12;
group per sex per 2) 26, 29, 29, 33 m and 36, 34, 34, 36 f;
exposure period); 3) tumours combined for all exposure periods:
6 h/d, 5d/week; bronchioloalveolar adenoma (#):1, 8, 10, 22 in m
none and 0, 4, 12, 26 in f; ASL: 0, 3, 7*, 13* in m
and 0, 0, 16*, 13* in f; extrahepatic angiosarcoma:
0, 5*, 2, 0 in m and 0, 1, 3, 9* in f; mammary
tumours(#): 0, 9, 3, 13 in f; incidences exposure
time-dependent
Table 32. (cont'd)
Species; strain; age Doses in mg/m3; 1) Effects other than tumours Reference
at start of exposure period; (dose in mg/m3);
experiment; initial frequency of treatment; 2) number of animals for
number of animals per post-exposure histopathological evaluation of
dose per exposure observation period neoplastic effects;
period; type of 3) type of tumour: number of animals
control; with this tumour/dose group (unless
otherwise given)
Hamster; Syrian 0, 130, 650, 1300, 1) > 130: survival ¡#; Maltoni et
golden; 11 weeks; 6500, 15 600, 2) see initial number of hamsters; al. (1981,
30 m, 60 m in 26 000; 30 weeks; 3) (#): ASL: 0, 0, 0, 2, 0, 1, 0; 1984)
control; 4 h/d, 5 d/week; acoustic duct tumour: 0, 0, 0, 3, 1, 2, 1; [BT8]b
untreated control 79 weeks c melanoma: 0, 1, 1, 0, 0, 1, 2, 1;
forestomach tumour: 3, 3, 4, 9, 17, 10, 10;
skin epithelial tumour: 3, 9, 3, 7, 3, 1, 7;
Hamster; Syrian 0 or 520; 6, 12, 1) survival ¡* (all exposure periods); Drew et al.
golden; 8-9 weeks, 18 mo, 2nd 2) see tumour incidences; (1983)
2nd experiment: 2, 8, experiment 6 or 3) tumour incidences in control and at different
14, 20 mo (6 mo 12 mo exposure at exposure periods: angiosarcoma (all sites): 0/143
exposure) or 2, 8, different age; (control), 13/88*, 4/52*, 2/103; mammary
14 mo (12 mo 6 h/d, 5 d/week; carcinoma: 0/143, 28/87*, 31/52*, 47/102*;
exposure); n.g. life span stomach adenoma: 5/138, 23/88*, 3/50*, 20/101*;
(only f, see number skin carcinoma: 0/133, 2/80; 9/48*, 3/90; 2nd
of hamster for experiment 6 mo exposure: angiosarcoma (all
evaluation); n.g. sites): 0/143 (control), 13/88*, 3/53*, 0/50,
0/52; mammary carcinoma: 0/143, 28/87*, 2/52*,
0/50, 1/52; stomach adenoma: 5/138, 23/88*,
15/53*, 6/49*, 0/52; skin carcinoma: 0/133, 2/80;
0/49, 0/46, 0/50; 2nd experiment 12 mo exposure:
angiosarcoma (all sites): 0/143 (control),
4/52*, 1/44, 0/43;
mammary carcinoma: 0/143, 31/52*, 6/44*, 0/42;
stomach adenoma: 5/138, 3/50*, 10/44*, 3/41;
skin carcinoma: 0/133, 2/80; 0/38, 0/30;
Table 32. (cont'd)
Species; strain; age Doses in mg/m3; 1) Effects other than tumours Reference
at start of exposure period; (dose in mg/m3);
experiment; initial frequency of treatment; 2) number of animals for
number of animals per post-exposure histopathological evaluation of
dose per exposure observation period neoplastic effects;
period; type of 3) type of tumour: number of animals
control; with this tumour/dose group (unless
otherwise given)
Rabbit; n.g.; n.g.; 0, 26 000; 1) n.g.; Caputo
40 exposed to VC, 15 mo; 4 h/d, 2) see tumour incidences; et al.
20 controls (no data 5 d/week; n.g. 3) tumour incidences: skin acanthoma: 0/20 (1974)
about sex); (control), 12/40*; lung adenocarcinoma 0/20, 6/40
treated control
a * = significant at P < 0.05; #: no statistical evaluation; f = females; m = males; mo = months; n.g. = not given
b Study number in experiments done by Maltoni and coworkers
c Animals were kept until spontaneous death or sacrificed at the end of given post exposure observation period
d Presumably significant in all exposed groups
Table 33. Summary of tumour types induced by long-term inhalation exposure to vinyl chloride
Lowest reported dose that significantly increased tumour incidences by tumour type (dose in mg/m3)
Species Liver Angiosarcoma Other Lung Mammary Nephro- Skin Neuro- Stomach Zymbal
angiosarcoma (other sites) liver carcinoma gland blastoma tumour blastoma tumour gland
tumours carcinoma tumour
Rat 130 in f c extrahepatic neoplastic 13 in 260 in 6500 in fore- 26 000
520 in m c 260 in f b,d nodules f h m b,g f b,k stomach in f + m f
lung 260 in f 650 in papilloma
2600 in f e b,d f g 78 000 in
hepato- f + m i
cellular
carcinoma
260 in
f d
Mouse 650 in 130 in m b,e 130 in 130 in
f + m e f b,d f b,d
all sites
130 in f b,d
Rabbit acanthoma
26 000,
n.d.
about sex
Hamster all sites 520 in b,j adenoma
520 in f b,d carcinoma 520 in
f b,d 520 in f f b,d
b,d
a Exposure in all studies 4-7 h/day, 5 days/week, exposure period at least 6 months
f = females; m = males; n.d. = no data
b The lowest dose tested with the study design described in the cited study
c Maltoni et al. (1981; BT9) f Maltoni et al. (1981; BT1) i Maltoni et al. (1981; BT6)
d Drew et al. (1983) g Maltoni et al. (1981; BT1, BT2) j Caputo et al. (1974)
e Lee et al. (1978) h Maltoni et al. (1981; BT15) k Maltoni & Cotti (1988)
7.7.3 The effect of age on susceptibility to tumour induction
Recently there has been some concern about early-life sensitivity
to vinyl chloride (Hiatt et al., 1994; Cogliano et al., 1996).
However, there is contradictory evidence (Drew et al., 1983 versus
Groth et al., 1981) concerning the effects of age on ASL induction in
rats, but final conclusions on increased susceptibility in 6- to
8-weeks-old animals could not be drawn from these data. The
discrepancy in results with rats is probably due to differences in
strain and/or experimental design. However, there is evidence from
other studies that there is possibly a higher sensitivity to liver
tumour induction in different rat strains in the first weeks of life,
a life-phase much earlier than that studied by Drew et al. (1983).
Studies on DNA adduct formation support these results.
F-344 rats, hamsters and mice (Swiss and B6C3F1) of different
age at the beginning of the exposure period (2, 8, 14 or 20 (not mice)
months old) were exposed to VC using the same experimental design and
same exposure period (Drew et al., 1983; see Table 32 for details).
For ASL in rats, angiosarcoma at all sites and mammary carcinoma
in all three species, neoplastic nodules in rats and lung carcinoma in
Swiss mice, the tumour response was highest in young animals
(2-month-old) exposed for 6 or 12 months. The validity of the
demonstrated age-related effects is limited in this study because only
statistical evaluation in comparison to control or to all other
groups, but not related to each other exposure group, was performed.
In addition, exposure later in life automatically shortens the period
of follow-up, and thus tends to lead to an apparent elevated
sensitivity at young age.
The effect of age on the susceptibility to induction of ASL in
Sprague-Dawley rats was also studied by Groth et al. (1981; see Table
30 for details). Rats aged 6, 18, 32 or 52 weeks were exposed to VC at
the same dose level and exposure period. In contrast to Drew et al.
(1983), the results of this study demonstrated that the older the rats
were at the start of the exposure period, the greater was the tumour
incidence. The maximum incidence was observed in male rats 52 weeks
old at first exposure and in females 32 weeks old at first exposure
(significantly increased compared to 6- or 18-week-old females). For
males the effect of age was statistically significant.
Exposure of 1-day-old rats of the same strain to high VC
concentrations for 5 weeks (BT14, see Table 30) revealed a remarkably
higher incidence of ASL, extrahepatic angiosarcoma and hepatoma
compared with 11-week-old rats exposed within the same experimental
design (BT10; Maltoni et al., 1981; see Table 30). However, these
experiments were not concurrent, and the tumour response in different
series from this laboratory has been variable (Tables 31, 32).
Maltoni & Cotti (1988) exposed 13-weeks-old pregnant
Sprague-Dawley rats and their progeny from the 12th day of gestation
to VC (Table 32). Although no statistical evaluation was performed it
seems that there was no significant difference between the dams and
the progeny concerning incidences in ASL, mammary carcinoma, and
neuroblastoma. The incidence of hepatocellular carcinoma, however, was
9% in dams and 42% in male and 60% in female progeny. In this study
the duration of exposure (and also of latency) was up to 69 weeks for
the progeny, but 56 weeks for the dams.
Laib et al. (1985a; see Table 30) presented evidence for
dose-related increased induction of ATPase-deficient liver foci in
Wistar and Sprague-Dawley rats, discussed as preneoplastic
hepatocellular lesions, after a 3-week exposure to low concentrations
(> 52 mg/m3). The increased induction of these foci was restricted
to a well-defined period of highest sensitivity beginning with rapid
liver growth in 7- to 21-day-old rats. No induction of these foci was
reported in adult rats exposed to 5200 mg/m3 for 70 days after
partial hepatectomy (no further details) (Laib et al., 1985b).
Comparative investigations on the alkylation of liver DNA in
young and adult Wistar rats exposed under the same exposure conditions
confirmed the age-related sensitivity of rats to VC (section 6.5.1 and
Table 26.)
7.7.4 The effect of gender on susceptibility to tumour induction
There is evidence that female rats of various strains are more
susceptible to liver tumour induction than males. Maltoni et al.
(1981, 1984) reported in all studies on Sprague-Dawley rats (BT1, BT2,
BT9, BT15; see Table 32) a higher incidence of ASL in female rats
compared with males after long-term inhalation. Similar results were
presented by Feron et al. (1979a,b) on Wistar rats, Lee et al. (1978)
on CD rats (Table 32) and Groth et al. (1981, Table 30) on
Sprague-Dawley rats. In inhalation experiments with pregnant
Sprague-Dawley rats, the investigators observed a higher incidence of
ASL and hepatocellular carcinoma in the female progeny than in the
males (Maltoni & Cotti, 1988; Table 32).
After long-term oral administration of VC (Table 29), incidences
of ASL (BT11, Sprague-Dawley rats; Maltoni et al., 1981), neoplastic
liver nodules and hepatocellular carcinoma (Wistar rats; Feron et al.,
1981 (not ASL) and Til et al., 1991) were higher in females than in
males. Interestingly, preneoplastic alterations in the liver, like
increased basophilic foci (Til et al., 1991; Table 29) or
ATPase-deficient foci (Laib et al., 1985a; Table 30), were observed in
female rats at lower doses than in males.
Although statistical analysis of sex differences was not
performed, in rats there is a tendency towards a higher susceptibility
to VC-induced ASL in female animals. Data on species other than rats
are not sufficient for an assessment of sex differences (Table 30,
32).
7.7.5 Carcinogenicity of metabolites
CAA was reported to induce hepatocellular tumours in B6C3F1 mice
when administered orally in drinking-water (Daniel et al., 1992). CEO
caused local tumours after repeated subcutaneous injection and skin
tumours in mice in classical initiation-promotion experiments (CEO
used as an initiator and 12-O- n-tetradecanoylphorbol-13-acetate as a
promoter), whereas CAA did not under comparable conditions (Zajdela et
al., 1980).
7.8 Genotoxicity
Genotoxicity studies on VC in vitro and in vivo in laboratory
animals are given in sections 7.8.1 and 7.8.2, respectively.
Genotoxicity studies on the metabolites of VC are described in section
7.8.3, and the mutagenic/promutagenic properties of DNA adducts formed
by the reactive VC metabolites CEO and CAA are discussed in section
7.8.4. Data on gene mutation and cytogenetic damage in humans exposed
to VC are given in section 8.4. A summary on the genotoxicity of VC
in vitro and in vivo, including human data, is presented in Table
36.
7.8.1 In vitro studies
Relevant studies on the genotoxicity of VC in vitro are
presented in Table 34. Genotoxic activity of VC has been detected in
several in vitro test systems, predominantly after metabolic
activation.
VC is mutagenic in the Ames test in the presence of metabolic
activation in Salmonella typhimurium strains TA100, TA1530 and
TA1535 but not in TA98, TA1537 and TA1538 (Rannug et al., 1974;
Bartsch et al., 1975; McCann et al., 1975; De Meester et al., 1980;
Shimada et al., 1985) indicating that the mutations are the result of
base-pair substitutions (transversion and transition) rather than
frameshift mutations. This is in agreement with the finding that
etheno-DNA adducts formed by the reactive metabolites CEO and CAA (see
section 6.5.1) are converted to actual mutations by base-pair
substitutions (see section 7.11.2 and 8.4). Barbin et al. (1997)
examined p53 mutations in VC-induced rat liver tumours and detected in
12 samples (11 ASL, 1 HCC) only one deletion but 12 base-pair
substitutions (transversion and transition).
In some studies, VC has been shown to also exert mutagenic
activity in S. typhimurium without addition of S9-mix (McCann et
al., 1975; De Meester et al., 1980; Shimada et al., 1985), but the
mutagenic effect was enhanced by addition of a metabolic activation
system (De Meester et al., 1980; Shimada et al., 1985; Victorin &
Ståhlberg, 1988). This increase was more pronounced when liver
extracts were derived from animals pretreated with an enzyme inducer
(Aroclor 1254) (De Meester et al., 1980). The reason for the mutagenic
Table 34. Genotoxicity of vinyl chloride in vitroa
Test type Test organism; Exposure conditions; Results Results Reference
species strain comments with MA without MA
Ames test Bact.; 20% VC in atmosphere; Rannug et al.
Salmonella up to 90 min + - (1974)
typhimurium - -
TA1535 - -
TA1536 - -
TA1537
TA1538
Ames test Bact.; 0.2, 2, 20% VC in atmosphere; n.g. Bartsch et al.
S. typhimurium 1.5-48 h; dose- and + (1975)
TA1530 time-dependent effect +
TA1535 -
TA1538 -
G-46
Ames test Bact.; 20% VC in atmosphere; McCann et al.
S. typhimurium 3, 6, 9 h; - - (1975)
TA98 time-dependent effect + +
TA100 + +
TA1535 - -
TA1538
Ames test Bact.; 1) 2-20% VC in De Meester et
S. typhimurium atmosphere; 16 h; + + al. (1980)
TA1530 dose-dependent effect
Ames test Bact.; 0.1-10% VC in atmosphere; Shimada et al.
S. typhimurium 18 h; - - (1985)
TA98 dose-dependent effect + +
TA100 + +
TA1535 - -
TA1537 - -
TA1538
Table 34. (cont'd)
Test type Test organism; Exposure conditions; Results Results Reference
species strain comments with MA without MA
Ames test Bact.; VC in DMSO added to soft Laumbach et
S. typhimurium agar and bacteria n.g. - al. (1977)
TA100
Ames test Bact.; 83 mM VC in liquid n.g. Bartsch et al.
S. typhimurium suspension; 30 min - (1975)
TA1530 -
TA1535 -
G-46
Gene Bact.; 10.6 mM VC in medium; + - Greim et al.
mutation Escherichia 2 h (1975)
assay coli
K12
Forward Yeast 16, 32, 48 mM VC in + - Loprieno et
mutation cells; medium; 1 h; al. (1977)
assay Schizo- dose-dependent effect
saccharomyces
pombe P1
Forward Yeast 16 or 48 mM VC in medium; + - Loprieno et
mutation cells; 5-240 min; time-dependent al. (1976)
assay S. pombe effect
SP.198
Table 34. (cont'd)
Test type Test organism; Exposure conditions; Results Results Reference
species strain comments with MA without MA
Reverse Yeast cells; incubated with 0.275 n.g. - Shahin (1976)
mutation Saccharomyces or 0.55% VC in DMSO;
assay cerevisiae 4-48 h
XV185-14C
Forward Fungi; VC solution in ethanol - - Drozdowicz &
mutation Neurospora for 3-4 h or 25, 50% Huang (1977)
assay crassa VC in atmosphere for
Ema 5297 3.5 or 24 h
Gene Plant; plant cutings exposed n.g. + Van't Hof &
mutation/ Tradescantia to VC in atmosphere; Schairer (1982)
deletion spec. 6 h; positive at
assay clone 4430 > 195 mg/m3
Cell mammalian 5, 10, 20, 30% VC in + - Drevon &
gene cells; atmosphere; 5 h; Kuroki (1979)
mutation Chinese dose-dependent effect
assay hamster
V79
HGPRT human cells; 25-400 µM VC in + n.g. Weisman (1992)
gene B-lymphoblastoid medium for 24 h;
mutation line cells with
assay metabolizing system
Gene yeast cells; 48 mM VC in incubation + - Loprieno et al.
conversion S. cerevisiae medium; 180-360 min (1976)
assay D4
Gene yeast cells; incubation in 0.275 n.g. - Shahin (1976)
conversion S. cerevisiae or 0.55% VC in DMSO;
assay D5 4-48 h
Table 34. (cont'd)
Test type Test organism; Exposure conditions; Results Results Reference
species strain comments with MA without MA
Gene yeast cells; 2.5% VC in atmosphere + - Eckardt
conversion S. cerevisiae for 1 h et al. (1981)
assay D7RAD
Cell mammalian 10, 20, 30, 40, 50% VC + n.g. Styles (1980)
trans- cells; in atmosphere; no data
formation BHK C1 13 about exposure period
assay
Cell mammalian 18, 180, 1315, 2662 n.g. + Tu et al. (1985)
trans- cells; mg/m3 VC in atmosphere;
formation BALB/c-3T3 24 h; dose-dependent
assay C1 1-13 effect
Rec-assay Bact.; initial 22 mM; 24 h n.g. - Elmore et al.
(DNA repair) B. subtilis (1976)
168M or MC-1
Unscheduled mammalian 5.0, 7.5 or 10% VC in + n.g. Shimada et
DNA cells; rat atmosphere; al. (1985)
synthesis hepatocytes 18 h; dose-dependent
effects
SCE assay human cells; 10, 25, 50, 75, 100% VC + - Anderson et
stimulated in atmosphere; 3 h; al. (1981)
lymphocytes dose-dependent effect
a Bact. = bacteria; DMSO = dimethylsulfoxide; MA = metabolic activation; n.g. = not given; SCE = sister-chromatid
exchange; + = positive; - = negative
activity in the Ames test in the absence of S9-mix has been suggested
to be a result of non-enzymic breakdown of VC or an internal bacterial
metabolism (Bartsch et al., 1976; Shimada et al., 1985), but the
origin of the direct mutagenic effect remains unclear.
Positive results in the Ames test were also observed with
metabolic activation by extracts prepared from human liver biopsies
(Bartsch et al., 1975). Mutagenicity of VC was dependent on
concentration (Bartsch et al., 1975; De Meester et al., 1980; Shimada
et al., 1985) and exposure duration (Bartsch et al., 1975; McCann et
al., 1975). VC was mutagenic when plates were exposed to a VC
atmosphere in a closed (Bartsch et al., 1975 and Table 34) or in a
dynamic flow-through system (Victorin & Ståhlberg, 1988). No mutagenic
effect was observed when VC was dissolved in aqueous solution with
(Bartsch et al., 1975) or without (Rannug et al., 1974; Laumbach et
al., 1977) metabolic activation, probably due to rapid loss of VC in
aqueous solution by evaporation (Bartsch et al., 1975).
Other gene mutation assays in bacteria (Greim et al., 1975),
yeast cells (Loprieno et al., 1976, 1977) and mammalian cells (Drevon
& Kuroki, 1979) revealed positive results exclusively in the presence
of metabolic activation. Mutagenic effects were also reported in a
human cell line containing cloned cytochrome P450IIE1, which is
capable of metabolizing VC (Weisman, 1992). Gene mutation was also
detected in plant cuttings (Tradescantia) exposed to VC (Van't Hof &
Schairer, 1982). No mutagenicity was observed in Neurospora crassa
with or without addition of exogenic activation system (Drozdowicz &
Huang, 1977) but the validity of this study is limited by
contradictory documentation.
In gene conversion assays, positive results were reported with
Saccharomyces cerevisiae in the presence of a metabolic activation
system (Loprieno et al., 1976; Eckardt et al., 1981). No mutagenic
effects were observed without metabolic activation (Shahin, 1976).
VC exposure induced unscheduled DNA synthesis in rat hepatocytes
(Shimada et al., 1985) and increased sister-chromatid exchange in
human lymphocytes after addition of an exogenic activation system
(Anderson et al., 1981). No growth inhibition was detected in DNA
repair-deficient bacteria without metabolic activation (Elmore et al.,
1976).
Cell transformation assays revealed positive results with
(Styles, 1980) or without (Tu et al., 1985) metabolic activation.
7.8.2 In vivo studies
Key studies on the genotoxicity of VC in vivo are documented in
Table 35. VC exposure induced gene mutation and mitotic recombination
in Drosophila melanogaster but not gene mutation in mammalian germ
cells. VC showed in vivo clastogenic effects, increased sister
chromatid exchanges and induced DNA breaks. VC induced also gene
conversion and forward mutations in host-mediated assays.
Mutagenic activity of VC was reported in the mitotic
recombination assay (Vogel & Nivard, 1993) and the sex-linked
recessive lethal (SLRL) assay (Magnusson & Ramel, 1976; Verburgt &
Vogel, 1977; Magnusson & Ramel, 1978) on D. melanogaster. The lowest
effective concentration in the SLRL assay was 2210 mg/m3 with a 2-day
exposure period and 78 mg/m3 after 17 days of exposure (Verburgt &
Vogel, 1977). In the same study, negative results were observed at
higher exposure concentrations with a 2-day exposure period in assays
on D. melanogaster for dominant lethals, translocations (not
tabulated) and sex chromosome loss. These results were discussed by
the authors as a consequence of a saturation effect observed in the
SLRL test (Verburgt & Vogel, 1977). However, sex chromosome loss was
observed in studies by Ballering et al. (1996) in D. melanogaster
exposed to higher concentrations.
No mutagenic activity was detected in the dominant lethal assay
with mice (Anderson et al., 1976; Himeno et al., 1983) and rats (Short
et al., 1977). No mutagenicity was reported in the mouse spot test
(Peter & Ungváry, 1980). Chromosomal aberrations in rats (Anderson &
Richardson, 1981) and hamsters (Basler & Röhrborn, 1980) were reported
and mouse bone marrow micronucleus tests (Jenssen & Ramel, 1980;
Richardson et al., 1983) gave positive results.
VC exposure (260, 650 and 1300 mg/m3) induced single-strand
breaks dose-dependently in the liver DNA of NMRI mice (Walles &
Holmberg, 1984; Walles et al., 1988). Increased frequencies of
sister-chromatid exchange and chromosome aberrations were observed in
the bone marrow of Chinese hamsters after exposure to 1.25, 2.5 or 5%
(v/v) for 24 h (Basler & Röhrborn, 1980).
7.8.3 Genotoxicity of VC metabolites
Metabolic activation of VC is necessary to form the genotoxic
metabolites. The metabolites themselves are genotoxic in the absence
of metabolic activation (see also section 6.3). The VC metabolites,
chloroethylene oxide (chloro-oxirane), chloroacetaldehyde, and
chloroacetic acid were investigated for genotoxicity. In vitro
studies discussed in this section were performed without metabolic
activation unless otherwise stated. Information on the mechanism of
mutagenesis is presented in section 6.5 and 7.11.2.
Chloroethylene oxide (CEO) was found to be the most effective VC
metabolite regarding forward mutation and gene conversion in yeast
(Loprieno et al., 1977), gene mutation in mammalian cells (Huberman et
al., 1975) and reverse mutation in bacteria (Malaveille et al., 1975;
Rannug et al., 1976; Hussain & Osterman-Golkar, 1976). The mutational
specificity of CEO was investigated in Escherichia coli, using trpA
mutant strains. In this system, CEO induced all types of base-pair
substitutions (except one, which was not tested) (Barbin et al.,
1985b). GC -> AT transitions were the most frequent, followed by
AT -> TA transversions. This metabolite inhibited growth in DNA
repair-deficient bacteria (Elmore et al., 1976; Laumbach et al.,
1977).
Table 35. Genotoxicity of vinyl chloride in vivoa
Species/Strain/Sex Test type Test conditions; comments Results References
Gene mutation
Mouse/CD-1/m dominant lethal 20 mice/group exposed to 0, 7800, - Anderson et
assay 26 000 or 78 000 mg/m3 for 6 h/d for al. (1976, 1977)
5 d before 8 wk mating; survival 100,
90, 95 and 45%
Rat/CD/m dominant lethal 12 mice/group exposed to 0, 130, 650, - Short et al.
assay or 2600 mg/m3 for 6 h/d, 5 d/wk; one (1977)
mating during week 11 of exposure;
no mortality
Mouse/CD-1/m dominant lethal a) 13 m exposed to 26 000 mg/m3 for - Himeno et al.
assay 4 h/d for 5 d (11 controls) before 7 wk (1983)
mating; b) 20 m exposed 4 h/d, 5 d/wk
to 13 000 mg/m3 for 10 wk before 3 wk
mating
Mouse/C57BL/f mouse spot test 44 pregnant mice exposed to 12 000 mg/m3 - Peter &
for 5 h on gestation day 10 (51 controls) Ungváry (1980)
Mouse/Swiss/n.d. host-mediated 4-6 mice exposed orally to 700 mg/kg bw. + Loprieno et al.
forward mutation in olive oil; yeast cells (S. pombe (1976)
assay SP.198) inoculated in peritoneum for
3, 6 or 12 h
D. melanogaster/ dominant lethal m exposed to 0 or 78 000 mg/m3 for 2 d; - Verburgt &
Berlin K/m assay total number of eggs per group at Vogel (1977)
least 6950; increase not significant; no
differences in hatchability (> 80%)
D. melanogaster/ Drosophila a) m exposed to 0, 1, 10, 20% VC in air + Magnusson &
Karsnäs/m SLRL test for 3 h (at least 491 chromosomes tested); Ramel (1978)
b) m exposed to 0, 1, or 10% VC for 3 h
after pretreatment with 1% phenobarbiturate
solution for 24 h;
a) positive at > 1% VC; no dose dependency;
pretreatment in b) increased mutagenicity
D. melanogaster/ Drosophila 50 m/group exposed continuously a) to 0, + Verburgt &
Berlin K/m SLRL test 78, 520, 2210, 26 000, 78 000, 130 000 mg/m3 Vogel (1977)
for 2 d or b) to 0, 78, 2210 mg/m3 for 17 d;
a) positive at > 2210 mg/m3, no clear dose
response;
b) positive at > 78 mg/m3
Mitotic
recombination
D. melanogaster/ mitotic eye mosaic assay; 48- to 72-h-old larvae + Vogel &
LS/f + m recombination exposed to 5200 mg/m3 for 17 h; light spots Nivard (1993)
assay of at least 500 eyes scored in adult f
(control 250 eyes scored); survival not
reduced
Chromosomal
abnormalities
Rat/Wistar/m cytogenetic 24 m/group exposed to 3900 mg/m3 for + Anderson &
assay a) 5 d (6 h/d) or b) 3 mo (6 h/d, 5 d/wk); Richardson
bone marrow sampled 24 h after exposure (1981)
period; increased number of cells with any
abnormality, significant in a)
Hamster/Chinese/ cytogenetic 2 m + 2 f per group exposed to 2.5% for 6, + Basler &
f+ m assay 12, or 24 h; 5 m + 5 f exposed to 5% VC Röhrborn
for 24 h; bone marrow samples prepared 26 h (1980)
after start of exposure; control 7 m + 7 f;
increased aberrations at > 6 h (gaps
excluded), effect dose related
Table 35. (cont'd)
Species/Strain/Sex Test type Test conditions; comments Results References
Mouse/CBA/m micronucleus 3 m exposed to 0 or 5% VC for 4 h; bone + Jenssen &
assay marrow examined 30 h after exposure Ramel (1980)
Mouse micronucleus 0, 260, 860 or 2600 mg/m3 for 2 × 4 h + Rodics et al.
CFLP assay (1981)
Mouse/ micronucleus 10 f and 10 m exposed to 0 or 130 000 mg/m3 + Richardson
C57BI/6J/f + m assay for 6 h; bone marrow examined 24 or 48 h et al. (1983)
after exposure
D. melanogaster/ sex m exposed to 0 or 78 000 mg/m3 for 2 d and - Verburgt &
Berlin K/m chromosome chromosomes analysed in progeny (at least Vogel (1977)
loss 6725 m + f per group)
D. melanogaster/ sex m exposed to 0 or 126 000 mg/m3 for 48 h; + Ballering et
ring-X/m chromosome chromosome loss determined in F1 (at al. (1996)
loss least n = 428; 3 broods)
Other effects
Rat/Wistar/m host mediated 20-30 rats exposed to 0 or 1% VC for 24 + Eckardt et al.
gene conversion h, starting 1 h after yeast cell (1981)
assay (S. cerevisiae D7RAD) injection (i.v.)
Mouse/NMRI/f alkaline elution 3-5 f per group; exposure to 1300 mg/m3 + Walles &
assay in liver for 39, 60, 117, 234 h (6 h/d, 5 d/ wk) Holmberg
DNA and sacrificed a) 2 h or b) 18 h (exposed (1984)
for 36, 114, 231 h) after exposure period;
concurrent control sacrificed after 36 or
231 h; positive in a) at 39 h, in b) at 114 h
Table 35. (cont'd)
Species/Strain/Sex Test type Test conditions; comments Results References
Hamster/Chinese SCE assay 2 m + 2 f per group exposed to 1.25 or 2.5% + Basler &
f + m VC for 6, 12, or 24 h; bone marrow samples Röhrborn
prepared 26 h after start of exposure; (1980)
control 4 m + 4 f; dose- and time-dependent
effect
a d = day; f = females; m = males; mo = months; n.d. = no data; SCE = sister-chromatid exchange;
SLRL = sex-linked recessive lethals; wk = weeks; + = positive; - = negative
CAA was 450 times less mutagenic than CEO in the Ames test but
more active than the concurrent positive control ethylene oxide
(Rannug et al., 1976). Positive results were reported with CAA in gene
mutation assays in bacteria (Malaveille et al., 1975; Bartsch et al.,
1975; McCann et al., 1975; Hussain & Osterman-Golkar, 1976; Elmore et
al., 1976; Laumbach et al., 1977; Perrard, 1985), yeast cells
(Loprieno et al., 1977), mammalian cells (Huberman et al., 1975), in a
human lymphoblast cell line (Sanchez & Recio, 1991) and in human cells
using shuttle vectors (Matsuda et al., 1995).
With chloroacetic acid no enhancement of the mutation frequency
could be detected in bacteria (Bartsch et al., 1975; Malaveille et
al., 1975; Rannug et al., 1976) or mammalian cells (Huberman et al.,
1975).
Evidence for mutagenic activity of photoreaction products formed
from VC was presented by Victorin & Ståhlberg (1991). Mixtures of VC
(up to 260 mg/m3) and nitrogen dioxide (but not VC alone) were
mutagenic in S. typhimurium TA100 after 40 min UV irradiation of the
gas mixture before exposure of bacteria.
CEO but not CAA showed a similar toxicity/mutagenic profile to VC
in the hprt locus in a metabolically competent human
B-lymphoblastoid cell line (Chiang et al., 1997; see also section
8.4.2).
7.8.4 Other toxic effects of VC metabolites
In the above studies on the genotoxic effects of CAA (see section
7.8.3), this compound appeared to be highly cytotoxic in various
cellular systems. CAA has also a high acute toxicity in animals, with
a LD50 value of 0.15 mmol/kg body weight. Kandala et al. (1990)
showed in vitro the concentration-dependant reversible inhibition of
DNA synthesis by CAA in rat and mouse cells without a reduction in
thymidine uptake or formation of nucleotides. In isolated rat
hepatocytes, CAA stimulates lipid peroxidation (Sood & O'Brien, 1993).
7.8.5 Mutagenic and promutagenic properties of DNA adducts formed by
VC metabolites
The major DNA adduct of VC, 7-(2'-oxoethyl)guanine (7-OEG), lacks
miscoding properties (Barbin et al., 1985a). In contrast,
1, N6-ethenoadenine (Epsilon A), 3, N4-ethenocytosine (Epsilon C)
and N2,3-ethenoguanine (Epsilon G) showed miscoding properties
(Singer, 1996; see Table 37). The promutagenic properties of the
etheno adducts involve mainly base-pair substitution mutations
(Grollman & Shibutani, 1994). Site-specific mutagenesis studies in E.
coli and in mammalian cell lines have shown that both Epsilon G and
Epsilon C can induce G:C -> A:T transitions; Epsilon C can also lead
to C:G -> A:T transversions (Cheng et al., 1991; Moriya et al.,
1994). Epsilon A can induce misincorporation of G, C, or A during
replication, thus inducing the base-pair substitutions A:T -> C:G,
A:T -> G:C or A:T -> TA (Basu et al., 1993; Pandya & Moriya, 1996).
7.8.6 Mutations in VC-induced tumours
Barbin et al. (1997) examined the presence of p53 gene
mutations (the function of the p53 gene is described in
section 8.4.2) in ASL and HCC tumours induced by VC in Sprague-Dawley
rats. Mutations were found in 11/25 ASL and 1/8 HCC. A twelve-base
deletion was found in one tumour; all others were base-pair
substitutions. Nine of the point mutations were observed at A:T base
pairs and of three G:C -> A:T transitions (Table 43).
Mutations of the p53 gene were also found in tumours from vinyl
chloride-exposed autoclave workers with liver angiosarcoma (ASL) and
hepatocellular carcinoma (HCC) (Hollstein et al., 1994; Boivin et al.,
1997; see also section 8.4 and Table 43). To date (1998) 11 out of 15
(73%) ASL from VC-exposed workers have been shown
immunohistochemically to have mutant p53 protein. Furthermore, a
statistically significant trend for mutant p53 protein has been found
in the serum of VC-exposed workers (Smith et al., 1998). In contrary
to studies in humans, no mutations were found in codons 12, 13 and 61
of the Ki- ras gene in rat liver tumours induced by VC, but mutations
were found involving codon 61 of the Ha- ras proto-oncogene (Froment
et al., 1994; Boivin-Angèle et al., in press) (see Table 44 and
section 8.4.2).
Connexin genes have been shown to restore normal cell growth when
transfected into certain tumorigenic cells and thus are considered to
form a family of tumour suppressor genes. Mutations of the connexin
37 (Cx37) gene in rat liver tumours (22 hepaticangiosarcomas and 3
hepatocellular carcinomas) induced by VC were analysed by
PCR-single-strand conformation polymorphism analysis and DNA
sequencing. The results suggested that Cx37-mediated gap junctional
intercellular communication may be disturbed in most of these
angiosarcomas but mutation of the Cx37 gene is rare (Saito et al.,
1997).
7.9 Factors modifying toxicity
Concurrent administration of ethanol (5% in drinking-water until
termination at month 30) and VC (1560 mg/m3, inhalation 4 h/day,
5 days/week for 1 year) to male Sprague-Dawley rats (80 rats per group
for histopathological evaluation) resulted in increased incidences of
ASL from 23% after exposure to VC alone to 50% in rats exposed to VC
and ethanol versus 0% in ethanol-treated rats and 0% in concurrent
controls. Ethanol had an additive effect on the incidence of
hepatocellular carcinoma (VC 43%, VC-ethanol 60%, ethanol 10%, control
1.3%) and lymphosarcoma (VC 7.5%, VC-ethanol 14%, ethanol 5%, control
2.5%) (Radike et al., 1981). This effect may be due to the interaction
of ethanol with VC metabolism.
Induction of certain enzymes of the mixed-function oxidase system
by pretreatment with phenobarbital (Jaeger et al., 1974, 1977;
Reynolds et al., 1975a,b,c) or the mixture of polychlorinated
Table 36. Summary of genotoxic effects induced by exposure to vinyl chloride in vitro and in vivoa
In vitro In vivo
Bacteria Yeast Plants Mammalian Human Insects Mammalia Humansc
GM DD GM GC GM GM DD CT GM SCE GM AN MR GM CA MN DD SCE GM CA MN SCE
+ -b + + + + + + + + + - + - + + + + + + + +
a AN = aneuploidy; CA = chromosomal aberration; CT = cell transformation; GM = gene mutation; GC = gene conversion;
DD = DNA damage; MN = micronuclei; MR = mitotic recombination; SCE = sister-chromatid exchange
b Single study, only tested without metabolic activation
c These results are described in Table 42 (section 8.4.1)
Table 37. Evidence for base-pair substitutions caused by etheno-DNA adducts
Ethenobase Base Base-pair System used Reference
incorporated substitution for study
opposite
adducta
1,N6-ethenoadenine C AT -> GC in vivo bacteriophage M13-Nhei in E. coli Basu et al.
(epsilon-A) transition (1993)
A AT -> TA in vivo single-strand vector shuttle in Pandya & Moriya
C AT -> CG E. coli or simian kidney (COS) cells; (1996)
transversions
3,N4-ethenocytosine A CG -> TA in vivo single-strand vector shuttle in Moriya et al.
(epsilon-C) transition E. coli or simian kidney (1994)
T CG -> AT in vitro (COS) cells; Zhang et al.
transversion E. coli DNA polymerase (1995)
I system
in vivo M13AB28 in SOS-(UV)-induced Jacobsen &
E. coli Humayun (1990)
in vivo M13 glyU phage transfection Borys et al.
of E. coli tester strain (1994)
in vitro E. coli DNA polymerase I system Simha et al.
(1991);
Palejwala et al.
(1991)
N2,3-ethenoguanine T GC -> AT in vivo E. coli DNA synthesis by M13G*1 Cheng et al. (1991)
(epsilon-G) transition assay
in vitro E. coli DNA polymerase I (Klenow Singer et al. (1991)
fragment); exonuclease-free Klenow;
Drosophila melanogaster polymerase
alpha-primer compex;
human immunodeficient virus-I
reverse transcriptase (HIV-RT)
in vitro E. coli DNA-dependent RNA Mroczkowska &
polymerase Kusmierek (1991)
Table 37. (cont'd)
Ethenobase Base Base-pair System used Reference
incorporated substitution for study
opposite
adducta
1,N2-ethenoguanine A GC -> TA in vitro E. coli DNA polymerase I Langouet et al.
G GC -> CG (exonuclease-free Klenow) (1997)
transversions
a A = adenine; C = cytosine; G = guanine; T = thymine
biphenyls (Arochlor 1254) (Reynolds et al., 1975a,b; Conolly et al.,
1978) enhanced acute hepatotoxicity in rats as measured by increased
activity of hepatic enzymes and/or focal hepatic necrosis.
Administration of SKF-525A, an inhibitor of the mixed-function oxidase
system, 30 min prior to VC exposure in phenobarbital-pretreated rats,
inhibited the enhancing effect of phenobarbital on VC hepatotoxicity
(Jaeger et al., 1977). Application of cysteine, a rate-limiting
precursor in hepatic glutathione (detoxification of reactive
chemicals), via drinking-water prior to VC exposure protected Arochlor
1254-pretreated rats against acute VC hepatotoxicity (Conolly &
Jaeger, 1979).
7.10 Mechanisms of toxicity - mode of action
7.10.1 Mechanisms of VC disease
Based on evidence of immunological abnormalities, such as
hyperimmunoglobinaemia and circulating immune complexes in workers
with "vinyl chloride disease" (section 8.3), Ward et al. (1976) have
proposed a possible mechanism for the vascular changes associated with
this disease. Reactive VC metabolite(s) bind(s) to a protein,
resulting in a structurally abnormal protein. This protein would react
as an antigen and initiate an immune response with B-cell
proliferation and hyperimmunoglobinaemia. Circulating immune complexes
formed by interaction of this antigen and antibodies would precipitate
in the extremities of exposed humans in response to the cold and
activate the complement sequence. The cryoprecipitates and reactions
secondary to the complement activation are proposed to produce
vascular occlusion and fibrinogen/fibrin conversion. The mechanism is
supported by findings of IgG deposition with associated complement C3
and fibrin in histological lesions (Grainger et al., 1980). The
resulting vascular insufficiency would explain the observed clinical,
radiological and histological findings in skin, skeletal and soft
tissues, and lungs (section 8.3) in workers occupationally exposed to
high concentrations of VC (Ward et al., 1976). No further studies were
identified that would directly confirm this mechanism, and the degree
to which it has been accepted is not clear. The available evidence
does not seem sufficient to suggest an autoimmune disease as a
pathogenetic mechanism. Further studies will be needed to establish
the true significance of the possibly transient immunological
abnormalities in VC-induced disorders.
7.10.2 Mechanism of carcinogenesis
There is a large body of data showing that VC acts as a genotoxic
carcinogen. After metabolic activation to CEO by CYP2E1, VC exerts
various genotoxic effects (including gene mutations and chromosomal
aberrations) in different organisms, including bacteria, yeasts,
mammalian cells in culture, Drosophila, rodents and humans (Table 36).
Among the mutagenic events induced by VC, base-pair substitutions
appear, so far, to be the most frequent. VC in the presence of an
activation system has a transforming activity on mammalian (rodent)
cells in culture (see Table 34).
Studies in vitro have demonstrated that metabolically activated
VC and its electrophilic metabolites CEO and CAA can alkylate nucleic
acid bases. 7-OEG, the major DNA adduct formed by VC and CEO does not
exhibit promutagenic properties. In contrast, four minor adducts,
Epsilon A, Epsilon C, N2,3-Epsilon G and 1,N2-Epsilon G, show
promutagenic properties, inducing mainly base-pair substitution
mutations and a low level of frameshift mutations.
7-OEG and three etheno adducts (Epsilon A, Epsilon C,
N2,3-Epsilon G) have been detected in DNA from rats and mice exposed
to VC. Highly variable background levels of Epsilon A and Epsilon C
were found in all the tissues examined. Following exposure of rats to
VC, significantly elevated levels of Epsilon A and Epsilon C were
measured in most tissues, except the brain; there was also no
significant increase of Epsilon A levels in the kidney and spleen.
The liver is one of the primary targets for VC-induced
carcinogenesis in rats and humans. It is also, by far, the major
tissue involved in VC activation in rats. Following exposure of rats
to VC, the distribution of etheno adduct levels (induced by the
exposure) is rather homogeneous within the organism. Epsilon C was
shown to accumulate as a function of length of exposure in at least
three organs (liver, kidney and lung). In contrast, Epsilon A
accumulated in the liver but not in the kidney and lung. In addition,
adduct levels (Epsilon A, Epsilon C) did not decrease in the liver,
for at least two months following the end of exposure. Etheno adducts
are formed as endogenous background levels in various tissues in
humans; no data are available on etheno adduct levels in humans
exposed to VC.
Mutations have been found in liver tumours associated with
exposure to VC. In human ASL, the Ki-ras gene is activated through a
GC -> AT mutation at base 2 of codon 13. Mutations, all AT -> TA
transversions, have been described in the p53 gene in three human
ASL. The Ki-ras gene activation is not found in rat ASL. However, 44%
of rat ASL were found to contain a mutated p53 gene: most mutations
were base-pair substitutions, involving mainly A:T base pairs. The
data suggest the existence of hot spots for mutations in the p53
gene, and one mutation found in two rat ASL was equivalent to the
same mutation characterized in one human ASL associated with VC
exposure. The Ha-ras gene is activated in rat HCC induced by VC,
through an AT -> TA transversion in codon 61.
The mutation spectra observed in liver tumours (ASL and/or HCC)
associated with VC exposure in humans and rats are clearly distinct
from those observed in sporadic liver tumours or in hepatic tumours
associated with other exposures. In rats, the substitution mutations
found at A:T base pairs in the ras and p53 genes are consistent
with the promutagenic properties of Epsilon A and with the
accumulation and persistence of this lesion in hepatic DNA.
Altogether, available data suggest that etheno adducts could be
involved in the initiation of hepatocarcinogenesis by VC. However,
they cannot explain the observed tissue- and cell-specificity.
More studies on the formation and repair of etheno adducts at the
cellular level (cell specificity), as well as at the molecular (gene
and DNA sequence) level, are warranted. Carcinogenesis is a multi-step
process and, obviously, there is a need for quantitative evaluation of
other critical biological end-points, such as effects of VC on
apoptosis, cell proliferation or intercellular communication in
vivo.
8. EFFECTS ON HUMANS
Only reports on effects for humans of exposure to VC have been
considered here and not reports where exposure has been to a number of
chemicals, e.g., at landfills, in factories manufacturing a number of
chemicals or in the PVC processing industry.
8.1 General population exposure
After an accident in Schönebeck, Germany in June 1996, involving
the derailment of a train carrying VC and subsequent fire, 325 persons
were documented as having acute symptoms but these correlated with
exposure to the pyrolytic products (e.g., HCl) and not to VC itself.
But a study on 29 persons exposed as a result of this accident showed
a significant increase in chromosomal aberrations compared to an
unexposed control group (Hüttner & Nikolova, 1998; see also Table 42
and section 3.2.3).
A case has been reported of epithelioid haemangioendothelioma
involving liver, bone and lungs in a man living for over 8 years
several hundred metres from a toxic waste dump next to a chemical
plant producing VC (Shin et al., 1991).
There have been several reports on the possible increased
prevalence of congenital malformations in populations exposed to
emissions from polymerization facilities (Edmonds et al., 1975, 1978;
Infante, 1976; Thériault et al., 1983; Rosenman et al., 1989) but none
of these studies showed a statistically significant correlation
between developmental toxicity and proximity to the facility
(Clemmesen, 1982; Hemminki & Vineis, 1985). A number of studies (e.g.,
Goldberg et al., 1995; Dolk et al., 1998) examined risk for cancer and
for adverse reproductive outcome in relation to proximity to
landfills. Although VC is one of the potential emissions from the
landfills, these studies do not directly address the population
exposure to VC and were not further considered.
In England and Wales, from 1975-1987 data, there were no
confirmed non-occupationally exposed cases of ASL (Elliott &
Kleinschmidt, 1997). In the USA, five non-occupational cases were
reported living within 1.6 km of a VC plant (Brady et al., 1977).
8.2 Controlled human studies
Three men and three women were exposed twice daily with a 6-h
interval for three successive days to 0, 0.4, 0.8, 1.2, 1.6 or 2.0%
VC. The NOEL was between 0.8 and 1.2%. Above this, dizziness, nausea,
dulling of vision and auditory cues were reported (Lester et al.,
1963).
Thirteen male volunteers were exposed to 130, 650 and 1300 mg/m3
for 7.5 h and subjective and neurological responses were measured
before the subject entered the chamber, 15 min after entrance and at
1-h intervals thereafter; 24-h post-exposure urine and blood samples
were taken and tested. No significant adverse effects were noted with
the exception of some dryness of eyes and nose at 1300 mg/m3. The
exposure had no noticeable effect on neurological responses nor did it
produce significant changes in the results of mental, coordination or
manual dexterity tests conducted during the exposure period. All
clinical laboratory studies performed in the post-exposure period were
normal and not significantly different from pre-exposure values
(Baretta et al., 1969).
8.3 Occupational exposure
8.3.1 Overview
VC was first produced commercially in the late 1920s. Various
effects caused by exposure to VC were reported: cardiac arrhythmia in
experimental animals (Oster et al., 1947); hepatic abnormalities in VC
workers (Tribukh et al., 1949; Filatova et al., 1958), acroosteolysis,
Raynaud-type-phenomenon and sclerodermoid skin lesions (Lelbach &
Marsteller, 1981). But it was not until it was found that VC could
cause cancer in animals (Viola et al., 1971; Maltoni et al., 1974) and
humans (Creech & Johnson, 1974) that levels of VC in the workplace
were drastically reduced. Some VC workers, in particular autoclave
cleaners, were estimated to have been exposed to as much as
2600 mg/m3 (1000 ppm) in the 1950s or earlier, reducing to a tenth of
this level by the mid-1970s (Table 20). After 1975 levels were usually
2.6-13 mg/m3 (1-5 ppm) in many countries, but in some countries where
production plants were not modernized, workers were or are exposed to
high levels of VC (Fucic et al., 1990a; Gáliková et al., 1994; Hozo et
al., 1996, 1997; see also Table 21).
It should be noted that the non-neoplastic and neoplastic effects
described in the following sections are due in most cases to the high
exposure of workers to VC before 1974. No cases of ASL (section 8.3.1)
have been reported to the International Register of Cases among (West)
European workers first exposed after 1972 (Storm & Rozman, 1997). This
is not the case for Eastern European countries where the old
regulation of 194 mg/m3 (75 ppm) in the working environment was valid
until recently (Hozo et al., 1997).
8.3.2 Non-neoplastic effects
8.3.2.1 Acute toxicity
In acute VC intoxication, the symptoms described include vertigo,
nausea and headache. At higher concentrations, VC exerts narcotic
effects and at one time was considered as a possible anaesthetic
(Patty et al., 1930; Peoples & Leake, 1933; Oster et al., 1947).
VC caused almost immediate death of VC polymerization workers in
two incidents of accidental poisoning. No values were given but the
strong smell and the narcotic effects of VC were reported (Danziger,
1960).
Concentrations of VC of the order of 26 000 mg/m3 (1%) in the
air induce unconsciousness and cardiac arrhythmia. Exposure of two
workers to 2.5% VC for 3 min caused dizziness, disorientation and a
burning sensation in the soles of the feet. There was complete
recovery except for a slight headache lasting 30 min (Danziger, 1960).
A man whose hands were accidentally sprayed with VC developed
erythema and some second-degree burns which healed without
complication (Harris, 1953). A patient complaining of eye burns from
VC recovered 48 h after the eye was rinsed for 15 min with saline
(McLaughlin, 1946).
8.3.2.2 Effects of short- and long-term exposure
Concentrations of VC in the region of 2590 mg/m3 (1000 ppm),
which were not unusual prior to 1974, over periods ranging from
1 month to several years, have been reported to cause a specific
pathological syndrome found in VC workers called the "vinyl chloride
illness". Symptoms described were earache and headache, dizziness,
unclear vision, fatigue and lack of appetite, nausea, sleeplessness,
breathlessness, stomachache, pain in the liver/spleen area, pain and
tingling sensation in the arms/legs, cold sensation at the
extremities, loss of libido and weight loss (Thiess & Versen, 1974).
Clinical findings included scleroderma-like changes in the fingers
with subsequent bony changes in the tips of the fingers described as
acroosteolysis, peripheral circulatory changes similar to Raynaud's
disease, and enlargement of the liver and spleen with a specific
histological appearance, and respiratory manifestations (Lange et al.,
1974; Suciu et al., 1975; Veltmann et al., 1975; Lelbach & Marsteller,
1981).
8.3.2.3 Organ effects
a) Skin and skeletal tissues
Occupational acroosteolysis has largely affected the most highly
exposed workers involved in scraping the insides of autoclaves in the
PVC production process (Harris & Adams, 1967; section 3). It is a rare
bone disease resulting in de-calcification of the terminal phalanges
of the hands and other extremities (Cordier et al., 1966; Wilson et
al., 1967; Markowitz et al., 1972). In these cases, acroosteolysis was
often preceded by soreness and tenderness, numbness, pallor and
cyanosis of the extremities especially the hands, a Raynaud-type
phenomenon caused by reversible constriction of the arterioles.
Sclerodermoid-like changes appear in the skin of the hands and
forearms and osteolytic and sclerotic lesions of the bones,
particularly the extremities and sacroiliac joints (Lange et al.,
1974). Most of the abnormalities disappeared and bones showed signs of
healing a year or two after the men had stopped work (Harris & Adams,
1967). A chronothermodynamic study of Raynaud's phenomenon secondary
to past exposure to VC showed that, although reduced, the symptoms
were still present after 8 years (Fontana et al., 1996).
A review of five studies from four countries involving 725
workers at risk from VC exposure showed that 3% developed
acroosteolysis; 10% Raynaud-type phenomenon and 6% sclerodermoid skin
lesions (Lelbach & Marsteller, 1981). Genetic susceptibility has been
suggested as a possible reason that not all workers who had been in
contact with VC developed symptoms (Black et al., 1983, 1986). The
scleroderma-like syndrome induced by VC exposure appears to be
different from that of other (systemic) scleroderma (Ostlere et al.,
1992). It has a shorter incubation period (1 month to 3 years compared
to 4 to 44 years) (Ishikawa et al., 1995) and there are immunological
differences (section 8.3.2.3f).
b) Hepatic effects
Exposure to VC is associated with hepatomegaly and/or
splenomegaly and with various histological lesions in the liver (Lange
et al., 1974; Ho et al., 1991). In one carefully described study,
advanced portal hypertension with histological findings of
non-cirrhotic fibrosis was diagnosed in 17 of 180 VC polymerization
workers (Lelbach & Marsteller, 1981).
Focal hepatocellular hyperplasia and focal mixed hyperplasia
(hyperplasia of sinusoidal cells along with hyperplasia of
hepatocytes) are early histological alterations indicative of VC
exposure (Tamburro et al., 1984). The precursor stage of ASL is
characterized by subcapsular fibrosis, progressive portal fibrosis and
a borderline increase of intralobular connective tissue, all
associated with focal stimulation and proliferation of sinusoidal
lining cells and hepatocytes. Transition to angiosarcoma is preceded
by focal dilatation of sinusoids with enlarged dedifferentiated lining
cells often containing peg-like hyperchromatic polymorphic nuclei
(Popper & Thomas, 1975; Gedigk et al., 1975).
There is a great similarity between the histological sequences in
the liver of rodents exposed by inhalation to VC (chapter 7; Spit et
al., 1981) and the lesions observed in VC-exposed workers (Popper et
al., 1981).
Twenty of the 39 "liver disease deaths" reported in an Italian
study of highly exposed PVC workers were due to liver cirrhosis, the
remainder being due to miscellaneous liver disorders. A statistical
evaluation was not possible (Pirastu et al., 1990). In another study,
two of 21 heavily exposed VC workers died from sequelae to
noncirrhotic portal fibrosis and portal hypertension (Lelbach, 1996).
In the USA and European cohort studies of VC-exposed workers (study
descriptions are given in section 8.3.3), deaths from chronic
non-malignant diseases of the liver had a low SMRs of 62 and 88; in
the European study this was significantly less than expected (Wong et
al., 1991; Simonato et al., 1991).
c) Cardiovascular effects
Some old studies reported statistically non-significant increased
mortality due to cardiovascular diseases among workers exposed to VC
(Ebihara, 1982; Greiser et al., 1982).
Laplanche et al. (1992) report an elevated incidence of
circulatory system diseases other than Raynaud's disease (RR 1.4, 95%
CI 1.0-1.8) in a 7-year follow-up of a cohort of 1100 VC workers. The
increased risk was mainly due to hypertension and "other circulatory
disorders". Likewise, another study with a 5-year follow-up on the
incidence of arterial hypertension (AH) and coronary heart disease
(CHD) in 105 VC and PVC workers exposed to VC at between 4 and
1036 mg/m3 showed that exposed workers had significantly higher blood
pressure than controls. The estimated relative risk (RR) for AH in
exposed workers was twice as high as in the controls, while there was
no significant difference regarding CHD. There was an
exposure-response relationship between the intensity of exposure and
the incidence of AH (Kotseva, 1996).
In contrast, both the large cohort studies reported a
statistically significant deficit in the mortality from cardiovascular
diseases, with SMRs of 87 and 81. In the USA study, the mortality was
even lower for the subcategory of arteriosclerotic heart disease (SMR
74.5, 95% CI 81.6-89.2 (Simonato et al., 1991; Wong et al., 1991). In
the Canadian study (Thériault & Allard, 1981), there was a
statistically non-significant 20% deficit in cardiovascular mortality,
based on 25 exposed cases.
d) Respiratory effects
Adverse respiratory effects reported in older case studies
included increased incidence of emphysema (Suciu et al., 1975),
decreased respiratory volume and vital capacity, respiratory
insufficiency (Suciu et al., 1975), decreased respiratory oxygen and
carbon dioxide transfer (Lloyd et al., 1984), pulmonary fibrosis of
the linear type (Suciu et al., 1975), abnormal chest X-rays (Lilis et
al., 1975) and dyspnoea (Walker, 1976). This is probably due in part
to confounding by smoking and presence of PVC-resin dust, which is
known to cause respiratory lesions (Mastrangelo et al., 1979; Lilis,
1981).
Both large cohorts (Wong et al., 1991; Simonato et al., 1991)
found a deficit in the mortality from non-malignant respiratory
diseases (SMR 81.6, and 77, respectively). Despite overall deficit in
mortality from respiratory diseases, Wong et al. (1991) found an
excess of emphysema/chronic obstructive pulmonary disease (COPD)
mortality, which, however, was highest among workers with a duration
of exposure less than 10 years.
e) Neurotoxicity
In patients with chronic occupational exposure, neurological
disturbances include sensory-motor polyneuropathy (Perticoni et al.,
1986; Podoll et al., 1990), trigeminal sensory neuropathy, slight
pyramidal signs and cerebellar and extrapyramidal motor disorders
(Langauer-Lewowicka et al., 1983). Psychiatric disturbances included
neurasthenic or depressive syndromes (Penin et al., 1975).
Sleeplessness (Gnesina et al., 1978; Gnesina & Pshenitsina, 1980;
Langauer-Lewowicka et al., 1983; Gnesina & Teklina, 1984) and loss of
sexual functions (see below) were frequently encountered. Pathological
EEG alterations were found in a high proportion of patients (Penin et
al., 1975; Stblová et al., 1981).
f) Immunotoxicity
The major immunological abnormalities reported in VC disease
patients include hyperimmunoglobulinaemia with a polyclonal increase
in IgG, cryoglobulinaemia, cryofibrinogenaemia, and in vivo
activation of complement (Ward et al., 1976). Immunofluorescent
examinations of skin and lung biopsies have demonstrated the
deposition of IgG with associated complements C3 and fibrin in
relation to the histological lesions described in small blood vessels
(Grainger et al., 1980). A statistically significant increase in
circulating immune complexes was observed in workers exposed to VC,
compared to unexposed workers (Ward et al, 1976) . The increase in
circulating immune complexes was greatest in women and in those with
duties involving exposure to relatively higher levels of VC
(Bogdanikowa & Zawilska, 1984).
Bencko et al. (1988) found significantly elevated IgA, IgG and
IgM levels in the serum of workers exposed to low levels of VC
(< 10 mg/m3), but found that in workers with excessive exposures to
VC (> 10 mg/m3) there was a significant drop in IgG level.
The antinuclear antibody (ANA) test is negative or at low titre
in VC disease (Ward et al., 1976), whereas it is often positive in
other scleroderma-like disorders.
There is much heterogeneity within VC disease, in terms of skin
involvement, severity, and organ involvement, and it is clear that
this could have an immunological background. Systemic sclerosis may be
a genetically linked autoimmune disease. Many autoimmune diseases show
statistically significant associations with certain
human-leukocyte-associated antigen (HLA) alleles. Black et al. (1983,
1986) compared the HLA frequencies and autoantibodies in workers with
VC disease ( n=44), asymptomatic workers ( n=30), systemic sclerosis
( n=50) and normal (blood donor) controls ( n=200). The HLA-DR5
allele occurred significantly more frequently in VC disease (36%) and
systemic sclerosis (30%) compared to asymptomatic workers (3%) and
normal controls (16%). Eleven out of 21 men with severe VC disease had
HLA markers B8, DR3 whereas none of the 23 workers with mild disease
were positive for these markers, suggesting that this haplotype
favours progression of the disease.
Splenomegaly has been detected in a number of VC-exposed workers
(Falk et al, 1974; Suciu et al., 1975; Veltmann et al., 1975; Makk et
al., 1976; Ho et al., 1991). Hyperplastic sinusoidal cells were found
in some specimens.
g) Reproductive toxicity
The reproductive effects of VC have been reviewed (Uzych, 1988;
Little, 1993; Olsen et al., 1995).
VC has been cited in several early case series from different
countries as being responsible for male sexual dysfunctions such as
potency troubles, "pathological changes in the ejaculates", decreased
androgen secretion or undefined sexual disorders (Suciu et al., 1975;
Veltman et al., 1975; Walker, 1976; Sanotsky et al., 1980; Makarov,
1984) in exposed workers, but there is a lack of controlled studies.
Similarly, various female sexual dysfunctions have been reported after
long term exposure to VC (Makarov et al., 1984).
The possible risk, arising from human male exposure to VC, for
pregnancy and fetal loss among the wives of workers was first raised
after a report by Infante et al. (1976a,b) suggesting that fetal loss
was significantly more among wives of exposed workers. Data for the
wives of 95 VC polymerization workers in the USA were contrasted with
data for the partners of PVC fabrication workers and rubber workers
(total 158). Prior to exposure, the fetal death rates for controls and
polymerization workers were 6.9 and 3.1% (age adjusted, respectively).
After exposure, the rates were 6.8 and 10.8%, respectively. Weaknesses
in the study were in the method of age adjustment, that information
was obtained from interviews with male workers only (and not their
wives), and that there were no data on maternal age or on smoking and
other abuses.
In a study on 534 VC production and polymerization workers whose
wives were under 45 on 1 January 1984, 82 men were exposed to VC
before their spouses' pregnancies. The crude rate of abortions (no. of
spontaneous abortions / no. of pregnancies) was 8.9 for exposed and
8.8 for unexposed (total number of pregnancies: 90 and 1027,
respectively). Adjustment for confounders did not suggest a
significant relationship between spontaneous abortion and paternal
exposure to VC (Mur et al., 1992).
No statistically significant correlation could be found between
exposure to VC and congenital central nervous system (CNS) defects
among the children of fathers working at VC polymerization facilities
in Kanawha County, USA (Edmonds et al., 1975, 1978), but the study
data were not sufficient for a clear evaluation (Uzych, 1988).
In a Chinese study of 236 female workers exposed for more than 1
year to VC and 239 controls using retrospective (levels 3.9 to
89.3 ppm) and prospective (0.2 to 130.7 ppm) epidemiology, VC did not
appear to influence involuntary infertility, pregnancy outcome or
course of parturition, although the incidence of toxaemia in pregnancy
was found to be higher among female workers exposed to higher
concentrations in the prospective study (Bao et al., 1988; Jiangl,
1990).
An investigation into reproductive function in 2736 workers
exposed to VC in 13 PVC factories and 3442 workers in other factories
not exposed to VC showed no significant differences in reproductive
outcome. However, in the female exposed group, the incidence of
pregnancy complication was significantly higher than that of the
control group, suggesting that VC may effect the pregnancy process in
female workers (Huang, 1994).
There have been studies on female workers in the PVC processing
industry (Lindbohm et al., 1985; Ahlborg et al., 1987), but, as VC was
not the only source of exposure, these studies were not considered
here.
8.3.3 Neoplastic effects
After the case series published in 1974 (Creech & Johnson, 1974)
on hepatic angiosarcoma among workers exposed to vinyl chloride,
several further case series and small epidemiological studies, mainly
with emphasis on hepatic tumours, were published in the 1970s and
1980s. They show that VC can cause the rare liver tumour (ASL). Other
(non-ASL) cancer sites/types reported for which some of these studies
have indicated an association with VC exposure are: liver (e.g.,
non-angiosarcoma) tumours, particularly hepatocellular carcinoma;
respiratory system; digestive system other than the liver; lymph and
haematopoietic tissue; brain and other central nervous system;
malignant melanoma.
The early studies, together with a prepublication report of the
USA cohort study (see below) were summarized by Sir Richard Doll
(Doll, 1988). More recently, the studies performed in Sweden, Italy,
the United Kingdom and Norway were updated and analysed together
(L'Abbé et al., 1989; Simonato et al., 1991). Parts of the European
study have also been published separately. An updated American study
(Wong et al., 1991) combined many smaller studies conducted earlier in
the USA. These two studies provide the most informative data on the
health effects associated with exposure to VC. Four smaller
prospective studies on VC exposure workers have been conducted in
Canada (Thériault & Allard, 1981), Germany (Weber et al., 1981;
Greiser et al., 1982), France (Laplanche, 1987, 1992) and the
former-USSR (Smulevich et al., 1988). The complete description of two
additional studies (Frentzel-Beyme et al., 1978; Huang, 1993a,b) was
not available to the Task Group and were not further considered.
The epidemiological studies on VC/PVC workers are described as
follows:
* Simonato et al. (1991) (European/IARC study), which incorporates
populations reported by: Byrén et al. (1976), Molina et al. (1981),
Hagmar et al. (1990) (Sweden); Fox & Collier (1977), Jones et al.
(1988) (United Kingdom); Heldaas et al. (1984, 1987) (Norway); Belli
et al. (1987), Pirastu et al. (1990, 1991, 1998) (Italy);
* Wong et al. (1991) (USA study)a, which incorporates populations
reported by Tabershaw & Gaffey (1974), Monson et al. (1975); Nicholson
et al. (1975), Ott et al. (1975), Waxweiler et al. (1976), Buffler et
al. (1979), Cooper (1981), Dahar et al. (1988), Wu et al. (1989);
* Thériault & Allard (1981) Canada;
* Weber et al. (1981), Greiser et al. (1982) Germany;
* Laplanche et al. (1987, 1992) France;
* Smulevich et al. (1988) former-USSR
The USA study consisted of 10 173 men who had worked for at least
one year in jobs involving exposure to VC prior to January 1973 in 37
plants in the USA. Observation covered the years 1942-1982, and the
observed mortality was compared with the expected rates, based on USA
national rates for white males, standardized for age, and calendar
time. The race was actually known for only 666 workers, among which 3%
were African. Altogether 1536 members of the cohort were identified as
having died, the vital status of 725 individuals (7.88%) remained
unknown, and death certificates were not obtained for 97 (6.3%). These
deaths were included in the calculation of the overall SMR, but not in
any cause-specific SMRs (which leads to an underestimation of all
disease-specific SMRs by 6.3%).The deaths were coded using the 7th ICD
revision, where the numbers 155 and 156 comprise liver (155, both
primary and secondary) and bile duct and gall bladder tumours (156).
The average length of employment was approximately 16 years, and 46%
of the cohort were employed before 1955, thus providing a possible
latency of more than 27 years for this group. The levels of exposure
were not known. The all-causes SMR was 90.1 (95% CI 85.6-94.7), based
on a total of 1536 deaths. The SMR for all cancers was 105.1
(94.4-116.5).
The European/IARC study comprised a total of 14 351 subjects from
19 factories. After exclusion of short-term employees (< 1 year),
females, deaths outside the observation period, members of more than
one cohort, 12 706 subjects remained for the analysis. The follow-up
a After the Task Group meeting, a report of an update of the USA
study became available. A summary of it is presented in Appendix
3.
was 97.7% complete, the average length of follow up 17 years, and the
total number of person-years 222 746. National rates of mortality,
specific for age and five-year calendar periods, were used for the
comparison. The observation period was different for different
factories, starting for most from 1955, and extended until 1986.
Calendar period-specific job exposure matrices (JEMs) were developed
for 13 of the 19 factories. These JEMs were developed using job title
as a basic unit in which exposure is assessed. Estimates of exposure
were assigned, a priori, by a group of industrial hygienists on the
basis of the historical information available from the companies.
These estimates have been used both for qualitative and
quantitative dose-response analysis. In particular, a cumulative dose
was computed for each subject by multiplying the number of years spent
in a specific job by the time-specific estimates in ppm and then
summing up all the periods.
The 7th (for liver tumours, codes 155-156), 8th and 9th revisions
(155) of ICD were used (thus combining primary and secondary tumours
of the liver and tumours of extrahepatic bile ducts). A statistically
significant deficit in over-all mortality was observed (SMR 88, 95% CI
83-93), while for all malignant neoplasms, the SMR was 104 (CI
95-114), with 445 cancer deaths observed.
The Canadian study (Thériault & Allard, 1981) studied the
mortality of 1659 workers in a chemical industry complex in Schwanigan
who had worked for no less than 5 years in the company between 1943
and 1972. Out of the total, 48 (2.9%) could not be traced. The
observation period was 20 years or more for 315 workers (70%), and the
duration of exposure no less than 10 years for 340 (75%). Since
several episodes of unconsciousness among workers were reported, the
exposure to VC had probably been high: no measurement data were
available. The causes of death during 1948-1972 of the cohort was
determined from death certificates. In the analysis, those exposed to
VC for more than 5 years (451) were compared with those exposed for
less than 6 months (870). In addition, the mortality of the exposed
subcohort was compared to the age-standardized mortality expected for
Quebec in the year 1971. For cancer cases, medical records were
consulted for the verification of the diagnosis. The overall mortality
was similar in the exposed and non-exposed cohorts, and gave an SMR of
0.83 for the VC-exposed cohort in comparison to the Quebec figures.
Smulevich et al. (1988) reported results from a retrospective
cohort study of workers employed in some of the oldest Soviet chemical
plants producing VC and PVC. The study comprised 2195 men and 1037
women employed for at least one month in the plants. Subjects were
followed from 1939 to 1977. Information on specific occupation within
the plant, duration of exposure, date and cause of death, and
postmortem results (for cancer deaths) were collected for all member
of the cohort. No information was provided on the means for doing the
follow-up nor on losses to follow-up. Workers were classified into
those with high level exposure to VC (> 300 mg/m3), moderate levels
(30-300 mg/m3) and low levels (< 30 mg/m3). Mortality for the
cohorts was compared with rates of the city of the plant for the years
1959, 1969 and 1975. There were 288 deaths registered in the cohort,
including 63 from cancer. The SMR for all cancers was 1.06.
A prospective cohort study of 1100 workers exposed to VC in
various French plants and 1100 unexposed subjects was initiated in
1980 (Laplanche et al., 1987, 1992). Unexposed controls were matched
to cases by age, plant and physician. Subjects were followed until
December 1988 for vital status, other health outcomes and occupation.
Information was collected on complete occupational history, smoking
and drinking habits, and medical history (see also section 8.3.2.3c
and Table 39).
Weber et al. (1981) and Greiser et al. (1982) reported a historic
prospective cohort study of 7021 male workers who had been or were
working on 31 December 1974 in VC/PVC plants in Germany and Austria
(follow-up 10.5 years). The control cohort consisted of 4910 Germans
and Austrians from the same chemical companies but not having contact
with VC (follow-up 15.5 years). SMR for liver tumours was 1523, with
no details of the number of ASL. For lymphatic tumours and leukaemia
there was a SMR of 214. No information was given on subtypes. A
summary of findings on selected neoplasms for the epidemiological
studies on workers exposed to VC is given in Tables 39 and 40.
8.3.3.1 Liver and biliary tract cancers
a) Features of angiosarcoma
Angiosarcoma of the liver (ASL), also known as
haemangioendothelial sarcoma, is an extremely rare liver tumour and is
difficult to diagnose. ASL constitutes only 2% of all primary tumours
of the liver in the general population. It has been associated only
with exposure to VC, Thorotrast (a contrast medium used in X-ray
radiography in the 1930s-1950s) and arsenic (Creech & Johnson, 1974;
Falk et al., 1974). In England and Wales, from 1975 to 1987, there was
an annual incidence of 1.4 cases per 10 million population (Elliott &
Kleinschmidt, 1997). Regular international surveillance of cases of
ASL from VC exposure show that 118 cases were registered by 1985
(Forman et al., 1985), 173 by 1993 (Lee et al., 1996), and 197 by 1999
(Association of Plastics Manufacturers in Europe, 1999) (Table 38).
The most prominent clinical symptoms are abdominal pain,
weakness, fatigue and weight loss with hepatosplenomegaly, ascites and
jaundice being the common clinical signs. It is suggested that
patients with non-cirrhotic portal fibrosis and a history of VC
exposure should be followed-up for likely ASL (Lee et al., 1996).
However, Lelbach (1996) noted that, strikingly, except for the final
stages, there was little impairment of hepatic function in ASL
patients. The average latent period between starting work in an
occupation involving VC exposure and ASL diagnosis/death for 99 cases
was 22 years (Purchase et al., 1987; Lelbach, 1996).
Table 38. Number of vinyl chloride-associated ASL cases
reported per country in 1972
Country ASL cases ASL cases ASL cases up to 1999
up to 1985a up to 1993b [changes since 1993] c
USA 35 44 50 [+6]
Germany 26 (West) 41 40 [-1]
France 18 28 31 [+3]
United Kingdom 9 20 21 [+1]
Canada 10 13 13
Croatia 4 4 12 [+8]
Slovakia 2 2 6 [+4]
Italy 4 8 8
Sweden 5 5 5
Japan 2 3 4 [+1]
Belgium 2 2 2
Norway 1 1 1
Spain 1 1
Australia 1 1
Brazil 1 [+1]
Israel 1 [+1]
Total d 118 173 197
a From: Forman et al. (1985)
b From: Lee et al. (1996)
c Association of Plastics Manufacturers in Europe (1999)
d It should be noted that from several countries known
to be producers of PVC, there is no information regarding
ASL cases.
Any treatment is generally unsuccessful and survival after
diagnosis usually averages less than 12 months. Hepatic failure and
intra-abdominal haemorrhage are the usual terminal effects (Riordan et
al., 1991; Lee et al., 1996). Liver transplantation might be the only
chance of survival (Hayashi et al., 1990). If ASL is detected early
enough, surgical resection and adjuvant chemotherapy is a possibility;
subsequent hepatic recurrence could be treated by radiation therapy
and chemoembolization (Paliard et al., 1991; Neshiwat et al., 1992;
Hozo et al., 1997).
b) Results of epidemiological studies
In addition to the index tumour, ASL, several case series have
been published on VC-exposed workers who had liver tumours other than
ASL, in particular hepatocellular carcinoma (HCC) (Koischwitz et al.,
1981; Evans et al., 1983; Dietz et al., 1985; Pirastu et al., 1990,
1991; Lelbach, 1996; Makita et al., 1997; Saurin et al., 1997). In the
USA study, statistically significantly elevated relative risk was
observed for the cancers of liver and biliary tract (SMR 641, CI
450-884). Mortality was related to the duration of exposure (SMR 182,
1235, and 1284 for workers with exposure duration of < 10, 10-20 and
> 20 years, respectively), and to the latency period (SMR 386, 590,
and 1218 for latency periods of < 20, 20-30 and > 30 years,
respectively. The risk was higher for workers hired before 1950 (SMR
780) than in for those hired 1950-1959 (SMR 440) or since 1960 (SMR
419). The risk was highest among workers hired at an early age (SMR
1611, 813 and 346 for those hired at < 25, 25-34 and > 35 years of
age. The authors noted, however, that those hired early and young were
likely to have longer duration of exposure and latency.
A total of 15 angiosarcomas were recorded on the death
certificates. Although no expected numbers were calculated, this must
be in a very marked excess, since the annual incidence of liver
angiosarcoma is approximately 2 per 10 million, which would in this
cohort lead to an expected number of approx. 0.05. The authors noted
that, when the 15 angiosarcomas were excluded, there remained 22 other
liver and biliary tract tumours; this represents an excess (5.7
expected, SMR 386, P < 0.02). This is likely to be an overestimate,
since some of these other liver cancers were likely to be
angiosarcomas.
In the European study, the SMR for liver neoplasms was 286 (95%
CI 183-425, and it increased with increasing latency (SMR 0, 253, 388
and 561 for latencies < 10, 10-19, 20-29 and > 30 years,
respectively), duration of exposure (SMR 94, 327, 310, 714 and 1111
for duration of employment of < 10, 10-14, 15-19, 20-24 and
> 25 years, respectively), ranked level of exposure, and cumulative
exposure (SMR 348, 400, 1429 and 1667 for < 2000, 2000-6000,
6000-10 000 and > 10 000 ppm-years, allowing for a 15-year
latency). A clear exposure-response relationship was seen between
ranked level of exposure (ppm) as well as cumulative exposure
(ppm-years) to VC and liver cancer mortality, with autoclave workers
having the highest risk (Table 41; Simonato et al., 1991). This
relationship is even more evident taking only those liver cancers
defined histologically as ASL. In addition, for those VC workers not
defined as autoclave workers, an exposure-response relationship could
be seen for those diagnosed as having ASL and non-ASL liver cancers
(Pirastu et al., 1990, 1991; Simonato et al., 1991).
Table 39. Summary of findings on selected neoplasms for the epidemiological
studies on workers exposed to VC
Cause of European/IARC USA study Weber et Smulevich et Thériault Laplanche All
mortality study (Simonato (Wong et al. (1981) al. (1988) & Allardt et al. studies d
et al., 1991) al., 1991) (1981) (1992)
Obs/Exp Obs/Exp Obs/Exp Obs/Exp Obs/Exp Exposed/ Obs/Exp
SMR SMR SMR SMR SMR Non-expos. RR SMR
(95% CI) (95% CI) (95% CI) (95% CI) (95% CI) (95% CI) (95% CI)
All causes 1438/1636.4 1536/1705.27 414/434.7 - 59/71.07 40/43
88 90 95 83 1.0
(83-93) (86-95) (0.6-1.5)
All malignant 445/427.8 359/341.7 79/82.9 63/58.88 20/16.37 966/927.65
neoplasms 104 105 103 b 107 122 1.3 104
(95-114) (94-116) (0.7-2.3) (98-111)
Liver cancer/ 24/8.4 37/5.77 12/0.9 0/n.a 8/0.14 3 81/19.21
angiosarcoma 286 641 1523 5714 533
(ASL) (183-425) (450-884) 8 ASL (+2 ASL 3 ASL (423-622)
16 ASL out of 15 ASL in undiagnosed)
17 liver cancer death
deaths with certificates.
histopathology 21 ASL in
international
register
Brain 14/13.1 23/12.76 2/1.3 4/2.61 0/0.6 43/30.37
107 181 162 153 0 142
(59-180) (114-271) (103-191)
Lung 144/148.3 111/115.87 24/26.6 1/1.2 2/5.78 c 282/297.75
97 96 96 83 34 95
(82-114) (79-116) (84-106)
Table 39.
Cause of European/IARC USA study Weber et Smulevich et Thériault Laplanche All
mortality study (Simonato (Wong et al. (1981) al. (1988) & Allardt et al. studies d
et al., 1991) al., 1991) (1981) (1992)
Obs/Exp Obs/Exp Obs/Exp Obs/Exp Obs/Exp Exposed/ Obs/Exp
SMR SMR SMR SMR SMR Non-expos. RR SMR
(95% CI) (95% CI) (95% CI) (95% CI) (95% CI) (95% CI) (95% CI)
Lymphatic and 29/32.7 37/36.28 15/7.7 10/2.2 1/1.67 92/80.55
haematopoietic 89 102 214 454 60 114
(72-141) (92-140)
Lymphomas 18/19.3 a 24/21.8 a 5/1.2
93 110 417
Stomach 49/45.1 10/16.01 18/14.4 21/24.7 - 98/100.21
109 63 138 85 98
(80-144) (30-115) (79-119)
a Lymphoma and malignant myeloma
b As reported in original paper
c All respiratory neoplasms
d Calculated by the Task Group. All studies except Laplanche et al. (1992) who did not provide observed and expected values
Table 40. Analysis of liver, brain and lung cancer mortality in the USA
and European cohort by duration of exposure
Organ Study < 10 years 10-19 years 20 + years
SMR (observed) SMR (observed) SMR (observed)
Liver USA (Wong 182 (6) 1236 (20) 1285 (11)
et al., 1991)
Europe (Simonato 0 (0) 253 (8) 432 (16)
et al., 1991)
Brain USA 165 (13) 121 (4) 386 (6)
Europe 59 (2) 113 (6) 136 (6)
Lung USA 93 (59) 114 (37) 75 (15)
Europe 95 (73) 107 (51) 83 (20)
Of the 17 liver cancer deaths, for which histopathological data
were available, 16 were classified as angiosarcomas. For the remaining
seven, the histological type remained unknown. The Task Group noted
that even if they all had been cancers other than angiosarcoma, the
relative risk of liver tumours other than angiosarcoma would not have
been elevated (expected number equiv. 8).
For 2643 workers from Norway and Sweden, cancer incidence
information was also available. The only site for which a
statistically significantly elevated risk was observed was the liver
(SIR 303, CI 122-623).
In the Canadian study, there was an excess of digestive tract
cancers (14 cases, SMR 259, P < 0.01) that was fully accounted for
by cancers of the liver (8 cases). All liver cancers were
angiosarcomas.
Hospital admission diagnoses were studied among 714 present and
1575 previous workers, identified from national Labour Insurance
Bureau records, with exposure to VC (Du & Wang, 1998). The frequencies
of different diagnoses were compared with similar data on workers in
the manufacture of optical instruments or of motorcycles. Eight cases
of primary liver cancer were observed among the VC-exposed workers
(out of 1044 admissions), while the number was 9/3667 for the optical
workers, and 9/5861 for motorcycle manufacturers, which gave 4.5 (95%
CI 1.5 to 13.3) and 6.5 (2.3-18.4) as age-adjusted morbidity odds
ratios (MOR), calculated from a linear regression model, for the
VC-exposed workers, compared to the two comparison groups. Increased
MORs (of borderline significant) were also observed for haematopoietic
cancer, chronic liver disease and liver cirrhosis, as well as other
chronic diseases, and accidents.
Two updates of parts of the Italian cohort (Pirastu et al., 1991)
have recently appeared. An elevated mortality of liver cancer (11
observed, 5.7 expected cases, SMR 193) was observed in Marghera. In an
extended survey for hepatic cancers, four further cases were
uncovered. There were 5 angiosarcomas, 5 hepatocellular carcinomas, 3
cirrhotic carcinomas, and in two cases the histology was not known
(Pirastu et al., 1997). In three other subcohorts (Pirastu et al.,
1998), the pooled SMR for liver cancer was 364 (7 observed cases, 90%
CI 108-390).
A survey of 5291 workers from 13 PVC manufacturing plants in 12
cities in China using 6276 workers unexposed to VC as controls was
carried out between 1982 and 1989. Although no cases of ASL were
reported, the mortality from liver cancer in male workers exposed to
VC was significantly higher than those of the control group and of the
male general population in the middle cities in China. Among malignant
tumours, liver cancer ranked first in exposed males. The authors found
that the average age for persons who were diagnosed with liver cancer
was significantly lower than that of the control group (Huang,
1993a,b).
Table 41. Mortality data for liver cancer according to the exposure
variables a
Exposure variable O SMR 95% CI 15 years of latency
SMR 95% CI
Job title
Ever autoclave worker 11 896 447-1603 1358 678-2430
Never autoclave worker b 13 181 97-130 284 151-485
Duration of
employment (years)
1-9 4 94 26-239 205 56-525
10-14 5 327 106-763 602 196-1406
15-19 4 310 84-794 310 84-794
20-24 6 714 262-1555 714 162-1555
> 25 5 1111 361-2593 1111 361-2593
Ranked level of
exposure (ppm)
Low (< 50) 3 (4) 119 25-347 227 (244) 47-664 (67-625)
Intermediate (50-499) 3 (7) 161 33-471 250 (551) 52-731 (222-1136)
High (> 500) 12 (12) 567 293-991 719 (719) 371-1255 (371-1255)
Unknown 6 (1) 317 177-691 486 (125) 182-1079 (3-697)
Cumulative exposure
(ppm-years)
0-1999 4 (9) 99 27-254 191 (348) 52-490 (159-662)
2000-5999 4 (4) 351 96-898 460 (400) 125-1177 (109-1024)
6000-9999 4 (7) 800 218-2048 851 (1429) 232-2179 (574-2943)
> 10 000 3 (3) 1429 295-4175 1667 (1667) 344-4871 (344-4871)
Unknown 9 (1) 357 163-678 536 (100) 245-1017 (2-557)
Table 41. (cont'd)
Whole cohort 24 286 183-425 445 285-663
a From: Simonato et al. (1991); O = observed number of deaths;
SMR = standardized mortality ratio; Cl = confidence interval.
The values in parentheses were determined in analyses including
the estimated job-exposure matrices
b In Norway, the longest-held job was used. In Sweden job rotation
was practiced, and no one was classified as an autoclave worker
8.3.3.2 Brain and central nervous system (CNS)
Statistically significant increases of brain and CNS tumours
after occupational exposure to VC were reported in USA surveys by
Waxweiler et al. (1977) and Cooper (1981), as well as in Sweden (Byrén
et al., 1976) and Germany (Greiser et al., 1982).
In the USA (study of Wong et al., 1991), the relative risk was
elevated for cancers of the brain and other CNS cancers (SMR 180.2, CI
114.1-270.6). Mortality was highest in the group with the longest
duration of exposure (SMR 164.7, 120.8 and 385.9, for workers with the
duration of exposure < 10 years, 10-20 years and > 20 years,
respectively), while no trend with latency was observed (SMR 183.8,
158.3 and 210.7, for latencies of < 20, 20-30 and > 30 years).
Workers hired early did not exhibit higher risk than those hired late
(SMR 156, 164 and 256, for employees hired before 1950, 1950-1960 and
after 1960, respectively. It was noted that most of the cases came
from the two plants with the highest rate of ASL.
In the European study, the SMR for brain cancer was 107
(CI 59-180). The small number of cases (total 14) made the assessment
of the relationship between duration of exposure and latency
difficult, but the risk was highest after longest latency (SMR 59,
113, 59 and 407 after a latency of < 10, 10-20, 20-30 and
> 30 years) and after longest duration of exposure (SMR 106, 78 and
183 for a duration of exposure of < 10, 10-20 and > 20 years).
There were even fewer cases for the assessment of the relationship
between estimated exposure and cancer risk (8 cases altogether), but
the point estimates were higher for higher estimated exposures (SMR
91, 42 and 128 for low, intermediate and high exposure), while little
covariation between estimated cumulative exposure and relative brain
cancer risk was observed (SMR 0, 162 and 120 for cumulative exposures
of < 50, 50-499 and > 500 ppm-years).
In the cancer incidence aspect of the European study, a
non-significantly elevated SIR was observed for cancer of the brain
(SIR 159 CI 68-312).
8.3.3.3 Respiratory tract
The study by Monson et al. (1974) first suggested an association
between VC and lung cancer.
In the USA study, the SMR for lung cancer was 95.8
(CI 78.7-115.5).
In the European study, the SMR for lung cancer was 97
(CI 82-114). It did not show any consistent relationship with duration
of employment, latency, ranked level of exposure or cumulative
exposure; the SMR was similar for ever- and never-autoclave workers.
In the cancer incidence aspect of the European study, a
non-significantly elevated SIR was observed for cancer of the lung
(SIR 152, CI 95-230).
In the Canadian study, the mortality from respiratory cancer was
low (relative risk in comparison to the unexposed cohort 0.36, and SMR
based on Quebec data, 34, based on two cases). Smoking habits, as
studied by a questionnaire, were similar in the exposed and
non-exposed cohorts.
8.3.3.4 Lymphatic and haematopoietic cancers
There were indications of increased risk of cancer of the
lymphatic and haematopoietic systems in several early studies (Monson
et al., 1975; Waxweiler et al., 1976; Greiser et al., 1982; Smulevich
et al., 1988).
In the Wong et al. (1991) USA study, the SMR for lymphatic and
haematopoietic cancer was 102 (CI 71.6-140.7, while that for lymphoma
and reticulosarcoma was 102.0 (CI 71.6-140.7).
In the European study, the overall SMR for lymphosarcoma was 170
(CI 69-351), based on seven cases. No cases were observed in the
groups with a latency of 20-30 or > 30 years, and six out of the
seven subjects had worked less than 10 years in VC-exposure
conditions.
8.3.3.5 Malignant melanoma
There was a relationship between VC exposure and malignant
melanoma of the skin in PVC production workers in the early Norwegian
study (Heldaas et al., 1984, 1987).
In the European study, a non-significant, elevated risk was
observed for melanoma, and the risk was higher in the incidence part
of the study (SIR 184, CI 79-302). However, the risk was limited to
one country only (Norway).
8.3.3.6 Breast cancer
Infante & Pesák (1994) noted that there had been no follow-up
study to that of Chiazze et al. (1977), who observed an excess in
breast cancer mortality. Although a subsequent study among these
researchers (Chiazze et al., 1980) did not identify a significantly
elevated odds ratio in relation to VC exposure, the statistical power
used in the study has been questioned (Infante & Pesák, 1994). There
have been no other reports on breast cancer in humans, but in most
Western countries women have not been working in jobs involving VC
exposure. In some countries, however, e.g., China and Eastern Europe,
women are/were probably exposed to higher VC levels than in Western
countries (see Table 21) but epidemiological studies are not
available. Mammary carcinoma has been reported in animal studies with
inhalative and oral exposure to VC (see section 7.7.2).
No breast cancer was observed in the study by Smulevich et al.
(1988).
8.3.3.7 Other cancer sites
In the European study, a non-significantly elevated risk was also
observed for urinary bladder (21 deaths observed, SMR 146, CI 91-224)
and, in the incidence part, for cancer of the stomach (SIR 150,
CI 80-256).
8.4 Genotoxicity studies
8.4.1 Cytogenetic studies of VC-exposed workers
Table 42 summarizes the results of cytogenetic studies of
chromosomal aberrations (CA), micronucleus formation (MN) and
sister-chromatid exchanges (SCEs) in the peripheral blood lymphocytes
of VC workers compared to controls. Studies on the genotoxicity of VC
have been reviewed (Uzych, 1988; Giri, 1995). Although in many studies
the exposure concentrations and duration of exposure were only
estimated, a dose-response relationship and a "normalization" of
genotoxic levels with time after reduction of exposure can be seen.
There was a clear relationship between incidence of CA and the
exposure concentrations/type of work (Purchase et al., 1978). The same
groups of workers showed lower or no effects after the exposure
concentration was reduced to < 13 mg/m3 (< 5 ppm) (Hansteen et al.,
1978; Anderson et al., 1980; Fucic et al., 1996). That such levels do
not cause an increase in CA was confirmed by several studies (Picciano
et al., 1977; Rössner et al., 1980; de Jong et al., 1988).
SCEs increased with the level of exposure to VC of workers
(Sinués et al., 1991). SCEs were generally not detected in blood
samples from workers exposed to VC at < 5 ppm.
In a micronucleus assay, workers exposed to VC had a much higher
frequency of micronucleated lymphocytes than the controls. However, a
significant decrease in the number of micronucleated lymphocytes was
found within 15 days after the last exposure, but even 90 days after
the last exposure (peak of 780 mg/m3) the frequency, although
decreased, was still above the control value (Fucic et al., 1994). A
similar effect had been shown with SCE frequencies (Fucic et al.,
1992).
Chromosomal aberrations were measured in peripheral blood
lymphocytes from 29 subjects potentially exposed to VC after an
accidental release into the environment (Hüttner & Nikolova, 1998).
The control group consisted of 29 non-exposed people, matched
regarding age, gender and smoking habits. The exposed group showed a
statistically significant increase in the mean frequency of aberrant
cells (1.47% versus 1.07% in the control group).
Table 42. Chromosome analysis in human T-lymphocytes (in vivo)
after vinyl chloride exposure during work at PVC polymerization
plants or due to other accidental exposure a
Test No. workers; Additional Duration Exposure Result Reference
No. controls information of exposure level
(years) (mg/m3)
HPRT mutation 24; 23 worked on post- average 7 average < 13 - Hüttner &
using T-cell chlorination of PVC Holzapfel (1996)
cloning assay in an open system
where free VC
evaporated
DNA single 16 3 groups of VC/PVC average 13 average 150 - Du et al. (1995)
strand breakage 104 workers average 16 5 -
122 average 16 2 -
Chromosomal 11; 10 PVC workers 4-28 peaks >1300 + Ducatman et
aberrations (average 15) al. (1975)
7; 3 9-29 50-75 + Funes-Cravioto
et al. (1975)
45; 93 0.5-12 n.g. + Szentesi et al. (1976)
57; 24 VC/PVC workers 10.7 (average 10-100 + Purchase et al. (1978)
classified according for autoclave
21; 6 to type of work workers) < 13 + Anderson et al. (1980)
23; 8 (1974) < 13 -
same as Purchase
et al. (1978)
Chromosomal 20; 20 with clinical + Fleig & Thiess (1974,
aberrations symptoms; without - 1978)
clinical symptoms
39; 16 1974 > 10 > 65 + Hansteen et al. (1978)
37; 32 same workers 1977 2.6 -
3; 9 10-27 50-400 + Kucerová et al. (1979)
37; 12 n.g. 2-111 + Katosova & Pavlenko (1985)
43; 22 average 11.2 2-41 + Hrivnak et al. (1990)
Table 42. (cont'd)
Test No. workers; Additional Duration Exposure Result Reference
No. controls information of exposure level
(years) (mg/m3)
19; 20 average 15 130; peaks of + Fucic et al. (1990b)
67; n.g. breaks occurring at average 15 5000 13; peaks + Fucic et al. (1990a)
non-random sites of 5000
10; n.g. 8-25; av 13; short + Garaj-Vrhovac et al. (1990)
average 15 peaks higher
n.g PVC workers 5 26 + Zhao et al. (1994)
PVC workers 2 26 -
PVC workers > 7 4 -
residents on site > 8 0.2 -
209; 295 3 groups average 4 < 2 to > 13 - Picciano et al. (1977)
31; 35 1977 2-3 < 10 - Rössner et al. (1980)
same group 1978 peak < 30 -
66; 39 1976-1977 1-11; average 6 3 peaks up - de Jong et al. (1988)
29; 29 exposure of general for short time to 26 unknown + Hüttner & Nikolova
population after (approx. 1 h) concentration (1998)
accidental release
of VC
Micronucleus 19; 20 average 15 130; peaks of 5000 +Fucic et al. (1990b)
assay 32 24 h to 90 days 10 peak of 780 + Fucic et al. (1994)
following a peak 5 mg/m3 every +
exposure of 3 months
780 mg/m3
Table 42. (cont'd)
Test No. workers; Additional Duration Exposure Result Reference
No. controls information of exposure level
(years) (mg/m3)
Sister-chromatid 9; 8 10-27 50-400 + Kucerová et al. (1979)
exchange 16, 16 > 10 2.6 - Hansteen et al. (1978)
21; 6 3.5 < 13 (-) Anderson et al. (1981)
31; 35 2-3 < 10 - Rössner et al. (1980)
31; 41 > 2 3-44; peaks of + Sinués et al. (1991)
21; 41 130
1-20
15; 10 1.5-35 13; peaks of + Fucic et al. (1992)
5000
0.3; peaks of - Fucic et al. (1996)
130
a HPRT = hypoxanthine guanine phosphoribosyltransferase; n.g. = not given
8.4.2 Mutations at the hypoxanthine guanine phosphoribosyltransferase
(hprt) locus
With the use of the hprt lymphocyte clonal assay it is possible
to determine the mutation frequency of the hprt gene and to
characterize its mutant spectra. Induced mutagenesis in the
lymphocytes of PVC production workers was measured by selecting mutant
T-cells with the hprt gene in medium containing 6-thioguanine. The
exposed workers had similar mutation frequencies (about 8 × 10-6) as
controls (Hüttner & Holzapfel, 1996).
The types and frequencies of mutations caused by VC in a human
B-lymphoblastoid line expressing cytochrome P-450 2EI (H2E1 cells)
were investigated by Chiang et al. (1997). VC was found to be toxic
and mutagenic to H2E1 cells as a function of incubation time when they
were exposed to 7.5% VC in air. This exposure resulted in 75% survival
and an hprt mutant frequency of 42 × 10-6 after 48 h, compared to
6 × 10-6 for unexposed cells. Exposure to 0.8 to 15.0% VC in air
produced similar mutation frequencies but without a clear
dose-response relationship, perhaps due to saturation of metabolic
activation. Ten percent (5/50) of VC-induced mutations showed
detectable deletions. Both CEO and CAA showed dose-dependent increases
in cell killing and mutant frequency. VC and CEO displayed similar
toxicity/mutation profiles and a similar frequency of large deletions,
whereas CAA showed greater toxicity and a larger frequency of deletion
mutations.
8.4.3 Mutations in ASL from VC-exposed workers
8.4.3.1 p53 gene
Normal and cancer cells appear to differ through discrete changes
in specific genes controlling proliferation and tissue homoeostasis.
The p53 tumour suppressor gene is present in every cell of the human
body and is located on the short arm of the human chromosome 17 p.
(Lane, 1994). It is mutated in about half of all cancer types arising
from a wide spectrum of tissues (Harris, 1996).
The encoded wild-type p53 protein, a 393 amino acid nuclear
phosphoprotein, activates the transcription of several genes,
including some involved in cell cycle control (e.g., p21), and has
been implicated in DNA repair processes, DNA replication and apoptosis
(Lane, 1994; Semenza & Weasel, 1997). The majority of cancer-related
mutations in p53 cluster in several "hot-spot" regions of the
protein that have been highly conserved through evolution. These
regions occur in the sequence-specific DNA-binding core domain of the
protein between amino acid residues 102 and 292 (Lane, 1994). The
mutations in p53 found in malignancies could thus result in
substitutions of amino acid residues in these regions that are
critical for the determination of its structure and function (Cho et
al., 1994; Brandt-Rauf et al., 1996).
The p53 gene was examined in tumours from four highly exposed
(before 1974) autoclave workers with ASL and one other VC worker with
hepatocellular carcinoma (HCC) (Hollstein et al., 1994). Two A to T
mutations in a highly conserved domain of the coding sequence (codon
249: AGG to TGG, Arg to Trp; and codon 255: ATC to TTC, Ile to Phe)
were found one each in the tumour but not in other normal cells of two
of the ASL patients (both smokers). A further A to T missense p53
mutation (codon 179: CAT to CTT) was found in a cell line from a
further VC-associated liver tumour patient (Boivin et al., 1997; see
Table 43). Such mutations are uncommon in human cancers (2.7% of a
total of 5085 cancers and 8% of a total of 290 primary liver cancers;
Hollstein et al., 1996). Furthermore, p53 mutations are uncommon in
sporadic (non-VC-induced) ASL (2/21 cases, 9%), supporting the
evidence linking VC exposure and ASL containing an increased frequency
of p53 mutations with a mutational spectrum (A to T) (Soini et al.,
1995). To date (1998) 11 out of 15 (73%) ASL from VC-exposed workers
have been shown immunohistochemically to have mutant p53 protein.
Further, a statistically significant trend for mutant p53 protein has
been found in the serum of VC-exposed workers (Smith et al., 1998);
this study is continuing (Li et al., 1998a,b).
p53 gene mutations were also found in 11 out of 25 (44%) ASL in
Sprague-Dawley rats induced by VC and 1 in 8 HCCs (Barbin et al.,
1997; see Table 43). This is a higher mutation rate than that found in
humans but again the majority of missense mutations involved were A:T
base pairs. The A:T -> T:A transversion observed in the first
nucleotide of codon 253 in two rat ASL is equivalent to the same
transversion characterized previously in codon 255 in one human ASL
associated with VC exposure (Trivers et al., 1995).
8.4.3.2 ras genes
The ras gene family, Ha- ras, Ki- ras and N- ras, are genes
coding for p21 and are frequently activated by point mutations in
codons 12, 13 and 61. These activated genes seem to play a key role in
the development of spontaneous or carcinogen-induced mammalian tumours
(Barbacid, 1987). In a study of ras oncogene mutations in tumours of
VC-exposed workers, 15 of 18 ASLs were found to contain a G to A
transition at the second base of codon 13 (GGC to GAC) of the
Ki- ras-2 gene (Marion et al., 1991, 1996; see Table 44). This
mutation leads to the substitution of glycine by aspartic acid at
amino acid residue 13 in the encoded p21 protein.
Ki- ras-2 gene mutations were found in codon 12 in 5/19 sporadic
ASL and in 2/5 thorotrast-induced ASL in humans. All seven mutated
tumours contained a G -> A transition at base 2. In addition, 4 of
these tumours (3 sporadic, 1 thorotrast-induced) contained a second
mutation in the Ki- ras gene, a G -> T transversion of the first
base of codon 12 (Przygodzki et al., 1997).
Table 43. Comparison of mutation spectra in the p53 gene in
liver tumours in humans and rats a
Species Tumour origin No. mutations/ No. and types References
no. of cases of mutations
Humans VC-associated 3/6 3 A:T -> T:A Hollstein
ASL et al. (1994)
cells cultured [CAT -> CTT] Boivin
from (codon 179) et al. (1997)
VC-associated
liver tumour
Rats VC-associated 11/25 5 A:T -> T:A Barbin et al.
ASL 2 A:T -> G:C (1997)
2 A:T -> C:G
3 G:C -> A:T
one 12 base-pair
deletion
1 deletion
HCC 1/8 1 A:T -> T:A
a Adapted from Barbin et al. (1997); ASL = angiosarcoma of the liver;
HCC = hepatocellular carcinoma
Table 44. Mutagenesis of ras proto-oncogenes in VC-associated
liver tumours in humans and rats
Tumour Gene Codon No. Base pair References
origin a involved mutations/ change/
No. tumours Codon
change
Human Ki-ras 2 Codon 15/18 G -> A/ Marion et al.
ASL gene 13 GGC -> GAC (1991, 1996);
DeVivo et al.
(1994)
Rat Ki-ras 2 0/10 Froment et al.
ASL gene (1994);
Boivin-Angèle
et al. (in press)
Rat Ha-ras Codon 5/8 AT -> TA/ Froment et al.
HCC gene 61 AT -> TA (1994);
Boivin-Angèle
et al. (in press)
a ASL = angiosarcoma of the liver; HCC = hepatocellular carcinoma
In studies in VC-induced ASL and HCC tumours in rats, no
mutations were found in Ki- ras genes but there were mutations at
codon 61 of the Ha- ras proto-oncogene (Froment et al., 1994;
Boivin-Angèle et al., in press).
8.5 Studies on biological markers
8.5.1 Excretion of metabolites
The concentration of the VC metabolite thiodiglycolic acid in the
urine has been found to correlate with VC exposure in workers and has
been suggested as a biological marker for VC (Müller et al., 1978)
(see also section 5.3).
8.5.2 Genetic assays
Increased levels of chromosomal abnormalities, compared to
control populations, were found in workers exposed to high levels of
VC but not in workers exposed to less than 13 mg/m3 (5 ppm) (see
Table 42). The micronucleus assay and mitotic activity have been
suggested as methods for determining recent VC exposure (Fucic &
Garaj-Vrhovac, 1997; see section 8.4).
8.5.3 Enzyme studies
Ever since the 1970s there has been disagreement as to the value
of monitoring levels of enzymes as a measure of VC exposure or as a
sign of VC disease. Liver function tests may be of limited value in
detecting VC-induced liver disease because, although hepatocytes are
the primary site of VC metabolism, they are not the target of toxicity
(Lelbach & Marsteller, 1981). Individual studies of VC-exposed
populations have reported abnormalities in one or more liver function
tests, such as aspartate aminotransferase (ASAT), alanine
aminotransferase (ALAT) and alkaline phosphatase (AP) (Lilis et al.,
1975; Waxweiler et al., 1977). However, Sugita et al. (1986) concluded
that VC probably had not affected these enzymes after adjusting for
confounding factors. This was confirmed by Du et al. (1995) who showed
that ASAT, ALAT and AP were mainly affected by the presence of
hepatitis B surface antigen (HBsAg) or anti-hepatitis C virus
(anti-HCV).
Gamma glutamyl transpeptidase (GGT) is thought to be associated
with VC exposure (Langauer-Lewowicka et al., 1983; Du et al., 1995)
and, according to the latter, this is the only enzyme that correlates
with VC exposure levels. It should be noted that GGT is readily
elevated in alcohol drinkers, so the value of this enzyme as a
biomarker of VC-induced liver injury becomes questionable. In workers
showing VC-induced liver dysfunction (VC levels 2.6-54 mg/m3; 1 to
21 ppm), ALAT was the earliest parameter raised, then serum GGT (Ho et
al., 1991). Ho et al. (1991) also reported that 10 of 12 workers with
VC-induced liver dysfunction showed improvements in liver function
tests after removal from exposure. There are case reports of
individuals whose angiosarcomas were first detected by abnormal liver
function tests (Makk et al., 1976), but liver function abnormalities
appear to be a relatively late finding (Falk & Steenland, 1998). No
published studies have been identified that evaluate the effectiveness
of medical surveillance in occupational groups with VC exposures below
2.6 mg/m3 (1 ppm).
8.5.4 von Willebrand factor
The immunoquantitation of the level of the von Willebrand factor
(vWf; factor VIII-related antigen, a large multimeric plasma
glycoprotein in the blood clotting system) in 107 VC exposed workers
was performed using an enzyme-linked immunosorbent assay (ELISA)
(Froment et al., 1991). The vWf level was slightly but significantly
higher in the VC-exposed workers than in the control group. The vWf
serum level of three patients with hepatic angiosarcoma was markedly
elevated.
8.5.5 p53 and ras proteins
Mutant p53 and p21 proteins or their antibodies can be detected
in sera from healthy workers exposed to VC (Marion et al., 1996;
Semenza & Weasel, 1997; Smith et al., 1998; Li et al., 1998a,b). A
dose-response relationship between estimated cumulative exposure and
the frequency of these onco-proteins has been reported (Table 45).
Statistically significantly elevated frequencies were observed even in
the group with lowest estimated exposure (cumulative exposure
< 40 ppm-years) (Li et al., 1998b). However, exposure estimations
were crude. In addition, similar findings have not been reported for
other VC-exposed groups.
8.6 Susceptible subpopulations
8.6.1 Age susceptibility
Evidence from animal studies suggests that there may be
early-life sensitivity to VC (section 7.7.3), but there is also
contradictory evidence. There has been concern that children living
near landfill sites might be particularly susceptible to VC formed and
released there (Hiatt et al., 1994; Cogliano et al., 1996). However,
there is no epidemiological evidence to support this.
8.6.2 Immunological susceptibility
Individual susceptibility may influence the development of VC
disease, since not all highly exposed workers developed clinical
symptoms or signs (Wilson et al., 1967; Black et al., 1986). Studies
conducted in a single population of VC-exposed workers have shown that
susceptibility for the disorder seems to be increased in the presence
of the human-leukocyte-associated antigen HLA-DR 5 or a gene in
linkage disequilibrium with it and an antigen associated with the
haplotype A1, B8, while DR3 seems to favour progression of the disease
(section 8.3.2.3e). The significance of this finding is unclear
without replication in another VC-exposed population.
8.6.3 Polymorphic genes in VC metabolism
Levels of CYP2E1 can vary considerably among individual humans
(Guengerich et al., 1991) and are a possible cause of differences of
susceptibility to VC.
El Ghissassi et al. (1995b) analysed the GSTM1 genotype in 133
Caucasian individuals who had had a high exposure to VC for several
years and had (or had not) clinical signs of chronic injury (see also
Froment et al., 1991, section 8.5.4). Of the 133 individuals, 62
workers had non-neoplastic symptoms, while 8 had liver cancer. The
frequency of GSTM1 null genotype in the whole group of exposed
workers was 61.7% (82 of 133) and 62.5% in patients with liver cancer.
In case-control studies reported by others, the frequency of GSTM1
null genotype ranged from 41 to 53% for Caucasians.
Table 45. p53 and p21 mutations (as detected by serum biomarkers analysis)
in relation to estimated vinyl chloride exposure (Li et al., 1998a)
Estimated Total no. of Both biomarkers No. of workers with No. of workers with Adjusted OR 95% CI
exposure workers negative mutation in mutation in
(ppm-years) either p53 or p21 both p21 and p53
0 43 38 5 0 1
< 500 42 20 21 1 11.1 3.3-37.5
501-2500 45 20 21 4 12.8 4.1-40.2
2501-5000 31 6 19 6 29.9 9.0-99.1
> 5000 54 15 22 17 31.2 10.4-94.2
9. EFFECTS ON OTHER ORGANISMS IN THE LABORATORY AND FIELD
9.1 Laboratory experiments
VC has been shown to be mutagenic in several in vitro and in
vivo test systems derived from organisms belonging to different
taxonomic levels. The details referring to bacterial, fungal and
mammalian cell lines or to whole organisms like insects or plants are
discussed in section 7.6. The carcinogenic effects of VC are addressed
in section 7.7. The following chapter focuses on investigations of
other signs of toxicity relevant to organisms that may be exposed to
VC in the environment. Standard tests on survival and reproduction
were not available. Care must be taken when interpreting the toxicity
results available as many were obtained from static tests using
nominal exposure concentrations. Such tests will have large losses of
VC due to volatilization, thus reducing the actual exposure to VC.
9.1.1 Microorganisms
9.1.1.1 Water
A consortium of anaerobic microorganisms (species not identified;
initially obtained from municipal sludge) was used for testing VC
toxicity. Both batch and semi-continuous assays were conducted. VC had
an inhibitory effect on the total gas production, beginning at a
concentration of 5.4 mg/litre and resulting in an EC50 value of
approximately 40 mg/litre, as seen in the batch assay over 3.5 days.
In the semi-continuous assay lasting 15 days, the threshold was
greater than 64 mg/litre (the highest concentration tested), probably
due to volatilization of VC (Stuckey et al., 1980).
The growth of five mixed bacterial populations (isolated from
natural aquatic systems) was not affected, as compared to controls, in
liquid cultures (closed flasks; 21°C; over 5 weeks) containing up to
900 mg VC/litre (Hill et al., 1976b).
The toxicity of waste effluents of a VC production plant to the
green alga Chlorella sp. was tested before and after the wastes were
treated by neutralization procedures (addition of aqueous solutions of
NaOH and catalysts). The crude effluents consisted of VC (15-18%,
weight), di-, tri- and pentachloroethanes (45%), dichloroethylene
(27%), dichloropropane (6%), ethyl chloride (1%) and unidentified
substances (1%). This mixture led to a weak inhibition of algal growth
(measured by changes in optical density at 678 nm), with a 72-h EC50
value of 1495 mg/litre (which corresponds to a VC concentration of
224 mg/litre). It should be noted that other compounds present in the
effluent may also have contributed to the toxicity. The corresponding
EC50 values of the neutralized waste samples (filtrates and extracts
of precipitates; composition not analysed) ranged from 4000 to
> 100 000 mg/litre (Demkowicz-Dobrzanski et al., 1993).
9.1.1.2 Soil
Soil (aquifer) microcosms enriched for methanotrophic activity
transformed up to 90% of the 1-17 mg/litre influent VC with no
apparent toxic effects (Dolan & McCarty, 1995a,b). However, when both
VC and 1,1-dichloroethylene were present, about 75% less
transformation of VC and a marked decrease in methane oxidation rate
was observed (Dolan & McCarty, 1995a). Toxic effects, measured as
decreased methane uptake, were seen during degradation of VC (VC
concentrations: up to saturated solutions; solubility: 2.7 mg/ml) by a
culture of mixed methanotrophs (seeded with soil from a defunct
landfill). A mixture of VC and trichloroethylene (TCE) and a triple
mixture of VC, cis-dichloroethylene (c-DCE) and TCE showed cumulative
toxicity (Chang & Alvarez-Cohen, 1996).
The nitrifying soil bacterium Nitrosomonas europaea had a
turnover-dependent loss of (ammonia-dependent) O2 uptake activity
after co-metabolic transformation of VC (Rasche et al., 1991).
9.1.2 Aquatic organisms
9.1.2.1 Invertebrates
VC reduced the population doubling time of a ciliated protozoon,
Tetrahymena pyriformis, population cultured in vitro (Sauvant et
al., 1995). The IC50 value was 540 mg/litre (8.6 mmol/litre).
During a 96-h assay VC had no effect on the survival of the
free-living nematode Panagrellus redivivus at concentrations ranging
from 10-3 to 10 -8 mol/litre (0.6-62 500 µg/litre), but it reduced the
developmental success of this species. The molting rate from the
fourth larval stage to adult (determined on progeny of 150 to 300
gravid females per assay; three replications) was significantly
decreased relative to controls, and that primarily at VC
concentrations of 6.3-6300 µg/litre (Samoiloff et al., 1980).
The effects of waste effluents from a VC plant described in
section 9.1.1.1 (test with Chlorella sp.) were also tested with the
crustacean Daphnia magna (n = 3 × 10 per experiment; age: 6-24 h).
The test parameter for the latter was lethality, which was determined
by procedures based on ISO 6341-1982 (ISO, 1989). Crude waste produced
a 24-h LC50 value of 80.7 mg/litre (related to VC: 12 mg/litre). The
neutralized wastes (filtrates and extracts of precipitates) were less
toxic, reflected by the higher LC50 values ranging from 445 to
> 100 000 mg/litre (Demkowicz-Dobrzanski et al., 1993).
9.1.2.2 Vertebrates
The acute toxicity of VC to fish was examined with a few
freshwater species; 96-h LC50 values of 1.22 g VC/litre and 1.06 g
VC/litre were reported for bluegill (Lepomis macrochirus) and
largemouth bass (Micropterus salmoides), respectively, but without
giving further details (Hann & Jensen, 1974). Brown et al. (1977)
exposed Northern pikes (Esox lucius) to 388 mg VC/litre; 10 days
after exposure all test animals (n=15) were dead (versus 1 of 20 in
controls during 120 days of observation). However, the test conditions
(e.g., handling of controls, water quality) were not sufficiently
described in this study. Tests of zebra fish ( Brachydanio rerio;
n = 10 per group) according to OECD guideline 203 (OECD, 1984), which
was adapted to volatile chemicals, resulted in LC50 values (based on
mean measured test concentrations) of 240 mg/litre (24 h) or
210 mg/litre (48 h, 72 h and 96 h). The no-observed-effect
concentration (NOEC) regarding mortality was 128 mg/litre (Groeneveld
et al., 1993).
Estimated benchmark values, concentrations believed to be
non-hazardous, to freshwater fish derived by several methods ranged
from 87.8 to 28 879 µg/litre (Suter, 1996). The Tier II secondary
acute value and secondary chronic value were reported to be
1570 µg/litre and 87.8 µg/litre, respectively. The lowest chronic
benchmark for fish was estimated to be 28 879 µg/litre, with a
corresponding fish EC20 of 14 520 µg/litre.
9.2 Field observations
9.2.1 Aquatic organisms
A field study of benthic invertebrates (Dickman et al., 1989;
Dickman & Rygiel, 1993) was carried out during 1986-1991 at 15 sites
of the Niagara River watershed (Canada) near a PVC plant. The VC
discharge in 1986 was estimated to be more than 32 kg (accompanied by
unknown quantities of organotin). The density and diversity of several
invertebrate groups (Amphipoda, Culicoidae, Chironomidae, Isopoda,
Hirudinea, Oligochaeta, Gastropoda, Trichoptera) was found to be low
as compared to a reference site. Chironomids (Diptera) turned out to
be the best indicators. They were absent at the sampling site closest
to the factory's discharge pipe, and their numbers increased with
increasing distances. Those collected nearest to the discharge site
primarily belonged to the genus Polypedilum, which is known to be
pollution tolerant. Nevertheless, individuals of this genus showed a
high proportion of larval mentum (labial plate) deformities (38%
versus 1.7-5.7% at reference sites). Altogether, the frequencies of
deformities at the discharge site fell significantly (P = 0.05) from
47% measured in 1986-1989 to 25% in 1991, which correlated with the
lower levels of VC (figures not given) being released into the river
in 1990-1991 (Dickman & Rygiel, 1993).
10. EVALUATION OF HUMAN HEALTH RISKS AND EFFECTS ON THE ENVIRONMENT
10.1 Evaluation of human health effects
10.1.1 Hazard identification
10.1.1.1 Non-neoplastic effects
a) Human data
i) Skin, connective tissue and bone
Among the non-neoplastic effects of vinyl chloride (VC) exposure,
disorders of the skin, connective tissue and bone are the most
specific and well-characterized. These include acroosteolysis,
Raynaud's phenomenon and sclerodermoid skin lesions. These conditions
were quite common in early studies, which may have involved exposures
to high levels of several hundred ppm. Acroosteolysis occurred
primarily in PVC production workers who had been involved in reactor
cleaning. Data on mortality from connective tissue disorders, a
relatively rare category of death, have not been provided in published
studies, nor are there sufficient data to estimate an
exposure-response relationship.
ii) Non-neoplastic liver disease
Non-neoplastic liver disease was also well-documented in early
studies of workers with high levels of exposure to VC. In one
carefully described study, "advanced portal hypertension" with
histological findings of non-cirrhotic fibrosis was diagnosed in 17
out of 180 VC polymerization workers. Histological alterations
reported in liver biopsy specimens obtained from VC monomer workers in
another series included focal hepatocytic hyperplasia and focal mixed
hyperplasia; these lesions were infrequent in the comparison group.
Reversible changes in liver function tests have been reported in VC
workers exposed to 2.6-54 mg/m3 (1-21 ppm). Despite evidence for the
induction of non-malignant liver disease from clinical studies, there
has been no evidence of excess mortality from studies of large
cohorts. There are also insufficient data to estimate exposure
response.
iii) Respiratory disease
There is some evidence for respiratory effects from both
morbidity and mortality studies of VC workers, but this may be related
to PVC-resin dust rather than VC monomer. Positive findings from
mortality studies are limited to one cohort. Therefore, the literature
does not clearly implicate respiratory disease as an effect of
exposure to VC monomer.
iv) Reproductive effects
Although animal studies implicate the reproductive system as a
potential target organ for vinyl chloride toxicity, studies in humans
have not been adequate to confirm these effects or to rule them out.
v) Cardiovascular disease
A few morbidity studies have reported an elevated incidence of
circulatory diseases among VC-exposed workers. Cardiovascular disease
mortality is significantly lower than expected in the two large
cohorts.
vi) Susceptible subpopulations
There is no strong evidence for the existence of susceptible
sub-populations in humans. It has been suggested that specific HLA
types and genetic polymorphisms in VC-metabolizing enzymes may be
factors in human susceptibility to VC-related diseases.
b) Experimental animal data
VC has been tested in several animals species and strains for
acute, short-term and long-term effects. Signs of acute toxicity
include congestion of internal organs, neurotoxic effects and
circulatory disturbances. In short-term studies VC caused mainly liver
damage, degenerative changes in the testes, degeneration and
inflammation in the lungs and degenerative lesion in the kidneys.
Immunological alterations in connection with VC exposure have also
been described. Reproductive performance or fertility were not
affected.
10.1.1.2 Neoplastics effects
a) Human data
In 1974, a case report associated exposure to VC with the
occurrence of angiosarcoma of the liver (ASL). Further case series and
small epidemiological studies were reported shortly thereafter. At a
later stage two studies, one in the USA and one in Europe, combined
data from those studies and updated the mortality follow-up. Apart
from these two large studies, four more smaller studies have been
conducted and fully reported and were also considered by the Task
Group.
There is a five-fold excess risk for liver cancer among workers
exposed to VC, mostly in PVC polymerization plants where the highest
exposures to VC occurs. The largest part of this excess risk is due to
the excess risk for ASL. In the European study, there was a 45-fold
excess risk for ASL in workers exposed to more than 10 000 ppm-years
compared to those exposed to less than 2000 ppm-years. In the European
study histopathological confirmation was available for 17 out of 24
liver cancers; of those confirmed 16 were ASL. In the USA study 21 out
of 37 liver cancer deaths were registered as ASL. In the Canadian
study all 8 liver cancer deaths were ASL, as was the case for the 3
liver cancer deaths in the French study. No information is available
concerning ASLs in the German study while no liver cancers were
diagnosed in the Russian study. It is probable that some ASLs
occurring among VC-exposed workers have remained undiagnosed and have
been coded in the death certificates as liver cancers unspecified or
as other liver-associated disease.
Several studies examined the risk specifically for hepatocellular
carcinoma (HCC) of the liver. No excess was observed in the European
study, nor in the Canadian or French studies. Data from the USA study
and also from the update of the Italian component of the European
cohort seem, however, to indicate that there is also an excess for
HCC. This is of smaller magnitude than that observed for ASL. The
interpretation of the results for HCC is complicated because of the
uncertainties concerning the accuracy of diagnosis of angiosarcomas
and HCC using death certificate information and the absence of compete
histopathological confirmation of the diagnoses of liver cancer in the
studies. Although the results are not fully consistent between
studies, the data suggest that there may be a small excess risk for
HCC.
Four out of five studies reporting results for brain tumours
identified a moderate excess risk with an SMR of 1.42 for the combined
data from five studies (43 observed, 95% CI 1.03-1.91). The risk
tended to increase with duration of exposure/employment in the
European and USA study. Furthermore, in the USA study, the highest
risk occurred in the two plants where most ASL cases had been
diagnosed, and where presumably the highest VC exposure had occurred.
In the European study where the dose-response relationship was
examined, no association was seen with cumulative exposure to VC. The
overall epidemiological evidence is suggestive of a possible risk
among VC workers.
Some of the smaller early studies had reported an increase in
lung cancer among VC workers. There was no indication of an excess
risk, however, in the two largest studies (European and USA) or in the
other four smaller studies. There was no association either with
duration of exposure/employment in the USA or the European study, nor
with cumulative exposure to VC in the European study.
Excess risk for malignant lymphomas had been reported in some
early small studies. No excess risk, however, was observed in the two
largest cohorts (USA and European) or in the Canadian cohort. An
excess risk for the combined category leukaemia and lymphoma was
observed in the Russian and the German study. In interpreting these
results it should be noted that the studies used different disease
classification and in some occasions grouped lymphomas with malignant
myeloma. The overall results exclude the presence of any large
increased risk for lymphomas or leukaemia.
b) Experimental animal data
VC is a multispecies, multiorgan carcinogen. It induces benign
and malignant tumours in several organs of various species, both in
males and females. The most prominent feature is the induction of the
rare angiosarcomas, especially in the liver. This phenomenon has been
demonstrated in different strains of mice and rats, as well as in
hamsters. Other tumours include nephroblastomas and neuroblastomas,
hepatocellular carcinomas, mammary gland carcinomas and lung adenomas.
VC is genotoxic, causing alterations and damage to DNA. VC
metabolites bind to the DNA yielding promutagenic DNA adducts.
Mutations and chromosomal abnormalities have been described in many
in vitro and in vivo systems. These genotoxic processes play a
significant role in tumorigenesis, while cell proliferation, secondary
to the VC-induced cell toxicity, may also be involved in the process.
10.1.2 Dose-response analysis
10.1.2.1 Non-neoplastic effects
a) Human data
The published studies examining non-neoplastic diseases in
VC-exposed subjects do not provide dose-response information.
b) Experimental animal data
Several short-term and long-term studies have been assessed for
establishing the NOAEL of VC. Liver cell polymorphism (variation in
size and shape of hepatocytes and their nuclei) reported in the
feeding study by Til et al. (1991) is recommended for quantifying
non-cancer risks from oral exposure to VC. The severity and incidences
of this liver lesion were statistically significantly increased at a
daily dose of 1.3 mg/kg body weight but not at 0.13 mg/kg per day
(Table 46), the no-observed-adverse-effect level (NOAEL) therefore
being 0.13 mg/kg per day. Basophilic liver cell foci induction in the
study of Til et al. (1991) occurred at lower concentrations, but it
was not considered by the Task Group to be a manifestation of
hepatocytic toxicity.
A lowest-observed-adverse-effect level (LOAEL) of 26 mg/m3 was
established based on a subchronic (3-12 months) inhalation toxicity
study in male rats by Bi et al. (1985). The basis for the estimation
was the increased relative liver weight and mild testicular
degeneration (Table 47), both effects being more pronounced in a
dose-related manner at the two higher dose levels tested (260 and
7800 mg/m3). A recent inhalation two-generation reproduction study
with VC in rats, using exposure concentrations of 26, 260 and 2860
mg/m3 (Shah, 1998), yielded the same LOAEL value. In this study,
increased relative liver weights and hypertrophy of centrilobular
hepatocytes were found in parental animals at all dose levels and a
dose-dependent manner.
10.1.2.2 Neoplastic effects
a) Human data
In most epidemiological studies dose-response analyses were not
available. Of the two largest studies conducted in the USA and Europe,
the USA study examined only duration of exposure and did not examine
dose-response. In the European study a calendar period-, job- and
plant-specific job exposure matrix was used. Expert judgement was used
to estimate exposures for the early time periods when no measurements
were available. The use of such a matrix implies that, when assigning
the exposure index to individuals in the cohorts, a certain degree of
misclassification occurs tending to underestimate the strength of a
dose-response relationship.
All of the results in this section are from the European study
(Simonato et al., 1991) unless otherwise noted. A clear dose-response
relationship was found only for ASL alone or in combination with other
primary liver cancers.
In all the analyses of liver cancer in the European cohort there
is a clear relationship with increasing estimated doses. The effect is
most evident for ASL, for which the risk estimate in the highest
exposure groups is two and half times larger than for all liver
cancers (Tables 48 and 49). These results are consistent with the
analyses published by Wu and colleagues in the updating of one of the
USA subcohorts. In this study, the average cumulative exposure score
to VC was 6 times higher for ASL cases than for other liver cancer
cases and control subjects.
Results from the regression models in the European study
indicated that cumulative dose appears as the most powerful
determinant of ASL, followed by time since first exposure. This can
also be seen from Table 50. The results from the regression models
indicated that there was only a small or no effect of calendar period
and age of hire. Both duration and intensity of exposure correlated
with liver cancer risk (see Table 41).
Dose-response analyses for lung cancer, brain tumours and
lymphomas are reported in section 8.3.
b) Experimental animals data
Studies using short- and long-term oral and inhalation exposure
were carried out to assess the carcinogenic activity of VC (section
7.7.) For assessing carcinogenic risk, pivotal studies have been
selected where the study design, exposure levels, observation period
and histopathological examinations are regarded as suitable for
assessing risk.
Table 46. Type and incidence of treatment-related liver-cell polymorphism
of rats orally exposed to vinyl chloride (from Til et al., 1991)
Males Females
N (total/group) 100 100 100 50 100 100 100 50
Dose mg/kg 0 0.014 0.13 1.3 0 0.014 0.13 1.3
per day
Liver cell No of animals with liver cell polymorphism of
polymorphisms varying severity
Slight 27 23 26 19 46 41 49 23
Moderate 4 4 7 10b 14 13 8 15a
Severe 1 1 1 3 2 3 4 9c
a P < 0.05
b P < 0.01
c P < 0.001
Table 47. Damage of testes induced by different concentration
of vinyl chloride (from Bi et al., 1985)
Group No. of Slight Moderate Severe Total %
animals damage damage damage
Control 74 9 3 2 14 18.9
10 ppm a 74 14 5 3 22 29.7
100 ppm b 74 19 5 3 27 36.5 d
3000 ppm c 75 29 8 5 42 56.0 e
a 26 mg/m3
b 260 mg/m3
c 7770 mg/m3
d P < 0.05
e P < 0.001
Table 48. Maximum likelihood estimates for final model with
cumulative exposure and years since first employment
for deaths from liver cancer (N = 24) a
Exposure variable Relative risk 95% confidence interval
Cumulative exposure
(ppm-years)
< 500 1.0
500-1999 1.2 0.1-11.4
2000-5999 4.6 1.0-21.0
6000-9999 12.2 2.5-59.6
> 10 000 17.1 3.1-93.6
Years since first employment
0-19 1.0
20-24 5.6 1.4-22.4
> 25 6.8 1.7-27.4
a From: Simonato et al. (1991)
Table 49. Maximum likelihood estimates for final model with
cumulative exposure and years since first employment
for deaths from angiosarcoma of the liver (N = 22) a
Exposure variable Relative risk 95% confidence interval
Cumulative exposure (ppm-years)
< 2000 1.0
2000-5999 6.8 1.1-41.7
6000-9999 24.7 4.1-150.1
>10 000 45.4 7.3-281.1
Years since first employment
0-19 1.0
20-24 4.7 1.0-22.8
>25 6.2 1.4-29.0
a From: Simonato et al. (1991)
Table 50. Absolute risk of angiosarcoma of the liver
per 100 000 (Simonato et al., 1991)
Cumulative exposure (ppm-years)
Years since < 2000 2000-5999 6000-9999 >10 000
first employment
0-19 1.0 6.8 24.4 44.8
20-24 4.7 32.0 115.6 212.5
>25 6.2 42.2 152.3 280.0
For oral exposure, it is recommended that female rats from the
Feron et al. (1981) study be used to quantify risk for VC, because the
incidence of liver tumour-bearing animals is greater than in males,
giving a more conservative estimate. Rats with either neoplastic liver
nodules, hepatocellular carcinomas and/or ASLs should be included for
quantification. These lesions are all dose-related. The neoplastic
nodules, while not malignant, have the potential to progress to
malignancy. While it is uncertain that VC induces tumour types other
than ASL in humans, tumour type concordance is not considered to be
necessary, and thus inclusion of all liver tumours is desirable as a
conservative approach.
If quantification is desired for ASL only, then the male rat is
recommended because the incidences for this end-point are higher in
males (Table 51). Including animals with angiosarcomas in other organs
is unnecessary because the only other site with significantly
increased incidences of this tumour type is the lung, and, with one
exception, all animals with ASL also had liver tumours.
For inhalation exposure, several studies have been conducted in
different species (section 7.7). Four experiments with similar design
and experimental conditions, carried out by Maltoni et al. (1981,
1984) in rats, were used for assessing carcinogenic risk after VC
inhalation exposure. Out of the treatment-related carcinogenic
responses, ASL, all vascular tumours of the liver and all liver
tumours combined were considered to be suitable for assessing risk
(Table 52). Induction of ASL is a special feature of VC and since this
tumour type was also observed in exposed humans, this end-point was
considered especially relevant for quantification of risk.
For ASL, increased incidence was observed in both males and
females. A clear dose-response relationship could be established in
female rats, and the effect was also suggestive in males. A similar
trend could be observed if all vascular tumours or all liver tumours
were considered. The other tumours, however, occurred only at low
incidences. The lowest dose at which ASL and other vascular tumours
were detected was 26 mg/m3 in females and 65 mg/m3 in males.
Table 51. Tumour incidences in oral feed study with vinyl chloride
in Wistar rats (from Feron et al., 1981)
Dose Neoplastic Hepato-cellular Liver Liver Mammary
(mg/kg liver carcinomas angio-sarcomas tumour-bearing adeno-carcinomas
bw) nodules rats
Females
0 2/57 0/57 0/57 2/57 3/57
1.7 26/57 4/58 0/57 28/58 2/58
5.0 39/59 19/59 2/59 49/59 4/59
14.1 44/57 29/57 9/57 56/57 7/57
Males
0 0/55 0/55 0/55 0/55
1.7 1/58 1/58 0/58 2/58
5.0 7/56 2/56 6/56 11/56
14.1 23/59 8/59 27/59 41/59
Table 52. Liver tumours in inhalation studies on rats
(from Maltoni et al., 1981, 1984)
Exposure Exposure Number of % ASL % liver % all liver
(mg/m3, (mg/m3, animals vascular tumours b
4 h/d, continuous) a tumours b
5 d/w)
Males
0 0 222 0 0 0
2.6 0.3 58 0 0 0
13 2 59 0 0 0
26 3 59 0 0 0
65 8 60 1.7 1.7 1.7
130 15 174 1.1 2.3 2.3
260 31 60 0 1.7 1.7
390 46 60 1.7 1.7 1.7
520 62 60 11.7 15.0 16.0
650 77 29 3.4 3.4 4.4
1300 155 30 0 0 0
6500 774 30 20.0 20.0 20.0
15 600 1857 29 10.3 10.3 10.3
26 000 3095 30 10.0 10.0 11.0
Females
0 0 239 0 0 0
2.6 0.3 60 0 0 0
13 2 60 0 0 0
26 3 60 6.7 6.7 6.7
65 8 60 6.7 8.3 8.3
130 15 180 7.2 10.6 10.6
260 31 60 1.7 1.7 1.7
390 46 60 8.3 8.3 8.3
520 62 60 8.3 11.7 15.0
650 77 30 6.7 10.0 10.0
1300 155 30 20.0 20.0 36.7
6500 774 30 23.3 23.3 30.0
15 600 1857 30 33.3 40.0 43.3
26 000 3095 30 13.3 13.3 13.3
a Exposure calculated to correspond to 24 h/day, 7 days/week
b % of animals with tumours were added, because no individual
animal data were available.
Nephroblastomas and neuroblastomas have also been observed in
some studies in rats after VC exposure. These rare tumours appeared in
a dose- and time-dependent manner; the doses required for tumour
induction were, however, higher than those for ASL.
Several inhalation studies of VC in mice have been carried out.
The induced spectrum of tumours was similar to that in rats:
angiosarcomas and angiomas of the liver and other organs, mammary
gland and lung tumours. In one study (Maltoni et al., 1981, 1984), a
clear dose-response relationship was observed for ASL as well as for
all vascular tumours in the female mice (Table 53). The lowest
carcinogenic dose was 130 mg/m3, higher than that in rats.
Mammary gland adenocarcinomas were observed in several VC
studies. Four inhalation studies with VC in Sprague-Dawley rats
(Maltoni et al., 1981, 1984) were evaluated (Table 54). A
statistically significantly increased incidence in mammary gland
carcinoma was observed in three of the four studies. However, in none
of these studies was a clear dose-response relationship observed.
Increased incidences of mammary carcinomas, without a dose-response
relationship, were also observed in Swiss mice after inhalation
exposure (Maltoni et al., 1981, 1984). Drew et al. (1983) exposed
Fischer-344 rats to 0 or 261 mg/m3 either for 6 or 12 months.
Increased incidences of mammary adenocarcinomas were observed in the
animals exposed at different ages but, since only one exposure level
was used, a dose-response relationship could not be established. In
Syrian hamsters, using similar study design but an exposure level of
520 mg/m3 (Drew et al., 1983), a high incidence of mammary tumours
was observed in animals exposed early in life.
In two other mouse studies (Lee et al., 1978; Drew et al., 1983),
elevated incidences of both mammary gland carcinomas and other mammary
tumours were observed. However, these studies were also inadequate for
risk assessment. In summary, increased incidence of mammary gland
carcinomas were observed in several experiments where animals were
exposed to VC, either orally or by inhalation, indicating
treatment-related carcinogenic effect. However, the lack of
dose-response or the use of a single dose excluded the use of these
data for risk assessment.
10.1.3 Human exposure
The aim of this section is to estimate present levels of exposure
to VC of the general population and of subpopulations with special
exposure, such as those living near point sources of VC and those
exposed at work. The purpose is to indicate worst-case scenarios
rather than average or most likely exposures.
The data available are very limited both in time and space. Often
the distance of the sampler from the point source or the sampling and
averaging times have not been reported. This makes the comparison of
results from different studies difficult.
Table 53. Incidences of different tumour types in Swiss mice after inhalation exposure to vinyl chloride
(from Maltoni et al., 1981, 1984 BT4)
Exposure Effective ASL Angiosarcoma Angioma Angioma Total Mammary Lung
(mg/m3) number (other organs) (liver) (other vascular gland adenoma
organs) tumours
Males
0 80 0 0 0 1 1 0 8
130 30 1 1 0 1 3 0 3
650 30 9 2 6 2 17 0 24
1300 30 6 2 1 1 10 1 24
6500 29 6 4 2 0 12 0 18
15 600 30 2 0 2 2 6 0 23
26 000 26 1 0 1 2 4 0 20
Females
0 70 0 1 0 0 1 1 7
130 30 0 0 1 4 5 13 3
650 30 9 1 5 1 17 12 17
1300 30 8 5 4 2 10 9 26
6500 30 10 4 3 1 12 10 22
15 600 30 11 1 5 1 6 9 24
26 000 30 9 1 5 2 4 14 26
Table 54. Incidence of mammary gland carcinomas in Sprague-Dawley
rats and Swiss mice after inhalation exposure to vinyl
chloride (from Maltoni et al., 1981, 1984) a
Exposure BT15 BT1 BT2 BT9 BT4
(mg/m3) (rat) (rat) (rat) (rat) (mouse)
0 6/60 (10) 0/29 1/100 (1) 5/50 (10) 1/70 (1.4)
2.6 12/60 (20)
13 22/60 (36.6) d
26 21/60 (35) c
65 15/60 (25)
130 2/30 (6.6) 59/150 (39) d 13/30 (43.3) d
260 3/60 (5)
390 6/60 (10) c
520 5/60 (8) b
650 2/30 (6.6) 12/30 (40) d
1300 1/30 (3.3) 9/30 (30) d
6500 2/30 (6.6) 10/30 (33.3) d
15 600 0/30 9/30 (30) d
26 000 3/30 (10) 14/30 (46.6) c
a Values are no. of tumour / no. of animals, with percentages in parentheses; statistical
significance compared to the concurrent control group (two tailed Fischer)
b P < 0.05
c P < 0.01
d P < 0.001
10.1.3.1 General population
The main sources of VC exposure for the general population are
VC/PVC plants, biodegradation of chlorinated solvents in landfills,
spills of chlorinated solvents from, for instance, dry cleaning shops,
and the metal and electronic industry. In addition, small amounts of
VC may be liberated from PVC.
There are no known natural sources of VC. Concentrations of VC in
ambient air samples were reported to range from < 0.013 to 26 µg/m3.
These data were collected in the 1970s and 1980s from rural, remote,
suburban and urban sites. The concentrations measured in rural and
suburban/urban sites were similar. There is a lack of recent data on
ambient air, making it difficult to conclude whether there has been a
historical reduction in ambient concentrations. The exposure to VC via
inhalation by the general population (non-occupational exposure) is
estimated not to exceed 10 µg/kg body weight per day, using the
derivation method described in Environmental Health Criteria 170:
Assessing Human Health Risks of Chemicals: Derivation of Guidance
Values for Health-based Exposure Limits (IPCS, 1994).
Concentrations (probably daily means) of VC measured in ambient
air from industrial areas were reported to be < 8000 µg/m3 in the
1970s and < 200 µg/m3 and 4800 µg/m3 in the 1980s in different
geographical areas. Monthly mean concentrations measured in the
vicinity of one plant during the 1990s ranged from 0.1 to 1.3 µg/m3.
Exposure of the general population living in the vicinity of
industrial areas was estimated not to exceed 3000 µg/kg per day in the
1970s, 70-1800 µg/kg per day in the 1980s and 0.5 µg/kg per day in the
1990s.
VC concentrations in air sampled from the vicinity of waste sites
ranged from < 0.08 to 104 µg/m3. These concentrations were reported
in air sampled during the 1980s and 1990s; no historical trend could
be established. Exposure of the general population living in the
vicinity of landfills was estimated to be < 300 µg/kg per day.
There are no data on VC concentrations in air from other areas
contaminated with chlorinated hydrocarbons.
Sources of VC in indoor air have included PVC products, smoking,
paints and aerosol propellants. Up to the 1970s, the VC concentration
in PVC exceeded 1000 mg/kg, and consequently VC was detected in indoor
air and cars. Since the late 1970s the VC content in PVC has been
regulated to approximately 1 mg/kg, therefore VC concentrations are
now unlikely to be significant in indoor air, except where there are
nearby external sources such as landfills, etc.
The general population may be exposed to VC in drinking-water. In
the majority of water samples analysed, VC was not present at
detectable concentrations. The maximum VC concentration reported in
drinking-water was 10 µg/litre, leading to a maximum exposure of
0.2 µg/kg per day. There is a lack of recent data on concentrations in
drinking-water, but these levels are expected to be below 10 µg/litre.
In groundwater near point sources, very high (up to 200 mg/litre)
concentrations of VC have been observed and well water nearby may be a
very significant source of VC: daily intakes may reach 5000 µg/kg per
day. Some recent studies have identified VC in PVC-bottled
drinking-water at levels below 1 µg/litre. The frequency of occurrence
of VC in such water is expected to be higher than in tap water.
Dietary exposure to VC from PVC packages used for food has been
calculated by several agencies, and, based upon estimated average
intakes in the United Kingdom and USA, an exposure of < 0.0004 µg/kg
per day was estimated for the late 1970s and early 1980s.
Exposure of the general population may be higher in situations
where large amounts of VC are accidentally released to the
environment, such as a spill during transportation. However, such
exposure is likely to be transient.
10.1.3.2 Occupational exposure
Staff working in plants where VC is manufactured or polymerized
into PVC or at landfills may be occupationally exposed to VC.
Occupational exposure to VC has continually decreased since the 1940s.
Since the 1970s, occupational exposure level (OEL) values have been
set at 2.6 to 18 mg/m3 in most European countries and the USA. Few
data are available to determine how frequently the actual workplace
exposures are within the OEL. Where data are available, compliance
during the 1990s was reported in some European countries. Based upon
an OEL of 18 mg/m3, an occupational exposure of 3000 µg/kg per day
can be calculated. The most recently reported average occupational
exposure in these countries was 0.3 mg/m3. This gives a daily dose of
50 µg/kg per day. However, concurrent exposure levels of more than
300 mg/m3 have also recently been reported in some countries.
Exposure to VC in PVC processing plants has been historically, and is
probably currently, much lower than in PVC/VC production plants.
10.1.4 Risk characterization
VC has been shown to be carcinogenic and toxic in both oral and
inhalation experimental bioassays, as well as in human epidemiological
studies. Experimental studies have also shown that it is genotoxic and
must be activated through a rate-limited pathway. In order to perform
quantitative risk assessment using animal bioassays, it will be
necessary to utilize a physiologically based pharmacokinetic model
(PBPK) to derive the concentration of active metabolite at the
critical target site, the liver, as well as to extrapolate dose from
animals to humans. The PBPK model should be validated, taking into
account the known metabolic pathways and using metabolic constants
determined experimentally, and should not introduce too many unknown
parameters. The Task Group was not in a position to evaluate
accurately the available pharmacokinetic models for their suitability
for risk assessment. Furthermore, in order to estimate the risk
predictions from epidemiological studies, original data on exposed
human populations, at the level of the individual, are required.
Some of the published PBPK models and risk assessments are
reviewed in Annex 2.
10.2 Evaluation of effects on the environment
VC is released to the environment from plants where it is
manufactured and/or used in the production of PVC. The most important
releases are emissions to the atmosphere, but VC may also be released
in waste water. It may also be formed in the environment as a product
of the biodegradation of chlorinated C2 solvents. This route of VC
formation may lead to emissions of VC from areas contaminated with
chlorinated C2 solvents, such as landfills.
The maximum VC concentration reported in surface water is
0.6 mg/litre. VC is unlikely to be bioaccumulated to a significant
extent in aquatic organisms.
The only toxicity study which was deemed of sufficient quality to
evaluate the effects of VC on aquatic organisms gave a 48-h LC50 of
210 mg/litre for a freshwater fish. There is a paucity of chronic
toxicity data for aquatic organisms. Despite these limitations, owing
to the rapid volatilization of VC, the low concentrations in surface
water and the low acute toxicity to fish, VC is not expected to
present a hazard to aquatic organisms.
There is a paucity of data for terrestrial organisms.
11. RECOMMENDATIONS FOR PROTECTION OF HUMAN HEALTH
11.1 Public health
The following measures should be taken:
* worldwide application of production technologies leading to low
residual VC levels in PVC;
* implementation and enforcement worldwide of steps that guarantee
minimal emissions of VC at production sites;
* identification, surveillance and emission and exposure control of
contaminated areas such as landfill sites.
11.2 Occupational health
Since VC is a genotoxic carcinogen, exposures should be kept as
low as possible, using the best available technology worldwide.
More information, education and training of workers potentially
exposed to VC regarding the risks involved and safe working procedures
and habits is required.
Monitoring and record-keeping of exposure and record-keeping of
exposed workers are needed.
12. FURTHER RESEARCH
The following topics need further research:
* follow-up of existing VC cohorts with quantitative exposure
assessment;
* epidemiological studies of populations with defined and low
exposure to VC;
* evaluation of the efficacy and usefulness of the medical
surveillance of VC-exposed workers;
* validation of effects and susceptibility biomarkers for VC;
* the role of post-DNA-damage events in the carcinogenicity of VC
and their relationships with target organ or cell type
specificity;
* evaluation of the sensitivity to VC in early childhood and
determination of possible mechanisms involved;
* the possibility of carcinogenicity of VC in mammary gland tumours
and the mechanism involved;
* optimization of remediation of contaminated sites;
* studies on the toxicity of VC to aquatic and terrestrial
organisms.
13. PREVIOUS EVALUATIONS BY INTERNATIONAL BODIES
Vinyl chloride was evaluated by the International Agency for
Research on Cancer (IARC, 1979) and the evaluation was updated in
Supplement 7 (IARC, 1987). There was sufficient evidence for
carcinogenicity of vinyl chloride in humans and sufficient evidence
for carcinogenicity in animals; the overall evaluation was that vinyl
chloride is carcinogenic to humans.
In the WHO Guidelines for Drinking-water Quality (WHO, 1996),
using results from the rat bioassay of Til et al. (1983) and applying
the linearized multistage model, the human lifetime exposure for a
10-5 excess risk of ASL was calculated to be 20 µg/day. It was also
assumed that in humans the number of cancers at other sites may equal
that of ASL, so that a correction (factor of 2) for cancers other than
ASL is justified. VC concentrations in drinking-water of 5 µg/litre
were calculated as being associated with excess risks of 10-5.
REFERENCES
ACGIH (1999) Documentation of the threshold limit values and exposure
indices. Cincinnati, Ohio, American Conference of Governmental and
Industrial Hygienists Inc.
Adkins B, Van Stee EW, Simmons JE, & Eustis SL (1986) Oncogenic
response of strain A/J mice to inhaled chemicals. J Toxicol Environ
Health, 17: 311-322.
Ahlborg G, Bjerkedal T, &Eg J (1987) Delivery outcome among women
employed in the plastics industry in Sweden and Norway. Am J Ind Med,
12: 507-517.
Albanus L, Gawell B-M, Larsson B, & Slorach SA (1979) [The Swedish
National Food Administration's decision to reduce the maximum
permitted level of vinyl chloride monomer in foodstuffs.] Var Foeda,
31: 161-168 (in Swedish).
Allen MR, Braithwaite A, & Hills CC (1997) Trace organic compounds in
landfill gas at seven UK waste disposal sites. Environ Sci Technol,
31: 1054-1061.
Allsopp MW & Vianello G (1992) Poly(vinyl)chloride. In: Elvers B,
Hawkins S, & Schulz G ed. Ullmann's encyclopedia of industrial
chemistry, 5th completely revised. -- Volume A21: Plastics, properties
and testing to polyvinyl compounds. Weinheim, Germany, VCH
Verlagsgesellschaft mbH, pp 717-742.
Andersen M, Clewell H, Gargas M, Smith FA, & Reitz RH (1987)
Physiologically based pharmacokinetics and the risk assessment process
for methylene chloride. Toxicol Appl Pharmacol, 87: 185-205.
Anderson D & Richardson CR (1981) Issues relevant to the assessment of
chemically induced chromosome damage in vivo and their relationship
to chemical mutagenesis. Mutat Res, 90: 261-272.
Anderson D, Hodge MCE, & Purchase IFH (1976) Vinyl chloride: Dominant
lethal studies in male CD-1 mice. Mutat Res, 40: 359-370.
Anderson D, Hodge MCE, & Purchase IFH (1977) Dominant lethal studies
with the halogenated olefins vinyl chloride and vinylidene dichloride
in male CD-1 mice. Environ Health Perspect, 21: 71-78.
Anderson D, Richardson CR, Weight TM, Purchase IFH, & Adams WGF (1980)
Chromosomal analyses in vinyl chloride exposed workers. Results from
analysis 18 and 42 months after initial sampling. Mutat Res, 79:
151-162.
Anderson D, Richardson CR, Purchase IFH, Evans HJ, & O'Riordan ML
(1981) Chromosomal analysis in vinyl chloride exposed workers:
Comparison of the standard technique with the sister-chromatid
exchange technique. Mutat Res, 83: 137-144.
Andersson Y & Ljungström E (1989) Gas phase reaction of the NO3
radical with organic compounds in the dark. Atmos Environ, 23:
1153-1155.
Ando M & Sayato Y (1984) Studies on vinyl chloride migrating into
drinking water from polyvinyl chloride pipe and reaction between vinyl
chloride and chlorine. Water Res, 18: 315-318.
Andreoli A (1985) Hazardous waste site investigations: Vinyl chloride
contamination of groundwater. In: Suffolk County Department of Health
Services ed. Proceedings of The Second Annual Eastern Regional Ground
Water Conference. New York, Suffolk County Department of Health
Services, pp 482-489.
Anghelescu F, Otoiu M, Dobrinescu E, Hagi-Paraschiv-Dossios L,
Dobrinescu G, & Ganea V (1969) Clinico-pathogenic considerations on
Raynaud`s phenomenon in the employees of the vinyl polychloride
industry. Med Interna, 21: 473-482.
Anna K & Alberti J (1978) [Source and use of organic halogenated
compounds and their distribution in water and wastewater.] In: Anna K
& Alberti J ed. [Water '77.] Berlin, Kongressvorträge Colloquium
Verlag, pp 154-158 (in German)
Anon (1973) FDA to propose ban on use of PVC for liquor use. Food Chem
News, 14: 3-4.
Anon (1981) [Measurement of hydrocarbon levels in ambient air in
Cologne: Report from the Environmental Office of the City of Cologne.]
Cologne, Germany, City of Cologne, pp 35-56 (in German).
Anon (1993) Quality standards for foods with no identity standards:
bottled water. Fed Reg, 58: 378-381.
Anon (1996) [ Dangerous substance transport accident -- train -- on
1st June in Schönbeck/Elbe. Train accident with resulting fire and
danger to the neighbourhood.] Heyrothsberge, Germany, Sachsen-Anhalt,
200 pp (Unpublished Conference report) (in German).
Arbin A, Östelius J, Callmer K, Sroka J, Hänninen K, & Axelsson S
(1983) Migration of chemicals from soft PVC bags into intravenous
solutions. Acta Pharm Suec, 3: 20-33.
Arneth JD, Schleyer R, Kerndorff H, & Milde G (1988) [Standardised
evaluation of groundwater contaminants from existing chemical
pollutants.] Bundesgesundheitsblatt, 31: 117-123 (in German).
Ashworth RA, Howe GB, Mullins ME, & Roger TN (1988) Air-water
partitioning coefficients of organics in dilute aqueous solutions. J
Hazard Mater, 18: 25-36.
Association of Plastics Manufacturers in Europe (1999) Management
report from the APME register of cases of angiosarcoma related to PVC
production. Brussels, Association of Plastics Manufacturers in Europe,
33 pp (Unpublished report).
ASTM (1993) Standard test method for vinyl chloride in workplace
atmospheres (charcoal tube method): D/4766-88. Philadelphia,
Pennsylvania, American Society for Testing and Materials, pp 323-328.
ASTM (1985) Analysis for determining residual vinyl chloride in ppb
range in vinyl chloride homo- and copolymers by headspace gas
chromatography. In: ASTM method D/4443-84. Philadelphia, Pennsylvania,
American Society for Testing and Materials.
Atkinson R (1985) Kinetics and mechanisms of the gas-phase reactions
of the hydroxyl radical with organic compounds under atmospheric
conditions. Chem Rev, 84: 69-201.
Atkinson R (1987) A structure-activity relationship for the estimation
of rate constants for the gas-phase reactions of OH radicals with
organic compounds. Int J Chem Kinet, 19: 799-828.
Atkinson R & Aschmann SM (1987) Kinetics of the gas-phase reactions of
Cl atoms with chloroethenes at 298 +/- 2 K and atmospheric pressure.
Int J Chem Kinet, 19: 1097-1105.
Atkinson R & Carter WPL (1984) Kinetics and mechanisms of the
gas-phase reactions of ozone with organic compounds under atmospheric
conditions. Chem Rev, 84: 437-470.
Atkinson R & Pitts JN (1977) Rate constants for the reaction of
O(3 P) atoms with CH2=CHF, CH2=CHCL, and CH2=CHBr over the
temperature range 298-442°K. J Chem Phys, 67: 2488-2491.
Atkinson R, Darnall KR, Lloyd AC, Winer AM, & Pitts JN (1979) Kinetics
and mechanisms of the reactions of the hydroxyl radical with organic
compounds in the gas phase. In: Pitts JN, Hammond GS, Gollnick K, &
Grosjean D ed. Advances in photochemistry. New York, John Wiley &
Sons, vol 11, pp 375-488.
Atkinson R, Aschmann SM, & Goodman MA (1987) Kinetics of the gas-phase
reactions of NO3 radicals with a series of alkynes, haloalkenes, and
alpha, beta-unsaturated aldehydes. Int J Chem Kinet, 19: 299-307.
Atri FR (1985) [Vinyl chloride.] In: Barndt D ed. [Chlorinated
hydrocarbons in the environment III.] Berlin, Technical University, pp
134-189 (Landscape Development and Environmental Research Series No.
34) (in German).
ATSDR (Agency for Toxic Substances and Disease Registry) (1990) ATSDR
case studies in environmental medicine: Vinyl chloride toxicity. Clin
Toxicol, 28: 267-285.
ATSDR (Agency for Toxic Substances and Disease Registry) (1997)
Toxicological profile for vinyl chloride - Update. Atlanta, Georgia,
Agency for Toxic Substances and Disease Registry, 239 pp.
Baek NH, Jaffé PR, & Shingal N (1990) Stimulating the degradation of
TCE under methanogenesis. J Environ Sci Health Environ Sci Eng, 25:
987-1005.
Baker LW & MacKay KP (1985) Hazardous waste management: Screening
models for estimating toxic air pollution near a hazardous waste
landfill. J Air Pollut Control Assoc, 35: 1190-1195.
Baker MT & Ronnenberg WC (1993) Contrasting effects of
1,1,1-trichloroethane on [14C]vinyl chloride metabolism and
activation in hepatic microsomes from phenobarbital- and
isoniazid-treated rats. Toxicol Appl Pharmacol, 119: 17-22.
Ballering LAP, Nivard MJM, & Vogel EW (1996) Characterization by
two-endpoint comparisons of the genetic toxicity profiles of vinyl
chloride and related etheno-adduct forming carcinogens in Drosophila.
Carcinogenesis, 17: 1083-1092.
Banzer JD (1979) The migration of vinyl chloride monomer from PVC pipe
into water. J Vinyl Technol, 1: 164-167.
Bao YS, Jiang H, & Liu J (1988) [The effects of vinyl chloride on
pregnancy parturition and fetal development among female workers.]
Chin J Prev Med, 22: 343-346 (in Chinese).
Barbacid M (1987) Ras genes. Ann Rev Biochem, 56: 779-827.
Barbin A (1998) Formation of DNA etheno adducts in rodents and humans
and their role in carcinogens. Acta Biochim Pol, 45: 145-161.
Barbin A (in press) The role of etheno DNA adducts in carcinogenesis
induced by vinyl chloride in rats. In: Singer B & Bartsch H ed.
Exocyclic DNA adducts in mutagenesis and carcinogenesis. Lyon,
International Agency for Research on Cancer, pp 1-29 (IARC Scientific
Publications No. 150).
Barbin A, Brésil H, Croisy A, Jacquignon P, Malaveille C, Montesano R,
& Bartsch H (1975) Liver microsome-mediated formation of alkylating
agents from vinyl bromide and vinyl chloride. Biochem Biophys Res
Commun, 67: 596-603.
Barbin A, Laib RJ, & Bartsch H (1985a) Lack of miscoding properties of
7-(2-oxoethyl)guanine, the major vinyl chloride-DNA adduct. Cancer
Res, 45: 2440-2444.
Barbin A, Besson F, Perrard MH, Béréziat JC, Kaldor J, Michel G, &
Bartsch H (1985b) Induction of specific base-pair substitutions in
E. coli trpA mutants by chloroethylene oxide, a carcinogenic vinyl
chloride metabolite. Mutat Res, 152: 147-156.
Barbin A, Forment O, Boivin S, Marion M-J, Belpoggi F, Maltoni C, &
Montesano R (1997) p53 Gene mutation pattern in rat liver tumors
induced by vinyl chloride. Cancer Res, 57: 1695-1698.
Baretta ED, Stewart RD, & Mutchler JE (1969) Monitoring exposures to
vinyl chloride vapor: Breath analysis and continuous air sampling. Am
Ind Hyg Assoc J, 30: 537-544.
Barnes AW (1976) Vinyl chloride and the production of PVC. Proc R Soc
Med, 69: 277-281.
Barr DB & Ashley DL (1998) A rapid, sensitive method for the
quantitation of N-acetyl-S-(2-hydroxyethyl)-L-cysteine in human urine
using isotope-dilution HPLC-MS-MS. J Anal Toxicol, 22: 96-104.
Barrio-Lage GA, Parsons FZ, Narbaitz RM, & Lorenzo PA (1990) Enhanced
anaerobic biodegradation of vinyl chloride in ground water. Environ
Toxicol Chem, 9: 403-415.
Barton HA, Creech JR, Go CS, Randall GM, & Seckel CS (1995)
Chloroethylene mixtures: Pharmacokinetic modeling and in vitro
metabolism of vinyl chloride, trichloroethylene, and
trans-1,2-dichloroethylene in rat. Toxicol Appl Pharmacol, 130:
237-247.
Bartsch H, Malaveille C, & Montesano R (1975) Human rat and mouse
liver-mediated mutagenicity of vinyl chloride in S. typhimurium
strains. Int J Cancer, 15: 429-437.
Bartsch H, Malaveille C, Barbin A, Bresil H, Tomatis L, & Montesano R
(1976) Mutagenicity and metabolism of vinyl chloride and related
compounds. Environ Health Perspect, 17: 193-198.
Bartsch H, Malaveille C, Barbin A, & Planche G (1979) Mutagenic and
alkylating metabolites of halo-ethylenes, chlorobutadienes and
dichlorobutenes produced by rodent or human liver tissues. Arch
Toxicol, 41: 249-277.
Bartsch H, Barbin A, Marion MJ, Nair J, & Gu Y (1994) Formation,
detection and role in carcinogenesis of ethenobases in DNA. Drug Metab
Rev, 26: 349-371.
Bartsch H, Nair J, & Velic I (1997) Etheno-DNA base adducts as tools
in human cancer aetiology and chemoprevention. Eur J Cancer Prev, 6:
529-534.
Basler A & Röhrborn G (1980) Vinyl chloride: An example for evaluating
mutagenic effects in mammals in vivo after exposure to inhalation.
Arch Toxicol, 45: 1-7.
Basu AK, Wood ML, Niedernhofer LJ, Ramos LA, & Essigmann JM (1993)
Mutagenic and genotoxic effects of three vinyl chloride-induced DNA
lesions: 1,N6-ethenoadenine, 3,N4-ethenocytosine, and
4-amino-5-(imidazol-2-yl)imidazole. Biochemistry, 32: 12793-12801.
Bauer U (1981) [Human exposure to environmental chemicals
-- investigations on volatile organic halogenated compounds in water,
air, food, and human tissues.] Zent.bl Bakteriol Hyg I Abt Orig B,
174: 1-237 (in German).
Bauer J & Sabel A (1975) [The influence of oxygen on the
polymerisation of vinyl chloride with the example of suspension
polymerisation.] Angew Makromol Chem, 47: 15-27 (in German).
Becker KH, Biehl HM, Bruckmann P, Fink EH, Führ F, Klöpffer W, Zellner
R, & Zetzsch C (1984) [Methods of the ecotoxicological evaluation of
chemicals: Photochemical degradation in the gas phase.] In: [Volume 6:
OH reaction rate constants and tropospheric lifetimes of selected
environmental chemicals -- Report 1980-1983 of the Atomic Research
Unit, Jülich.] Bonn, Germany, Ministry for Research & Technology, pp
5-18 (in German).
Belli S, Bertazzi PA, Comba P, Foa V, Maltoni C, Masina A, Pirastu R,
Reggiani A, & Vigotti MA (1987) A cohort study on vinyl chloride
manufactures in Italy: Study design and preliminary results. Cancer
Lett, 35: 253-261.
Bencko V & Ungváry G (1994) Risk assessment of chemicals - a central
European perspective. Cent Eur J Public Health, 2: 70-72.
Bencko V, Wagner V, Wagnerová M, Bátora J, & Hrebacka J (1988)
Immunobiochemical profiles of workers differing in the degree of
occupational exposure to vinyl chloride. J Hyg Epidemiol Microbiol
Immunol, 32: 375-384.
Benfenati E, Natangelo M, Davoli E, & Fanelli R (1991) Migration of
vinyl chloride into PVC-bottled drinking-water assessed by gas
chromatography-mass spectrometry. Food Chem Toxicol, 29: 131-134.
Berens AR, Crider LB, Tomanek CL, & Whitney J (1975) Analysis in vinyl
chloride in PVC powders by head-space gas chromatography. Appl Polym
Sci, 19: 3169-3172.
Bergert KH & Betz V (1976) [Organic contaminants in urban air
- Results of a GC/MS analysis.] Chem Ing Tech, 48: 47-48 (in German).
Berman E & Dong J (1994) Photocatalytic decomposition of organic
pollutants in gas streams. Chem Oxid, 3: 183-189.
Besemer AC, Hollander JC, Huldy HJ, Laurier MD, Maas RJ, & Mulder HC
(1984) [Criteria document on vinyl chloride.] The Hague, The
Netherlands, Ministry of Housing.
Bi W, Wang Y, Huang M, & Meng D (1985) Effect of vinyl chloride on
testis in rats. Ecotoxicol Environ Saf, 10: 281-289.
BIA (German Professional Associations' Institute for Occupational
Safety) (1996) [Report on the occupational exposure to cancer-inducing
substances.] Sankt Augustin, Germany, Professional Association Office
(HVBG), pp 1-68 (in German).
BIA (German Professional Associations' Institute for Occupational
Safety) (1997) [List of hazardous substances, 1997 -- Safety and
health protection at work.] Sankt Augustin, Germany, Professional
Association Office (HVBG), 112 pp (BIA Report 10/97) (in German).
Black CM, Welsh KI, Walker AE, Catoggio LJ, Bernstein RM, McGregor AR,
& Lloyd Jones JK (1983) Genetic susceptibility to scleroderma-like
syndrome induced by vinyl chloride. Lancet, 1: 53-55.
Black C, Pereira S, McWhirter A, Welsh K, & Laurent R (1986) Genetic
susceptibility to scleroderma-like syndrome in symptomatic and a
symptomatic workers exposed to vinyl chloride. J Rheumatol, 13:
1059-1062.
Boettner EA, Ball G, & Weiss B (1969) Analysis of the volatile
combustion products of vinyl plastics. J Appl Polym Sci, 13: 377-391.
Bogdanikowa B & Zawilska J (1984) [Immune complexes in the serum of
patients occupationally exposed to vinyl chloride.] Przeglad Lek, 41:
253-257 (in Russian)
Boivin S, Pedron S, Bertrand S, Desmouliere A, Martel-Planche G,
Bancel B, Montesano R, Trepo C, & Marion MJ (1997) Myofibroblast-like
cells exhibiting a mutated p53 gene isolated from liver human tumor of
a worker exposed to vinyl chloride. In: Wisse E, Knook DL, & Balabaud
C ed. Cells of the hepatic sinusoid. Leiden, The Netherlands, Kupffer
Cell Foundation, pp 449-451.
Boivin-Angèle S, Lefrancois L, Froment O, Spiethoff A, Bogdanffy MS,
Wegener K, Wesch H, Barbin A, Bancel B, Trépo C, Bartsch H, Swenberg
J, & Marion MJ (in press) Ras gene mutations in vinyl
chloride-induced liver tumors are carcinogen-specific but vary with
cell type and species. Int J Cancer.
Bol'shakov AM (1969) [Working conditions in the production of
synthetic leather.] Gig Vopr Proizvod Primen Polim Mater, 1969: 47-52
(in Russian)
Bolt HM (1986) Metabolic activation of vinyl chloride, formation of
nucleic acid adducts and relevance to carcinogenesis. In: Singer B &
Bartsch H ed. The role of cyclic nucleic acid adducts in
carcinogenesis and mutagenesis. Lyon, International Agency for
Research on Cancer, pp 261-268 (IARC Scientific Publications No. 70).
Bolt HM (1994) Vinyl halides, haloaldehydes and monohaloalkanes. In:
Hemmininki K, Dipple A, Shuker DEG, Kadlubar FF, Segerbäck D, &
Bartsch H ed. DNA adducts: Identification and biological significance.
Lyon, International Agency for Research on Cancer pp 141-150 (IARC
Scientific Publications No. 125).
Bolt HM, Kappus H, Buchter A, & Bolt W (1976) Disposition of
[1,2-14C] vinyl chloride in the rat. Arch Toxicol, 35: 153-162.
Bolt HM, Laib RJ, Kappus H, & Buchter A (1977) Pharmacokinetics of
vinyl chloride in the rat. Toxicology, 7: 179-188.
Bolt HM, Filser JG, Laib RJ, & Ottenwälder H (1980) Binding kinetics
of vinyl chloride and vinyl bromide at very low doses. Arch Toxicol,
3(suppl): 129-142.
Bönnighausen KH (1986) [Production and properties of vinyl chloride.]
In: Becker CW & Braun A ed. [Plastics handbook.] Munich, Wien, Carl
Hanser Verlag, pp 38-57 (in German).
Borys E, Mroczkowska-Slupska MM, & Kusmierek JT (1994) The induction
of adaptive response to alkylating agents in Escherichia coli
reduced the frequency of specific C->T mutations in
chloroacetaldehyde-treated M13 glyU phage. Mutagenesis, 9: 407-410.
Bouscaren R, Frank R, & Veldt C (1987) Environment and quality of
life: Hydrocarbons identification of air quality problems in member
states of the European communities (Contract No.
84-B-6642-11-015-11-N) -- Final report. Brussels, Commission of the
European Communities, pp 39-173.
Boyden BH, Banh DT, Huckabay HK, & Fernandes JB (1992) Using inclined
cascade aeration to strip chlorinated VOCs from drinking water. Am
Water Waste Assoc, 84: 62-69.
Bradley PM & Chapelle FH (1996) Anaerobic mineralization of vinyl
chloride in Fe(III)-reducing, aquifer sediments. Environ Sci Technol
30: 2084-2086.
Bradley PM & Chapelle FH (1998a) Effect of contaminant concentration
on aerobic microbial mineralization of DCE and VC in stream-bed
sediments. Environ Sci Technol, 32: 553-557.
Bradley PM & Chapelle FH (1998b) Microbial mineralization of VC and
DCE under different terminal electron accepting conditions. Anaerob
Clin Microbiol, 4: 81-87.
Brady J, Liberatore F, Harper P, Greenwald P, Burnett W, Davies JNP,
Bishop M, Polan A, & Viana N (1977) Angiosarcoma of the liver: An
epidemiologic survey. J Natl Cancer Inst, 59: 1383-1385.
Brandt-Rauf PW, Chen JM, Marion MJ, Smith SJ, Luo JC, Carney W, &
Pincus MR (1996) Conformational effects in the p53 protein of
mutations induced during chemical carcinogenesis: Molecular dynamic
and immunologic analyses. J Protein Chem, 15: 367-375.
Brauch H-J, Kühn W, & Werner P (1987) [Vinyl chloride in contaminated
groundwater.] Vom Wasser, 68: 23-32 (in German).
Breder CV, Dennison JL, & Brown ME (1975) Gas-liquid chromatographic
determination of vinyl chloride in vinyl chloride polymers,
food-simulating solvents, and other samples. J Assoc Off Anal Chem,
58: 1214-1220.
Brown KW & Donnelly KC (1988) An estimation of the risk associated
with the organic constituents of hazardous and municipal waste
landfill leachates. Hazard Waste Hazard Mater, 5: 1-30.
Brown ER, Sinclair T, Keith L, Beamer P, Hazdra JJ, Nair V, &
Callaghan O (1977) Chemical pollutants in relation to diseases in
fish. Ann NY Acad Sci, 298: 535-546
Bruckmann P & Mülder W (1982) [The content of organic trace substances
in landfill gases.] Müll Abfall, 12: 339-346 (in German).
Bruckmann P, Kersten W, Funcke W, Balfanz E, König J, Theisen J, Ball
M, & Päpke O (1988) The occurrence of chlorinated and other organic
trace compounds in urban air. Chemosphere, 17: 2363-2380.
BUA (German Chemical Society Advisory Committee on Existing Chemicals
of Environmental Relevance) (1989) [Vinyl chloride.] Weinheim,
Germany, VCH Verlagsgesellschaft mbH, pp 1-63 (BUA Report 35) (in
German).
BUA (German Chemical Society Advisory Committee on Existing Chemicals
of Environmental Relevance) (1993) [OH-Radicals in the troposphere:
Concentration and effects.] Stuttgart, S. Hirzel Wissenschaftliche
Verlagsgesellschaft, pp 1-163 (BUA Report 100) (in German).
Buchter A, Bolt HM, & Kappus H (1977) [The distribution of
1,2-14C-vinyl chloride in rat tissue.] Int Arch Occup Environ Health,
39: 27-32 (in German).
Buchter A, Bolt HM, Filser J, Georgens HW, Laib RJ, & Bolt W (1978)
[Pharmacokinetics and carcinogenesis of vinyl chloride: occupational
risk evaluation.] Verh Dtsch Ges Arbeitsmed, 18: 111-124 (in German).
Buchter A, Filser JG, Peter H, & Bolt HM (1980) Pharmacokinetics of
vinyl chloride in the Rhesus monkey. Toxicol Lett, 6: 33-36.
Buffler PA, Wood S, Eifler C, Suarez L, & Kilian DJ (1979) Mortality
experience of workers in a vinyl chloride monomer production plant. J
Occup Med, 41: 195-203.
Burmaster DE (1982) The new pollution: Groundwater contamination.
Environment, 24: 33-36.
Burns and Roe Industrial Services Organization Paramas (1982) Fate and
priority pollutants in publicly owned treatment works. In : Fate and
priority pollutants in publicly owned treatment works -- Final Report,
Volumes I and II. Washington, DC, US Environmental Protection Agency,
pp 1-88 (EPA 440/1-82/303).
Byron D, Engholm G, Englund A, & Westerholm P (1976) Mortality and
cancer morbidity in a group of Swedish VCM and PVC production workers.
Environ Health Perspect, 17: 167-170.
Callahan M, Slimak M, Gabel N, May I, Fowler C, Freed R, Jennings P,
Durfee R, Whitmore F, Maesstri B, Mabey W, Hoit B, & Gould C (1979)
Water-related environmental fate of 129 priority pollutants. In:
Water-related environmental fate of 129 priority pollutants -- Volume
II: Halogenated aliphatic hydrocarbons, halogenated ethers, monocyclic
aromatics, phthalate esters, polycyclic aromatic hydrocarbons,
nitrosamines, and miscellaneous compounds. Springfield, Virginia,
Versar Inc., pp 49/1-49/10 (EPA/440/4-79/029b).
Camarena E & Coy CA (1984) Regulation of toxic or hazardous air
contaminants from land disposal sites. In: Regulation of toxic or
hazardous air contaminants from land disposal sites. For presentation
at the 77th Annual Meeting of the Air Pollution Control Association,
San Francisco, California, 24-29 June 1984. Pittsburgh, Pennsylvania,
Air Pollution Control Association, pp 1-15.
Caputo A, Viola PL, & Bigott A (1974) Oncogenicity of vinyl chloride
at low concentrations in rats and rabbits. Med Sci, 2: 1582.
Carassiti V, Chiorboli C, Bignozzi CA, Ferri A, & Maldotti A (1977)
Atmospheric photo-oxidation of vinyl chloride: A generalized treatment
of the relative rates of product formation. Ann Chim, 67: 499-512.
Carr CJ, Burgison RM, Vitcha JF, & Krantz JC (1949) Chemical
constitution of hydrocarbons and cardiac automaticity. J Pharmacol Exp
Ther, 97: 1-3.
Carroll WF, Borrelli FE, Garrity PJ, Jacobs RA, Lewis JW, McCreedy RL,
& Weston AF (1996) Characterization of emissions of dioxins and furans
from ethylene dichloride (EDC), vinyl chloride (VCM) and
polyvinylchloride (PVC) facilities in the United States: I. Resin,
treated wastewater, and ethylene dichloride. Organohalog Compd, 27:
62-67.
Carter SR & Jewell WJ (1993) Biotransformation of tetrachloroethylene
by anaerobic attached-films at low temperatures. Water Res, 27:
607-615.
Castro CE (1993) Biodehalogenation: The kinetics and rates of the
microbial cleavage of carbon-halogen bonds. Environ Toxicol Chem, 12:
1609-1618.
Castro CE, Riebeth DM, & Belser NO (1992a) Biodehalogenation: The
metabolism of vinyl chloride by Methylosinus trichosporium OB-3b. A
sequential oxidative and reductive pathway through chloroethylene
oxide. Environ Toxicol Chem, 11: 749-755.
Castro CE, Wade RS, Riebeth DM, Bartnicki EW, & Belser NO (1992b)
Biodehalogenation: Rapid metabolism of vinyl chloride by a soil
Pseudomonas sp. direct hydrolysis of a vinyl C-Cl bond. Environ
Toxicol Chem, 11: 757-764.
Castro CE, Helvenston MC, & Belser NO (1994) Biodehalogenation,
reductive dehalogenation by Methanobacterium thermoautotrophicum.
Comparison with nickel(I)octaethylisobacteriochlorin anion. An F-430
model. Environ Toxicol Chem, 13: 429-433.
CEC (1978) Council directive of 29 June 1978 on the approximation of
the laws, regulations and administrative provisions of the Member
States on the protection of the health of workers exposed to vinyl
chloride monomer. Off J Eur Communities, L197: 12-18.
CEQ (Council on Environmental Quality) (1981) Contamination of ground
water by toxic organic chemicals. In: Contamination of ground water by
toxic organic chemicals. Washington, DC, US Government Printing
Office, pp 1-107.
Chan RKS, Anselmo KJ, Reynolds CE, & Worman CH (1978) Diffusion of
vinyl chloride from PVC packaging material into food simulating
solvents. Polym Eng Sci, 18: 601-607.
Chang H-L & Alvarez-Cohen L (1996) Biodegradation of individual and
multiple chlorinated aliphatic hydrocarbons by methane-oxidizing
cultures. Appl Environ Microbiol, 62: 3371-3377.
Charlton J, Chow A, & Gesser HD (1983) Accidental release of vinyl
chloride: The train derailment near MacGregor, Manitoba. In: Saxena J
ed. Hazard assessment of chemicals: Current developments. New York,
London, Academic Press, vol 2, pp 245-267.
CHEM-FACTS (1992) PVC. In: Chemical Intelligence Services ed.
Chem-Facts PVC. Dunstable, United Kingdom, UK Reed Telepublishing Ltd,
pp 1-224.
Chen CW & Blancato JN (1989) Incorporation of biological information
in cancer risk assessment: Example-vinyl chloride. Cell Biol Toxicol,
5: 417-444.
Chen CS & Zoltek J (1995) Organic priority pollutants in
wetland-treated leachates at a landfill in central Florida.
Chemosphere, 31: 3455-3464.
Cheng KC, Preston BD, Cahill DS, Dosanjh MK, Singer B, & Loeb LA
(1991) The vinyl chloride DNA derivative N2,3-ethenoguanine produces
G -> A transitions in Escherichia coli. Proc Natl Acad Sci (USA),
88: 9974-9978.
Chiang S-Y, Swenberg JA, Weisman WH, & Skopek TR (1997) Mutagenicity
of vinyl chloride and its reactive metabolites, chloroethylene oxide
and chloroacetaldehyde, in a metabolically competent human
B-lymphoblatoid line. Carcinogenesis, 18: 31-36.
Chiazze L, Nichols WE, & Wong O (1977) Mortality among employees of
PVC fabricators. J Occup Med, 19: 623-628.
Chiazze L, Wong O, Nichols W, & Ference L (1980) Breast cancer
mortality among PVC fabricators. J Occup Med, 22: 677-679.
Chilton J & Chilton K (1992) A critique of risk modeling and risk
assessment of municipal landfills based on US Environmental Protection
Agency techniques. Waste Manage Res, 10: 505-516.
Chiou CT, Peters LJ, & Freed VH (1979) A physical concept of
soil-water equilibria for nonionic organic compounds. Science, 206:
831-832.
Chitgopekar NP, Reible DD, & Thibodeaux LJ (1990) Modeling short range
air dispersion from area sources of non-buoyant toxics. J Air Waste
Manage Assoc, 40: 1121-1128.
Cho Y, Gorina S, Jeffrey PD, & Pavletich NP (1994) Crystal structure
of a p53 tumor suppressor-DNA complex: Understanding tumorigenic
mutations. Science, 265: 346-355.
Chung FL, Chen HJC, & Nath RG (1996) Lipid peroxidation as a potential
endogenous source for the formation of exocyclic DNA adducts.
Carcinogenesis, 17: 2105-2111.
Ciroussel F, Barbin A, Eberle G, & Bartsch H (1990) Investigations on
the relationship between DNA ethenobase adduct levels in several
organs of vinyl chloride-exposed rats and cancer susceptibility.
Biochem Pharmacol, 39: 1109-1113.
Clement International Corporation (1990) Development and validation of
methods for applying pharmacokinetic data in risk assessment. Ruston,
Louisiana, Clement International Corporation, vol 5, pp 1-103
(AAMRL-TR-90-072).
Clemmesen J (1982) Mutagenicity and teratogenicity of vinyl chloride
monomer (VCM). Mutat Res, 98: 97-100.
Clewell HJ, Gentry PR, Gearhart JM, Allen BC, & Andersen ME (1995a)
The development and validation of a physiologically based
pharmacokinetic model for vinyl chloride and its application in a
carcinogenic risk assessment for vinyl chloride. Washington, DC, US
Environmental Protection Agency, Office of Health and Environmental
Assessment/Occupational Safety and Health Administration, Directorate
of Health Standards Programs, 110 pp (Unpublished report prepared by
ACF Kayser).
Clewell HJ, Gentry PR, Gearhart JM, Allen BC, & Andersen ME (1995b)
Considering pharmacokinetic and mechanistic information in cancer risk
assessments for environmental contaminants: Examples with vinyl
chloride and trichloroethylene. Chemosphere, 31: 2561-2578.
Cline PV & Viste DR (1985) Migration and degradation patterns of
volatile organic compounds. Waste Manage Res, 3: 351-360.
Cocciarini D & Campaña H (1992) Biotreatment of EDC -- VCM wastewater
effluent. Water Sci Technol, 25: 259-261.
Cochran JW (1988) A rapid, sensitive method for the analysis of
halogenated gases in water. J High Resol Chromatogr Chromatogr Commun,
11: 663-665.
Cochran JW & Henson JM (1988) Analysis of volatile organic chemicals
in aqueous samples by purge/GC with selective water removal. J High
Resol Chromatogr Chromatogr Commun, 11: 869-873.
Codex Committee (1984) Report to the Codex Committee on Food Additives
in responses to circular letter CL 1983/20-FA, May 1983, on the
subject of packaging materials (Report cx/FA 84/11). Canada, Codex
Committee, pp 1-24.
Coenen W (1986) [Concentration of carcinogenic substances at the
workplace: An analysis of results from the BIA data bank.]
Berufsgenoss Informationsz Arb.sicherh Unvallversicher, 1: 1-5 ( in
German).
Cogliano VJ, Hiatt GFS, & Den A (1996) Quantitative cancer assessment
for vinyl chloride: Indications of early-life sensitivity. Toxicology,
111: 21-28.
Cole RH, Frederick RE, Healy RP, & Rolan RG (1984) Preliminary
findings of the priority pollutant monitoring project of the
nationwide urban runoff program. J Water Pollut Control Fed, 56:
898-908.
Conkle JP, Camp BJ, & Welch BE (1975) Trace composition of human
respiratory gas. Arch Environ Health, 30: 290-29.5
Connor MS (1984) Comparison of the carcinogenic risks from fish versus
groundwater contamination by organic compounds. Environ Sci Technol,
18: 628-631.
Conolly RB & Jaeger RJ (1979) Acute hepatoxicity of vinyl chloride and
ethylene: Modification by trichloropropene oxide, diethylmaleate, and
cysteine. Toxicol Appl Pharmacol, 50: 523-531.
Conolly RB, Jaeger RJ, & Szabo S (1978) Acute hepatotoxicity of
ethylene, vinyl fluoride, vinyl chloride, and vinyl bromide after
Aroclor 1254 pretreatment. Exp Mol Pathol, 28: 25-33.
Cooper WC (1981) Epidemiologic study of vinyl chloride workers:
Mortality through December. Environ Health Perspect, 41: 101-106.
Cordier JM, Fievez C, Lefevre MJ, & Sevrin A (1966) Acroostérolyse et
lésions cutanées associées chez deux ouvriers affectés au nettoyage
d'autoclaves. Cah Méd Trav, 4: 3-39.
Cotruvo JA, Goldhaber S, & Vogt C (1986) Regulatory significance of
organic contamination in the decade of the 1980s. In: Ram NR ed.
Organic carcinogens in drinking water. New York, Wiley, pp 511-530.
Cox RA, Eggleton AEJ, & Sandalls FJ (1974) Photochemical reactivity of
vinyl chloride. London, Her Majesty's Stationery Office (Report No.
AERE-R-7820).
Cox E, Edwards E, Lehmicke L, & Major D (1995) Intrinsic
biodegradation of trichloroethene and trichloroethane in a sequential
anaerobic-aerobic aquifer. In: Hinchee RE, Wilson JT, & Downey DC ed.
Intrinsic bioremediation. Columbus, Ohio, Battelle Press, pp 223-231.
Craun GF (1984) Health aspects of groundwater pollution. In: Bitton G
& Gerba CP ed. Groundwater pollution microbiology. New York,
Chichester, Brisbane, Toronto, John Wiley & Sons, pp 135-179.
Creech JL & Johnson MN (1974) Angiosarcoma of liver in the manufacture
of polyvinyl chloride. J Occup Med, 16: 150-151.
Crump KS (1984) An improved procedure for low-dose carcinogenic risk
assessment from animal data. J Environ Pathol Toxicol, 5: 339-348.
Crutzen PJ (1982) The global distribution of hydroxyl. In: Goldberg ED
ed. Atmospheric chemistry. Berlin, Heidelberg, New York, Springer
Verlag, pp 313-328.
Dahar WS, Bond GG, McLaren EA, Sabel FL, Lipps TE, & Cook RR (1988)
Update to vinyl chloride mortality study. J Occup Med, 30: 648-649.
Daniel FB, Deangelo AB, Stober JA, Olson GR, & Page NP (1992)
Hepatocarcinogenicity of chloral hydrate, 2-chloracetaldehyde, and
dichloroacetic acid in the male B6C3F1 mouse. Fundam Appl Toxicol, 19:
159-168.
Daniels GA & Proctor DE (1975) VCM extraction from PVC bottles. Mod
Packag, April: 45-48.
Dann T & Wang D (1992) Volatile organic compound measurements in
Canadian urban and rural areas: 1989-1990. Presented at the 85th
Annual Meeting of the Air and Waste Management Association, Kansas
City, Missouri, 21-26 June 1992. Pittsburgh, Pennsylvania, Air and
Waste Management Association, pp 1-16 (Paper 92-75.16).
Danziger H (1960) Accidental poisoning by vinyl chloride: Report of
two cases. Can Med Assoc J, 82: 828-830.
Davies IW & Perry R (1975) Vinyl chloride monomer's effect on liquids
in PVC bottles. Environ Pollut Manage, 5: 22-23.
Davis JW & Carpenter CL (1990) Aerobic biodegradation of vinyl
chloride in groundwater samples. Appl Environ Microbiol, 56:
3878-3880.
De Bruin WP, Kotterman MJJ, Posthumus MA, Schraa G, & Zehnder AJB
(1992) Complete biological reductive transformation of
tetrachloroethene to ethane. Appl Environ Microbiol, 58: 1996-2000.
Deipser A (1998) Biodegradation of volatile CFCs, H-CFCs and VC in
compost and marl. Waste Manage Res, 16: 330-341
De Jong G, Van Sittert NJ, & Natarajan AT (1988) Cytogenetic
monitoring of industrial populations potentially exposed to genotoxic
chemicals and of control populations. Mutat Res, 204: 451-464.
DeLassus PT & Schmidt DD (1981) Solubilities of vinyl chloride and
vinylidene chloride in water. J Chem Eng Data, 26: 274-276.
De Meester C, Duverger van Bogaert M, Lambotte-Vandepaer M, Roberfroid
M, Poncelet F, & Mercier M (1980) Mutagenicity of vinyl chloride in
the Ames test: Possible artifacts related to experimental conditions.
Mutat Res, 77: 175-179.
Demkowicz-Dobrzanski K, Nalecz-Jawecki G, Wawer Z, Bargiel T, Zak M, &
Sawicki J (1993) Toxicity of neutralized wastes following a synthesis
of chlorinated hydrocarbons to Daphina magna and Chlorella sp.
Sci Total Environ, 1993(suppl): 1151-1158.
Depret P & Bindelle JP (1998) Vinyl chloride leak detection and
fugitive emission monitoring experience from the west-European PVC
industry. In: 1st International Conference on Protecting the
Environment: Controlling leakages in process industry, Rome, Italy,
11-12 May 1998. Cranfield, United Kingdom, BHR Publishers, pp 167-188
(BHR Series No. 30).
DeVivo I, Marion M-J, Smith SJ, Carney WP, & Brandt-Rauf PW (1994)
Mutant c-Ki- ras p 21 protein in chemical carcinogenesis in humans
exposed to vinyl chloride. Cancer Causes Control, 5: 273-278.
Diachenko GW, Breder CV, Brown ME, & Dennison JL (1977) Gas-liquid
chromatographic headspace technique for determination of vinyl
chloride in corn oil and three food-simulating solvents. J Assoc Off
Anal Chem, 60: 570-575.
Dickman MD & Rygiel G (1993) Density, diversity and incidence of
deformities of benthic invertebrates near a vinyl chloride discharge
point source in the Niagara River watershed. In: Gorsuch JW, Dwyer FJ,
Ingersoll CG, & La Point TW ed. Environmental toxicology and risk
assessment. Philadelphia, Pennsylvania, American Society for Testing
and Materials, vol 2, pp 663-680.
Dickman M, Lan Q, & Matthews B (1989) Teratogens in the Niagara River
watershed as reflected by chironomid (Diptera: Chironomidae) labial
plate deformities. Water Pollut Res J Can, 24: 47-79
Dieter HH & Kerndorff H (1993) Presence and importance of
organochlorine solvents and other compounds in Germany's groundwater
and drinking water. Ann Ist Super Sanita, 29: 263-277.
Dietz A, Langbein G, & Permanetter W (1985) [Vinylchloride-induced
hepato-cellular carcinoma.] Klin Wochenschr, 63: 325-331 (in German).
Dilling WL (1977) Interphase transfer processes: II. Evaporation rates
of chloromethanes, ethanes, ethylenes, propanes and propylenes from
dilute aqueous solutions -- Comparisons with theoretical predictions.
Environ Sci Technol, 11: 405-409.
Dilling WL, Tefertiller NB, & Kallos GJ (1975) Evaporation rates and
reactivities of methylene chloride, chloroform, 1,1,1-trichloroethane,
trichloroethylene, tetrachloroethylene, and other chlorinated
compounds in dilute aqueous solutions. Environ Sci Technol, 9:
833-838.
Dilling WL, Bredeweg CJ, & Tefertiller NB (1976) Organic
photochemistry. Simulated atmospheric photodecomposition rates of
methylene chloride, 1,1,1-trichloroethane, trichloroethylene,
tetrachloroethylene, and other compounds. Environ Sci Technol, 10:
351-356.
Dimmick WF (1981) EPA programs of vinyl chloride monitoring in ambient
air. Environ Health Perspect, 41: 203-206.
DiStefano TD, Gossett JM, & Zinder SH (1991) Reductive dechlorination
of high concentrations of tetrachloroethene to ethene by an anaerobic
enrichment culture in the absence of methanogenesis. Appl Environ
Microbiol, 57: 2287-2292.
Dobecki M & Romaniwicz B (1993) [Occupational exposure to toxic
substances during the production of vinyl chloride and chlorinated
organic solvents.] Med Prac, 44: 99-102 (in Polish).
Dolan ME & McCarty PL (1995a) Methanotrophic chloroethene
transformation capacities and 1,1-dichloroethene transformation
product toxicity. Environ Sci Technol, 29: 2741-2747.
Dolan ME & McCarty PL (1995b) Small-column microcosm for assessing
methane-stimulated vinyl chloride transformation in aquifer samples.
Environ Sci Technol, 29: 1892-1897.
Dolk H, Vrijheid M, Armstrong B, Abramsky L, Bianchi F, Garne E, Nelen
V, Robert E, Scott JES, Stone D, & Tenconi R (1998) Risk of congenital
anomalies near hazardous-waste landfill sites in Europe: The
EUROHAZCON study. Lancet, 352: 423-427.
Doll R (1988) Effects of exposure to vinyl chloride. Scand J Work
Environ Health, 14: 61-78.
Dosanjh MK, Chenna A, Kim E, Fraenkel-Conrat H, Samson L, & Singer B
(1994) All four known cyclic adducts formed in DNA by the vinyl
chloride metabolite chloroacetaldehyde are released by a human DNA
glycosylase. Proc Natl Acad Sci (USA), 91: 1024-1028.
Draminski W & Trojanowska B (1981) Chromatographic determination of
thiodiglycolic acid - a metabolite of vinyl chloride. Arch Toxicol,
48: 289-292.
Dreher EL (1986) Chloroethylenes. In: Gerhartz W, Yamamoto YS,
Campbell FT, Pfefferkorn R, & Rounsaville JF ed. Ullmann's
encyclopedia of industrial chemistry, 5th completely revised.
-- Volume A6: Ceramics to chlorohydrins. Weinheim, Germany, VCH
Verlagsgesellschaft mbH, pp 283-294.
Dressman RC & McFarren EF (1978) Determination of vinyl chloride
migration from polyvinyl chloride pipe into water. Am Water Works
Assoc J, 70: 29-30.
Drevon C & Kuroki T (1979) Mutagenicity of vinyl chloride, vinylidene
chloride and chloroprene in V79 Chinese hamster cells. Mutat Res, 67:
173-182.
Drew RT, Boorman GA, Haseman JK, McConnell EE, Busey WM, & Moore JA
(1983) The effect of age and exposure duration on cancer induction by
a known carcinogen in rats, mice and hamsters. Toxicol Appl Pharmacol,
68: 120-130.
Driscoll JN, Duffy M, Pappas S, & Webb M (1987) Analysis for purgeable
organics in water by capillary GC/PID-ElCD. J Chromatogr Sci, 25:
369-375.
Drozdowicz BZ & Huang PC (1977) Lack of mutagenicity of vinyl chloride
in two strains of Neurospora Crassa. Mutat Res, 48: 43-50.
D'Souza RW & Andersen ME (1988) Physiologically based pharmacokinetic
model for vinylidene chloride. Toxicol Appl Pharmacol, 95: 230-240
Du CL & Wang JD (1998) Increased morbidity odds ratio of primary liver
cancer and cirrhosis of the liver among vinyl chloride monomer
workers. Occup Environ Med, 55: 528-532.
Du JT, Tseng MT, & Tamburro CH (1982) The effect of repeated vinyl
chloride exposure on rat hepatic metabolizing enzymes. Toxicol Appl
Pharmacol, 62: 1-10.
Du C-L, Kuo M-L, Chang H-L, Sheu T-J, & Wang J-D (1995) Changes in
lymphocyte single strand breakage and liver function of workers
exposed to vinyl chloride monomer. Toxicol Lett, 77: 379-385.
Du C-L, Chan C-C, & Wang J-D (1996) Comparison of personal and area
sampling strategies in assessing workers' exposure to vinyl chloride
monomer. Bull Environ Contam Toxicol, 56: 534-542.
Ducatman A, Hirschhorn K, & Selikoff IJ (1975) Vinyl chloride exposure
and human chromosome aberrations. Mutat Res, 31: 163-168.
Dulson W (1978) In: Heller A ed. [Organic chemical pollutants in the
atmosphere.] Stuttgart, Gustav Fischer Verlag, pp 84-85 (Reports
Series on Water, Soil, and Air) (in German).
Dummer G & Schmidhammer L (1983) [A new process for the removal of
aliphatic chlorinated hydrocarbons from wastewater using absorber
resins.] VDI-Ber, 545: 885-901 (in German).
Dummer G & Schmidhammer L (1984) [A new process for the removal of
aliphatic chlorinated hydrocarbons from wastewater using absorber
resins.] Chem Ing Tech, 56: 242-243 (in German).
Duprat F, Fabry JP, Gradiski D, & Magadur JL (1977) Metabolic approach
to industrial poisoning: Blood kinetics and distribution of
[14C]-vinyl chloride monomer (VCM). Acta Pharmacol Toxicol, 41(suppl):
142-143.
Dyksen JE & Hess AF (1982) Alternatives for controlling organics in
groundwater supplies. J Am Water Works Assoc, 74: 394-403.
Eberle G, Barbin A, Laib RJ, Ciroussel F, Thomale J, Bartsch H, &
Rajewsky MF (1989) 1,N6-etheno-2'-deoxyadenosine and
3,N4-eteno-2'-deoxycytidine detected by monoclonal antibodies in lung
and liver DNA of rats exposed to vinyl chloride. Carcinogenesis, 10:
209-212.
Ebihara J (1982) [Cause-specific mortality study on VCM workers.] J
Sci Lab, 58: 383-389 (in Chinese).
ECETOC (1983) Experimental assessment of the phototransformation of
chemicals in the atmosphere. Brussels, European Centre for
Ecotoxicology and Toxicology of Chemicals, pp 1-36 (ECETOC Monograph
No. 7).
ECETOC (1988) The mutagenicity and carcinogenicity of vinyl chloride:
A historical review and assessment. Brussels, European Centre for
Ecotoxicology and Toxicology of Chemicals, pp 1-147 (ECETOC Technical
Report No. 31).
Eckardt F, Muliawan H, De Ruiter N, & Kappus H (1981) Rat hepatic
vinyl chloride metabolites induce gene conversion in the yeast strain
d7 rad in vitro and in vivo. Mutat Res, 91: 381-390.
Edmonds LD, Falk H, & Nissim JE (1975) Congenital malformations and
vinyl chloride. Lancet, 29: 1098.
Edmonds LD, Anderson C, Flynt JW, & James LM (1978) Congenital central
nervous system malformations and vinyl chloride monomer exposure: A
community study. Teratology, 17: 137-142.
Ehtesham-Ud Din AF-M, Nordbo E, & Underdal B (1977) The content of
vinylchloride monomer (VCM) in foods packed in polyvinylchloride
(PVC). Lebensm.wiss Technol, 10: 33-35.
Eisenreich SJ, Looney BB, & Thornton JD (1981) Airborne organic
contaminants in the Great Lakes ecosystem. Environ Sci Technol, 15:
30-38.
El Ghissassi FE, Barbin A, Nair J, & Bartsch H (1995a) Formation of
1,N6-ethenoadenine and 3,N4-ethenocytosine by lipid peroxidation
products and nucleic acid bases. Chem Res Toxicol, 8: 278-283.
El Ghissassi F, Biovin S, Lefrançois L, Barbin A, & Marion M-J (1995b)
Glutathione transferase Mu1-1 (GSTM1) genotype in individuals exposed
to vinyl chloride: Relation to "vinyl chloride disease" and liver
angiosarcoma. Clin Chem, 41: 1922-1924.
El Ghissassi F, Barbin A, & Bartsch H (1998) Metabolic activation of
vinyl chloride by rat liver microsomes: Low-dose kinetics and
involvement of cytochrome P450 2E1. Biochem Pharmacol, 55: 1445-1452.
Elliott P & Kleinschmidt I (1997) Angiosarcoma of the liver in Great
Britain in proximity to vinyl chloride sites. Occup Environ Med, 54:
14-18.
Ellis DE, Lutz EJ, Klecka GM, Pardiek DL, Salvo JJ, Heitkamp MA,
Gannon DJ, Mikula CC, Vogel CM, Sayles GD, Kampbell DH, Wilson JT, &
Maiers DT (1996) Remediation technology development forum intrinsic
remediation project at Dover Air Force base, Delaware. In: Proceedings
of the Symposium on Natural Attenuation of Chlorinated Organics in
Ground Water, 11-13 September 1996. Washington, DC, US Environmental
Protection Agency, pp 93-97 (EPA 540/R-96/509).
Elmore JD, Wong JL, Laumbach AD, & Streips UN (1976) Vinyl chloride
mutagenicity via the metabolites chlorooxirane and chloroacetaldehyde
monomer hydrate. Biochim Biophys Acta, 442: 405-419.
EMO (1992) Field investigation report. In: EMO field investigation
report -- Volume 5. Richland, Washington, Environmental Management
Operations, pp 6.1-7.6 (EMO Report No. 1028).
Ensign SA, Hyman MR, & Arp DJ (1992) Cometabolic degradation of
chlorinated alkenes by alkene monooxygenase in a propylene-grown
Xanthobacter strain. Appl Environ Microbiol, 58: 3038-3046.
Ensley BD (1991) Biochemical diversity of trichloroethylene
metabolism. Annu Rev Microbiol, 45: 283-299.
Euro Chlor (1999) Euro Chlor risk assessment for the marine
environment, OSPARCOM Region -- North Sea: Vinylchoride. Brussels,
Euro Chlor, pp 1-25.
European Council of Vinyl Manufacturers (1994) On the environmental
impact of the manufacture of poly vinyl chloride (PVC): A description
of best available techniques. Brussels, European Council of Vinyl
Manufacturers, 50 pp.
Evans DMD, Williams WJ, & Kung ITM (1983) Angiosarcoma and
hepatocellular carcinoma in vinyl chloride workers. Histopathology, 7:
377-388.
Ewers J, Freier-Schröder D, & Knackmuss H-J (1990) Selection of
trichloroethene (TCE) degrading bacteria that resist inactivation by
TCE. Arch Mikrobiol, 154: 410-413.
Fairley P (1997) Greenpeace plans earth day blitz against vinyl
industry. Chem Week, April 16: 8.
Falk H & Steenland NK (1998) Vinyl chloride and polyvinyl chloride.
In: Rom WN ed. Environmental and occupational medicine, 3rd ed.
Philadelphia, Pennsylvania, Lippincou-Raven Publishers, pp 1251-1261.
Falk H, Creech JLJ, Heath CWJ, Johnson MN, & Key MM (1974) Hepatic
disease among workers at a vinyl chloride polymerisation plant. J Am
Med Assoc, 230: 59-63.
Fathepure BZ & Tiedje JM (1994) Reductive dechlorination of
tetrachloroethylene by a chlorobenzoate-enriched biofilm reactor.
Environ Sci Technol, 28: 746-752.
Fayad NM, Sheikheldin SY, Al-Malack MH, El-Mubarak AH, & Khaja N
(1997) Migration of vinyl chloride monomer (VCM) and additives into
PVC bottled drinking water. Environ Sci Eng, 32: 1065-1083.
Fedtke N, Walker VE, & Swenberg JA (1989) Determination of
7-(2-oxoethyl)guanine and N2,3-ethenoguanine in DNA hydrolysates by
HPLC. Arch Toxicol, 13(suppl): 214-218.
Fedtke N, Boucheron JA, Turner MJ, & Swenberg JA (1990a) Vinyl
chloride-induced DNA adducts. I. Quantitative determination of
N2,3-ethenoguanine based on electrophore labeling. Carcinogenesis,
11: 1279-1285.
Fedtke N, Boucheron JA, Walker VE, & Swenberg JA (1990b) Vinyl
chloride-induced DNA adducts: II. Formation and persistence of
7-(2'-oxoethyl)guanine and N2,3-ethenoguanine in rat tissue DNA.
Carcinogenesis, 11: 1287-1292.
Fernando RC, Nair J, Barbin A, Miller JA, & Bartsch H (1996) Detection
of 1,N6-ethenodeoxyadenosine and 3,N4-ethenodeoxycytidine by
immunoaffinity/32P-postlabelling in liver and lung DNA of mice treated
with ethyl carbamate (urethane) or its metabolites. Carcinogenesis,
17: 1711-1718.
Feron VJ & Kroes P (1979) One-year time-sequence inhalation toxicity
study of vinyl chloride in rats. II. Morphological changes in the
respiratory tract, ceruminous gland, brain, kidneys, heart and spleen.
Toxicology, 13: 131-141.
Feron VJ, Speek AJ, Willems MI, Van Battum D, & De Groot AP (1975)
Observations on the oral administration and toxicity of vinyl chloride
in rats. Food Cosmet Toxicol, 13: 633-638.
Feron VJ, Kruysse A, & Til HP (1979a) One-year time sequence
inhalation toxicity study of vinyl chloride in rats: I. Growth,
mortality, haematology, clinical chemistry, and organ weights.
Toxicology, 13: 25-28.
Feron VJ, Spit BJ, Immel HR, & Kroes R (1979b) One-year time-sequence
inhalation toxicity study of vinyl chloride in rats: III.
Morphological changes in the liver. Toxicology, 13: 143-154.
Feron VJ, Hendriksen CFM, Speek AJ, Til HP, & Spit BJ (1981) Lifespan
oral toxicity study of vinyl chloride in rats. Food Cosmet Toxicol,
19: 317-333.
Filatova VS & Gronsberg ES (1957) [Sanitary-hygienic conditions of
work in the production of polychlorvinylic tar and measures of
improvement.] Gig i Sanit, 22: 38-42 (in Russian).
Filatova VS, Balakonova PH, & Gronsberg ES (1958) [Hygienic
characteristics of vinyl chloride.] Gig Tr Prof Zabol, 2: 6-9 (in
Russian).
Filser JG & Bolt HM (1979) Pharmacokinetics of halogenated ethylenes
in rats. Arch Toxicol, 42: 123-136.
Finnish Government (1992) [Resolution 919 on work with vinyl
chloride], 4 pp (in Finnish).
Fiorenza S, Hockman ELr, Szojka S, Woeller RM, & Wigger JW (1994)
Natural anaerobic degradation of chlorinated solvents at a Canadian
manufacturing plant. In: Hinchee RE, Leeson A, Semprini L, & Ong SK
ed. Bioremediation of chlorinated and polycyclic aromatic hydrocarbon
compounds. Boca Raton, Florida, Lewis Publishers, Inc., pp 277-286.
Fishbein L (1979) Potential halogenated industrial carcinogenic and
mutagenic chemicals: I.Halogenated unsaturated hydrocarbons. Sci Total
Environ, 11: 111-161.
Fleig I & Thiess AM (1974) [Chromosome tests in vinyl chloride exposed
workers.] Arbeitsmed Sozialmed Präventivmed, 12: 280-283 (in German).
Fleig I & Thiess AM (1978) Mutagenicity of vinyl chloride. J Occup
Med, 20: 557-561.
Fogel MM, Taddeo AR, & Fogel S (1986) Biodegradation of chlorinated
ethenes by a methane-utilizing mixed culture. Appl Environ Microbiol,
51: 720-724.
Fogel S, Findlay M, Moore A, & Leahy M (1987) Biodegradation of
chlorinated chemicals in groundwater by methane oxidizing bacteria
-- Proceedings of the National Water Well Association/ American
Petroleum Industry (NWWA/API) Conference on Petroleum Hydrocarbons and
Organic Chemicals in Ground Water: Prevention, detection and
restoration, Houston, Texas, 17-19 November 1987. Dublin, Ohio,
National Water Well Association, pp 187-206.
Fontana L, Gautherie M, Albuisson E, Fleury-Duhamel N, Meyer S, &
Catlina P (1996) Etude chronothermobiologique de phénomènes de Raynaud
secondaires à une exposition ancienne au chlorure de vinyle monomère.
Arch Mal Prof, 51: 9-18.
Forman D, Bennett B, Stafford J, & Doll R (1985) Exposure to vinyl
chloride and angiosarcoma of the liver: A report of the register of
cases. Br J Ind Med, 42: 750-753.
Fox AJ & Collier PF (1977) Mortality experience of workers exposed to
vinyl chloride monomer in the manufacture of polyvinyl chloride in
Great Britain. Br J Ind Med, 34: 1-10.
Fredrickson JK, Bolton H, & Brockman FJ (1993) In situ and on-site
bioreclamation. Environ Sci Technol, 27: 1711-1716.
Freedman DL & Gossett JM (1989) Biological reductive dechlorination of
tetrachloroethylene and trichloroethylene to ethylene under
methanogenic conditions. Appl Environ Microbiol, 55: 2144-2151.
Freitag D, Geyer H, Kraus A, Viswanathan R, Kotzias D, Attar A, Klein
W, & Korte F (1982) Ecotoxicological profile analysis: VII. Screening
chemicals for their environmental behavior by comparative evaluation.
Ecotoxicol Environ Saf, 6: 60-81.
Freitag D, Ballhorn L, Geyer H, & Korte F (1985) Environmental hazard
profile of organic chemicals. Chemosphere, 14: 1589-1616.
Frentzel-Beyme R, Schmitz T, & Thiess AM (1978) [Mortality study on
VC/PVC workers of BASF, Ludwigshafen, Rhine.] Arbeitsmed Sozialmed
Präventivmed, 130: 218-228 (in German).
Fricke C, Clarkson C, Lomnitz E, & O'Farrell T (1985) Comparing
priority pollutants in municipal sludges. BioCycle, Jan/Feb: 35-37.
Froment O, Boivin S, Barbin A, Bancel B, Trepo C, & Marion M-J (1994)
Mutagenesis of ras proto-oncogenes in rat liver tumors induced by
vinyl chloride. Cancer Res, 54: 5340-5345.
Froment O, Marion M-J, Lepot D, Contassot J-C, & Trepo C (1991)
Immunoquantitation of Von Willebrand factor (factor VIII-related
antigen) in vinyl chloride exposed workers. Cancer Lett, 61(3):
201-206.
Froneberg B, Johnson PL ,& Landrigan PJ (1982) Respiratory illness
caused by overheating of polyvinyl chloride. Br J Ind Med, 39:
239-243.
Fuchs G, Gawell BM, Albanus L, & Storach S (1975) [Vinyl chloride
monomer levels in edible fats.] Var Foeda, 27: 134-145 (in Swedish).
Fucic A & Garaj-Vrhovac V (1997) Micronucleus assay and mitotic
activity -- the methods for determination of recent vinyl chloride
monomer exposure. Environ Mol Mutagen, 29: 19-23.
Fucic A, Horvat D, & Dimitrovic B (1990a) Localization of breaks
induced by vinyl chloride in the human chromosomes of lymphocytes.
Mutat Res, 243: 95-99.
Fucic A, Horvat D, & Dimitrovic B (1990b) Mutagenicity of vinyl
chloride in man: Comparison of chromosome aberrations with
micronucleus and sister-chromatid exchange frequencies. Mutat Res,
242: 265-270.
Fucic A, Garaj-Vrhovac V, Dimitrovic B, & Skara M (1992) The
persistence of sister-chromatid exchange frequencies in men
occupationally exposed to vinyl chloride monomer. Mutat Res, 281:
129-132.
Fucic A, Garaj-Vrhovac V, Barkovic D, & Kubelka D (1994) The
sensitivity of the micronucleus assay for the detection of
occupational exposure to vinyl chloride monomer. Mutat Res, 325:
53-56.
Fucic A, Barkovic D, Garaj-Vrhovac V, Kubelka D, Ivanic B, Dabo T, &
Mijic A (1996) A nine-year follow up study of a population
occupationally exposed to vinyl chloride monomer. Mutat Res, 361:
49-53.
Fujimoto T, Rennert AM, & Wijnen MHJ (1970) Primary steps in the
photolysis of vinyl chloride. Ber Bunsenges Phys Chem, 74: 282-286.
Funes-Cravioto F, Lambert B, Lindsten J, Ehrenberg L, Natarajan AT, &
Osterman-Golkar S (1975) Chromosome aberrations in workers exposed to
vinyl chloride. Lancet, 1: 459.
Gáliková E, Tomíková K, Zigová A, Mesko D, Buchancová J, Petrisková J,
L'uptáková M, Ja, M, & Karaffová N (1994) [General health of workers
in the Nováky chemical works exposed to vinyl chloride.] Prac Lék, 46:
251-256 (in Czech).
Garaj-Vrhovac V, Fucic A, & Horvat D (1990) Comparison of chromosome
aberration and micronucleus induction in human lymphocytes after
occupational exposure to vinyl chloride monomer and microwave
radiation. Period Biol, 4: 411-416.
Gargas ML, Burgess RJ, Voisard DE, Cason GH, & Andersen ME (1989)
Partition coefficients of low-molecular-weight volatile chemicals in
various liquids and tissues. Toxicol Appl Pharmacol, 98: 87-99.
Gargas ML, Clewell HJ, & Andersen ME (1990) Gas uptake inhalation
techniques and the rates of metabolism of chloromethanes,
chloroethanes, and chloroethylenes in the rat. Inhal Toxicol, 2:
295-319.
Gay BW, Lonnemann WA, Bridbord K, & Moran JB (1975) Measurements of
vinyl chloride from aerosol sprays. Ann NY Acad Sci, 246: 286-295.
Gay BW, Hanst PL, Bufalini JJ, & Noonan RC (1976) Atmospheric
oxidations of chlorinated ethylenes. Environ Sci Technol, 10: 58-67.
Gedigk P, Müller R, & Bechtelsheimer H (1975) Morphology of liver
damage among polyvinyl chloride production workers. A report on 51
cases. Ann NY Acad Sci, 246: 278-285
Gehring PJ, Watanabe PG, & Park CN (1978) Resolution of dose-response
toxicity data for chemicals requiring metabolic activation:
Example-vinyl chloride. Toxicol Appl Pharmacol, 44: 581-591.
German Environmental Office (UBA) (1978) [Air contamination with vinyl
chloride (VC) from PVC-Products.] Umweltbundesamt Ber, 5: 1-23 (in
German).
German Expert Group on the Environment (1988) [Report on the
environment, 1987.] Stuttgart, W. Kohlhammer, pp 229-231.
German Federal Health Office (1998) [New directive on articles for
personal use.] Bundesgesetzblatt, 1: 5-36 (in German).
German Federal Health Office (BGA) (1992) [Recommendation on measures
in case of occurrence of vinyl chloride in ground water used for
drinking water production.] Bundesgesundheitsblatt, 35: 364 (in
German).
Gilbert SG, Giacin JR, Morano JR, & Rosen JD (1975) Detecting small
quantities of residual vinyl chloride monomer. Package Dev, July/Aug:
20-24.
Giri AK (1995) Genetic toxicology of vinyl chloride -- a review. Mutat
Res, 339: 1-14.
Gnesina EA & Pshenitsina AI (1980) [Time course of the heart rate and
galvanic skin reflex during night sleep in patients with chronic
occupational intoxication by vinyl chloride.] Gig Tr Prof Zabol, 5:
39-42 (in Russian).
Gnesina EA & Teklina LG (1984) [Characteristics of night sleep of
patients with chronic occupational vinyl chloride.] Gig Tr Prof Zabol,
6: 34-36 (in Russian).
Gnesina EA, Antonyuzhenko VA, Mashikhin EN, & Rostovtseva GG (1978)
[On the nature and frequency of sleep disorders in persons exposed to
the effect of vinylchloride during work.] Gig Tr Prof Zabol, 11: 16-19
(in Russian).
Going JE (1976) Sampling and analysis of selected toxic substances:
Task III -- Vinyl chloride, secondary sources. Washington, DC, US
Environmental Protection Agency, pp 1-30 (EPA 560/6-76-002).
Goldberg MS, Al-Homsi N, Goulet L, & Riberdy H (1995) Incidence of
cancer among persons living near a municipal solid waste landfill site
in Montreal, Quebec. Arch Environ Health, 50(6): 416-424.
Goodenkauf O & Atkinson JC (1986) Occurrence of volatile organic
chemicals in Nebraska ground water. Ground Water, 24: 231-233.
Gordon SJ & Meeks SA (1977) A study of gaseous pollutants in the
Houston, Texas area. AICHE Symp Ser, 73: 84-94.
Gossett JM (1987) Measurement of Henry's law constants for C1 and C2
chlorinated hydrocarbons. Environ Sci Technol, 21: 202-208.
Gossett RW, Brown DA, & Young DR (1983) Predicting the bioaccumulation
of organic compounds in marine organisms using octanol/water partition
coefficients. Mar Pollut Bull, 14: 387-392.
Grainger RG, Walker AE, & Ward AM (1980) Vinyl chloride
monomer-induced disease: Clinical, radiological and immunological
aspects. In: Preger L ed. Induced disease, drug irradiation,
occupation. New York, Grune and Stratton, pp 191-214.
Green T & Hathway DE (1975) The biological fate in rats of vinyl
chloride in relation to its oncogenicity. Chem-Biol Interact, 11:
545-562.
Green T & Hathway DE (1977) The chemistry and biogenesis of the
S-containing metabolites of vinyl chloride in rats. Chem-Biol
Interact, 17: 137-150.
Green T & Hathway DE (1978) Interactions of vinyl chloride with
rat-liver DNA in vitro. Chem-Biol Interact, 22: 211-224.
Greim H, Bonse G, Radwan Z, Reichert D, & Henschler D (1975)
Mutagenicity in vitro and potential carcinogenicity of chlorinated
ethylenes as a function of metabolic oxirane formation. Biochem
Pharmacol, 24: 2013-2017.
Greiser E, Reinl W, & Weber H (1982) [Vinyl chloride exposure and
mortality of German chemical workers in comparison to comparison of
non-exposed chemical workers and PVC workers.] Zent.bl Arbeitsmed
Arbeitsschutz Prophyl, 32: 44-62 (in German).
Grimsrud EP & Rasmussen RA (1975) Survey and analysis of halocarbons
in the atmosphere by gas chromatography-mass spectrometry. Atmos
Environ, 9: 1014-1017.
Groeneveld AHC, Zijlstra AG, Feenstra AFM, & Berends AG (1993) The
acute toxicity of vinylchloride to the Zebra fish ( Brachydanio
rerio ). Graveland, The Netherlands, Solvay Duphar BV, pp 1-19.
Grollman AP & Shibutani S (1994) Mutagenic specifity of chemical
carcinogens as determined by studies of single DNA adducts. In:
Hemmininki K, Dipple A, Shuker DEG, Kadlubar FF, Segerbäck D, &
Bartsch H ed. DNA adducts: Identification and biological significance.
Lyon, International Agency for Research on Cancer, pp 385-397 (IARC
Scientific Publications No 125).
Grosjean D & Williams EL (1992) Environmental persistence of organic
compounds estimated from structure-reactivity and linear free-energy
relationships. Unsaturated aliphatics. Atmos Environ, 26A: 1395-1405.
Groth DH, Coate WB, Ulland BM, & Hornung RW (1981) Effects of aging on
the induction of angiosarcoma. Environ Health Perspect, 41: 53-57.
Gryder-Boutet DE & Kennish JM (1988) Using headspace sampling with
capillary column GC-MS to analyze trace volatile organics in water and
wastewater. Am Water Works Assoc J, 80: 52-55.
Guengerich FP (1992) Roles of the vinyl chloride oxidation products
2-chlorooxirane and 2-chloroacetaldehyde in the in vitro formation
of etheno adducts of nucleic acid bases. Chem Res Toxicol, 5: 2-5.
Guengerich FP & Strickland TW (1977) Metabolism of vinyl chloride:
Destruction of the heme of highly purified liver microsomal cytochrome
P-450 by a metabolite. Mol Pharmacol, 13: 993-1004.
Guengerich FP & Watanabe PG (1979) Metabolism of [14C]- and
[36Cl]-labeled vinyl chloride in vivo and in vitro. Biochem
Pharmacol, 28: 589-596.
Guengerich FP, Crawford WMJ, & Watanabe PG (1979) Activation of vinyl
chloride to covalently bound metabolites: Roles of 2-chloroethylene
oxide and 2-chloroacetaldehyde. Biochemistry, 18: 5177-5182.
Guengerich FP, Mason PS, Stott WJ, Fox TR, & Watanabe PG (1981) Roles
of 2-haloethylene oxide and 2-haloacetaldehydes derived from vinyl
bromide and vinyl chloride in irreversible binding to protein and DNA.
Cancer Res, 41: 4391-4398.
Guengerich FP, Kim D-H, & Iwasaki M (1991) Role of human cytochrome
P-450 IIE1 in the oxidation of many low molecular weight cancer
suspects. Chem Res Toxicol, 4: 168-179.
Guichard Y, El Ghissassi F, Nair J, Bartsch H, & Barbin A (1996)
Formation and accumulation of DNA ethenobases in adult Sprague-Dawley
rats exposed to vinyl chloride. Carcinogenesis, 17: 1553-1559.
Guicherit R & Schulting FL (1985) The occurrence of organic chemicals
in the atmosphere of the Netherlands. Sci Total Environ, 43: 193-219.
Gürtler R, Möller U, Sommer S, Müller H, & Kleinermanns K (1994)
Photooxidation of exhaust pollutants. Chemosphere, 29: 1671-1682.
Gwinner LM, Laib RJ, Filser JG, & Bolt HM (1983) Evidence of
chloroethylene oxide being the reactive metabolite of vinyl chloride
towards DNA: Comparative studies with 2,2'-dichlorodiethylether.
Carcinogenesis, 4: 1483-1486.
Haag WR, Johnson MD, & Scofield R (1996) Direct photolysis of
trichloroethene in air: Effect of cocontaminants, toxicity of
products, and hydrothermal treatment of products. Environ Sci Technol,
30: 414-421.
Hagmar L, Akesson B, Nielsen J, Andersson C, Lindén K, Attewell R, &
Möller T (1990) Mortality and cancer morbidity in workers exposed to
low levels of vinyl chloride monomer at a polyvinyl chloride
processing plant. Am J Ind Med, 17: 553-565.
Haguenoer JM, Frimat P, Cantineau A, Pollard F, & Bobowski R (1979)
Les risques liés au chlorure de vinyle (CVM) et leur prévention dans
une usine de polymérisation. Arch Mal Prof Méd Travail Séc Soc, 40:
1115-1130.
Hahn A, Michalak H, Begemann K, Heinemeyer G, & Gundert-Remy U (1998)
[Transportation accident with vinyl chloride: Health effects in 325
victims.] Umweltmed Forsch Prax, 3: 144-155 (in German).
Hallbourg RR, Delfino JJ, & Miller WL (1992) Organic priority
pollutants in groundwater and surface water at three landfills in
North Central Florida. Water Air Soil Pollut, 65: 307-322.
Hann RW & Jensen PA (1974) Water quality characteristics of hazardous
materials. In : Hann RW & Jensen PA ed. Water quality characteristics
of hazardous materials. College Station, Texas, Environmental
Engineering Information Center, pp 1-21.
Hansen OG (1991) PVC in the health care sector. Med Device Technol, 2:
18-23.
Hansteen I-L, Hillestad L, Thiis-Evensen E, & Heldaas SS (1978)
Effects of vinyl chloride in man: A cytogenetic follow-up study. Mutat
Res, 51: 271-278.
Harkov R, Kebbekus B, Bozzelli W, & Lioy PJ (1983) Measurement of
selected volatile organic compounds at three locations in New Jersey
during the summer season. J Air Pollut Control Assoc, 33: 1177-1183.
Harkov R, Kebbekus B, Bozelli JW, Lioy JW, & Daisey J (1984)
Comparison of selected volatile organic compounds during summer and
winter at urban sites in New Jersey. Sci Total Environ, 38: 259-274.
Harris DK (1953) Health problems in the manufacture and use of
plastics. Br J Ind Med, 10: 255-268.
Harris CC (1996) p53 Tumor suppressor gene: At the crossroads of
molecular carcinogenesis, molecular epidemiology, and cancer risk
assessment. Environ Health Perspect, 104: 435-439.
Harris DK & Adams WGF (1967) Acro-osteolysis occurring in men engaged
in the polymerization of vinyl chloride. Br Med J, 3: 712-714.
Harris LR & Sarvadi DG (1994) Synthetic polymers. In: Clayton GD &
Clayton FE ed. Patty's industrial hygiene and toxicology, 4th ed. New
York, John Wiley & Sons, Inc., vol 2, part E, pp 3673-4005.
Hartmans S (1995) Microbial degradation of vinyl chloride. In: Singh
VP ed. Biotransformations: Microbial degradation of health risk
compounds. Amsterdam, Oxford, New York, Elsevier Science Publishers,
pp 239-248.
Hartmans S & De Bont JAM (1992) Aerobic vinyl chloride metabolism in
Mycobacterium aurum L1. Appl Environ Microbiol, 58: 1220-1226.
Hartmans S, De Bont JAM, Tramper J, & Luyben KCAM (1985) Bacterial
degradation of vinyl chloride. Biotechnol Lett, 7: 383-388.
Hartmans S, Kaptein A, Tramper J, & De Bont JAM (1992)
Characterization of a Mycobacterium sp. and a Xanthobacter sp. for
the removal of vinyl chloride and 1,2-dichloroethane from waste gases.
Appl Microbiol Biotechnol, 37: 796-801.
Hauschild I, Schröer A, Siedersleben M, & Starnick J (1994) The
microbial growth of Mycobacterium aurum L1 on vinyl chloride with
respect to inhibitory and limiting influence of substrate and oxygen.
Water Sci Technol, 30: 125-132.
Hayashi H, Sakamuto N, Aoi T, Fukumura A, Mano H, & Inagaki T (1990)
Invasive character of malignant endothelial cells in
vinyl-chloride-induced liver angiosarcoma. Nagoya J Med Sci, 52:
13-17.
Hedley WH, Cheng JT, McCormick R, & Lewis WA (1976) Sampling of
automobile interiors for vinyl chloride monomer. Research Triangle
Park, North Carolina, US Environmental Protection Agency, 34 pp
(EPA-600/2-76-124; prepared by Monsanto Research Corporation, Dayton,
Ohio).
Hefner RE, Watanabe PG, & Gehring PJ (1975a) Preliminary studies on
the fate of inhaled vinyl chloride monomer (VCM) in rats. Environ
Health Perspect, 11: 85-95.
Hefner RE, Watanabe PG, & Gehring PJ (1975b) Short communication:
Percutaneous absorption of vinyl chloride. Toxicol Appl Pharmacol, 34:
529-532.
Hefner RE, Watanabe PG, & Gehring PJ (1975c) Preliminary studies of
the fate of inhaled vinyl chloride monomer in rats. Ann NY Acad Sci,
246: 135-148.
Heger M, Müller G, & Norpoth K (1981) [Investigations on the
correlation between vinyl chloride (=VCM)-uptake and excretion of its
metabolites by 15 VCM-exposed workers.] Int Arch Occup Environ Health,
48: 205-210 (in German).
Hehir RM, McNamara BP, McLaughlin J, Willigan DA, Bierbower G, &
Hardisty JF (1981) Cancer induction following single and multiple
exposure to a constant amount of vinyl chloride monomer. Environ
Health Perspect, 41: 63-72.
Heldaas SS, Langard S, & Andersen A (1984) Incidence of cancer among
vinyl chloride and polyvinyl chloride workers. Br J Ind Med, 41:
25-30.
Heldaas SS, Langard SL, & Andersen AA (1987) Incidence of cancer among
vinyl chloride and polyvinyl chloride workers: Further evidence for an
association with malignant melanoma. Br J Ind Med, 44: 278-280.
Helfgott TB, Hart FL, & Bedard RG (1977) An index of refractory
organics.. Ada, Oklahoma, US Environmental Protection Agency, pp 1-143
(EPA-600/2-77-174; prepared by the Environmental Engineering Program,
University of Connecticut, Storrs, Connecticut).
Helmig D, Pollock W, Greenberg J, & Zimmerman P (1996) Gas
chromatography mass spectrometry analysis of volatile organic trace
gases at Mauna Loa Observatory, Hawaii. J Geophys Res Atmos, D101:
14,697-14,710.
Hemminki K & Vineis P (1985) Extrapolation of the evidence on
teratogenicity of chemicals between humans and experimental animals:
Chemicals other than drugs. Teratogen Carcinogen Mutagen, 5: 251-318.
Hendry DG & Kenley RA (1979) Atmospheric reaction products of organic
compounds. Washington, DC, US Environmental Protection Agency, pp 1-80
(EPA-560/12-79-001; prepared by SRI International, Menlo Park,
California).
Henning KD & Richter E (1985) [Waste gas energy -- Treatment of gas
from sewage sludge and landfill sites using activated charcoal and
carbon-molecular sieves/methane enrichment.] Entsorgungspraxis, 5:
268-272 (in German).
Hesstvedt E, Hov O, & Isaksen ISA (1976) Photochemical lifetime of
vinylchloride in the atmosphere. In: Photochemical lifetime of
vinylchloride in the atmosphere. Oslo, Sweden, University of Oslo,
Institute for Geophysics, pp1-12 (Report No. 17).
Hiatt GFS, Cogliano VJ, Becker RA, Siegel DM, & Den A (1994) Vinyl
chloride action levels: Indoor air exposures at a superfund site. In:
Andrews JS, Frumkin H, Johnson BL, Mehlman MA, Xintaras C, & Bucsela
JA ed. Hazardous waste and public health: International Congress on
the Health Effects of Hazardous Waste. Princeton, New Jersey,
Princeton Scientific Publishing, pp 525-529 .
Hill RH, McCammon CS, Saalwaechter AT, Teass AW, & Woodfin WJ (1976a)
Gas-chromatographic determination of vinyl chloride in air samples
collected on charcoal. Anal Chem, 48: 1395-1398.
Hill J, Kollig HP, Paris DF, Wolfe NL, & Zepp RG (1976b) Dynamic
behavior of vinyl chloride in aquatic ecosystems. Athens, Georgia, US
Environmental Protection Agency, Environmental Research Laboratory, pp
1-43 (EPA 600/3-76-001).
Himeno S, Okuda H, & Suzuki T (1983) Lack of dominant lethal effects
in male CD-1 mice after short-term and long-term exposures to vinyl
chloride monomer. Toxicol Lett, 16: 47-53.
HMIP (Her Majesty's Inspectorate of Pollution) (1996) The chemical
release inventory 1994. London, Her Majesty's Stationary Office, pp
1-198.
Ho JSY (1989) A sequential analysis for volatile organics in water by
purge-and-trap capillary column gas chromatography with
photoionization and electrolytic conductivity detectors in series. J
Chromatogr Sci, 27: 91-98.
Ho SF, Phoon WH, Gan SL, & Chan YK (1991) Persistent liver dysfunction
among workers at a vinyl chloride monomer polymerization plant. J Soc
Occup Med, 41: 10-16.
Hocking C (1975) PVC and other plastics in food packaging. Aust
Packag, 23: 30-37.
Hodgson AT, Garbesi K, Sextro RG, & Daisey JM (1992) Soil-gas
contamination and entry of volatile organic compounds into a house
near a landfill. J Air Waste Manage Assoc, 42: 277-283.
Hoffmann D, Patrianakos C, Brunnemann KD, & Gori GB (1976)
Chromatographic determination of vinyl chloride in tobacco smoke. Anal
Chem, 48: 47-50.
Höfler F, Schneider J, & Möckel HJ (1986) Analytical investigations on
the composition of gaseous effluents from a garbage dump. Fresenius Z
Anal Chem, 325: 365-368.
Holliger C (1995) The anaerobic microbiology and biotreatment of
chlorinated ethenes. Curr Opin Biotechnol, 6: 347-351.
Hollstein M, Marion M-J, Lehman T, Welsh J, Harris CC, Martel-Planche
G, Kusters I, & Montesano R (1994) p53 Mutations at A:T base pairs in
angiosarcomas of vinyl chloride-exposed factory workers.
Carcinogenesis, 15: 1-3.
Hollstein M, Shomer G, Greenblatt M, Soussi T, Hovig E, Montesano R, &
Harris CC (1996) Somatic point mutations in the p53 gene of human
tumors and cell lines: Updated compilation. Nucleic Acids Res, 24:
141-146.
Holm L, Westlin A, & Holmberg B (1982) Technical control measures in
the prevention of occupational cancer: An example from the PVC
industry. In: Proceedings of the International Symposium on Prevention
of Occupational Cancer. Geneva, International Labour Office, pp
538-546.
Holmberg B, Kronevi T, & Winelli M (1979) The pathology of vinyl
chloride exposed mice. Acta Vet Scand, 17: 328-342.
Hong CB, Winston JM, Thornburg LP, Lee CC, & Woods JS (1981) Follow-up
study on the carcinogenicity of vinyl chloride and vinylidene chloride
in rats and mice: Tumor incidence and mortality subsequent to
exposure. J Toxicol Environ Health, 7: 909-924.
Hori M, Kobayashi Y, & Ota Y (1972) Vinyl chloride monomer odor
concentration. Plast Ind News, 18: 164-168.
Howard CJ (1976) Rate constants for the gas-phase reactions of OH
radicals with ethylene and halogenated ethylene compounds. J Chem
Phys, 65: 4771-4777.
Howard PH (1989) Vinyl chloride. In: Howard PH ed. Handbook of
environmental fate and exposure data for organic chemicals -- Volume
I: Large production and priority pollutants. Chelsea, Michigan, Lewis
Publishers, Inc., pp 551-560.
Howard PH, Boethling RS, Jarvis WF, Meylan WM, & Michalenko EM (1991)
Vinyl chloride. In: Printup HT ed. Handbook of environmental
degradation rates. Chelsea, Michigan, Lewis Publishers, Inc., pp
138-139.
Hozo I (1998) Croatian cases of ASL. Presented at the Meeting of the
Association of Plastics Manufacturers in Europe, Oslo, 4-5 June 1998
(Unpublished report).
Hozo I, Andelinovic S, Ljutic D, Miric D, Bojic L, & Gasperic I (1996)
Vinyl chloride monomer exposure by the plastic industry workers
-- basic condition for liver angiosarcoma appearance. Med Arhiv, 50:
9-14.
Hozo I, Andelinovic S, Ljutic D, Bojic L, Miric D, & Giunio L (1997)
Two new cases of liver angiosarcoma: History and perspectives of liver
angiosarcoma among plastic industry workers. Toxicol Ind Health, 13:
639-647.
Hrivnak L, Rozinova Z, Korony S, & Fabianova E (1990) Cytogenetic
analysis of peripheral blood lymphocytes in workers exposed to vinyl
chloride. Mutat Res, 240: 83-85.
HSC (Health and Safety Commission, UK) (1995) Control of vinyl
chloride at work: Approved code of practice. London, Health and Safety
Executive, 10 pp.
HSE (Health and Safety Executive) (1987) Vinyl chloride in air. In:
Vinyl chloride in air: Laboratory method using charcoal adsorbent
tubes, solvent desorption and gas chromatography -- MDHS 24: Methods
for the determination of hazardous substances. London, Health and
Safety Executive, pp 1-4.
Huang M (1993a) Prospective epidemiological investigation on
occupational malignant tumor in workers exposed to vinyl chloride. J
Hyg Res, 22: 193-196.
Huang M-Y (1993b) Epidemiological investigation on malignant tumors in
workers exposed to vinyl chloride. Chin J Ind Hyg Occup Dis, 11:
147-151.
Huang M-Y (1994) An investigation on the effect of reproductive
function in workers occupationally exposed to vinyl chloride. Chin J
Ind Hyg Occup Dis, 12: 12-15.
Huberman E, Bartsch H, & Sachs L (1975) Mutation induction in Chinese
hamster V79 cells by two vinyl chloride metabolites, chloroethylene
oxide and 2-chloroacetaldehyde. Int J Cancer, 16: 639-644.
Hung IF, Huang KC, Hung CK, & Shih TS (1996) A thermal desorption gas
chromatographic method for sampling and analysis of airborne vinyl
chloride. Chemosphere, 32: 661-666.
Hussain S & Osterman-Golkar S (1976) Comment on the mutagenic
effectiveness of vinyl chloride metabolites. Chem-Biol Interact, 12:
265-267.
Hüttner E & Holzapfel B (1996) HPRT mutant frequencies and detection
of large deletions by multiplex-PCR in human lymphocytes of vinyl
chloride exposed and non-exposed populations. Toxicol Lett, 88:
175-183.
Hüttner E & Nikolova T (1998) Cytogenetic analysis of peripheral
lymphocytes in a population exposed to vinyl chloride through an
accidental release into the environment. Toxicol Lett, 96/97: 143-148.
Hwang Y-L, Olson JD, & Keller GE (1992) Steam stripping for removal of
organic pollutants from water: 2. Vapor-liquid equilibrium data. Ind
Eng Chem Res, 31: 1759-1768.
IARC (1978) Environmental carcinogenesis selected methods of analysis.
In: Environmental carcinogenesis selected methods of analysis
-- Volume 2: Methods for the measurement of vinyl chloride in poly
(vinyl chloride), air, water and foodstuffs. Lyon, International
Agency for Research on Cancer, pp 19-126 (IARC Scientific Publications
No. 22).
IARC (1979) Vinyl chloride, polyvinyl chloride and vinyl
chloride-vinyl acetate copolymers. Lyon, International Agency for
Research on Cancer, pp 377-438 (IARC Monographs on the Evaluation of
Carcinogenic Risk of Chemicals to Humans, Volume 19).
IARC (1987) Overall evaluations of carcinogenicity: An updating of
IARC monographs, volumes 1 to 42. Lyon, International Agency for
Research on Cancer, pp 373-376 (IARC Monographs on the Evaluation of
the Carcinogenic Risk of Chemicals to Humans, Supplement 7).
IARC (1997) Polychlorinated dibenzo-para-dioxins and polychlorinated
dibenzofurans. Lyon, International Agency for Research on Cancer, pp
1-666 (IARC Monographs on the Evaluation of the Carcinogenic Risk of
Chemicals to Humans, Volume 69).
Infante PF (1976) Oncogenic and mutagenic risks in communities with
polyvinyl chloride production facilities. Ann NY Acad Sci, 271: 49-57.
Infante PF & Pesák J (1994) A historical perspective of some
occupationally related diseases of women. J Occup Med, 36: 826-831.
Infante PF, Wagoner JK, McMichael AJ, Waxweiler RJ, & Falk H (1976a)
Genetic risks of vinyl chloride. Lancet, April: 734-735.
Infante PF, Wagoner JK, McMichael AJ, Waxweiler RJ, & Falk H (1976b)
Genetic risks of vinyl chloride. Lancet, June: 1289-1290.
IPCS (1989) Environmental health criteria 88: Polychlorinated
dibenzo-para-dioxins and dibenzofurans. Geneva, World Health
Organization, International Programme on Chemical Safety.
IPCS (1994) Environmental health criteria 170: Assessing human health
risks of chemicals: Derivation of guidance values for health-based
exposure limits. Geneva, World Health Organization, International
Programme on Chemical Safety.
Ishikawa O, Weerite S, Tamura A, & Miyachi Y (1995) Occupational
scleroderma: A 17-year follow-up study. Br J Dermatol, 133: 786-789.
ISO (1989) Water quality -- determination of the inhibition of the
mobility of Daphnia magna Straus (Cladocera, Crustacea). In:
International standard ISO-6341, 2nd ed. Geneva, International
Standard Organization, pp 1-7.
Jacobsen JS & Humayun MZ (1990) Mechanisms of mutagenesis by the vinyl
chloride metabolite chloroacetaldehyde. Effect of gene-targeted in
vitro adduction of M13 DNA on DNA template activity in vivo and
in vitro. Biochemistry, 29: 496-504.
Jaeger RJ, Reynolds ES, Conolly RB, Moslen MT, Szabo S, & Murphy SD
(1974) Acute hepatic injury by vinyl chloride in rats pretreated with
phenobarbital. Nature (Lond), 252: 724-726.
Jaeger RJ, Murphy SD, Reynolds ES, Szabo S, & Moslen MT (1977)
Chemical modification of acute hepatotoxicity of vinyl chloride
monomer in rats. Toxicol Appl Pharmacol, 41: 597-607.
Jain MK & Criddle CS (1995) Metabolism and cometabolism of halogenated
C-1 and C-2 hydrocarbons. Prog Ind Microbiol, 32: 65-102.
Janson O (1989) [Characterization of landfill gases by chosen trace
substances in particular through sulphur compounds.] Müll Abfall 4:
198-208 (in German).
Jedrychowski RA, Sokal JA, & Chmielnicka J (1984) Influence of
exposure mode on vinyl chloride action. Arch Toxicol, 55: 195-198.
Jeffers PM & Wolfe NL (1996) Homogeneous hydrolysis rate constants
-- Part II: Additions, corrections and halogen effects. Environ
Toxicol Chem, 15: 1066-1070.
Jenssen D & Ramel C (1980) The micronucleus test as part of a
short-term mutagenicity test program for the prediction of
carcinogenicity evaluated by 143 agents tested. Mutat Res, 75:
191-202.
Jiangl H (1990) The effects of vinyl chloride on pregnancy,
parturition and fetal development among female workers. Abstr Pap Am
Chem Soc, 199: 1-2.
John JA, Smith FA, Leong BKJ & Schwetz BA (1977) The effects of
maternally inhaled vinyl chloride on embryonal and fetal development
in mice, rats and rabbits. Toxicol Appl Pharmacol, 39: 497-513.
John AJ, Smith FA, & Schwetz BA (1981) Vinyl chloride: Inhalation
teratology study in mice, rats and rabbits. Environ Health Perspect,
41: 171-177.
Jones JH (1981) Worker exposure to vinyl chloride and poly(vinyl
chloride). Environ Health Perspect, 41: 129-136.
Jones RD, Smith DM, & Thomas PG (1988) A mortality study of vinyl
chloride monomer workers employed in the United Kingdom in 1940-1974.
Scand J Work Environ Health, 14: 153-160.
Jury WA, Russo D, Streile G, & El Abd H (1992) Correction to
"evaluation of volatilization by organic chemicals residing below the
soil surface". Water Resour Res, 28: 607-608.
Kagiya T, Takemoto K, & Uyama Y (1975) [Photo-oxidative decomposition
of vinyl chloride in the air.] Nippon Kagaku Kaishi, 11: 1922-1925 (in
Japanese).
Kandala JC, Mrema JEK, DeAngelo A, Daniel FB, & Guntaka RV (1990)
2-chloroacetaldehyde and 2-chloroacetal are potent inhibitors of DNA
synthesis in animal cells. Biochem Biophys Res Commun, 167: 457-463.
Kanno S, Nojima K, Takahaski K, Iseki H, Takamori S, & Takamatsu K
(1977) Studies on the photochemistry of aliphatic halogenated
hydrocarbons II. Chemosphere, 8: 509-514.
Kappus H, Bolt HM, Buchter HA, & Bolt W (1975) Rat liver microsomes
catalyse covalent binding of [14C]-vinyl chloride to macromolecules.
Nature (Lond), 257: 134-135.
Kappus H, Bolt HM, Buchter HA, & Bolt W (1976) Liver microsomal uptake
of [14C] vinyl chloride and transformation to protein alkylating
metabolites in vitro. Toxicol Appl Pharmacol, 37: 461-471.
Kästner M (1991) Reductive dechlorination of tri-and
tetrachloroethylenes depends on transition from aerobic to anaerobic
conditions. Appl Environ Microbiol, 57: 2039-2046.
Katosova LD & Pavlenko GI (1985) Cytogenetic examination of the
workers of chemical industry. Mutat Res, 147: 301-302.
Kenaga EE & Goring CAI (1980) Relationship between water solubility,
soil sorption, octanol-water partitioning, and concentration of
chemicals in biota. In: Eaton JG, Parrish PR, & Hendricks AC ed.
Aquatic toxicology. Philadelphia, Pennsylvania, American Society for
Testing and Materials, pp 78-115 (ASTM STP 707).
Keplinger ML, Goode JW, Gordon DE, & Calandra JC (1975) Interim
results of exposure of rats, hamsters, and mice to vinyl chloride. Ann
NY Acad Sci, 246: 219-224.
Kinnunen J (1996) [Ambient air quality in the surroundings of
Kilpilahti, 1995 -- Part 2: Volatile organic compounds.] Porvoo,
Finland, Neste Environmental and Industrial Hygiene Institute, pp 1-40
(Report No. 960024Y) (in Finnish).
Kinnunen J (1997) [Ambient air quality in the surroundings of
Kilpilahti, 1996 -- Part 2: Volatile organic compounds].Porvoo,
Finland, Neste Environmental and Industrial Hygiene Institute, pp 1-34
(Report No. 97003Y) (in Finnish).
Kirchner K, Helf D, Ott P, & Vogt S (1990) The reaction of OH radicals
with 1,1-di-, tri-, and tetrachloroethylene. Ber Bunsenges Phys Chem,
94: 77-83.
Klamt A (1993) Estimation of gas-phase hydroxyl radical rate constants
of organic compounds from molecular orbital calculations. Chemosphere,
26: 1273-1289.
Klein W, Herrchen M, & Scheunert I (1989) [The problem of bound
residues in soil.] VDI-Ber, 745: 451-465 (in German).
Klöpffer W, Haag F, Kohl E-G, & Frank R (1988) Testing of the abiotic
degradation of chemicals in the atmosphere: The smog chamber approach.
Ecotoxicol Environ Saf, 15: 298-319.
Klöpffer W, Kaufmann G, & Frank R (1985) [Phototransformation of air
pollutants: Rapid test for the determination of KOH.] Z Naturforsch,
40A: 686-692 (in German).
Knight KR & Gibbons R (1987) Increased collagen synthesis and
cross-link formation in the skin of rats exposed to vinyl chloride
monomer. Clin Sci, 72: 673-678.
Koischwitz D, Lelbach WK, Lackner K, & Hermanutz D (1981) Angiosarcoma
of the liver and hepatocellular carcinomas induced by vinyl chloride.
Fortschr Röntgenstr, 134: 283-290.
Kolb B (1982) Multiple headspace extraction -- a procedure for
eliminating the influence of the sample matrix in quantitative
headspace gas chromatography. Chromatographia, 15: 587-594.
Kollar V, Kemka R, & Misianik J (1988) Determination of vinyl chloride
and vinyl acetate in working atmosphere. Chem Pap, 42: 147-160.
Kontominas MG, Gupta C, & Gilbert SG (1985) Interaction of
vinylchloride with polyvinylchloride in model food systems by
HPLC-chromatography: Migration aspects. Lebensm.wiss Technol, 18:
353-356.
Kopetz K, Mürmann P, Tenkhoff N, & Terwiesch B (1986) [Aspects of
worker, environment and consumer protection in the production,
processing and use of VC and PVC.] In: Becker CW & Braun A ed.
[Plastics Handbook -- Volume 2/2: Polyvinyl chloride.] Munich, Wien,
Carl Hanser Verlag, pp 1437-1465 (in German)
Köster M (1989) [Occurrence, evaluation and analysis of vinyl chloride
as a degradation product of chlorinated ethenes in contaminated
areas.] GWF-Wasser/Abwasser, 130: 694-697 (in German).
Kotseva K (1996) Five year follow-up study of the incidence of
arterial hypertension and coronary heart disease in vinyl chloride
monomer and polyvinyl chloride production workers. Int Arch Occup
Environ Health, 68: 377-379.
Krajewski J & Dobecki M (1978) Determination of vinyl chloride in the
air by gas chromatographic method. Med Prac, 29: 403-410.
Krajewski J, Dobecki M & Gromiec J (1980) Retention of vinyl chloride
in the human lung. Br J Ind Med, 37: 373-374.
Kraybill HF (1983) Assessment of human exposure and health risk to
environmental contaminants in the atmosphere and water with special
reference to cancer. J Environ Sci Health, C1: 175-232.
Kromann A, Ludvigsen L, Albrechtsen H-J, Christensen TH, Ejlertsson J,
& Svensson BH (1998) Degradability of chlorinated aliphatic compounds
in methanogenic leachates sampled at eight landfills. Waste Manage
Res, 16: 54-62.
Kruschel BD, Bell RW, Chapman RE, Spencer MJ, & Smith KV (1994)
Analysis of ambient polar and non polar volatile organic compounds
(VOCs) by thermal desorption, high resolution gas chromatography-mass
spectrometry (TD/HRGC/MS). J High Resol Chromatogr, 17: 187-190.
Kucerová M, Polívková Z, & Bátora J (1979) Comparative evaluation of
the frequency of chromosomal aberrations and the SCE numbers in
peripheral lymphocytes of workers occupationally exposed to vinyl
chloride monomer. Mutat Res, 67: 97-100.
Kuchenmeister F, Wang M, Klein RG, & Schmezer P (1996) Transport of
reactive metabolites of procarcinogens between different liver cell
types, as demonstrated by the single cell microgel electrophoresis
assay. Toxicol Lett, 88: 29-34.
Kudo YY, Yamada S, Nakamura I, Sakaguchi T, Sakaguchi S, Ohe T,
Kamagata H, Naito A, & Nakazawa S (1990) On the changes in peripheral
red cells in mice exposed to vinyl chloride monomer. Pol J Occup Med,
3: 301-310.
Kurlyandski BA, Stovbur NN, & Turusov VS (1981) [About hygienic
regimentation of vinyl chloride.] Gig i Sanit, 3: 74-75 (in Russian).
Kusmierek JT & Singer B (1992) 1,N2-ethenodeoxyguanosine: Properties
and formation in chloroacetaldehyde-treated polynucleotides and DNA.
Chem Res Toxicol, 5: 634-638.
L'Abbé KA, Ferro G, Winkelman R, Saracci R, & Simonato L (1989)
Mortality and cancer incidence results of the European multi-centric
cohort study of workers employed in the vinyl chloride industry. Lyon,
International Agency for Research on Cancer (IARC Internal Report
89/007).
Lackey LW, Phelps TJ, Korde V, Nold S, Ringelberg D, Bienkowski PR ,&
White DC (1994) Feasibility testing for the on-site bioremediation of
organic wastes by native microbial consortia. Int Biodeterior
Biodegrad, 33: 41-59.
Lahl U, Zeschmar-Lahl B, & Jager J (1991) [Release of pollutants from
municipal waste deponies.] Wasser Luft Boden, 1: 64-66 ( in German).
Lahmann E (1980) [Measurements of air pollutants in Berlin: Literature
study on measurement programmes and their results.] WaBoLu- Berichte,
1: 4.
Laib RJ & Bolt HM (1977) Alkylation of RNA by vinyl chloride
metabolites in vitro and in vivo: Formation of
1- N6-etheno-adenosine. Toxicology, 8: 185-195.
Laib RJ & Bolt HM (1978) Formation of 3,N4-ethenocytidine moieties in
RNA by vinyl chloride metabolites in vitro and in vivo. Arch
Toxicol, 39: 235-240.
Laib RJ & Bolt HM (1980) Trans-membrane alkylation: a new method for
studying irreversible binding of reactive metabolites to nucleic
acids. Biochem Pharmacol, 29: 449-452.
Laib RJ, Gwinner LM, & Bolt HM (1981) DNA alkylation by vinyl chloride
metabolites: Etheno derivatives or 7-alkylation of guanine? Chem-Biol
Interact, 37: 219-231.
Laib RJ, Pellio T, Wünschel UM, Zimmermann N, & Bolt HM (1985a) The
rat liver foci bioassay: II. Investigations on the dose-dependant
induction of the ATPase-deficient foci by vinyl chloride at very low
doses. Carcinogenesis, 6: 69-72.
Laib RJ, Klein KP, & Bolt HM (1985b) The rat liver foci bioassay: I.
Age-dependence of induction by vinyl chloride of ATPase-deficient
foci. Carcinogenesis, 6: 65-68.
Laib RJ, Bolt HM, Cartier R, & Bartsch H (1989) Increased alkylation
of liver DNA and cell turnover in young versus old rats exposed to
vinyl chloride correlated with cancer susceptibility. Toxicol Lett,
45: 231-239.
LAI (German State Committee for Air Pollution Control) (1992) [Cancer
risk through air pollution: Development of "criteria for carcinogenic
air pollution" assigned by the Environment Ministers Conference.]
Düsseldorf, Ministry for the Environment, Area and Agriculture, 330 pp
(in German).
Lakhanisky T, Bazzoni D, Jadot P, Joris I, Laurent C, Ottogali M, Pays
A, Planard C, Ros Y, & Vleminckx C (1993) Cytogenetic monitoring of a
village population potentially exposed to a low level of environmental
pollutants -- Phase 1: SCE analysis. Mutat Res, 319: 317-323.
Lane DP (1994) p53 and human cancer. Br Med Bull, 50: 582-599.
Langauer-Lewowicka H, Kurzbauer H, Byczkowska Z, & Wocka-Marek T
(1983) Vinyl chloride disease -- neurological disturbances. Int Arch
Occup Environ Health, 52: 151-157.
Lange C-E, Jühe S, Stein G, & Veltman G (1974) [The so-called vinyl
chloride disease: An occupationally caused systemic sclerosis.] Int
Arch Arbeitsmed, 32: 1-32 (in German).
Langouet S, Müller M, & Guengerich FP (1997) Misincorporation of dNTPs
opposite 1,N2-ethenoguanine and
5,6,7,8-tetrahydro-7-hydroxy-9-oxoimidazo[1,2-a]purine in
oligonucleotides by Echerichia coli polymerases I exo- and II exo-, T7
polymerase exo-, human immunodeficiency virus-1 reverse transcriptase,
and rat polymerase ß. Biochemistry, 36: 6069-6079.
Laplanche A, Clavel F, Contassot JC, & Lanouziere C (1987) Exposure to
vinyl chloride monomer: report on a cohort study. Br J Ind Med, 44:
711-715.
Laplanche A, Clavel-Chapelon F, Contassot JC, & Lanouziére C (1992)
Exposure to vinyl chloride monomer: Results of a cohort study after a
seven year follow up. Br J Ind Med, 49: 134-137.
Laumbach AD, Lee S, Wong J, & Streips UN (1977) Studies on the
mutagenicity of vinyl chloride metabolites and related chemicals. In:
Nienburg HE ed. Prevention and detection of cancer. New York, Basel,
Marcel Dekker Inc., vol 2, part 1, pp 155-170.
Lederer M (1959) [A vinyl chloride peroxide.] Angew Chem, 71: 162 (in
German).
Lee CC, Bhandaari JC, Winston JM, House WB, Peters PJ, Dixon RL, &
Woods JS (1977) Inhalation toxicity of vinyl chloride and vinylidene
chloride. Environ Health Perspect, 21: 25-32.
Lee CC, Bhandari JC, Winston JM, House WB, Dixon RL, & Woods JS (1978)
Carcinogenicity of vinyl chloride and vinylidene chloride. J Toxicol
Environ Health, 4: 15-30.
Lee MD, Mazierski PF, Buchanan RJ, Ellis DE, & Sehayek LS (1995)
Intrinsic in situ anaerobic biodegradation of chlorinated solvents at
an industrial landfill. In: Hinchee RE, Wilson JT, & Downey DC ed.
Intrinsic bioremediation. Columbus, Ohio, Battelle Press, pp 205-222.
Lee FI, Smith PM, Bennett B, & Williams DMJ (1996) Occupationally
related angiosarcoma of the liver in the United Kingdom 1972-1994.
Gut, 39: 312-318.
Leisinger T (1992) Microorganisms for the introduction of chlorinated
aliphatic hydrocarbons into the carbon cycle. In: Mongkolsuk Sea ed.
Biotechnology and environmental science: Molecular approaches. New
York, London, Plenum Press, pp 143-147.
Lelbach WK (1996) A 25-year follow-up study of heavily exposed vinyl
chloride workers in Germany. Am J Ind Med, 29: 446-458.
Lelbach WK & Marsteller HJ (1981) Vinyl chloride -- associated
disease. Adv Intern Med Pediatr, 47: 1-110.
Lesage S, Jackson RE, Priddle MW, & Riemann PG (1990) Occurrence and
fate of organic solvent residues in anoxic groundwater at the
Gloucester Landfill, Canada. Environ Sci Technol, 24: 559-566.
Lesage S, McBride RA, Cureton PM, & Brown S (1993) Fate of organic
solvents in landfill leachates under simulated field conditions and in
anaerobic microcosms. Waste Manage Res, 11: 215-226.
Leschber R, Mergler-Voelkl R, & Nerger M (1990) Soil and groundwater
contamination by low boiling chlorinated hydrocarbons in Berlin. Int J
Environ Anal Chem, 39: 159-164.
Lester D, Greenberg LA, & Adams WR (1963) Effects of single and
repeated exposures of humans and rats to vinyl chloride. Am Ind Hyg
Assoc J, 24: 265-275.
Li Y, Asherova M, Marion M-J, & Brandt-Rauf PW (1998a) Mutant
oncoprotein biomarkers in chemical carcinogenesis. In : Mendelsohn ML,
Mohr LC, & Peeters J ed. Biomarkers: Medical and workplace
applications. Washington, DC, National Academy Press, pp 345-353.
Li Y, Marion MJ, Asherova M, Coulibaly D, Smith SJ, Do T, Carney WP, &
Brandt-Rauf PW (1998b) Mutant p21 ras in vinyl chloride-exposed
workers. Biomarkers, 3: 433-439.
Lilis R (1981) Review of pulmonary effects of poly(vinyl chloride) and
vinyl chloride exposure. Environ Health Perspect, 40: 167-169.
Lilis R, Anderson H, Nicholson WJ, Daum S, Fishbein AS, & Selikoff IR
(1975) Prevalence of disease among vinyl chloride and polyvinyl
chloride workers. Ann NY Acad Sci, 246: 22-41.
Lillian D, Singh HB, Appleby A, Lobban L, Arnts R, Gumpert R, Hague R,
Toomey J, Kazazis J, Antell M, Hansen D, & Scott B (1975) Atmospheric
fates of halogenated compounds. Environ Sci Technol, 9: 1042-1048.
Lindbohm M-L, Hemminki K, & Kyyrönen P (1985) Spontaneous abortions
among women employed in the plastics industry. Am J Ind Med, 8:
579-586.
Lipsky D & Jacot B (1985) Hazardous emissions from sanitary landfills.
In: Hazardous emissions from sanitary landfills. For presentation at
the 78th Annual Meeting of the Air Pollution Control Association,
Detroit, Michigan, 16-21 June 1985. New York, Fred C. Hart Associates,
pp 1-13.
Little J (1993) Are occupational carcinogens teratogenic ? In:
Richardson M ed. Reproductive toxicology. Weinheim, Germany, VCH
Verlagsgesellschaft mbH pp 3-43.
Little JC, Daisey JM, & Nazaroff WW (1992) Transport of subsurface
contaminants into buildings. Environ Sci Technol, 26: 2058-2066.
Lloyd MH, Gauld S, Copland L, & Soutar CA (1984) Epidemiological study
of the lung function of workers at a factory manufacturing polyvinyl
chloride. Br J Ind Med, 41: 328-333.
Lopez-Avila V, Heath N, & Hu A (1987a) Determination of purgeable
halocarbons and aromatics by photoionization and hall electrolytic
conductivity detectors connected in series. J Chromatogr Sci, 25:
356-363.
Lopez-Avila V, Wood R, Flanagan M, & Scott R (1987b) Determination of
volatile priority pollutants in water by purge and trap and capillary
column gas chromatography/mass spectrometry. J Chromatogr Sci, 25:
286-291.
Loprieno N, Barale R, Baroncelli S, Bauer C, Bronzetti G, Cammellini
A, Cercignani G, Corsi C, Gervasi G, Leporini C, Nieri R, Rossi AM,
Stretti G, & Turchi G (1976) Evaluation of the genetic effects induced
by vinyl chloride monomer (VCM) under mammalian metabolic activation:
Studies in vitro and in vivo. Mutat Res, 40: 85-95.
Loprieno N, Barale R, Baroncelli S, Bartsch H, Bronzetti G, Cammellini
A, Corsi C, Frezza D, Nieri R, Leporini C, Rosellini D, & Rossi AM
(1977) Induction of gene mutations and gene conversions by vinyl
chloride metabolites in yeast. Cancer Res, 36: 253-257.
Lu P-Y, Metcalf RL, Plummer N, & Mandel D (1977) The environmental
fate of three carcinogens: Benzo-(alpha)-pyrene, benzidine, and vinyl
chloride evaluated in laboratory model ecosystems. Arch Environ Contam
Toxicol, 6: 129-142.
Lundberg I, Gustavsson A, Holmberg B, Molina G, & Westerholm P (1993)
Mortality and cancer incidence among PVC-processing workers in Sweden.
Am J Ind Med, 23: 313-319.
Lutz W-K & Schlatter J (1993) The relative importance of mutagens and
carcinogens in the diet. Pharmacol Toxicol, 72: 104-107.
Lyman WJ, Reehl WF, & Rosenblatt DH (1982) Handbook of chemical
property estimation methods: Environmental behavior of organic
compounds. New York, McGraw-Hill Book Company, pp 4-25.
Lyman WJ, Reehl WF, & Rosenblatt DH (1990) Handbook of chemical
property estimation methods. Environmental behavior of organic
compounds. Washington, DC, American Chemical Society, pp 15.4-15.20.
Mabey WR, Smith JH, Podoll RT, Johnson HL, Mill T, Chou TW, Gates J,
Partiridge IW, & Vandenberg D (1982) Aquatic fate process data for
organic priority pollutants. Washington, DC, US Environmental
Protection Agency, pp 1-407 (EPA-440/4-81-014; prepared by SRI
International, Menlo Park, California).
McCann J, Simmon V, Streitwieser D, & Ames BN (1975) Mutagenicity of
chloroacetaldehyde, a possible metabolic product of 1,2-dichloroethane
(ethylene dichloride), chloroethanol (ethylene chlorohydrin), vinyl
chloride, and cyclophosphamide. Proc Natl Acad Sci (USA), 72:
3190-3193.
McCarty PL (1993) In situ bioremediation of chlorinated solvents. Curr
Opin Biotechnol, 4: 323-330.
McCarty PL (1996) Biotic and abiotic transformations of chlorinated
solvents in ground water. In: Proceedings of the Symposium on Natural
Attenuation of Chlorinated Organics in Ground Water, Dallas, Texas,
11-13 September 1996. Washington, DC, US Environmental Protection
Agency, pp 5-9 (EPA 540/R-96/509).
McCarty PL (1997) Breathing with chlorinated solvents. Science, 276:
1521-1522.
McCarty PL & Reinhard M (1993) Biological and chemical transformations
of halogenated aliphatic compounds in aquatic and terrestrial
environments. In: Oremland RS ed. Biogeochemistry of global change:
Radioactively active trace gases. New York, Chapman & Hall, pp
839-852.
McLaughlin RS (1946) Chemical burns of the human cornea. Am J
Ophthalmol, 29: 1355-1362.
McMurry JR & Tarr J (1978) Vinyl chloride in Texas` ambient air. Proc
Annu Tech Meet Inst Environ Sci, 24: 149-153.
Magnusson J & Ramel C (1976) Mutagenic effects of vinyl chloride in
Drosophila melanogaster. Mutat Res, 38: 115.
Magnusson J & Ramel C (1978) Mutagenic effects of vinyl chloride on
Drosophila melanogaster with and without pretreatment with sodium
phenobarbiturate. Mutat Res, 57: 307-312.
Major DW, Hodgins WW, & Butler BJ (1991) Field and laboratory evidence
of in situ biotransformation of tetrachloroethene to ethene and ethane
at a chemical transfer facility in North Toronto. In: Hinchee RE &
Otfenbuttel RF eds. Onsite bioreclamation. Stoneham, Massachusetts,
Butterworth-Heinemann, pp 147-171.
Major D, Cox E, Edwards E, & Hare P (1995) Intrinsic dechlorination of
trichloroethene to ethene in a bedrock aquifer. In: Hinchee RE, Wilson
JT, & Downey DC ed. Intrinsic bioremediation. Columbus, Ohio, Battelle
Press, pp 197-203.
Makarov IA (1984) [Sexual disorders in male workers occupationally
exposed to methylmethacrylate and vinyl chloride.] Gig Tr Prof Zabol,
6: 19-23 (in Russian).
Makarov IA, Solovyeva MS, & Gnelitsky GI (1984) [On sexual disorders
in women chronically exposed to methylmethacrylate and vinyl
chloride.] Gig Tr Prof Zabol, 3: 22-27 (in Russian).
Makita O, Ono K, Sakurai T, Tanigawa H, Ando Y, Yamashita K,
Yoshimatsu M, & Takahashi M (1997) [A case of hepatocellular carcinoma
related with exposure to vinyl chloride monomer.] Nippon Shokakibyo
Gakkai Zasshi, 94 : 215-219 (in Japanese).
Makk L, Delmore F, Creech JL, Ogden LL, Fadell EH, Songster CL,
Clanton J, Johnson MN, & Christopherson WM (1976) Clinical and
morphologic features of hepatic angiosarcoma in vinyl chloride
workers. Cancer, 37: 149-163.
Malachowsky K, Phelps TJ, & White DC (1991) Aerobic mineralization of
vinyl chloride by branch-shaped gram-positive bacteria. Abstr Gen Meet
Am Soc Microbiol, 91: 277.
Malachowsky KJ, Phelps TJ, Teboli AB, Minnikin DE, & White DC (1994)
Aerobic mineralization of trichloroethylene, vinyl chloride, and
aromatic compounds by Rhodococcus species. Appl Environ Microbiol,
60: 542-548.
Malaveille C, Bartsch H, Barbin A, Camus AM, & Montesano R (1975)
Mutagenicity of vinyl chloride, chloroethylene oxide,
chloroacetaldehyde and chloroethanol. Biochem Biophys Res Commun, 63:
363-370.
Malle K-G (1984) [The implications for water conservation of the EU
priority list of 129 dangerous substances.] Z Wasser Abwasser Forsch,
17: 75-81 (in German).
Maltoni C & Cotti G (1988) Carcinogenicity of vinyl chloride in
Sprague-Dawley rats after prenatal and postnatal exposure. Ann NY Acad
Sci, 534: 145-159.
Maltoni C & Lefemine G (1975) Carcinogenicity bioassays of vinyl
chloride: Current results. Ann NY Acad Sci, 246: 195-218.
Maltoni C, Lefemine G, Chieco P, & Carretti D (1974) Vinyl chloride
carcinogenesis: Current results and perspectives. Med Lav, 65:
421-444.
Maltoni C, Lefemine C, Ciliberti A, Cotti G, & Carretti D (1979) Vinyl
chloride carcinogenicity bioassays (BT project) as an experimental
model for risk identification and assessment in environmental and
occupational carcinogenesis. In: Institut du Radium, Fondation Curie
ed. Epidémiologie animale et épidémiologie humaine: Le cas du chlorure
de vinyle monomère. Paris, Publications Essentielles, pp 17-20.
Maltoni C, Lefemine G, Ciliberti A, Cotti G, & Caretti D (1981)
Carcinogenicity bioassays of vinyl chloride monomer: A model of risk
assessment on an experimental basis. Environ Health Perspect, 41:
3-29.
Maltoni C, Lefemine G, Ciliberrti A, Cotti G, & Carretti D (1984) In :
Maltoni C, Lefemine G, Ciliberrti A, Cotti G, & Carretti D ed.
Experimental research on vinyl chloride carcinogenesis -- Volume II:
Archives of research on industrial carcinogenesis. Princeton, New
Jersey, Princeton Scientific Publisher Inc., 533 pp.
Marion M-J, De VI, Smith S, Luo JC, & Brandt RP (1996) The molecular
epidemiology of occupational carcinogenesis in vinyl chloride exposed
workers. Int Arch Occup Environ Health, 68: 394-398.
Marion M-J, Froment O, & Trépo C (1991) Activation of Ki- ras gene by
point mutation in human liver angiosarcoma associated with vinyl
chloride exposure. Mol Carcinogen, 4: 450-454.
Markowitz SS, McDonald CJ, Fethiere W, & Kerzner MS (1972)
Occupational acroosteolysis. Arch Dermatol, 106: 219-223.
Mastrangelo G, Manno M, Marcer G, Bartolucci GB, Gemignani C, Saladino
G, Simonato L, & Saia B (1979) Polyvinyl chloride pneumoconiosis:
Epidemiological study of exposed workers. J Occup Med, 21: 540-542.
Mastromatteo E, Fisher AM, Christie H, & Danziger H (1960) Acute
inhalation toxicity of vinyl chloride to laboratory animals. Am Ind
Hyg Assoc J, 21: 394-398.
Matsuda T, Yagi T, Kawanishi M, Matsui S, & Takebe H (1995) Molecular
analysis of mutations induced by 2-chloroacetaldehyde, the ultimate
carcinogenic form of vinyl chloride, in human cells using shuttle
vectors. Carcinogenesis, 16: 2389-2394.
Maymo-Gatell X, Tandoi V, Gossett JM, & Zinder SH (1995)
Characterization of an H2-utilizing enrichment culture that
reductively dechlorinates tetrachloroethene to vinyl chloride and
ethene in the absence of methanogenesis and acetogenesis. Appl Environ
Microbiol, 61: 3928-3933.
Maymó-Gatell X, Chien YT, Gossett JM, & Zinder SH (1997) Isolation of
a bacterium that reductively dechlorinates tetrachloroethene to
ethene. Science, 276: 1568-1571.
Meier T (1994) [Small helpers with a big impact: Bacteria for cleaning
waste gases containing vinyl chloride.] Entsorgungs-Technik, 6: 41-44
(in German).
Meier T (1996) [Biological treatment process for exhaust air
containing hydrogen sulphide and vinyl chloride.] Chem Techn, 25: 40
(in German) .
Meylan WM & Howard PH (1993) Computer estimation of the atmospheric
gas-phase reaction rate of organic compounds with hydroxyl radicals
and ozone. Chemosphere, 26: 2293-2299.
Middeldorp PJM, Van Aalst MA, Rijnaarts HHM, Stams FJM, De Kreuk HF,
Schraa G, & Bosma TNP (1998) Stimulation of reductive dechlorination
for in situ bioremediation of a soil contaminated with chlorinated
ethenes. Water Sci Technol, 37: 105-110.
Milde G, Nerger M, & Mergler R (1988) Biological degradation of
volatile chlorinated hydrocarbons in groundwater. Water Sci Technol,
20: 67-73.
Miller NL (1992) Stability analysis of toxic substances within aquatic
ecosystems and their effect on aquatic populations. Ecol Model, 60:
151-165.
Miller A (1993) Dioxin emissions from EDC/VCM plants. Environ Sci
Technol, 27: 1014-1015.
Mirkova E, Mihailova A, & Nosko M (1978) [Embryotoxic and teratogenic
effects in vinyl chloride.] Khig Zdraveopaz, 21: 440-443 (in Russian).
Molina G, Holmberg B, Elofsson S, Holmlund L, Moosing R, & Westerholm
P (1981) Mortality and cancer rates among workers in the Swedish PVC
processing industry. Environ Health Perspect, 41: 145-151.
Monson RR, Peters JM, & Johnson MN (1975) Proportional mortality among
vinyl chloride workers. Environ Health Perspect, 11: 75-77.
Moriya M, Zhang W, Johnson F, & Grollman AP (1994) Mutagenic potency
of exocyclic DNA adducts: Marked differences between Escherichia coli
and simian kidney cells. Proc Natl Acad Sci (USA), 91: 11899-11903.
Mroczkowska MM & Kusmierek JT (1991) Miscoding potential of
N2,3-ethenoguanine studied in an Escherichia coli DNA-dependent
RNA polymerase in vitro system and possible role of this adduct in
vinyl chloride-induced mutagenesis. Mutagenesis, 6: 385-390.
Müller JPH & Korte F (1977) [Contributions to ecological chemistry
-- report 136: Brief report on the nonsensitized photooxidation of
chloroethylenes.] Chemosphere, 6: 341-346 (in German).
Müller G, Norpoth K, & Eckard R (1976) Identification of two urine
metabolites of vinyl chloride by GC-MS-investigations. Int Arch Occup
Environ Health, 38: 69-75.
Müller G, Norpoth K, Kusters E, Herweg K, & Versin E (1978)
Determination of thiodiglycolic acid in urine specimens of vinyl
chloride exposed workers. Int Arch Occup Environ Health, 41: 199-205.
Müller G, Norpoth K, & Wicktramasinghe RH (1979) An analytical method
using GC-MS for the quantitative determination of urinary
thiodiglycolic acid. Int Arch Occup Environ Health, 44: 185-191.
Müller M, Belas FJ, Blair IA, & Guengerich FP (1997) Analysis of 1,
N2-ethenoguanine and 5,6,7,9-tetrahydro-7-hydroxy-9-oxoimidazo
(1,2-a) purine in DNA treated with 2-chlorooxirane by high performance
of amounts to other DNA adducts. Chem Res Toxicol, 10: 242-247.
Mur JM, Mandereau L, Deplan F, Paris A, Richard A, & Hemon D (1992)
Spontaneous abortion and exposure to vinyl chloride. Lancet, 339:
127-128.
Murdoch IA & Hammond AR (1977) A practical method for the measurement
of vinyl chloride monomer (VCM) in air. Ann Occup Hyg, 20: 55-61.
Murray WD & Richardson M (1993) Progress toward the biological
treatment of C1 and C2 halogenated hydrocarbons. Crit Rev Environ Sci
Technol, 23: 195-217.
Nair J, Barbin A, Guichard Y, & Bartsch MH (1995)
1,N6-ethenodeoxyadenosine and 3,N4-ethenodeoxycytidine in liver DNA
from humans and untreated rodents detected by
immunoaffinity/32P-postlabelling. Carcinogenesis, 16: 613-617.
Nair J, Sone H, Nagao M, Barbin A, & Bartsch H (1996) Copper-dependent
formation of miscoding etheno-DNA adducts in the liver of Long Evans
Cinnamon (LEC) rats developing hereditary hepatitis and hepatocellular
carcinoma. Cancer Res, 56: 1267-1271.
Nair J, Vaca CE, Velic I, Mutanen M, Valasta LM, & Bartsch H (1997)
High dietary omega-6 polyunsaturated fatty acids drastically
increase the formation of etheno-DNA base adducts in white blood cells
of female subjects. Cancer Epidemiol Biomarkers Prev, 6: 597-601.
Nair J, Carmichael PL, Fernando RC, Phillips DH, Strain AJ, & Bartsch
H (1998) Lipid peroxidation-induced etheno-DNA adducts in the liver of
patients with the genetic metal storage disorders Wilson's disease and
primary hemochromatosis. Cancer Epidemiol Biomarkers Prev, 7: 435-440.
Nakamura H & Mimura S (1979) Hygienic studies of materials for water
supplies I: Residual vinyl chloride monomer in commercial PVC-pipe and
water extractability of residual vinyl chloride monomer. Sewage Ind
Wastes, 91: 315.
Nakano N, Yamamoto A, & Nagashima K (1996) Development of a monitoring
system for vinyl chloride gas in air by using an HCl monitoring tape
and pyrolyzer. Talanta (Lond), 43: 459-463.
Narayanan B, Suidan MT, Gelderloos AB, & Brenner RC (1995) Anaerobic
treatment of volatile and semivolatile organic compounds in municipal
wastewater. Water Environ Res, 67: 46-56.
Nathan MF (1978) Choosing a process for chloride removal. Chem Eng,
January: 93-100.
Naylor LM & Loehr RC (1982) Priority pollutants in municipal sewage
sludge. BioCycle, July/Aug: 18-22.
Nelms LH, Reisszner KD, & West (1977) Personal vinyl chloride
monitoring device with permeation technique for sampling. Anal Chem,
49: 994-998.
Nelson YM & Jewell WJ (1993) Vinyl chloride biodegradation with
methanotrophic attached films. J Environ Eng, 119: 890-907.
Nelson NA, Robins TG, Garrison RP, Schuman M, & White RF (1993)
Historical characterization of exposure to mixed solvents for an
epidemiologic study of automotive assembly plant workers. Appl Occup
Environ Hyg, 8: 693-702.
Nerger M & Mergler-Völkl R (1988) [Biological degradation of volatile
chlorinated hydrocarbons in groundwater and sewage.] Z Wasser Abwasser
Forsch, 21: 16-19 (in German).
Neshiwat LF, Friedland ML, Schorr-Lesnick B, Feldman S, Glucksman WJ,
& Russo RD (1992) Hepatic angiosarcoma. Am J Med, 93: 219-222.
Neuhoff S (1988) [Safety planning in a city with chemical industry
- Cologne.] Brandschutz/ Dtsch Feuerw Ztg, 4: 168-175 (in German).
Nicholson WJ, Hammond EC, Seidman H, & Selikoff IJ (1975) Mortality
experience of a cohort of vinyl chloride-polyvinyl chloride workers.
Ann NY Acad Sci, 246: 225-230.
NIOSH (1994) Vinyl chloride. In: Eller PM & Cassinelli ME ed. NIOSH
manual of analytical methods, 4th ed. Cincinnati, Ohio, National
Institute for Occupational Safety and Health, pp 1-4.
NIOSH (1997) Pocket guide to chemical hazards. Cincinnati, Ohio,
National Institute for Occupational Safety and Health, pp 330-331.
Nishikawa H, Katami T, Takahara Y, Sumida H, & Yasuhara A (1992)
Emission of organic compounds by combustion of waste plastics
involving vinyl chloride polymer. Chemosphere, 25: 1953-1960.
Nystén T (1988) [Groundwater pollution cases in Uusima county.]
Helsinki, Finnish National Water Board, pp 1-46 (in Finnish).
OECD (1981a) Combined chronic toxicity/carcinogenicity studies. In:
OECD guidelines for testing of chemicals -- Section 4: Health effects
(No. 453). Paris, Organisation for Economic Co-operation and
Development, pp 1-15.
OECD (1981b) Carcinogenicity studies. In: OECD guidelines for testing
of chemicals -- Section 4: Health effects (No. 451). Paris,
Organisation for Economic Co-operation and Development, pp 1-15.
OECD (1983a) One-generation reproduction toxicity study. In: OECD
guidelines for testing of chemicals -- Section 4: Health effects (No.
415). Paris, Organisation for Economic Co-operation and Development,
pp 1-8 .
OECD (1983b) Two-generation reproduction toxicity study. In: OECD
guidelines for testing of chemicals -- Section 4: Health effects (No.
416). Paris, Organisation for Economic Co-operation and Development,
pp 1-8.
OECD (1984) OECD guideline for testing of chemicals No. 203: Fish,
acute toxicity test. Paris, Organisation for Economic Co-operation and
Development, pp 1-12.
Oesch F & Doerjer G (1982) Detection of N2,3-ethenoguanine in DNA
after treatment with chloroacetaldehyde in vitro. Carcinogenesis, 3:
663-665.
Olsen GW, Ramlow JM, & Hearn S (1995) Paternal occupational exposure
and spontaneous abortions: A closer look at paternal recall and vinyl
chloride. Am J Ind Med, 27: 611-613.
Orusev T, Popovski P, Bauer S, & Nikolova K (1976) Occupational risk
in the production of poly(vinyl chloride . God Zb Med Fak Skopje, 22:
33-38.
OSHA (Occupational Safety and Health Administration, US Department of
Labour) (1998) Title 29. Code Fed Regul, Part 1910(1910.1000):
138-143.
Oster RH, Carr CJ, Krantz JC, & Sauerwald MJ (1947) Anesthesia: XXVII.
Narcosis with vinyl chloride. Anesthesiology, 8: 359-361.
Osterman-Golkar S, Hultmark D, Segerbäck D, Calleman CJ, Göthe R,
Ehrenberg L, & Wachtmeister CA (1977) Alkylation of DNA and proteins
in mice exposed to vinyl chloride. Biochem Biophys Res Commun, 76:
259-266.
Ostlere LS, Harris D, Buckley C, Black C, & Rustin MHA (1992) Atypical
systemic sclerosis following exposure to vinyl chloride monomer: A
case report and review of the cutaneous aspects of vinyl chloride
disease. Clin Exp Dermatol, 17: 208-210.
Ott MG, Langner RR, & Holder BH (1975) Vinyl chloride exposure in a
controlled industrial environment. Arch Environ Health, 30: 333-339.
Ottenwälder H & Bolt HM (1980) Metabolic activation of vinyl chloride
and vinyl bromide by isolated hepatocytes by isolated hepatocytes and
hepatic sinusoidal cells. J Environ Pathol Toxicol, 4: 411-417.
Page GW (1981) Comparison of groundwater and surface water for
patterns and levels of contamination by toxic substances. Environ Sci
Technol, 15: 1475-1481.
Page BD & O'Grady R (1977) Gas-solid chromatographic confirmation of
vinyl chloride levels in oils and vinegars by using electrolytic
conductivity detection. J Assoc Off Anal Chem, 60: 576-578.
Palejwala VA, Simha D, & Humayun MZ (1991) Mechanisms of mutagenesis
by exocyclic DNA adducts. Transfection of m-viral DNA bearing a
site-specific adduct shows that ethenocytosine is a highly efficient
RecA-independent mutagenic noninstructional lesion. Biochemistry, 30:
8736-8743.
Paliard P, Valette PJ, Berger F, Contassot JC, & Partensky C (1991)
Péliose hépatique tardive après traitement d'un angiosarcome hépatique
chez un sujet exposé au chlorure de vinyl monomère. Gastroenterol Clin
Biol, 15: 445-448.
Pandya GA & Moriya M (1996) 1,N6-ethenodeoxyadenosine, a DNA adduct
highly mutagenic in mammalian cells. Biochemistry, 35: 11487-11492.
Pankow JF & Rosen ME (1988) Determination of volatile compounds in
water by purging directly to a capillary column with whole column
cryotrapping. Environ Sci Technol, 22: 398-405.
Pankow JF, Johnson RL, & Cherry JA (1993) Air sparging in gate wells
in cutoff walls and trenches for control of plumes of volatile organic
compounds (VOCs). Ground Water, 31: 654-663.
Park K-K, Leim A, Stewart BC, & Miller JA (1993) Vinyl carbamate
epoxide, a major strong electrophilic, mutagenic and carcinogenic
metabolite of vinyl carbamate and ethyl carbamate (urethane).
Carcinogenesis, 14: 441-450.
Parsons F, Wood PR, & DeMarco J (1984) Transformations of
tetrachloroethene and trichloroethene in microcosms and groundwater. J
Am Water Works Assoc, 76: 56-59.
Patty FA (1963) Vinyl chloride. In: Patty FA ed. Industrial hygiene
and toxicology. New York, Interscience Publishers, pp 1303-1305.
Patty FA, Yant WP, & Waite CP (1930) Acute response of Guinea pigs to
vapors of some new commercial organic compounds: V. Vinyl chloride.
Public Health Rep, 45: 1963-1971.
Pau JC, Knoll JE, & Midgett MR (1988) Evaluation of a process gas
chromatograph as a continuos emission monitor for benzene and vinyl
chloride. Int J Air Pollut Control Waste Manage, 38: 1528-1529.
Pearson CR & McConnell G (1975) Chlorinated C1 and C2 hydrocarbons in
the marine environment. Proc R Soc (Lond), B189: 305-332.
Pellizzari ED, Erickson MD, & Zweidinger RA (1979) Formulation of a
preliminary assessment of halogenated organic compounds in man and
environmental media. Washington, DC, US Environmental Protection
Agency (EPA-560/13-79-006; prepared by the Research Triangle
Institute, Research Triangle Park, North Carolina.).
Penin H, Sargar G, Lange C-E & Veltman G (1975) [Neurological,
psychiatrical and electroencephalographical findings in patients with
vinyl chloride disease.] Verh Dtsch Ges Arbeitsmed, 15: 299-304 (in
German).
Peoples AS & Leake CD (1933) The anesthetic action of vinyl chloride.
J Pharmacol Exp Ther, 48: 284-285.
Perrard M-H (1985) Mutagenicity and toxicity of chloroethylene oxide
and chloroacetaldehyde. Experientia (Basel), 41: 676-677.
Perry RA, Atkinson R, & Pitts JN (1977) Rate constants for the
reaction of OH radicals with CH2=CHF, CH2=CHCl, and CH2=CHBr over the
temperature range 299-426°K. J Chem Phys, 67: 458-462.
Perticoni GF, Abbritti G, Cantisani TA, Bondi L, & Mauro L (1986)
Polineuropathy in workers with long exposure to vinyl chloride
electrophysiological study. Electromyogr Clin Neurophysiol, 26: 41-47.
Pessayre D, Wandscheer JC, Descatoire V, Artigou JY, & Benhamou JP
(1979) Formation and inactivation of a chemically reactive metabolite
of vinyl chloride. Toxicol Appl Pharmacol, 49: 505-515.
Peter S & Ungváry G (1980) Lack of mutagenic effect of vinyl chloride
monomer in the mammalian spot test. Mutat Res, 77: 193-196.
Pettit BR (1986) The analysis of thiodiglycollic acid by selected ion
monitoring. Clin Chim Acta, 156: 85-90.
Pfab W & Mücke G (1977) [Migration of chosen monomers in food
simulants.] Verpackungsrundschau, 6: 49-52 (in German).
Phelps TJ, Malachowsky K, Schram RM, & White DC (1991a) Aerobic
mineralization of vinyl chloride by a bacterium of the order
Actinomycetales. Appl Environ Microbiol, 57: 1252-1254.
Phelps TJ, Niedzielski JJ, Malachowsky KJ, Schram RM, Herbes SE, &
White DC (1991b) Biodegradation of mixed-organic wastes by microbial
consortia in continuous-recycle expanded-bed bioreactors. Environ Sci
Technol, 25: 1461-1465.
Picciano DJ, Flake RE, Gay PC, & Kilian DJ (1977) Vinyl chloride
cytogenetics. J Occup Med, 19: 527-530.
Pirastu R, Comba P, Reggiani A, Foa V, Masina A, & Maltoni C (1990)
Mortality from liver disease among Italian vinyl chloride
monomer/polyvinyl chloride manufacturers. Am J Ind Med, 17: 155-161.
Pirastu R, Bellis S, Bruno C, Maltoni C, Masina A, & Reggiani A (1991)
[The mortality among the workers of vinyl chloride in Italy.] Med Lav,
82: 388-423 (in Italian).
Pirastu R, Chellini E, Carnevale F, De Santis M, Bracci C, & Comba P
(1997) [Cohort mortality study of vinyl chloride exposed workers in
Terrara, Rosignano and Ravenna.] Rome, Istituto Superiore di Sanità
(in Italian).
Pirastu R, Bruno C, De Santis M, & Comba P (1998) Cohort mortality
study of vinyl chloride exposed workers in Ferrara, Rosignano and
Ravenna. Epidemiol Prevenz, 22: 226-236.
Pitts JN (1993) Atmospheric formation and fates of toxic ambient air
pollutants. Occup Med, 3: 621-662.
Platt UF, Winer AM, Biermann HW, Atkinson R, & Pitts JN (1984)
Measurement of nitrate radical concentrations in continental air.
Environ Sci Technol, 18: 365-369.
Pleil JD & Lindstrom AB (1997) Exhaled human breath measurement
methods for assessing exposure to halogenated volatile organic
compounds. Clin Chem, 43: 723-730.
Plugge H & Safe S (1977) Vinyl chloride metabolism -- a review.
Chemosphere, 6: 309-325.
Podoll K, Berg-Dammer E, & Noth J (1990) [Neurological and
psychiatrical disorders in vinyl chloride disease.] Fortschr Neurol
Psychiatr, 58: 439-443 (in German).
Popper H & Thomas LB (1975) Alterations of liver and spleen among
workers exposed to vinyl chloride. Ann NY Acad Sci, 246: 172-194.
Popper H, Maltoni C, & Selikoff IJ (1981) Vinyl chloride-induced
hepatic lesions in man and rodents: A comparison. Liver, 1: 7-20.
Portier R, Miller G, Hoover D, & Simar R (1992) Field applications of
biological reactors removing ethylene dichloride, trichloroethylene,
and vinyl chloride from contaminated groundwaters. Abstr Gen Meet Am
Soc Microbiol, 92: 382.
Portier RJ, Shane BS, Walsh MM, & Williams MB (1993) In situ
remediation of EDC contaminated vadose soil: A toxicological
assessment. Symposium on Emerging Technologies: Metals, Oxidation and
Separation, Belmont, Texas, 25-26 February. Waste Manage, 13(5-7):
532-533.
Poy F, Cobelli L, Banfi S, & Fossati F (1987) Determination of vinyl
chloride monomer residue in poly(vinyl chloride) at the
parts-per-billion level with an automatic purge- and - trap technique.
J Chromatogr, 395: 281-289.
Prodan L, Suciu I, Pîslaru V, Ilea E, & Pascu L (1975) Experimental
acute toxicity of vinyl chloride (monochloroethene). Ann NY Acad Sci,
246: 154-158.
Przygodzki RM, Finkelstein SD, Keohavong P, Zhu D, Bakker A, Swalsky
PA, Soini Y, Ishak KG, & Bennet WP (1997) Sporadic and
thorotrast-induced angiosarcomas of the liver manifest frequent and
multiple point mutations in K- ras-2. Lab Invest, 76: 153-159.
Pudill R (1993) [Comparative toxicological and ecotoxicological
evaluation of volatile emissions from waste deponies and
incinerators.] Umweltwiss Schadst -Forsch, 5: 223-227 (in German).
Purchase IFH, Richardson CR, Anderson D, Paddle GM, & Adams WGF (1978)
Chromosomal analyses in vinyl chloride-exposed workers. Mutat Res, 57:
325-334.
Purchase IFH, Stafford J, & Paddle GM (1987) Vinyl chloride: An
assessment of risk of occupational exposure. Food Chem Toxicol, 25:
187-202.
Puschmann J (1975) [Determination of vinyl chloride in PVC and food
simulants.] Angew Makromol Chem ,47: 29-41 (in German).
Radike MJ, Stemmer KL, & Bingham E (1981) Effect of ethanol on vinyl
chloride carcinogenesis. Environ Health Perspect, 41: 59-62.
Rakus J, Brantley T, & Szabo E (1991) Environmental concerns with
vinyl medical devices. J Vinyl Technol, 13: 195-198.
Ramsey JC & Andersen ME (1984) A physiologically based description of
the inhalation pharmacokinetics of styrene in rats and humans. Toxicol
Appl Pharmacol, 73: 159-175.
Randall PM (1994) Pollution prevention strategies for the minimization
of industrial wastes in the VCM-PVC industry. Environ Prog, 13:
269-277.
Rannug U, Johansson A, Ramel C, & Wachtmeister CA (1974) The
mutagenicity of vinyl chloride after metabolic activation. Ambio, 3:
194-197.
Rannug U, Göthe R, & Wachtmeister CA (1976) The mutagenicity of
chloroethylene oxide, chloroacetaldehyde, 2-chloroethanol and
chloroacetic acid, conceivable metabolites of vinyl chloride.
Chem-Biol Interact, 12: 251-263.
Rappoport Z & Gal A (1969) Vinylic cations from solvolysis: I. The
trianisylvinyl halide system. J Am Chem Soc, 91: 5246-5254.
Rasche ME, Hyman MR, & Arp DJ (1991) Factors limiting aliphatic
chlorocarbon degradation by Nitrosomonas europaea: Cometabolic
inactivation of ammonia monooxygenase and substrate specificity. Appl
Environ Microbiol, 57: 2986-2994
Rashad MM, El-Belbessy SF, Hussein NG, Helmey MH, & El-Toukhy MA
(1994) Effect on some enzyme activities of occupational exposure to
vinyl chloride monomer for five consecutive years. Med Sci Res, 22:
289-290.
Rehm T & Werner R (1996) [Polyvinyl chloride.] Kunststoffe, 86:
1474-1476 (in German).
Reitz RH, Gargas ML, Andersen ME, Provan WM, & Green TL (1996)
Predicting cancer risk from vinyl chloride exposure with a
physiologically based pharmacokinetic model. Toxicol Appl Pharmacol,
137: 253-267.
Reynolds ES, Jaeger RJ, & Murphy SD (1975a) Acute liver injury by
vinyl chloride: Involvement of endoplastic reticulum in
phenobarbital-pretreated rats. Environ Health Perspect, 11: 227-233.
Reynolds ES, Moslen MT, Szabo S, & Jaeger RJ (1975b) Vinyl
chloride-induced deactivation of cytochrome P-450 and other components
of the liver mixed function oxidase system: An in vivo study. Res
Commun Chem Pathol Pharmacol, 12: 685-694.
Reynolds ES, Moslen MT, Szabo S, Jaeger RJ, & Murphy SD (1975c)
Hepatotoxicity of vinyl chloride and 1,1-dichloroethylene. Am J
Pathol, 81: 219-231.
Richardson CR, Styles JA, & Bennett IP (1983) Activity of vinyl
chloride monomer in the mouse micronucleus assay. Mutat Res, 122:
139-142.
Riordan SM, Loo CK, Haber RW, & Thomas MC (1991) Vinyl chloride
related hepatic angiosarcoma in a polyvinyl chloride autoclave cleaner
in Australia. Med J Aust, 155: 125-128.
Rippen G (1995) Handbook of environmental chemicals. Landsberg,
Germany, Ecomed Verlag.
Roberts PV, Simprini L, Hopkins GD, Grbic-Galic D, McCarty PL, &
Reinhard M (1989) In-situ aquifer restoration of the chlorinated
aliphatics by methanotrophic bacteria. Washington, DC, US
Environmental Protection Agency, pp 1-232 (EPA/600/2-89/033; prepared
by Stanford University, Department of Civil Engineering, Stanford,
California.).
Rodics K, Barcza G, & Ungváry G (1981) Mutagenicity of VC exposition:
Studies of the dose-response relationship in the micronucleus test.
Egeszsegtudomany, 25: 313-416.
Rohrschneider L, Jaeschke A, & Kubik W (1971) [Investigation into air
pollution at heights of up to 500 m above an industrial site.] Chem
Ing Tech, 43: 1010-1017 (in German).
Rosenman KD, Rizzo JE, Conomos MG, & Halpin GJ (1989) Central nervous
system malformations in relation to two polyvinyl chloride production
facilities. Arch Environ Health, 44: 279-282.
Rösli M, Zimmerli B, & Marek B (1975) Residues of vinyl chloride
monomer in edible oils. Mitt Geb Lebensm.unters Hyg, 66: 507-511.
Rossberg M, Lendle W, Tögel A, Dreher EL, Langer E, Rassaerts H,
Kleinschmidt P, Strack H, Beck U, Lipper KA, Torkelson TR, Löser E, &
Beutel KK (1986) Chlorinated hydrocarbons. In: Gerhartz W, Yamamoto
YS, Campbell FT, Pfefferkorn R, & Rounsaville JF ed. Ullmann's
encyclopedia of industrial chemistry, 5th completely revis ed.
-- Volume A6: Ceramics to chlorohydrins. Weinheim, Germany, VCH
Verlagsgesellschaft mbH, pp 283-297.
Rössner P, Srám RJ, Nováková J, & Lambl V (1980) Cytogenetic analysis
in workers occupationally exposed to vinyl chloride. Mutat Res, 73:
425-427.
Rowe VK (1975) Experience in industrial exposure control. Ann NY Acad
Sci, 246: 306-310.
Ryan JA, Bell RM, Davidson JM, & O'Connor GA (1988) Plant uptake of
non-ionic organic chemicals from soils. Chemosphere, 17: 2299-2323.
Sabadie N, Malaveille C, Camus AM, & Bartsch H (1980) Comparison of
the hydroxylation of benzo(a)pyrene with the metabolism of vinyl
chloride, N-nitrosomorpholine, and N-nitroso-N'-methylpiperazine to
mutagens by human and rat liver microsomal fractions. Cancer Res, 40:
119-126.
Sabel GV & Clark TP (1984) Volatile organic compounds as indicators of
municipal solid waste leachate contamination. Waste Manage Res, 32:
119-130.
Sabljic A (1984) Prediction of the nature and strength of soil
sorption of organic pollutants by molecular topology. J Agric Food
Chem, 32: 243-246.
Saito T, Barbin A, Omori Y, & Yamasaki H (1997) Connexion 37 mutation
in rat hepatic angiosarcomas induced by vinyl chloride. Cancer Res,
57: 375-377.
Salkinoja-Salonen M, Uotila J, Jokela J, Laine M, & Saski E (1995)
Organic halogens in the environment: Studies of environmental
biodegradability and human exposure. Environ Health Perspect, 103:
63-69.
Samoiloff MR, Schulz S, Jordan Y, Denich K, & Arnott E (1980) A rapid
simple long-term toxicity assay for aquatic contaminants using the
nematode Panagrellus redivivus. Can J Fish Aquat Sci, 37: 1167-1174
Sanchez JH & Recio L (1991) Mutagenicity of chloroacetaldehyde in
human TK6 lymphoblasts. Environ Mol Mutagen, 17(suppl 19): 64.
Sanhueza E & Heicklen J (1975) Oxidation of chloroethylene. J Phys
Chem, 79: 677-681.
Sanhueza E, Hisatsune IC, & Heicklen J (1976) Oxidation of
haloethylenes. Chem Rev, 76: 801-826.
Sanotsky IV, Davtian RM, & Glushchenko VJ (1980) [Study of the
reproductive function in men exposed to chemicals.] Gig Tr Prof Zabol,
6: 28-32 (in Russian)
Saparbaev M & Laval J (1998) 3,N4-ethenocytosine, a highly mutagenic
adduct, is a primary substrate for Escherichia coli double-stranded
uracil-DNA glycosylase and human mismatch-specific thymine-DNA
glycosylas. Proc Natl Acad Sci (USA), 95: 8508-8513.
Saparbaev M, Kleibl K, & Laval J (1995) Eschericia coli,
Saccharomyces cerevisiae, rat and human 3-methyladenine DNA
glycosylases repair 1,N6-ethenoademine when present in DNA. Nucleic
Acids Res, 23: 3750-3755.
Saurin JC, Taniere P, Mion F, Jacob P, Partensky C, Paliard P, &
Berger F (1997) Primary hepatocellular carcinoma in workers exposed to
vinyl chloride: a report of two cases. Cancer, 79: 1671-1677.
Sauvant MP, Pépin D, & Bohatier J, & Groliere CA (1995) Comparison of
six bioassays for assessing in vitro acute toxicity and
structure-activity relationships for vinyl chloride monomer, its main
metabolites and derivates. Sci Total Environ, 172: 79-92.
Schaffner F (1979) Effect of long-term vinyl chloride exposure in
mouse liver structure. In: Remmer H, Bolt HM, Bannasch P, & Popper H
ed. Proceedings of the Symposium on Primary Liver Tumors, Titisee,
Germany, 3-5 October 1977. New York, MTP Press Limited, International
Medical Publishers, pp 189-199.
Scherb K (1978) [Investigations into the evaporation of low molecular
chlorinated hydrocarbons from running water.] Münch Beitr
Abwasser- Fisch- Flussbiol, 30: 235-248 (in German).
Scherer E, Van der Laken CJ, Gwinner LM, Laib RJ, & Emmelot P (1981)
Modification of deoxyguanosine by chloroethylene oxide.
Carcinogenesis, 2: 671-677.
Schippert E (1987) [Purification of water contaminated with
chlorinated of hydrocarbons - comparison of direct absorption to
activated charcoal and stripping with air by purification of the air
by activated charcoal.] In: Thome-Kozmiensky KJ ed. [Existing
chemicals.] Berlin, Publishers for Energy and Environmental Technics,
pp 674-680 (in German).
Schlett C & Pfeifer B (1993) [Gas chromatographic determination of
vinyl chloride after purge-and trap concentration and mass
spectrometric detection.] Vom Wasser, 81: 1-6 (in German).
Schleyer R, Arneth J-D, Kerndorff H, & Milde G (1988) [Main priority
contaminants in waste dump sites: Selection criteria for evaluation
with respect to ground water path.] In: Wolf K, Van den Brink WJ, &
Colon FJ ed. [Cleaning of contaminated sites '88: Second TNO/BMFT
International Congress on Sanitation of Contaminated Sites, Hamburg,
11-15 April 1988.] Berlin, Federal Environmental Agency, vol 1, pp
249-253 (in German).
Schulz-Berendt V (1993) Soil bio remediation in practice: Problems and
results. In: Eijsackers HJP & Hamers T ed. Integrated soil and
sediment research: A basis for proper protection. Dordrecht, The
Netherlands, Kluwer Academic Publishers, pp 559-575.
Seiber JN (1996) Toxic air contaminants in urban atmospheres:
Experience in California. Atmos Environ, 30: 751-756.
Semenza JC & Weasel LH (1997) Molecular epidemiology in environmental
health: The potential of tumor suppressor gene p53 as a biomarker.
Environ Health Perspect, 105: 155-163.
Semprini L (1995) In situ bioremediation of chlorinated solvents.
Environ Health Perspect, 103: 101-105.
Semprini L, Roberts PV, Hopkins GD, & McCarty PL (1990) A field
evaluation of in-situ biodegradation of chlorinated ethenes -- Part 2:
Results of biostimulation and biotransformation experiments. Ground
Water, 28: 715-727.
Semprini L, Hopkins GD, Roberts PV, Grbic-Galic D, & McCarty PL (1991)
A field evaluation of in situ biodegradation of chlorinated ethenes
-- Part 3: Studies of competitive inhibition. Ground Water, 29:
239-250.
Semprini L, Kitanidis PK, Kampbell DH, & Wilson JT (1995) Anaerobic
transformation of chlorinated aliphatic hydrocarbons in a sand aquifer
based on spatial chemical distributions. Water Resour Res, 31:
1051-1062.
Shah HC (1998) Vinyl chloride combined inhalation two-generation
reproduction and developmental toxicity study in CD rats: Reproduction
study -- Final report (Study No. 96-4080). East Millstone, Huntingdon
Life Sciences Inc., vol I, pp 1-1726 (Unpublished report).
Shah JJ & Singh HB (1988) Distribution of volatile organic chemicals
in outdoor and indoor air. Environ Sci Technol, 22: 1381-1388.
Shahin MM (1976) The non-mutagenicity and -- recombinogenicity of
vinyl chloride in the absence of metabolic activation. Mutat Res, 40:
269-272.
Sharma RP & Gehring PJ (1979) Immunologic effects of vinyl chloride in
mice. Ann NY Acad Sci, 320: 551-563.
Sharma PK & McCarty PL (1996) Isolation and characterization of a
facultatively aerobic bacterium that reductively dehalogenates
tetrachloroethene to cis-1,2-dichloroethene. Appl Environ Microbiol,
62: 761-765.
Shechter M (1985) An anatomy of a groundwater contamination episode. J
Environ Econ Manage, 12: 72-88.
Sheldon LS & Hites RA (1978) Organic compounds in the Delaware river.
Environ Sci Technol, 12: 1188-1194.
Shimada T, Swanson AF, Leber P, & Williams GM (1985) Activities of
chlorinated ethane and ethylene compounds in the Salmonella/rat
microsome mutagenesis and rat hepatocyte/DNA repair assays under vapor
phase exposure conditions. Cell Biol Toxicol, 1: 159-179.
Shimp JF, Tracy JC, Davis LC, Lee E, Huang W, & Erickson LE (1993)
Beneficial effects of plants in the remediation of soil and
groundwater contaminated with organic materials. Crit Rev Environ Sci
Technol, 23: 41-77.
Shin MS, Carpenter JT, & Ho K-J (1991) Epithelioid
hemangioendothelioma: CT manifestations and possible linkage to vinyl
chloride exposure. J Comput Assist Tomogr, 15: 505-507.
Shirey RE (1995) Rapid analysis of environmental samples using
solid-phase microextraction (SPME) and narrow bore capillary columns.
J High Resol Chromatogr, 18: 495-499.
Short RD, Minor JL, Winston JM, & Lee C-C (1977) A dominant lethal
study in male rats after repeated exposures to vinyl chloride or
vinylidene chloride. J Toxicol Environ Health, 3: 965-968.
Simha D, Palejwala VA, & Humayun MZ (1991) Mechanisms of mutagenesis
by exocyclic DNA adducts: Construction and in vitro template
characteristics of an oligonucleotide bearing a single site-specific
ethenocytosine. Biochemistry, 30: 8727-8735.
Simonato L, L'Abbé KA, Andersen A, Belli S, Comba P, Engholm G, Ferro
G, Hagmar L, Langard S, Lundberg I, Pirastu R, Thomas P, Winkelmann R,
& Saracci R (1991) A collaborative study of cancer incidence and
mortality among vinyl chloride workers. Scand J Work Environ Health,
17: 159-169.
Simpson G, Klasmeier M, Hill H, Atkinson D, Radolovich G, Lopez-Avila
V, & Jones TL (1996) Evaluation of gas chromatography coupled with ion
mobility spectrometry for monitoring Vinyl Chloride and other
chlorinated and aromatic compounds in air samples. J High Resol
Chromatogr, 19: 301-312.
Singer B (1996) DNA damage: Chemistry, repair, and mutagenic
potential. Regul Toxicol Pharmacol, 23: 2-13.
Singer B, Kusmierek JT, Folkman W, Chavez F, & Dosanjh MK (1991)
Evidence for the mutagenic potential of the vinyl chloride induced
adduct, N2,3-etheno-deoxyguanosine, using a site-directed kinetic
assay. Carcinogenesis, 12: 745-747.
Singh HB, Ludwig FL, & Johnson WB (1978) Tropospheric ozone:
Concentrations and variabilities in clean remote atmospheres. Atmos
Environ, 12: 2185-2196.
Singh HB, Jaber HM, & Davenport JE (1984) Reactivity/volatility
classification of selected organic chemicals: existing data. Research
Triangle Park, North Carolina, US Environmental Protection Agency, pp
1-190 (EPA-600/3-84-082; prepared by SRI International, Menlo Park,
California).
Sinués B, Sanz A, Bernal ML, Tres A, Alcala A, Lanuza J, Ceballos C, &
Sáenz MA (1991) Sister chromatid exchanges, proliferating rate index,
and micronuclei in biomonitoring of internal exposure to vinyl
chloride monomer in plastic industry workers. Toxicol Appl Pharmacol,
108: 37-45.
Sittig M (1985) Vinylchloride. In: Sittig M ed. Handbook of toxic and
hazardous chemicals and carcinogens, 2nd ed. Park Ridge, New Jersey,
Noyes Publications, pp 918-921.
Skeen RS, Gao J, & Hooker BS (1995) Kinetics of chlorinated ethylene
dehalogenation under methanogenic conditions. Biotechnol Bioeng, 48:
659-666.
Smerasta P, Srivburuang P, Tongtan N, Ishiwata H, & Yoshihira K (1991)
Migration and material tests of some food-contact plastic wares made
in Thailand. Bull Natl Inst Hyg Sci, 109: 105-106.
Smith LR & Dragun J (1984) Degradation of volatile chlorinated
aliphatic priority pollutants in groundwater. Environ Int, 10:
291-298.
Smith SJ, Li Y, Whitley R, Marion MJ, Partilo S, Carney WP, &
Brandt-Rauf PW (1998) Molecular epidemiology of p53 protein mutations
in workers exposed to vinyl chloride. Am J Epidemiol, 147: 302-308.
Smulevich VB, Fedotova IV, & Filatova VS (1988) Increasing evidence of
the risk of cancer in workers exposed to vinyl chloride. Br J Ind Med,
45: 93-97.
Soini Y, Welsh JA, Ishak KG, & Bennett WP (1995) p53 mutations in
primary hepatic angiosarcomas not associated with vinyl chloride
exposure. Carcinogenesis, 16: 2879-2881.
Sokal JA, Baranski B, Majka J, Rolecki R, Stetkiewicz J,
Ivanova-Chemishanska L, Vergieva T, Antonov G, Mirkova E, Kolakowski
A, Szendzikowski S, & Wróblewska K (1980) Experimental studies on the
chronic toxic effects of vinyl chloride in rats. J Hyg Epidemiol
Microbiol Immunol, 24: 285-294.
Solionova LG, Smulevich VB, Turbin EV, Krivosheyeva LV, & Plotnikov JV
(1992) Carcinogens in rubber production in the Soviet Union. Scand J
Work Environ Health, 18: 120-123.
Sood C & O'Brien PJ (1993) Molecular mechanisms of
chloroacetaldehyde-induced cytotoxicity in isolated rat hepatocytes.
Biochem Pharmacol, 46: 1621-1626.
Soule R, Symonik D, Jones D, Turgeon D, & Gerbec B (1996) Vinyl
Chloride loss during laboratory holding time. Regul Toxicol Pharmacol,
23: 209-212.
Spit BJ, Feron VJ, & Hendriksen CFM (1981) Ultrastructure of hepatic
angiosarcoma in rats induced by vinyl chloride. Exp Mol Pathol, 35:
277-284.
Staples CA, Werner AF, & Hoogheem TJ (1985) Assessment of priority
pollutant concentrations in the United States using STORET database.
Environ Toxicol Chem, 4: 131-142.
Stareczek T (1988) [Differences in content of free monomer during
storage and processing of mono- and copolymer vinyl chloride.] Polim
Tworz Wielk 33: 145-147 (in Polish).
Stephens RD, Ball NB, & Mar DM (1986) A multimedia study of hazardous
waste landfill gas migration. In: Cohen Y ed. Pollutants in a
multimedia environment. New York, London, Plenum Press, pp 265-287.
Storm JE & Rozman KK (1997) Evaluation of alternative methods for
establishing safe levels of occupational exposure to vinyl halides.
Regul Toxicol Pharmacol, 25: 240-255.
Strandberg GW, Donaldson TL, & Farr LL (1989) Degradation of
trichloroethylene and trans-1,2-dichloroethylene by a methanotrophic
consortium in a fixed-film, packed-bed bioreactor. Environ Sci
Technol, 23: 1422-1425.
Stringer RL, Costner P, & Johnston PA (1995) PVC manufacture as a
source of PCDD/Fs. Organohalog Compd, 24: 119-123.
Stuart JD (1983) Organics transported thru selected geologic media:
Assessment of organics transported away from industrial waste disposal
sites. Springfield, Virginia, US Department of the Interior, pp 1-10
(Document No. A-089 CONN, prepared by the University's Institute of
Water Resources, Storrs, Connecticut, USA).
Stuckey DC, Owen WF, & McCarty PL (1980) Anaerobic toxicity evaluation
by batch and semi-continuous assays. J Water Pollut Control Fed, 52:
720-729.
Studniarek M, Durski K, Liniecki J, Brykalski D, Poznanska A, &
Gluszcz M (1989) Effects of vinyl chloride on liver function of
exposed workers, evaluated by measurements of plasma clearance of the
99mTc-N-2,4-dimethylacetanilido-iminoiacetate complex. J Appl Toxicol,
9: 213-218.
Stblová V, Lambl V, Chumcal O, Kellerová V, Pasková V, Vítovcocá J, &
Zlab L (1981) Neurological changes in vinyl chloride-exposed workers.
J Hyg Epidemiol Microbiol Immunol, 25: 233-243.
Styles JA (1980) Studies on the detection of carcinogens using a
mammalian cell transformation assay with liver homogenate activation.
In: Norpoth KH & Garner RC ed. Short-term test systems for detecting
carcinogens. Berlin, Heidelberg, New York, Springer Verlag, pp
226-238.
Suciu I, Prodan L, Ilea E, Paduraru A, & Pascu L (1975) Clinical
manifestations in vinyl chloride poisoning. Ann NY Acad Sci, 246:
53-69.
Sugita M, Masuda Y, & Tschuiya K (1986) Early detection and signs of
hepatoangiosarcoma among vinyl chloride workers. Am J Ind Med, 10:
411-417.
Suter GW (1996) Toxicological benchmarks for screening contaminants of
potential concern for effects on freshwater biota. Environ Toxicol
Chem, 15: 1232-1241.
Suzuki Y (1978) Pulmonary tumors induced in mice by vinyl chloride
monomer. Environ Res, 16: 285-301.
Suzuki Y (1980) Nonneoplastic effects of vinyl chloride in mouse lung.
Environ Res, 21: 235-253.
Suzuki Y (1981) Neoplastic and nonneoplastic effects of vinylchloride
in mouse lung. Environ Health Perspect, 41:31-52.
Suzuki Y (1983) Neoplastic effect of vinyl chloride in mouse lung --
lower doses and short-term exposure. Environ Res, 32: 91-103.
Svensson K (1994) Legislation, control and research in the Nordic
countries on plastics for packaging food. Food Addit Contam, 11:
241-248.
Swenberg JA, Fedtke N, Ciroussel F, Barbin A, & Bartsch H (1992)
Etheno adducts formed in DNA of vinyl chloride-exposed rats are highly
persistent in liver. Carcinogenesis, 13: 727-729.
Swenberg JA, La KD, Scheller NA, & Kuen-Yuh W (1995) Dose-response
relationships for carcinogens. Toxicol Lett, 82/83: 751-756.
Szadkowski D & Lehnert G (1982) [Vinyl chloride as a cause of disease
- a bibliography.] In: Szadkowski D & Lehnert G ed. [On the subject of
VC/PVC.] Frankfurt am Main, Germany, Henssler KG, pp 1-20 (in German).
Szentesi I, Hornyák É, Ungváry G, Czeizel A, Bognár Z, & Timar M
(1976) High rate of chromosomal aberration in PVC workers. Mutat Res,
37: 313-316.
Tabershaw IR & Gaffey WR (1974) Mortality study of workers in the
manufacture of vinyl chloride and its polymers. J Occup Med, 16:
509-518.
Tamburro CH, Makk L, & Popper H (1984) Early hepatic histologic
alterations among chemical(vinyl monomer) workers. Hepatology, 4:
413-418.
Tandoi V, DiStefano TD, Bowser PA, Gossett JM, & Zinder SH (1994)
Reductive dehalogenation of chlorinated ethenes and halogenated
ethanes by a high-rate anaerobic enrichment culture. Environ Sci
Technol, 28: 973-979.
Tátrai E & Ungváry G (1981) On the acute hepatotoxicity of inhaled
vinyl chloride. Acta Morphol Acad Sci Hung, 29: 221-226.
Terwiesch B (1982) Vinyl chloride peroxide explosion in a vinyl
chloride recovery plant. J Macromol Sci Chem, A17: 1081-1092.
Tester DA (1976) The extraction of vinyl chloride from PVC containers.
J Soc Cosmet Chem, 49: 459-466.
Theisen J, Funcke W, Balfanz E, & König J (1989) Determination of
PCDFs and PCDDs in fire accidents and laboratory combustion tests
involving PVC-containing materials. Chemosphere, 19: 423-428.
Theisen J, Funcke W, & Hamm S (1991) [Investigation of possible risks
to the environment from burning plastics.] In: [Investigation of
possible risks to the environment from burning plastics.] Münster,
Germany, Society for Occupational and Environmental Analytic (GFA), pp
91-131 (in German).
Thériault G & Allard P (1981) Cancer mortality of a group of Canadian
workers exposed to vinyl chloride monomer. J Occup Med, 23: 671-676.
Thériault G, Iturra H, & Gingras S (1983) Evaluation of the
association between birth defects and exposures to ambient vinyl
chloride. Teratology, 27: 359-370.
Thiess AM & Versen P (1974) [Occupational medical thoughts on the
so-called 'vinyl chloride disease'.] Arbeitsmed Sozialmed
Präventivmed, 7: 146-148 (in German).
Thomas VN & Ramstad T (1992) A dynamic headspace GC method for the
determination of vinyl chloride monomer in stored solutions of
cefmetazole sodium in PVC bags. Acta Pharm Nord, 4: 97-104.
Tie X, Kao C-Y, Mroz EJ, Cicerone RJ, Alyea FN, & Cunnold DM (1992)
Three-dimensional simulations of atmospheric methyl chloroform: Effect
of an ocean sink. J Geophys Res Atmos, D97: 20.751-20.769.
Til HP, Feron VJ, & Immel HR (1991) Lifetime (149-week) oral
carcinogenicity study of vinyl chloride in rats. Food Chem Toxicol,
29: 713-718.
Til HP, Immel HP, & Feron FJ (1983) Lifespan oral carcinogenicity
study of vinyl chloride in rats. Zeist, The Netherlands, Organization
for Applied Scientific Research (TNO), pp 1-29 (TNO Report No.
V-83.285/291099).
Topudurti K (1992) A UV/oxidation technology demonstration to treat
groundwater contaminated with VOCs. Water Sci Technol, 25: 347-354.
Torkelson TR (1994) Halogenated aliphatic hydrocarbons containing
chlorine, bromine, and iodine.. In: Clayton GD & Clayton FE ed.
Patty's industrial hygiene and toxicology, 4th ed. New York, John
Wiley & Sons, Inc., vol 2, part E, pp 4007-4251.
Torkelson TR, Oyen F, & Rowe VK (1961) The toxicity of vinyl chloride
as determined by repeated exposure of laboratory animals. Am Ind Hyg
Assoc J, 22: 354-361.
Tribukh SL, Tikhomirova NP, Levin SV, & Kozlov LA (1949) Working
conditions and measures for their sanitation in the production and
utilization of vinyl chloride plastics. Gig i Sanit, 10: 38-45.
Trivers GE, Cawley HL, DeBenedetti VMG, Hollstein M, Marion MJ, Benett
WP, Hoover ML, Prives CC, Tamburro CC, & Harris CC (1995) Anti-p53
antibodies in sera of workers occupationally exposed to vinyl
chloride. J Natl Cancer Inst, 87: 1400-1407.
Tsien H-C, Brusseau GA, Hanson RS, & Wackett LP (1989) Biodegradation
of trichloroethylene by Methylosinus trichosporium OB3b. Appl
Environ Microbiol, 55: 3155-3161.
Tsivoglou EC (1967) Tracer measurement of stream reaeration.
Washington, DC, Federal Water Pollution Control Administration (PB-229
923).
Tu AS, Murray TA, Hatch KM, Sivak A, & Milman HA (1985) In vitro
transformation of BALB/c-3T3 cells by chlorinated ethanes and
ethylenes. Cancer Lett, 28: 85-92.
Tuazon EC, Atkinson R, Aschmann SM, Goodman MA, & Winer AM (1988)
Atmospheric reactions of chloroethenes with the OH radical. Int J Chem
Kinet, 20: 241-265.
Turner AC, Payne AP, & Bushby BR (1984) Determination of ambient
levels of vinyl chloride monomer (VCM) around manufacturers in the UK,
Part 7. Warren Spring Laboratory, pp 1-26 (Report No. LR 514[AP]).
Uchiyama H, Nakajima T, Yagi O, & Tabuchi T (1989) Aerobic degradation
of trichloroethylene at high concentration by a methane-utilizing
mixed culture. Agric Biol Chem, 53: 1019-1024.
UK MAFF (Ministry of Agriculture Fisheries and Food) (1978) Working
Party on Vinyl Chloride -- Survey of vinyl chloride content of
polyvinyl chloride for food contact and of foods: Second report of the
Steering Group on Food Surveillance. London, Her Majesty's Stationary
Office, pp 1-16 (Food Surveillance Paper No. 2).
UK MAFF (Ministry of Agriculture Fisheries and Food) (1984) Steering
Group on Food Surveillance -- Progress report 1984: Fourteenth report
of the Steering Group on Food Surveillance. London, Her Majesty's
Stationery Office, pp 11-15, 46 (Food Surveillance Paper No. 14).
Ungváry G, Hudák A, Tátrai E, Lörincz M, & Folly G (1978) Effects of
vinyl chloride exposure alone and in combination with trypan blue --
applied systematically during all thirds of pregnancy on the fetuses
of CFY rats. Toxicology, 11: 45-54.
US EPA (1975) Scientific and technical assessment report on vinyl
chloride and polyvinyl chloride. Research Triangle Park, North
Carolina, US Environmental Protection Agency, Office of Research and
Development, Environmental Research Center (EPA 600/6-75-004).
US EPA (1982) Vinyl chloride -- a review of national emission
standards. Research Triangle Park, North Carolina, US Environmental
Protection Agency, pp 1-37 (EPA 450/3-82-003).
US EPA (1985a) Health and environmental effects profile for
chloroethene. Cincinnati, Ohio, US Environmental Protection Agency, pp
1-176 (EPA 600/x-85/374; PB88-174529).
US EPA (1985b) Drinking water criteria document for vinyl chloride.
Washington, DC, US Environmental Protection Agency, pp 1-18 (Report
TR-540-162; prepared by ICAIR Life Systems, Inc., Cleveland, Ohio).
US EPA (1987) Incorporation of biological information in cancer risk
assessment: Example -- vinyl chloride. Washington, DC, US
Environmental Protection Agency, pp 1-51 (EPA 600/21).
US EPA (1992) Superfund record of decision, Hagen farm, WI: Second
remedial action- final. Washington, DC, US Environmental Protection
Agency, pp 1-40 (EPA/ROD/RO5-92/216; prepared by the Hagen Farm Site,
Groundwater Control Operable Unit, Dane Country, Wisconsin).
US EPA (1995a) Health effects assessment summary tables (HEAST).
Washington, DC, US Environmental Protection Agency, Office of Solid
Waste and Emergency Response (EPA 540 R-95/036).
US EPA (1995b) Drinking water standards and health advisories table.
San Francisco, California, US Environmental Protection Agency, Region
IX, 26 pp.
US EPA (1996) Proposed guidelines cancer risk assessment. Fed Reg,
61(79): 177960-180011.
US FDA (Food and Drug Administration) (1973) Prior-sanctioned
polyvinyl chloride resin. Fed Reg, 38: 12931.
US FDA (Food and Drug Administration) (1986) Vinyl chloride polymers:
Withdrawal of proposal. Fed Reg, 51: 4173-4188.
Uzych L (1988) Human male exposure to vinyl chloride and possible
teratogenic and mutagenic risks: A review. Hum Toxicol, 7: 517-527.
Van der Meer JR, Bosma TNP, De Bruin WP, Harms H, Holliger C,
Rijnaarts HHM, Tros ME, Schraa G, & Zehnder AJB (1992) Versatility of
soil column experiments to study biodegradation of halogenated
compounds under environmental conditions. Biodegradation, 3: 265-284.
Van Duuren BL (1975) On the possible mechanism of carcinogenic action
of vinyl chloride. Ann NY Acad Sci, 246: 258-267.
Van Lierop JBH (1979) Determination of potentially carcinogenic
compounds in food. In: Van Lierop JBH ed. Determination of potentially
carcinogenic compounds in food:Trace analysis of vinylchloride,
vinylidenechloride, acrylonitrile, epichlorohydrin and
diethylpyrocarbonate. Utrecht, The Netherlands, University of Utrecht,
pp 1-187.
van Sittert NJ & De Jong G (1985) Biomonitoring of exposure to
potential mutagens and carcinogens in industrial populations. Food
Chem Toxicol, 23: 23-31.
Van't Hof J & Schairer LA (1982) Tradescantia assay system for gaseous
mutagens. A report of the US Environmental Protection Agency Gene-Tox
Program. Mutat Res, 99: 303-315.
Vannelli T, Logan M, Arciero DM, & Hooper AB (1990) Degradation of
halogenated aliphatic compounds by the ammonia-oxidizing bacterium
Nitrosomonas europaea. Appl Environ Microbiol, 56: 1169-1171.
Veltman G, Lange CE, Jühe S, Stein G, & Bachner U (1975) Clinical
manifestations and course of vinyl chloride disease. Ann NY Acad Sci,
246: 6-17.
Verburgt FG & Vogel E (1977) Vinyl chloride mutagenesis in Drosophila
melanogaster. Mutat Res, 48: 327-337.
Victorin K & Ståhlberg M (1988) A method for studying the mutagenicity
of some gaseous compounds in Salmonella typhimurium. Environ Mol
Mutagen, 11: 65-77.
Victorin K & Ståhlberg M (1991) Mutagenic activity of photoreaction
products formed from chlorinated ethenes and nitrogen dioxide. Environ
Res, 55: 178-187.
Viinanen R (1993) Monitoring results of VC in a PVC plant in
1981-1993. Porvoo, Finland, Neste Environmental and Industrial Hygiene
Institute, pp 1-13 (Research Report No. 93025T).
Viola PL (1970) Pathology of vinyl chloride. Med Lav, 61: 174-180.
Viola PL, Bigotti A, & Caputo A (1971) Oncogenic response of rat skin,
lungs and bones to vinyl chloride. Cancer Res, 31: 516-522.
Vogel TM & McCarty PL (1985) Biotransformation of tetrachloroethylene
to trichloroethylene, dichloroethylene, vinyl chloride, and carbon
dioxide under methanogenic conditions. Appl Environ Microbiol, 49:
1080-1083.
Vogel EW & Nivard MJM (1993) Performance of 181 chemicals in a
Drosophila assay predominantly monitoring interchromosomal mitotic
recombination. Mutagenesis, 8: 57-81.
Vogel TM, Criddle CS, & McCarty PL (1987) Transformations of
halogenated aliphatic compounds. Environ Sci Technol, 21: 722-736.
Vom Bruck CG, Rudolph FB, Figge K, & Eckert WR (1979) Application of
the diffusion theory to migration of plastics components into packed
goods: Survey of recent migration studies. Food Cosmet Toxicol, 17:
153-157.
Wackett LP, Brusseau GA, Householder SR, & Hanson RS (1989) Survey of
microbial oxygenases: Trichloroethylene degradation by
propane-oxidizing bacteria. Appl Environ Microbiol, 55: 2960-2964.
Wagenaar H, Langeland K, Hardman R, Sergeant Y, Brenner K, Sandra P,
Rappe C, Fernandes A, & Tiernan T (1998) Analysis of PCDDs and PCDFs
in virgin suspension PVC resin. Chemosphere, 36: 1-12.
Wakeman IB & Johnson HR (1978) Vinyl chloride formation from the
thermal degradation of poly(vinyl chloride). Polymer, 18: 404-407.
Walker AE (1976) Clinical aspects of vinyl chloride disease: Skin.
Proc R Soc Med, 69: 286-289.
Walles SA & Holmberg B (1984) Induction of single-strand breaks in DNA
of mice after inhalation of vinyl chloride. Cancer Lett, 25: 13-18.
Walles SA, Holmberg B, Svensson K, Osterman-Golkar S, & Sigvardsson K
(1988) Induction of single-strand breaks in liver DNA of mice after
inhalation of vinylchloride. In: Bartsch H, Hemminki K, & O-Neill IK
ed. Methods for detecting DNA damaging agents in humans: Applications
in cancer epidemiology and prevention. Lyon, International Agency for
Research on Cancer, pp 227-231 (IARC Scientific Publications No. 89).
Wang T, Lenahan R, & Kanik M (1985) Impact of trichlorethylene
contaminated groundwater discharged to the main canal and Indian River
Lagoon, Vero Beach, Florida. Bull Environ Contam Toxicol, 34: 578-586.
Wang YL & Zhao XH (1987) Occupational health of working women in
China. Asia-Pac J Public Health, 1(4): 66-71.
Ward AM, Udnoon S, Watkins J, Walker AE, & Darke CS (1976)
Immunological mechanisms in the pathogenesis of vinyl chloride
disease. Br Med J, 1: 936-938.
Ward RS, Williams GM, & Hills CC (1996) Changes in major and trace
components of landfill gas during subsurface migration. Waste Manage
Res, 14: 243-261.
Watanabe PG & Gehring PJ (1976) Dose-dependent fate of vinyl chloride
and its possible relationship to oncogenicity in rats. Environ Health
Perspect, 17: 145-152.
Watanabe PG, McGowan GR, & Gehring PJ (1976a) Fate of [14C]vinyl
chloride after single oral administration in rats. Toxicol Appl
Pharmacol, 36: 339-352.
Watanabe PG, McGowan RG, Madrid EO, & Gehring PJ (1976b) Fate of
[14C]vinyl chloride following inhalation exposure in rats. Toxicol
Appl Pharmacol, 37: 49-59.
Watanabe PG, Hefner RE, & Gehring PJ (1976c) Vinyl chloride-induced
depression of hepatic non-protein sulfhydryl content and effects on
bromosulphalein (BSP) clearance in rats. Toxicology, 6: 1-8.
Watson JR, Lawrence RC, & Lovering EG (1977) A GLC method for
monitoring vinyl chloride in drugs and in polyvinyl chloride drug
containers. Can J Pharmacol Sci, 12: 94-97.
Watanabe PG, Zempel JA, Pegg DG, & Gehring PJ (1978a) Hepatic
macromolecular binding following exposure to vinyl chloride. Toxicol
Appl Pharmacol, 44: 571-579.
Watanabe PG, Zempel JA, & Gehring PJ (1978b) Comparison of the fate of
vinyl chloride following single and repeated exposure in rats. Toxicol
Appl Pharmacol, 44: 391-399.
Watson JR, Lawrence RC, & Lovering EG (1979) A GC headspace method for
monitoring vinyl chloride in liquid drug and cosmetic products. Can J
Pharmacol Sci, 14: 57-60.
Waxweiler RJ, Stringer W, Wagoner JK, Jones J, Falk H, & Carter C
(1976) Neoplastic risk among workers exposed to vinyl chloride. Ann NY
Acad Sci 271: 40-48.
Waxweiler J, Falk H, McMichael A, Mallov JS, Grivas AS, & Stringer WT
(1977) A cross-sectional epidemiologic survey of vinyl chloride
workers. Cincinnati, Ohio, National Institute of Occupational Safety
and Health, pp 1-43 (DHEW (NIOSH) Publication No.77-177).
Weaver JW, Wilson JT, & Kampbell DH (1996) Extraction of degradation
rate constants from the St Joseph, Michigan, trichloroethene site. In:
Proceedings of the Symposium on Natural Attenuation of Chlorinated
Organics in Ground Water, 11-13 September 1996. Washington, DC, US
Environmental Protection Agency, pp 69-73 (EPA 540/R-96/509).
Weber H, Reinl W, & Greiser E (1981) German investigations on
morbidity and mortality of workers exposed to vinyl chloride. Environ
Health Perspect, 41: 95-99.
Weisman WH (1992) Toxicity, mutagenicity, and mutational spectra of
vinyl chloride, 2-chloroethylene oxide, and chloroacetaldehyde in a
human lymphoblastoid line expressing cytochrome P 450IIE1. Chapel
Hill, North Carolina, University of North Carolina, pp 1-85
(AFIT/CI/CIA-92-030).
Westrick JJ, Mello JW, & Thomas RF (1984) The ground water supply
survey. J Am Water Works Assoc, 76: 52-59.
Wheeler RN (1981) Poly(vinyl Chloride) processes and products. Environ
Health Perspect, 41: 123-128.
WHO (1987) Vinyl chloride. In: Air quality guidelines for Europe.
Copenhagen, World Health Organization, Regional Office for Europe, pp
158-169 (WHO Regional Publications, European Series No. 23).
WHO (1996) Vinyl chloride. In: Guidelines for drinking-water quality
-- Volume 2: Health criteria and other supporting information. Geneva,
World Health Organization, pp 424-431.
Wild SR & Jones KC (1992) Organic chemicals entering agricultural
soils in sewage sludges: screening for their potential to transfer to
crop plants and livestock. Sci Total Environ, 119: 85-119.
Williams DT (1976a) Gas-liquid chromatographic headspace method for
vinyl chloride in vinegars and alcoholic beverages. J Assoc Off Anal
Chem, 59: 30-31.
Williams DT (1976b) Confirmation of vinyl chloride in food by
conversion to 1-chloro-1,2-dibromoethane. J Assoc Off Anal Chem, 59:
32-34.
Williams DT & Miles WF (1975) Gas-liquid chromatographic determination
of vinyl chloride in alcoholic beverages, vegetable oils, and
vinegars. J Am Optom Assoc, 58: 272-275.
Williamson J & Kavanagh B (1987) Vinyl chloride monomer and other
contaminants in PVC welding fumes. Am Ind Hyg Assoc J, 48: 432-436.
Wilson RH, McCormick WE, Tatum CF, & Creech JL (1967) Occupational
acroosteolysis: Report of 31 cases. J Am Med Assoc, 201: 577-580.
Wisniewska-Knypl JM, Klimczak J, & Kolakowski J (1980) Monooxygenase
activity and ultrastructural changes of liver in the course of chronic
exposure of rats to vinyl chloride. Int Arch Occup Environ Health, 46:
241-249.
Withey JR (1976) Pharmacodynamics and uptake of vinyl chloride monomer
administered by various routes to rats. J Toxicol Environ Health, 1:
381-394.
Wittsiepe J (1990) Occurrence of vinyl chloride and other halogenated
C1- and C2- hydrocarbons in German surface water. Organohalog Compd,
4: 425-434.
Wittsiepe J, Selenka F, & Jackwerth E (1990) Gas-chromatographic
determination of trace amounts of vinyl chloride in water and air
after derivatization to 1,2-dibromochloroethane. Fresenius J Anal
Chem, 336: 322-327.
Wittsiepe J, Wallschläger D, Selenka F, & Jackwerth E (1993)
Gas-chromatographic determination of trace amounts of vinyl chloride
and dichloroethenes in water after purge-and-trap preconcentration and
conversion into their dibrominated derivatives. Fresenius J Anal Chem,
346: 1028-1034.
Wittsiepe J, Selenka F, & Jackwerth E (1996) Gas chromatographic
determination of trace amounts of vinyl chloride and dichloroethenes
in landfill-gas. Fresenius J Anal Chem, 354: 910-914.
Woldbaek T & Klaboe P (1978) The photochemical reaction of
vinylchloride with oxygen studied by ir spectroscopy. Spectrochim
Acta, A34: 481-487.
Wolf K, Holland R, & Rjaratnam A (1987) Vinyl chloride contamination:
The hidden threat. J Hazard Mater, 15: 163-184.
Wong O, Whorton MD, Foliart DE, & Ragland D (1991) An industry-wide
epidemiologic study of vinyl chloride workers, 1942-1982. Am J Ind
Med, 20: 317-334.
Wood JA & Porter ML (1987) Hazardous pollutants in class II landfills.
Int J Air Pollut Control Hazard Waste Manage, 37: 609-615.
Wrede F (1995) [Polyvinyl chloride (PVC).] Kunststoffe, 85: 1515-1521
(in German).
Wright DK, Mahler KO, & Davis J (1992) The application of
multidimensional GC techniques to the analysis of sub-PPB levels of
vinyl chloride monomer in polyvinylchloride. J Chromatogr Sci, 30:
291-295.
Wu W, Steenland K, Brown D, Wells V, Jones J, Schulte P, & Halperin W
(1989) Cohort and case-control analyses of workers exposed to vinyl
chloride: An update. J Occup Med, 31: 518-523.
Yamamoto K, Fukushima M, Kakutani N, & Kuroda K (1997) Volatile
organic compounds in urban rivers and their estuaries in Osaka, Japan.
Environ Pollut, 95: 135-143.
Zajdela F, Croisy A, Barbin A, Malaveille C, Tomatis L, & Bartsch H
(1980) Carcinogenicity of chloroethylene oxide, an ultimate reactive
metabolite of vinyl chloride, and bis(chloromethyl)ether after
subcutaneous administration and in initiation-promotion experiments in
mice. Cancer Res, 40: 352-356.
Zeff JD & Barich JT (1992) UV/Oxidation of organic contaminants in
ground, waste and leachate waters. Water Pollut Res J Can, 27:139-150.
Zhang J, Hatakeyama S, & Akimoto H (1983) Rate constants of the
reaction of ozone with trans-1,2-dichloroethene and vinyl chloride
in air. Int J Chem Kinet, 15: 655-668.
Zhang W, Johnson F, Grollman AP, & Shibutani S (1995) Miscoding by the
exocyclic and related DNA adducts 3,N4-etheno-2'deoxycytidine, 3,
N4-ethano-2'deoxycytidine, and 3-(2-hydroxyethyl)-2'deoxyuridine.
Chem Res Toxicol, 8: 157-163.
Zhao M-Y, Ying C-J, Shao N, Yang Y, Yang C-F, Shi L, & Liu W-Q (1994)
The study of health effects of vinyl chloride air pollution on
population. Biomed Environ Sci, 7: 136-143.
Zuccato E, Marcucci F, Fanelli R ,& Mussini E (1979) Head-space
gas-chromatographic analysis of vinyl chloride monomer in rat blood
and tissues. Xenobiotica, 9: 27-31.
ANNEX 1. REGULATIONS CONCERNING VINYL CHLORIDE
1. Regulations regarding VC levels in PVC materials and
food and drink
According to EC regulations PVC materials intended to come in
contact with foodstuffs must not contain VC at levels > 1 mg/kg; the
limit in the packed food is 10 µg/kg (CEC, 1978). The corresponding
German regulation additionally includes commodities, toys and tricks,
and articles which are for toiletry use (German Federal Health Office,
1998). The US FDA proposed to restrict the VC content of polymers used
for food contact to 5-50 µg/kg (US FDA, 1986).
A limit value of 2 µg/litre was issued by the US EPA for
drinking-water (US EPA, 1995b) and by the US FDA for bottled water
(Anon, 1993). The Maximum Contaminant Level of the Californian
Department of Health Services (DOHS) is 0.5 µg/litre (US EPA, 1995b).
In Germany, there are recommendations to tolerate 2 µg/litre in
drinking-water (German Federal Health Office, 1992).
2. Occupational exposure limit values for vinyl
chloride
Some examples of present exposure limit values are given in
Table 55.
Table 55. Occupational exposure limit values for vinyl chloride
Country/organization Exposure limit Value Value Reference
description (ppm) (mg/m3)
Europe; United Kingdom; personal 8-h TWA 7 18.2 CEC (1978); HSC (1995);
Germany; Finland BIA (1997)
5 12.8 Finnish Government
(1992)
Europe; United Kingdom; working area 3 8 CEC (1978); Finnish
Germany; Finland (annual) Government (1992);
HSC (1995); BIA (1997)
Czech Republic, Slovakia, MACKa 10 Bencko & Ungváry
Hungary and Poland (1994)
USA 15 min 5 12.8 OSHA (1998)
8 h 1 2.6 OSHA (1998)
TLV 8-h TWA 1 2.6 ACGIH (1999)
0b NIOSH (1997)
a Maximal allowable concentration (K indicates the carcinogenic properties of VC)
b No detectable exposure
TLV = threshold limit value; TWA = time weighted average;
OSHA = US Occupational Safety and Health Administration;
ACGIH = US American Conference of Governmental and Industrial
Hygienists Inc.; HSC = UK Health and Safety Commission;
BIA = German Professional Associations
Institute for Occupational Safety; Finnish Government = Government
Resolution 919/92, Finland
ANNEX 2. PHYSIOLOGICAL MODELLING AND RECENT RISK ASSESSMENTS
1. Physiological modelling of toxicokinetic data for vinyl chloride
Physiologically based toxicokinetic (PBTK) models intend to
predict the dose of active metabolites reaching target tissues in
different species, including humans, and thereby to improve the
toxicokinetic extrapolation in cancer risk assessments. PBTK models
have also been used as a tool to examine the behaviour of VC in
mammalian systems.
The model of Chen & Blancato (1989), also cited as a US EPA
(1987) model, proposed a PBTK model based on the styrene model of
Ramsey & Andersen (1984) with four compartments, representing the
liver, a fat compartment that includes all of the fatty tissues, a
richly-perfused tissue compartment that includes all organs except the
liver, and a slowly perfused compartment that includes muscular and
skin tissues. Liver was assumed to be the sole metabolizing organ, and
the metabolism of VC was assumed to occur via one pathway following
Michaelis-Menten-kinetics and saturable at high concentration levels.
Physiological parameters (e.g., body weight, lung weight, ventilation
rate, tissue volume and blood flows to various tissue groups) were
reference values from US EPA. This model was validated by Clement
International Corporation (1990) with gas uptake experiments in rats
and with data sets available in the literature (Gehring et al., 1978).
At concentrations < 65 mg/m3 the model fitted observed data, but
at higher concentrations the model predicted a greater amount of VC
metabolism than observed.
A second model (Gargas et al., 1990) was a generic model of
volatile chemical kinetics in a recirculated closed chamber, which was
used to identify global metabolic parameters in the rat for a number
of chemicals including VC. It incorporated a second, linear metabolic
pathway (presumed to be glutathione conjugation) in parallel with the
saturable (oxidative) pathway.
Continuing on from these above models, Clement International
Corporation (1990) re-fitted the one- and two-pathway descriptions to
gas uptake data and then compared their predictions to measurements of
total metabolism by Gehring et al. (1978) and Watanabe et al. (1976b).
Two refinements to the model were investigated: 1) reaction of VC with
glutathione (GSH), or 2) the products of both the saturable and the
linear pathways were assumed to react with GSH. Neither description
fitted both total metabolism and GSH depletion data and the authors
suggested a new PBTK model featuring two saturable oxidative pathways,
both producing reactive metabolites. Clewell et al. (1995a) developed
a more elaborate model of VC based on the suggestions from Clement
International Corporation (1990). The initial metabolism of VC was
hypothesized to occur via two saturable pathways (one representing
high affinity, low capacity oxidation by CYP2E1 and one representing
low affinity, high capacity oxidation by other P-450 isozymes),
producing in both cases chloroethylene oxide (CEO) as an intermediate
product. CAA (from CEO) was modelled as the major substrate in GSH
conjugation, with a lesser amount of CEO as the glutathione
transferase substrate. The model was similar to that proposed by Chen
& Blancato (1989) with regard to number and type of compartments,
physiological parameters and the assumption that metabolism of VC
takes place solely in the liver. Partition coefficients taken from
literature data (Gargas et al., 1989) were used. The metabolic
parameters for the two oxidative pathways were estimated from gas
uptake experiments conducted by Clement International Corporation
(1990). In this model it is assumed that the reactive VC metabolites
were further degraded to carbon dioxide, or reacted with GSH or with
cellular material. Parameters for subsequent metabolism were taken
from the PBTK model for vinylidene chloride (D'Souza & Andersen, 1988)
and were also used for species other than the rat after appropriate
allometric scaling, i.e., metabolic capacity scales approximately
according to body weight raised to 3/4 power (Andersen et al.,
1987). Support for the use of this principle for VC comes from data on
the metabolism of VC in non-human primates (Buchter et al., 1980). In
humans there is no evidence of a low-affinity high-capacity oxidation
by other P-450 isozymes so this parameter was set at zero for humans.
There is a wide variability of human CYP2E1 activity of about an order
of magnitude, whereas in inbred strains typically used in animal
studies the variability in CYP2E1 activity is small. The Clewell model
was then used to calculate dose metrics for ASL in animal bioassays
(Maltoni et al., 1981, 1984; Feron et al., 1981) as well as for human
inhalation exposure (see Table 56).
Reitz et al. (1996) developed a PBTK model for VC similar to that
proposed by Clewell et al. (1995a) with a single saturable pathway
based on a model for methylene chloride (Andersen et al., 1987) with
modifications on the size of liver compartment and allometric
constants. Partition coefficients were obtained from Gargas et al.
(1989). Metabolic parameters were calculated from gas uptake
experiments with male and female Sprague-Dawley rats. Metabolic
parameters for mice and humans were estimated from these experimental
data on rats and interspecies ratios obtained from literature
(Andersen et al., 1987). The model has been validated against data on
total metabolism in the rat (Watanabe et al., 1976b), gas uptake data
in the mouse, and inhalation data in the human on exhaled
concentrations of VC following VC exposure (Baretta et al., 1969).
2. Recent risk assessments of VC
2.1 Simonato et al. (1991)
Simonato et al. (1991) performed a regression analysis from
epidemiological data (see sections 8.2 and 8.3) to assess the relative
risk of liver cancer and ASL in occupational exposure to VC using the
variables cumulative exposure (ppm-years) and years since first
employment (Tables 48 and 49). On the basis of this regression
analysis and including cumulative exposure and years since first
exposure in the model, the absolute risk of ASL per 100 000 was
calculated (Table 50).
2.2 Clewell et al. (1995b)
A PBTK model was used to predict the total production of reactive
metabolites from VC in the animal bioassays and in human exposure
scenarios. These measures of internal exposure were then used in the
linearized multistage model (LMS) (Crump, 1984) to predict the risk
associated with lifetime exposure to VC in air or drinking-water.
The most appropriate toxicokinetic dose metric for a reactive
metabolite was considered to be the total amount of the metabolite
generated divided by the volume of the tissue in which it is produced
(Andersen et al., 1987), which, in the case of VC and ASL, would be
the total amount of metabolism divided by the volume of the liver.
Specifically, the average amount generated in a single day is used,
averaged over the lifetime (i.e., the lifetime average daily dose, or
LADD). The use of a dose rate such as LADD, rather than total lifetime
dose, has been reported to provide a better cross-species
extrapolation of chemical carcinogenic potency (D'Souza & Andersen,
1988). According to the authors, a body surface area adjustment is no
longer necessary when using a LADD.
The animal bioassays used are shown in Table 56. The 95% upper
confidence limits on the human risk estimates for lifetime exposure to
2.6 µg VC/m3 (1 ppb) were then calculated on the basis of each of the
sets of bioassay data, using the LMS model. The risk estimates on
inhalation studies in rats agreed with those in mice and were also
comparable with those from studies on rat diet. The estimates on oral
gavage were much higher (× 6), which the authors suggested might be
due to the use of corn oil.
Table 56. Human risk estimates (per million) for lifetime exposure to
2.6 µg VC/m3 (1 ppb) air based on the incidence
of ASL in animal bioassay a
Animal bioassay 95% UCL risk/million Reference
study per ppb
Males Females
Mouse inhalation 1.52 3.27 Maltoni et al. (1981, 1984)
Rat inhalation 5.17 2.24 Maltoni et al. (1981, 1984)
Rat diet 3.05 1.10 Feron et al. (1981)
Rat gavage 8.68 15.70 Maltoni et al. (1981, 1984)
a From: Clewell et al. (1995b); UCL = the 95% upper confidence limit of the
human risk estimates for lifetime exposure to 2.6 µg VC/m3 (1 ppb)
Risk calculations were also carried out on the basis of
epidemiological data using a linear relative risk dose-response model
(see Table 57). A PBTK model was run for the exposure scenario
appropriate to each of the selected subcohorts from each of the
studies.
Table 57. Human risk estimates (per million) for lifetime inhalation
of 2.6 µg VC/m3 (1 ppb) air based on the incidence of
ASL in human epidemiological studies a
95% UCL risk/million per ppb b Epidemiological study
0.71-4.22 Fox & Collier (1977)
0.97-3.60 Jones et al. (1988)
0.40-0.79 Simonato et al. (1991)
a From: Clewell et al. (1995b)
b The range of risk estimates reflects uncertainty in
the appropriate value for P0, the background
probability of death from liver cancer. The lower
risk estimate was calculated using the value
of P0 used in the study of Fox & Collier (1977),
while the higher risk estimate was calculated using
an estimate of the lifetime liver cancer mortality
in the USA population (Chen & Blancato, 1989).
Therefore there is not a true range but a
reflection of two assumptions about the background
rate of liver cancer in humans.
To obtain an estimate of the carcinogenic potency, the resulting
internal dose metrics were multiplied by the appropriate durations to
obtain the cumulative internal doses, which were then input into the
relative risk model together with observed and expected liver cancer
deaths for each subcohort. A 95% upper confidence limit on the
lifetime risk per ppb of VC was estimated to compare results with the
animal-based results obtained with the LMS model. The estimates from
the three epidemiological studies compared well with those from the
toxicokinetic animal-based studies. From the Simonato data, a 0.4 to
0.8 × 10-6 risk per ppm VC was estimated, which Clewell et al.
(1995b) noted is about 30 below the published inhalation unit risk of
8.4 × 10-6 (µg/m3)-1. A carcinogenic risk estimate of ASL from a
lifetime exposure to 1 µg VC/litre in human drinking-water was
1.14 × 10-6 (µg/litre)-1 and was derived from Feron's study on male
rats after dietary administration of VC. This value was compared with
the published unit risk of 5.4 × 10-5 (µg/litre)-1 (US EPA, 1995a).
2.3 Reitz et al. (1996)
Reitz et al. (1996) used a PBTK model to calculate LADDs and
these were fit to a LMS model. The data generated in rats were used to
predict ASL incidence in mice and were found to fit with the data from
Maltoni's experiment BT 4 and those of Lee et al. (1978).
The PBTK model was used to calculate LADDs for humans: mg VC
metabolites formed/day per litre of liver tissue for conditions
thought likely to have been present in the workplace in past years
(i.e., VC concentrations 50-2000 ppm; 8 h/day; 5 days/week and 10 or
20 years/70 years). From these estimates of dose the LMS model was
used to estimate the likelihood that tumours would be produced based
on the rat model. These results were then compared with the actual
data from Simonato et al. (1991). Predictions using this PBTK model
were substantially higher (10-35 fold) than actually observed in
humans (see Table 58). But the PBTK-based value for the 95% UCL for
excess lifetime risk associated with continuous inhalation of 1 µg/m3
was 5.7 × 10-7, compared with the much higher risk, 8.4 × 10-5, using
conventional risk estimates (US EPA, 1995a). Reitz et al. (1996)
concluded that even using the PBTK model, the risk of ASL is
overestimated and suggested that the livers of humans are less
sensitive to the carcinogenic effect of reactive VC metabolites than
the livers of the commonly used inbred laboratory rodents.
2.4 Storm & Rozman (1997)
Storm & Rozman (1997) use a no-threshold (LMS model and benchmark
dose approach with linear extrapolation) and threshold (NOEL/LOEL and
benchmark dose uncertainty factor approaches) models and compare them
with the present occupational exposure limits in the 0.5 to 5 ppm
range (Table 59). Although VC is a genotoxic carcinogen, the authors
argued that for VC a threshold exists because DNA adducts formed by
reaction with vinyl chloride metabolites are repaired by DNA
glycosylase in both rat and human liver in vitro (Dosanjh et al.,
1994; Saparbaev et al., 1995), and only when repair capability is
exceeded does cancer develop (Swenberg et al., 1995).
Storm & Rozman (1997) used Maltoni's inhalation study with
Sprague-Dawley rats (Maltoni et al., 1981) and, after adjusting all
exposures to reflect equivalent continuous human exposures and then to
LADD in mg/kg-day, used these dose measures in the low-dose
extrapolation models. (Occupational exposures were derived by
converting appropriately derived LADDs to equivalent occupational
exposures, assuming an inhalation rate of 10 m3/working day and that
exposures occurred 8 h/day, 50 weeks/year for 45 years).
No-threshold models evaluated were the LMS model for
extrapolation of genetic carcinogen dose-response curves to low doses,
and the benchmark dose approach (BMD) with linear extrapolation (US
EPA, 1996). Maximum likelihood estimates (MLE) and 95% UCL (qi*) on
Table 58. Predicted versus observed ASL incidence for humans
assuming occupational exposure to VC a
Ppm Years Ppm-years Linearized multistage Human observed
exposed prediction based on (incidence/100 000)
animal studies
(incidence/100 000)
100 10 1000 374 b 6.2 e
50 20 1000 376 b 6.2 e
200 20 4000 1465 b 42.2 f
1 45 45 21 c 0 g
6-11 d
a From: Storm & Rozman (1997)
b Derived by Reitz et al. (1996) assuming an occupational exposure of
5 days/week, 50 weeks/year
c Calculated from unit risk derived by Reitz et al. (1996), assuming
occupational rather than continuous exposures
d Calculated from human risk/million per ppb based on Simonato et al. (1991)
derived by Clewell et al. (1995b), assuming occupational rather than
continuous exposures
e Derived by Simonato et al. (1991) for exposure < 2000 ppm-years
and > 25 years since first employment
f Derived by Simonato et al. (1991) for exposure 2000-5999 ppm-years
and > 25 years since first employment
g No ASL reported among workers first exposed in or after 1974 when the
OSHA permissible exposure limit (PEL) of 1 ppm was promulgated.
the linear coefficient (q i) were derived using a LMS model. The
resultant linear coefficients from these and the results from Reitz et
al. (1996) described above using the PBTK model were then used to
derive safe levels of occupational exposure. For the BMD approach, a
computer programme was used to derive MLEs as well as 95% lower bound
(LB) estimates for specified risk levels along with X2
goodness-of-fit statistics, in this case P > 0.01. The MLE and LB
of both the exposure associated with 10% incidence (ED10 and LED10)
and the exposure associated with 1% incidence (ED01 and LED01),
respectively, were used for linear extrapolation to zero (US EPA,
1996). "Safe" levels of occupational exposure were derived from the
slopes of these lines.
Threshold models evaluated included a) the LOEL/NOEL
uncertainty factor approach traditionally used for non-carcinogenic
effects and b) the BMD and uncertainty factor approach being developed
as a replacement for the former. A NOEL of 13 mg/m3 (5 ppm) was
derived from the Maltoni et al. (1981) bioassay in rats. An
uncertainty factor of 2 was applied to account for the half-lifetime
exposure duration of the VC study, a standard uncertainty factor of 10
to account for potential intraspecies variability in susceptibility,
an interspecies uncertainty factor of 3 or 0.3 to cover the assumption
that humans were more or less sensitive than rats in this study giving
total uncertainty factors of 60 or 6, respectively. The same factors
were applied to MLE and LB estimates of dose using the BMD
curve-fitting approach.
The X2 goodness-of-fit P value for the BMD curve was only
acceptable when the apparent outliers at 100 and 250 ppm and high-dose
responses at 500, 2500 and 6000 ppm were eliminated, and these values
were therefore also eliminated in the LMS model for purpose of
comparison.
Both no-threshold models based on administered dose provide
estimates of occupational safe levels of about 0.004 ppm, at least two
orders of magnitude lower than the present accepted "safety" limit
value of 0.5-5 ppm when using an acceptable level of risk of 1 in
100 000 (1 × 10-5). With the PBTK model, estimates were about
0.04 ppm. For threshold models, using the BMD curve-fitting approach,
with an ED01 and LED01 point of departure, occupational levels of 0.1
to 0.7 ppm, respectively, were estimated for humans less or more
sensitive than rats. Estimations based on the rat NOEL approach gave
values of 0.4 to 4 ppm, respectively, for humans more and less
sensitive than rats.
Table 59. Comparison of estimated safe levels for occupational
exposure to VC (from Storm & Rozman, 1997)
No-threshold Slope Risk 1/100 000
models (mg/kg-day)-1 assuming
occupational
exposure a (ppb)
Linearized Administered dose b MLE 1.4 × 10-2 4.3
multistage UCL 1.8 × 10-2 3.4
PBTK dose c MLE 1.6 × 10-3 39.4
UCL 2.0 × 10-3 31.2
US EPA d UCL 2.9 × 10-1 0.2
Benchmark ED01-Point of MLE 1.4 × 10-2 4.3
dose departure LB 1.8 × 10-2 3.4
Threshold BMD b Uncertainty Risk 1/100 000
models (mg/kg- factor assuming
day) occupational
exposure a (ppm)
Benchmark Humans less MLE 0.7 6 e 0.7
dose-ED01 sensitive LB 0.5 6 e 0.7
Human = rat MLE 0.7 20 f 0.2
sensitivity LB 0.5 20 f 0.2
Humans more MLE 0.7 60 g 0.1
sensitive LB 0.5 60 g 0.1
Table 59. (cont'd)
LADD Uncertainty Risk 1/100 000
(mg/kg- factor assuming
day) occupational
exposure a (ppm)
Rat NOEL Humans less 3.6 6 e 3.8
(5 ppm) sensitive
Human = rat 3.6 20 f 1.1
sensitivity
Humans more 3.6 60 g 0.4
sensitive
a Occupational exposures assume 10 m3/day inhalation rate, 5 days/week, 50 weeks/year,
for 45 years averaged over a lifetime of 70 years (Note: Levels are ppb for
no-threshold models, ppm for threshold models)
b Derived using a computer programme
c Slope from Reitz et al. (1996)
d Slope from US EPA Health Effects Assessment Summary Tables (US EPA, 1995)
e Safety factors of 10 for intraspecies, 0.3 for interspecies extrapolation and
2 for half-lifetime exposure
f Safety factors of 10 for intraspecies extrapolation and 2 for
half-lifetime exposure
g Safety factors of 10 for intraspecies, 3 for interspecies extrapolation, and
2 for half-lifetime exposure
MLE = maximum likelihood estimate;
UCL = 95% upper confidence limit;
LB = 95% lower bound on dose
ANNEX 3. EXECUTIVE SUMMARY OF VINYL CHLORIDE PANEL REPORT
Executive summary of
Mundt KA, Dell LD, Austin RA, Luippold RS, Noess R & Bigelow C.
Epidemiological study of men employed in the vinyl chloride industry
between 1942 and 1972: I. Re-analysis of mortality through December
31, 1982; and II. Update of mortality through December 31, 1995.
Final Report. Arlington, Virginia: The Vinyl Chloride Panel, Chemical
Manufacturers Association, January 8, 1999. 178 pagesa
A cohort of 10 109 men who were employed for at least one year in
vinyl chloride exposed jobs between 1942 and 1972 at any one of
37 facilities in the USA or Canada was followed for mortality. Through
December 31, 1995, a total of 3191 deaths had occurred among cohort
members. A re-analysis of mortality through December 1982, at which
time 1569 deaths had occurred, was conducted to establish baseline
results against which the updated mortality results could be compared.
The re-analysis of mortality through December 1982 was necessary for
four reasons: (1) the cohort was reduced from 10 173 to 10 109 after
the removal of study subjects who were duplicates or were not eligible
for cohort inclusion; (2) a substantial proportion of cohort members
previously lost to follow-up were identified and restored to the
cohort; (3) an appropriate referent population, based on regional
mortality rates, was constructed and used for the mortality update
through 1995; and (4) the number of categories of death that could be
evaluated in the analysis increased from 61 to 92 causes of death.
Through 1995, mortality from all causes of death was 17% lower
than state mortality weighted according to person-years accumulated
among employees in the state where the plant was located. This result
was similar to the 16% deficit seen for mortality from all causes of
death through 1982. Mortality from all cancers, however, showed a
deficit of 4% through 1995, in contrast with the 2% excess seen as of
1982. Specific cancers showing meaningful excesses both through 1982
and through 1995 included cancers of the liver and biliary tract,
brain, and connective and soft tissues. However, several causes of
death previously believed to be related to vinyl chloride exposure
were not seen in excess, including lung cancer, cancers of lymphatic
and haematopoietic tissue, emphysema and pneumoconioses and other lung
diseases, including chronic obstructive pulmonary disease (COPD).
a This summary is reprinted by permission of the Vinyl Chloride
Health Committee. This study, which is an update of the Wong et
al. (1991) study, was not available at the Task Group meeting and
thus did not affect the evaluation of the health risks of vinyl
chloride. It is printed here as additional information to the
reader.
Through 1995, mortality from liver and biliary tract cancer,
including angiosarcoma of the liver, was significantly increased
(SMR=359; 95% CI: 284-446) although the excess was smaller than that
observed through 1982 (SMR=428; 95% CI: 304-585). The SMR for liver
and biliary tract cancer increased with duration of exposure from 83
(95% CI: 33-171) to 215 (95% CI: 103-396) to 679 (95% CI: 483-929) to
688 (95% CI: 440-1023) for those exposed from 1 to 4 years, 5 to 9
years, 10 to 19 years and 20 years or more, respectively. The SMR for
liver and biliary tract cancer also increased with time since first
exposure from 0 (no deaths observed) to 287 (95% CI: 131-544) to 323
(95% CI: 200-493) to 434 (95% CI: 322-572) for those 1 to 9 years, 10
to 19 years, 20 to 29 years, and 30 or more years since first
exposure, respectively. Results of a Cox proportional hazards model
that fitted age at first exposure, duration of exposure, and year of
first exposure indicated that duration of exposure was most strongly
associated with increased risk of mortality from liver and biliary
tract cancer. The adjusted hazards ratio for 1 to 4 years, 5 to 9
years, 10 to 19 years and 20 years or more duration of exposure were
1.0 (referent group), 2.8 (95% CI: 1.0-7.3), 9.0 (95% CI: 4.0-20.7)
and 6.0 (95% CI: 2.5-14.4), respectively. This confirms the recognized
association between vinyl chloride exposure and liver and biliary
tract cancer, mainly due to a large excess of angiosarcoma of the
liver.
Mortality from brain cancer showed an excess (SMR=142; 95% CI:
100-197) that was slightly smaller than that seen in the re-analysis
through 1982 (SMR=169; 95% CI: 105-255). Since 1982, an additional 14
brain cancer deaths were observed (12.2 expected). The SMRs for brain
cancer were elevated among those exposed for 5 to 9 years (SMR=193;
95% CI: 96-346) and those exposed for 20 years or more (SMR=290; 95%
CI: 132-551). Results of a Cox proportional hazards model adjusted for
duration of employment showed that age at employment of 35 years or
older and longer duration of employment were both associated with
increased risk of mortality from brain cancer, and these associations
were not confounded. The association of brain cancer deaths with vinyl
chloride exposure is uncertain, as the older age at first employment
in the vinyl chloride industry suggests that these cohort members
might have sustained exposure to some other carcinogen prior to
employment in vinyl chloride.
Mortality from cancers of the connective and soft tissues also
showed an excess (SMR=270; 95% CI: 139-472), based on 12 deaths.
Through 1982, mortality from connective and soft tissue cancers was
also elevated (SMR=367; 95% CI: 147-755), based on seven observed
deaths. This result was not reported in earlier investigations of this
cohort because mortality for this cause of death category had not been
previously evaluated. Although the total number of deaths due to this
rare cause was small, the observed excess is potentially important for
two reasons. First, very little is known about the risk factors for
connective and soft tissue cancers, a category including numerous
histological types of cancer. Second, some of these cancers may have
been mis-diagnosed or mis-reported angiosarcomas of the liver, one
form of soft tissue sarcoma known to be related to vinyl chloride
exposure.
No excesses in mortality were seen for lung cancer or cancers of
the lymphatic and haematopoietic system. In contrast to the previous
report which showed an excess of mortality from emphysema and chronic
obstructive disease, mortality from emphysema and mortality from
pneumoconioses and other respiratory diseases (PORD), the category
which included chronic obstructive pulmonary disease, each showed
deficits (SMR=61; 95% CI: 40-89 for emphysema and SMR=73; 95% CI:
60-88 for PORD). The previously published findings with respect to
mortality due to these causes of death may have resulted from
methodological errors.
In conclusion, this update of the industry-wide cohort confirmed
a strong association between duration of employment in the vinyl
chloride industry prior to 1974 and cancers of the liver and biliary
tract, mostly resulting from a large excess of deaths due to
angiosarcoma of the liver. This association has been observed in all
similar studies and represents one of the most consistent effects
observed in the occupational health literature. Cancers of the
connective and soft tissues, though occurring in relatively small
numbers, also appear to be related to employment in the industry. The
elucidation of the actual mechanism for this observed association will
require further investigation. The brain cancer mortality excess
observed in the analyses through 1982 is less clearly associated with
vinyl chloride, and may reflect risk factors other than vinyl chloride
present in the early years of the industry or reflecting exposures
sustained prior to employment in this industry. Since 1982, no such
excess has occurred.
This study represents one of the largest and longest-followed
cohorts potentially exposed to substantial levels of vinyl chloride.
The brain cancer and emphysema excesses previously reported are not
sustained, however, the brain cancer excess remains unexplained.
Except for deaths due [to] cancers of the liver and biliary tract, and
to a lesser extent cancers of connective and soft tissues, the
mortality patterns for this cohort remain at or below that expected in
the general population.
RESUME
La présente monographie traite du chlorure de vinyle monomère
lui-même, à l'exclusion de son polymère, le chlorure de polyvinyle
(PVC). Le problème de l'exposition à des mélanges contenant du
chlorure de vinyle n'est pas abordé.
1. Identité, propriétés physiques et chimiques et méthodes d'analyse
Dans les conditions ambiantes, le chlorure de vinyle se présente
sous la forme d'un gaz incolore inflammable à l'odeur douceâtre. Sa
tension de vapeur est élevée, de même que la valeur de sa constante
pour la loi d'Henry; sa solubilité dans l'eau est relativement faible.
Il est plus lourd que l'air et il est soluble dans presque tous les
solvants organiques. On le transporte à l'état liquide sous pression.
A la température ambiante et en l'absence d'air, le chlorure de
vinyle pur et sec est très stable et non corrosif. Toutefois,
au-dessus de 450°C ou en présence d'hydroxyde de sodium ou de
potassium, il peut subir une décomposition partielle. Sa combustion
dans l'air donne naissance à du dioxyde de carbone et à du chlorure
d'hydrogène. En présence d'air et d'oxygène, des peroxydes très
explosifs peuvent se former, ce qui exige une surveillance permanente
et la limitation de la teneur en oxygène, en particulier dans les
installations de récupération du chlorure de vinyle. En présence
d'eau, il se forme de l'acide chlorhydrique.
Du point de vue industriel, ce sont les réactions de
polymérisation conduisant au chlorure de polyvinyle qui sont
techniquement les plus importantes, mais les réactions d'addition des
halogènes sur la double liaison, qui conduisent au
1,1,2-trichloréthane ou au 1,1-dichloréthane, ont aussi leur
importance.
On peut surveiller la concentration de chlorure de vinyle dans
l'atmosphère en piégeant le composé sur un support adsorbant et en
procédant à une analyse par chromatographie en phase gazeuse après
désorption liquide ou thermique. Pour le dosage dans l'air ambiant, il
peut être nécessaire d'utiliser plusieurs supports adsorbants ou
pièges froids disposés en série pour mieux capter le composé. Les pics
de concentration sur les lieux de travail peuvent se mesurer au moyen
d'instruments à lecture directe utilisant notamment des détecteurs à
ionisation de flamme ou à photo-ionisation. Pour le contrôle en
continu, ont utilise des analyseurs basés sur la spectrophotométrie
infrarouge ou sur la chromatographie en phase gazeuse avec détection
par ionisation de flamme et qui sont munis d'un système
d'enregistrement et de traitement des données. Pour doser le chlorure
de vinyle dans les liquides et les solides, on a recours à l'injection
directe, à l'extraction et de plus en plus, à la technique de l'espace
de tête ou à celle de purgeage-piégeage. Pour l'analyse de ces
échantillons, on utilise aussi la chromatographie en phase gazeuse
avec détection par ionisation de flamme ou spectrométrie de masse.
2. Sources d'exposition humaine dans l'environnement
Le chlorure de vinyle n'existe pas à l'état naturel, même si on
en a décelé la présence dans les gaz qui s'échappent des décharges et
dans les eaux souterraines, par suite de la décomposition de rejets de
solvants constitués d'hydrocarbures chlorés, ou encore dans
l'environnement des lieux de travail où on fait usage de ce genre de
solvants. Il y a également du chlorure de vinyle dans la fumée de
cigarette.
La production industrielle du chlorure de vinyle utilise deux
réactions principales: a) l'hydrochloration de l'acétylène et b) le
craquage thermique (à environ 500°C) du 1,2-dichloréthane produit par
chloration ou oxychloration directes de l'éthylène par le chlore ou
par un mélange HCl + air/O2. Ce procédé est actuellement le plus
couramment utilisé.
La production mondiale de PVC et par conséquent celle de chlorure
de vinyle monomère a été d'environ 27 millions de tonnes en 1998.
Vingt pour cent des matières plastiques utilisées dans le monde sont à
base de PVC et ce polymère se retrouve dans presque tous les secteurs
de l'industrie. Environ 95% de la production mondiale de chlorure de
vinyle sert à la fabrication de PVC. Le reste est utilisé pour la
production de solvants chlorés, essentiellement du
1,1,1-trichloréthane (10 000 tonnes par an).
Trois procédés principaux sont utilisés pour la production
industrielle du PVC: la polymérisation en suspension (80% de la
production mondiale), la polymérisation en émulsion (12%) et la
polymérisation en masse (8%). La plupart des études de cas faisant
état des effets nocifs du chlorure de vinyle concernent des usines
produisant du PVC par polymérisation en suspension (on dit aussi
polymérisation en perles).
On a signalé des émissions de chlorure de vinyle lors d'accidents
survenus dans des usines produisant du PVC ou pendant le transport.
Dans de nombreux pays, on récupère le chlorure de vinyle résiduel non
polymérisé lors de la production de PVC ou qui se trouve dans les
rejets gazeux ou les eaux usées. Faute de certaines précautions, il
est possible de retrouver du chlorure de vinyle dans les résines et
autres produits à base de PVC.
Le taux de chlorure de vinyle résiduel présent dans le PVC est
réglementé dans de nombreux pays depuis la fin des années 1970. Depuis
lors, les émissions de chlorure de vinyle dues à la décomposition
thermique du PVC sont soit indétectables, soit très faibles.
Des dioxines peuvent se former lors de la production de chlorure
de vinyle. Les quantités qui sont répandues dans l'environnement sont
controversées.
3. Transport, distribution et transformation dans l'environnement
En raison de sa forte tension de vapeur, le chlorure de vinyle
libéré dans l'environnement devrait exister presque uniquement en
phase gazeuse. Il existe des indices de dépôts sous forme liquide.
Le chlorure de vinyle est relativement peu soluble dans l'eau et
sa capacité d'adsorption aux particules et aux solides en suspension
est faible. C'est par volatilisation que le composé s'élimine le plus
rapidement des eaux de surface. La demi-vie de volatilisation à partir
des eaux de surface varie d'environ 1h à 40 h.
A partir du sol, la demi-vie de volatilisation est, selon les
calculs, égale à 0,2-0,5 jours. On estime que les pertes de chlorure
de vinyle (au bout d'un an sous un mètre de terre) vont de 0,1 à 45%,
selon le type de sol. Le coefficient de sorption pédologique tiré des
données physico-chimiques indique que le potentiel de sorption du
composé est faible et que par conséquent il est très mobile dans le
sol. Il existe une autre voie importante de distribution dans
l'environnement, à savoir le lessivage qui peut entraîner le chlorure
de vinyle jusqu'aux nappes d'eau souterraines où il est susceptible de
persister pendant des années.
Les études de laboratoire portant sur des organismes aquatiques
révèlent une certaine tendance à la bioaccumulation, mais pas de
bioamplification le long de la chaîne alimentaire.
A quelques exceptions près, le chlorure de vinyle ne se laisse
pas facilement dégrader par les groupements de microorganismes
inadaptés dans les conditions ambiantes. On estime que la demi-vie de
biodégradation par des micro-organismes non acclimatés est de l'ordre
de plusieurs mois ou années. Toutefois, certaines cultures
microbiennes enrichies ou pures (par exemple Mycobacterium sp.) sont
capables de décomposer le chlorure de vinyle lorsque les conditions
culturales sont optimales. Les principaux produits de dégradation sont
l'acide glycolique ou le dioxyde de carbone après conversion aérobie
et l'éthane, l'éthylène, le méthane ou le chlorométhane après
transformation anaérobie. Souvent, les microorganismes aérobies
décomposent plus rapidement le chlorure de vinyle que les
microorganismes anaérobies.
Le principal processus atmosphérique est la réaction avec les
radicaux OH produits par voie photochimique; la demi-vie
troposphérique résultant de ce processus est de 1 à 4 jours. Les
réactions de photolyse réalisées dans des conditions expérimentales
donnent naissance à plusieurs composés importants, comme le
chloracétaldéhyde, le formaldéhyde et le chlorure de formyle.
On pense que les réactions de photolyse, de même que l'hydrolyse
chimique, sont de peu d'importance en milieu aqueux. Toutefois, la
présence de photosensibilisateurs peut faciliter la transformation du
chlorure de vinyle.
On a des raisons de penser que le chlorure de vinyle réagit avec
le chlore ou les chlorures utilisés pour la désinfection de l'eau pour
donner du chloracétaldéhyde et autres composés indésirables. Il existe
d'autres interactions possibles, notamment avec les sels, dont un
grand nombre sont susceptibles de former des complexes avec le
chlorure de vinyle, ce qui peut en accroître la solubilité.
Parmi les méthodes utilisées (avec des succès divers) pour
éliminer le chlorure de vinyle présent dans l'eau, on peut citer le
lavage, l'extraction, l'adsorption et l'oxydation. Certaines méthodes
de biopurification (applicables aux eaux souterraines et aux sols)
associent l'évaporation ou d'autres techniques à un traitement
microbien. Le chlorure de vinyle présent dans les rejets gazeux peut
être recyclé, incinéré ou décomposé par voie microbienne. La majeure
partie du chlorure de vinyle produit industriellement se retrouve à
l'état lié dans des articles en PVC. L'incinération de ces produits
comporte un risque de formation de PCDD, de PCDF et d'autres dérivés
chlorés indésirables.
4. Concentrations dans l'environnement et exposition humaine
La population dans son ensemble est très peu exposée au chlorure
de vinyle.
La concentration de chlorure de vinyle dans l'air ambiant est
faible, généralement inférieure à 3 µg/m3. La population peut être
davantage exposée lorsque de grandes quantités de chlorure de vinyle
sont libérées accidentellement dans l'environnement, par exemple en
cas de déversement pendant le transport. Il s'agit cependant d'une
exposition qui a toutes chances d'être passagère. A proximité de sites
industriels où l'on produit du chlorure de vinyle et du PVC ou près de
décharges, on a enregistré des concentrations beaucoup plus élevées
(pouvant aller respectivement jusqu'à 8000 µg/m3 et 100 µg/m3).
La principale voie d'exposition professionnelle est la voie
respiratoire et alle intervient principalement dans les usines qui
produisent du chlorure de vinyle et du PVC. Dans les années 1940 et
1950, l'exposition professionnelle au chlorure de vinyle était de
plusieurs milliers de mg/m3; elle atteignait encore plusieurs
centaines de mg/m3 dans les années 1960 et au début des années
1970.Une fois reconnu le risque cancérogène inhérent au chlorure de
vinyle, la norme d'exposition à ce composé a été fixée au cours des
années 1970 à environ 13-26 mg/m3 (5-10 ppm) dans la plupart des
pays. Le respect de ces directives a fait fortement chuter la
concentration du chlorure de vinyle sur les lieux de travail, mais au
cours des années 1990 on a tout de même fait état de concentrations
plus élevées et ce pourrait encore être le cas actuellement dans
quelques pays.
Il est arrivé que l'on trouve du chlorure de vinyle dans les eaux
de surface, dans les sédiments et dans les boues d'égout, avec des
concentrations maximales respectivement égales à 570 µg/litre,
580 µg/kg et 62 000 µg/litre. Des échantillons de sol prélevés près
d'une teinturerie abandonnée se sont révélés présenter une forte
teneur en chlorure de vinyle (jusqu'à 900 mg/kg). On a mis en évidence
des concentrations de l'ordre de 60 000 µg/litre ou davantage dans des
eaux souterraines ou dans des eaux de lessivage provenant de zones
contaminées par des hydrocarbures chlorés. Des teneurs élevées
(jusqu'à 200 mg/litre) ont également été mises en évidence, 10 ans
après le repérage des premières fuites, dans l'eau de puits situés à
proximité d'une usine de PVC.
Les quelques données disponibles montrent que les tissus des
petits vertébrés aquatiques et des poissons peuvent contenir du
chlorure de vinyle.
Dans la majorité des échantillons d'eau de boisson analysés, le
chlorure de vinyle n'était pas présent à des concentrations
décelables. La concentration maximale dont il ait été fait état était
de 10 µg/litre dans de l'eau prête à être consommée. On manque de
données récentes sur la concentration du composé dans l'eau de
boisson, mais elle devrait être inférieure à 10 µg/litre. Si la source
utilisée pour la boisson est contaminée, l'exposition peut être plus
importante. Des études récentes ont permis de déceler la présence de
chlorure de vinyle dans de l'eau minérale en bouteilles de PVC à une
concentration inférieure à 1 µg/litre. On peut penser que la présence
de chlorure de vinyle est plus fréquente dans ces eaux en bouteilles
que dans l'eau du robinet.
L'utilisation de PVC pour l'emballage peut entraîner la présence
de chlorure de vinyle dans les denrées alimentaires, les produits
pharmaceutiques et les cosmétiques. On en a décelé jusqu'à 20 mg/kg
dans des liqueurs, jusqu'à 18 mg/kg dans des huiles végétales et
jusqu'à 9,8 mg/kg dans du vinaigre. Depuis le début des années 1970,
le nombre d'échantillons contrôlés positifs pour le chlorure de vinyle
est moindre et la concentration est plus faible qu'auparavant, grâce
aux mesures législatives prises par un certain nombre de pays.
Plusieurs organismes ont calculé l'exposition au chlorure de
vinyle due à la contamination des denrées alimentaires par leur
emballage de PVC. En se basant sur valeur estimative moyenne de la
dose ingérée au Royaume-Uni et aux Etats-Unis, on a calculé qu'à la
fin des années 1970 et au début des années 1980, l'exposition devait
être inférieure à 0,0004 µg/kg par jour. Une étude ancienne a mis
évidence du chlorure de vinyle dans la fumée de tabac à des
concentrations de quelques ng par cigarette.
5. Cinétique et métabolisme chez les animaux de laboratoire et
l'Homme
Après inhalation ou ingestion, le chlorure de vinyle est
facilement et rapidement absorbé. Des études effectuées sur des
animaux et des sujets humains, dans des conditions d'équilibre
dynamique, ont montré qu'environ 40% de la dose inhalée étaient
absorbés après exposition par la voie respiratoire. L'expérimentation
animale montre que l'absorption peut dépasser 95% après ingestion.
L'absorption percutanée de chlorure de vinyle à l'état gazeux est peu
importante.
Les données fournies par les études sur l'inhalation et
l'ingestion de chlorure de vinyle par des rats montrent que le composé
se répartit rapidement et largement dans l'organisme. Par suite d'une
métabolisation et d'une excrétion rapides, son accumulation est
limitée. Chez le rat, le passage transplacentaire se produit
rapidement. On n'a pas connaissance d'études consacrées à la
répartition du composé dans l'organisme après exposition par voie
percutanée.
Après inhalation ou ingestion, la principale voie métabolique
consiste en une oxydation par le cytochrome P-450 (CYP2E1) qui conduit
à la formation d'oxyde de chloréthylène, un époxyde à courte vie
extrêmement réactif, qui se transpose très vite en chloracétaldéhyde.
La principale réaction de détoxication de ces deux métabolites
réactifs ainsi que de l'acide chloracétique, produit de
déshydrogénation du chloracétaldéhyde, consiste en une conjugaison
avec le glutathion catalysée par la glutathion- S-transférase. Les
conjugués sont ensuite transformés en dérivés de la cystéine
[ S-(2-hydroxyéthyl)cystéine, N-acétyl- S-
(2-hydroxyéthyl)cystéine, S-carboxyméthylcystéine et acide
thiodiglycolique] et ils sont excrétés dans l'urine. Un autre
métabolite, le dioxyde de carbone, est excrété dans l'air exhalé.
Les isozymes de la CYP2E1 et de la glutathion- S-transférase
présentent d'importantes variations d'activité interspécifiques et
interindividuelles.
Après exposition par inhalation ou ingestion de faibles doses de
chlorure de vinyle, le composé est éliminé par métabolisation et ses
métabolites non volatils sont principalement excrétés par la voie
urinaire. L'étude comparée de l'absorption après inhalation montre que
relativement au poids corporel, le chlorure de vinyle est métabolisé
moins rapidement par l'organisme humain que par l'organisme animal.
Toutefois, si on tient compte de la superficie du corps, on constate
que la clairance métabolique est comparable chez l'Homme et chez les
autres mammifères. Lorsque l'exposition - toujours par inhalation ou
ingestion - est plus importante, la principale voie d'excrétion chez
l'animal consiste dans l'élimination du composé initial dans l'air
expiré, ce qui témoigne de la saturation des voies métaboliques.
Quelle que soit la dose, l'excrétion par la voie fécale est
secondaire. On n'a pas trouvé d'étude qui soit spécialement consacrée
à l'excrétion par la voie biliaire.
On estime que l'oxyde de chloréthylène est le métabolite le plus
important in vivo, pour ce qui est des effets cancérogènes et
mutagènes du chlorure de vinyle. Cet époxyde réagit sur d'ADN pour
former un certain nombre d'adduits, principalement de la
7-(2'-oxoéthyl)guanine (7-OEG) et, en moindre quantité, des
éthénoadduits exocycliques comme la 1, N 6-éthénoadénine (Epsilon
A), la 3, N 4-éthénocytosine (Epsilon C) et la N2,3-éthénoguanine
(Epsilon G). Contrairement à l'adduit principal, la 7-OEG, ces
éthénoadduits avec l'ADN ont des propriétés promutagènes. On a pu
doser la 7-OEG, la Epsilon A, la Epsilon C et la Epsilon G dans les
divers tissus de rongeurs exposés à du chlorure de vinyle. On a mis au
point des modèles toxicocinétiques à base physiologique pour rendre
compte de la relation entre la dose aux tissus cibles et les points
d'aboutissement toxicologique du chlorure de vinyle.
6. Effets sur les mammifères de laboratoire et les systèmes d'épreuve
in vitro
Administré par inhalation à des animaux de plusieurs espèces, le
chlorure de vinyle s'est révélé d'une faible toxicité aiguë. La CL50
à 2 h pour le rat, la souris, le cobaye et le lapin a été trouvée
respectivement égale à 390 000, 293 000, 595 000 et 295 000 mg/m3. On
ne possède aucune donnée sur la toxicité aiguë du composé par voie
orale ou percutanée. L'inhalation brutale de chlorure de vinyle a un
effet stuporeux. Chez des rats, des souris et des hamsters, la mort a
été précédée d'un accroissement de l'activité motrice, d'ataxie et de
convulsions suivies d'une défaillance respiratoire. Chez des chiens
plongés dans un état stuporeux après exposition brutale à une
concentration de 260 000 mg/m3, on a noté de graves arythmies
cardiaques. Des rats, exposés par la voie respiratoire à du chlorure
de vinyle, ont présenté un certain nombre d'anomalies
anatomopathologiques telles que congestion des organes internes,
notamment des poumons, du foie et des reins, et oedème pulmonaire.
On ne dispose pas d'études ni de données appropriées qui
permettent d'évaluer les effets de l'exposition par voie percutanée ou
encore le pouvoir irritant ou sensibilisateur au niveau cutané.
Une exposition de courte durée (13 semaines) au chlorure de
vinyle par la voie orale a permis d'obtenir une valeur de 30 mg/kg
pour la NOEL, c'est-à-dire la dose sans effet observable, le critère
retenu étant l'augmentation du poids du foie.
Chez plusieurs espèces, le principal organe cible après
exposition de courte durée (jusqu'à 6 mois) par voie respiratoire se
révèle être le foie. Chez des rats soumis à une dose de 26 mg/m3 (la
dose la plus faible utilisée) on a constaté une augmentation du poids
relatif du foie et des modification hépatocellulaires; à dose plus
élevée (> 260 mg/m3), les anomalies hépatiques étaient plus
prononcées et dépendaient de la dose. Les autres organes cibles
étaient le rein, le poumon et le testicule. Les rats, les souris et
les lapins se sont révélés plus sensibles que les cobayes et les
chiens.
Une exposition de longue durée par la voie respiratoire a
entraîné une augmentation statistiquement significative de la
mortalité chez certaines souches de rats à des doses ne dépassant pas
260 mg/m3, chez des souris à la dose de 130 mg/m3 et chez des
hamsters à la dose de 520 mg/m3 sur des durées d'exposition
variables. Des rats exposés à une dose de 130 mg/m3 ont présenté une
diminution du poids corporel et une augmentation du poids relatif de
la rate ainsi qu'une dégénérescence hépatocellulaire et une
prolifération des cellules pariétales des capillaires sinusoïdes. A
dose plus élevée, on a noté une dégénérescence testiculaire, une
néphrose tubulaire et des foyers de dégénérescence au niveau du
myocarde. Chez des rats et des souris exposés par la voie respiratoire
la dose sans effet nocif observable (NOAEL) est inférieure à 130
mg/m3 pour ce qui est des effets cancérogènes.
Des études d'alimentation de longue durée ont fait ressortir une
augmentation de la mortalité, un accroissement du poids du foie et une
modification de la morphologie de cet organe.
Après exposition par la voie orale, on a observé un polymorphisme
hépatocellulaire (variation de la taille et de la forme des
hépatocytes et de leur noyau) chez des rats soumis à des doses de
chlorure de vinyle ne dépassant pas 1,3 mg/kg de poids corporel. La
NOAEL était égale à 0,13 mg/kg de poids corporel.
Après administration prolongée à des rats de chlorure de vinyle
dans des granulés de PVC par la voie alimentaire, on a observé un
accroissement significatif de l'incidence des tumeurs au niveau du
foie. Il s'agissait d'angiosarcomes à la dose quotidienne de 5,0 mg/kg
p.c., et de nodules néoplasiques (femelles) ou de carcinomes
hépatocellulaires (mâles) à la dose de 1,3 mg/kg p.c.
Des études au cours desquelles on a fait inhaler du chlorure de
vinyle à des rats Sprague-Dawley ont fait ressortir une relation
dose-réponse dans le cas des angiosarcomes du foie et, à forte
concentration, pour les carcinomes de la glande de Zymbal. En
revanche, on n'a pas observé de relation dose-réponse bien nette dans
le cas des hépatomes ou des angiosarcomes extrahépatiques, des
néphroblastomes, des neuroblastomes ou des tumeurs des glandes
mammaires. Chez la souris, le spectre tumoral induit par une
exposition respiratoire de longue durée est analogue à celui que l'on
observe chez le rat, mais on a aussi noté une augmentation des tumeurs
pulmonaires propre à la souris. Chez des hamsters, on a relevé une
augmentation de l'incidence des angiosarcomes hépatiques, des tumeurs
des glandes mammaires et du conduit auditif, des mélanomes et des
épithéliomas gastriques et cutanés.
Un certain nombre de systèmes d'épreuve in vitro ont permis de
mettre en évidence les effets mutagènes et génotoxiques du chlorure de
vinyle, surtout après activation métabolique. Le composé s'est révélé
mutagène dans le test d'Ames sur les souches TA100, TA1530 et TA1535
de S. typhimurium, à l'exclusion des souches TA98, TA1537 et TA1538,
ce qui dénote des mutations par substitution de paires de bases
(transversion et transition) plutôt que des mutations par décalage du
cadre de lecture. Ces résultats concordent avec une autre observation,
à savoir que les éthénoadduits qui se forment lors de l'attaque de
l'ADN par l'oxyde de chloréthylène et par le chloracétaldéhyde
aboutissent effectivement à des mutations par substitution de paires
de bases.
D'autres tests de mutation génique effectués sur des bactéries,
des levures et des cellules mammaliennes ont donné des résultats
positifs, mais seulement en présence d'activation métabolique. Des
effets mutagènes ont également été observés dans des lignées
cellulaires humaines contenant le cytochrome P-450IIE1 obtenu par
clonage, qui est capable de métaboliser le chlorure de vinyle. On a
aussi décelé des mutations dans des fragments d'un végétal
(Tradescantia) mis en présence de chlorure de vinyle. Des tests de
conversion génique ont donné des résultats positifs dans le cas de
Saccharomyces cerevisiae en présence d'un système d'activation
métabolique. En présence de chlorure de vinyle, des hépatocytes de rat
ont été le siège d'une synthèse non programmée de l'ADN et un
accroissement des échanges entre chromatides-soeurs a été observé dans
des lymphocytes humains après addition d'un système activateur
exogène. Chez des bactéries dont le système de réparation de l'ADN
était défectueux, on n'a pas décelé d'inhibition de la croissance en
l'absence de système activateur. Les tests de transformation
cellulaire ont donné des résultats positifs avec ou sans activation
métabolique.
Chez Drosophila melanogaster, l'exposition au chlorure de
vinyle a provoqué des mutations géniques et des recombinaisons
mitotiques, mais ces effets n'ont pas été observés sur des cellules
germinales de mammifères. Le composé a des effets clastogènes chez des
rongeurs, il augmente les échanges entre chromatides-soeurs chez le
hamster et provoque la rupture des brins de l'ADN chez la souris. Des
tests par passage sur hôte (rat) ont montré que le chlorure de vinyle
provoquait une conversion génique et les mutations directes chez des
levures.
L'oxyde de chloréthylène et le chloracétaldéhyde se sont révélés
mutagènes dans un certain nombre de systèmes. L'oxyde de chloréthylène
est un mutagène puissant, alors que le chloracétaldéhyde est fortement
toxique. Ces deux composés sont cancérogènes pour la souris, l'oxyde
de chloréthylène étant de loin le plus actif.
Les mutations observées au niveau des gènes ras et p53 ont
été analysées sur des tumeurs hépatiques induites chez des rats
Sprague-Dawley par du chlorure de vinyle: dans les carcinomes, on a
constaté la présence de substitutions de paires de bases au niveau du
gène Ha- ras; dans les angiosarcomes, ces substitutions intéressaient
le gène p53. La présence de ces mutations concorde avec la
formation, observée après exposition au chlorure de vinyle,
d'éthénoadduits persistants dans l'ADN des hépatocytes, éthénoadduits
dont on connaît le caractère promutagène.
L'étude du mécanisme par lequel s'exerce l'effet cancérogène du
chlorure de vinyle donne à penser que l'intermédiaire réactif que
constitue l'oxyde de chloréthylène attaque l'ADN pour former des
éthénoadduits, ce qui conduit à la substitution de paires de bases et
à la transformation néoplasique.
7. Effets sur l'Homme
L'exposition à des concentrations de l'ordre de 2590 mg/m3
(1000 ppm), qui n'étaient pas rares avant 1974, pendant des périodes
de 1 mois à plusieurs années, seraient à l'origine d'un syndrome
pathologique particulier observé chez des ouvriers travaillant sur le
chlorure de vinyle et appelé "maladie du chlorure de vinyle". Les
symptômes évoqués consistaient en douleurs auriculaires et céphalées,
étourdissements, troubles visuels, fatigue et perte d'appétit,
nausées, insomnies, essoufflement, douleurs au niveau de l'estomac et
dans la région du foie et de la rate, douleurs et picotements dans les
membres, sensation de froid aux extrémités, diminution de la libido et
perte de poids. Sur le plan clinique, on a relevé au niveau des doigts
des modifications à type de sclérodermie évoluant vers des anomalies
osseuses qualifiées d'acro-ostéolyse, avec des anomalies de la
circulation périphérique identiques à celles qui sont caractéristiques
de la maladie de Raynaud, une hypertrophie du foie et de la rate
d'histologie particulière et des manifestations respiratoires.
Les études sur l'Homme ne sont pas suffisantes pour pouvoir
confirmer la présence d'effets sur la fonction de reproduction. Un
certain nombre d'études font état d'une augmentation de l'incidence
des affections de l'appareil circulatoire chez des ouvriers
travaillant sur le chlorure de vinyle. Toutefois, les études
effectuées sur des cohortes d'effectif plus important ont mis en
évidence une diminution de la mortalité due aux maladies
cardiovasculaires.
Les études épidémiologiques fournissent une argumentation solide
et cohérente tendant à prouver que l'exposition au chlorure de vinyle
provoque une forme rare de cancer, l'angiosarcome du foie. On a
également établi un lien entre certaines tumeurs cérébrales ou
carcinomes hépatocellulaires et le chlorure de vinyle, sans qu'on
puisse considérer les données obtenues à cet égard comme définitives.
Les autres localisations où l'on a observé une augmentation des
cancers sont le poumon, les tissus lymphatiques et hématopoïétiques et
la peau.
Le chlorure de vinyle a des effets mutagènes et clastogènes chez
l'Homme. On a ainsi constaté que chez des ouvriers exposés à de fortes
concentrations de chlorure de vinyle, la fréquence des aberrations
chromosomiques, des micronoyaux et des échanges entre
chromatides-soeurs dans les lymphocytes du sang périphériques, était
plus élevée que chez les témoins. Dans de nombreuses études,
l'intensité et la durée de l'exposition ne sont qu'estimatives, mais
on peut néanmoins observer l'existence d'une relation dose-réponse et
une "normalisation" des effets génotoxiques avec le temps lorsque
l'exposition diminue.
Des mutations ponctuelles ont été décelées au niveau des gènes
p53 et ras dans des tumeurs (angiosarcomes du foie) prélevées sur
des ouvriers très exposés travaillant sur des autoclaves (avant 1974)
ainsi que dans un carcinome hépatocellulaire dont était porteur un
autre ouvrier également exposé au chlorure de vinyle.
Parmi les marqueurs biologiques dont on a étudié la possibilité
d'utilisation comme indicateurs d'une exposition au chlorure de
vinyle, on peut citer a) l'excrétion de métabolites (par exemple
l'acide thiodiglycolique), b) des marqueurs génétiques comme la
présence d'anomalies chromosomiques ou de micronoyaux, c) le taux de
certaines enzymes (par exemple celles qui sont mesurées dans les tests
de la fonction hépatique), d) les oncoprotéines sériques et leurs
anticorps en tant que biomarqueurs des effets induits par le chlorure
de vinyle.
Les enfants qui vivent à proximité de décharges et autres sources
ponctuelles d'exposition au chlorure de vinyle pourraient courir un
risque accru, compte tenu de la sensibilité plus forte constatée chez
les jeunes animaux. On n'a toutefois pas de preuves directes d'une
telle sensibilité chez l'Homme.
C'est seulement dans le cas de l'angiosarcome du foie-seul ou en
association avec d'autres tumeurs hépatiques - qu'une relation
dose-réponse claire ressort des études épidémiologiques. Il n'existe
qu'une seule étude épidémiologique qui ait produit des données
suffisantes pour une estimation quantitative de la relation
dose-réponse.
8. Effets sur les autres êtres vivants au laboratoire et dans leur
milieu naturel
On manque des données toxicologiques habituelles au sujet de la
survie et de la reproduction des organismes aquatiques exposés au
chlorure de vinyle. Il faut interpréter avec prudence les données
disponibles car pour la plupart, elles proviennent de tests au cours
desquels on n'a pas mesuré les concentrations auxquelles ces
organismes étaient exposés et il n'a donc pas été tenu compte des
pertes par volatilisation.
La concentration la plus faible de chlorure de vinyle qui
produise un effet sur des microorganismes a été trouvée égale à 40
mg/litre. Il s'agit d'une valeur de la CE50 obtenue lors d'un test
statique de 3,5 jours sur des microorganismes anaérobies pour lequel
le critère retenu était l'inhibition de la respiration.
La concentration la plus faible qui produise un effet sur des
organismes supérieurs a été trouvée égale à 210 mg/litre (CL50 à 48 h
pour un poisson d'eau douce); avec une concentration sans effet nocif
observable (NOAEC) de 128 mg/litre. Chez d'autres espèces, on a
constaté des effets à plus faible concentration, mais la portée
écologique de ces effets n'a pas été vérifiée.
On peut prévoir que la concentration de chlorure de vinyle sans
effet nocif pour les poissons d'eau douce se situe entre 0,088 et
29 mg/litre.
On manque de données concernant des effets du chlorure de vinyle
sur les organismes terrestres.
RESUMEN
Esta monografía se ocupa exclusivamente del cloruro de vinilo
(VC) como monómero y no es una evaluación del cloruro de polivinilo
(PVC), polímero del VC. No se tratan las exposiciones a mezclas con
VC.
1. Identidad, propiedades físicas y químicas y métodos analíticos
En condiciones normales, el VC es un gas incoloro, inflamable,
con un olor ligeramente dulce. Tiene una presión de vapor alta, un
valor elevado para la constante de la Ley de Henry y una solubilidad
en agua relativamente baja. Es más pesado que el aire y soluble en
casi todos los disolventes orgánicos. Se transporta en forma líquida
bajo presión.
A temperatura ambiente en ausencia de aire, el VC purificado seco
es muy estable y no corrosivo, pero por encima de 450°C, o en
presencia de hidróxido sódico o potásico, se puede producir una
descomposición parcial. La combustión del VC en el aire produce
anhídrido carbónico y cloruro de hidrógeno. En presencia de aire y de
oxígeno, se pueden formar peróxidos muy explosivos, por lo que hay que
mantener una vigilancia constante y limitar el contenido de oxígeno,
en particular en las plantas de recuperación de VC. En presencia de
agua se forma ácido clorhídrico.
Desde el punto de vista industrial, las reacciones de
polimerización para obtener PVC son técnicamente las más importantes,
aunque también lo son las reacciones de adición con otros halógenos en
el doble enlace, por ejemplo, para obtener 1,1,2-tricloroetano o
1,1-dicloroetano.
La concentración de VC en el aire se puede vigilar mediante su
retención en adsorbentes y, tras la desorción líquida o térmica, el
análisis por cromatografía de gases. En las mediciones del aire
ambiente, se pueden necesitar varios adsorbentes en serie o colectores
refrigerados para aumentar la eficacia de la retención. Las
concentraciones máximas en los lugares de trabajo se pueden medir con
instrumentos de lectura directa, por ejemplo basados en la detección
por FID o la PID. En la vigilancia continua se han utilizado
analizadores de rayos infrarrojos y de cromatografía de
gases/detección de ionización de llama combinados con el registro y
procesamiento de datos. En el análisis del VC en líquidos y sólidos se
utilizan la inyección directa, la extracción y cada vez más las
técnicas del espacio libre superior y de purga y retención. En estas
muestras también se analiza el VC mediante cromatografía de gases,
combinada, por ejemplo, con detectores de ionización de llama o de
espectrometría de masas.
2. Fuentes de exposición humana y ambiental
No se tiene conocimiento de que el VC se produzca de forma
natural, aunque se ha encontrado en los gases de vertedero y en el
agua freática como producto de la degradación de hidrocarburos
clorados depositados como residuos de disolventes en los vertederos o
en el entorno de lugares de trabajo en los que se utilizan dichos
disolventes. El VC también está presente en el humo de los
cigarrillos.
La producción industrial de VC se lleva a cabo mediante dos
reacciones principales: a) hidrocloración del acetileno; y b)
descomposición térmica (a unos 500°C) del 1,2-dicloroetano producido
mediante cloración directa (etileno y cloro) o la oxicloración
(etileno, ClH y aire/O2) de etileno en el "proceso equilibrado". En
la actualidad se utiliza más el segundo proceso.
La producción mundial de PVC (y por consiguiente de VC) en 1998
fue de unos 27 millones de toneladas. El PVC representa el 20% del
material plástico utilizado y se emplea en la mayoría de los sectores
industriales. Alrededor del 95% de la producción mundial de VC se
utiliza para la fabricación de PVC. El resto se destina a la
producción de disolventes clorados, fundamentalmente de
1,1,1-tricloroetano (10 000 toneladas/año).
Son tres los procesos principales que se utilizan en la
fabricación comercial de PVC: suspensión (equivalente al 80% de la
producción mundial), emulsión (12%) y en masa o a granel (8%). La
mayoría de los estudios monográficos en los que se describen efectos
adversos del VC se refieren a instalaciones que utilizan el proceso de
suspensión (llamado también de dispersión).
Hay varios informes de liberación de VC a causa de accidentes en
instalaciones de fabricación de PVC o durante el transporte. En
numerosos países se ha introducido la recuperación del VC no
convertido residual procedente de la polimerización y de otras fuentes
del proceso, como por ejemplo los efluentes de gases residuales y de
agua. Cuando no se toman precauciones especiales, se puede detectar VC
en resinas y productos de PVC.
La concentración residual de VC en el PVC está reglamentada en
muchos países desde finales de los años setenta. Desde entonces, la
liberación de VC a partir de la degradación térmica del PVC no es
detectable o se produce a niveles muy bajos.
En la producción de VC se pueden formar dioxinas como
contaminantes. Las concentraciones de dioxinas liberadas al medio
ambiente son un tema polémico.
3. Transporte, distribución y transformación en el medio ambiente
Debido a su alta presión de vapor, cabe esperar que el VC que se
libera a la atmósfera se mantenga casi totalmente en la fase de vapor.
Hay indicios de deposición húmeda.
La solubilidad del VC en agua es relativamente baja y su
capacidad de adsorción a la materia particulada y los sedimentos
escasa. La volatilización es el proceso más rápido de eliminación del
VC que se incorpora al agua superficial. Se han notificado semividas
para la volatilización del agua superficial que oscilan entre
alrededor de una y 40 horas.
Las semividas de volatilización a partir del suelo se calcularon
en 0,2-0,5 días. Las pérdidas estimadas de VC (tras un año bajo una
cubierta de suelo de 1 m) oscilaron entre el 0,1% y el 45%, en función
del tipo de suelo. Los coeficientes de sorción en el suelo estimados a
partir de datos fisicoquímicos indican un potencial escaso y, por
consiguiente, una movilidad alta en el suelo. Otra vía importante de
distribución es la lixiviación a través del suelo hacia el agua
freática, donde el VC puede persistir durante años.
En experimentos de laboratorio con organismos acuáticos se
observó una cierta bioacumulación, pero no hubo bioamplificación en la
cadena alimentaria.
Salvo en un pequeño número de excepciones, las agrupaciones
microbianas no adaptadas no degradan fácilmente el VC en condiciones
normales. Se estimó que las semividas de biodegradación máxima sin
adaptación del VC eran del orden de varios meses o años. Sin embargo,
los cultivos enriquecidos especiales o puros (por ejemplo,
Mycobacterium spp.) son capaces de degradar el CV en condiciones de
cultivo óptimas. Los productos principales de degradación fueron el
ácido glicólico o el anhídrido carbónico tras la conversión aerobia y
etano, eteno, metano o clorometano mediante transformación anaerobia.
Con frecuencia, la reacción de degradación del CV es más rápida por
vía aerobia que anaerobia.
El proceso predominante de transformación en la atmósfera es la
reacción con radicales OH producidos por vía fotoquímica, dando lugar
a semividas en la troposfera estimadas en 1-4 días. Durante las
reacciones experimentales de fotolisis se generan varios compuestos
críticos, como cloroacetaldehído, formaldehído y cloruro de formilo.
Se considera que las relaciones fotolíticas, así como la
hidrólisis química, tienen escasa importancia en los medios acuosos.
Sin embargo, la presencia de fotosensibilizadores puede potenciar la
transformación del VC.
Hay indicios de que el VC reacciona con el cloro o el cloruro
utilizado en la desinfección del agua, produciendo de esta manera
cloroacetaldehído y otros compuestos no deseados. Otra posibilidad de
interacción es con las sales, muchas de las cuales tienen la capacidad
de formar complejos con el VC, aumentando tal vez su solubilidad.
Los métodos utilizados (con diferente éxito) para la eliminación
del VC de las aguas contaminadas son la separación, la extracción, la
adsorción y la oxidación. Algunas técnicas de biocorrección in situ
(para el agua freática o el suelo) combinan la evaporación y otros
métodos con el tratamiento microbiano. El VC de los gases de desecho
se puede reciclar, incinerar o degradar por medios microbiológicos. La
mayor parte del CV de producción industrial se encuentra en los
artículos de PVC. Con su incineración se corre el riesgo de que se
formen dibenzodioxinas policloradas/dibenzofuranos policlorados y
otros compuestos orgánicos clorados perjudiciales.
4. Niveles medioambientales y exposición humana
La exposición de la población general al VC es muy pequeña.
Las concentraciones atmosféricas de VC en el aire ambiente son
bajas, normalmente inferiores a 3 µg/m3. La exposición de la
población general puede ser mayor en situaciones en las cuales se haya
producido una liberación accidental de grandes cantidades de VC a la
atmósfera, por ejemplo por un escape durante el transporte. Sin
embargo, esta exposición probablemente es transitoria. Cerca de zonas
industriales y de eliminación de desechos de VC/PVC se han registrado
concentraciones mucho más altas (hasta 8000 µg/m3 y 100 µg/m3,
respectivamente).
Las concentraciones en el aire de los espacios cerrados en casas
adyacentes a vertederos alcanzaron concentraciones máximas de
1000 µg/m3.
La vía más importante de exposición ocupacional es la inhalación
y se produce fundamentalmente en instalaciones de VC/PVC. La
exposición ocupacional al VC ascendió a varios miles de mg/m3 en los
años cuarenta y cincuenta y fue de varios cientos de mg/m3 en los
sesenta y comienzo de los setenta. Tras el reconocimiento del peligro
carcinogénico del VC, en los años setenta se establecieron en la
mayoría de los países normas de exposición ocupacional de alrededor de
13-26 mg/m3 (5-10 ppm). El cumplimiento de estas directrices ha
reducido considerablemente las concentraciones de VC en los lugares de
trabajo, pero incluso en los años noventa se han notificado
concentraciones más altas, que se pueden encontrar todavía en algunos
países.
Ocasionalmente se ha detectado VC en aguas superficiales,
sedimento y fangos cloacales, con máximos de 570 µg/litro, 580 µg/kg y
62 000 µg/litro, respectivamente. Las muestras de suelo recogidas
cerca de una tienda de productos químicos de limpieza abandonada
contenían concentraciones muy elevadas de VC (hasta 900 mg/kg). Las
concentraciones máximas de VC en el agua freática o lixiviada de zonas
contaminadas por hidrocarburos clorados ascendieron a 60 000 µg/litro
(o más). Se detectaron concentraciones altas (hasta 200 mg/litro) en
el agua de un pozo cercano a una instalación de PVC 10 años después de
las filtraciones.
Los escasos datos disponibles ponen de manifiesto que el VC puede
estar presente en los tejidos de pequeños invertebrados acuáticos y de
peces.
En la mayoría de las muestras de agua de bebida analizadas, no
había VC presente en concentraciones detectables. La concentración
máxima de VC notificada en agua de bebida tratada fue de 10 µg/litro.
No se dispone de datos recientes sobre las concentraciones de VC en el
agua de bebida, pero cabe prever que sean inferiores a 10 µg/litro. Si
se utiliza agua contaminada como fuente de agua de bebida, se podrían
producir exposiciones más elevadas. En algunos estudios recientes se
ha identificado VC en agua de bebida embotellada en envases de PVC en
concentraciones inferiores a 1 µg/litro. Probablemente sea más
frecuente la presencia de VC en este tipo de agua que en la de grifo.
El envasado con ciertos materiales de PVC puede producir la
contaminación por CV de productos alimenticios, farmacéuticos o
cosméticos, incluso licores (hasta 20 mg/kg), aceites vegetales (hasta
18 mg/kg), vinagres (hasta 9,8 mg/kg) y colutorios (hasta 7,9 mg/kg).
Gracias a las medidas legislativas adoptadas por numerosos países,
desde comienzos de los años setenta se ha logrado una reducción
significativa de las concentraciones de VC y/o en el número de
muestras positivas.
Varios organismos han calculado la exposición al VC a través de
los envases de PVC utilizados para productos alimenticios y, teniendo
cuenta el promedio de ingesta estimado en el Reino Unido y en los
Estados Unidos, se calculó una exposición < 0,0004 µg/kg para finales
de los años setenta y comienzos de los ochenta. En un estudio inicial
se identificó VC en el humo del tabaco en concentraciones del orden de
ng/cigarrillo.
5. Cinética y metabolismo en animales de laboratorio y en el ser
humano
Tras las exposición por inhalación o por vía oral, el VC se
absorbe con rapidez y facilidad. La vía primaria de exposición al VC
es la inhalación. En estudios realizados con animales y con personas
en condiciones estables, después de la exposición por inhalación se
absorbe aproximadamente el 40% del VC inspirado. En estudios de
exposición oral con animales se observó una absorción de más del 95%.
La absorción cutánea al VC en estado gaseoso no es significativa.
Los datos obtenidos en estudios de administración oral y por
inhalación en ratas indican una distribución rápida y generalizada del
VC. La rapidez del metabolismo y la excreción limita la acumulación de
VC en el organismo. En las ratas se produce un desplazamiento rápido
del VC a través de la placenta. No se ha informado de estudios sobre
distribución tras la exposición cutánea.
La principal ruta de metabolismo del CV después de la inhalación
o la ingestión oral consiste en la oxidación por el citocromo P-450
(CYP2E1) para formar óxido de cloroetileno (CEO), epóxido muy reactivo
de vida breve que reacciona de nuevo rápidamente para formar
cloroacetaldehído (CAA). La reacción primaria de desintoxicación de
estos dos metabolitos reactivos, así como del ácido cloroacético,
producto de la deshidrogenación del CAA, es la conjugación con el
glutatión en una reacción catalizada por la glutatión- S-transferasa.
Los productos de la conjugación sufren ulteriores modificaciones para
formar derivados de la cisteína con sustituciones
(S-(2-hidroxietil)-cisteína, N-acetil- S-(2-hidroxietil)
cisteína, S-carboximetil cisteína y ácido tiodiglicólico) y se
excretan en la orina. Otro metabolito, el anhídrido carbónico, se
exhala en el aire.
Se sabe que el CYP2E1 y las isoenzimas de la
glutatión-S-transferasa tienen variaciones individuales importantes de
actividad interespecíficas e intraespecíficas.
Tras la exposición por inhalación o por vía oral a dosis bajas,
el CV se elimina metabólicamente y los metabolitos no volátiles se
excretan fundamentalmente en la orina. En investigaciones comparativas
de la absorción de VC por inhalación se puso de manifiesto una
velocidad de eliminación metabólica en el ser humano menor que en los
animales de laboratorio, en función del peso corporal. Sin embargo,
una vez corregida con arreglo a la superficie corporal, la eliminación
metabólica del VC en el ser humano es comparable a la observada en
otras especies de mamíferos. Al aumentar la exposición oral o por
inhalación, la vía más importante de excreción en los animales es la
exhalación de VC inalterado, lo cual indica que hay una saturación de
las rutas metabólicas. Con independencia de la dosis aplicada, la
excreción de metabolitos por las heces es sólo una vía secundaria. No
se encontraron estudios en los que se investigase específicamente la
excreción por la bilis.
Se considera que el CEO es el metabolito más importante in vivo
con respecto a los efectos mutagénicos y carcinogénicos del VC. El
CEO reacciona con el ADN para producir el aducto principal
7-(2'-oxoetil)guanina (7-OEG) y, a niveles más bajos, los aductos
exocíclicos de eteno, 1, N 6-etenoadenina (Epsilon A),
3, N4-etenocitosina (Epsilon C) y N 2,3-etenoguanina (Epsilon G).
Los aductos de eteno del ADN tienen propiedades promutagénicas, a
diferencia del aducto principal 7-OEG. Se han medido las
concentraciones de 7-OEG, Epsilon A, Epsilon C y Epsilon G en diversos
tejidos de roedores expuestos al VC. Se han creado modelos
toxicocinéticos con una base fisiológica para describir la relación
entre la concentración en el tejido destinatario y los efectos finales
tóxicos del VC.
6. Efectos en mamíferos de laboratorio y en sistemas de prueba
in vitro
La toxicidad aguda del VC administrado por inhalación a diversas
especies parece ser baja. Se notificaron CL50 a las 2 horas para
ratas, ratones, cobayas y conejos de 390 000, 293 000, 595 000 y
295 000 mg/m3, respectivamente. No hay datos disponibles sobre la
toxicidad aguda tras la administración oral o la aplicación cutánea.
El VC tiene un efecto estupefaciente después de la administración
aguda por inhalación. En ratas, ratones y hámsteres, la muerte estuvo
precedida por un aumento de la actividad motora, ataxia y
convulsiones, y a continuación colapso respiratorio. En perros se
produjo una arritmia cardíaca grave en estado de narcosis tras la
exposición por inhalación a 260 000 mg/m3. Después de la exposición
aguda de ratas al VC por inhalación se observaron diversos efectos
patológicos, entre ellos congestión de los órganos internos, sobre
todo los pulmones, el hígado y los riñones, así como edema pulmonar.
No hay estudios o datos importantes disponibles para la
evaluación de los efectos de la exposición cutánea, la irritación de
la piel o la propiedad de sensibilización del VC.
De la exposición oral de ratas al VC durante un período breve de
13 semanas se obtuvo una concentración sin efectos observados (NOEL),
basada en el aumento de peso del hígado, de 30 mg/kg.
En diversas especies, el principal órgano destinatario en la
exposición breve (hasta 6 meses) al VC por inhalación fue el hígado.
Con una concentración de 26 mg/m3 (la dosis más baja utilizada) se
observaron en ratas un aumento del peso relativo del hígado y cambios
hepatocelulares; a concentraciones superiores (> 260 mg/m3) se
produjeron también cambios hepáticos más acentuados dependientes de la
dosis. Otros órganos destinatarios fueron los riñones, los pulmones y
los testículos. Las ratas, los ratones y los conejos parecen ser más
sensibles que los cobayas y los perros.
La exposición prolongada al VC por inhalación produjo un aumento
estadísticamente significativo de la mortalidad en algunas estirpes de
ratas con concentraciones de sólo 260 mg/m3, en ratones con 130
mg/m3 y en hámsteres con 520 mg/m3 para diversos períodos de
exposición. Las ratas expuestas a 130 mg/m3 mostraron una reducción
del peso corporal y un aumento del peso relativo del bazo,
degeneración hepatocelular y proliferación de las células de
revestimiento de los sinusoides del hígado. La exposición de ratas a
concentraciones más elevadas produjo una alteración degenerativa de
los testículos, nefrosis tubular y degeneración focal del miocardio.
En ratas y ratones expuestos por inhalación, la concentración sin
efectos adversos observados (NOAEL) para los efectos no neoplásicos es
inferior a 130 mg/m3.
En estudios de alimentación crónica se puso de manifiesto un
aumento de la mortalidad, un peso mayor del hígado y una alteración
morfológica del hígado.
Tras la exposición oral de ratas a concentraciones de sólo
1,3 mg/kg de peso corporal se pudo observar polimorfismo de las
células hepáticas (variación del tamaño y la forma de los hepatocitos
y sus núcleos). La NOAEL fue de 0,13 mg/kg de peso corporal.
En estudios prolongados de alimentación realizados en ratas con
gránulos de VC y PVC se produjo un aumento significativo de la
incidencia del angiosarcoma hepático (ASH) con 5,0 mg/kg de peso
corporal al día y nódulos neoplásicos en el hígado (hembras) y
carcinoma hepatocelular (CHC) (machos) con 1,3 mg/kg de peso corporal
al día.
En estudios de inhalación de VC en ratas Sprague-Dawley se
observó una relación dosis-respuesta en el caso del ASH y, a
concentraciones más altas, carcinoma de las glándulas de Zymbal. No se
observó una dependencia clara de la dosis para el hepatoma o el
angiosarcoma extrahepático, los nefroblastomas, los neuroblastomas o
los tumores mamarios malignos. En ratones, el espectro de tumores
inducidos por la exposición prolongada mediante inhalación es
semejante al observado en ratas, pero se detectó sólo en ratones un
aumento de los tumores pulmonares. En hámsteres se notificó un aumento
de la incidencia de tumores de ASH, tumores de las glándulas mamarias
y el conducto acústico, melanomas, tumores de estómago y del epitelio
cutáneo.
Se han detectado efectos mutagénicos y genotóxicos del VC en
varios sistemas de prueba in vitro, fundamentalmente después de la
activación metabólica. El VC tiene un efecto mutagénico en la prueba
de Ames con las cepas TA100, TA1530 y TA1535 de S. typhimurium, pero
no con las cepas TA98, TA1537 y TA1538, lo cual indica que las
mutaciones son el resultado de la sustitución de pares de bases
(transversión y transición) más que de mutaciones por desfase. Esto
está en consonancia con el resultado de que los aductos de eteno del
ADN formados por los metabolitos reactivos CEO y CAA se convierten en
mutaciones reales mediante las sustituciones de pares de bases.
Otras valoraciones de mutaciones genéticas en bacterias,
levaduras y células de mamíferos han puesto de manifiesto resultados
positivos exclusivamente en presencia de una activación metabólica. Se
notificaron asimismo efectos mutagénicos en una línea de células
humanas con citocromo P-450IIE1 clonado, que es capaz de metabolizar
el VC. También se detectó una mutación genética en esquejes de plantas
(Tradescantia) expuestos al VC. En las valoraciones de la conversión
genética, se notificaron resultados positivos con Saccharomyces
cerevisiae en presencia de un sistema de activación metabólica. La
exposición al VC indujo síntesis no programada de ADN en hepatocitos
de rata y un aumento del intercambio de crómatidas hermanas en
linfocitos humanos tras la adición del sistema de activación exógeno.
No se detectó inhibición del crecimiento en bacterias deficientes en
enzimas de reparación del ADN sin activación metabólica. En
valoraciones de la transformación celular se obtuvieron resultados
positivos con activación metabólica y sin ella.
La exposición al VC indujo mutaciones genéticas y recombinación
mitótica en Drosophila melanogaster, pero no produjo ninguna
mutación genética en células germinales de mamíferos. El VC mostró
efectos clastogénicos en roedores, aumentó el intercambio de
cromátidas hermanas de hámster e indujo el fraccionamiento del ADN en
ratones. En valoraciones mediadas por huéspedes (ratas), el VC indujo
una conversión genética y mutaciones adaptativas en levaduras.
Se observó un efecto mutagénico del CEO y el CAA en diferentes
sistemas de prueba. El CEO es un mutágeno potente, mientras que el CAA
es muy tóxico. Ambos mostraron efectos carcinogénicos en ratones,
siendo el CEO mucho más activo que el CAA.
Se analizaron las mutaciones de los genes ras y p53 en
tumores de hígado inducidos por el VC en ratas Sprague-Dawley: se
encontraron sustiticiones de pares de bases en el gen Ha- ras en el
carcinoma hepatocelular y en el gen p-53 en el ASH. Estas mutaciones
coinciden con la formación observada y la persistencia de aductos de
eteno en el ADN del hígado, tras la exposición de ratas a VC, y con
las propiedades promutagénicas conocidas de los aductos de eteno.
Los estudios sobre los mecanismos de la carcinogenicidad del VC
parecen indicar que el epóxido intermedio reactivo CEO tiene una
interacción con el ADN para formar aductos de eteno, que dan lugar a
una sustitución de pares de bases que lleva a una transformación
neoplásica.
7. Efectos en el ser humano
Se ha notificado que concentraciones de VC del orden de
2590 mg/m3 (1000 ppm), que no eran raras antes de 1974, durante
periodos comprendidos entre un mes y varios años provocaban un
síndrome patológico específico observado en los trabajadores del VC,
llamado "enfermedad del cloruro de vinilo". Los síntomas descritos
fueron dolor de oídos y de cabeza, vértigo, visión borrosa, cansancio
y falta de apetito, náuseas, insomnio, dificultad respiratoria, dolor
de estómago, dolor en la zona del hígado/bazo, dolor y sensación de
hormigueo en los brazos y las piernas, sensación de frío en las
extremidades, pérdida de la libido y disminución del peso. Entre los
resultados clínicos figuraron cambios en los dedos del tipo del
escleroderma, con modificaciones óseas posteriores en la punta de los
dedos descritas como acroosteólisis, cambios en la circulación
periférica idénticos a los clásicos de la enfermedad de Raynaud y
agrandamiento del hígado y del bazo con una aspecto histológico
específico, así como manifestaciones respiratorias.
Los estudios en seres humanos no han sido adecuados para
confirmar los efectos en el sistema reproductor. En un pequeño número
de estudios de morbilidad se ha notificado una elevada incidencia de
enfermedades circulatorias entre los trabajadores relacionados con el
cloruro de vinilo. Sin embargo, en estudios de cohortes amplios se ha
observado una mortalidad más baja que la debida a enfermedades
cardiovasculares.
Hay pruebas manifiestas y convincentes obtenidas de estudios
epidemiológicos de que la exposición al VC produce un tumor raro, el
angiosarcoma hepático. También pueden asociarse con el VC casos de
tumores cerebrales y carcinoma hepatocelular, aunque las pruebas no
pueden considerarse definitivas. Otros puntos notificados como de una
mayor incidencia de cáncer, pero de manera menos convincente, son el
pulmón, los tejidos linfático y hematopoyético y la piel.
El VC es mutagénico y clastogénico en el ser humano. Se ha
observado un aumento de la frecuencia de aberraciones cromosómicas,
micronúcleos e intercambio de cromátidas hermanas en los linfocitos de
la sangre periférica de los trabajadores expuestos a concentraciones
elevadas de VC en comparación con los testigos. Aunque en muchos
estudios solamente se estimaron las concentraciones y la duración de
la exposición, se puede observar una relación dosis-respuesta y la
"normalización" de los efectos genotóxicos con el paso del tiempo
después de la reducción de la exposición.
Se han detectado mutaciones puntuales en los genes p 53 y ras
en tumores de personas que trabajaban con autoclaves y que estaban
muy expuestas (antes de 1974) afectadas de angiosarcoma hepático y en
otro trabajador relacionado con el CV con carcinoma hepatocelular.
Los marcadores biológicos investigados como indicadores de la
exposición al VC o de los efectos inducidos por el VC son los
siguientes: a) excreción de metabolitos del VC (por ejemplo, ácido
tiodiglicólico), b) valoraciones genéticas (por ejemplo, anomalías
cromosómicas o valoración de micronúcleos), c) concentraciones de
enzimas (por ejemplo, en pruebas de la función hepática), d)
oncoproteínas séricas (p21 y p53) y/o sus anticuerpos como
biomarcadores de los efectos inducidos por el VC.
Los niños que viven en lugares cercanos a vertederos y otras
fuentes puntuales pueden correr un riesgo mayor, tomando como base las
pruebas que parecen derivarse de la sensibilidad en las primeras fases
de la vida en estudios realizados con animales. Sin embargo, no hay
pruebas directas en el ser humano.
En los estudios epidemiológicos solamente hay una relación
dosis-respuesta clara para el angiocarcinoma hepático solo o en
combinación con otros tumores del hígado. Sólo en un estudio
epidemiológico hay datos suficientes para una estimación cuantitativa
de la relación dosis-respuesta.
8. Efectos en otros organismos en el laboratorio y en el medio
ambiente
Se carece de datos normalizados de toxicidad relativos a la
supervivencia y la reproducción de los organismos acuáticos expuestos
al VC. Hay que interpretar con cautela los datos disponibles, porque
la mayoría de ellos se obtuvieron en pruebas en las cuales no se midió
la concentración de la exposición, por lo que no se tuvieron en cuenta
las pérdidas debidas a la volatilización.
La concentración más baja de VC que produjo un efecto en los
microorganismos fue de 40 mg/litro. Fue un valor de la CE50 basado en
la inhibición de la respiración de microorganismos anaerobios en la
valoración de un lote durante 3,5 días.
La concentración más baja que produjo un efecto en organismos
superiores fue de 210 mg/litro (CL50 a las 48 horas para un pez de
agua dulce), con una concentración sin efectos adversos observados
(NOAEC) correspondiente de 128 mg/litro. Se han notificado efectos en
otras especies debidos a concentraciones más bajas de VC, pero no se
comprobó la importancia ecológica de dichos efectos.
Las concentraciones de VC estimadas como no peligrosas para los
peces de agua dulce se calculó que oscilaban entre 0,088 y 29
mg/litro.
Hay pocos datos sobre los efectos del VC en los organismos
terrestres.
carcinoma in female F-344 rats. This exposure duration-related effect
was not observed in mice and hamsters, except angiosarcoma at all
sites in hamsters.
The lowest reported dose (13 mg/m3) to cause a statistically
significant increase in tumour incidences was demonstrated for mammary
adenocarcinoma in female rats (Maltoni et al. 1981; BT15). Although
this tumour type is common in untreated rats and no clear
dose-response relationship was shown (probably due to reduced survival
in the high-dose groups, Maltoni et al., 1984; BT1, BT2, BT9, BT15),
the increase in tumour rate was considered by the Task Group to be of
toxicological relevance, since the incidence was increased compared to
concurrent and historical controls of the same colony (Maltoni et al.,
1981, 1984) and similar results on mammary gland tumours were
presented in further studies on rats (Drew et al., 1983) and other
species (Lee et al., 1978; Maltoni et al., 1981, 1984; Drew et al.,
1983) (see also Table 33; Fig. 4).
b) Mice
In mice, the spectrum of tumours induced by long-term inhalation
exposure is similar to that observed in rats, but an increase in lung
tumours was only observed in mice (Lee et al., 1978; Maltoni et al.,
1981; Drew et al., 1983, see also Table 32). At a dose level of
130 mg/m3, incidences in angiosarcoma (liver, extrahepatic or all
sites combined), lung carcinoma, and mammary gland carcinoma showed a
statistically significant increase (Lee et al., 1978; Drew et al.,
1983). Doses lower than 130 mg/m3 were not investigated, but at the
dose levels investigated, the angiosarcoma frequencies were higher in
mice than in rats (Lee et al., 1978). No clear-cut relationship with
time of exposure (6-18 months) and incidence of ASL was observed; this
could, however, have been due to decreased survival and shorter
follow-up after longer exposure time (Drew et al., 1983).
c) Other species
Limited data are available on other species. In experiment BT8
with male Syrian golden hamsters, low frequencies of ASL, acoustic
duct tumours and melanomas were observed. In addition, an increased
incidence in forestomach and skin epithelial tumours was reported, but
these tumours were also observed in controls, there was no
dose-response relationship and a statistical evaluation was not
presented (Maltoni et al., 1981).
Female hamsters were exposed to a single dose for different
exposure periods (Drew et al., 1983). Angiosarcomas (all sites), skin
tumours and increased incidences in mammary gland carcinoma and
stomach adenomas (glandular portion of the stomach) were reported
(Drew et al., 1983).
In a study reported only as an abstract, Caputo et al. (1974)
reported increased incidences in skin acanthoma and lung
adenocarcinoma in rabbits exposed to VC.
Table 32. Inhalation studies on the long-term toxicity/carcinogenicity of vinyl chloride in experimental animalsa
Species; strain; age Doses in mg/m3; 1) Effects other than tumours Reference
at start of exposure period; (dose in mg/m3);
experiment; initial frequency of treatment; 2) number of animals for
number of animals per post-exposure histopathological evaluation of
dose per exposure observation period neoplastic effects;
period; type of 3) type of tumour: number of animals
control; with this tumour/dose group (unless
otherwise given)
Rat; 0, 130, 650, 1300, 1) > 650: survival ¡# (f, in m at > 1300); Maltoni et
Sprague-Dawley; 13 6500, 15 600, > 15 600: body weight ¡# (m, in f at 26 000); al. (1981,
weeks; 30 rats/sex; 26 000; 52 weeks; 2) 29-30 m and 29-30 f per group; 1984)
untreated control 4 h/d, 5 d/week; 3) ASL in m: 0, 0, 1, 0, 6*, 3, 3 and in [BT1]b
83 weeks c f : 0, 1, 2, 6*, 7*, 10*, 4; Zymbal gland
carcinoma in m: 0, 0, 0, 3, 1, 3, 10* and in f:
0, 0, 0, 1, 1, 4, 6*; hepatoma in f:
0, 0, 0, 5, 2, 1, 0;
nephroblastoma in m: 0, 0, 1, 2, 5*, 4, 3
and in f: 0, 1, 4*, 4*, 1, 1, 2;
neuroblastoma in m: 0, 0, 0, 0, 2, 2, 2 and in f:
0, 0, 0, 0, 2, 1, 5*;
mammary adenocarcinoma in f: 0, 2, 2, 1, 2, 0, 3
Rat; 0, 260, 390, 520; 1) > 130: survival ¡# (m, in f at 520); Maltoni et
Sprague-Dawley; 52 weeks; 4 h/d, 2) 59-60 rats per treatment group, control al. (1981,
13 weeks; 60 5 d/week; 91 weeks c 65 m and 100 f; 1984)
rats/sex, control 3) ASL in m: 0, 0, 1, 7* and in f: 0, 1, 5*, 5*; [BT2]b
85 m and 100 f; mammary adenocarcinoma in f: 1, 3, 6*, 5;
untreated control nephroblastoma in m: 0, 8*, 8*, 5* and in f:
0, 2, 3, 2
Table 32. (cont'd)
Species; strain; age Doses in mg/m3; 1) Effects other than tumours Reference
at start of exposure period; (dose in mg/m3);
experiment; initial frequency of treatment; 2) number of animals for
number of animals per post-exposure histopathological evaluation of
dose per exposure observation period neoplastic effects;
period; type of 3) type of tumour: number of animals
control; with this tumour/dose group (unless
otherwise given)
Rat; Sprague-Dawley; 0, 130; 52 weeks; 1) survival ¡# (f); Maltoni et
13 weeks; 150 rats 4 h/d, 5 d/week; 2) 144 m and 150 f, control 48 m and 50 f; al. (1981,
per sex, control 50 m 90 weeks c 3) ASL in m: 0, 2 and in f: 0, 12*; 1984)
and 50 f; mammary adenocarcinoma in f: 5, 59*; [BT9]b
untreated control no significant effects on incidences of other
tumour types
Rat; 0, 2.6, 13, 26, 65; 1) > 2.6 survival ¡# (m+f); Maltoni et
Sprague-Dawley; 52 weeks; 4 h/d, 2) 58-60 m and 60 f per group; al. (1981,
13 weeks; 60 rats/sex; 5 d/week; 3) ASL in m: 0, 0, 0, 0, 1 and in f: 0, 0, 0, 1, 1984)
untreated control 95 weeks c 4; mammary adenocarcinoma in f: 6, 12, 22*, [BT15]b
21*, 15*; nephroblastoma in m: 0, 0, 0, 0, 1;
hepatoma in f: 0, 0, 0, 0, 1
Rat; Spague-Dawley; 0, 6500; life time 1) survival & body weight ¡# Maltoni &
13 weeks (pregnant (up to 69 weeks); 2) 60, 54 dams, progeny 158, 63 m and 149, 64 f Cotti (1988)
rats) or gestation day 4 h/d, 5 d/week 3) tumour incidences in % (#):
12 (embryos); see for 7 weeks, than ASLd : 0, 50.0 in dams and 0, 56.2 in m and
number of animals for 7 h/d, 5 d/week; 0, 73.0 in f progeny; hepatocellular carcinoma:
for evaluation; none 0, 9.2 in dams, 0.6, 42.2 in m and 0, 60.3 in f
untreated control progeny mammary carcinoma: 7.4, 7.4 in dams, 5.4,
4.7 in f progeny; neuroblastomad: 0, 59.2 in dams,
0, 48.4 in m and 0, 42.8 in f progeny
Table 32. (cont'd)
Species; strain; age Doses in mg/m3; 1) Effects other than tumours Reference
at start of exposure period; (dose in mg/m3);
experiment; initial frequency of treatment; 2) number of animals for
number of animals per post-exposure histopathological evaluation of
dose per exposure observation period neoplastic effects;
period; type of 3) type of tumour: number of animals
control; with this tumour/dose group (unless
otherwise given)
Rat; Fischer-344; 0 or 260; 6, 12, 1) survival ¡* (exposure periods > 6 mo); Drew et al.
8-9 weeks, 2nd 18, 24 mo, 2nd 2) 112 (control), 76, 55, 55, 55 per exposure (1983)
experiment: 2, 8, experiment 6 or period; 2nd experiment 6 mo exposure: 112
14, 20 mo (6 mo 12 mo exposure (control), 76, 52, 51, 53 per age group; 2nd
exposure) or 2, 8, at different age; experiment 12 mo exposure: 112 (control), 55,
14 mo (12 mo 6 h/d, 5 d/week; 54, 49 per age group;
exposure); n.g. (only life span 3) ASL: 1, 4*, 11*,13*, 19*; angiosarcoma all
f, see number of sites: 2, 4, 12*, 15*, 24*; mammary fibroadenoma:
rats for evaluation); 24, 28*, 28*, 24*, 26*; mammary adenocarcinoma:
n.g. 5, 6, 11*, 9*, 5*; neoplastic liver nodules: 4,
15*, 20*, 7*, 6*; hepatocellular carcinoma: 1, 3,
4*, 8*, 9*; 2nd experiment 6 mo exposure: ASL: 1,
4*, 2, 0, 0; angiosarcoma all sites: 2, 4, 2, 0,
0; mammary fibroadenoma: 24, 28*, 23*, 17, 20;
mammary adenocarcinoma: 5, 6, 2, 3, 2; neoplastic
liver nodules: 4, 15*, 10*, 2, 4; hepatocellular
carcinoma: 1, 3, 6*, 0, 1;
2nd experiment 12 mo exposure: ASL: 1, 11*, 5*,
2; angiosarcoma all sites: 2, 12*, 5*, 2; mammary
fibroadenoma: 24, 28*, 16*, 15; mammary
adenocarcinoma: 5, 11*, 4, 0; neoplastic liver
nodules: 4, 20*, 4, 4; hepatocellular carcinoma:
1, 4*, 1, 0;
Table 32. (cont'd)
Species; strain; age Doses in mg/m3; 1) Effects other than tumours Reference
at start of exposure period; (dose in mg/m3);
experiment; initial frequency of treatment; 2) number of animals for
number of animals per post-exposure histopathological evaluation of
dose per exposure observation period neoplastic effects;
period; type of 3) type of tumour: number of animals
control; with this tumour/dose group (unless
otherwise given)
Rat; Wistar; 11 0, 130, 650, 1300, 1) > 130: survival & body weight ¡# Maltoni et
weeks; control 40 m, 6500, 15 600, 2) 38, 28, 28, 27, 25, 26, 27; al. (1981,
other groups 30 m; 26 000; 52 weeks; 3) (#):ASL: 0, 0, 1, 3, 3, 3, 8; hepatoma: 0, 0, 1984)
untreated control 4 h/d, 5 d/week; 0, 0, 1, 2, 0; nephroblastoma: 0, 1, 0, 2, 0, [BT17]b
113 weeks c 2, 1; neuroblastoma: 0, 0, 0, 0, 1, 1, 3;
Zymbal gland carcinoma: 0, 0, 0, 0, 0, 2, 2
Rat; Wistar; newly 0 or 13 000; 1) mortality !# (9 m and 10 f still alive at week Feron et al.
weaned; 62 rats/sex; 52 weeks, 10 rats 52); body weights ¡* (f+m); blood clotting time (1979a,b)
treated control per dose per sex ¡#; liver function ¡# (BSB-retention test); Feron &
sacrificed at week relative liver, kidney and spleen weight !*; Kroes
4, 13, 26 (see degree of tubular nephrosis !# (f+m); focal (1979)
section 7.2); 7 h/d, degeneration of myocardium and thickened walls
5 d/week; none of arteries # (f+m); haematopoietic activity
in the spleen !# (f+m); distended liver
sinusoids # (f+m);
2) 62 m and 62 f per group;
3) cumulative tumours (#):ASL: 0, 3 in m and 0, 6
in f ; Zymbal gland tumour: 0, 7 in m and 0, 4
in f; tumour of nasal cavity: 0, 10 in m and
0, 10 in f;
Table 32. (cont'd)
Species; strain; age Doses in mg/m3; 1) Effects other than tumours Reference
at start of exposure period; (dose in mg/m3);
experiment; initial frequency of treatment; 2) number of animals for
number of animals per post-exposure histopathological evaluation of
dose per exposure observation period neoplastic effects;
period; type of 3) type of tumour: number of animals
control; with this tumour/dose group (unless
otherwise given)
Rat; CD; 2 mo; 0, 130, 650, 2600; 1) > 650: survival ¡# (m+f); Lee et al.
36 rats/sex; 12 mo, 4 rats/dose 2) 35, 36, 36, 34 m and 35, 36, 34, 36 f; (1978)
treated control per sex terminated 3) tumours combined for all exposure periods:
at month 1, 2, 3, 6, ASL: 0, 0, 2, 6 in m and 0, 0, 10*, 15* in f;
9; 6 h/d, 5 d/week; lung angiosarcoma: 0, 0, 0, 4 in m and 0, 0,
none 3, 9* in f; other tumours not related to VC
treatment
Rat [no further data 0, 14, 25, 266, 1) no data Kurlyandski
provided] 3690; 52 weeks; 2) 70, 50, 39, 43, 51 m et al.
4.5 h/d, 5 d/week 3) ASL: 0, 0, 7.7, 9.3, 11.8%; (1981)
other angiosarcomas: 0, 1.0, 2.5, 0, 3.9%
other liver tumours: 2.8, 2.0, 2.6, 11.6, 13.7%
haemoblastoma: 4.3, 14.0, 15.4, 34.9, 2.0%
other tumours: 21.5, 28.1, 15.4, 9.3, 23.5%
Mouse; Swiss; 0, 130, 650, 1300, 1) > 130: survival ¡# (m+f); > 1300: body weight ¡# Maltoni et
11 weeks; 30 mice 6500, 15 600, (m, in f at > 650); al. (1981,
per sex, control 26 000; 30 weeks; 2) 80, 30, 30, 30, 29, 30, 26 m and 70, 30, 30, 1984)
80 m and 70 f; 4 h/d, 5 d/week; 30, 30, 30, 30 f; [BT4]b
untreated control 51 weeks c 3) combined (f+m) tumours (#): ASL: 0, 1, 18, 14,
16, 13, 10; liver angioma: 0,1, 11, 5, 5, 7, 6;
extrahepatic angiosarcoma: 1, 1, 3, 7, 8, 1, 1;
lung tumour: 15, 6, 41, 50, 40, 47, 46;
mammary carcinoma in f : 1, 13, 12, 9, 10, 9, 14
Table 32. (cont'd)
Species; strain; age Doses in mg/m3; 1) Effects other than tumours Reference
at start of exposure period; (dose in mg/m3);
experiment; initial frequency of treatment; 2) number of animals for
number of animals per post-exposure histopathological evaluation of
dose per exposure observation period neoplastic effects;
period; type of 3) type of tumour: number of animals
control; with this tumour/dose group (unless
otherwise given)
Mouse; Swiss CD-1; 0 or 130; 6, 12, 1) survival ¡* (all exposure periods); Drew et
8-9 weeks, 2nd 18 mo, 2nd 2) 71 (control), 67, 47, 45 per exposure period; al. (1983)
experiment: 2, 8, experiment 6 or 12 mo 2nd experiment 6 mo exposure: 71 (control), 67,
14 mo (6 or 12 mo exposure at 49, 53 per age group; 2nd experiment 12 mo
exposure); n.g. (only different age; exposure: 71 (control), 47, 46, 50 per age group;
f, see number of 6 h/d, 5 d/week; 3) angiosarcoma (all sites): 1, 29*, 30*, 20*;
mice for evaluation); lifespan mammary gland carcinoma: 2, 33*, 22*, 22*; lung
n.g. carcinoma: 9, 18*, 15*, 11*; 2nd experiment 6 mo
exposure: angiosarcoma (all sites): 1, 29*, 11*,
5; mammary gland carcinoma: 2, 33*, 13*, 2; lung
carcinoma: 9, 18*, 13*, 7;
2nd experiment 12 mo exposure: angiosarcoma
(all sites): 1, 30*, 17*, 3;
mammary gland carcinoma: 2, 22*, 8*, 0; lung
carcinoma: 9, 15*, 9*, 3;
Mouse; CD-1; 2 mo; 0, 130, 650, 2600; 1) > 650: survival ¡# (tumour development), all Lee et al.
total 36 mice/sex; 1, 2, 3, 6, 9, and mice in high-dose group and females in mid-dose (1978)
treated control 12 mo (4 mice per group died or were terminated at mo 10-12;
group per sex per 2) 26, 29, 29, 33 m and 36, 34, 34, 36 f;
exposure period); 3) tumours combined for all exposure periods:
6 h/d, 5d/week; bronchioloalveolar adenoma (#):1, 8, 10, 22 in m
none and 0, 4, 12, 26 in f; ASL: 0, 3, 7*, 13* in m
and 0, 0, 16*, 13* in f; extrahepatic angiosarcoma:
0, 5*, 2, 0 in m and 0, 1, 3, 9* in f; mammary
tumours(#): 0, 9, 3, 13 in f; incidences exposure
time-dependent
Table 32. (cont'd)
Species; strain; age Doses in mg/m3; 1) Effects other than tumours Reference
at start of exposure period; (dose in mg/m3);
experiment; initial frequency of treatment; 2) number of animals for
number of animals per post-exposure histopathological evaluation of
dose per exposure observation period neoplastic effects;
period; type of 3) type of tumour: number of animals
control; with this tumour/dose group (unless
otherwise given)
Hamster; Syrian 0, 130, 650, 1300, 1) > 130: survival ¡#; Maltoni et
golden; 11 weeks; 6500, 15 600, 2) see initial number of hamsters; al. (1981,
30 m, 60 m in 26 000; 30 weeks; 3) (#): ASL: 0, 0, 0, 2, 0, 1, 0; 1984)
control; 4 h/d, 5 d/week; acoustic duct tumour: 0, 0, 0, 3, 1, 2, 1; [BT8]b
untreated control 79 weeks c melanoma: 0, 1, 1, 0, 0, 1, 2, 1;
forestomach tumour: 3, 3, 4, 9, 17, 10, 10;
skin epithelial tumour: 3, 9, 3, 7, 3, 1, 7;
Hamster; Syrian 0 or 520; 6, 12, 1) survival ¡* (all exposure periods); Drew et al.
golden; 8-9 weeks, 18 mo, 2nd 2) see tumour incidences; (1983)
2nd experiment: 2, 8, experiment 6 or 3) tumour incidences in control and at different
14, 20 mo (6 mo 12 mo exposure at exposure periods: angiosarcoma (all sites): 0/143
exposure) or 2, 8, different age; (control), 13/88*, 4/52*, 2/103; mammary
14 mo (12 mo 6 h/d, 5 d/week; carcinoma: 0/143, 28/87*, 31/52*, 47/102*;
exposure); n.g. life span stomach adenoma: 5/138, 23/88*, 3/50*, 20/101*;
(only f, see number skin carcinoma: 0/133, 2/80; 9/48*, 3/90; 2nd
of hamster for experiment 6 mo exposure: angiosarcoma (all
evaluation); n.g. sites): 0/143 (control), 13/88*, 3/53*, 0/50,
0/52; mammary carcinoma: 0/143, 28/87*, 2/52*,
0/50, 1/52; stomach adenoma: 5/138, 23/88*,
15/53*, 6/49*, 0/52; skin carcinoma: 0/133, 2/80;
0/49, 0/46, 0/50; 2nd experiment 12 mo exposure:
angiosarcoma (all sites): 0/143 (control),
4/52*, 1/44, 0/43;
mammary carcinoma: 0/143, 31/52*, 6/44*, 0/42;
stomach adenoma: 5/138, 3/50*, 10/44*, 3/41;
skin carcinoma: 0/133, 2/80; 0/38, 0/30;
Table 32. (cont'd)
Species; strain; age Doses in mg/m3; 1) Effects other than tumours Reference
at start of exposure period; (dose in mg/m3);
experiment; initial frequency of treatment; 2) number of animals for
number of animals per post-exposure histopathological evaluation of
dose per exposure observation period neoplastic effects;
period; type of 3) type of tumour: number of animals
control; with this tumour/dose group (unless
otherwise given)
Rabbit; n.g.; n.g.; 0, 26 000; 1) n.g.; Caputo
40 exposed to VC, 15 mo; 4 h/d, 2) see tumour incidences; et al.
20 controls (no data 5 d/week; n.g. 3) tumour incidences: skin acanthoma: 0/20 (1974)
about sex); (control), 12/40*; lung adenocarcinoma 0/20, 6/40
treated control
a * = significant at P < 0.05; #: no statistical evaluation; f = females; m = males; mo = months; n.g. = not given
b Study number in experiments done by Maltoni and coworkers
c Animals were kept until spontaneous death or sacrificed at the end of given post exposure observation period
d Presumably significant in all exposed groups
Table 33. Summary of tumour types induced by long-term inhalation exposure to vinyl chloride
Lowest reported dose that significantly increased tumour incidences by tumour type (dose in mg/m3)
Species Liver Angiosarcoma Other Lung Mammary Nephro- Skin Neuro- Stomach Zymbal
angiosarcoma (other sites) liver carcinoma gland blastoma tumour blastoma tumour gland
tumours carcinoma tumour
Rat 130 in f c extrahepatic neoplastic 13 in 260 in 6500 in fore- 26 000
520 in m c 260 in f b,d nodules f h m b,g f b,k stomach in f + m f
lung 260 in f 650 in papilloma
2600 in f e b,d f g 78 000 in
hepato- f + m i
cellular
carcinoma
260 in
f d
Mouse 650 in 130 in m b,e 130 in 130 in
f + m e f b,d f b,d
all sites
130 in f b,d
Rabbit acanthoma
26 000,
n.d.
about sex
Hamster all sites 520 in b,j adenoma
520 in f b,d carcinoma 520 in
f b,d 520 in f f b,d
b,d
a Exposure in all studies 4-7 h/day, 5 days/week, exposure period at least 6 months
f = females; m = males; n.d. = no data
b The lowest dose tested with the study design described in the cited study
c Maltoni et al. (1981; BT9) f Maltoni et al. (1981; BT1) i Maltoni et al. (1981; BT6)
d Drew et al. (1983) g Maltoni et al. (1981; BT1, BT2) j Caputo et al. (1974)
e Lee et al. (1978) h Maltoni et al. (1981; BT15) k Maltoni & Cotti (1988)
7.7.3 The effect of age on susceptibility to tumour induction
Recently there has been some concern about early-life sensitivity
to vinyl chloride (Hiatt et al., 1994; Cogliano et al., 1996).
However, there is contradictory evidence (Drew et al., 1983 versus
Groth et al., 1981) concerning the effects of age on ASL induction in
rats, but final conclusions on increased susceptibility in 6- to
8-weeks-old animals could not be drawn from these data. The
discrepancy in results with rats is probably due to differences in
strain and/or experimental design. However, there is evidence from
other studies that there is possibly a higher sensitivity to liver
tumour induction in different rat strains in the first weeks of life,
a life-phase much earlier than that studied by Drew et al. (1983).
Studies on DNA adduct formation support these results.
F-344 rats, hamsters and mice (Swiss and B6C3F1) of different
age at the beginning of the exposure period (2, 8, 14 or 20 (not mice)
months old) were exposed to VC using the same experimental design and
same exposure period (Drew et al., 1983; see Table 32 for details).
For ASL in rats, angiosarcoma at all sites and mammary carcinoma
in all three species, neoplastic nodules in rats and lung carcinoma in
Swiss mice, the tumour response was highest in young animals
(2-month-old) exposed for 6 or 12 months. The validity of the
demonstrated age-related effects is limited in this study because only
statistical evaluation in comparison to control or to all other
groups, but not related to each other exposure group, was performed.
In addition, exposure later in life automatically shortens the period
of follow-up, and thus tends to lead to an apparent elevated
sensitivity at young age.
The effect of age on the susceptibility to induction of ASL in
Sprague-Dawley rats was also studied by Groth et al. (1981; see Table
30 for details). Rats aged 6, 18, 32 or 52 weeks were exposed to VC at
the same dose level and exposure period. In contrast to Drew et al.
(1983), the results of this study demonstrated that the older the rats
were at the start of the exposure period, the greater was the tumour
incidence. The maximum incidence was observed in male rats 52 weeks
old at first exposure and in females 32 weeks old at first exposure
(significantly increased compared to 6- or 18-week-old females). For
males the effect of age was statistically significant.
Exposure of 1-day-old rats of the same strain to high VC
concentrations for 5 weeks (BT14, see Table 30) revealed a remarkably
higher incidence of ASL, extrahepatic angiosarcoma and hepatoma
compared with 11-week-old rats exposed within the same experimental
design (BT10; Maltoni et al., 1981; see Table 30). However, these
experiments were not concurrent, and the tumour response in different
series from this laboratory has been variable (Tables 31, 32).
Maltoni & Cotti (1988) exposed 13-weeks-old pregnant
Sprague-Dawley rats and their progeny from the 12th day of gestation
to VC (Table 32). Although no statistical evaluation was performed it
seems that there was no significant difference between the dams and
the progeny concerning incidences in ASL, mammary carcinoma, and
neuroblastoma. The incidence of hepatocellular carcinoma, however, was
9% in dams and 42% in male and 60% in female progeny. In this study
the duration of exposure (and also of latency) was up to 69 weeks for
the progeny, but 56 weeks for the dams.
Laib et al. (1985a; see Table 30) presented evidence for
dose-related increased induction of ATPase-deficient liver foci in
Wistar and Sprague-Dawley rats, discussed as preneoplastic
hepatocellular lesions, after a 3-week exposure to low concentrations
(> 52 mg/m3). The increased induction of these foci was restricted
to a well-defined period of highest sensitivity beginning with rapid
liver growth in 7- to 21-day-old rats. No induction of these foci was
reported in adult rats exposed to 5200 mg/m3 for 70 days after
partial hepatectomy (no further details) (Laib et al., 1985b).
Comparative investigations on the alkylation of liver DNA in
young and adult Wistar rats exposed under the same exposure conditions
confirmed the age-related sensitivity of rats to VC (section 6.5.1 and
Table 26.)
7.7.4 The effect of gender on susceptibility to tumour induction
There is evidence that female rats of various strains are more
susceptible to liver tumour induction than males. Maltoni et al.
(1981, 1984) reported in all studies on Sprague-Dawley rats (BT1, BT2,
BT9, BT15; see Table 32) a higher incidence of ASL in female rats
compared with males after long-term inhalation. Similar results were
presented by Feron et al. (1979a,b) on Wistar rats, Lee et al. (1978)
on CD rats (Table 32) and Groth et al. (1981, Table 30) on
Sprague-Dawley rats. In inhalation experiments with pregnant
Sprague-Dawley rats, the investigators observed a higher incidence of
ASL and hepatocellular carcinoma in the female progeny than in the
males (Maltoni & Cotti, 1988; Table 32).
After long-term oral administration of VC (Table 29), incidences
of ASL (BT11, Sprague-Dawley rats; Maltoni et al., 1981), neoplastic
liver nodules and hepatocellular carcinoma (Wistar rats; Feron et al.,
1981 (not ASL) and Til et al., 1991) were higher in females than in
males. Interestingly, preneoplastic alterations in the liver, like
increased basophilic foci (Til et al., 1991; Table 29) or
ATPase-deficient foci (Laib et al., 1985a; Table 30), were observed in
female rats at lower doses than in males.
Although statistical analysis of sex differences was not
performed, in rats there is a tendency towards a higher susceptibility
to VC-induced ASL in female animals. Data on species other than rats
are not sufficient for an assessment of sex differences (Table 30,
32).
7.7.5 Carcinogenicity of metabolites
CAA was reported to induce hepatocellular tumours in B6C3F1 mice
when administered orally in drinking-water (Daniel et al., 1992). CEO
caused local tumours after repeated subcutaneous injection and skin
tumours in mice in classical initiation-promotion experiments (CEO
used as an initiator and 12-O- n-tetradecanoylphorbol-13-acetate as a
promoter), whereas CAA did not under comparable conditions (Zajdela et
al., 1980).
7.8 Genotoxicity
Genotoxicity studies on VC in vitro and in vivo in laboratory
animals are given in sections 7.8.1 and 7.8.2, respectively.
Genotoxicity studies on the metabolites of VC are described in section
7.8.3, and the mutagenic/promutagenic properties of DNA adducts formed
by the reactive VC metabolites CEO and CAA are discussed in section
7.8.4. Data on gene mutation and cytogenetic damage in humans exposed
to VC are given in section 8.4. A summary on the genotoxicity of VC
in vitro and in vivo, including human data, is presented in Table
36.
7.8.1 In vitro studies
Relevant studies on the genotoxicity of VC in vitro are
presented in Table 34. Genotoxic activity of VC has been detected in
several in vitro test systems, predominantly after metabolic
activation.
VC is mutagenic in the Ames test in the presence of metabolic
activation in Salmonella typhimurium strains TA100, TA1530 and
TA1535 but not in TA98, TA1537 and TA1538 (Rannug et al., 1974;
Bartsch et al., 1975; McCann et al., 1975; De Meester et al., 1980;
Shimada et al., 1985) indicating that the mutations are the result of
base-pair substitutions (transversion and transition) rather than
frameshift mutations. This is in agreement with the finding that
etheno-DNA adducts formed by the reactive metabolites CEO and CAA (see
section 6.5.1) are converted to actual mutations by base-pair
substitutions (see section 7.11.2 and 8.4). Barbin et al. (1997)
examined p53 mutations in VC-induced rat liver tumours and detected in
12 samples (11 ASL, 1 HCC) only one deletion but 12 base-pair
substitutions (transversion and transition).
In some studies, VC has been shown to also exert mutagenic
activity in S. typhimurium without addition of S9-mix (McCann et
al., 1975; De Meester et al., 1980; Shimada et al., 1985), but the
mutagenic effect was enhanced by addition of a metabolic activation
system (De Meester et al., 1980; Shimada et al., 1985; Victorin &
Ståhlberg, 1988). This increase was more pronounced when liver
extracts were derived from animals pretreated with an enzyme inducer
(Aroclor 1254) (De Meester et al., 1980). The reason for the mutagenic
Table 34. Genotoxicity of vinyl chloride in vitroa
Test type Test organism; Exposure conditions; Results Results Reference
species strain comments with MA without MA
Ames test Bact.; 20% VC in atmosphere; Rannug et al.
Salmonella up to 90 min + - (1974)
typhimurium - -
TA1535 - -
TA1536 - -
TA1537
TA1538
Ames test Bact.; 0.2, 2, 20% VC in atmosphere; n.g. Bartsch et al.
S. typhimurium 1.5-48 h; dose- and + (1975)
TA1530 time-dependent effect +
TA1535 -
TA1538 -
G-46
Ames test Bact.; 20% VC in atmosphere; McCann et al.
S. typhimurium 3, 6, 9 h; - - (1975)
TA98 time-dependent effect + +
TA100 + +
TA1535 - -
TA1538
Ames test Bact.; 1) 2-20% VC in De Meester et
S. typhimurium atmosphere; 16 h; + + al. (1980)
TA1530 dose-dependent effect
Ames test Bact.; 0.1-10% VC in atmosphere; Shimada et al.
S. typhimurium 18 h; - - (1985)
TA98 dose-dependent effect + +
TA100 + +
TA1535 - -
TA1537 - -
TA1538
Table 34. (cont'd)
Test type Test organism; Exposure conditions; Results Results Reference
species strain comments with MA without MA
Ames test Bact.; VC in DMSO added to soft Laumbach et
S. typhimurium agar and bacteria n.g. - al. (1977)
TA100
Ames test Bact.; 83 mM VC in liquid n.g. Bartsch et al.
S. typhimurium suspension; 30 min - (1975)
TA1530 -
TA1535 -
G-46
Gene Bact.; 10.6 mM VC in medium; + - Greim et al.
mutation Escherichia 2 h (1975)
assay coli
K12
Forward Yeast 16, 32, 48 mM VC in + - Loprieno et
mutation cells; medium; 1 h; al. (1977)
assay Schizo- dose-dependent effect
saccharomyces
pombe P1
Forward Yeast 16 or 48 mM VC in medium; + - Loprieno et
mutation cells; 5-240 min; time-dependent al. (1976)
assay S. pombe effect
SP.198
Table 34. (cont'd)
Test type Test organism; Exposure conditions; Results Results Reference
species strain comments with MA without MA
Reverse Yeast cells; incubated with 0.275 n.g. - Shahin (1976)
mutation Saccharomyces or 0.55% VC in DMSO;
assay cerevisiae 4-48 h
XV185-14C
Forward Fungi; VC solution in ethanol - - Drozdowicz &
mutation Neurospora for 3-4 h or 25, 50% Huang (1977)
assay crassa VC in atmosphere for
Ema 5297 3.5 or 24 h
Gene Plant; plant cutings exposed n.g. + Van't Hof &
mutation/ Tradescantia to VC in atmosphere; Schairer (1982)
deletion spec. 6 h; positive at
assay clone 4430 > 195 mg/m3
Cell mammalian 5, 10, 20, 30% VC in + - Drevon &
gene cells; atmosphere; 5 h; Kuroki (1979)
mutation Chinese dose-dependent effect
assay hamster
V79
HGPRT human cells; 25-400 µM VC in + n.g. Weisman (1992)
gene B-lymphoblastoid medium for 24 h;
mutation line cells with
assay metabolizing system
Gene yeast cells; 48 mM VC in incubation + - Loprieno et al.
conversion S. cerevisiae medium; 180-360 min (1976)
assay D4
Gene yeast cells; incubation in 0.275 n.g. - Shahin (1976)
conversion S. cerevisiae or 0.55% VC in DMSO;
assay D5 4-48 h
Table 34. (cont'd)
Test type Test organism; Exposure conditions; Results Results Reference
species strain comments with MA without MA
Gene yeast cells; 2.5% VC in atmosphere + - Eckardt
conversion S. cerevisiae for 1 h et al. (1981)
assay D7RAD
Cell mammalian 10, 20, 30, 40, 50% VC + n.g. Styles (1980)
trans- cells; in atmosphere; no data
formation BHK C1 13 about exposure period
assay
Cell mammalian 18, 180, 1315, 2662 n.g. + Tu et al. (1985)
trans- cells; mg/m3 VC in atmosphere;
formation BALB/c-3T3 24 h; dose-dependent
assay C1 1-13 effect
Rec-assay Bact.; initial 22 mM; 24 h n.g. - Elmore et al.
(DNA repair) B. subtilis (1976)
168M or MC-1
Unscheduled mammalian 5.0, 7.5 or 10% VC in + n.g. Shimada et
DNA cells; rat atmosphere; al. (1985)
synthesis hepatocytes 18 h; dose-dependent
effects
SCE assay human cells; 10, 25, 50, 75, 100% VC + - Anderson et
stimulated in atmosphere; 3 h; al. (1981)
lymphocytes dose-dependent effect
a Bact. = bacteria; DMSO = dimethylsulfoxide; MA = metabolic activation; n.g. = not given; SCE = sister-chromatid
exchange; + = positive; - = negative
activity in the Ames test in the absence of S9-mix has been suggested
to be a result of non-enzymic breakdown of VC or an internal bacterial
metabolism (Bartsch et al., 1976; Shimada et al., 1985), but the
origin of the direct mutagenic effect remains unclear.
Positive results in the Ames test were also observed with
metabolic activation by extracts prepared from human liver biopsies
(Bartsch et al., 1975). Mutagenicity of VC was dependent on
concentration (Bartsch et al., 1975; De Meester et al., 1980; Shimada
et al., 1985) and exposure duration (Bartsch et al., 1975; McCann et
al., 1975). VC was mutagenic when plates were exposed to a VC
atmosphere in a closed (Bartsch et al., 1975 and Table 34) or in a
dynamic flow-through system (Victorin & Ståhlberg, 1988). No mutagenic
effect was observed when VC was dissolved in aqueous solution with
(Bartsch et al., 1975) or without (Rannug et al., 1974; Laumbach et
al., 1977) metabolic activation, probably due to rapid loss of VC in
aqueous solution by evaporation (Bartsch et al., 1975).
Other gene mutation assays in bacteria (Greim et al., 1975),
yeast cells (Loprieno et al., 1976, 1977) and mammalian cells (Drevon
& Kuroki, 1979) revealed positive results exclusively in the presence
of metabolic activation. Mutagenic effects were also reported in a
human cell line containing cloned cytochrome P450IIE1, which is
capable of metabolizing VC (Weisman, 1992). Gene mutation was also
detected in plant cuttings (Tradescantia) exposed to VC (Van't Hof &
Schairer, 1982). No mutagenicity was observed in Neurospora crassa
with or without addition of exogenic activation system (Drozdowicz &
Huang, 1977) but the validity of this study is limited by
contradictory documentation.
In gene conversion assays, positive results were reported with
Saccharomyces cerevisiae in the presence of a metabolic activation
system (Loprieno et al., 1976; Eckardt et al., 1981). No mutagenic
effects were observed without metabolic activation (Shahin, 1976).
VC exposure induced unscheduled DNA synthesis in rat hepatocytes
(Shimada et al., 1985) and increased sister-chromatid exchange in
human lymphocytes after addition of an exogenic activation system
(Anderson et al., 1981). No growth inhibition was detected in DNA
repair-deficient bacteria without metabolic activation (Elmore et al.,
1976).
Cell transformation assays revealed positive results with
(Styles, 1980) or without (Tu et al., 1985) metabolic activation.
7.8.2 In vivo studies
Key studies on the genotoxicity of VC in vivo are documented in
Table 35. VC exposure induced gene mutation and mitotic recombination
in Drosophila melanogaster but not gene mutation in mammalian germ
cells. VC showed in vivo clastogenic effects, increased sister
chromatid exchanges and induced DNA breaks. VC induced also gene
conversion and forward mutations in host-mediated assays.
Mutagenic activity of VC was reported in the mitotic
recombination assay (Vogel & Nivard, 1993) and the sex-linked
recessive lethal (SLRL) assay (Magnusson & Ramel, 1976; Verburgt &
Vogel, 1977; Magnusson & Ramel, 1978) on D. melanogaster. The lowest
effective concentration in the SLRL assay was 2210 mg/m3 with a 2-day
exposure period and 78 mg/m3 after 17 days of exposure (Verburgt &
Vogel, 1977). In the same study, negative results were observed at
higher exposure concentrations with a 2-day exposure period in assays
on D. melanogaster for dominant lethals, translocations (not
tabulated) and sex chromosome loss. These results were discussed by
the authors as a consequence of a saturation effect observed in the
SLRL test (Verburgt & Vogel, 1977). However, sex chromosome loss was
observed in studies by Ballering et al. (1996) in D. melanogaster
exposed to higher concentrations.
No mutagenic activity was detected in the dominant lethal assay
with mice (Anderson et al., 1976; Himeno et al., 1983) and rats (Short
et al., 1977). No mutagenicity was reported in the mouse spot test
(Peter & Ungváry, 1980). Chromosomal aberrations in rats (Anderson &
Richardson, 1981) and hamsters (Basler & Röhrborn, 1980) were reported
and mouse bone marrow micronucleus tests (Jenssen & Ramel, 1980;
Richardson et al., 1983) gave positive results.
VC exposure (260, 650 and 1300 mg/m3) induced single-strand
breaks dose-dependently in the liver DNA of NMRI mice (Walles &
Holmberg, 1984; Walles et al., 1988). Increased frequencies of
sister-chromatid exchange and chromosome aberrations were observed in
the bone marrow of Chinese hamsters after exposure to 1.25, 2.5 or 5%
(v/v) for 24 h (Basler & Röhrborn, 1980).
7.8.3 Genotoxicity of VC metabolites
Metabolic activation of VC is necessary to form the genotoxic
metabolites. The metabolites themselves are genotoxic in the absence
of metabolic activation (see also section 6.3). The VC metabolites,
chloroethylene oxide (chloro-oxirane), chloroacetaldehyde, and
chloroacetic acid were investigated for genotoxicity. In vitro
studies discussed in this section were performed without metabolic
activation unless otherwise stated. Information on the mechanism of
mutagenesis is presented in section 6.5 and 7.11.2.
Chloroethylene oxide (CEO) was found to be the most effective VC
metabolite regarding forward mutation and gene conversion in yeast
(Loprieno et al., 1977), gene mutation in mammalian cells (Huberman et
al., 1975) and reverse mutation in bacteria (Malaveille et al., 1975;
Rannug et al., 1976; Hussain & Osterman-Golkar, 1976). The mutational
specificity of CEO was investigated in Escherichia coli, using trpA
mutant strains. In this system, CEO induced all types of base-pair
substitutions (except one, which was not tested) (Barbin et al.,
1985b). GC -> AT transitions were the most frequent, followed by
AT -> TA transversions. This metabolite inhibited growth in DNA
repair-deficient bacteria (Elmore et al., 1976; Laumbach et al.,
1977).
Table 35. Genotoxicity of vinyl chloride in vivoa
Species/Strain/Sex Test type Test conditions; comments Results References
Gene mutation
Mouse/CD-1/m dominant lethal 20 mice/group exposed to 0, 7800, - Anderson et
assay 26 000 or 78 000 mg/m3 for 6 h/d for al. (1976, 1977)
5 d before 8 wk mating; survival 100,
90, 95 and 45%
Rat/CD/m dominant lethal 12 mice/group exposed to 0, 130, 650, - Short et al.
assay or 2600 mg/m3 for 6 h/d, 5 d/wk; one (1977)
mating during week 11 of exposure;
no mortality
Mouse/CD-1/m dominant lethal a) 13 m exposed to 26 000 mg/m3 for - Himeno et al.
assay 4 h/d for 5 d (11 controls) before 7 wk (1983)
mating; b) 20 m exposed 4 h/d, 5 d/wk
to 13 000 mg/m3 for 10 wk before 3 wk
mating
Mouse/C57BL/f mouse spot test 44 pregnant mice exposed to 12 000 mg/m3 - Peter &
for 5 h on gestation day 10 (51 controls) Ungváry (1980)
Mouse/Swiss/n.d. host-mediated 4-6 mice exposed orally to 700 mg/kg bw. + Loprieno et al.
forward mutation in olive oil; yeast cells (S. pombe (1976)
assay SP.198) inoculated in peritoneum for
3, 6 or 12 h
D. melanogaster/ dominant lethal m exposed to 0 or 78 000 mg/m3 for 2 d; - Verburgt &
Berlin K/m assay total number of eggs per group at Vogel (1977)
least 6950; increase not significant; no
differences in hatchability (> 80%)
D. melanogaster/ Drosophila a) m exposed to 0, 1, 10, 20% VC in air + Magnusson &
Karsnäs/m SLRL test for 3 h (at least 491 chromosomes tested); Ramel (1978)
b) m exposed to 0, 1, or 10% VC for 3 h
after pretreatment with 1% phenobarbiturate
solution for 24 h;
a) positive at > 1% VC; no dose dependency;
pretreatment in b) increased mutagenicity
D. melanogaster/ Drosophila 50 m/group exposed continuously a) to 0, + Verburgt &
Berlin K/m SLRL test 78, 520, 2210, 26 000, 78 000, 130 000 mg/m3 Vogel (1977)
for 2 d or b) to 0, 78, 2210 mg/m3 for 17 d;
a) positive at > 2210 mg/m3, no clear dose
response;
b) positive at > 78 mg/m3
Mitotic
recombination
D. melanogaster/ mitotic eye mosaic assay; 48- to 72-h-old larvae + Vogel &
LS/f + m recombination exposed to 5200 mg/m3 for 17 h; light spots Nivard (1993)
assay of at least 500 eyes scored in adult f
(control 250 eyes scored); survival not
reduced
Chromosomal
abnormalities
Rat/Wistar/m cytogenetic 24 m/group exposed to 3900 mg/m3 for + Anderson &
assay a) 5 d (6 h/d) or b) 3 mo (6 h/d, 5 d/wk); Richardson
bone marrow sampled 24 h after exposure (1981)
period; increased number of cells with any
abnormality, significant in a)
Hamster/Chinese/ cytogenetic 2 m + 2 f per group exposed to 2.5% for 6, + Basler &
f+ m assay 12, or 24 h; 5 m + 5 f exposed to 5% VC Röhrborn
for 24 h; bone marrow samples prepared 26 h (1980)
after start of exposure; control 7 m + 7 f;
increased aberrations at > 6 h (gaps
excluded), effect dose related
Table 35. (cont'd)
Species/Strain/Sex Test type Test conditions; comments Results References
Mouse/CBA/m micronucleus 3 m exposed to 0 or 5% VC for 4 h; bone + Jenssen &
assay marrow examined 30 h after exposure Ramel (1980)
Mouse micronucleus 0, 260, 860 or 2600 mg/m3 for 2 × 4 h + Rodics et al.
CFLP assay (1981)
Mouse/ micronucleus 10 f and 10 m exposed to 0 or 130 000 mg/m3 + Richardson
C57BI/6J/f + m assay for 6 h; bone marrow examined 24 or 48 h et al. (1983)
after exposure
D. melanogaster/ sex m exposed to 0 or 78 000 mg/m3 for 2 d and - Verburgt &
Berlin K/m chromosome chromosomes analysed in progeny (at least Vogel (1977)
loss 6725 m + f per group)
D. melanogaster/ sex m exposed to 0 or 126 000 mg/m3 for 48 h; + Ballering et
ring-X/m chromosome chromosome loss determined in F1 (at al. (1996)
loss least n = 428; 3 broods)
Other effects
Rat/Wistar/m host mediated 20-30 rats exposed to 0 or 1% VC for 24 + Eckardt et al.
gene conversion h, starting 1 h after yeast cell (1981)
assay (S. cerevisiae D7RAD) injection (i.v.)
Mouse/NMRI/f alkaline elution 3-5 f per group; exposure to 1300 mg/m3 + Walles &
assay in liver for 39, 60, 117, 234 h (6 h/d, 5 d/ wk) Holmberg
DNA and sacrificed a) 2 h or b) 18 h (exposed (1984)
for 36, 114, 231 h) after exposure period;
concurrent control sacrificed after 36 or
231 h; positive in a) at 39 h, in b) at 114 h
Table 35. (cont'd)
Species/Strain/Sex Test type Test conditions; comments Results References
Hamster/Chinese SCE assay 2 m + 2 f per group exposed to 1.25 or 2.5% + Basler &
f + m VC for 6, 12, or 24 h; bone marrow samples Röhrborn
prepared 26 h after start of exposure; (1980)
control 4 m + 4 f; dose- and time-dependent
effect
a d = day; f = females; m = males; mo = months; n.d. = no data; SCE = sister-chromatid exchange;
SLRL = sex-linked recessive lethals; wk = weeks; + = positive; - = negative
CAA was 450 times less mutagenic than CEO in the Ames test but
more active than the concurrent positive control ethylene oxide
(Rannug et al., 1976). Positive results were reported with CAA in gene
mutation assays in bacteria (Malaveille et al., 1975; Bartsch et al.,
1975; McCann et al., 1975; Hussain & Osterman-Golkar, 1976; Elmore et
al., 1976; Laumbach et al., 1977; Perrard, 1985), yeast cells
(Loprieno et al., 1977), mammalian cells (Huberman et al., 1975), in a
human lymphoblast cell line (Sanchez & Recio, 1991) and in human cells
using shuttle vectors (Matsuda et al., 1995).
With chloroacetic acid no enhancement of the mutation frequency
could be detected in bacteria (Bartsch et al., 1975; Malaveille et
al., 1975; Rannug et al., 1976) or mammalian cells (Huberman et al.,
1975).
Evidence for mutagenic activity of photoreaction products formed
from VC was presented by Victorin & Ståhlberg (1991). Mixtures of VC
(up to 260 mg/m3) and nitrogen dioxide (but not VC alone) were
mutagenic in S. typhimurium TA100 after 40 min UV irradiation of the
gas mixture before exposure of bacteria.
CEO but not CAA showed a similar toxicity/mutagenic profile to VC
in the hprt locus in a metabolically competent human
B-lymphoblastoid cell line (Chiang et al., 1997; see also section
8.4.2).
7.8.4 Other toxic effects of VC metabolites
In the above studies on the genotoxic effects of CAA (see section
7.8.3), this compound appeared to be highly cytotoxic in various
cellular systems. CAA has also a high acute toxicity in animals, with
a LD50 value of 0.15 mmol/kg body weight. Kandala et al. (1990)
showed in vitro the concentration-dependant reversible inhibition of
DNA synthesis by CAA in rat and mouse cells without a reduction in
thymidine uptake or formation of nucleotides. In isolated rat
hepatocytes, CAA stimulates lipid peroxidation (Sood & O'Brien, 1993).
7.8.5 Mutagenic and promutagenic properties of DNA adducts formed by
VC metabolites
The major DNA adduct of VC, 7-(2'-oxoethyl)guanine (7-OEG), lacks
miscoding properties (Barbin et al., 1985a). In contrast,
1, N6-ethenoadenine (Epsilon A), 3, N4-ethenocytosine (Epsilon C)
and N2,3-ethenoguanine (Epsilon G) showed miscoding properties
(Singer, 1996; see Table 37). The promutagenic properties of the
etheno adducts involve mainly base-pair substitution mutations
(Grollman & Shibutani, 1994). Site-specific mutagenesis studies in E.
coli and in mammalian cell lines have shown that both Epsilon G and
Epsilon C can induce G:C -> A:T transitions; Epsilon C can also lead
to C:G -> A:T transversions (Cheng et al., 1991; Moriya et al.,
1994). Epsilon A can induce misincorporation of G, C, or A during
replication, thus inducing the base-pair substitutions A:T -> C:G,
A:T -> G:C or A:T -> TA (Basu et al., 1993; Pandya & Moriya, 1996).
7.8.6 Mutations in VC-induced tumours
Barbin et al. (1997) examined the presence of p53 gene
mutations (the function of the p53 gene is described in
section 8.4.2) in ASL and HCC tumours induced by VC in Sprague-Dawley
rats. Mutations were found in 11/25 ASL and 1/8 HCC. A twelve-base
deletion was found in one tumour; all others were base-pair
substitutions. Nine of the point mutations were observed at A:T base
pairs and of three G:C -> A:T transitions (Table 43).
Mutations of the p53 gene were also found in tumours from vinyl
chloride-exposed autoclave workers with liver angiosarcoma (ASL) and
hepatocellular carcinoma (HCC) (Hollstein et al., 1994; Boivin et al.,
1997; see also section 8.4 and Table 43). To date (1998) 11 out of 15
(73%) ASL from VC-exposed workers have been shown
immunohistochemically to have mutant p53 protein. Furthermore, a
statistically significant trend for mutant p53 protein has been found
in the serum of VC-exposed workers (Smith et al., 1998). In contrary
to studies in humans, no mutations were found in codons 12, 13 and 61
of the Ki- ras gene in rat liver tumours induced by VC, but mutations
were found involving codon 61 of the Ha- ras proto-oncogene (Froment
et al., 1994; Boivin-Angèle et al., in press) (see Table 44 and
section 8.4.2).
Connexin genes have been shown to restore normal cell growth when
transfected into certain tumorigenic cells and thus are considered to
form a family of tumour suppressor genes. Mutations of the connexin
37 (Cx37) gene in rat liver tumours (22 hepaticangiosarcomas and 3
hepatocellular carcinomas) induced by VC were analysed by
PCR-single-strand conformation polymorphism analysis and DNA
sequencing. The results suggested that Cx37-mediated gap junctional
intercellular communication may be disturbed in most of these
angiosarcomas but mutation of the Cx37 gene is rare (Saito et al.,
1997).
7.9 Factors modifying toxicity
Concurrent administration of ethanol (5% in drinking-water until
termination at month 30) and VC (1560 mg/m3, inhalation 4 h/day,
5 days/week for 1 year) to male Sprague-Dawley rats (80 rats per group
for histopathological evaluation) resulted in increased incidences of
ASL from 23% after exposure to VC alone to 50% in rats exposed to VC
and ethanol versus 0% in ethanol-treated rats and 0% in concurrent
controls. Ethanol had an additive effect on the incidence of
hepatocellular carcinoma (VC 43%, VC-ethanol 60%, ethanol 10%, control
1.3%) and lymphosarcoma (VC 7.5%, VC-ethanol 14%, ethanol 5%, control
2.5%) (Radike et al., 1981). This effect may be due to the interaction
of ethanol with VC metabolism.
Induction of certain enzymes of the mixed-function oxidase system
by pretreatment with phenobarbital (Jaeger et al., 1974, 1977;
Reynolds et al., 1975a,b,c) or the mixture of polychlorinated
Table 36. Summary of genotoxic effects induced by exposure to vinyl chloride in vitro and in vivoa
In vitro In vivo
Bacteria Yeast Plants Mammalian Human Insects Mammalia Humansc
GM DD GM GC GM GM DD CT GM SCE GM AN MR GM CA MN DD SCE GM CA MN SCE
+ -b + + + + + + + + + - + - + + + + + + + +
a AN = aneuploidy; CA = chromosomal aberration; CT = cell transformation; GM = gene mutation; GC = gene conversion;
DD = DNA damage; MN = micronuclei; MR = mitotic recombination; SCE = sister-chromatid exchange
b Single study, only tested without metabolic activation
c These results are described in Table 42 (section 8.4.1)
Table 37. Evidence for base-pair substitutions caused by etheno-DNA adducts
Ethenobase Base Base-pair System used Reference
incorporated substitution for study
opposite
adducta
1,N6-ethenoadenine C AT -> GC in vivo bacteriophage M13-Nhei in E. coli Basu et al.
(epsilon-A) transition (1993)
A AT -> TA in vivo single-strand vector shuttle in Pandya & Moriya
C AT -> CG E. coli or simian kidney (COS) cells; (1996)
transversions
3,N4-ethenocytosine A CG -> TA in vivo single-strand vector shuttle in Moriya et al.
(epsilon-C) transition E. coli or simian kidney (1994)
T CG -> AT in vitro (COS) cells; Zhang et al.
transversion E. coli DNA polymerase (1995)
I system
in vivo M13AB28 in SOS-(UV)-induced Jacobsen &
E. coli Humayun (1990)
in vivo M13 glyU phage transfection Borys et al.
of E. coli tester strain (1994)
in vitro E. coli DNA polymerase I system Simha et al.
(1991);
Palejwala et al.
(1991)
N2,3-ethenoguanine T GC -> AT in vivo E. coli DNA synthesis by M13G*1 Cheng et al. (1991)
(epsilon-G) transition assay
in vitro E. coli DNA polymerase I (Klenow Singer et al. (1991)
fragment); exonuclease-free Klenow;
Drosophila melanogaster polymerase
alpha-primer compex;
human immunodeficient virus-I
reverse transcriptase (HIV-RT)
in vitro E. coli DNA-dependent RNA Mroczkowska &
polymerase Kusmierek (1991)
Table 37. (cont'd)
Ethenobase Base Base-pair System used Reference
incorporated substitution for study
opposite
adducta
1,N2-ethenoguanine A GC -> TA in vitro E. coli DNA polymerase I Langouet et al.
G GC -> CG (exonuclease-free Klenow) (1997)
transversions
a A = adenine; C = cytosine; G = guanine; T = thymine
biphenyls (Arochlor 1254) (Reynolds et al., 1975a,b; Conolly et al.,
1978) enhanced acute hepatotoxicity in rats as measured by increased
activity of hepatic enzymes and/or focal hepatic necrosis.
Administration of SKF-525A, an inhibitor of the mixed-function oxidase
system, 30 min prior to VC exposure in phenobarbital-pretreated rats,
inhibited the enhancing effect of phenobarbital on VC hepatotoxicity
(Jaeger et al., 1977). Application of cysteine, a rate-limiting
precursor in hepatic glutathione (detoxification of reactive
chemicals), via drinking-water prior to VC exposure protected Arochlor
1254-pretreated rats against acute VC hepatotoxicity (Conolly &
Jaeger, 1979).
7.10 Mechanisms of toxicity - mode of action
7.10.1 Mechanisms of VC disease
Based on evidence of immunological abnormalities, such as
hyperimmunoglobinaemia and circulating immune complexes in workers
with "vinyl chloride disease" (section 8.3), Ward et al. (1976) have
proposed a possible mechanism for the vascular changes associated with
this disease. Reactive VC metabolite(s) bind(s) to a protein,
resulting in a structurally abnormal protein. This protein would react
as an antigen and initiate an immune response with B-cell
proliferation and hyperimmunoglobinaemia. Circulating immune complexes
formed by interaction of this antigen and antibodies would precipitate
in the extremities of exposed humans in response to the cold and
activate the complement sequence. The cryoprecipitates and reactions
secondary to the complement activation are proposed to produce
vascular occlusion and fibrinogen/fibrin conversion. The mechanism is
supported by findings of IgG deposition with associated complement C3
and fibrin in histological lesions (Grainger et al., 1980). The
resulting vascular insufficiency would explain the observed clinical,
radiological and histological findings in skin, skeletal and soft
tissues, and lungs (section 8.3) in workers occupationally exposed to
high concentrations of VC (Ward et al., 1976). No further studies were
identified that would directly confirm this mechanism, and the degree
to which it has been accepted is not clear. The available evidence
does not seem sufficient to suggest an autoimmune disease as a
pathogenetic mechanism. Further studies will be needed to establish
the true significance of the possibly transient immunological
abnormalities in VC-induced disorders.
7.10.2 Mechanism of carcinogenesis
There is a large body of data showing that VC acts as a genotoxic
carcinogen. After metabolic activation to CEO by CYP2E1, VC exerts
various genotoxic effects (including gene mutations and chromosomal
aberrations) in different organisms, including bacteria, yeasts,
mammalian cells in culture, Drosophila, rodents and humans (Table 36).
Among the mutagenic events induced by VC, base-pair substitutions
appear, so far, to be the most frequent. VC in the presence of an
activation system has a transforming activity on mammalian (rodent)
cells in culture (see Table 34).
Studies in vitro have demonstrated that metabolically activated
VC and its electrophilic metabolites CEO and CAA can alkylate nucleic
acid bases. 7-OEG, the major DNA adduct formed by VC and CEO does not
exhibit promutagenic properties. In contrast, four minor adducts,
Epsilon A, Epsilon C, N2,3-Epsilon G and 1,N2-Epsilon G, show
promutagenic properties, inducing mainly base-pair substitution
mutations and a low level of frameshift mutations.
7-OEG and three etheno adducts (Epsilon A, Epsilon C,
N2,3-Epsilon G) have been detected in DNA from rats and mice exposed
to VC. Highly variable background levels of Epsilon A and Epsilon C
were found in all the tissues examined. Following exposure of rats to
VC, significantly elevated levels of Epsilon A and Epsilon C were
measured in most tissues, except the brain; there was also no
significant increase of Epsilon A levels in the kidney and spleen.
The liver is one of the primary targets for VC-induced
carcinogenesis in rats and humans. It is also, by far, the major
tissue involved in VC activation in rats. Following exposure of rats
to VC, the distribution of etheno adduct levels (induced by the
exposure) is rather homogeneous within the organism. Epsilon C was
shown to accumulate as a function of length of exposure in at least
three organs (liver, kidney and lung). In contrast, Epsilon A
accumulated in the liver but not in the kidney and lung. In addition,
adduct levels (Epsilon A, Epsilon C) did not decrease in the liver,
for at least two months following the end of exposure. Etheno adducts
are formed as endogenous background levels in various tissues in
humans; no data are available on etheno adduct levels in humans
exposed to VC.
Mutations have been found in liver tumours associated with
exposure to VC. In human ASL, the Ki-ras gene is activated through a
GC -> AT mutation at base 2 of codon 13. Mutations, all AT -> TA
transversions, have been described in the p53 gene in three human
ASL. The Ki-ras gene activation is not found in rat ASL. However, 44%
of rat ASL were found to contain a mutated p53 gene: most mutations
were base-pair substitutions, involving mainly A:T base pairs. The
data suggest the existence of hot spots for mutations in the p53
gene, and one mutation found in two rat ASL was equivalent to the
same mutation characterized in one human ASL associated with VC
exposure. The Ha-ras gene is activated in rat HCC induced by VC,
through an AT -> TA transversion in codon 61.
The mutation spectra observed in liver tumours (ASL and/or HCC)
associated with VC exposure in humans and rats are clearly distinct
from those observed in sporadic liver tumours or in hepatic tumours
associated with other exposures. In rats, the substitution mutations
found at A:T base pairs in the ras and p53 genes are consistent
with the promutagenic properties of Epsilon A and with the
accumulation and persistence of this lesion in hepatic DNA.
Altogether, available data suggest that etheno adducts could be
involved in the initiation of hepatocarcinogenesis by VC. However,
they cannot explain the observed tissue- and cell-specificity.
More studies on the formation and repair of etheno adducts at the
cellular level (cell specificity), as well as at the molecular (gene
and DNA sequence) level, are warranted. Carcinogenesis is a multi-step
process and, obviously, there is a need for quantitative evaluation of
other critical biological end-points, such as effects of VC on
apoptosis, cell proliferation or intercellular communication in
vivo.
8. EFFECTS ON HUMANS
Only reports on effects for humans of exposure to VC have been
considered here and not reports where exposure has been to a number of
chemicals, e.g., at landfills, in factories manufacturing a number of
chemicals or in the PVC processing industry.
8.1 General population exposure
After an accident in Schönebeck, Germany in June 1996, involving
the derailment of a train carrying VC and subsequent fire, 325 persons
were documented as having acute symptoms but these correlated with
exposure to the pyrolytic products (e.g., HCl) and not to VC itself.
But a study on 29 persons exposed as a result of this accident showed
a significant increase in chromosomal aberrations compared to an
unexposed control group (Hüttner & Nikolova, 1998; see also Table 42
and section 3.2.3).
A case has been reported of epithelioid haemangioendothelioma
involving liver, bone and lungs in a man living for over 8 years
several hundred metres from a toxic waste dump next to a chemical
plant producing VC (Shin et al., 1991).
There have been several reports on the possible increased
prevalence of congenital malformations in populations exposed to
emissions from polymerization facilities (Edmonds et al., 1975, 1978;
Infante, 1976; Thériault et al., 1983; Rosenman et al., 1989) but none
of these studies showed a statistically significant correlation
between developmental toxicity and proximity to the facility
(Clemmesen, 1982; Hemminki & Vineis, 1985). A number of studies (e.g.,
Goldberg et al., 1995; Dolk et al., 1998) examined risk for cancer and
for adverse reproductive outcome in relation to proximity to
landfills. Although VC is one of the potential emissions from the
landfills, these studies do not directly address the population
exposure to VC and were not further considered.
In England and Wales, from 1975-1987 data, there were no
confirmed non-occupationally exposed cases of ASL (Elliott &
Kleinschmidt, 1997). In the USA, five non-occupational cases were
reported living within 1.6 km of a VC plant (Brady et al., 1977).
8.2 Controlled human studies
Three men and three women were exposed twice daily with a 6-h
interval for three successive days to 0, 0.4, 0.8, 1.2, 1.6 or 2.0%
VC. The NOEL was between 0.8 and 1.2%. Above this, dizziness, nausea,
dulling of vision and auditory cues were reported (Lester et al.,
1963).
Thirteen male volunteers were exposed to 130, 650 and 1300 mg/m3
for 7.5 h and subjective and neurological responses were measured
before the subject entered the chamber, 15 min after entrance and at
1-h intervals thereafter; 24-h post-exposure urine and blood samples
were taken and tested. No significant adverse effects were noted with
the exception of some dryness of eyes and nose at 1300 mg/m3. The
exposure had no noticeable effect on neurological responses nor did it
produce significant changes in the results of mental, coordination or
manual dexterity tests conducted during the exposure period. All
clinical laboratory studies performed in the post-exposure period were
normal and not significantly different from pre-exposure values
(Baretta et al., 1969).
8.3 Occupational exposure
8.3.1 Overview
VC was first produced commercially in the late 1920s. Various
effects caused by exposure to VC were reported: cardiac arrhythmia in
experimental animals (Oster et al., 1947); hepatic abnormalities in VC
workers (Tribukh et al., 1949; Filatova et al., 1958), acroosteolysis,
Raynaud-type-phenomenon and sclerodermoid skin lesions (Lelbach &
Marsteller, 1981). But it was not until it was found that VC could
cause cancer in animals (Viola et al., 1971; Maltoni et al., 1974) and
humans (Creech & Johnson, 1974) that levels of VC in the workplace
were drastically reduced. Some VC workers, in particular autoclave
cleaners, were estimated to have been exposed to as much as
2600 mg/m3 (1000 ppm) in the 1950s or earlier, reducing to a tenth of
this level by the mid-1970s (Table 20). After 1975 levels were usually
2.6-13 mg/m3 (1-5 ppm) in many countries, but in some countries where
production plants were not modernized, workers were or are exposed to
high levels of VC (Fucic et al., 1990a; Gáliková et al., 1994; Hozo et
al., 1996, 1997; see also Table 21).
It should be noted that the non-neoplastic and neoplastic effects
described in the following sections are due in most cases to the high
exposure of workers to VC before 1974. No cases of ASL (section 8.3.1)
have been reported to the International Register of Cases among (West)
European workers first exposed after 1972 (Storm & Rozman, 1997). This
is not the case for Eastern European countries where the old
regulation of 194 mg/m3 (75 ppm) in the working environment was valid
until recently (Hozo et al., 1997).
8.3.2 Non-neoplastic effects
8.3.2.1 Acute toxicity
In acute VC intoxication, the symptoms described include vertigo,
nausea and headache. At higher concentrations, VC exerts narcotic
effects and at one time was considered as a possible anaesthetic
(Patty et al., 1930; Peoples & Leake, 1933; Oster et al., 1947).
VC caused almost immediate death of VC polymerization workers in
two incidents of accidental poisoning. No values were given but the
strong smell and the narcotic effects of VC were reported (Danziger,
1960).
Concentrations of VC of the order of 26 000 mg/m3 (1%) in the
air induce unconsciousness and cardiac arrhythmia. Exposure of two
workers to 2.5% VC for 3 min caused dizziness, disorientation and a
burning sensation in the soles of the feet. There was complete
recovery except for a slight headache lasting 30 min (Danziger, 1960).
A man whose hands were accidentally sprayed with VC developed
erythema and some second-degree burns which healed without
complication (Harris, 1953). A patient complaining of eye burns from
VC recovered 48 h after the eye was rinsed for 15 min with saline
(McLaughlin, 1946).
8.3.2.2 Effects of short- and long-term exposure
Concentrations of VC in the region of 2590 mg/m3 (1000 ppm),
which were not unusual prior to 1974, over periods ranging from
1 month to several years, have been reported to cause a specific
pathological syndrome found in VC workers called the "vinyl chloride
illness". Symptoms described were earache and headache, dizziness,
unclear vision, fatigue and lack of appetite, nausea, sleeplessness,
breathlessness, stomachache, pain in the liver/spleen area, pain and
tingling sensation in the arms/legs, cold sensation at the
extremities, loss of libido and weight loss (Thiess & Versen, 1974).
Clinical findings included scleroderma-like changes in the fingers
with subsequent bony changes in the tips of the fingers described as
acroosteolysis, peripheral circulatory changes similar to Raynaud's
disease, and enlargement of the liver and spleen with a specific
histological appearance, and respiratory manifestations (Lange et al.,
1974; Suciu et al., 1975; Veltmann et al., 1975; Lelbach & Marsteller,
1981).
8.3.2.3 Organ effects
a) Skin and skeletal tissues
Occupational acroosteolysis has largely affected the most highly
exposed workers involved in scraping the insides of autoclaves in the
PVC production process (Harris & Adams, 1967; section 3). It is a rare
bone disease resulting in de-calcification of the terminal phalanges
of the hands and other extremities (Cordier et al., 1966; Wilson et
al., 1967; Markowitz et al., 1972). In these cases, acroosteolysis was
often preceded by soreness and tenderness, numbness, pallor and
cyanosis of the extremities especially the hands, a Raynaud-type
phenomenon caused by reversible constriction of the arterioles.
Sclerodermoid-like changes appear in the skin of the hands and
forearms and osteolytic and sclerotic lesions of the bones,
particularly the extremities and sacroiliac joints (Lange et al.,
1974). Most of the abnormalities disappeared and bones showed signs of
healing a year or two after the men had stopped work (Harris & Adams,
1967). A chronothermodynamic study of Raynaud's phenomenon secondary
to past exposure to VC showed that, although reduced, the symptoms
were still present after 8 years (Fontana et al., 1996).
A review of five studies from four countries involving 725
workers at risk from VC exposure showed that 3% developed
acroosteolysis; 10% Raynaud-type phenomenon and 6% sclerodermoid skin
lesions (Lelbach & Marsteller, 1981). Genetic susceptibility has been
suggested as a possible reason that not all workers who had been in
contact with VC developed symptoms (Black et al., 1983, 1986). The
scleroderma-like syndrome induced by VC exposure appears to be
different from that of other (systemic) scleroderma (Ostlere et al.,
1992). It has a shorter incubation period (1 month to 3 years compared
to 4 to 44 years) (Ishikawa et al., 1995) and there are immunological
differences (section 8.3.2.3f).
b) Hepatic effects
Exposure to VC is associated with hepatomegaly and/or
splenomegaly and with various histological lesions in the liver (Lange
et al., 1974; Ho et al., 1991). In one carefully described study,
advanced portal hypertension with histological findings of
non-cirrhotic fibrosis was diagnosed in 17 of 180 VC polymerization
workers (Lelbach & Marsteller, 1981).
Focal hepatocellular hyperplasia and focal mixed hyperplasia
(hyperplasia of sinusoidal cells along with hyperplasia of
hepatocytes) are early histological alterations indicative of VC
exposure (Tamburro et al., 1984). The precursor stage of ASL is
characterized by subcapsular fibrosis, progressive portal fibrosis and
a borderline increase of intralobular connective tissue, all
associated with focal stimulation and proliferation of sinusoidal
lining cells and hepatocytes. Transition to angiosarcoma is preceded
by focal dilatation of sinusoids with enlarged dedifferentiated lining
cells often containing peg-like hyperchromatic polymorphic nuclei
(Popper & Thomas, 1975; Gedigk et al., 1975).
There is a great similarity between the histological sequences in
the liver of rodents exposed by inhalation to VC (chapter 7; Spit et
al., 1981) and the lesions observed in VC-exposed workers (Popper et
al., 1981).
Twenty of the 39 "liver disease deaths" reported in an Italian
study of highly exposed PVC workers were due to liver cirrhosis, the
remainder being due to miscellaneous liver disorders. A statistical
evaluation was not possible (Pirastu et al., 1990). In another study,
two of 21 heavily exposed VC workers died from sequelae to
noncirrhotic portal fibrosis and portal hypertension (Lelbach, 1996).
In the USA and European cohort studies of VC-exposed workers (study
descriptions are given in section 8.3.3), deaths from chronic
non-malignant diseases of the liver had a low SMRs of 62 and 88; in
the European study this was significantly less than expected (Wong et
al., 1991; Simonato et al., 1991).
c) Cardiovascular effects
Some old studies reported statistically non-significant increased
mortality due to cardiovascular diseases among workers exposed to VC
(Ebihara, 1982; Greiser et al., 1982).
Laplanche et al. (1992) report an elevated incidence of
circulatory system diseases other than Raynaud's disease (RR 1.4, 95%
CI 1.0-1.8) in a 7-year follow-up of a cohort of 1100 VC workers. The
increased risk was mainly due to hypertension and "other circulatory
disorders". Likewise, another study with a 5-year follow-up on the
incidence of arterial hypertension (AH) and coronary heart disease
(CHD) in 105 VC and PVC workers exposed to VC at between 4 and
1036 mg/m3 showed that exposed workers had significantly higher blood
pressure than controls. The estimated relative risk (RR) for AH in
exposed workers was twice as high as in the controls, while there was
no significant difference regarding CHD. There was an
exposure-response relationship between the intensity of exposure and
the incidence of AH (Kotseva, 1996).
In contrast, both the large cohort studies reported a
statistically significant deficit in the mortality from cardiovascular
diseases, with SMRs of 87 and 81. In the USA study, the mortality was
even lower for the subcategory of arteriosclerotic heart disease (SMR
74.5, 95% CI 81.6-89.2 (Simonato et al., 1991; Wong et al., 1991). In
the Canadian study (Thériault & Allard, 1981), there was a
statistically non-significant 20% deficit in cardiovascular mortality,
based on 25 exposed cases.
d) Respiratory effects
Adverse respiratory effects reported in older case studies
included increased incidence of emphysema (Suciu et al., 1975),
decreased respiratory volume and vital capacity, respiratory
insufficiency (Suciu et al., 1975), decreased respiratory oxygen and
carbon dioxide transfer (Lloyd et al., 1984), pulmonary fibrosis of
the linear type (Suciu et al., 1975), abnormal chest X-rays (Lilis et
al., 1975) and dyspnoea (Walker, 1976). This is probably due in part
to confounding by smoking and presence of PVC-resin dust, which is
known to cause respiratory lesions (Mastrangelo et al., 1979; Lilis,
1981).
Both large cohorts (Wong et al., 1991; Simonato et al., 1991)
found a deficit in the mortality from non-malignant respiratory
diseases (SMR 81.6, and 77, respectively). Despite overall deficit in
mortality from respiratory diseases, Wong et al. (1991) found an
excess of emphysema/chronic obstructive pulmonary disease (COPD)
mortality, which, however, was highest among workers with a duration
of exposure less than 10 years.
e) Neurotoxicity
In patients with chronic occupational exposure, neurological
disturbances include sensory-motor polyneuropathy (Perticoni et al.,
1986; Podoll et al., 1990), trigeminal sensory neuropathy, slight
pyramidal signs and cerebellar and extrapyramidal motor disorders
(Langauer-Lewowicka et al., 1983). Psychiatric disturbances included
neurasthenic or depressive syndromes (Penin et al., 1975).
Sleeplessness (Gnesina et al., 1978; Gnesina & Pshenitsina, 1980;
Langauer-Lewowicka et al., 1983; Gnesina & Teklina, 1984) and loss of
sexual functions (see below) were frequently encountered. Pathological
EEG alterations were found in a high proportion of patients (Penin et
al., 1975; Stblová et al., 1981).
f) Immunotoxicity
The major immunological abnormalities reported in VC disease
patients include hyperimmunoglobulinaemia with a polyclonal increase
in IgG, cryoglobulinaemia, cryofibrinogenaemia, and in vivo
activation of complement (Ward et al., 1976). Immunofluorescent
examinations of skin and lung biopsies have demonstrated the
deposition of IgG with associated complements C3 and fibrin in
relation to the histological lesions described in small blood vessels
(Grainger et al., 1980). A statistically significant increase in
circulating immune complexes was observed in workers exposed to VC,
compared to unexposed workers (Ward et al, 1976) . The increase in
circulating immune complexes was greatest in women and in those with
duties involving exposure to relatively higher levels of VC
(Bogdanikowa & Zawilska, 1984).
Bencko et al. (1988) found significantly elevated IgA, IgG and
IgM levels in the serum of workers exposed to low levels of VC
(< 10 mg/m3), but found that in workers with excessive exposures to
VC (> 10 mg/m3) there was a significant drop in IgG level.
The antinuclear antibody (ANA) test is negative or at low titre
in VC disease (Ward et al., 1976), whereas it is often positive in
other scleroderma-like disorders.
There is much heterogeneity within VC disease, in terms of skin
involvement, severity, and organ involvement, and it is clear that
this could have an immunological background. Systemic sclerosis may be
a genetically linked autoimmune disease. Many autoimmune diseases show
statistically significant associations with certain
human-leukocyte-associated antigen (HLA) alleles. Black et al. (1983,
1986) compared the HLA frequencies and autoantibodies in workers with
VC disease ( n=44), asymptomatic workers ( n=30), systemic sclerosis
( n=50) and normal (blood donor) controls ( n=200). The HLA-DR5
allele occurred significantly more frequently in VC disease (36%) and
systemic sclerosis (30%) compared to asymptomatic workers (3%) and
normal controls (16%). Eleven out of 21 men with severe VC disease had
HLA markers B8, DR3 whereas none of the 23 workers with mild disease
were positive for these markers, suggesting that this haplotype
favours progression of the disease.
Splenomegaly has been detected in a number of VC-exposed workers
(Falk et al, 1974; Suciu et al., 1975; Veltmann et al., 1975; Makk et
al., 1976; Ho et al., 1991). Hyperplastic sinusoidal cells were found
in some specimens.
g) Reproductive toxicity
The reproductive effects of VC have been reviewed (Uzych, 1988;
Little, 1993; Olsen et al., 1995).
VC has been cited in several early case series from different
countries as being responsible for male sexual dysfunctions such as
potency troubles, "pathological changes in the ejaculates", decreased
androgen secretion or undefined sexual disorders (Suciu et al., 1975;
Veltman et al., 1975; Walker, 1976; Sanotsky et al., 1980; Makarov,
1984) in exposed workers, but there is a lack of controlled studies.
Similarly, various female sexual dysfunctions have been reported after
long term exposure to VC (Makarov et al., 1984).
The possible risk, arising from human male exposure to VC, for
pregnancy and fetal loss among the wives of workers was first raised
after a report by Infante et al. (1976a,b) suggesting that fetal loss
was significantly more among wives of exposed workers. Data for the
wives of 95 VC polymerization workers in the USA were contrasted with
data for the partners of PVC fabrication workers and rubber workers
(total 158). Prior to exposure, the fetal death rates for controls and
polymerization workers were 6.9 and 3.1% (age adjusted, respectively).
After exposure, the rates were 6.8 and 10.8%, respectively. Weaknesses
in the study were in the method of age adjustment, that information
was obtained from interviews with male workers only (and not their
wives), and that there were no data on maternal age or on smoking and
other abuses.
In a study on 534 VC production and polymerization workers whose
wives were under 45 on 1 January 1984, 82 men were exposed to VC
before their spouses' pregnancies. The crude rate of abortions (no. of
spontaneous abortions / no. of pregnancies) was 8.9 for exposed and
8.8 for unexposed (total number of pregnancies: 90 and 1027,
respectively). Adjustment for confounders did not suggest a
significant relationship between spontaneous abortion and paternal
exposure to VC (Mur et al., 1992).
No statistically significant correlation could be found between
exposure to VC and congenital central nervous system (CNS) defects
among the children of fathers working at VC polymerization facilities
in Kanawha County, USA (Edmonds et al., 1975, 1978), but the study
data were not sufficient for a clear evaluation (Uzych, 1988).
In a Chinese study of 236 female workers exposed for more than 1
year to VC and 239 controls using retrospective (levels 3.9 to
89.3 ppm) and prospective (0.2 to 130.7 ppm) epidemiology, VC did not
appear to influence involuntary infertility, pregnancy outcome or
course of parturition, although the incidence of toxaemia in pregnancy
was found to be higher among female workers exposed to higher
concentrations in the prospective study (Bao et al., 1988; Jiangl,
1990).
An investigation into reproductive function in 2736 workers
exposed to VC in 13 PVC factories and 3442 workers in other factories
not exposed to VC showed no significant differences in reproductive
outcome. However, in the female exposed group, the incidence of
pregnancy complication was significantly higher than that of the
control group, suggesting that VC may effect the pregnancy process in
female workers (Huang, 1994).
There have been studies on female workers in the PVC processing
industry (Lindbohm et al., 1985; Ahlborg et al., 1987), but, as VC was
not the only source of exposure, these studies were not considered
here.
8.3.3 Neoplastic effects
After the case series published in 1974 (Creech & Johnson, 1974)
on hepatic angiosarcoma among workers exposed to vinyl chloride,
several further case series and small epidemiological studies, mainly
with emphasis on hepatic tumours, were published in the 1970s and
1980s. They show that VC can cause the rare liver tumour (ASL). Other
(non-ASL) cancer sites/types reported for which some of these studies
have indicated an association with VC exposure are: liver (e.g.,
non-angiosarcoma) tumours, particularly hepatocellular carcinoma;
respiratory system; digestive system other than the liver; lymph and
haematopoietic tissue; brain and other central nervous system;
malignant melanoma.
The early studies, together with a prepublication report of the
USA cohort study (see below) were summarized by Sir Richard Doll
(Doll, 1988). More recently, the studies performed in Sweden, Italy,
the United Kingdom and Norway were updated and analysed together
(L'Abbé et al., 1989; Simonato et al., 1991). Parts of the European
study have also been published separately. An updated American study
(Wong et al., 1991) combined many smaller studies conducted earlier in
the USA. These two studies provide the most informative data on the
health effects associated with exposure to VC. Four smaller
prospective studies on VC exposure workers have been conducted in
Canada (Thériault & Allard, 1981), Germany (Weber et al., 1981;
Greiser et al., 1982), France (Laplanche, 1987, 1992) and the
former-USSR (Smulevich et al., 1988). The complete description of two
additional studies (Frentzel-Beyme et al., 1978; Huang, 1993a,b) was
not available to the Task Group and were not further considered.
The epidemiological studies on VC/PVC workers are described as
follows:
* Simonato et al. (1991) (European/IARC study), which incorporates
populations reported by: Byrén et al. (1976), Molina et al. (1981),
Hagmar et al. (1990) (Sweden); Fox & Collier (1977), Jones et al.
(1988) (United Kingdom); Heldaas et al. (1984, 1987) (Norway); Belli
et al. (1987), Pirastu et al. (1990, 1991, 1998) (Italy);
* Wong et al. (1991) (USA study)a, which incorporates populations
reported by Tabershaw & Gaffey (1974), Monson et al. (1975); Nicholson
et al. (1975), Ott et al. (1975), Waxweiler et al. (1976), Buffler et
al. (1979), Cooper (1981), Dahar et al. (1988), Wu et al. (1989);
* Thériault & Allard (1981) Canada;
* Weber et al. (1981), Greiser et al. (1982) Germany;
* Laplanche et al. (1987, 1992) France;
* Smulevich et al. (1988) former-USSR
The USA study consisted of 10 173 men who had worked for at least
one year in jobs involving exposure to VC prior to January 1973 in 37
plants in the USA. Observation covered the years 1942-1982, and the
observed mortality was compared with the expected rates, based on USA
national rates for white males, standardized for age, and calendar
time. The race was actually known for only 666 workers, among which 3%
were African. Altogether 1536 members of the cohort were identified as
having died, the vital status of 725 individuals (7.88%) remained
unknown, and death certificates were not obtained for 97 (6.3%). These
deaths were included in the calculation of the overall SMR, but not in
any cause-specific SMRs (which leads to an underestimation of all
disease-specific SMRs by 6.3%).The deaths were coded using the 7th ICD
revision, where the numbers 155 and 156 comprise liver (155, both
primary and secondary) and bile duct and gall bladder tumours (156).
The average length of employment was approximately 16 years, and 46%
of the cohort were employed before 1955, thus providing a possible
latency of more than 27 years for this group. The levels of exposure
were not known. The all-causes SMR was 90.1 (95% CI 85.6-94.7), based
on a total of 1536 deaths. The SMR for all cancers was 105.1
(94.4-116.5).
The European/IARC study comprised a total of 14 351 subjects from
19 factories. After exclusion of short-term employees (< 1 year),
females, deaths outside the observation period, members of more than
one cohort, 12 706 subjects remained for the analysis. The follow-up
a After the Task Group meeting, a report of an update of the USA
study became available. A summary of it is presented in Appendix
3.
was 97.7% complete, the average length of follow up 17 years, and the
total number of person-years 222 746. National rates of mortality,
specific for age and five-year calendar periods, were used for the
comparison. The observation period was different for different
factories, starting for most from 1955, and extended until 1986.
Calendar period-specific job exposure matrices (JEMs) were developed
for 13 of the 19 factories. These JEMs were developed using job title
as a basic unit in which exposure is assessed. Estimates of exposure
were assigned, a priori, by a group of industrial hygienists on the
basis of the historical information available from the companies.
These estimates have been used both for qualitative and
quantitative dose-response analysis. In particular, a cumulative dose
was computed for each subject by multiplying the number of years spent
in a specific job by the time-specific estimates in ppm and then
summing up all the periods.
The 7th (for liver tumours, codes 155-156), 8th and 9th revisions
(155) of ICD were used (thus combining primary and secondary tumours
of the liver and tumours of extrahepatic bile ducts). A statistically
significant deficit in over-all mortality was observed (SMR 88, 95% CI
83-93), while for all malignant neoplasms, the SMR was 104 (CI
95-114), with 445 cancer deaths observed.
The Canadian study (Thériault & Allard, 1981) studied the
mortality of 1659 workers in a chemical industry complex in Schwanigan
who had worked for no less than 5 years in the company between 1943
and 1972. Out of the total, 48 (2.9%) could not be traced. The
observation period was 20 years or more for 315 workers (70%), and the
duration of exposure no less than 10 years for 340 (75%). Since
several episodes of unconsciousness among workers were reported, the
exposure to VC had probably been high: no measurement data were
available. The causes of death during 1948-1972 of the cohort was
determined from death certificates. In the analysis, those exposed to
VC for more than 5 years (451) were compared with those exposed for
less than 6 months (870). In addition, the mortality of the exposed
subcohort was compared to the age-standardized mortality expected for
Quebec in the year 1971. For cancer cases, medical records were
consulted for the verification of the diagnosis. The overall mortality
was similar in the exposed and non-exposed cohorts, and gave an SMR of
0.83 for the VC-exposed cohort in comparison to the Quebec figures.
Smulevich et al. (1988) reported results from a retrospective
cohort study of workers employed in some of the oldest Soviet chemical
plants producing VC and PVC. The study comprised 2195 men and 1037
women employed for at least one month in the plants. Subjects were
followed from 1939 to 1977. Information on specific occupation within
the plant, duration of exposure, date and cause of death, and
postmortem results (for cancer deaths) were collected for all member
of the cohort. No information was provided on the means for doing the
follow-up nor on losses to follow-up. Workers were classified into
those with high level exposure to VC (> 300 mg/m3), moderate levels
(30-300 mg/m3) and low levels (< 30 mg/m3). Mortality for the
cohorts was compared with rates of the city of the plant for the years
1959, 1969 and 1975. There were 288 deaths registered in the cohort,
including 63 from cancer. The SMR for all cancers was 1.06.
A prospective cohort study of 1100 workers exposed to VC in
various French plants and 1100 unexposed subjects was initiated in
1980 (Laplanche et al., 1987, 1992). Unexposed controls were matched
to cases by age, plant and physician. Subjects were followed until
December 1988 for vital status, other health outcomes and occupation.
Information was collected on complete occupational history, smoking
and drinking habits, and medical history (see also section 8.3.2.3c
and Table 39).
Weber et al. (1981) and Greiser et al. (1982) reported a historic
prospective cohort study of 7021 male workers who had been or were
working on 31 December 1974 in VC/PVC plants in Germany and Austria
(follow-up 10.5 years). The control cohort consisted of 4910 Germans
and Austrians from the same chemical companies but not having contact
with VC (follow-up 15.5 years). SMR for liver tumours was 1523, with
no details of the number of ASL. For lymphatic tumours and leukaemia
there was a SMR of 214. No information was given on subtypes. A
summary of findings on selected neoplasms for the epidemiological
studies on workers exposed to VC is given in Tables 39 and 40.
8.3.3.1 Liver and biliary tract cancers
a) Features of angiosarcoma
Angiosarcoma of the liver (ASL), also known as
haemangioendothelial sarcoma, is an extremely rare liver tumour and is
difficult to diagnose. ASL constitutes only 2% of all primary tumours
of the liver in the general population. It has been associated only
with exposure to VC, Thorotrast (a contrast medium used in X-ray
radiography in the 1930s-1950s) and arsenic (Creech & Johnson, 1974;
Falk et al., 1974). In England and Wales, from 1975 to 1987, there was
an annual incidence of 1.4 cases per 10 million population (Elliott &
Kleinschmidt, 1997). Regular international surveillance of cases of
ASL from VC exposure show that 118 cases were registered by 1985
(Forman et al., 1985), 173 by 1993 (Lee et al., 1996), and 197 by 1999
(Association of Plastics Manufacturers in Europe, 1999) (Table 38).
The most prominent clinical symptoms are abdominal pain,
weakness, fatigue and weight loss with hepatosplenomegaly, ascites and
jaundice being the common clinical signs. It is suggested that
patients with non-cirrhotic portal fibrosis and a history of VC
exposure should be followed-up for likely ASL (Lee et al., 1996).
However, Lelbach (1996) noted that, strikingly, except for the final
stages, there was little impairment of hepatic function in ASL
patients. The average latent period between starting work in an
occupation involving VC exposure and ASL diagnosis/death for 99 cases
was 22 years (Purchase et al., 1987; Lelbach, 1996).
Table 38. Number of vinyl chloride-associated ASL cases
reported per country in 1972
Country ASL cases ASL cases ASL cases up to 1999
up to 1985a up to 1993b [changes since 1993] c
USA 35 44 50 [+6]
Germany 26 (West) 41 40 [-1]
France 18 28 31 [+3]
United Kingdom 9 20 21 [+1]
Canada 10 13 13
Croatia 4 4 12 [+8]
Slovakia 2 2 6 [+4]
Italy 4 8 8
Sweden 5 5 5
Japan 2 3 4 [+1]
Belgium 2 2 2
Norway 1 1 1
Spain 1 1
Australia 1 1
Brazil 1 [+1]
Israel 1 [+1]
Total d 118 173 197
a From: Forman et al. (1985)
b From: Lee et al. (1996)
c Association of Plastics Manufacturers in Europe (1999)
d It should be noted that from several countries known
to be producers of PVC, there is no information regarding
ASL cases.
Any treatment is generally unsuccessful and survival after
diagnosis usually averages less than 12 months. Hepatic failure and
intra-abdominal haemorrhage are the usual terminal effects (Riordan et
al., 1991; Lee et al., 1996). Liver transplantation might be the only
chance of survival (Hayashi et al., 1990). If ASL is detected early
enough, surgical resection and adjuvant chemotherapy is a possibility;
subsequent hepatic recurrence could be treated by radiation therapy
and chemoembolization (Paliard et al., 1991; Neshiwat et al., 1992;
Hozo et al., 1997).
b) Results of epidemiological studies
In addition to the index tumour, ASL, several case series have
been published on VC-exposed workers who had liver tumours other than
ASL, in particular hepatocellular carcinoma (HCC) (Koischwitz et al.,
1981; Evans et al., 1983; Dietz et al., 1985; Pirastu et al., 1990,
1991; Lelbach, 1996; Makita et al., 1997; Saurin et al., 1997). In the
USA study, statistically significantly elevated relative risk was
observed for the cancers of liver and biliary tract (SMR 641, CI
450-884). Mortality was related to the duration of exposure (SMR 182,
1235, and 1284 for workers with exposure duration of < 10, 10-20 and
> 20 years, respectively), and to the latency period (SMR 386, 590,
and 1218 for latency periods of < 20, 20-30 and > 30 years,
respectively. The risk was higher for workers hired before 1950 (SMR
780) than in for those hired 1950-1959 (SMR 440) or since 1960 (SMR
419). The risk was highest among workers hired at an early age (SMR
1611, 813 and 346 for those hired at < 25, 25-34 and > 35 years of
age. The authors noted, however, that those hired early and young were
likely to have longer duration of exposure and latency.
A total of 15 angiosarcomas were recorded on the death
certificates. Although no expected numbers were calculated, this must
be in a very marked excess, since the annual incidence of liver
angiosarcoma is approximately 2 per 10 million, which would in this
cohort lead to an expected number of approx. 0.05. The authors noted
that, when the 15 angiosarcomas were excluded, there remained 22 other
liver and biliary tract tumours; this represents an excess (5.7
expected, SMR 386, P < 0.02). This is likely to be an overestimate,
since some of these other liver cancers were likely to be
angiosarcomas.
In the European study, the SMR for liver neoplasms was 286 (95%
CI 183-425, and it increased with increasing latency (SMR 0, 253, 388
and 561 for latencies < 10, 10-19, 20-29 and > 30 years,
respectively), duration of exposure (SMR 94, 327, 310, 714 and 1111
for duration of employment of < 10, 10-14, 15-19, 20-24 and
> 25 years, respectively), ranked level of exposure, and cumulative
exposure (SMR 348, 400, 1429 and 1667 for < 2000, 2000-6000,
6000-10 000 and > 10 000 ppm-years, allowing for a 15-year
latency). A clear exposure-response relationship was seen between
ranked level of exposure (ppm) as well as cumulative exposure
(ppm-years) to VC and liver cancer mortality, with autoclave workers
having the highest risk (Table 41; Simonato et al., 1991). This
relationship is even more evident taking only those liver cancers
defined histologically as ASL. In addition, for those VC workers not
defined as autoclave workers, an exposure-response relationship could
be seen for those diagnosed as having ASL and non-ASL liver cancers
(Pirastu et al., 1990, 1991; Simonato et al., 1991).
Table 39. Summary of findings on selected neoplasms for the epidemiological
studies on workers exposed to VC
Cause of European/IARC USA study Weber et Smulevich et Thériault Laplanche All
mortality study (Simonato (Wong et al. (1981) al. (1988) & Allardt et al. studies d
et al., 1991) al., 1991) (1981) (1992)
Obs/Exp Obs/Exp Obs/Exp Obs/Exp Obs/Exp Exposed/ Obs/Exp
SMR SMR SMR SMR SMR Non-expos. RR SMR
(95% CI) (95% CI) (95% CI) (95% CI) (95% CI) (95% CI) (95% CI)
All causes 1438/1636.4 1536/1705.27 414/434.7 - 59/71.07 40/43
88 90 95 83 1.0
(83-93) (86-95) (0.6-1.5)
All malignant 445/427.8 359/341.7 79/82.9 63/58.88 20/16.37 966/927.65
neoplasms 104 105 103 b 107 122 1.3 104
(95-114) (94-116) (0.7-2.3) (98-111)
Liver cancer/ 24/8.4 37/5.77 12/0.9 0/n.a 8/0.14 3 81/19.21
angiosarcoma 286 641 1523 5714 533
(ASL) (183-425) (450-884) 8 ASL (+2 ASL 3 ASL (423-622)
16 ASL out of 15 ASL in undiagnosed)
17 liver cancer death
deaths with certificates.
histopathology 21 ASL in
international
register
Brain 14/13.1 23/12.76 2/1.3 4/2.61 0/0.6 43/30.37
107 181 162 153 0 142
(59-180) (114-271) (103-191)
Lung 144/148.3 111/115.87 24/26.6 1/1.2 2/5.78 c 282/297.75
97 96 96 83 34 95
(82-114) (79-116) (84-106)
Table 39.
Cause of European/IARC USA study Weber et Smulevich et Thériault Laplanche All
mortality study (Simonato (Wong et al. (1981) al. (1988) & Allardt et al. studies d
et al., 1991) al., 1991) (1981) (1992)
Obs/Exp Obs/Exp Obs/Exp Obs/Exp Obs/Exp Exposed/ Obs/Exp
SMR SMR SMR SMR SMR Non-expos. RR SMR
(95% CI) (95% CI) (95% CI) (95% CI) (95% CI) (95% CI) (95% CI)
Lymphatic and 29/32.7 37/36.28 15/7.7 10/2.2 1/1.67 92/80.55
haematopoietic 89 102 214 454 60 114
(72-141) (92-140)
Lymphomas 18/19.3 a 24/21.8 a 5/1.2
93 110 417
Stomach 49/45.1 10/16.01 18/14.4 21/24.7 - 98/100.21
109 63 138 85 98
(80-144) (30-115) (79-119)
a Lymphoma and malignant myeloma
b As reported in original paper
c All respiratory neoplasms
d Calculated by the Task Group. All studies except Laplanche et al. (1992) who did not provide observed and expected values
Table 40. Analysis of liver, brain and lung cancer mortality in the USA
and European cohort by duration of exposure
Organ Study < 10 years 10-19 years 20 + years
SMR (observed) SMR (observed) SMR (observed)
Liver USA (Wong 182 (6) 1236 (20) 1285 (11)
et al., 1991)
Europe (Simonato 0 (0) 253 (8) 432 (16)
et al., 1991)
Brain USA 165 (13) 121 (4) 386 (6)
Europe 59 (2) 113 (6) 136 (6)
Lung USA 93 (59) 114 (37) 75 (15)
Europe 95 (73) 107 (51) 83 (20)
Of the 17 liver cancer deaths, for which histopathological data
were available, 16 were classified as angiosarcomas. For the remaining
seven, the histological type remained unknown. The Task Group noted
that even if they all had been cancers other than angiosarcoma, the
relative risk of liver tumours other than angiosarcoma would not have
been elevated (expected number equiv. 8).
For 2643 workers from Norway and Sweden, cancer incidence
information was also available. The only site for which a
statistically significantly elevated risk was observed was the liver
(SIR 303, CI 122-623).
In the Canadian study, there was an excess of digestive tract
cancers (14 cases, SMR 259, P < 0.01) that was fully accounted for
by cancers of the liver (8 cases). All liver cancers were
angiosarcomas.
Hospital admission diagnoses were studied among 714 present and
1575 previous workers, identified from national Labour Insurance
Bureau records, with exposure to VC (Du & Wang, 1998). The frequencies
of different diagnoses were compared with similar data on workers in
the manufacture of optical instruments or of motorcycles. Eight cases
of primary liver cancer were observed among the VC-exposed workers
(out of 1044 admissions), while the number was 9/3667 for the optical
workers, and 9/5861 for motorcycle manufacturers, which gave 4.5 (95%
CI 1.5 to 13.3) and 6.5 (2.3-18.4) as age-adjusted morbidity odds
ratios (MOR), calculated from a linear regression model, for the
VC-exposed workers, compared to the two comparison groups. Increased
MORs (of borderline significant) were also observed for haematopoietic
cancer, chronic liver disease and liver cirrhosis, as well as other
chronic diseases, and accidents.
Two updates of parts of the Italian cohort (Pirastu et al., 1991)
have recently appeared. An elevated mortality of liver cancer (11
observed, 5.7 expected cases, SMR 193) was observed in Marghera. In an
extended survey for hepatic cancers, four further cases were
uncovered. There were 5 angiosarcomas, 5 hepatocellular carcinomas, 3
cirrhotic carcinomas, and in two cases the histology was not known
(Pirastu et al., 1997). In three other subcohorts (Pirastu et al.,
1998), the pooled SMR for liver cancer was 364 (7 observed cases, 90%
CI 108-390).
A survey of 5291 workers from 13 PVC manufacturing plants in 12
cities in China using 6276 workers unexposed to VC as controls was
carried out between 1982 and 1989. Although no cases of ASL were
reported, the mortality from liver cancer in male workers exposed to
VC was significantly higher than those of the control group and of the
male general population in the middle cities in China. Among malignant
tumours, liver cancer ranked first in exposed males. The authors found
that the average age for persons who were diagnosed with liver cancer
was significantly lower than that of the control group (Huang,
1993a,b).
Table 41. Mortality data for liver cancer according to the exposure
variables a
Exposure variable O SMR 95% CI 15 years of latency
SMR 95% CI
Job title
Ever autoclave worker 11 896 447-1603 1358 678-2430
Never autoclave worker b 13 181 97-130 284 151-485
Duration of
employment (years)
1-9 4 94 26-239 205 56-525
10-14 5 327 106-763 602 196-1406
15-19 4 310 84-794 310 84-794
20-24 6 714 262-1555 714 162-1555
> 25 5 1111 361-2593 1111 361-2593
Ranked level of
exposure (ppm)
Low (< 50) 3 (4) 119 25-347 227 (244) 47-664 (67-625)
Intermediate (50-499) 3 (7) 161 33-471 250 (551) 52-731 (222-1136)
High (> 500) 12 (12) 567 293-991 719 (719) 371-1255 (371-1255)
Unknown 6 (1) 317 177-691 486 (125) 182-1079 (3-697)
Cumulative exposure
(ppm-years)
0-1999 4 (9) 99 27-254 191 (348) 52-490 (159-662)
2000-5999 4 (4) 351 96-898 460 (400) 125-1177 (109-1024)
6000-9999 4 (7) 800 218-2048 851 (1429) 232-2179 (574-2943)
> 10 000 3 (3) 1429 295-4175 1667 (1667) 344-4871 (344-4871)
Unknown 9 (1) 357 163-678 536 (100) 245-1017 (2-557)
Table 41. (cont'd)
Whole cohort 24 286 183-425 445 285-663
a From: Simonato et al. (1991); O = observed number of deaths;
SMR = standardized mortality ratio; Cl = confidence interval.
The values in parentheses were determined in analyses including
the estimated job-exposure matrices
b In Norway, the longest-held job was used. In Sweden job rotation
was practiced, and no one was classified as an autoclave worker
8.3.3.2 Brain and central nervous system (CNS)
Statistically significant increases of brain and CNS tumours
after occupational exposure to VC were reported in USA surveys by
Waxweiler et al. (1977) and Cooper (1981), as well as in Sweden (Byrén
et al., 1976) and Germany (Greiser et al., 1982).
In the USA (study of Wong et al., 1991), the relative risk was
elevated for cancers of the brain and other CNS cancers (SMR 180.2, CI
114.1-270.6). Mortality was highest in the group with the longest
duration of exposure (SMR 164.7, 120.8 and 385.9, for workers with the
duration of exposure < 10 years, 10-20 years and > 20 years,
respectively), while no trend with latency was observed (SMR 183.8,
158.3 and 210.7, for latencies of < 20, 20-30 and > 30 years).
Workers hired early did not exhibit higher risk than those hired late
(SMR 156, 164 and 256, for employees hired before 1950, 1950-1960 and
after 1960, respectively. It was noted that most of the cases came
from the two plants with the highest rate of ASL.
In the European study, the SMR for brain cancer was 107
(CI 59-180). The small number of cases (total 14) made the assessment
of the relationship between duration of exposure and latency
difficult, but the risk was highest after longest latency (SMR 59,
113, 59 and 407 after a latency of < 10, 10-20, 20-30 and
> 30 years) and after longest duration of exposure (SMR 106, 78 and
183 for a duration of exposure of < 10, 10-20 and > 20 years).
There were even fewer cases for the assessment of the relationship
between estimated exposure and cancer risk (8 cases altogether), but
the point estimates were higher for higher estimated exposures (SMR
91, 42 and 128 for low, intermediate and high exposure), while little
covariation between estimated cumulative exposure and relative brain
cancer risk was observed (SMR 0, 162 and 120 for cumulative exposures
of < 50, 50-499 and > 500 ppm-years).
In the cancer incidence aspect of the European study, a
non-significantly elevated SIR was observed for cancer of the brain
(SIR 159 CI 68-312).
8.3.3.3 Respiratory tract
The study by Monson et al. (1974) first suggested an association
between VC and lung cancer.
In the USA study, the SMR for lung cancer was 95.8
(CI 78.7-115.5).
In the European study, the SMR for lung cancer was 97
(CI 82-114). It did not show any consistent relationship with duration
of employment, latency, ranked level of exposure or cumulative
exposure; the SMR was similar for ever- and never-autoclave workers.
In the cancer incidence aspect of the European study, a
non-significantly elevated SIR was observed for cancer of the lung
(SIR 152, CI 95-230).
In the Canadian study, the mortality from respiratory cancer was
low (relative risk in comparison to the unexposed cohort 0.36, and SMR
based on Quebec data, 34, based on two cases). Smoking habits, as
studied by a questionnaire, were similar in the exposed and
non-exposed cohorts.
8.3.3.4 Lymphatic and haematopoietic cancers
There were indications of increased risk of cancer of the
lymphatic and haematopoietic systems in several early studies (Monson
et al., 1975; Waxweiler et al., 1976; Greiser et al., 1982; Smulevich
et al., 1988).
In the Wong et al. (1991) USA study, the SMR for lymphatic and
haematopoietic cancer was 102 (CI 71.6-140.7, while that for lymphoma
and reticulosarcoma was 102.0 (CI 71.6-140.7).
In the European study, the overall SMR for lymphosarcoma was 170
(CI 69-351), based on seven cases. No cases were observed in the
groups with a latency of 20-30 or > 30 years, and six out of the
seven subjects had worked less than 10 years in VC-exposure
conditions.
8.3.3.5 Malignant melanoma
There was a relationship between VC exposure and malignant
melanoma of the skin in PVC production workers in the early Norwegian
study (Heldaas et al., 1984, 1987).
In the European study, a non-significant, elevated risk was
observed for melanoma, and the risk was higher in the incidence part
of the study (SIR 184, CI 79-302). However, the risk was limited to
one country only (Norway).
8.3.3.6 Breast cancer
Infante & Pesák (1994) noted that there had been no follow-up
study to that of Chiazze et al. (1977), who observed an excess in
breast cancer mortality. Although a subsequent study among these
researchers (Chiazze et al., 1980) did not identify a significantly
elevated odds ratio in relation to VC exposure, the statistical power
used in the study has been questioned (Infante & Pesák, 1994). There
have been no other reports on breast cancer in humans, but in most
Western countries women have not been working in jobs involving VC
exposure. In some countries, however, e.g., China and Eastern Europe,
women are/were probably exposed to higher VC levels than in Western
countries (see Table 21) but epidemiological studies are not
available. Mammary carcinoma has been reported in animal studies with
inhalative and oral exposure to VC (see section 7.7.2).
No breast cancer was observed in the study by Smulevich et al.
(1988).
8.3.3.7 Other cancer sites
In the European study, a non-significantly elevated risk was also
observed for urinary bladder (21 deaths observed, SMR 146, CI 91-224)
and, in the incidence part, for cancer of the stomach (SIR 150,
CI 80-256).
8.4 Genotoxicity studies
8.4.1 Cytogenetic studies of VC-exposed workers
Table 42 summarizes the results of cytogenetic studies of
chromosomal aberrations (CA), micronucleus formation (MN) and
sister-chromatid exchanges (SCEs) in the peripheral blood lymphocytes
of VC workers compared to controls. Studies on the genotoxicity of VC
have been reviewed (Uzych, 1988; Giri, 1995). Although in many studies
the exposure concentrations and duration of exposure were only
estimated, a dose-response relationship and a "normalization" of
genotoxic levels with time after reduction of exposure can be seen.
There was a clear relationship between incidence of CA and the
exposure concentrations/type of work (Purchase et al., 1978). The same
groups of workers showed lower or no effects after the exposure
concentration was reduced to < 13 mg/m3 (< 5 ppm) (Hansteen et al.,
1978; Anderson et al., 1980; Fucic et al., 1996). That such levels do
not cause an increase in CA was confirmed by several studies (Picciano
et al., 1977; Rössner et al., 1980; de Jong et al., 1988).
SCEs increased with the level of exposure to VC of workers
(Sinués et al., 1991). SCEs were generally not detected in blood
samples from workers exposed to VC at < 5 ppm.
In a micronucleus assay, workers exposed to VC had a much higher
frequency of micronucleated lymphocytes than the controls. However, a
significant decrease in the number of micronucleated lymphocytes was
found within 15 days after the last exposure, but even 90 days after
the last exposure (peak of 780 mg/m3) the frequency, although
decreased, was still above the control value (Fucic et al., 1994). A
similar effect had been shown with SCE frequencies (Fucic et al.,
1992).
Chromosomal aberrations were measured in peripheral blood
lymphocytes from 29 subjects potentially exposed to VC after an
accidental release into the environment (Hüttner & Nikolova, 1998).
The control group consisted of 29 non-exposed people, matched
regarding age, gender and smoking habits. The exposed group showed a
statistically significant increase in the mean frequency of aberrant
cells (1.47% versus 1.07% in the control group).
Table 42. Chromosome analysis in human T-lymphocytes (in vivo)
after vinyl chloride exposure during work at PVC polymerization
plants or due to other accidental exposure a
Test No. workers; Additional Duration Exposure Result Reference
No. controls information of exposure level
(years) (mg/m3)
HPRT mutation 24; 23 worked on post- average 7 average < 13 - Hüttner &
using T-cell chlorination of PVC Holzapfel (1996)
cloning assay in an open system
where free VC
evaporated
DNA single 16 3 groups of VC/PVC average 13 average 150 - Du et al. (1995)
strand breakage 104 workers average 16 5 -
122 average 16 2 -
Chromosomal 11; 10 PVC workers 4-28 peaks >1300 + Ducatman et
aberrations (average 15) al. (1975)
7; 3 9-29 50-75 + Funes-Cravioto
et al. (1975)
45; 93 0.5-12 n.g. + Szentesi et al. (1976)
57; 24 VC/PVC workers 10.7 (average 10-100 + Purchase et al. (1978)
classified according for autoclave
21; 6 to type of work workers) < 13 + Anderson et al. (1980)
23; 8 (1974) < 13 -
same as Purchase
et al. (1978)
Chromosomal 20; 20 with clinical + Fleig & Thiess (1974,
aberrations symptoms; without - 1978)
clinical symptoms
39; 16 1974 > 10 > 65 + Hansteen et al. (1978)
37; 32 same workers 1977 2.6 -
3; 9 10-27 50-400 + Kucerová et al. (1979)
37; 12 n.g. 2-111 + Katosova & Pavlenko (1985)
43; 22 average 11.2 2-41 + Hrivnak et al. (1990)
Table 42. (cont'd)
Test No. workers; Additional Duration Exposure Result Reference
No. controls information of exposure level
(years) (mg/m3)
19; 20 average 15 130; peaks of + Fucic et al. (1990b)
67; n.g. breaks occurring at average 15 5000 13; peaks + Fucic et al. (1990a)
non-random sites of 5000
10; n.g. 8-25; av 13; short + Garaj-Vrhovac et al. (1990)
average 15 peaks higher
n.g PVC workers 5 26 + Zhao et al. (1994)
PVC workers 2 26 -
PVC workers > 7 4 -
residents on site > 8 0.2 -
209; 295 3 groups average 4 < 2 to > 13 - Picciano et al. (1977)
31; 35 1977 2-3 < 10 - Rössner et al. (1980)
same group 1978 peak < 30 -
66; 39 1976-1977 1-11; average 6 3 peaks up - de Jong et al. (1988)
29; 29 exposure of general for short time to 26 unknown + Hüttner & Nikolova
population after (approx. 1 h) concentration (1998)
accidental release
of VC
Micronucleus 19; 20 average 15 130; peaks of 5000 +Fucic et al. (1990b)
assay 32 24 h to 90 days 10 peak of 780 + Fucic et al. (1994)
following a peak 5 mg/m3 every +
exposure of 3 months
780 mg/m3
Table 42. (cont'd)
Test No. workers; Additional Duration Exposure Result Reference
No. controls information of exposure level
(years) (mg/m3)
Sister-chromatid 9; 8 10-27 50-400 + Kucerová et al. (1979)
exchange 16, 16 > 10 2.6 - Hansteen et al. (1978)
21; 6 3.5 < 13 (-) Anderson et al. (1981)
31; 35 2-3 < 10 - Rössner et al. (1980)
31; 41 > 2 3-44; peaks of + Sinués et al. (1991)
21; 41 130
1-20
15; 10 1.5-35 13; peaks of + Fucic et al. (1992)
5000
0.3; peaks of - Fucic et al. (1996)
130
a HPRT = hypoxanthine guanine phosphoribosyltransferase; n.g. = not given
8.4.2 Mutations at the hypoxanthine guanine phosphoribosyltransferase
(hprt) locus
With the use of the hprt lymphocyte clonal assay it is possible
to determine the mutation frequency of the hprt gene and to
characterize its mutant spectra. Induced mutagenesis in the
lymphocytes of PVC production workers was measured by selecting mutant
T-cells with the hprt gene in medium containing 6-thioguanine. The
exposed workers had similar mutation frequencies (about 8 × 10-6) as
controls (Hüttner & Holzapfel, 1996).
The types and frequencies of mutations caused by VC in a human
B-lymphoblastoid line expressing cytochrome P-450 2EI (H2E1 cells)
were investigated by Chiang et al. (1997). VC was found to be toxic
and mutagenic to H2E1 cells as a function of incubation time when they
were exposed to 7.5% VC in air. This exposure resulted in 75% survival
and an hprt mutant frequency of 42 × 10-6 after 48 h, compared to
6 × 10-6 for unexposed cells. Exposure to 0.8 to 15.0% VC in air
produced similar mutation frequencies but without a clear
dose-response relationship, perhaps due to saturation of metabolic
activation. Ten percent (5/50) of VC-induced mutations showed
detectable deletions. Both CEO and CAA showed dose-dependent increases
in cell killing and mutant frequency. VC and CEO displayed similar
toxicity/mutation profiles and a similar frequency of large deletions,
whereas CAA showed greater toxicity and a larger frequency of deletion
mutations.
8.4.3 Mutations in ASL from VC-exposed workers
8.4.3.1 p53 gene
Normal and cancer cells appear to differ through discrete changes
in specific genes controlling proliferation and tissue homoeostasis.
The p53 tumour suppressor gene is present in every cell of the human
body and is located on the short arm of the human chromosome 17 p.
(Lane, 1994). It is mutated in about half of all cancer types arising
from a wide spectrum of tissues (Harris, 1996).
The encoded wild-type p53 protein, a 393 amino acid nuclear
phosphoprotein, activates the transcription of several genes,
including some involved in cell cycle control (e.g., p21), and has
been implicated in DNA repair processes, DNA replication and apoptosis
(Lane, 1994; Semenza & Weasel, 1997). The majority of cancer-related
mutations in p53 cluster in several "hot-spot" regions of the
protein that have been highly conserved through evolution. These
regions occur in the sequence-specific DNA-binding core domain of the
protein between amino acid residues 102 and 292 (Lane, 1994). The
mutations in p53 found in malignancies could thus result in
substitutions of amino acid residues in these regions that are
critical for the determination of its structure and function (Cho et
al., 1994; Brandt-Rauf et al., 1996).
The p53 gene was examined in tumours from four highly exposed
(before 1974) autoclave workers with ASL and one other VC worker with
hepatocellular carcinoma (HCC) (Hollstein et al., 1994). Two A to T
mutations in a highly conserved domain of the coding sequence (codon
249: AGG to TGG, Arg to Trp; and codon 255: ATC to TTC, Ile to Phe)
were found one each in the tumour but not in other normal cells of two
of the ASL patients (both smokers). A further A to T missense p53
mutation (codon 179: CAT to CTT) was found in a cell line from a
further VC-associated liver tumour patient (Boivin et al., 1997; see
Table 43). Such mutations are uncommon in human cancers (2.7% of a
total of 5085 cancers and 8% of a total of 290 primary liver cancers;
Hollstein et al., 1996). Furthermore, p53 mutations are uncommon in
sporadic (non-VC-induced) ASL (2/21 cases, 9%), supporting the
evidence linking VC exposure and ASL containing an increased frequency
of p53 mutations with a mutational spectrum (A to T) (Soini et al.,
1995). To date (1998) 11 out of 15 (73%) ASL from VC-exposed workers
have been shown immunohistochemically to have mutant p53 protein.
Further, a statistically significant trend for mutant p53 protein has
been found in the serum of VC-exposed workers (Smith et al., 1998);
this study is continuing (Li et al., 1998a,b).
p53 gene mutations were also found in 11 out of 25 (44%) ASL in
Sprague-Dawley rats induced by VC and 1 in 8 HCCs (Barbin et al.,
1997; see Table 43). This is a higher mutation rate than that found in
humans but again the majority of missense mutations involved were A:T
base pairs. The A:T -> T:A transversion observed in the first
nucleotide of codon 253 in two rat ASL is equivalent to the same
transversion characterized previously in codon 255 in one human ASL
associated with VC exposure (Trivers et al., 1995).
8.4.3.2 ras genes
The ras gene family, Ha- ras, Ki- ras and N- ras, are genes
coding for p21 and are frequently activated by point mutations in
codons 12, 13 and 61. These activated genes seem to play a key role in
the development of spontaneous or carcinogen-induced mammalian tumours
(Barbacid, 1987). In a study of ras oncogene mutations in tumours of
VC-exposed workers, 15 of 18 ASLs were found to contain a G to A
transition at the second base of codon 13 (GGC to GAC) of the
Ki- ras-2 gene (Marion et al., 1991, 1996; see Table 44). This
mutation leads to the substitution of glycine by aspartic acid at
amino acid residue 13 in the encoded p21 protein.
Ki- ras-2 gene mutations were found in codon 12 in 5/19 sporadic
ASL and in 2/5 thorotrast-induced ASL in humans. All seven mutated
tumours contained a G -> A transition at base 2. In addition, 4 of
these tumours (3 sporadic, 1 thorotrast-induced) contained a second
mutation in the Ki- ras gene, a G -> T transversion of the first
base of codon 12 (Przygodzki et al., 1997).
Table 43. Comparison of mutation spectra in the p53 gene in
liver tumours in humans and rats a
Species Tumour origin No. mutations/ No. and types References
no. of cases of mutations
Humans VC-associated 3/6 3 A:T -> T:A Hollstein
ASL et al. (1994)
cells cultured [CAT -> CTT] Boivin
from (codon 179) et al. (1997)
VC-associated
liver tumour
Rats VC-associated 11/25 5 A:T -> T:A Barbin et al.
ASL 2 A:T -> G:C (1997)
2 A:T -> C:G
3 G:C -> A:T
one 12 base-pair
deletion
1 deletion
HCC 1/8 1 A:T -> T:A
a Adapted from Barbin et al. (1997); ASL = angiosarcoma of the liver;
HCC = hepatocellular carcinoma
Table 44. Mutagenesis of ras proto-oncogenes in VC-associated
liver tumours in humans and rats
Tumour Gene Codon No. Base pair References
origin a involved mutations/ change/
No. tumours Codon
change
Human Ki-ras 2 Codon 15/18 G -> A/ Marion et al.
ASL gene 13 GGC -> GAC (1991, 1996);
DeVivo et al.
(1994)
Rat Ki-ras 2 0/10 Froment et al.
ASL gene (1994);
Boivin-Angèle
et al. (in press)
Rat Ha-ras Codon 5/8 AT -> TA/ Froment et al.
HCC gene 61 AT -> TA (1994);
Boivin-Angèle
et al. (in press)
a ASL = angiosarcoma of the liver; HCC = hepatocellular carcinoma
In studies in VC-induced ASL and HCC tumours in rats, no
mutations were found in Ki- ras genes but there were mutations at
codon 61 of the Ha- ras proto-oncogene (Froment et al., 1994;
Boivin-Angèle et al., in press).
8.5 Studies on biological markers
8.5.1 Excretion of metabolites
The concentration of the VC metabolite thiodiglycolic acid in the
urine has been found to correlate with VC exposure in workers and has
been suggested as a biological marker for VC (Müller et al., 1978)
(see also section 5.3).
8.5.2 Genetic assays
Increased levels of chromosomal abnormalities, compared to
control populations, were found in workers exposed to high levels of
VC but not in workers exposed to less than 13 mg/m3 (5 ppm) (see
Table 42). The micronucleus assay and mitotic activity have been
suggested as methods for determining recent VC exposure (Fucic &
Garaj-Vrhovac, 1997; see section 8.4).
8.5.3 Enzyme studies
Ever since the 1970s there has been disagreement as to the value
of monitoring levels of enzymes as a measure of VC exposure or as a
sign of VC disease. Liver function tests may be of limited value in
detecting VC-induced liver disease because, although hepatocytes are
the primary site of VC metabolism, they are not the target of toxicity
(Lelbach & Marsteller, 1981). Individual studies of VC-exposed
populations have reported abnormalities in one or more liver function
tests, such as aspartate aminotransferase (ASAT), alanine
aminotransferase (ALAT) and alkaline phosphatase (AP) (Lilis et al.,
1975; Waxweiler et al., 1977). However, Sugita et al. (1986) concluded
that VC probably had not affected these enzymes after adjusting for
confounding factors. This was confirmed by Du et al. (1995) who showed
that ASAT, ALAT and AP were mainly affected by the presence of
hepatitis B surface antigen (HBsAg) or anti-hepatitis C virus
(anti-HCV).
Gamma glutamyl transpeptidase (GGT) is thought to be associated
with VC exposure (Langauer-Lewowicka et al., 1983; Du et al., 1995)
and, according to the latter, this is the only enzyme that correlates
with VC exposure levels. It should be noted that GGT is readily
elevated in alcohol drinkers, so the value of this enzyme as a
biomarker of VC-induced liver injury becomes questionable. In workers
showing VC-induced liver dysfunction (VC levels 2.6-54 mg/m3; 1 to
21 ppm), ALAT was the earliest parameter raised, then serum GGT (Ho et
al., 1991). Ho et al. (1991) also reported that 10 of 12 workers with
VC-induced liver dysfunction showed improvements in liver function
tests after removal from exposure. There are case reports of
individuals whose angiosarcomas were first detected by abnormal liver
function tests (Makk et al., 1976), but liver function abnormalities
appear to be a relatively late finding (Falk & Steenland, 1998). No
published studies have been identified that evaluate the effectiveness
of medical surveillance in occupational groups with VC exposures below
2.6 mg/m3 (1 ppm).
8.5.4 von Willebrand factor
The immunoquantitation of the level of the von Willebrand factor
(vWf; factor VIII-related antigen, a large multimeric plasma
glycoprotein in the blood clotting system) in 107 VC exposed workers
was performed using an enzyme-linked immunosorbent assay (ELISA)
(Froment et al., 1991). The vWf level was slightly but significantly
higher in the VC-exposed workers than in the control group. The vWf
serum level of three patients with hepatic angiosarcoma was markedly
elevated.
8.5.5 p53 and ras proteins
Mutant p53 and p21 proteins or their antibodies can be detected
in sera from healthy workers exposed to VC (Marion et al., 1996;
Semenza & Weasel, 1997; Smith et al., 1998; Li et al., 1998a,b). A
dose-response relationship between estimated cumulative exposure and
the frequency of these onco-proteins has been reported (Table 45).
Statistically significantly elevated frequencies were observed even in
the group with lowest estimated exposure (cumulative exposure
< 40 ppm-years) (Li et al., 1998b). However, exposure estimations
were crude. In addition, similar findings have not been reported for
other VC-exposed groups.
8.6 Susceptible subpopulations
8.6.1 Age susceptibility
Evidence from animal studies suggests that there may be
early-life sensitivity to VC (section 7.7.3), but there is also
contradictory evidence. There has been concern that children living
near landfill sites might be particularly susceptible to VC formed and
released there (Hiatt et al., 1994; Cogliano et al., 1996). However,
there is no epidemiological evidence to support this.
8.6.2 Immunological susceptibility
Individual susceptibility may influence the development of VC
disease, since not all highly exposed workers developed clinical
symptoms or signs (Wilson et al., 1967; Black et al., 1986). Studies
conducted in a single population of VC-exposed workers have shown that
susceptibility for the disorder seems to be increased in the presence
of the human-leukocyte-associated antigen HLA-DR 5 or a gene in
linkage disequilibrium with it and an antigen associated with the
haplotype A1, B8, while DR3 seems to favour progression of the disease
(section 8.3.2.3e). The significance of this finding is unclear
without replication in another VC-exposed population.
8.6.3 Polymorphic genes in VC metabolism
Levels of CYP2E1 can vary considerably among individual humans
(Guengerich et al., 1991) and are a possible cause of differences of
susceptibility to VC.
El Ghissassi et al. (1995b) analysed the GSTM1 genotype in 133
Caucasian individuals who had had a high exposure to VC for several
years and had (or had not) clinical signs of chronic injury (see also
Froment et al., 1991, section 8.5.4). Of the 133 individuals, 62
workers had non-neoplastic symptoms, while 8 had liver cancer. The
frequency of GSTM1 null genotype in the whole group of exposed
workers was 61.7% (82 of 133) and 62.5% in patients with liver cancer.
In case-control studies reported by others, the frequency of GSTM1
null genotype ranged from 41 to 53% for Caucasians.
Table 45. p53 and p21 mutations (as detected by serum biomarkers analysis)
in relation to estimated vinyl chloride exposure (Li et al., 1998a)
Estimated Total no. of Both biomarkers No. of workers with No. of workers with Adjusted OR 95% CI
exposure workers negative mutation in mutation in
(ppm-years) either p53 or p21 both p21 and p53
0 43 38 5 0 1
< 500 42 20 21 1 11.1 3.3-37.5
501-2500 45 20 21 4 12.8 4.1-40.2
2501-5000 31 6 19 6 29.9 9.0-99.1
> 5000 54 15 22 17 31.2 10.4-94.2
9. EFFECTS ON OTHER ORGANISMS IN THE LABORATORY AND FIELD
9.1 Laboratory experiments
VC has been shown to be mutagenic in several in vitro and in
vivo test systems derived from organisms belonging to different
taxonomic levels. The details referring to bacterial, fungal and
mammalian cell lines or to whole organisms like insects or plants are
discussed in section 7.6. The carcinogenic effects of VC are addressed
in section 7.7. The following chapter focuses on investigations of
other signs of toxicity relevant to organisms that may be exposed to
VC in the environment. Standard tests on survival and reproduction
were not available. Care must be taken when interpreting the toxicity
results available as many were obtained from static tests using
nominal exposure concentrations. Such tests will have large losses of
VC due to volatilization, thus reducing the actual exposure to VC.
9.1.1 Microorganisms
9.1.1.1 Water
A consortium of anaerobic microorganisms (species not identified;
initially obtained from municipal sludge) was used for testing VC
toxicity. Both batch and semi-continuous assays were conducted. VC had
an inhibitory effect on the total gas production, beginning at a
concentration of 5.4 mg/litre and resulting in an EC50 value of
approximately 40 mg/litre, as seen in the batch assay over 3.5 days.
In the semi-continuous assay lasting 15 days, the threshold was
greater than 64 mg/litre (the highest concentration tested), probably
due to volatilization of VC (Stuckey et al., 1980).
The growth of five mixed bacterial populations (isolated from
natural aquatic systems) was not affected, as compared to controls, in
liquid cultures (closed flasks; 21°C; over 5 weeks) containing up to
900 mg VC/litre (Hill et al., 1976b).
The toxicity of waste effluents of a VC production plant to the
green alga Chlorella sp. was tested before and after the wastes were
treated by neutralization procedures (addition of aqueous solutions of
NaOH and catalysts). The crude effluents consisted of VC (15-18%,
weight), di-, tri- and pentachloroethanes (45%), dichloroethylene
(27%), dichloropropane (6%), ethyl chloride (1%) and unidentified
substances (1%). This mixture led to a weak inhibition of algal growth
(measured by changes in optical density at 678 nm), with a 72-h EC50
value of 1495 mg/litre (which corresponds to a VC concentration of
224 mg/litre). It should be noted that other compounds present in the
effluent may also have contributed to the toxicity. The corresponding
EC50 values of the neutralized waste samples (filtrates and extracts
of precipitates; composition not analysed) ranged from 4000 to
> 100 000 mg/litre (Demkowicz-Dobrzanski et al., 1993).
9.1.1.2 Soil
Soil (aquifer) microcosms enriched for methanotrophic activity
transformed up to 90% of the 1-17 mg/litre influent VC with no
apparent toxic effects (Dolan & McCarty, 1995a,b). However, when both
VC and 1,1-dichloroethylene were present, about 75% less
transformation of VC and a marked decrease in methane oxidation rate
was observed (Dolan & McCarty, 1995a). Toxic effects, measured as
decreased methane uptake, were seen during degradation of VC (VC
concentrations: up to saturated solutions; solubility: 2.7 mg/ml) by a
culture of mixed methanotrophs (seeded with soil from a defunct
landfill). A mixture of VC and trichloroethylene (TCE) and a triple
mixture of VC, cis-dichloroethylene (c-DCE) and TCE showed cumulative
toxicity (Chang & Alvarez-Cohen, 1996).
The nitrifying soil bacterium Nitrosomonas europaea had a
turnover-dependent loss of (ammonia-dependent) O2 uptake activity
after co-metabolic transformation of VC (Rasche et al., 1991).
9.1.2 Aquatic organisms
9.1.2.1 Invertebrates
VC reduced the population doubling time of a ciliated protozoon,
Tetrahymena pyriformis, population cultured in vitro (Sauvant et
al., 1995). The IC50 value was 540 mg/litre (8.6 mmol/litre).
During a 96-h assay VC had no effect on the survival of the
free-living nematode Panagrellus redivivus at concentrations ranging
from 10-3 to 10 -8 mol/litre (0.6-62 500 µg/litre), but it reduced the
developmental success of this species. The molting rate from the
fourth larval stage to adult (determined on progeny of 150 to 300
gravid females per assay; three replications) was significantly
decreased relative to controls, and that primarily at VC
concentrations of 6.3-6300 µg/litre (Samoiloff et al., 1980).
The effects of waste effluents from a VC plant described in
section 9.1.1.1 (test with Chlorella sp.) were also tested with the
crustacean Daphnia magna (n = 3 × 10 per experiment; age: 6-24 h).
The test parameter for the latter was lethality, which was determined
by procedures based on ISO 6341-1982 (ISO, 1989). Crude waste produced
a 24-h LC50 value of 80.7 mg/litre (related to VC: 12 mg/litre). The
neutralized wastes (filtrates and extracts of precipitates) were less
toxic, reflected by the higher LC50 values ranging from 445 to
> 100 000 mg/litre (Demkowicz-Dobrzanski et al., 1993).
9.1.2.2 Vertebrates
The acute toxicity of VC to fish was examined with a few
freshwater species; 96-h LC50 values of 1.22 g VC/litre and 1.06 g
VC/litre were reported for bluegill (Lepomis macrochirus) and
largemouth bass (Micropterus salmoides), respectively, but without
giving further details (Hann & Jensen, 1974). Brown et al. (1977)
exposed Northern pikes (Esox lucius) to 388 mg VC/litre; 10 days
after exposure all test animals (n=15) were dead (versus 1 of 20 in
controls during 120 days of observation). However, the test conditions
(e.g., handling of controls, water quality) were not sufficiently
described in this study. Tests of zebra fish ( Brachydanio rerio;
n = 10 per group) according to OECD guideline 203 (OECD, 1984), which
was adapted to volatile chemicals, resulted in LC50 values (based on
mean measured test concentrations) of 240 mg/litre (24 h) or
210 mg/litre (48 h, 72 h and 96 h). The no-observed-effect
concentration (NOEC) regarding mortality was 128 mg/litre (Groeneveld
et al., 1993).
Estimated benchmark values, concentrations believed to be
non-hazardous, to freshwater fish derived by several methods ranged
from 87.8 to 28 879 µg/litre (Suter, 1996). The Tier II secondary
acute value and secondary chronic value were reported to be
1570 µg/litre and 87.8 µg/litre, respectively. The lowest chronic
benchmark for fish was estimated to be 28 879 µg/litre, with a
corresponding fish EC20 of 14 520 µg/litre.
9.2 Field observations
9.2.1 Aquatic organisms
A field study of benthic invertebrates (Dickman et al., 1989;
Dickman & Rygiel, 1993) was carried out during 1986-1991 at 15 sites
of the Niagara River watershed (Canada) near a PVC plant. The VC
discharge in 1986 was estimated to be more than 32 kg (accompanied by
unknown quantities of organotin). The density and diversity of several
invertebrate groups (Amphipoda, Culicoidae, Chironomidae, Isopoda,
Hirudinea, Oligochaeta, Gastropoda, Trichoptera) was found to be low
as compared to a reference site. Chironomids (Diptera) turned out to
be the best indicators. They were absent at the sampling site closest
to the factory's discharge pipe, and their numbers increased with
increasing distances. Those collected nearest to the discharge site
primarily belonged to the genus Polypedilum, which is known to be
pollution tolerant. Nevertheless, individuals of this genus showed a
high proportion of larval mentum (labial plate) deformities (38%
versus 1.7-5.7% at reference sites). Altogether, the frequencies of
deformities at the discharge site fell significantly (P = 0.05) from
47% measured in 1986-1989 to 25% in 1991, which correlated with the
lower levels of VC (figures not given) being released into the river
in 1990-1991 (Dickman & Rygiel, 1993).
10. EVALUATION OF HUMAN HEALTH RISKS AND EFFECTS ON THE ENVIRONMENT
10.1 Evaluation of human health effects
10.1.1 Hazard identification
10.1.1.1 Non-neoplastic effects
a) Human data
i) Skin, connective tissue and bone
Among the non-neoplastic effects of vinyl chloride (VC) exposure,
disorders of the skin, connective tissue and bone are the most
specific and well-characterized. These include acroosteolysis,
Raynaud's phenomenon and sclerodermoid skin lesions. These conditions
were quite common in early studies, which may have involved exposures
to high levels of several hundred ppm. Acroosteolysis occurred
primarily in PVC production workers who had been involved in reactor
cleaning. Data on mortality from connective tissue disorders, a
relatively rare category of death, have not been provided in published
studies, nor are there sufficient data to estimate an
exposure-response relationship.
ii) Non-neoplastic liver disease
Non-neoplastic liver disease was also well-documented in early
studies of workers with high levels of exposure to VC. In one
carefully described study, "advanced portal hypertension" with
histological findings of non-cirrhotic fibrosis was diagnosed in 17
out of 180 VC polymerization workers. Histological alterations
reported in liver biopsy specimens obtained from VC monomer workers in
another series included focal hepatocytic hyperplasia and focal mixed
hyperplasia; these lesions were infrequent in the comparison group.
Reversible changes in liver function tests have been reported in VC
workers exposed to 2.6-54 mg/m3 (1-21 ppm). Despite evidence for the
induction of non-malignant liver disease from clinical studies, there
has been no evidence of excess mortality from studies of large
cohorts. There are also insufficient data to estimate exposure
response.
iii) Respiratory disease
There is some evidence for respiratory effects from both
morbidity and mortality studies of VC workers, but this may be related
to PVC-resin dust rather than VC monomer. Positive findings from
mortality studies are limited to one cohort. Therefore, the literature
does not clearly implicate respiratory disease as an effect of
exposure to VC monomer.
iv) Reproductive effects
Although animal studies implicate the reproductive system as a
potential target organ for vinyl chloride toxicity, studies in humans
have not been adequate to confirm these effects or to rule them out.
v) Cardiovascular disease
A few morbidity studies have reported an elevated incidence of
circulatory diseases among VC-exposed workers. Cardiovascular disease
mortality is significantly lower than expected in the two large
cohorts.
vi) Susceptible subpopulations
There is no strong evidence for the existence of susceptible
sub-populations in humans. It has been suggested that specific HLA
types and genetic polymorphisms in VC-metabolizing enzymes may be
factors in human susceptibility to VC-related diseases.
b) Experimental animal data
VC has been tested in several animals species and strains for
acute, short-term and long-term effects. Signs of acute toxicity
include congestion of internal organs, neurotoxic effects and
circulatory disturbances. In short-term studies VC caused mainly liver
damage, degenerative changes in the testes, degeneration and
inflammation in the lungs and degenerative lesion in the kidneys.
Immunological alterations in connection with VC exposure have also
been described. Reproductive performance or fertility were not
affected.
10.1.1.2 Neoplastics effects
a) Human data
In 1974, a case report associated exposure to VC with the
occurrence of angiosarcoma of the liver (ASL). Further case series and
small epidemiological studies were reported shortly thereafter. At a
later stage two studies, one in the USA and one in Europe, combined
data from those studies and updated the mortality follow-up. Apart
from these two large studies, four more smaller studies have been
conducted and fully reported and were also considered by the Task
Group.
There is a five-fold excess risk for liver cancer among workers
exposed to VC, mostly in PVC polymerization plants where the highest
exposures to VC occurs. The largest part of this excess risk is due to
the excess risk for ASL. In the European study, there was a 45-fold
excess risk for ASL in workers exposed to more than 10 000 ppm-years
compared to those exposed to less than 2000 ppm-years. In the European
study histopathological confirmation was available for 17 out of 24
liver cancers; of those confirmed 16 were ASL. In the USA study 21 out
of 37 liver cancer deaths were registered as ASL. In the Canadian
study all 8 liver cancer deaths were ASL, as was the case for the 3
liver cancer deaths in the French study. No information is available
concerning ASLs in the German study while no liver cancers were
diagnosed in the Russian study. It is probable that some ASLs
occurring among VC-exposed workers have remained undiagnosed and have
been coded in the death certificates as liver cancers unspecified or
as other liver-associated disease.
Several studies examined the risk specifically for hepatocellular
carcinoma (HCC) of the liver. No excess was observed in the European
study, nor in the Canadian or French studies. Data from the USA study
and also from the update of the Italian component of the European
cohort seem, however, to indicate that there is also an excess for
HCC. This is of smaller magnitude than that observed for ASL. The
interpretation of the results for HCC is complicated because of the
uncertainties concerning the accuracy of diagnosis of angiosarcomas
and HCC using death certificate information and the absence of compete
histopathological confirmation of the diagnoses of liver cancer in the
studies. Although the results are not fully consistent between
studies, the data suggest that there may be a small excess risk for
HCC.
Four out of five studies reporting results for brain tumours
identified a moderate excess risk with an SMR of 1.42 for the combined
data from five studies (43 observed, 95% CI 1.03-1.91). The risk
tended to increase with duration of exposure/employment in the
European and USA study. Furthermore, in the USA study, the highest
risk occurred in the two plants where most ASL cases had been
diagnosed, and where presumably the highest VC exposure had occurred.
In the European study where the dose-response relationship was
examined, no association was seen with cumulative exposure to VC. The
overall epidemiological evidence is suggestive of a possible risk
among VC workers.
Some of the smaller early studies had reported an increase in
lung cancer among VC workers. There was no indication of an excess
risk, however, in the two largest studies (European and USA) or in the
other four smaller studies. There was no association either with
duration of exposure/employment in the USA or the European study, nor
with cumulative exposure to VC in the European study.
Excess risk for malignant lymphomas had been reported in some
early small studies. No excess risk, however, was observed in the two
largest cohorts (USA and European) or in the Canadian cohort. An
excess risk for the combined category leukaemia and lymphoma was
observed in the Russian and the German study. In interpreting these
results it should be noted that the studies used different disease
classification and in some occasions grouped lymphomas with malignant
myeloma. The overall results exclude the presence of any large
increased risk for lymphomas or leukaemia.
b) Experimental animal data
VC is a multispecies, multiorgan carcinogen. It induces benign
and malignant tumours in several organs of various species, both in
males and females. The most prominent feature is the induction of the
rare angiosarcomas, especially in the liver. This phenomenon has been
demonstrated in different strains of mice and rats, as well as in
hamsters. Other tumours include nephroblastomas and neuroblastomas,
hepatocellular carcinomas, mammary gland carcinomas and lung adenomas.
VC is genotoxic, causing alterations and damage to DNA. VC
metabolites bind to the DNA yielding promutagenic DNA adducts.
Mutations and chromosomal abnormalities have been described in many
in vitro and in vivo systems. These genotoxic processes play a
significant role in tumorigenesis, while cell proliferation, secondary
to the VC-induced cell toxicity, may also be involved in the process.
10.1.2 Dose-response analysis
10.1.2.1 Non-neoplastic effects
a) Human data
The published studies examining non-neoplastic diseases in
VC-exposed subjects do not provide dose-response information.
b) Experimental animal data
Several short-term and long-term studies have been assessed for
establishing the NOAEL of VC. Liver cell polymorphism (variation in
size and shape of hepatocytes and their nuclei) reported in the
feeding study by Til et al. (1991) is recommended for quantifying
non-cancer risks from oral exposure to VC. The severity and incidences
of this liver lesion were statistically significantly increased at a
daily dose of 1.3 mg/kg body weight but not at 0.13 mg/kg per day
(Table 46), the no-observed-adverse-effect level (NOAEL) therefore
being 0.13 mg/kg per day. Basophilic liver cell foci induction in the
study of Til et al. (1991) occurred at lower concentrations, but it
was not considered by the Task Group to be a manifestation of
hepatocytic toxicity.
A lowest-observed-adverse-effect level (LOAEL) of 26 mg/m3 was
established based on a subchronic (3-12 months) inhalation toxicity
study in male rats by Bi et al. (1985). The basis for the estimation
was the increased relative liver weight and mild testicular
degeneration (Table 47), both effects being more pronounced in a
dose-related manner at the two higher dose levels tested (260 and
7800 mg/m3). A recent inhalation two-generation reproduction study
with VC in rats, using exposure concentrations of 26, 260 and 2860
mg/m3 (Shah, 1998), yielded the same LOAEL value. In this study,
increased relative liver weights and hypertrophy of centrilobular
hepatocytes were found in parental animals at all dose levels and a
dose-dependent manner.
10.1.2.2 Neoplastic effects
a) Human data
In most epidemiological studies dose-response analyses were not
available. Of the two largest studies conducted in the USA and Europe,
the USA study examined only duration of exposure and did not examine
dose-response. In the European study a calendar period-, job- and
plant-specific job exposure matrix was used. Expert judgement was used
to estimate exposures for the early time periods when no measurements
were available. The use of such a matrix implies that, when assigning
the exposure index to individuals in the cohorts, a certain degree of
misclassification occurs tending to underestimate the strength of a
dose-response relationship.
All of the results in this section are from the European study
(Simonato et al., 1991) unless otherwise noted. A clear dose-response
relationship was found only for ASL alone or in combination with other
primary liver cancers.
In all the analyses of liver cancer in the European cohort there
is a clear relationship with increasing estimated doses. The effect is
most evident for ASL, for which the risk estimate in the highest
exposure groups is two and half times larger than for all liver
cancers (Tables 48 and 49). These results are consistent with the
analyses published by Wu and colleagues in the updating of one of the
USA subcohorts. In this study, the average cumulative exposure score
to VC was 6 times higher for ASL cases than for other liver cancer
cases and control subjects.
Results from the regression models in the European study
indicated that cumulative dose appears as the most powerful
determinant of ASL, followed by time since first exposure. This can
also be seen from Table 50. The results from the regression models
indicated that there was only a small or no effect of calendar period
and age of hire. Both duration and intensity of exposure correlated
with liver cancer risk (see Table 41).
Dose-response analyses for lung cancer, brain tumours and
lymphomas are reported in section 8.3.
b) Experimental animals data
Studies using short- and long-term oral and inhalation exposure
were carried out to assess the carcinogenic activity of VC (section
7.7.) For assessing carcinogenic risk, pivotal studies have been
selected where the study design, exposure levels, observation period
and histopathological examinations are regarded as suitable for
assessing risk.
Table 46. Type and incidence of treatment-related liver-cell polymorphism
of rats orally exposed to vinyl chloride (from Til et al., 1991)
Males Females
N (total/group) 100 100 100 50 100 100 100 50
Dose mg/kg 0 0.014 0.13 1.3 0 0.014 0.13 1.3
per day
Liver cell No of animals with liver cell polymorphism of
polymorphisms varying severity
Slight 27 23 26 19 46 41 49 23
Moderate 4 4 7 10b 14 13 8 15a
Severe 1 1 1 3 2 3 4 9c
a P < 0.05
b P < 0.01
c P < 0.001
Table 47. Damage of testes induced by different concentration
of vinyl chloride (from Bi et al., 1985)
Group No. of Slight Moderate Severe Total %
animals damage damage damage
Control 74 9 3 2 14 18.9
10 ppm a 74 14 5 3 22 29.7
100 ppm b 74 19 5 3 27 36.5 d
3000 ppm c 75 29 8 5 42 56.0 e
a 26 mg/m3
b 260 mg/m3
c 7770 mg/m3
d P < 0.05
e P < 0.001
Table 48. Maximum likelihood estimates for final model with
cumulative exposure and years since first employment
for deaths from liver cancer (N = 24) a
Exposure variable Relative risk 95% confidence interval
Cumulative exposure
(ppm-years)
< 500 1.0
500-1999 1.2 0.1-11.4
2000-5999 4.6 1.0-21.0
6000-9999 12.2 2.5-59.6
> 10 000 17.1 3.1-93.6
Years since first employment
0-19 1.0
20-24 5.6 1.4-22.4
> 25 6.8 1.7-27.4
a From: Simonato et al. (1991)
Table 49. Maximum likelihood estimates for final model with
cumulative exposure and years since first employment
for deaths from angiosarcoma of the liver (N = 22) a
Exposure variable Relative risk 95% confidence interval
Cumulative exposure (ppm-years)
< 2000 1.0
2000-5999 6.8 1.1-41.7
6000-9999 24.7 4.1-150.1
>10 000 45.4 7.3-281.1
Years since first employment
0-19 1.0
20-24 4.7 1.0-22.8
>25 6.2 1.4-29.0
a From: Simonato et al. (1991)
Table 50. Absolute risk of angiosarcoma of the liver
per 100 000 (Simonato et al., 1991)
Cumulative exposure (ppm-years)
Years since < 2000 2000-5999 6000-9999 >10 000
first employment
0-19 1.0 6.8 24.4 44.8
20-24 4.7 32.0 115.6 212.5
>25 6.2 42.2 152.3 280.0
For oral exposure, it is recommended that female rats from the
Feron et al. (1981) study be used to quantify risk for VC, because the
incidence of liver tumour-bearing animals is greater than in males,
giving a more conservative estimate. Rats with either neoplastic liver
nodules, hepatocellular carcinomas and/or ASLs should be included for
quantification. These lesions are all dose-related. The neoplastic
nodules, while not malignant, have the potential to progress to
malignancy. While it is uncertain that VC induces tumour types other
than ASL in humans, tumour type concordance is not considered to be
necessary, and thus inclusion of all liver tumours is desirable as a
conservative approach.
If quantification is desired for ASL only, then the male rat is
recommended because the incidences for this end-point are higher in
males (Table 51). Including animals with angiosarcomas in other organs
is unnecessary because the only other site with significantly
increased incidences of this tumour type is the lung, and, with one
exception, all animals with ASL also had liver tumours.
For inhalation exposure, several studies have been conducted in
different species (section 7.7). Four experiments with similar design
and experimental conditions, carried out by Maltoni et al. (1981,
1984) in rats, were used for assessing carcinogenic risk after VC
inhalation exposure. Out of the treatment-related carcinogenic
responses, ASL, all vascular tumours of the liver and all liver
tumours combined were considered to be suitable for assessing risk
(Table 52). Induction of ASL is a special feature of VC and since this
tumour type was also observed in exposed humans, this end-point was
considered especially relevant for quantification of risk.
For ASL, increased incidence was observed in both males and
females. A clear dose-response relationship could be established in
female rats, and the effect was also suggestive in males. A similar
trend could be observed if all vascular tumours or all liver tumours
were considered. The other tumours, however, occurred only at low
incidences. The lowest dose at which ASL and other vascular tumours
were detected was 26 mg/m3 in females and 65 mg/m3 in males.
Table 51. Tumour incidences in oral feed study with vinyl chloride
in Wistar rats (from Feron et al., 1981)
Dose Neoplastic Hepato-cellular Liver Liver Mammary
(mg/kg liver carcinomas angio-sarcomas tumour-bearing adeno-carcinomas
bw) nodules rats
Females
0 2/57 0/57 0/57 2/57 3/57
1.7 26/57 4/58 0/57 28/58 2/58
5.0 39/59 19/59 2/59 49/59 4/59
14.1 44/57 29/57 9/57 56/57 7/57
Males
0 0/55 0/55 0/55 0/55
1.7 1/58 1/58 0/58 2/58
5.0 7/56 2/56 6/56 11/56
14.1 23/59 8/59 27/59 41/59
Table 52. Liver tumours in inhalation studies on rats
(from Maltoni et al., 1981, 1984)
Exposure Exposure Number of % ASL % liver % all liver
(mg/m3, (mg/m3, animals vascular tumours b
4 h/d, continuous) a tumours b
5 d/w)
Males
0 0 222 0 0 0
2.6 0.3 58 0 0 0
13 2 59 0 0 0
26 3 59 0 0 0
65 8 60 1.7 1.7 1.7
130 15 174 1.1 2.3 2.3
260 31 60 0 1.7 1.7
390 46 60 1.7 1.7 1.7
520 62 60 11.7 15.0 16.0
650 77 29 3.4 3.4 4.4
1300 155 30 0 0 0
6500 774 30 20.0 20.0 20.0
15 600 1857 29 10.3 10.3 10.3
26 000 3095 30 10.0 10.0 11.0
Females
0 0 239 0 0 0
2.6 0.3 60 0 0 0
13 2 60 0 0 0
26 3 60 6.7 6.7 6.7
65 8 60 6.7 8.3 8.3
130 15 180 7.2 10.6 10.6
260 31 60 1.7 1.7 1.7
390 46 60 8.3 8.3 8.3
520 62 60 8.3 11.7 15.0
650 77 30 6.7 10.0 10.0
1300 155 30 20.0 20.0 36.7
6500 774 30 23.3 23.3 30.0
15 600 1857 30 33.3 40.0 43.3
26 000 3095 30 13.3 13.3 13.3
a Exposure calculated to correspond to 24 h/day, 7 days/week
b % of animals with tumours were added, because no individual
animal data were available.
Nephroblastomas and neuroblastomas have also been observed in
some studies in rats after VC exposure. These rare tumours appeared in
a dose- and time-dependent manner; the doses required for tumour
induction were, however, higher than those for ASL.
Several inhalation studies of VC in mice have been carried out.
The induced spectrum of tumours was similar to that in rats:
angiosarcomas and angiomas of the liver and other organs, mammary
gland and lung tumours. In one study (Maltoni et al., 1981, 1984), a
clear dose-response relationship was observed for ASL as well as for
all vascular tumours in the female mice (Table 53). The lowest
carcinogenic dose was 130 mg/m3, higher than that in rats.
Mammary gland adenocarcinomas were observed in several VC
studies. Four inhalation studies with VC in Sprague-Dawley rats
(Maltoni et al., 1981, 1984) were evaluated (Table 54). A
statistically significantly increased incidence in mammary gland
carcinoma was observed in three of the four studies. However, in none
of these studies was a clear dose-response relationship observed.
Increased incidences of mammary carcinomas, without a dose-response
relationship, were also observed in Swiss mice after inhalation
exposure (Maltoni et al., 1981, 1984). Drew et al. (1983) exposed
Fischer-344 rats to 0 or 261 mg/m3 either for 6 or 12 months.
Increased incidences of mammary adenocarcinomas were observed in the
animals exposed at different ages but, since only one exposure level
was used, a dose-response relationship could not be established. In
Syrian hamsters, using similar study design but an exposure level of
520 mg/m3 (Drew et al., 1983), a high incidence of mammary tumours
was observed in animals exposed early in life.
In two other mouse studies (Lee et al., 1978; Drew et al., 1983),
elevated incidences of both mammary gland carcinomas and other mammary
tumours were observed. However, these studies were also inadequate for
risk assessment. In summary, increased incidence of mammary gland
carcinomas were observed in several experiments where animals were
exposed to VC, either orally or by inhalation, indicating
treatment-related carcinogenic effect. However, the lack of
dose-response or the use of a single dose excluded the use of these
data for risk assessment.
10.1.3 Human exposure
The aim of this section is to estimate present levels of exposure
to VC of the general population and of subpopulations with special
exposure, such as those living near point sources of VC and those
exposed at work. The purpose is to indicate worst-case scenarios
rather than average or most likely exposures.
The data available are very limited both in time and space. Often
the distance of the sampler from the point source or the sampling and
averaging times have not been reported. This makes the comparison of
results from different studies difficult.
Table 53. Incidences of different tumour types in Swiss mice after inhalation exposure to vinyl chloride
(from Maltoni et al., 1981, 1984 BT4)
Exposure Effective ASL Angiosarcoma Angioma Angioma Total Mammary Lung
(mg/m3) number (other organs) (liver) (other vascular gland adenoma
organs) tumours
Males
0 80 0 0 0 1 1 0 8
130 30 1 1 0 1 3 0 3
650 30 9 2 6 2 17 0 24
1300 30 6 2 1 1 10 1 24
6500 29 6 4 2 0 12 0 18
15 600 30 2 0 2 2 6 0 23
26 000 26 1 0 1 2 4 0 20
Females
0 70 0 1 0 0 1 1 7
130 30 0 0 1 4 5 13 3
650 30 9 1 5 1 17 12 17
1300 30 8 5 4 2 10 9 26
6500 30 10 4 3 1 12 10 22
15 600 30 11 1 5 1 6 9 24
26 000 30 9 1 5 2 4 14 26
Table 54. Incidence of mammary gland carcinomas in Sprague-Dawley
rats and Swiss mice after inhalation exposure to vinyl
chloride (from Maltoni et al., 1981, 1984) a
Exposure BT15 BT1 BT2 BT9 BT4
(mg/m3) (rat) (rat) (rat) (rat) (mouse)
0 6/60 (10) 0/29 1/100 (1) 5/50 (10) 1/70 (1.4)
2.6 12/60 (20)
13 22/60 (36.6) d
26 21/60 (35) c
65 15/60 (25)
130 2/30 (6.6) 59/150 (39) d 13/30 (43.3) d
260 3/60 (5)
390 6/60 (10) c
520 5/60 (8) b
650 2/30 (6.6) 12/30 (40) d
1300 1/30 (3.3) 9/30 (30) d
6500 2/30 (6.6) 10/30 (33.3) d
15 600 0/30 9/30 (30) d
26 000 3/30 (10) 14/30 (46.6) c
a Values are no. of tumour / no. of animals, with percentages in parentheses; statistical
significance compared to the concurrent control group (two tailed Fischer)
b P < 0.05
c P < 0.01
d P < 0.001
10.1.3.1 General population
The main sources of VC exposure for the general population are
VC/PVC plants, biodegradation of chlorinated solvents in landfills,
spills of chlorinated solvents from, for instance, dry cleaning shops,
and the metal and electronic industry. In addition, small amounts of
VC may be liberated from PVC.
There are no known natural sources of VC. Concentrations of VC in
ambient air samples were reported to range from < 0.013 to 26 µg/m3.
These data were collected in the 1970s and 1980s from rural, remote,
suburban and urban sites. The concentrations measured in rural and
suburban/urban sites were similar. There is a lack of recent data on
ambient air, making it difficult to conclude whether there has been a
historical reduction in ambient concentrations. The exposure to VC via
inhalation by the general population (non-occupational exposure) is
estimated not to exceed 10 µg/kg body weight per day, using the
derivation method described in Environmental Health Criteria 170:
Assessing Human Health Risks of Chemicals: Derivation of Guidance
Values for Health-based Exposure Limits (IPCS, 1994).
Concentrations (probably daily means) of VC measured in ambient
air from industrial areas were reported to be < 8000 µg/m3 in the
1970s and < 200 µg/m3 and 4800 µg/m3 in the 1980s in different
geographical areas. Monthly mean concentrations measured in the
vicinity of one plant during the 1990s ranged from 0.1 to 1.3 µg/m3.
Exposure of the general population living in the vicinity of
industrial areas was estimated not to exceed 3000 µg/kg per day in the
1970s, 70-1800 µg/kg per day in the 1980s and 0.5 µg/kg per day in the
1990s.
VC concentrations in air sampled from the vicinity of waste sites
ranged from < 0.08 to 104 µg/m3. These concentrations were reported
in air sampled during the 1980s and 1990s; no historical trend could
be established. Exposure of the general population living in the
vicinity of landfills was estimated to be < 300 µg/kg per day.
There are no data on VC concentrations in air from other areas
contaminated with chlorinated hydrocarbons.
Sources of VC in indoor air have included PVC products, smoking,
paints and aerosol propellants. Up to the 1970s, the VC concentration
in PVC exceeded 1000 mg/kg, and consequently VC was detected in indoor
air and cars. Since the late 1970s the VC content in PVC has been
regulated to approximately 1 mg/kg, therefore VC concentrations are
now unlikely to be significant in indoor air, except where there are
nearby external sources such as landfills, etc.
The general population may be exposed to VC in drinking-water. In
the majority of water samples analysed, VC was not present at
detectable concentrations. The maximum VC concentration reported in
drinking-water was 10 µg/litre, leading to a maximum exposure of
0.2 µg/kg per day. There is a lack of recent data on concentrations in
drinking-water, but these levels are expected to be below 10 µg/litre.
In groundwater near point sources, very high (up to 200 mg/litre)
concentrations of VC have been observed and well water nearby may be a
very significant source of VC: daily intakes may reach 5000 µg/kg per
day. Some recent studies have identified VC in PVC-bottled
drinking-water at levels below 1 µg/litre. The frequency of occurrence
of VC in such water is expected to be higher than in tap water.
Dietary exposure to VC from PVC packages used for food has been
calculated by several agencies, and, based upon estimated average
intakes in the United Kingdom and USA, an exposure of < 0.0004 µg/kg
per day was estimated for the late 1970s and early 1980s.
Exposure of the general population may be higher in situations
where large amounts of VC are accidentally released to the
environment, such as a spill during transportation. However, such
exposure is likely to be transient.
10.1.3.2 Occupational exposure
Staff working in plants where VC is manufactured or polymerized
into PVC or at landfills may be occupationally exposed to VC.
Occupational exposure to VC has continually decreased since the 1940s.
Since the 1970s, occupational exposure level (OEL) values have been
set at 2.6 to 18 mg/m3 in most European countries and the USA. Few
data are available to determine how frequently the actual workplace
exposures are within the OEL. Where data are available, compliance
during the 1990s was reported in some European countries. Based upon
an OEL of 18 mg/m3, an occupational exposure of 3000 µg/kg per day
can be calculated. The most recently reported average occupational
exposure in these countries was 0.3 mg/m3. This gives a daily dose of
50 µg/kg per day. However, concurrent exposure levels of more than
300 mg/m3 have also recently been reported in some countries.
Exposure to VC in PVC processing plants has been historically, and is
probably currently, much lower than in PVC/VC production plants.
10.1.4 Risk characterization
VC has been shown to be carcinogenic and toxic in both oral and
inhalation experimental bioassays, as well as in human epidemiological
studies. Experimental studies have also shown that it is genotoxic and
must be activated through a rate-limited pathway. In order to perform
quantitative risk assessment using animal bioassays, it will be
necessary to utilize a physiologically based pharmacokinetic model
(PBPK) to derive the concentration of active metabolite at the
critical target site, the liver, as well as to extrapolate dose from
animals to humans. The PBPK model should be validated, taking into
account the known metabolic pathways and using metabolic constants
determined experimentally, and should not introduce too many unknown
parameters. The Task Group was not in a position to evaluate
accurately the available pharmacokinetic models for their suitability
for risk assessment. Furthermore, in order to estimate the risk
predictions from epidemiological studies, original data on exposed
human populations, at the level of the individual, are required.
Some of the published PBPK models and risk assessments are
reviewed in Annex 2.
10.2 Evaluation of effects on the environment
VC is released to the environment from plants where it is
manufactured and/or used in the production of PVC. The most important
releases are emissions to the atmosphere, but VC may also be released
in waste water. It may also be formed in the environment as a product
of the biodegradation of chlorinated C2 solvents. This route of VC
formation may lead to emissions of VC from areas contaminated with
chlorinated C2 solvents, such as landfills.
The maximum VC concentration reported in surface water is
0.6 mg/litre. VC is unlikely to be bioaccumulated to a significant
extent in aquatic organisms.
The only toxicity study which was deemed of sufficient quality to
evaluate the effects of VC on aquatic organisms gave a 48-h LC50 of
210 mg/litre for a freshwater fish. There is a paucity of chronic
toxicity data for aquatic organisms. Despite these limitations, owing
to the rapid volatilization of VC, the low concentrations in surface
water and the low acute toxicity to fish, VC is not expected to
present a hazard to aquatic organisms.
There is a paucity of data for terrestrial organisms.
11. RECOMMENDATIONS FOR PROTECTION OF HUMAN HEALTH
11.1 Public health
The following measures should be taken:
* worldwide application of production technologies leading to low
residual VC levels in PVC;
* implementation and enforcement worldwide of steps that guarantee
minimal emissions of VC at production sites;
* identification, surveillance and emission and exposure control of
contaminated areas such as landfill sites.
11.2 Occupational health
Since VC is a genotoxic carcinogen, exposures should be kept as
low as possible, using the best available technology worldwide.
More information, education and training of workers potentially
exposed to VC regarding the risks involved and safe working procedures
and habits is required.
Monitoring and record-keeping of exposure and record-keeping of
exposed workers are needed.
12. FURTHER RESEARCH
The following topics need further research:
* follow-up of existing VC cohorts with quantitative exposure
assessment;
* epidemiological studies of populations with defined and low
exposure to VC;
* evaluation of the efficacy and usefulness of the medical
surveillance of VC-exposed workers;
* validation of effects and susceptibility biomarkers for VC;
* the role of post-DNA-damage events in the carcinogenicity of VC
and their relationships with target organ or cell type
specificity;
* evaluation of the sensitivity to VC in early childhood and
determination of possible mechanisms involved;
* the possibility of carcinogenicity of VC in mammary gland tumours
and the mechanism involved;
* optimization of remediation of contaminated sites;
* studies on the toxicity of VC to aquatic and terrestrial
organisms.
13. PREVIOUS EVALUATIONS BY INTERNATIONAL BODIES
Vinyl chloride was evaluated by the International Agency for
Research on Cancer (IARC, 1979) and the evaluation was updated in
Supplement 7 (IARC, 1987). There was sufficient evidence for
carcinogenicity of vinyl chloride in humans and sufficient evidence
for carcinogenicity in animals; the overall evaluation was that vinyl
chloride is carcinogenic to humans.
In the WHO Guidelines for Drinking-water Quality (WHO, 1996),
using results from the rat bioassay of Til et al. (1983) and applying
the linearized multistage model, the human lifetime exposure for a
10-5 excess risk of ASL was calculated to be 20 µg/day. It was also
assumed that in humans the number of cancers at other sites may equal
that of ASL, so that a correction (factor of 2) for cancers other than
ASL is justified. VC concentrations in drinking-water of 5 µg/litre
were calculated as being associated with excess risks of 10-5.
REFERENCES
ACGIH (1999) Documentation of the threshold limit values and exposure
indices. Cincinnati, Ohio, American Conference of Governmental and
Industrial Hygienists Inc.
Adkins B, Van Stee EW, Simmons JE, & Eustis SL (1986) Oncogenic
response of strain A/J mice to inhaled chemicals. J Toxicol Environ
Health, 17: 311-322.
Ahlborg G, Bjerkedal T, &Eg J (1987) Delivery outcome among women
employed in the plastics industry in Sweden and Norway. Am J Ind Med,
12: 507-517.
Albanus L, Gawell B-M, Larsson B, & Slorach SA (1979) [The Swedish
National Food Administration's decision to reduce the maximum
permitted level of vinyl chloride monomer in foodstuffs.] Var Foeda,
31: 161-168 (in Swedish).
Allen MR, Braithwaite A, & Hills CC (1997) Trace organic compounds in
landfill gas at seven UK waste disposal sites. Environ Sci Technol,
31: 1054-1061.
Allsopp MW & Vianello G (1992) Poly(vinyl)chloride. In: Elvers B,
Hawkins S, & Schulz G ed. Ullmann's encyclopedia of industrial
chemistry, 5th completely revised. -- Volume A21: Plastics, properties
and testing to polyvinyl compounds. Weinheim, Germany, VCH
Verlagsgesellschaft mbH, pp 717-742.
Andersen M, Clewell H, Gargas M, Smith FA, & Reitz RH (1987)
Physiologically based pharmacokinetics and the risk assessment process
for methylene chloride. Toxicol Appl Pharmacol, 87: 185-205.
Anderson D & Richardson CR (1981) Issues relevant to the assessment of
chemically induced chromosome damage in vivo and their relationship
to chemical mutagenesis. Mutat Res, 90: 261-272.
Anderson D, Hodge MCE, & Purchase IFH (1976) Vinyl chloride: Dominant
lethal studies in male CD-1 mice. Mutat Res, 40: 359-370.
Anderson D, Hodge MCE, & Purchase IFH (1977) Dominant lethal studies
with the halogenated olefins vinyl chloride and vinylidene dichloride
in male CD-1 mice. Environ Health Perspect, 21: 71-78.
Anderson D, Richardson CR, Weight TM, Purchase IFH, & Adams WGF (1980)
Chromosomal analyses in vinyl chloride exposed workers. Results from
analysis 18 and 42 months after initial sampling. Mutat Res, 79:
151-162.
Anderson D, Richardson CR, Purchase IFH, Evans HJ, & O'Riordan ML
(1981) Chromosomal analysis in vinyl chloride exposed workers:
Comparison of the standard technique with the sister-chromatid
exchange technique. Mutat Res, 83: 137-144.
Andersson Y & Ljungström E (1989) Gas phase reaction of the NO3
radical with organic compounds in the dark. Atmos Environ, 23:
1153-1155.
Ando M & Sayato Y (1984) Studies on vinyl chloride migrating into
drinking water from polyvinyl chloride pipe and reaction between vinyl
chloride and chlorine. Water Res, 18: 315-318.
Andreoli A (1985) Hazardous waste site investigations: Vinyl chloride
contamination of groundwater. In: Suffolk County Department of Health
Services ed. Proceedings of The Second Annual Eastern Regional Ground
Water Conference. New York, Suffolk County Department of Health
Services, pp 482-489.
Anghelescu F, Otoiu M, Dobrinescu E, Hagi-Paraschiv-Dossios L,
Dobrinescu G, & Ganea V (1969) Clinico-pathogenic considerations on
Raynaud`s phenomenon in the employees of the vinyl polychloride
industry. Med Interna, 21: 473-482.
Anna K & Alberti J (1978) [Source and use of organic halogenated
compounds and their distribution in water and wastewater.] In: Anna K
& Alberti J ed. [Water '77.] Berlin, Kongressvorträge Colloquium
Verlag, pp 154-158 (in German)
Anon (1973) FDA to propose ban on use of PVC for liquor use. Food Chem
News, 14: 3-4.
Anon (1981) [Measurement of hydrocarbon levels in ambient air in
Cologne: Report from the Environmental Office of the City of Cologne.]
Cologne, Germany, City of Cologne, pp 35-56 (in German).
Anon (1993) Quality standards for foods with no identity standards:
bottled water. Fed Reg, 58: 378-381.
Anon (1996) [ Dangerous substance transport accident -- train -- on
1st June in Schönbeck/Elbe. Train accident with resulting fire and
danger to the neighbourhood.] Heyrothsberge, Germany, Sachsen-Anhalt,
200 pp (Unpublished Conference report) (in German).
Arbin A, Östelius J, Callmer K, Sroka J, Hänninen K, & Axelsson S
(1983) Migration of chemicals from soft PVC bags into intravenous
solutions. Acta Pharm Suec, 3: 20-33.
Arneth JD, Schleyer R, Kerndorff H, & Milde G (1988) [Standardised
evaluation of groundwater contaminants from existing chemical
pollutants.] Bundesgesundheitsblatt, 31: 117-123 (in German).
Ashworth RA, Howe GB, Mullins ME, & Roger TN (1988) Air-water
partitioning coefficients of organics in dilute aqueous solutions. J
Hazard Mater, 18: 25-36.
Association of Plastics Manufacturers in Europe (1999) Management
report from the APME register of cases of angiosarcoma related to PVC
production. Brussels, Association of Plastics Manufacturers in Europe,
33 pp (Unpublished report).
ASTM (1993) Standard test method for vinyl chloride in workplace
atmospheres (charcoal tube method): D/4766-88. Philadelphia,
Pennsylvania, American Society for Testing and Materials, pp 323-328.
ASTM (1985) Analysis for determining residual vinyl chloride in ppb
range in vinyl chloride homo- and copolymers by headspace gas
chromatography. In: ASTM method D/4443-84. Philadelphia, Pennsylvania,
American Society for Testing and Materials.
Atkinson R (1985) Kinetics and mechanisms of the gas-phase reactions
of the hydroxyl radical with organic compounds under atmospheric
conditions. Chem Rev, 84: 69-201.
Atkinson R (1987) A structure-activity relationship for the estimation
of rate constants for the gas-phase reactions of OH radicals with
organic compounds. Int J Chem Kinet, 19: 799-828.
Atkinson R & Aschmann SM (1987) Kinetics of the gas-phase reactions of
Cl atoms with chloroethenes at 298 +/- 2 K and atmospheric pressure.
Int J Chem Kinet, 19: 1097-1105.
Atkinson R & Carter WPL (1984) Kinetics and mechanisms of the
gas-phase reactions of ozone with organic compounds under atmospheric
conditions. Chem Rev, 84: 437-470.
Atkinson R & Pitts JN (1977) Rate constants for the reaction of
O(3 P) atoms with CH2=CHF, CH2=CHCL, and CH2=CHBr over the
temperature range 298-442°K. J Chem Phys, 67: 2488-2491.
Atkinson R, Darnall KR, Lloyd AC, Winer AM, & Pitts JN (1979) Kinetics
and mechanisms of the reactions of the hydroxyl radical with organic
compounds in the gas phase. In: Pitts JN, Hammond GS, Gollnick K, &
Grosjean D ed. Advances in photochemistry. New York, John Wiley &
Sons, vol 11, pp 375-488.
Atkinson R, Aschmann SM, & Goodman MA (1987) Kinetics of the gas-phase
reactions of NO3 radicals with a series of alkynes, haloalkenes, and
alpha, beta-unsaturated aldehydes. Int J Chem Kinet, 19: 299-307.
Atri FR (1985) [Vinyl chloride.] In: Barndt D ed. [Chlorinated
hydrocarbons in the environment III.] Berlin, Technical University, pp
134-189 (Landscape Development and Environmental Research Series No.
34) (in German).
ATSDR (Agency for Toxic Substances and Disease Registry) (1990) ATSDR
case studies in environmental medicine: Vinyl chloride toxicity. Clin
Toxicol, 28: 267-285.
ATSDR (Agency for Toxic Substances and Disease Registry) (1997)
Toxicological profile for vinyl chloride - Update. Atlanta, Georgia,
Agency for Toxic Substances and Disease Registry, 239 pp.
Baek NH, Jaffé PR, & Shingal N (1990) Stimulating the degradation of
TCE under methanogenesis. J Environ Sci Health Environ Sci Eng, 25:
987-1005.
Baker LW & MacKay KP (1985) Hazardous waste management: Screening
models for estimating toxic air pollution near a hazardous waste
landfill. J Air Pollut Control Assoc, 35: 1190-1195.
Baker MT & Ronnenberg WC (1993) Contrasting effects of
1,1,1-trichloroethane on [14C]vinyl chloride metabolism and
activation in hepatic microsomes from phenobarbital- and
isoniazid-treated rats. Toxicol Appl Pharmacol, 119: 17-22.
Ballering LAP, Nivard MJM, & Vogel EW (1996) Characterization by
two-endpoint comparisons of the genetic toxicity profiles of vinyl
chloride and related etheno-adduct forming carcinogens in Drosophila.
Carcinogenesis, 17: 1083-1092.
Banzer JD (1979) The migration of vinyl chloride monomer from PVC pipe
into water. J Vinyl Technol, 1: 164-167.
Bao YS, Jiang H, & Liu J (1988) [The effects of vinyl chloride on
pregnancy parturition and fetal development among female workers.]
Chin J Prev Med, 22: 343-346 (in Chinese).
Barbacid M (1987) Ras genes. Ann Rev Biochem, 56: 779-827.
Barbin A (1998) Formation of DNA etheno adducts in rodents and humans
and their role in carcinogens. Acta Biochim Pol, 45: 145-161.
Barbin A (in press) The role of etheno DNA adducts in carcinogenesis
induced by vinyl chloride in rats. In: Singer B & Bartsch H ed.
Exocyclic DNA adducts in mutagenesis and carcinogenesis. Lyon,
International Agency for Research on Cancer, pp 1-29 (IARC Scientific
Publications No. 150).
Barbin A, Brésil H, Croisy A, Jacquignon P, Malaveille C, Montesano R,
& Bartsch H (1975) Liver microsome-mediated formation of alkylating
agents from vinyl bromide and vinyl chloride. Biochem Biophys Res
Commun, 67: 596-603.
Barbin A, Laib RJ, & Bartsch H (1985a) Lack of miscoding properties of
7-(2-oxoethyl)guanine, the major vinyl chloride-DNA adduct. Cancer
Res, 45: 2440-2444.
Barbin A, Besson F, Perrard MH, Béréziat JC, Kaldor J, Michel G, &
Bartsch H (1985b) Induction of specific base-pair substitutions in
E. coli trpA mutants by chloroethylene oxide, a carcinogenic vinyl
chloride metabolite. Mutat Res, 152: 147-156.
Barbin A, Forment O, Boivin S, Marion M-J, Belpoggi F, Maltoni C, &
Montesano R (1997) p53 Gene mutation pattern in rat liver tumors
induced by vinyl chloride. Cancer Res, 57: 1695-1698.
Baretta ED, Stewart RD, & Mutchler JE (1969) Monitoring exposures to
vinyl chloride vapor: Breath analysis and continuous air sampling. Am
Ind Hyg Assoc J, 30: 537-544.
Barnes AW (1976) Vinyl chloride and the production of PVC. Proc R Soc
Med, 69: 277-281.
Barr DB & Ashley DL (1998) A rapid, sensitive method for the
quantitation of N-acetyl-S-(2-hydroxyethyl)-L-cysteine in human urine
using isotope-dilution HPLC-MS-MS. J Anal Toxicol, 22: 96-104.
Barrio-Lage GA, Parsons FZ, Narbaitz RM, & Lorenzo PA (1990) Enhanced
anaerobic biodegradation of vinyl chloride in ground water. Environ
Toxicol Chem, 9: 403-415.
Barton HA, Creech JR, Go CS, Randall GM, & Seckel CS (1995)
Chloroethylene mixtures: Pharmacokinetic modeling and in vitro
metabolism of vinyl chloride, trichloroethylene, and
trans-1,2-dichloroethylene in rat. Toxicol Appl Pharmacol, 130:
237-247.
Bartsch H, Malaveille C, & Montesano R (1975) Human rat and mouse
liver-mediated mutagenicity of vinyl chloride in S. typhimurium
strains. Int J Cancer, 15: 429-437.
Bartsch H, Malaveille C, Barbin A, Bresil H, Tomatis L, & Montesano R
(1976) Mutagenicity and metabolism of vinyl chloride and related
compounds. Environ Health Perspect, 17: 193-198.
Bartsch H, Malaveille C, Barbin A, & Planche G (1979) Mutagenic and
alkylating metabolites of halo-ethylenes, chlorobutadienes and
dichlorobutenes produced by rodent or human liver tissues. Arch
Toxicol, 41: 249-277.
Bartsch H, Barbin A, Marion MJ, Nair J, & Gu Y (1994) Formation,
detection and role in carcinogenesis of ethenobases in DNA. Drug Metab
Rev, 26: 349-371.
Bartsch H, Nair J, & Velic I (1997) Etheno-DNA base adducts as tools
in human cancer aetiology and chemoprevention. Eur J Cancer Prev, 6:
529-534.
Basler A & Röhrborn G (1980) Vinyl chloride: An example for evaluating
mutagenic effects in mammals in vivo after exposure to inhalation.
Arch Toxicol, 45: 1-7.
Basu AK, Wood ML, Niedernhofer LJ, Ramos LA, & Essigmann JM (1993)
Mutagenic and genotoxic effects of three vinyl chloride-induced DNA
lesions: 1,N6-ethenoadenine, 3,N4-ethenocytosine, and
4-amino-5-(imidazol-2-yl)imidazole. Biochemistry, 32: 12793-12801.
Bauer U (1981) [Human exposure to environmental chemicals
-- investigations on volatile organic halogenated compounds in water,
air, food, and human tissues.] Zent.bl Bakteriol Hyg I Abt Orig B,
174: 1-237 (in German).
Bauer J & Sabel A (1975) [The influence of oxygen on the
polymerisation of vinyl chloride with the example of suspension
polymerisation.] Angew Makromol Chem, 47: 15-27 (in German).
Becker KH, Biehl HM, Bruckmann P, Fink EH, Führ F, Klöpffer W, Zellner
R, & Zetzsch C (1984) [Methods of the ecotoxicological evaluation of
chemicals: Photochemical degradation in the gas phase.] In: [Volume 6:
OH reaction rate constants and tropospheric lifetimes of selected
environmental chemicals -- Report 1980-1983 of the Atomic Research
Unit, Jülich.] Bonn, Germany, Ministry for Research & Technology, pp
5-18 (in German).
Belli S, Bertazzi PA, Comba P, Foa V, Maltoni C, Masina A, Pirastu R,
Reggiani A, & Vigotti MA (1987) A cohort study on vinyl chloride
manufactures in Italy: Study design and preliminary results. Cancer
Lett, 35: 253-261.
Bencko V & Ungváry G (1994) Risk assessment of chemicals - a central
European perspective. Cent Eur J Public Health, 2: 70-72.
Bencko V, Wagner V, Wagnerová M, Bátora J, & Hrebacka J (1988)
Immunobiochemical profiles of workers differing in the degree of
occupational exposure to vinyl chloride. J Hyg Epidemiol Microbiol
Immunol, 32: 375-384.
Benfenati E, Natangelo M, Davoli E, & Fanelli R (1991) Migration of
vinyl chloride into PVC-bottled drinking-water assessed by gas
chromatography-mass spectrometry. Food Chem Toxicol, 29: 131-134.
Berens AR, Crider LB, Tomanek CL, & Whitney J (1975) Analysis in vinyl
chloride in PVC powders by head-space gas chromatography. Appl Polym
Sci, 19: 3169-3172.
Bergert KH & Betz V (1976) [Organic contaminants in urban air
- Results of a GC/MS analysis.] Chem Ing Tech, 48: 47-48 (in German).
Berman E & Dong J (1994) Photocatalytic decomposition of organic
pollutants in gas streams. Chem Oxid, 3: 183-189.
Besemer AC, Hollander JC, Huldy HJ, Laurier MD, Maas RJ, & Mulder HC
(1984) [Criteria document on vinyl chloride.] The Hague, The
Netherlands, Ministry of Housing.
Bi W, Wang Y, Huang M, & Meng D (1985) Effect of vinyl chloride on
testis in rats. Ecotoxicol Environ Saf, 10: 281-289.
BIA (German Professional Associations' Institute for Occupational
Safety) (1996) [Report on the occupational exposure to cancer-inducing
substances.] Sankt Augustin, Germany, Professional Association Office
(HVBG), pp 1-68 (in German).
BIA (German Professional Associations' Institute for Occupational
Safety) (1997) [List of hazardous substances, 1997 -- Safety and
health protection at work.] Sankt Augustin, Germany, Professional
Association Office (HVBG), 112 pp (BIA Report 10/97) (in German).
Black CM, Welsh KI, Walker AE, Catoggio LJ, Bernstein RM, McGregor AR,
& Lloyd Jones JK (1983) Genetic susceptibility to scleroderma-like
syndrome induced by vinyl chloride. Lancet, 1: 53-55.
Black C, Pereira S, McWhirter A, Welsh K, & Laurent R (1986) Genetic
susceptibility to scleroderma-like syndrome in symptomatic and a
symptomatic workers exposed to vinyl chloride. J Rheumatol, 13:
1059-1062.
Boettner EA, Ball G, & Weiss B (1969) Analysis of the volatile
combustion products of vinyl plastics. J Appl Polym Sci, 13: 377-391.
Bogdanikowa B & Zawilska J (1984) [Immune complexes in the serum of
patients occupationally exposed to vinyl chloride.] Przeglad Lek, 41:
253-257 (in Russian)
Boivin S, Pedron S, Bertrand S, Desmouliere A, Martel-Planche G,
Bancel B, Montesano R, Trepo C, & Marion MJ (1997) Myofibroblast-like
cells exhibiting a mutated p53 gene isolated from liver human tumor of
a worker exposed to vinyl chloride. In: Wisse E, Knook DL, & Balabaud
C ed. Cells of the hepatic sinusoid. Leiden, The Netherlands, Kupffer
Cell Foundation, pp 449-451.
Boivin-Angèle S, Lefrancois L, Froment O, Spiethoff A, Bogdanffy MS,
Wegener K, Wesch H, Barbin A, Bancel B, Trépo C, Bartsch H, Swenberg
J, & Marion MJ (in press) Ras gene mutations in vinyl
chloride-induced liver tumors are carcinogen-specific but vary with
cell type and species. Int J Cancer.
Bol'shakov AM (1969) [Working conditions in the production of
synthetic leather.] Gig Vopr Proizvod Primen Polim Mater, 1969: 47-52
(in Russian)
Bolt HM (1986) Metabolic activation of vinyl chloride, formation of
nucleic acid adducts and relevance to carcinogenesis. In: Singer B &
Bartsch H ed. The role of cyclic nucleic acid adducts in
carcinogenesis and mutagenesis. Lyon, International Agency for
Research on Cancer, pp 261-268 (IARC Scientific Publications No. 70).
Bolt HM (1994) Vinyl halides, haloaldehydes and monohaloalkanes. In:
Hemmininki K, Dipple A, Shuker DEG, Kadlubar FF, Segerbäck D, &
Bartsch H ed. DNA adducts: Identification and biological significance.
Lyon, International Agency for Research on Cancer pp 141-150 (IARC
Scientific Publications No. 125).
Bolt HM, Kappus H, Buchter A, & Bolt W (1976) Disposition of
[1,2-14C] vinyl chloride in the rat. Arch Toxicol, 35: 153-162.
Bolt HM, Laib RJ, Kappus H, & Buchter A (1977) Pharmacokinetics of
vinyl chloride in the rat. Toxicology, 7: 179-188.
Bolt HM, Filser JG, Laib RJ, & Ottenwälder H (1980) Binding kinetics
of vinyl chloride and vinyl bromide at very low doses. Arch Toxicol,
3(suppl): 129-142.
Bönnighausen KH (1986) [Production and properties of vinyl chloride.]
In: Becker CW & Braun A ed. [Plastics handbook.] Munich, Wien, Carl
Hanser Verlag, pp 38-57 (in German).
Borys E, Mroczkowska-Slupska MM, & Kusmierek JT (1994) The induction
of adaptive response to alkylating agents in Escherichia coli
reduced the frequency of specific C->T mutations in
chloroacetaldehyde-treated M13 glyU phage. Mutagenesis, 9: 407-410.
Bouscaren R, Frank R, & Veldt C (1987) Environment and quality of
life: Hydrocarbons identification of air quality problems in member
states of the European communities (Contract No.
84-B-6642-11-015-11-N) -- Final report. Brussels, Commission of the
European Communities, pp 39-173.
Boyden BH, Banh DT, Huckabay HK, & Fernandes JB (1992) Using inclined
cascade aeration to strip chlorinated VOCs from drinking water. Am
Water Waste Assoc, 84: 62-69.
Bradley PM & Chapelle FH (1996) Anaerobic mineralization of vinyl
chloride in Fe(III)-reducing, aquifer sediments. Environ Sci Technol
30: 2084-2086.
Bradley PM & Chapelle FH (1998a) Effect of contaminant concentration
on aerobic microbial mineralization of DCE and VC in stream-bed
sediments. Environ Sci Technol, 32: 553-557.
Bradley PM & Chapelle FH (1998b) Microbial mineralization of VC and
DCE under different terminal electron accepting conditions. Anaerob
Clin Microbiol, 4: 81-87.
Brady J, Liberatore F, Harper P, Greenwald P, Burnett W, Davies JNP,
Bishop M, Polan A, & Viana N (1977) Angiosarcoma of the liver: An
epidemiologic survey. J Natl Cancer Inst, 59: 1383-1385.
Brandt-Rauf PW, Chen JM, Marion MJ, Smith SJ, Luo JC, Carney W, &
Pincus MR (1996) Conformational effects in the p53 protein of
mutations induced during chemical carcinogenesis: Molecular dynamic
and immunologic analyses. J Protein Chem, 15: 367-375.
Brauch H-J, Kühn W, & Werner P (1987) [Vinyl chloride in contaminated
groundwater.] Vom Wasser, 68: 23-32 (in German).
Breder CV, Dennison JL, & Brown ME (1975) Gas-liquid chromatographic
determination of vinyl chloride in vinyl chloride polymers,
food-simulating solvents, and other samples. J Assoc Off Anal Chem,
58: 1214-1220.
Brown KW & Donnelly KC (1988) An estimation of the risk associated
with the organic constituents of hazardous and municipal waste
landfill leachates. Hazard Waste Hazard Mater, 5: 1-30.
Brown ER, Sinclair T, Keith L, Beamer P, Hazdra JJ, Nair V, &
Callaghan O (1977) Chemical pollutants in relation to diseases in
fish. Ann NY Acad Sci, 298: 535-546
Bruckmann P & Mülder W (1982) [The content of organic trace substances
in landfill gases.] Müll Abfall, 12: 339-346 (in German).
Bruckmann P, Kersten W, Funcke W, Balfanz E, König J, Theisen J, Ball
M, & Päpke O (1988) The occurrence of chlorinated and other organic
trace compounds in urban air. Chemosphere, 17: 2363-2380.
BUA (German Chemical Society Advisory Committee on Existing Chemicals
of Environmental Relevance) (1989) [Vinyl chloride.] Weinheim,
Germany, VCH Verlagsgesellschaft mbH, pp 1-63 (BUA Report 35) (in
German).
BUA (German Chemical Society Advisory Committee on Existing Chemicals
of Environmental Relevance) (1993) [OH-Radicals in the troposphere:
Concentration and effects.] Stuttgart, S. Hirzel Wissenschaftliche
Verlagsgesellschaft, pp 1-163 (BUA Report 100) (in German).
Buchter A, Bolt HM, & Kappus H (1977) [The distribution of
1,2-14C-vinyl chloride in rat tissue.] Int Arch Occup Environ Health,
39: 27-32 (in German).
Buchter A, Bolt HM, Filser J, Georgens HW, Laib RJ, & Bolt W (1978)
[Pharmacokinetics and carcinogenesis of vinyl chloride: occupational
risk evaluation.] Verh Dtsch Ges Arbeitsmed, 18: 111-124 (in German).
Buchter A, Filser JG, Peter H, & Bolt HM (1980) Pharmacokinetics of
vinyl chloride in the Rhesus monkey. Toxicol Lett, 6: 33-36.
Buffler PA, Wood S, Eifler C, Suarez L, & Kilian DJ (1979) Mortality
experience of workers in a vinyl chloride monomer production plant. J
Occup Med, 41: 195-203.
Burmaster DE (1982) The new pollution: Groundwater contamination.
Environment, 24: 33-36.
Burns and Roe Industrial Services Organization Paramas (1982) Fate and
priority pollutants in publicly owned treatment works. In : Fate and
priority pollutants in publicly owned treatment works -- Final Report,
Volumes I and II. Washington, DC, US Environmental Protection Agency,
pp 1-88 (EPA 440/1-82/303).
Byron D, Engholm G, Englund A, & Westerholm P (1976) Mortality and
cancer morbidity in a group of Swedish VCM and PVC production workers.
Environ Health Perspect, 17: 167-170.
Callahan M, Slimak M, Gabel N, May I, Fowler C, Freed R, Jennings P,
Durfee R, Whitmore F, Maesstri B, Mabey W, Hoit B, & Gould C (1979)
Water-related environmental fate of 129 priority pollutants. In:
Water-related environmental fate of 129 priority pollutants -- Volume
II: Halogenated aliphatic hydrocarbons, halogenated ethers, monocyclic
aromatics, phthalate esters, polycyclic aromatic hydrocarbons,
nitrosamines, and miscellaneous compounds. Springfield, Virginia,
Versar Inc., pp 49/1-49/10 (EPA/440/4-79/029b).
Camarena E & Coy CA (1984) Regulation of toxic or hazardous air
contaminants from land disposal sites. In: Regulation of toxic or
hazardous air contaminants from land disposal sites. For presentation
at the 77th Annual Meeting of the Air Pollution Control Association,
San Francisco, California, 24-29 June 1984. Pittsburgh, Pennsylvania,
Air Pollution Control Association, pp 1-15.
Caputo A, Viola PL, & Bigott A (1974) Oncogenicity of vinyl chloride
at low concentrations in rats and rabbits. Med Sci, 2: 1582.
Carassiti V, Chiorboli C, Bignozzi CA, Ferri A, & Maldotti A (1977)
Atmospheric photo-oxidation of vinyl chloride: A generalized treatment
of the relative rates of product formation. Ann Chim, 67: 499-512.
Carr CJ, Burgison RM, Vitcha JF, & Krantz JC (1949) Chemical
constitution of hydrocarbons and cardiac automaticity. J Pharmacol Exp
Ther, 97: 1-3.
Carroll WF, Borrelli FE, Garrity PJ, Jacobs RA, Lewis JW, McCreedy RL,
& Weston AF (1996) Characterization of emissions of dioxins and furans
from ethylene dichloride (EDC), vinyl chloride (VCM) and
polyvinylchloride (PVC) facilities in the United States: I. Resin,
treated wastewater, and ethylene dichloride. Organohalog Compd, 27:
62-67.
Carter SR & Jewell WJ (1993) Biotransformation of tetrachloroethylene
by anaerobic attached-films at low temperatures. Water Res, 27:
607-615.
Castro CE (1993) Biodehalogenation: The kinetics and rates of the
microbial cleavage of carbon-halogen bonds. Environ Toxicol Chem, 12:
1609-1618.
Castro CE, Riebeth DM, & Belser NO (1992a) Biodehalogenation: The
metabolism of vinyl chloride by Methylosinus trichosporium OB-3b. A
sequential oxidative and reductive pathway through chloroethylene
oxide. Environ Toxicol Chem, 11: 749-755.
Castro CE, Wade RS, Riebeth DM, Bartnicki EW, & Belser NO (1992b)
Biodehalogenation: Rapid metabolism of vinyl chloride by a soil
Pseudomonas sp. direct hydrolysis of a vinyl C-Cl bond. Environ
Toxicol Chem, 11: 757-764.
Castro CE, Helvenston MC, & Belser NO (1994) Biodehalogenation,
reductive dehalogenation by Methanobacterium thermoautotrophicum.
Comparison with nickel(I)octaethylisobacteriochlorin anion. An F-430
model. Environ Toxicol Chem, 13: 429-433.
CEC (1978) Council directive of 29 June 1978 on the approximation of
the laws, regulations and administrative provisions of the Member
States on the protection of the health of workers exposed to vinyl
chloride monomer. Off J Eur Communities, L197: 12-18.
CEQ (Council on Environmental Quality) (1981) Contamination of ground
water by toxic organic chemicals. In: Contamination of ground water by
toxic organic chemicals. Washington, DC, US Government Printing
Office, pp 1-107.
Chan RKS, Anselmo KJ, Reynolds CE, & Worman CH (1978) Diffusion of
vinyl chloride from PVC packaging material into food simulating
solvents. Polym Eng Sci, 18: 601-607.
Chang H-L & Alvarez-Cohen L (1996) Biodegradation of individual and
multiple chlorinated aliphatic hydrocarbons by methane-oxidizing
cultures. Appl Environ Microbiol, 62: 3371-3377.
Charlton J, Chow A, & Gesser HD (1983) Accidental release of vinyl
chloride: The train derailment near MacGregor, Manitoba. In: Saxena J
ed. Hazard assessment of chemicals: Current developments. New York,
London, Academic Press, vol 2, pp 245-267.
CHEM-FACTS (1992) PVC. In: Chemical Intelligence Services ed.
Chem-Facts PVC. Dunstable, United Kingdom, UK Reed Telepublishing Ltd,
pp 1-224.
Chen CW & Blancato JN (1989) Incorporation of biological information
in cancer risk assessment: Example-vinyl chloride. Cell Biol Toxicol,
5: 417-444.
Chen CS & Zoltek J (1995) Organic priority pollutants in
wetland-treated leachates at a landfill in central Florida.
Chemosphere, 31: 3455-3464.
Cheng KC, Preston BD, Cahill DS, Dosanjh MK, Singer B, & Loeb LA
(1991) The vinyl chloride DNA derivative N2,3-ethenoguanine produces
G -> A transitions in Escherichia coli. Proc Natl Acad Sci (USA),
88: 9974-9978.
Chiang S-Y, Swenberg JA, Weisman WH, & Skopek TR (1997) Mutagenicity
of vinyl chloride and its reactive metabolites, chloroethylene oxide
and chloroacetaldehyde, in a metabolically competent human
B-lymphoblatoid line. Carcinogenesis, 18: 31-36.
Chiazze L, Nichols WE, & Wong O (1977) Mortality among employees of
PVC fabricators. J Occup Med, 19: 623-628.
Chiazze L, Wong O, Nichols W, & Ference L (1980) Breast cancer
mortality among PVC fabricators. J Occup Med, 22: 677-679.
Chilton J & Chilton K (1992) A critique of risk modeling and risk
assessment of municipal landfills based on US Environmental Protection
Agency techniques. Waste Manage Res, 10: 505-516.
Chiou CT, Peters LJ, & Freed VH (1979) A physical concept of
soil-water equilibria for nonionic organic compounds. Science, 206:
831-832.
Chitgopekar NP, Reible DD, & Thibodeaux LJ (1990) Modeling short range
air dispersion from area sources of non-buoyant toxics. J Air Waste
Manage Assoc, 40: 1121-1128.
Cho Y, Gorina S, Jeffrey PD, & Pavletich NP (1994) Crystal structure
of a p53 tumor suppressor-DNA complex: Understanding tumorigenic
mutations. Science, 265: 346-355.
Chung FL, Chen HJC, & Nath RG (1996) Lipid peroxidation as a potential
endogenous source for the formation of exocyclic DNA adducts.
Carcinogenesis, 17: 2105-2111.
Ciroussel F, Barbin A, Eberle G, & Bartsch H (1990) Investigations on
the relationship between DNA ethenobase adduct levels in several
organs of vinyl chloride-exposed rats and cancer susceptibility.
Biochem Pharmacol, 39: 1109-1113.
Clement International Corporation (1990) Development and validation of
methods for applying pharmacokinetic data in risk assessment. Ruston,
Louisiana, Clement International Corporation, vol 5, pp 1-103
(AAMRL-TR-90-072).
Clemmesen J (1982) Mutagenicity and teratogenicity of vinyl chloride
monomer (VCM). Mutat Res, 98: 97-100.
Clewell HJ, Gentry PR, Gearhart JM, Allen BC, & Andersen ME (1995a)
The development and validation of a physiologically based
pharmacokinetic model for vinyl chloride and its application in a
carcinogenic risk assessment for vinyl chloride. Washington, DC, US
Environmental Protection Agency, Office of Health and Environmental
Assessment/Occupational Safety and Health Administration, Directorate
of Health Standards Programs, 110 pp (Unpublished report prepared by
ACF Kayser).
Clewell HJ, Gentry PR, Gearhart JM, Allen BC, & Andersen ME (1995b)
Considering pharmacokinetic and mechanistic information in cancer risk
assessments for environmental contaminants: Examples with vinyl
chloride and trichloroethylene. Chemosphere, 31: 2561-2578.
Cline PV & Viste DR (1985) Migration and degradation patterns of
volatile organic compounds. Waste Manage Res, 3: 351-360.
Cocciarini D & Campaña H (1992) Biotreatment of EDC -- VCM wastewater
effluent. Water Sci Technol, 25: 259-261.
Cochran JW (1988) A rapid, sensitive method for the analysis of
halogenated gases in water. J High Resol Chromatogr Chromatogr Commun,
11: 663-665.
Cochran JW & Henson JM (1988) Analysis of volatile organic chemicals
in aqueous samples by purge/GC with selective water removal. J High
Resol Chromatogr Chromatogr Commun, 11: 869-873.
Codex Committee (1984) Report to the Codex Committee on Food Additives
in responses to circular letter CL 1983/20-FA, May 1983, on the
subject of packaging materials (Report cx/FA 84/11). Canada, Codex
Committee, pp 1-24.
Coenen W (1986) [Concentration of carcinogenic substances at the
workplace: An analysis of results from the BIA data bank.]
Berufsgenoss Informationsz Arb.sicherh Unvallversicher, 1: 1-5 ( in
German).
Cogliano VJ, Hiatt GFS, & Den A (1996) Quantitative cancer assessment
for vinyl chloride: Indications of early-life sensitivity. Toxicology,
111: 21-28.
Cole RH, Frederick RE, Healy RP, & Rolan RG (1984) Preliminary
findings of the priority pollutant monitoring project of the
nationwide urban runoff program. J Water Pollut Control Fed, 56:
898-908.
Conkle JP, Camp BJ, & Welch BE (1975) Trace composition of human
respiratory gas. Arch Environ Health, 30: 290-29.5
Connor MS (1984) Comparison of the carcinogenic risks from fish versus
groundwater contamination by organic compounds. Environ Sci Technol,
18: 628-631.
Conolly RB & Jaeger RJ (1979) Acute hepatoxicity of vinyl chloride and
ethylene: Modification by trichloropropene oxide, diethylmaleate, and
cysteine. Toxicol Appl Pharmacol, 50: 523-531.
Conolly RB, Jaeger RJ, & Szabo S (1978) Acute hepatotoxicity of
ethylene, vinyl fluoride, vinyl chloride, and vinyl bromide after
Aroclor 1254 pretreatment. Exp Mol Pathol, 28: 25-33.
Cooper WC (1981) Epidemiologic study of vinyl chloride workers:
Mortality through December. Environ Health Perspect, 41: 101-106.
Cordier JM, Fievez C, Lefevre MJ, & Sevrin A (1966) Acroostérolyse et
lésions cutanées associées chez deux ouvriers affectés au nettoyage
d'autoclaves. Cah Méd Trav, 4: 3-39.
Cotruvo JA, Goldhaber S, & Vogt C (1986) Regulatory significance of
organic contamination in the decade of the 1980s. In: Ram NR ed.
Organic carcinogens in drinking water. New York, Wiley, pp 511-530.
Cox RA, Eggleton AEJ, & Sandalls FJ (1974) Photochemical reactivity of
vinyl chloride. London, Her Majesty's Stationery Office (Report No.
AERE-R-7820).
Cox E, Edwards E, Lehmicke L, & Major D (1995) Intrinsic
biodegradation of trichloroethene and trichloroethane in a sequential
anaerobic-aerobic aquifer. In: Hinchee RE, Wilson JT, & Downey DC ed.
Intrinsic bioremediation. Columbus, Ohio, Battelle Press, pp 223-231.
Craun GF (1984) Health aspects of groundwater pollution. In: Bitton G
& Gerba CP ed. Groundwater pollution microbiology. New York,
Chichester, Brisbane, Toronto, John Wiley & Sons, pp 135-179.
Creech JL & Johnson MN (1974) Angiosarcoma of liver in the manufacture
of polyvinyl chloride. J Occup Med, 16: 150-151.
Crump KS (1984) An improved procedure for low-dose carcinogenic risk
assessment from animal data. J Environ Pathol Toxicol, 5: 339-348.
Crutzen PJ (1982) The global distribution of hydroxyl. In: Goldberg ED
ed. Atmospheric chemistry. Berlin, Heidelberg, New York, Springer
Verlag, pp 313-328.
Dahar WS, Bond GG, McLaren EA, Sabel FL, Lipps TE, & Cook RR (1988)
Update to vinyl chloride mortality study. J Occup Med, 30: 648-649.
Daniel FB, Deangelo AB, Stober JA, Olson GR, & Page NP (1992)
Hepatocarcinogenicity of chloral hydrate, 2-chloracetaldehyde, and
dichloroacetic acid in the male B6C3F1 mouse. Fundam Appl Toxicol, 19:
159-168.
Daniels GA & Proctor DE (1975) VCM extraction from PVC bottles. Mod
Packag, April: 45-48.
Dann T & Wang D (1992) Volatile organic compound measurements in
Canadian urban and rural areas: 1989-1990. Presented at the 85th
Annual Meeting of the Air and Waste Management Association, Kansas
City, Missouri, 21-26 June 1992. Pittsburgh, Pennsylvania, Air and
Waste Management Association, pp 1-16 (Paper 92-75.16).
Danziger H (1960) Accidental poisoning by vinyl chloride: Report of
two cases. Can Med Assoc J, 82: 828-830.
Davies IW & Perry R (1975) Vinyl chloride monomer's effect on liquids
in PVC bottles. Environ Pollut Manage, 5: 22-23.
Davis JW & Carpenter CL (1990) Aerobic biodegradation of vinyl
chloride in groundwater samples. Appl Environ Microbiol, 56:
3878-3880.
De Bruin WP, Kotterman MJJ, Posthumus MA, Schraa G, & Zehnder AJB
(1992) Complete biological reductive transformation of
tetrachloroethene to ethane. Appl Environ Microbiol, 58: 1996-2000.
Deipser A (1998) Biodegradation of volatile CFCs, H-CFCs and VC in
compost and marl. Waste Manage Res, 16: 330-341
De Jong G, Van Sittert NJ, & Natarajan AT (1988) Cytogenetic
monitoring of industrial populations potentially exposed to genotoxic
chemicals and of control populations. Mutat Res, 204: 451-464.
DeLassus PT & Schmidt DD (1981) Solubilities of vinyl chloride and
vinylidene chloride in water. J Chem Eng Data, 26: 274-276.
De Meester C, Duverger van Bogaert M, Lambotte-Vandepaer M, Roberfroid
M, Poncelet F, & Mercier M (1980) Mutagenicity of vinyl chloride in
the Ames test: Possible artifacts related to experimental conditions.
Mutat Res, 77: 175-179.
Demkowicz-Dobrzanski K, Nalecz-Jawecki G, Wawer Z, Bargiel T, Zak M, &
Sawicki J (1993) Toxicity of neutralized wastes following a synthesis
of chlorinated hydrocarbons to Daphina magna and Chlorella sp.
Sci Total Environ, 1993(suppl): 1151-1158.
Depret P & Bindelle JP (1998) Vinyl chloride leak detection and
fugitive emission monitoring experience from the west-European PVC
industry. In: 1st International Conference on Protecting the
Environment: Controlling leakages in process industry, Rome, Italy,
11-12 May 1998. Cranfield, United Kingdom, BHR Publishers, pp 167-188
(BHR Series No. 30).
DeVivo I, Marion M-J, Smith SJ, Carney WP, & Brandt-Rauf PW (1994)
Mutant c-Ki- ras p 21 protein in chemical carcinogenesis in humans
exposed to vinyl chloride. Cancer Causes Control, 5: 273-278.
Diachenko GW, Breder CV, Brown ME, & Dennison JL (1977) Gas-liquid
chromatographic headspace technique for determination of vinyl
chloride in corn oil and three food-simulating solvents. J Assoc Off
Anal Chem, 60: 570-575.
Dickman MD & Rygiel G (1993) Density, diversity and incidence of
deformities of benthic invertebrates near a vinyl chloride discharge
point source in the Niagara River watershed. In: Gorsuch JW, Dwyer FJ,
Ingersoll CG, & La Point TW ed. Environmental toxicology and risk
assessment. Philadelphia, Pennsylvania, American Society for Testing
and Materials, vol 2, pp 663-680.
Dickman M, Lan Q, & Matthews B (1989) Teratogens in the Niagara River
watershed as reflected by chironomid (Diptera: Chironomidae) labial
plate deformities. Water Pollut Res J Can, 24: 47-79
Dieter HH & Kerndorff H (1993) Presence and importance of
organochlorine solvents and other compounds in Germany's groundwater
and drinking water. Ann Ist Super Sanita, 29: 263-277.
Dietz A, Langbein G, & Permanetter W (1985) [Vinylchloride-induced
hepato-cellular carcinoma.] Klin Wochenschr, 63: 325-331 (in German).
Dilling WL (1977) Interphase transfer processes: II. Evaporation rates
of chloromethanes, ethanes, ethylenes, propanes and propylenes from
dilute aqueous solutions -- Comparisons with theoretical predictions.
Environ Sci Technol, 11: 405-409.
Dilling WL, Tefertiller NB, & Kallos GJ (1975) Evaporation rates and
reactivities of methylene chloride, chloroform, 1,1,1-trichloroethane,
trichloroethylene, tetrachloroethylene, and other chlorinated
compounds in dilute aqueous solutions. Environ Sci Technol, 9:
833-838.
Dilling WL, Bredeweg CJ, & Tefertiller NB (1976) Organic
photochemistry. Simulated atmospheric photodecomposition rates of
methylene chloride, 1,1,1-trichloroethane, trichloroethylene,
tetrachloroethylene, and other compounds. Environ Sci Technol, 10:
351-356.
Dimmick WF (1981) EPA programs of vinyl chloride monitoring in ambient
air. Environ Health Perspect, 41: 203-206.
DiStefano TD, Gossett JM, & Zinder SH (1991) Reductive dechlorination
of high concentrations of tetrachloroethene to ethene by an anaerobic
enrichment culture in the absence of methanogenesis. Appl Environ
Microbiol, 57: 2287-2292.
Dobecki M & Romaniwicz B (1993) [Occupational exposure to toxic
substances during the production of vinyl chloride and chlorinated
organic solvents.] Med Prac, 44: 99-102 (in Polish).
Dolan ME & McCarty PL (1995a) Methanotrophic chloroethene
transformation capacities and 1,1-dichloroethene transformation
product toxicity. Environ Sci Technol, 29: 2741-2747.
Dolan ME & McCarty PL (1995b) Small-column microcosm for assessing
methane-stimulated vinyl chloride transformation in aquifer samples.
Environ Sci Technol, 29: 1892-1897.
Dolk H, Vrijheid M, Armstrong B, Abramsky L, Bianchi F, Garne E, Nelen
V, Robert E, Scott JES, Stone D, & Tenconi R (1998) Risk of congenital
anomalies near hazardous-waste landfill sites in Europe: The
EUROHAZCON study. Lancet, 352: 423-427.
Doll R (1988) Effects of exposure to vinyl chloride. Scand J Work
Environ Health, 14: 61-78.
Dosanjh MK, Chenna A, Kim E, Fraenkel-Conrat H, Samson L, & Singer B
(1994) All four known cyclic adducts formed in DNA by the vinyl
chloride metabolite chloroacetaldehyde are released by a human DNA
glycosylase. Proc Natl Acad Sci (USA), 91: 1024-1028.
Draminski W & Trojanowska B (1981) Chromatographic determination of
thiodiglycolic acid - a metabolite of vinyl chloride. Arch Toxicol,
48: 289-292.
Dreher EL (1986) Chloroethylenes. In: Gerhartz W, Yamamoto YS,
Campbell FT, Pfefferkorn R, & Rounsaville JF ed. Ullmann's
encyclopedia of industrial chemistry, 5th completely revised.
-- Volume A6: Ceramics to chlorohydrins. Weinheim, Germany, VCH
Verlagsgesellschaft mbH, pp 283-294.
Dressman RC & McFarren EF (1978) Determination of vinyl chloride
migration from polyvinyl chloride pipe into water. Am Water Works
Assoc J, 70: 29-30.
Drevon C & Kuroki T (1979) Mutagenicity of vinyl chloride, vinylidene
chloride and chloroprene in V79 Chinese hamster cells. Mutat Res, 67:
173-182.
Drew RT, Boorman GA, Haseman JK, McConnell EE, Busey WM, & Moore JA
(1983) The effect of age and exposure duration on cancer induction by
a known carcinogen in rats, mice and hamsters. Toxicol Appl Pharmacol,
68: 120-130.
Driscoll JN, Duffy M, Pappas S, & Webb M (1987) Analysis for purgeable
organics in water by capillary GC/PID-ElCD. J Chromatogr Sci, 25:
369-375.
Drozdowicz BZ & Huang PC (1977) Lack of mutagenicity of vinyl chloride
in two strains of Neurospora Crassa. Mutat Res, 48: 43-50.
D'Souza RW & Andersen ME (1988) Physiologically based pharmacokinetic
model for vinylidene chloride. Toxicol Appl Pharmacol, 95: 230-240
Du CL & Wang JD (1998) Increased morbidity odds ratio of primary liver
cancer and cirrhosis of the liver among vinyl chloride monomer
workers. Occup Environ Med, 55: 528-532.
Du JT, Tseng MT, & Tamburro CH (1982) The effect of repeated vinyl
chloride exposure on rat hepatic metabolizing enzymes. Toxicol Appl
Pharmacol, 62: 1-10.
Du C-L, Kuo M-L, Chang H-L, Sheu T-J, & Wang J-D (1995) Changes in
lymphocyte single strand breakage and liver function of workers
exposed to vinyl chloride monomer. Toxicol Lett, 77: 379-385.
Du C-L, Chan C-C, & Wang J-D (1996) Comparison of personal and area
sampling strategies in assessing workers' exposure to vinyl chloride
monomer. Bull Environ Contam Toxicol, 56: 534-542.
Ducatman A, Hirschhorn K, & Selikoff IJ (1975) Vinyl chloride exposure
and human chromosome aberrations. Mutat Res, 31: 163-168.
Dulson W (1978) In: Heller A ed. [Organic chemical pollutants in the
atmosphere.] Stuttgart, Gustav Fischer Verlag, pp 84-85 (Reports
Series on Water, Soil, and Air) (in German).
Dummer G & Schmidhammer L (1983) [A new process for the removal of
aliphatic chlorinated hydrocarbons from wastewater using absorber
resins.] VDI-Ber, 545: 885-901 (in German).
Dummer G & Schmidhammer L (1984) [A new process for the removal of
aliphatic chlorinated hydrocarbons from wastewater using absorber
resins.] Chem Ing Tech, 56: 242-243 (in German).
Duprat F, Fabry JP, Gradiski D, & Magadur JL (1977) Metabolic approach
to industrial poisoning: Blood kinetics and distribution of
[14C]-vinyl chloride monomer (VCM). Acta Pharmacol Toxicol, 41(suppl):
142-143.
Dyksen JE & Hess AF (1982) Alternatives for controlling organics in
groundwater supplies. J Am Water Works Assoc, 74: 394-403.
Eberle G, Barbin A, Laib RJ, Ciroussel F, Thomale J, Bartsch H, &
Rajewsky MF (1989) 1,N6-etheno-2'-deoxyadenosine and
3,N4-eteno-2'-deoxycytidine detected by monoclonal antibodies in lung
and liver DNA of rats exposed to vinyl chloride. Carcinogenesis, 10:
209-212.
Ebihara J (1982) [Cause-specific mortality study on VCM workers.] J
Sci Lab, 58: 383-389 (in Chinese).
ECETOC (1983) Experimental assessment of the phototransformation of
chemicals in the atmosphere. Brussels, European Centre for
Ecotoxicology and Toxicology of Chemicals, pp 1-36 (ECETOC Monograph
No. 7).
ECETOC (1988) The mutagenicity and carcinogenicity of vinyl chloride:
A historical review and assessment. Brussels, European Centre for
Ecotoxicology and Toxicology of Chemicals, pp 1-147 (ECETOC Technical
Report No. 31).
Eckardt F, Muliawan H, De Ruiter N, & Kappus H (1981) Rat hepatic
vinyl chloride metabolites induce gene conversion in the yeast strain
d7 rad in vitro and in vivo. Mutat Res, 91: 381-390.
Edmonds LD, Falk H, & Nissim JE (1975) Congenital malformations and
vinyl chloride. Lancet, 29: 1098.
Edmonds LD, Anderson C, Flynt JW, & James LM (1978) Congenital central
nervous system malformations and vinyl chloride monomer exposure: A
community study. Teratology, 17: 137-142.
Ehtesham-Ud Din AF-M, Nordbo E, & Underdal B (1977) The content of
vinylchloride monomer (VCM) in foods packed in polyvinylchloride
(PVC). Lebensm.wiss Technol, 10: 33-35.
Eisenreich SJ, Looney BB, & Thornton JD (1981) Airborne organic
contaminants in the Great Lakes ecosystem. Environ Sci Technol, 15:
30-38.
El Ghissassi FE, Barbin A, Nair J, & Bartsch H (1995a) Formation of
1,N6-ethenoadenine and 3,N4-ethenocytosine by lipid peroxidation
products and nucleic acid bases. Chem Res Toxicol, 8: 278-283.
El Ghissassi F, Biovin S, Lefrançois L, Barbin A, & Marion M-J (1995b)
Glutathione transferase Mu1-1 (GSTM1) genotype in individuals exposed
to vinyl chloride: Relation to "vinyl chloride disease" and liver
angiosarcoma. Clin Chem, 41: 1922-1924.
El Ghissassi F, Barbin A, & Bartsch H (1998) Metabolic activation of
vinyl chloride by rat liver microsomes: Low-dose kinetics and
involvement of cytochrome P450 2E1. Biochem Pharmacol, 55: 1445-1452.
Elliott P & Kleinschmidt I (1997) Angiosarcoma of the liver in Great
Britain in proximity to vinyl chloride sites. Occup Environ Med, 54:
14-18.
Ellis DE, Lutz EJ, Klecka GM, Pardiek DL, Salvo JJ, Heitkamp MA,
Gannon DJ, Mikula CC, Vogel CM, Sayles GD, Kampbell DH, Wilson JT, &
Maiers DT (1996) Remediation technology development forum intrinsic
remediation project at Dover Air Force base, Delaware. In: Proceedings
of the Symposium on Natural Attenuation of Chlorinated Organics in
Ground Water, 11-13 September 1996. Washington, DC, US Environmental
Protection Agency, pp 93-97 (EPA 540/R-96/509).
Elmore JD, Wong JL, Laumbach AD, & Streips UN (1976) Vinyl chloride
mutagenicity via the metabolites chlorooxirane and chloroacetaldehyde
monomer hydrate. Biochim Biophys Acta, 442: 405-419.
EMO (1992) Field investigation report. In: EMO field investigation
report -- Volume 5. Richland, Washington, Environmental Management
Operations, pp 6.1-7.6 (EMO Report No. 1028).
Ensign SA, Hyman MR, & Arp DJ (1992) Cometabolic degradation of
chlorinated alkenes by alkene monooxygenase in a propylene-grown
Xanthobacter strain. Appl Environ Microbiol, 58: 3038-3046.
Ensley BD (1991) Biochemical diversity of trichloroethylene
metabolism. Annu Rev Microbiol, 45: 283-299.
Euro Chlor (1999) Euro Chlor risk assessment for the marine
environment, OSPARCOM Region -- North Sea: Vinylchoride. Brussels,
Euro Chlor, pp 1-25.
European Council of Vinyl Manufacturers (1994) On the environmental
impact of the manufacture of poly vinyl chloride (PVC): A description
of best available techniques. Brussels, European Council of Vinyl
Manufacturers, 50 pp.
Evans DMD, Williams WJ, & Kung ITM (1983) Angiosarcoma and
hepatocellular carcinoma in vinyl chloride workers. Histopathology, 7:
377-388.
Ewers J, Freier-Schröder D, & Knackmuss H-J (1990) Selection of
trichloroethene (TCE) degrading bacteria that resist inactivation by
TCE. Arch Mikrobiol, 154: 410-413.
Fairley P (1997) Greenpeace plans earth day blitz against vinyl
industry. Chem Week, April 16: 8.
Falk H & Steenland NK (1998) Vinyl chloride and polyvinyl chloride.
In: Rom WN ed. Environmental and occupational medicine, 3rd ed.
Philadelphia, Pennsylvania, Lippincou-Raven Publishers, pp 1251-1261.
Falk H, Creech JLJ, Heath CWJ, Johnson MN, & Key MM (1974) Hepatic
disease among workers at a vinyl chloride polymerisation plant. J Am
Med Assoc, 230: 59-63.
Fathepure BZ & Tiedje JM (1994) Reductive dechlorination of
tetrachloroethylene by a chlorobenzoate-enriched biofilm reactor.
Environ Sci Technol, 28: 746-752.
Fayad NM, Sheikheldin SY, Al-Malack MH, El-Mubarak AH, & Khaja N
(1997) Migration of vinyl chloride monomer (VCM) and additives into
PVC bottled drinking water. Environ Sci Eng, 32: 1065-1083.
Fedtke N, Walker VE, & Swenberg JA (1989) Determination of
7-(2-oxoethyl)guanine and N2,3-ethenoguanine in DNA hydrolysates by
HPLC. Arch Toxicol, 13(suppl): 214-218.
Fedtke N, Boucheron JA, Turner MJ, & Swenberg JA (1990a) Vinyl
chloride-induced DNA adducts. I. Quantitative determination of
N2,3-ethenoguanine based on electrophore labeling. Carcinogenesis,
11: 1279-1285.
Fedtke N, Boucheron JA, Walker VE, & Swenberg JA (1990b) Vinyl
chloride-induced DNA adducts: II. Formation and persistence of
7-(2'-oxoethyl)guanine and N2,3-ethenoguanine in rat tissue DNA.
Carcinogenesis, 11: 1287-1292.
Fernando RC, Nair J, Barbin A, Miller JA, & Bartsch H (1996) Detection
of 1,N6-ethenodeoxyadenosine and 3,N4-ethenodeoxycytidine by
immunoaffinity/32P-postlabelling in liver and lung DNA of mice treated
with ethyl carbamate (urethane) or its metabolites. Carcinogenesis,
17: 1711-1718.
Feron VJ & Kroes P (1979) One-year time-sequence inhalation toxicity
study of vinyl chloride in rats. II. Morphological changes in the
respiratory tract, ceruminous gland, brain, kidneys, heart and spleen.
Toxicology, 13: 131-141.
Feron VJ, Speek AJ, Willems MI, Van Battum D, & De Groot AP (1975)
Observations on the oral administration and toxicity of vinyl chloride
in rats. Food Cosmet Toxicol, 13: 633-638.
Feron VJ, Kruysse A, & Til HP (1979a) One-year time sequence
inhalation toxicity study of vinyl chloride in rats: I. Growth,
mortality, haematology, clinical chemistry, and organ weights.
Toxicology, 13: 25-28.
Feron VJ, Spit BJ, Immel HR, & Kroes R (1979b) One-year time-sequence
inhalation toxicity study of vinyl chloride in rats: III.
Morphological changes in the liver. Toxicology, 13: 143-154.
Feron VJ, Hendriksen CFM, Speek AJ, Til HP, & Spit BJ (1981) Lifespan
oral toxicity study of vinyl chloride in rats. Food Cosmet Toxicol,
19: 317-333.
Filatova VS & Gronsberg ES (1957) [Sanitary-hygienic conditions of
work in the production of polychlorvinylic tar and measures of
improvement.] Gig i Sanit, 22: 38-42 (in Russian).
Filatova VS, Balakonova PH, & Gronsberg ES (1958) [Hygienic
characteristics of vinyl chloride.] Gig Tr Prof Zabol, 2: 6-9 (in
Russian).
Filser JG & Bolt HM (1979) Pharmacokinetics of halogenated ethylenes
in rats. Arch Toxicol, 42: 123-136.
Finnish Government (1992) [Resolution 919 on work with vinyl
chloride], 4 pp (in Finnish).
Fiorenza S, Hockman ELr, Szojka S, Woeller RM, & Wigger JW (1994)
Natural anaerobic degradation of chlorinated solvents at a Canadian
manufacturing plant. In: Hinchee RE, Leeson A, Semprini L, & Ong SK
ed. Bioremediation of chlorinated and polycyclic aromatic hydrocarbon
compounds. Boca Raton, Florida, Lewis Publishers, Inc., pp 277-286.
Fishbein L (1979) Potential halogenated industrial carcinogenic and
mutagenic chemicals: I.Halogenated unsaturated hydrocarbons. Sci Total
Environ, 11: 111-161.
Fleig I & Thiess AM (1974) [Chromosome tests in vinyl chloride exposed
workers.] Arbeitsmed Sozialmed Präventivmed, 12: 280-283 (in German).
Fleig I & Thiess AM (1978) Mutagenicity of vinyl chloride. J Occup
Med, 20: 557-561.
Fogel MM, Taddeo AR, & Fogel S (1986) Biodegradation of chlorinated
ethenes by a methane-utilizing mixed culture. Appl Environ Microbiol,
51: 720-724.
Fogel S, Findlay M, Moore A, & Leahy M (1987) Biodegradation of
chlorinated chemicals in groundwater by methane oxidizing bacteria
-- Proceedings of the National Water Well Association/ American
Petroleum Industry (NWWA/API) Conference on Petroleum Hydrocarbons and
Organic Chemicals in Ground Water: Prevention, detection and
restoration, Houston, Texas, 17-19 November 1987. Dublin, Ohio,
National Water Well Association, pp 187-206.
Fontana L, Gautherie M, Albuisson E, Fleury-Duhamel N, Meyer S, &
Catlina P (1996) Etude chronothermobiologique de phénomènes de Raynaud
secondaires à une exposition ancienne au chlorure de vinyle monomère.
Arch Mal Prof, 51: 9-18.
Forman D, Bennett B, Stafford J, & Doll R (1985) Exposure to vinyl
chloride and angiosarcoma of the liver: A report of the register of
cases. Br J Ind Med, 42: 750-753.
Fox AJ & Collier PF (1977) Mortality experience of workers exposed to
vinyl chloride monomer in the manufacture of polyvinyl chloride in
Great Britain. Br J Ind Med, 34: 1-10.
Fredrickson JK, Bolton H, & Brockman FJ (1993) In situ and on-site
bioreclamation. Environ Sci Technol, 27: 1711-1716.
Freedman DL & Gossett JM (1989) Biological reductive dechlorination of
tetrachloroethylene and trichloroethylene to ethylene under
methanogenic conditions. Appl Environ Microbiol, 55: 2144-2151.
Freitag D, Geyer H, Kraus A, Viswanathan R, Kotzias D, Attar A, Klein
W, & Korte F (1982) Ecotoxicological profile analysis: VII. Screening
chemicals for their environmental behavior by comparative evaluation.
Ecotoxicol Environ Saf, 6: 60-81.
Freitag D, Ballhorn L, Geyer H, & Korte F (1985) Environmental hazard
profile of organic chemicals. Chemosphere, 14: 1589-1616.
Frentzel-Beyme R, Schmitz T, & Thiess AM (1978) [Mortality study on
VC/PVC workers of BASF, Ludwigshafen, Rhine.] Arbeitsmed Sozialmed
Präventivmed, 130: 218-228 (in German).
Fricke C, Clarkson C, Lomnitz E, & O'Farrell T (1985) Comparing
priority pollutants in municipal sludges. BioCycle, Jan/Feb: 35-37.
Froment O, Boivin S, Barbin A, Bancel B, Trepo C, & Marion M-J (1994)
Mutagenesis of ras proto-oncogenes in rat liver tumors induced by
vinyl chloride. Cancer Res, 54: 5340-5345.
Froment O, Marion M-J, Lepot D, Contassot J-C, & Trepo C (1991)
Immunoquantitation of Von Willebrand factor (factor VIII-related
antigen) in vinyl chloride exposed workers. Cancer Lett, 61(3):
201-206.
Froneberg B, Johnson PL ,& Landrigan PJ (1982) Respiratory illness
caused by overheating of polyvinyl chloride. Br J Ind Med, 39:
239-243.
Fuchs G, Gawell BM, Albanus L, & Storach S (1975) [Vinyl chloride
monomer levels in edible fats.] Var Foeda, 27: 134-145 (in Swedish).
Fucic A & Garaj-Vrhovac V (1997) Micronucleus assay and mitotic
activity -- the methods for determination of recent vinyl chloride
monomer exposure. Environ Mol Mutagen, 29: 19-23.
Fucic A, Horvat D, & Dimitrovic B (1990a) Localization of breaks
induced by vinyl chloride in the human chromosomes of lymphocytes.
Mutat Res, 243: 95-99.
Fucic A, Horvat D, & Dimitrovic B (1990b) Mutagenicity of vinyl
chloride in man: Comparison of chromosome aberrations with
micronucleus and sister-chromatid exchange frequencies. Mutat Res,
242: 265-270.
Fucic A, Garaj-Vrhovac V, Dimitrovic B, & Skara M (1992) The
persistence of sister-chromatid exchange frequencies in men
occupationally exposed to vinyl chloride monomer. Mutat Res, 281:
129-132.
Fucic A, Garaj-Vrhovac V, Barkovic D, & Kubelka D (1994) The
sensitivity of the micronucleus assay for the detection of
occupational exposure to vinyl chloride monomer. Mutat Res, 325:
53-56.
Fucic A, Barkovic D, Garaj-Vrhovac V, Kubelka D, Ivanic B, Dabo T, &
Mijic A (1996) A nine-year follow up study of a population
occupationally exposed to vinyl chloride monomer. Mutat Res, 361:
49-53.
Fujimoto T, Rennert AM, & Wijnen MHJ (1970) Primary steps in the
photolysis of vinyl chloride. Ber Bunsenges Phys Chem, 74: 282-286.
Funes-Cravioto F, Lambert B, Lindsten J, Ehrenberg L, Natarajan AT, &
Osterman-Golkar S (1975) Chromosome aberrations in workers exposed to
vinyl chloride. Lancet, 1: 459.
Gáliková E, Tomíková K, Zigová A, Mesko D, Buchancová J, Petrisková J,
L'uptáková M, Ja, M, & Karaffová N (1994) [General health of workers
in the Nováky chemical works exposed to vinyl chloride.] Prac Lék, 46:
251-256 (in Czech).
Garaj-Vrhovac V, Fucic A, & Horvat D (1990) Comparison of chromosome
aberration and micronucleus induction in human lymphocytes after
occupational exposure to vinyl chloride monomer and microwave
radiation. Period Biol, 4: 411-416.
Gargas ML, Burgess RJ, Voisard DE, Cason GH, & Andersen ME (1989)
Partition coefficients of low-molecular-weight volatile chemicals in
various liquids and tissues. Toxicol Appl Pharmacol, 98: 87-99.
Gargas ML, Clewell HJ, & Andersen ME (1990) Gas uptake inhalation
techniques and the rates of metabolism of chloromethanes,
chloroethanes, and chloroethylenes in the rat. Inhal Toxicol, 2:
295-319.
Gay BW, Lonnemann WA, Bridbord K, & Moran JB (1975) Measurements of
vinyl chloride from aerosol sprays. Ann NY Acad Sci, 246: 286-295.
Gay BW, Hanst PL, Bufalini JJ, & Noonan RC (1976) Atmospheric
oxidations of chlorinated ethylenes. Environ Sci Technol, 10: 58-67.
Gedigk P, Müller R, & Bechtelsheimer H (1975) Morphology of liver
damage among polyvinyl chloride production workers. A report on 51
cases. Ann NY Acad Sci, 246: 278-285
Gehring PJ, Watanabe PG, & Park CN (1978) Resolution of dose-response
toxicity data for chemicals requiring metabolic activation:
Example-vinyl chloride. Toxicol Appl Pharmacol, 44: 581-591.
German Environmental Office (UBA) (1978) [Air contamination with vinyl
chloride (VC) from PVC-Products.] Umweltbundesamt Ber, 5: 1-23 (in
German).
German Expert Group on the Environment (1988) [Report on the
environment, 1987.] Stuttgart, W. Kohlhammer, pp 229-231.
German Federal Health Office (1998) [New directive on articles for
personal use.] Bundesgesetzblatt, 1: 5-36 (in German).
German Federal Health Office (BGA) (1992) [Recommendation on measures
in case of occurrence of vinyl chloride in ground water used for
drinking water production.] Bundesgesundheitsblatt, 35: 364 (in
German).
Gilbert SG, Giacin JR, Morano JR, & Rosen JD (1975) Detecting small
quantities of residual vinyl chloride monomer. Package Dev, July/Aug:
20-24.
Giri AK (1995) Genetic toxicology of vinyl chloride -- a review. Mutat
Res, 339: 1-14.
Gnesina EA & Pshenitsina AI (1980) [Time course of the heart rate and
galvanic skin reflex during night sleep in patients with chronic
occupational intoxication by vinyl chloride.] Gig Tr Prof Zabol, 5:
39-42 (in Russian).
Gnesina EA & Teklina LG (1984) [Characteristics of night sleep of
patients with chronic occupational vinyl chloride.] Gig Tr Prof Zabol,
6: 34-36 (in Russian).
Gnesina EA, Antonyuzhenko VA, Mashikhin EN, & Rostovtseva GG (1978)
[On the nature and frequency of sleep disorders in persons exposed to
the effect of vinylchloride during work.] Gig Tr Prof Zabol, 11: 16-19
(in Russian).
Going JE (1976) Sampling and analysis of selected toxic substances:
Task III -- Vinyl chloride, secondary sources. Washington, DC, US
Environmental Protection Agency, pp 1-30 (EPA 560/6-76-002).
Goldberg MS, Al-Homsi N, Goulet L, & Riberdy H (1995) Incidence of
cancer among persons living near a municipal solid waste landfill site
in Montreal, Quebec. Arch Environ Health, 50(6): 416-424.
Goodenkauf O & Atkinson JC (1986) Occurrence of volatile organic
chemicals in Nebraska ground water. Ground Water, 24: 231-233.
Gordon SJ & Meeks SA (1977) A study of gaseous pollutants in the
Houston, Texas area. AICHE Symp Ser, 73: 84-94.
Gossett JM (1987) Measurement of Henry's law constants for C1 and C2
chlorinated hydrocarbons. Environ Sci Technol, 21: 202-208.
Gossett RW, Brown DA, & Young DR (1983) Predicting the bioaccumulation
of organic compounds in marine organisms using octanol/water partition
coefficients. Mar Pollut Bull, 14: 387-392.
Grainger RG, Walker AE, & Ward AM (1980) Vinyl chloride
monomer-induced disease: Clinical, radiological and immunological
aspects. In: Preger L ed. Induced disease, drug irradiation,
occupation. New York, Grune and Stratton, pp 191-214.
Green T & Hathway DE (1975) The biological fate in rats of vinyl
chloride in relation to its oncogenicity. Chem-Biol Interact, 11:
545-562.
Green T & Hathway DE (1977) The chemistry and biogenesis of the
S-containing metabolites of vinyl chloride in rats. Chem-Biol
Interact, 17: 137-150.
Green T & Hathway DE (1978) Interactions of vinyl chloride with
rat-liver DNA in vitro. Chem-Biol Interact, 22: 211-224.
Greim H, Bonse G, Radwan Z, Reichert D, & Henschler D (1975)
Mutagenicity in vitro and potential carcinogenicity of chlorinated
ethylenes as a function of metabolic oxirane formation. Biochem
Pharmacol, 24: 2013-2017.
Greiser E, Reinl W, & Weber H (1982) [Vinyl chloride exposure and
mortality of German chemical workers in comparison to comparison of
non-exposed chemical workers and PVC workers.] Zent.bl Arbeitsmed
Arbeitsschutz Prophyl, 32: 44-62 (in German).
Grimsrud EP & Rasmussen RA (1975) Survey and analysis of halocarbons
in the atmosphere by gas chromatography-mass spectrometry. Atmos
Environ, 9: 1014-1017.
Groeneveld AHC, Zijlstra AG, Feenstra AFM, & Berends AG (1993) The
acute toxicity of vinylchloride to the Zebra fish ( Brachydanio
rerio ). Graveland, The Netherlands, Solvay Duphar BV, pp 1-19.
Grollman AP & Shibutani S (1994) Mutagenic specifity of chemical
carcinogens as determined by studies of single DNA adducts. In:
Hemmininki K, Dipple A, Shuker DEG, Kadlubar FF, Segerbäck D, &
Bartsch H ed. DNA adducts: Identification and biological significance.
Lyon, International Agency for Research on Cancer, pp 385-397 (IARC
Scientific Publications No 125).
Grosjean D & Williams EL (1992) Environmental persistence of organic
compounds estimated from structure-reactivity and linear free-energy
relationships. Unsaturated aliphatics. Atmos Environ, 26A: 1395-1405.
Groth DH, Coate WB, Ulland BM, & Hornung RW (1981) Effects of aging on
the induction of angiosarcoma. Environ Health Perspect, 41: 53-57.
Gryder-Boutet DE & Kennish JM (1988) Using headspace sampling with
capillary column GC-MS to analyze trace volatile organics in water and
wastewater. Am Water Works Assoc J, 80: 52-55.
Guengerich FP (1992) Roles of the vinyl chloride oxidation products
2-chlorooxirane and 2-chloroacetaldehyde in the in vitro formation
of etheno adducts of nucleic acid bases. Chem Res Toxicol, 5: 2-5.
Guengerich FP & Strickland TW (1977) Metabolism of vinyl chloride:
Destruction of the heme of highly purified liver microsomal cytochrome
P-450 by a metabolite. Mol Pharmacol, 13: 993-1004.
Guengerich FP & Watanabe PG (1979) Metabolism of [14C]- and
[36Cl]-labeled vinyl chloride in vivo and in vitro. Biochem
Pharmacol, 28: 589-596.
Guengerich FP, Crawford WMJ, & Watanabe PG (1979) Activation of vinyl
chloride to covalently bound metabolites: Roles of 2-chloroethylene
oxide and 2-chloroacetaldehyde. Biochemistry, 18: 5177-5182.
Guengerich FP, Mason PS, Stott WJ, Fox TR, & Watanabe PG (1981) Roles
of 2-haloethylene oxide and 2-haloacetaldehydes derived from vinyl
bromide and vinyl chloride in irreversible binding to protein and DNA.
Cancer Res, 41: 4391-4398.
Guengerich FP, Kim D-H, & Iwasaki M (1991) Role of human cytochrome
P-450 IIE1 in the oxidation of many low molecular weight cancer
suspects. Chem Res Toxicol, 4: 168-179.
Guichard Y, El Ghissassi F, Nair J, Bartsch H, & Barbin A (1996)
Formation and accumulation of DNA ethenobases in adult Sprague-Dawley
rats exposed to vinyl chloride. Carcinogenesis, 17: 1553-1559.
Guicherit R & Schulting FL (1985) The occurrence of organic chemicals
in the atmosphere of the Netherlands. Sci Total Environ, 43: 193-219.
Gürtler R, Möller U, Sommer S, Müller H, & Kleinermanns K (1994)
Photooxidation of exhaust pollutants. Chemosphere, 29: 1671-1682.
Gwinner LM, Laib RJ, Filser JG, & Bolt HM (1983) Evidence of
chloroethylene oxide being the reactive metabolite of vinyl chloride
towards DNA: Comparative studies with 2,2'-dichlorodiethylether.
Carcinogenesis, 4: 1483-1486.
Haag WR, Johnson MD, & Scofield R (1996) Direct photolysis of
trichloroethene in air: Effect of cocontaminants, toxicity of
products, and hydrothermal treatment of products. Environ Sci Technol,
30: 414-421.
Hagmar L, Akesson B, Nielsen J, Andersson C, Lindén K, Attewell R, &
Möller T (1990) Mortality and cancer morbidity in workers exposed to
low levels of vinyl chloride monomer at a polyvinyl chloride
processing plant. Am J Ind Med, 17: 553-565.
Haguenoer JM, Frimat P, Cantineau A, Pollard F, & Bobowski R (1979)
Les risques liés au chlorure de vinyle (CVM) et leur prévention dans
une usine de polymérisation. Arch Mal Prof Méd Travail Séc Soc, 40:
1115-1130.
Hahn A, Michalak H, Begemann K, Heinemeyer G, & Gundert-Remy U (1998)
[Transportation accident with vinyl chloride: Health effects in 325
victims.] Umweltmed Forsch Prax, 3: 144-155 (in German).
Hallbourg RR, Delfino JJ, & Miller WL (1992) Organic priority
pollutants in groundwater and surface water at three landfills in
North Central Florida. Water Air Soil Pollut, 65: 307-322.
Hann RW & Jensen PA (1974) Water quality characteristics of hazardous
materials. In : Hann RW & Jensen PA ed. Water quality characteristics
of hazardous materials. College Station, Texas, Environmental
Engineering Information Center, pp 1-21.
Hansen OG (1991) PVC in the health care sector. Med Device Technol, 2:
18-23.
Hansteen I-L, Hillestad L, Thiis-Evensen E, & Heldaas SS (1978)
Effects of vinyl chloride in man: A cytogenetic follow-up study. Mutat
Res, 51: 271-278.
Harkov R, Kebbekus B, Bozzelli W, & Lioy PJ (1983) Measurement of
selected volatile organic compounds at three locations in New Jersey
during the summer season. J Air Pollut Control Assoc, 33: 1177-1183.
Harkov R, Kebbekus B, Bozelli JW, Lioy JW, & Daisey J (1984)
Comparison of selected volatile organic compounds during summer and
winter at urban sites in New Jersey. Sci Total Environ, 38: 259-274.
Harris DK (1953) Health problems in the manufacture and use of
plastics. Br J Ind Med, 10: 255-268.
Harris CC (1996) p53 Tumor suppressor gene: At the crossroads of
molecular carcinogenesis, molecular epidemiology, and cancer risk
assessment. Environ Health Perspect, 104: 435-439.
Harris DK & Adams WGF (1967) Acro-osteolysis occurring in men engaged
in the polymerization of vinyl chloride. Br Med J, 3: 712-714.
Harris LR & Sarvadi DG (1994) Synthetic polymers. In: Clayton GD &
Clayton FE ed. Patty's industrial hygiene and toxicology, 4th ed. New
York, John Wiley & Sons, Inc., vol 2, part E, pp 3673-4005.
Hartmans S (1995) Microbial degradation of vinyl chloride. In: Singh
VP ed. Biotransformations: Microbial degradation of health risk
compounds. Amsterdam, Oxford, New York, Elsevier Science Publishers,
pp 239-248.
Hartmans S & De Bont JAM (1992) Aerobic vinyl chloride metabolism in
Mycobacterium aurum L1. Appl Environ Microbiol, 58: 1220-1226.
Hartmans S, De Bont JAM, Tramper J, & Luyben KCAM (1985) Bacterial
degradation of vinyl chloride. Biotechnol Lett, 7: 383-388.
Hartmans S, Kaptein A, Tramper J, & De Bont JAM (1992)
Characterization of a Mycobacterium sp. and a Xanthobacter sp. for
the removal of vinyl chloride and 1,2-dichloroethane from waste gases.
Appl Microbiol Biotechnol, 37: 796-801.
Hauschild I, Schröer A, Siedersleben M, & Starnick J (1994) The
microbial growth of Mycobacterium aurum L1 on vinyl chloride with
respect to inhibitory and limiting influence of substrate and oxygen.
Water Sci Technol, 30: 125-132.
Hayashi H, Sakamuto N, Aoi T, Fukumura A, Mano H, & Inagaki T (1990)
Invasive character of malignant endothelial cells in
vinyl-chloride-induced liver angiosarcoma. Nagoya J Med Sci, 52:
13-17.
Hedley WH, Cheng JT, McCormick R, & Lewis WA (1976) Sampling of
automobile interiors for vinyl chloride monomer. Research Triangle
Park, North Carolina, US Environmental Protection Agency, 34 pp
(EPA-600/2-76-124; prepared by Monsanto Research Corporation, Dayton,
Ohio).
Hefner RE, Watanabe PG, & Gehring PJ (1975a) Preliminary studies on
the fate of inhaled vinyl chloride monomer (VCM) in rats. Environ
Health Perspect, 11: 85-95.
Hefner RE, Watanabe PG, & Gehring PJ (1975b) Short communication:
Percutaneous absorption of vinyl chloride. Toxicol Appl Pharmacol, 34:
529-532.
Hefner RE, Watanabe PG, & Gehring PJ (1975c) Preliminary studies of
the fate of inhaled vinyl chloride monomer in rats. Ann NY Acad Sci,
246: 135-148.
Heger M, Müller G, & Norpoth K (1981) [Investigations on the
correlation between vinyl chloride (=VCM)-uptake and excretion of its
metabolites by 15 VCM-exposed workers.] Int Arch Occup Environ Health,
48: 205-210 (in German).
Hehir RM, McNamara BP, McLaughlin J, Willigan DA, Bierbower G, &
Hardisty JF (1981) Cancer induction following single and multiple
exposure to a constant amount of vinyl chloride monomer. Environ
Health Perspect, 41: 63-72.
Heldaas SS, Langard S, & Andersen A (1984) Incidence of cancer among
vinyl chloride and polyvinyl chloride workers. Br J Ind Med, 41:
25-30.
Heldaas SS, Langard SL, & Andersen AA (1987) Incidence of cancer among
vinyl chloride and polyvinyl chloride workers: Further evidence for an
association with malignant melanoma. Br J Ind Med, 44: 278-280.
Helfgott TB, Hart FL, & Bedard RG (1977) An index of refractory
organics.. Ada, Oklahoma, US Environmental Protection Agency, pp 1-143
(EPA-600/2-77-174; prepared by the Environmental Engineering Program,
University of Connecticut, Storrs, Connecticut).
Helmig D, Pollock W, Greenberg J, & Zimmerman P (1996) Gas
chromatography mass spectrometry analysis of volatile organic trace
gases at Mauna Loa Observatory, Hawaii. J Geophys Res Atmos, D101:
14,697-14,710.
Hemminki K & Vineis P (1985) Extrapolation of the evidence on
teratogenicity of chemicals between humans and experimental animals:
Chemicals other than drugs. Teratogen Carcinogen Mutagen, 5: 251-318.
Hendry DG & Kenley RA (1979) Atmospheric reaction products of organic
compounds. Washington, DC, US Environmental Protection Agency, pp 1-80
(EPA-560/12-79-001; prepared by SRI International, Menlo Park,
California).
Henning KD & Richter E (1985) [Waste gas energy -- Treatment of gas
from sewage sludge and landfill sites using activated charcoal and
carbon-molecular sieves/methane enrichment.] Entsorgungspraxis, 5:
268-272 (in German).
Hesstvedt E, Hov O, & Isaksen ISA (1976) Photochemical lifetime of
vinylchloride in the atmosphere. In: Photochemical lifetime of
vinylchloride in the atmosphere. Oslo, Sweden, University of Oslo,
Institute for Geophysics, pp1-12 (Report No. 17).
Hiatt GFS, Cogliano VJ, Becker RA, Siegel DM, & Den A (1994) Vinyl
chloride action levels: Indoor air exposures at a superfund site. In:
Andrews JS, Frumkin H, Johnson BL, Mehlman MA, Xintaras C, & Bucsela
JA ed. Hazardous waste and public health: International Congress on
the Health Effects of Hazardous Waste. Princeton, New Jersey,
Princeton Scientific Publishing, pp 525-529 .
Hill RH, McCammon CS, Saalwaechter AT, Teass AW, & Woodfin WJ (1976a)
Gas-chromatographic determination of vinyl chloride in air samples
collected on charcoal. Anal Chem, 48: 1395-1398.
Hill J, Kollig HP, Paris DF, Wolfe NL, & Zepp RG (1976b) Dynamic
behavior of vinyl chloride in aquatic ecosystems. Athens, Georgia, US
Environmental Protection Agency, Environmental Research Laboratory, pp
1-43 (EPA 600/3-76-001).
Himeno S, Okuda H, & Suzuki T (1983) Lack of dominant lethal effects
in male CD-1 mice after short-term and long-term exposures to vinyl
chloride monomer. Toxicol Lett, 16: 47-53.
HMIP (Her Majesty's Inspectorate of Pollution) (1996) The chemical
release inventory 1994. London, Her Majesty's Stationary Office, pp
1-198.
Ho JSY (1989) A sequential analysis for volatile organics in water by
purge-and-trap capillary column gas chromatography with
photoionization and electrolytic conductivity detectors in series. J
Chromatogr Sci, 27: 91-98.
Ho SF, Phoon WH, Gan SL, & Chan YK (1991) Persistent liver dysfunction
among workers at a vinyl chloride monomer polymerization plant. J Soc
Occup Med, 41: 10-16.
Hocking C (1975) PVC and other plastics in food packaging. Aust
Packag, 23: 30-37.
Hodgson AT, Garbesi K, Sextro RG, & Daisey JM (1992) Soil-gas
contamination and entry of volatile organic compounds into a house
near a landfill. J Air Waste Manage Assoc, 42: 277-283.
Hoffmann D, Patrianakos C, Brunnemann KD, & Gori GB (1976)
Chromatographic determination of vinyl chloride in tobacco smoke. Anal
Chem, 48: 47-50.
Höfler F, Schneider J, & Möckel HJ (1986) Analytical investigations on
the composition of gaseous effluents from a garbage dump. Fresenius Z
Anal Chem, 325: 365-368.
Holliger C (1995) The anaerobic microbiology and biotreatment of
chlorinated ethenes. Curr Opin Biotechnol, 6: 347-351.
Hollstein M, Marion M-J, Lehman T, Welsh J, Harris CC, Martel-Planche
G, Kusters I, & Montesano R (1994) p53 Mutations at A:T base pairs in
angiosarcomas of vinyl chloride-exposed factory workers.
Carcinogenesis, 15: 1-3.
Hollstein M, Shomer G, Greenblatt M, Soussi T, Hovig E, Montesano R, &
Harris CC (1996) Somatic point mutations in the p53 gene of human
tumors and cell lines: Updated compilation. Nucleic Acids Res, 24:
141-146.
Holm L, Westlin A, & Holmberg B (1982) Technical control measures in
the prevention of occupational cancer: An example from the PVC
industry. In: Proceedings of the International Symposium on Prevention
of Occupational Cancer. Geneva, International Labour Office, pp
538-546.
Holmberg B, Kronevi T, & Winelli M (1979) The pathology of vinyl
chloride exposed mice. Acta Vet Scand, 17: 328-342.
Hong CB, Winston JM, Thornburg LP, Lee CC, & Woods JS (1981) Follow-up
study on the carcinogenicity of vinyl chloride and vinylidene chloride
in rats and mice: Tumor incidence and mortality subsequent to
exposure. J Toxicol Environ Health, 7: 909-924.
Hori M, Kobayashi Y, & Ota Y (1972) Vinyl chloride monomer odor
concentration. Plast Ind News, 18: 164-168.
Howard CJ (1976) Rate constants for the gas-phase reactions of OH
radicals with ethylene and halogenated ethylene compounds. J Chem
Phys, 65: 4771-4777.
Howard PH (1989) Vinyl chloride. In: Howard PH ed. Handbook of
environmental fate and exposure data for organic chemicals -- Volume
I: Large production and priority pollutants. Chelsea, Michigan, Lewis
Publishers, Inc., pp 551-560.
Howard PH, Boethling RS, Jarvis WF, Meylan WM, & Michalenko EM (1991)
Vinyl chloride. In: Printup HT ed. Handbook of environmental
degradation rates. Chelsea, Michigan, Lewis Publishers, Inc., pp
138-139.
Hozo I (1998) Croatian cases of ASL. Presented at the Meeting of the
Association of Plastics Manufacturers in Europe, Oslo, 4-5 June 1998
(Unpublished report).
Hozo I, Andelinovic S, Ljutic D, Miric D, Bojic L, & Gasperic I (1996)
Vinyl chloride monomer exposure by the plastic industry workers
-- basic condition for liver angiosarcoma appearance. Med Arhiv, 50:
9-14.
Hozo I, Andelinovic S, Ljutic D, Bojic L, Miric D, & Giunio L (1997)
Two new cases of liver angiosarcoma: History and perspectives of liver
angiosarcoma among plastic industry workers. Toxicol Ind Health, 13:
639-647.
Hrivnak L, Rozinova Z, Korony S, & Fabianova E (1990) Cytogenetic
analysis of peripheral blood lymphocytes in workers exposed to vinyl
chloride. Mutat Res, 240: 83-85.
HSC (Health and Safety Commission, UK) (1995) Control of vinyl
chloride at work: Approved code of practice. London, Health and Safety
Executive, 10 pp.
HSE (Health and Safety Executive) (1987) Vinyl chloride in air. In:
Vinyl chloride in air: Laboratory method using charcoal adsorbent
tubes, solvent desorption and gas chromatography -- MDHS 24: Methods
for the determination of hazardous substances. London, Health and
Safety Executive, pp 1-4.
Huang M (1993a) Prospective epidemiological investigation on
occupational malignant tumor in workers exposed to vinyl chloride. J
Hyg Res, 22: 193-196.
Huang M-Y (1993b) Epidemiological investigation on malignant tumors in
workers exposed to vinyl chloride. Chin J Ind Hyg Occup Dis, 11:
147-151.
Huang M-Y (1994) An investigation on the effect of reproductive
function in workers occupationally exposed to vinyl chloride. Chin J
Ind Hyg Occup Dis, 12: 12-15.
Huberman E, Bartsch H, & Sachs L (1975) Mutation induction in Chinese
hamster V79 cells by two vinyl chloride metabolites, chloroethylene
oxide and 2-chloroacetaldehyde. Int J Cancer, 16: 639-644.
Hung IF, Huang KC, Hung CK, & Shih TS (1996) A thermal desorption gas
chromatographic method for sampling and analysis of airborne vinyl
chloride. Chemosphere, 32: 661-666.
Hussain S & Osterman-Golkar S (1976) Comment on the mutagenic
effectiveness of vinyl chloride metabolites. Chem-Biol Interact, 12:
265-267.
Hüttner E & Holzapfel B (1996) HPRT mutant frequencies and detection
of large deletions by multiplex-PCR in human lymphocytes of vinyl
chloride exposed and non-exposed populations. Toxicol Lett, 88:
175-183.
Hüttner E & Nikolova T (1998) Cytogenetic analysis of peripheral
lymphocytes in a population exposed to vinyl chloride through an
accidental release into the environment. Toxicol Lett, 96/97: 143-148.
Hwang Y-L, Olson JD, & Keller GE (1992) Steam stripping for removal of
organic pollutants from water: 2. Vapor-liquid equilibrium data. Ind
Eng Chem Res, 31: 1759-1768.
IARC (1978) Environmental carcinogenesis selected methods of analysis.
In: Environmental carcinogenesis selected methods of analysis
-- Volume 2: Methods for the measurement of vinyl chloride in poly
(vinyl chloride), air, water and foodstuffs. Lyon, International
Agency for Research on Cancer, pp 19-126 (IARC Scientific Publications
No. 22).
IARC (1979) Vinyl chloride, polyvinyl chloride and vinyl
chloride-vinyl acetate copolymers. Lyon, International Agency for
Research on Cancer, pp 377-438 (IARC Monographs on the Evaluation of
Carcinogenic Risk of Chemicals to Humans, Volume 19).
IARC (1987) Overall evaluations of carcinogenicity: An updating of
IARC monographs, volumes 1 to 42. Lyon, International Agency for
Research on Cancer, pp 373-376 (IARC Monographs on the Evaluation of
the Carcinogenic Risk of Chemicals to Humans, Supplement 7).
IARC (1997) Polychlorinated dibenzo-para-dioxins and polychlorinated
dibenzofurans. Lyon, International Agency for Research on Cancer, pp
1-666 (IARC Monographs on the Evaluation of the Carcinogenic Risk of
Chemicals to Humans, Volume 69).
Infante PF (1976) Oncogenic and mutagenic risks in communities with
polyvinyl chloride production facilities. Ann NY Acad Sci, 271: 49-57.
Infante PF & Pesák J (1994) A historical perspective of some
occupationally related diseases of women. J Occup Med, 36: 826-831.
Infante PF, Wagoner JK, McMichael AJ, Waxweiler RJ, & Falk H (1976a)
Genetic risks of vinyl chloride. Lancet, April: 734-735.
Infante PF, Wagoner JK, McMichael AJ, Waxweiler RJ, & Falk H (1976b)
Genetic risks of vinyl chloride. Lancet, June: 1289-1290.
IPCS (1989) Environmental health criteria 88: Polychlorinated
dibenzo-para-dioxins and dibenzofurans. Geneva, World Health
Organization, International Programme on Chemical Safety.
IPCS (1994) Environmental health criteria 170: Assessing human health
risks of chemicals: Derivation of guidance values for health-based
exposure limits. Geneva, World Health Organization, International
Programme on Chemical Safety.
Ishikawa O, Weerite S, Tamura A, & Miyachi Y (1995) Occupational
scleroderma: A 17-year follow-up study. Br J Dermatol, 133: 786-789.
ISO (1989) Water quality -- determination of the inhibition of the
mobility of Daphnia magna Straus (Cladocera, Crustacea). In:
International standard ISO-6341, 2nd ed. Geneva, International
Standard Organization, pp 1-7.
Jacobsen JS & Humayun MZ (1990) Mechanisms of mutagenesis by the vinyl
chloride metabolite chloroacetaldehyde. Effect of gene-targeted in
vitro adduction of M13 DNA on DNA template activity in vivo and
in vitro. Biochemistry, 29: 496-504.
Jaeger RJ, Reynolds ES, Conolly RB, Moslen MT, Szabo S, & Murphy SD
(1974) Acute hepatic injury by vinyl chloride in rats pretreated with
phenobarbital. Nature (Lond), 252: 724-726.
Jaeger RJ, Murphy SD, Reynolds ES, Szabo S, & Moslen MT (1977)
Chemical modification of acute hepatotoxicity of vinyl chloride
monomer in rats. Toxicol Appl Pharmacol, 41: 597-607.
Jain MK & Criddle CS (1995) Metabolism and cometabolism of halogenated
C-1 and C-2 hydrocarbons. Prog Ind Microbiol, 32: 65-102.
Janson O (1989) [Characterization of landfill gases by chosen trace
substances in particular through sulphur compounds.] Müll Abfall 4:
198-208 (in German).
Jedrychowski RA, Sokal JA, & Chmielnicka J (1984) Influence of
exposure mode on vinyl chloride action. Arch Toxicol, 55: 195-198.
Jeffers PM & Wolfe NL (1996) Homogeneous hydrolysis rate constants
-- Part II: Additions, corrections and halogen effects. Environ
Toxicol Chem, 15: 1066-1070.
Jenssen D & Ramel C (1980) The micronucleus test as part of a
short-term mutagenicity test program for the prediction of
carcinogenicity evaluated by 143 agents tested. Mutat Res, 75:
191-202.
Jiangl H (1990) The effects of vinyl chloride on pregnancy,
parturition and fetal development among female workers. Abstr Pap Am
Chem Soc, 199: 1-2.
John JA, Smith FA, Leong BKJ & Schwetz BA (1977) The effects of
maternally inhaled vinyl chloride on embryonal and fetal development
in mice, rats and rabbits. Toxicol Appl Pharmacol, 39: 497-513.
John AJ, Smith FA, & Schwetz BA (1981) Vinyl chloride: Inhalation
teratology study in mice, rats and rabbits. Environ Health Perspect,
41: 171-177.
Jones JH (1981) Worker exposure to vinyl chloride and poly(vinyl
chloride). Environ Health Perspect, 41: 129-136.
Jones RD, Smith DM, & Thomas PG (1988) A mortality study of vinyl
chloride monomer workers employed in the United Kingdom in 1940-1974.
Scand J Work Environ Health, 14: 153-160.
Jury WA, Russo D, Streile G, & El Abd H (1992) Correction to
"evaluation of volatilization by organic chemicals residing below the
soil surface". Water Resour Res, 28: 607-608.
Kagiya T, Takemoto K, & Uyama Y (1975) [Photo-oxidative decomposition
of vinyl chloride in the air.] Nippon Kagaku Kaishi, 11: 1922-1925 (in
Japanese).
Kandala JC, Mrema JEK, DeAngelo A, Daniel FB, & Guntaka RV (1990)
2-chloroacetaldehyde and 2-chloroacetal are potent inhibitors of DNA
synthesis in animal cells. Biochem Biophys Res Commun, 167: 457-463.
Kanno S, Nojima K, Takahaski K, Iseki H, Takamori S, & Takamatsu K
(1977) Studies on the photochemistry of aliphatic halogenated
hydrocarbons II. Chemosphere, 8: 509-514.
Kappus H, Bolt HM, Buchter HA, & Bolt W (1975) Rat liver microsomes
catalyse covalent binding of [14C]-vinyl chloride to macromolecules.
Nature (Lond), 257: 134-135.
Kappus H, Bolt HM, Buchter HA, & Bolt W (1976) Liver microsomal uptake
of [14C] vinyl chloride and transformation to protein alkylating
metabolites in vitro. Toxicol Appl Pharmacol, 37: 461-471.
Kästner M (1991) Reductive dechlorination of tri-and
tetrachloroethylenes depends on transition from aerobic to anaerobic
conditions. Appl Environ Microbiol, 57: 2039-2046.
Katosova LD & Pavlenko GI (1985) Cytogenetic examination of the
workers of chemical industry. Mutat Res, 147: 301-302.
Kenaga EE & Goring CAI (1980) Relationship between water solubility,
soil sorption, octanol-water partitioning, and concentration of
chemicals in biota. In: Eaton JG, Parrish PR, & Hendricks AC ed.
Aquatic toxicology. Philadelphia, Pennsylvania, American Society for
Testing and Materials, pp 78-115 (ASTM STP 707).
Keplinger ML, Goode JW, Gordon DE, & Calandra JC (1975) Interim
results of exposure of rats, hamsters, and mice to vinyl chloride. Ann
NY Acad Sci, 246: 219-224.
Kinnunen J (1996) [Ambient air quality in the surroundings of
Kilpilahti, 1995 -- Part 2: Volatile organic compounds.] Porvoo,
Finland, Neste Environmental and Industrial Hygiene Institute, pp 1-40
(Report No. 960024Y) (in Finnish).
Kinnunen J (1997) [Ambient air quality in the surroundings of
Kilpilahti, 1996 -- Part 2: Volatile organic compounds].Porvoo,
Finland, Neste Environmental and Industrial Hygiene Institute, pp 1-34
(Report No. 97003Y) (in Finnish).
Kirchner K, Helf D, Ott P, & Vogt S (1990) The reaction of OH radicals
with 1,1-di-, tri-, and tetrachloroethylene. Ber Bunsenges Phys Chem,
94: 77-83.
Klamt A (1993) Estimation of gas-phase hydroxyl radical rate constants
of organic compounds from molecular orbital calculations. Chemosphere,
26: 1273-1289.
Klein W, Herrchen M, & Scheunert I (1989) [The problem of bound
residues in soil.] VDI-Ber, 745: 451-465 (in German).
Klöpffer W, Haag F, Kohl E-G, & Frank R (1988) Testing of the abiotic
degradation of chemicals in the atmosphere: The smog chamber approach.
Ecotoxicol Environ Saf, 15: 298-319.
Klöpffer W, Kaufmann G, & Frank R (1985) [Phototransformation of air
pollutants: Rapid test for the determination of KOH.] Z Naturforsch,
40A: 686-692 (in German).
Knight KR & Gibbons R (1987) Increased collagen synthesis and
cross-link formation in the skin of rats exposed to vinyl chloride
monomer. Clin Sci, 72: 673-678.
Koischwitz D, Lelbach WK, Lackner K, & Hermanutz D (1981) Angiosarcoma
of the liver and hepatocellular carcinomas induced by vinyl chloride.
Fortschr Röntgenstr, 134: 283-290.
Kolb B (1982) Multiple headspace extraction -- a procedure for
eliminating the influence of the sample matrix in quantitative
headspace gas chromatography. Chromatographia, 15: 587-594.
Kollar V, Kemka R, & Misianik J (1988) Determination of vinyl chloride
and vinyl acetate in working atmosphere. Chem Pap, 42: 147-160.
Kontominas MG, Gupta C, & Gilbert SG (1985) Interaction of
vinylchloride with polyvinylchloride in model food systems by
HPLC-chromatography: Migration aspects. Lebensm.wiss Technol, 18:
353-356.
Kopetz K, Mürmann P, Tenkhoff N, & Terwiesch B (1986) [Aspects of
worker, environment and consumer protection in the production,
processing and use of VC and PVC.] In: Becker CW & Braun A ed.
[Plastics Handbook -- Volume 2/2: Polyvinyl chloride.] Munich, Wien,
Carl Hanser Verlag, pp 1437-1465 (in German)
Köster M (1989) [Occurrence, evaluation and analysis of vinyl chloride
as a degradation product of chlorinated ethenes in contaminated
areas.] GWF-Wasser/Abwasser, 130: 694-697 (in German).
Kotseva K (1996) Five year follow-up study of the incidence of
arterial hypertension and coronary heart disease in vinyl chloride
monomer and polyvinyl chloride production workers. Int Arch Occup
Environ Health, 68: 377-379.
Krajewski J & Dobecki M (1978) Determination of vinyl chloride in the
air by gas chromatographic method. Med Prac, 29: 403-410.
Krajewski J, Dobecki M & Gromiec J (1980) Retention of vinyl chloride
in the human lung. Br J Ind Med, 37: 373-374.
Kraybill HF (1983) Assessment of human exposure and health risk to
environmental contaminants in the atmosphere and water with special
reference to cancer. J Environ Sci Health, C1: 175-232.
Kromann A, Ludvigsen L, Albrechtsen H-J, Christensen TH, Ejlertsson J,
& Svensson BH (1998) Degradability of chlorinated aliphatic compounds
in methanogenic leachates sampled at eight landfills. Waste Manage
Res, 16: 54-62.
Kruschel BD, Bell RW, Chapman RE, Spencer MJ, & Smith KV (1994)
Analysis of ambient polar and non polar volatile organic compounds
(VOCs) by thermal desorption, high resolution gas chromatography-mass
spectrometry (TD/HRGC/MS). J High Resol Chromatogr, 17: 187-190.
Kucerová M, Polívková Z, & Bátora J (1979) Comparative evaluation of
the frequency of chromosomal aberrations and the SCE numbers in
peripheral lymphocytes of workers occupationally exposed to vinyl
chloride monomer. Mutat Res, 67: 97-100.
Kuchenmeister F, Wang M, Klein RG, & Schmezer P (1996) Transport of
reactive metabolites of procarcinogens between different liver cell
types, as demonstrated by the single cell microgel electrophoresis
assay. Toxicol Lett, 88: 29-34.
Kudo YY, Yamada S, Nakamura I, Sakaguchi T, Sakaguchi S, Ohe T,
Kamagata H, Naito A, & Nakazawa S (1990) On the changes in peripheral
red cells in mice exposed to vinyl chloride monomer. Pol J Occup Med,
3: 301-310.
Kurlyandski BA, Stovbur NN, & Turusov VS (1981) [About hygienic
regimentation of vinyl chloride.] Gig i Sanit, 3: 74-75 (in Russian).
Kusmierek JT & Singer B (1992) 1,N2-ethenodeoxyguanosine: Properties
and formation in chloroacetaldehyde-treated polynucleotides and DNA.
Chem Res Toxicol, 5: 634-638.
L'Abbé KA, Ferro G, Winkelman R, Saracci R, & Simonato L (1989)
Mortality and cancer incidence results of the European multi-centric
cohort study of workers employed in the vinyl chloride industry. Lyon,
International Agency for Research on Cancer (IARC Internal Report
89/007).
Lackey LW, Phelps TJ, Korde V, Nold S, Ringelberg D, Bienkowski PR ,&
White DC (1994) Feasibility testing for the on-site bioremediation of
organic wastes by native microbial consortia. Int Biodeterior
Biodegrad, 33: 41-59.
Lahl U, Zeschmar-Lahl B, & Jager J (1991) [Release of pollutants from
municipal waste deponies.] Wasser Luft Boden, 1: 64-66 ( in German).
Lahmann E (1980) [Measurements of air pollutants in Berlin: Literature
study on measurement programmes and their results.] WaBoLu- Berichte,
1: 4.
Laib RJ & Bolt HM (1977) Alkylation of RNA by vinyl chloride
metabolites in vitro and in vivo: Formation of
1- N6-etheno-adenosine. Toxicology, 8: 185-195.
Laib RJ & Bolt HM (1978) Formation of 3,N4-ethenocytidine moieties in
RNA by vinyl chloride metabolites in vitro and in vivo. Arch
Toxicol, 39: 235-240.
Laib RJ & Bolt HM (1980) Trans-membrane alkylation: a new method for
studying irreversible binding of reactive metabolites to nucleic
acids. Biochem Pharmacol, 29: 449-452.
Laib RJ, Gwinner LM, & Bolt HM (1981) DNA alkylation by vinyl chloride
metabolites: Etheno derivatives or 7-alkylation of guanine? Chem-Biol
Interact, 37: 219-231.
Laib RJ, Pellio T, Wünschel UM, Zimmermann N, & Bolt HM (1985a) The
rat liver foci bioassay: II. Investigations on the dose-dependant
induction of the ATPase-deficient foci by vinyl chloride at very low
doses. Carcinogenesis, 6: 69-72.
Laib RJ, Klein KP, & Bolt HM (1985b) The rat liver foci bioassay: I.
Age-dependence of induction by vinyl chloride of ATPase-deficient
foci. Carcinogenesis, 6: 65-68.
Laib RJ, Bolt HM, Cartier R, & Bartsch H (1989) Increased alkylation
of liver DNA and cell turnover in young versus old rats exposed to
vinyl chloride correlated with cancer susceptibility. Toxicol Lett,
45: 231-239.
LAI (German State Committee for Air Pollution Control) (1992) [Cancer
risk through air pollution: Development of "criteria for carcinogenic
air pollution" assigned by the Environment Ministers Conference.]
Düsseldorf, Ministry for the Environment, Area and Agriculture, 330 pp
(in German).
Lakhanisky T, Bazzoni D, Jadot P, Joris I, Laurent C, Ottogali M, Pays
A, Planard C, Ros Y, & Vleminckx C (1993) Cytogenetic monitoring of a
village population potentially exposed to a low level of environmental
pollutants -- Phase 1: SCE analysis. Mutat Res, 319: 317-323.
Lane DP (1994) p53 and human cancer. Br Med Bull, 50: 582-599.
Langauer-Lewowicka H, Kurzbauer H, Byczkowska Z, & Wocka-Marek T
(1983) Vinyl chloride disease -- neurological disturbances. Int Arch
Occup Environ Health, 52: 151-157.
Lange C-E, Jühe S, Stein G, & Veltman G (1974) [The so-called vinyl
chloride disease: An occupationally caused systemic sclerosis.] Int
Arch Arbeitsmed, 32: 1-32 (in German).
Langouet S, Müller M, & Guengerich FP (1997) Misincorporation of dNTPs
opposite 1,N2-ethenoguanine and
5,6,7,8-tetrahydro-7-hydroxy-9-oxoimidazo[1,2-a]purine in
oligonucleotides by Echerichia coli polymerases I exo- and II exo-, T7
polymerase exo-, human immunodeficiency virus-1 reverse transcriptase,
and rat polymerase ß. Biochemistry, 36: 6069-6079.
Laplanche A, Clavel F, Contassot JC, & Lanouziere C (1987) Exposure to
vinyl chloride monomer: report on a cohort study. Br J Ind Med, 44:
711-715.
Laplanche A, Clavel-Chapelon F, Contassot JC, & Lanouziére C (1992)
Exposure to vinyl chloride monomer: Results of a cohort study after a
seven year follow up. Br J Ind Med, 49: 134-137.
Laumbach AD, Lee S, Wong J, & Streips UN (1977) Studies on the
mutagenicity of vinyl chloride metabolites and related chemicals. In:
Nienburg HE ed. Prevention and detection of cancer. New York, Basel,
Marcel Dekker Inc., vol 2, part 1, pp 155-170.
Lederer M (1959) [A vinyl chloride peroxide.] Angew Chem, 71: 162 (in
German).
Lee CC, Bhandaari JC, Winston JM, House WB, Peters PJ, Dixon RL, &
Woods JS (1977) Inhalation toxicity of vinyl chloride and vinylidene
chloride. Environ Health Perspect, 21: 25-32.
Lee CC, Bhandari JC, Winston JM, House WB, Dixon RL, & Woods JS (1978)
Carcinogenicity of vinyl chloride and vinylidene chloride. J Toxicol
Environ Health, 4: 15-30.
Lee MD, Mazierski PF, Buchanan RJ, Ellis DE, & Sehayek LS (1995)
Intrinsic in situ anaerobic biodegradation of chlorinated solvents at
an industrial landfill. In: Hinchee RE, Wilson JT, & Downey DC ed.
Intrinsic bioremediation. Columbus, Ohio, Battelle Press, pp 205-222.
Lee FI, Smith PM, Bennett B, & Williams DMJ (1996) Occupationally
related angiosarcoma of the liver in the United Kingdom 1972-1994.
Gut, 39: 312-318.
Leisinger T (1992) Microorganisms for the introduction of chlorinated
aliphatic hydrocarbons into the carbon cycle. In: Mongkolsuk Sea ed.
Biotechnology and environmental science: Molecular approaches. New
York, London, Plenum Press, pp 143-147.
Lelbach WK (1996) A 25-year follow-up study of heavily exposed vinyl
chloride workers in Germany. Am J Ind Med, 29: 446-458.
Lelbach WK & Marsteller HJ (1981) Vinyl chloride -- associated
disease. Adv Intern Med Pediatr, 47: 1-110.
Lesage S, Jackson RE, Priddle MW, & Riemann PG (1990) Occurrence and
fate of organic solvent residues in anoxic groundwater at the
Gloucester Landfill, Canada. Environ Sci Technol, 24: 559-566.
Lesage S, McBride RA, Cureton PM, & Brown S (1993) Fate of organic
solvents in landfill leachates under simulated field conditions and in
anaerobic microcosms. Waste Manage Res, 11: 215-226.
Leschber R, Mergler-Voelkl R, & Nerger M (1990) Soil and groundwater
contamination by low boiling chlorinated hydrocarbons in Berlin. Int J
Environ Anal Chem, 39: 159-164.
Lester D, Greenberg LA, & Adams WR (1963) Effects of single and
repeated exposures of humans and rats to vinyl chloride. Am Ind Hyg
Assoc J, 24: 265-275.
Li Y, Asherova M, Marion M-J, & Brandt-Rauf PW (1998a) Mutant
oncoprotein biomarkers in chemical carcinogenesis. In : Mendelsohn ML,
Mohr LC, & Peeters J ed. Biomarkers: Medical and workplace
applications. Washington, DC, National Academy Press, pp 345-353.
Li Y, Marion MJ, Asherova M, Coulibaly D, Smith SJ, Do T, Carney WP, &
Brandt-Rauf PW (1998b) Mutant p21 ras in vinyl chloride-exposed
workers. Biomarkers, 3: 433-439.
Lilis R (1981) Review of pulmonary effects of poly(vinyl chloride) and
vinyl chloride exposure. Environ Health Perspect, 40: 167-169.
Lilis R, Anderson H, Nicholson WJ, Daum S, Fishbein AS, & Selikoff IR
(1975) Prevalence of disease among vinyl chloride and polyvinyl
chloride workers. Ann NY Acad Sci, 246: 22-41.
Lillian D, Singh HB, Appleby A, Lobban L, Arnts R, Gumpert R, Hague R,
Toomey J, Kazazis J, Antell M, Hansen D, & Scott B (1975) Atmospheric
fates of halogenated compounds. Environ Sci Technol, 9: 1042-1048.
Lindbohm M-L, Hemminki K, & Kyyrönen P (1985) Spontaneous abortions
among women employed in the plastics industry. Am J Ind Med, 8:
579-586.
Lipsky D & Jacot B (1985) Hazardous emissions from sanitary landfills.
In: Hazardous emissions from sanitary landfills. For presentation at
the 78th Annual Meeting of the Air Pollution Control Association,
Detroit, Michigan, 16-21 June 1985. New York, Fred C. Hart Associates,
pp 1-13.
Little J (1993) Are occupational carcinogens teratogenic ? In:
Richardson M ed. Reproductive toxicology. Weinheim, Germany, VCH
Verlagsgesellschaft mbH pp 3-43.
Little JC, Daisey JM, & Nazaroff WW (1992) Transport of subsurface
contaminants into buildings. Environ Sci Technol, 26: 2058-2066.
Lloyd MH, Gauld S, Copland L, & Soutar CA (1984) Epidemiological study
of the lung function of workers at a factory manufacturing polyvinyl
chloride. Br J Ind Med, 41: 328-333.
Lopez-Avila V, Heath N, & Hu A (1987a) Determination of purgeable
halocarbons and aromatics by photoionization and hall electrolytic
conductivity detectors connected in series. J Chromatogr Sci, 25:
356-363.
Lopez-Avila V, Wood R, Flanagan M, & Scott R (1987b) Determination of
volatile priority pollutants in water by purge and trap and capillary
column gas chromatography/mass spectrometry. J Chromatogr Sci, 25:
286-291.
Loprieno N, Barale R, Baroncelli S, Bauer C, Bronzetti G, Cammellini
A, Cercignani G, Corsi C, Gervasi G, Leporini C, Nieri R, Rossi AM,
Stretti G, & Turchi G (1976) Evaluation of the genetic effects induced
by vinyl chloride monomer (VCM) under mammalian metabolic activation:
Studies in vitro and in vivo. Mutat Res, 40: 85-95.
Loprieno N, Barale R, Baroncelli S, Bartsch H, Bronzetti G, Cammellini
A, Corsi C, Frezza D, Nieri R, Leporini C, Rosellini D, & Rossi AM
(1977) Induction of gene mutations and gene conversions by vinyl
chloride metabolites in yeast. Cancer Res, 36: 253-257.
Lu P-Y, Metcalf RL, Plummer N, & Mandel D (1977) The environmental
fate of three carcinogens: Benzo-(alpha)-pyrene, benzidine, and vinyl
chloride evaluated in laboratory model ecosystems. Arch Environ Contam
Toxicol, 6: 129-142.
Lundberg I, Gustavsson A, Holmberg B, Molina G, & Westerholm P (1993)
Mortality and cancer incidence among PVC-processing workers in Sweden.
Am J Ind Med, 23: 313-319.
Lutz W-K & Schlatter J (1993) The relative importance of mutagens and
carcinogens in the diet. Pharmacol Toxicol, 72: 104-107.
Lyman WJ, Reehl WF, & Rosenblatt DH (1982) Handbook of chemical
property estimation methods: Environmental behavior of organic
compounds. New York, McGraw-Hill Book Company, pp 4-25.
Lyman WJ, Reehl WF, & Rosenblatt DH (1990) Handbook of chemical
property estimation methods. Environmental behavior of organic
compounds. Washington, DC, American Chemical Society, pp 15.4-15.20.
Mabey WR, Smith JH, Podoll RT, Johnson HL, Mill T, Chou TW, Gates J,
Partiridge IW, & Vandenberg D (1982) Aquatic fate process data for
organic priority pollutants. Washington, DC, US Environmental
Protection Agency, pp 1-407 (EPA-440/4-81-014; prepared by SRI
International, Menlo Park, California).
McCann J, Simmon V, Streitwieser D, & Ames BN (1975) Mutagenicity of
chloroacetaldehyde, a possible metabolic product of 1,2-dichloroethane
(ethylene dichloride), chloroethanol (ethylene chlorohydrin), vinyl
chloride, and cyclophosphamide. Proc Natl Acad Sci (USA), 72:
3190-3193.
McCarty PL (1993) In situ bioremediation of chlorinated solvents. Curr
Opin Biotechnol, 4: 323-330.
McCarty PL (1996) Biotic and abiotic transformations of chlorinated
solvents in ground water. In: Proceedings of the Symposium on Natural
Attenuation of Chlorinated Organics in Ground Water, Dallas, Texas,
11-13 September 1996. Washington, DC, US Environmental Protection
Agency, pp 5-9 (EPA 540/R-96/509).
McCarty PL (1997) Breathing with chlorinated solvents. Science, 276:
1521-1522.
McCarty PL & Reinhard M (1993) Biological and chemical transformations
of halogenated aliphatic compounds in aquatic and terrestrial
environments. In: Oremland RS ed. Biogeochemistry of global change:
Radioactively active trace gases. New York, Chapman & Hall, pp
839-852.
McLaughlin RS (1946) Chemical burns of the human cornea. Am J
Ophthalmol, 29: 1355-1362.
McMurry JR & Tarr J (1978) Vinyl chloride in Texas` ambient air. Proc
Annu Tech Meet Inst Environ Sci, 24: 149-153.
Magnusson J & Ramel C (1976) Mutagenic effects of vinyl chloride in
Drosophila melanogaster. Mutat Res, 38: 115.
Magnusson J & Ramel C (1978) Mutagenic effects of vinyl chloride on
Drosophila melanogaster with and without pretreatment with sodium
phenobarbiturate. Mutat Res, 57: 307-312.
Major DW, Hodgins WW, & Butler BJ (1991) Field and laboratory evidence
of in situ biotransformation of tetrachloroethene to ethene and ethane
at a chemical transfer facility in North Toronto. In: Hinchee RE &
Otfenbuttel RF eds. Onsite bioreclamation. Stoneham, Massachusetts,
Butterworth-Heinemann, pp 147-171.
Major D, Cox E, Edwards E, & Hare P (1995) Intrinsic dechlorination of
trichloroethene to ethene in a bedrock aquifer. In: Hinchee RE, Wilson
JT, & Downey DC ed. Intrinsic bioremediation. Columbus, Ohio, Battelle
Press, pp 197-203.
Makarov IA (1984) [Sexual disorders in male workers occupationally
exposed to methylmethacrylate and vinyl chloride.] Gig Tr Prof Zabol,
6: 19-23 (in Russian).
Makarov IA, Solovyeva MS, & Gnelitsky GI (1984) [On sexual disorders
in women chronically exposed to methylmethacrylate and vinyl
chloride.] Gig Tr Prof Zabol, 3: 22-27 (in Russian).
Makita O, Ono K, Sakurai T, Tanigawa H, Ando Y, Yamashita K,
Yoshimatsu M, & Takahashi M (1997) [A case of hepatocellular carcinoma
related with exposure to vinyl chloride monomer.] Nippon Shokakibyo
Gakkai Zasshi, 94 : 215-219 (in Japanese).
Makk L, Delmore F, Creech JL, Ogden LL, Fadell EH, Songster CL,
Clanton J, Johnson MN, & Christopherson WM (1976) Clinical and
morphologic features of hepatic angiosarcoma in vinyl chloride
workers. Cancer, 37: 149-163.
Malachowsky K, Phelps TJ, & White DC (1991) Aerobic mineralization of
vinyl chloride by branch-shaped gram-positive bacteria. Abstr Gen Meet
Am Soc Microbiol, 91: 277.
Malachowsky KJ, Phelps TJ, Teboli AB, Minnikin DE, & White DC (1994)
Aerobic mineralization of trichloroethylene, vinyl chloride, and
aromatic compounds by Rhodococcus species. Appl Environ Microbiol,
60: 542-548.
Malaveille C, Bartsch H, Barbin A, Camus AM, & Montesano R (1975)
Mutagenicity of vinyl chloride, chloroethylene oxide,
chloroacetaldehyde and chloroethanol. Biochem Biophys Res Commun, 63:
363-370.
Malle K-G (1984) [The implications for water conservation of the EU
priority list of 129 dangerous substances.] Z Wasser Abwasser Forsch,
17: 75-81 (in German).
Maltoni C & Cotti G (1988) Carcinogenicity of vinyl chloride in
Sprague-Dawley rats after prenatal and postnatal exposure. Ann NY Acad
Sci, 534: 145-159.
Maltoni C & Lefemine G (1975) Carcinogenicity bioassays of vinyl
chloride: Current results. Ann NY Acad Sci, 246: 195-218.
Maltoni C, Lefemine G, Chieco P, & Carretti D (1974) Vinyl chloride
carcinogenesis: Current results and perspectives. Med Lav, 65:
421-444.
Maltoni C, Lefemine C, Ciliberti A, Cotti G, & Carretti D (1979) Vinyl
chloride carcinogenicity bioassays (BT project) as an experimental
model for risk identification and assessment in environmental and
occupational carcinogenesis. In: Institut du Radium, Fondation Curie
ed. Epidémiologie animale et épidémiologie humaine: Le cas du chlorure
de vinyle monomère. Paris, Publications Essentielles, pp 17-20.
Maltoni C, Lefemine G, Ciliberti A, Cotti G, & Caretti D (1981)
Carcinogenicity bioassays of vinyl chloride monomer: A model of risk
assessment on an experimental basis. Environ Health Perspect, 41:
3-29.
Maltoni C, Lefemine G, Ciliberrti A, Cotti G, & Carretti D (1984) In :
Maltoni C, Lefemine G, Ciliberrti A, Cotti G, & Carretti D ed.
Experimental research on vinyl chloride carcinogenesis -- Volume II:
Archives of research on industrial carcinogenesis. Princeton, New
Jersey, Princeton Scientific Publisher Inc., 533 pp.
Marion M-J, De VI, Smith S, Luo JC, & Brandt RP (1996) The molecular
epidemiology of occupational carcinogenesis in vinyl chloride exposed
workers. Int Arch Occup Environ Health, 68: 394-398.
Marion M-J, Froment O, & Trépo C (1991) Activation of Ki- ras gene by
point mutation in human liver angiosarcoma associated with vinyl
chloride exposure. Mol Carcinogen, 4: 450-454.
Markowitz SS, McDonald CJ, Fethiere W, & Kerzner MS (1972)
Occupational acroosteolysis. Arch Dermatol, 106: 219-223.
Mastrangelo G, Manno M, Marcer G, Bartolucci GB, Gemignani C, Saladino
G, Simonato L, & Saia B (1979) Polyvinyl chloride pneumoconiosis:
Epidemiological study of exposed workers. J Occup Med, 21: 540-542.
Mastromatteo E, Fisher AM, Christie H, & Danziger H (1960) Acute
inhalation toxicity of vinyl chloride to laboratory animals. Am Ind
Hyg Assoc J, 21: 394-398.
Matsuda T, Yagi T, Kawanishi M, Matsui S, & Takebe H (1995) Molecular
analysis of mutations induced by 2-chloroacetaldehyde, the ultimate
carcinogenic form of vinyl chloride, in human cells using shuttle
vectors. Carcinogenesis, 16: 2389-2394.
Maymo-Gatell X, Tandoi V, Gossett JM, & Zinder SH (1995)
Characterization of an H2-utilizing enrichment culture that
reductively dechlorinates tetrachloroethene to vinyl chloride and
ethene in the absence of methanogenesis and acetogenesis. Appl Environ
Microbiol, 61: 3928-3933.
Maymó-Gatell X, Chien YT, Gossett JM, & Zinder SH (1997) Isolation of
a bacterium that reductively dechlorinates tetrachloroethene to
ethene. Science, 276: 1568-1571.
Meier T (1994) [Small helpers with a big impact: Bacteria for cleaning
waste gases containing vinyl chloride.] Entsorgungs-Technik, 6: 41-44
(in German).
Meier T (1996) [Biological treatment process for exhaust air
containing hydrogen sulphide and vinyl chloride.] Chem Techn, 25: 40
(in German) .
Meylan WM & Howard PH (1993) Computer estimation of the atmospheric
gas-phase reaction rate of organic compounds with hydroxyl radicals
and ozone. Chemosphere, 26: 2293-2299.
Middeldorp PJM, Van Aalst MA, Rijnaarts HHM, Stams FJM, De Kreuk HF,
Schraa G, & Bosma TNP (1998) Stimulation of reductive dechlorination
for in situ bioremediation of a soil contaminated with chlorinated
ethenes. Water Sci Technol, 37: 105-110.
Milde G, Nerger M, & Mergler R (1988) Biological degradation of
volatile chlorinated hydrocarbons in groundwater. Water Sci Technol,
20: 67-73.
Miller NL (1992) Stability analysis of toxic substances within aquatic
ecosystems and their effect on aquatic populations. Ecol Model, 60:
151-165.
Miller A (1993) Dioxin emissions from EDC/VCM plants. Environ Sci
Technol, 27: 1014-1015.
Mirkova E, Mihailova A, & Nosko M (1978) [Embryotoxic and teratogenic
effects in vinyl chloride.] Khig Zdraveopaz, 21: 440-443 (in Russian).
Molina G, Holmberg B, Elofsson S, Holmlund L, Moosing R, & Westerholm
P (1981) Mortality and cancer rates among workers in the Swedish PVC
processing industry. Environ Health Perspect, 41: 145-151.
Monson RR, Peters JM, & Johnson MN (1975) Proportional mortality among
vinyl chloride workers. Environ Health Perspect, 11: 75-77.
Moriya M, Zhang W, Johnson F, & Grollman AP (1994) Mutagenic potency
of exocyclic DNA adducts: Marked differences between Escherichia coli
and simian kidney cells. Proc Natl Acad Sci (USA), 91: 11899-11903.
Mroczkowska MM & Kusmierek JT (1991) Miscoding potential of
N2,3-ethenoguanine studied in an Escherichia coli DNA-dependent
RNA polymerase in vitro system and possible role of this adduct in
vinyl chloride-induced mutagenesis. Mutagenesis, 6: 385-390.
Müller JPH & Korte F (1977) [Contributions to ecological chemistry
-- report 136: Brief report on the nonsensitized photooxidation of
chloroethylenes.] Chemosphere, 6: 341-346 (in German).
Müller G, Norpoth K, & Eckard R (1976) Identification of two urine
metabolites of vinyl chloride by GC-MS-investigations. Int Arch Occup
Environ Health, 38: 69-75.
Müller G, Norpoth K, Kusters E, Herweg K, & Versin E (1978)
Determination of thiodiglycolic acid in urine specimens of vinyl
chloride exposed workers. Int Arch Occup Environ Health, 41: 199-205.
Müller G, Norpoth K, & Wicktramasinghe RH (1979) An analytical method
using GC-MS for the quantitative determination of urinary
thiodiglycolic acid. Int Arch Occup Environ Health, 44: 185-191.
Müller M, Belas FJ, Blair IA, & Guengerich FP (1997) Analysis of 1,
N2-ethenoguanine and 5,6,7,9-tetrahydro-7-hydroxy-9-oxoimidazo
(1,2-a) purine in DNA treated with 2-chlorooxirane by high performance
of amounts to other DNA adducts. Chem Res Toxicol, 10: 242-247.
Mur JM, Mandereau L, Deplan F, Paris A, Richard A, & Hemon D (1992)
Spontaneous abortion and exposure to vinyl chloride. Lancet, 339:
127-128.
Murdoch IA & Hammond AR (1977) A practical method for the measurement
of vinyl chloride monomer (VCM) in air. Ann Occup Hyg, 20: 55-61.
Murray WD & Richardson M (1993) Progress toward the biological
treatment of C1 and C2 halogenated hydrocarbons. Crit Rev Environ Sci
Technol, 23: 195-217.
Nair J, Barbin A, Guichard Y, & Bartsch MH (1995)
1,N6-ethenodeoxyadenosine and 3,N4-ethenodeoxycytidine in liver DNA
from humans and untreated rodents detected by
immunoaffinity/32P-postlabelling. Carcinogenesis, 16: 613-617.
Nair J, Sone H, Nagao M, Barbin A, & Bartsch H (1996) Copper-dependent
formation of miscoding etheno-DNA adducts in the liver of Long Evans
Cinnamon (LEC) rats developing hereditary hepatitis and hepatocellular
carcinoma. Cancer Res, 56: 1267-1271.
Nair J, Vaca CE, Velic I, Mutanen M, Valasta LM, & Bartsch H (1997)
High dietary omega-6 polyunsaturated fatty acids drastically
increase the formation of etheno-DNA base adducts in white blood cells
of female subjects. Cancer Epidemiol Biomarkers Prev, 6: 597-601.
Nair J, Carmichael PL, Fernando RC, Phillips DH, Strain AJ, & Bartsch
H (1998) Lipid peroxidation-induced etheno-DNA adducts in the liver of
patients with the genetic metal storage disorders Wilson's disease and
primary hemochromatosis. Cancer Epidemiol Biomarkers Prev, 7: 435-440.
Nakamura H & Mimura S (1979) Hygienic studies of materials for water
supplies I: Residual vinyl chloride monomer in commercial PVC-pipe and
water extractability of residual vinyl chloride monomer. Sewage Ind
Wastes, 91: 315.
Nakano N, Yamamoto A, & Nagashima K (1996) Development of a monitoring
system for vinyl chloride gas in air by using an HCl monitoring tape
and pyrolyzer. Talanta (Lond), 43: 459-463.
Narayanan B, Suidan MT, Gelderloos AB, & Brenner RC (1995) Anaerobic
treatment of volatile and semivolatile organic compounds in municipal
wastewater. Water Environ Res, 67: 46-56.
Nathan MF (1978) Choosing a process for chloride removal. Chem Eng,
January: 93-100.
Naylor LM & Loehr RC (1982) Priority pollutants in municipal sewage
sludge. BioCycle, July/Aug: 18-22.
Nelms LH, Reisszner KD, & West (1977) Personal vinyl chloride
monitoring device with permeation technique for sampling. Anal Chem,
49: 994-998.
Nelson YM & Jewell WJ (1993) Vinyl chloride biodegradation with
methanotrophic attached films. J Environ Eng, 119: 890-907.
Nelson NA, Robins TG, Garrison RP, Schuman M, & White RF (1993)
Historical characterization of exposure to mixed solvents for an
epidemiologic study of automotive assembly plant workers. Appl Occup
Environ Hyg, 8: 693-702.
Nerger M & Mergler-Völkl R (1988) [Biological degradation of volatile
chlorinated hydrocarbons in groundwater and sewage.] Z Wasser Abwasser
Forsch, 21: 16-19 (in German).
Neshiwat LF, Friedland ML, Schorr-Lesnick B, Feldman S, Glucksman WJ,
& Russo RD (1992) Hepatic angiosarcoma. Am J Med, 93: 219-222.
Neuhoff S (1988) [Safety planning in a city with chemical industry
- Cologne.] Brandschutz/ Dtsch Feuerw Ztg, 4: 168-175 (in German).
Nicholson WJ, Hammond EC, Seidman H, & Selikoff IJ (1975) Mortality
experience of a cohort of vinyl chloride-polyvinyl chloride workers.
Ann NY Acad Sci, 246: 225-230.
NIOSH (1994) Vinyl chloride. In: Eller PM & Cassinelli ME ed. NIOSH
manual of analytical methods, 4th ed. Cincinnati, Ohio, National
Institute for Occupational Safety and Health, pp 1-4.
NIOSH (1997) Pocket guide to chemical hazards. Cincinnati, Ohio,
National Institute for Occupational Safety and Health, pp 330-331.
Nishikawa H, Katami T, Takahara Y, Sumida H, & Yasuhara A (1992)
Emission of organic compounds by combustion of waste plastics
involving vinyl chloride polymer. Chemosphere, 25: 1953-1960.
Nystén T (1988) [Groundwater pollution cases in Uusima county.]
Helsinki, Finnish National Water Board, pp 1-46 (in Finnish).
OECD (1981a) Combined chronic toxicity/carcinogenicity studies. In:
OECD guidelines for testing of chemicals -- Section 4: Health effects
(No. 453). Paris, Organisation for Economic Co-operation and
Development, pp 1-15.
OECD (1981b) Carcinogenicity studies. In: OECD guidelines for testing
of chemicals -- Section 4: Health effects (No. 451). Paris,
Organisation for Economic Co-operation and Development, pp 1-15.
OECD (1983a) One-generation reproduction toxicity study. In: OECD
guidelines for testing of chemicals -- Section 4: Health effects (No.
415). Paris, Organisation for Economic Co-operation and Development,
pp 1-8 .
OECD (1983b) Two-generation reproduction toxicity study. In: OECD
guidelines for testing of chemicals -- Section 4: Health effects (No.
416). Paris, Organisation for Economic Co-operation and Development,
pp 1-8.
OECD (1984) OECD guideline for testing of chemicals No. 203: Fish,
acute toxicity test. Paris, Organisation for Economic Co-operation and
Development, pp 1-12.
Oesch F & Doerjer G (1982) Detection of N2,3-ethenoguanine in DNA
after treatment with chloroacetaldehyde in vitro. Carcinogenesis, 3:
663-665.
Olsen GW, Ramlow JM, & Hearn S (1995) Paternal occupational exposure
and spontaneous abortions: A closer look at paternal recall and vinyl
chloride. Am J Ind Med, 27: 611-613.
Orusev T, Popovski P, Bauer S, & Nikolova K (1976) Occupational risk
in the production of poly(vinyl chloride . God Zb Med Fak Skopje, 22:
33-38.
OSHA (Occupational Safety and Health Administration, US Department of
Labour) (1998) Title 29. Code Fed Regul, Part 1910(1910.1000):
138-143.
Oster RH, Carr CJ, Krantz JC, & Sauerwald MJ (1947) Anesthesia: XXVII.
Narcosis with vinyl chloride. Anesthesiology, 8: 359-361.
Osterman-Golkar S, Hultmark D, Segerbäck D, Calleman CJ, Göthe R,
Ehrenberg L, & Wachtmeister CA (1977) Alkylation of DNA and proteins
in mice exposed to vinyl chloride. Biochem Biophys Res Commun, 76:
259-266.
Ostlere LS, Harris D, Buckley C, Black C, & Rustin MHA (1992) Atypical
systemic sclerosis following exposure to vinyl chloride monomer: A
case report and review of the cutaneous aspects of vinyl chloride
disease. Clin Exp Dermatol, 17: 208-210.
Ott MG, Langner RR, & Holder BH (1975) Vinyl chloride exposure in a
controlled industrial environment. Arch Environ Health, 30: 333-339.
Ottenwälder H & Bolt HM (1980) Metabolic activation of vinyl chloride
and vinyl bromide by isolated hepatocytes by isolated hepatocytes and
hepatic sinusoidal cells. J Environ Pathol Toxicol, 4: 411-417.
Page GW (1981) Comparison of groundwater and surface water for
patterns and levels of contamination by toxic substances. Environ Sci
Technol, 15: 1475-1481.
Page BD & O'Grady R (1977) Gas-solid chromatographic confirmation of
vinyl chloride levels in oils and vinegars by using electrolytic
conductivity detection. J Assoc Off Anal Chem, 60: 576-578.
Palejwala VA, Simha D, & Humayun MZ (1991) Mechanisms of mutagenesis
by exocyclic DNA adducts. Transfection of m-viral DNA bearing a
site-specific adduct shows that ethenocytosine is a highly efficient
RecA-independent mutagenic noninstructional lesion. Biochemistry, 30:
8736-8743.
Paliard P, Valette PJ, Berger F, Contassot JC, & Partensky C (1991)
Péliose hépatique tardive après traitement d'un angiosarcome hépatique
chez un sujet exposé au chlorure de vinyl monomère. Gastroenterol Clin
Biol, 15: 445-448.
Pandya GA & Moriya M (1996) 1,N6-ethenodeoxyadenosine, a DNA adduct
highly mutagenic in mammalian cells. Biochemistry, 35: 11487-11492.
Pankow JF & Rosen ME (1988) Determination of volatile compounds in
water by purging directly to a capillary column with whole column
cryotrapping. Environ Sci Technol, 22: 398-405.
Pankow JF, Johnson RL, & Cherry JA (1993) Air sparging in gate wells
in cutoff walls and trenches for control of plumes of volatile organic
compounds (VOCs). Ground Water, 31: 654-663.
Park K-K, Leim A, Stewart BC, & Miller JA (1993) Vinyl carbamate
epoxide, a major strong electrophilic, mutagenic and carcinogenic
metabolite of vinyl carbamate and ethyl carbamate (urethane).
Carcinogenesis, 14: 441-450.
Parsons F, Wood PR, & DeMarco J (1984) Transformations of
tetrachloroethene and trichloroethene in microcosms and groundwater. J
Am Water Works Assoc, 76: 56-59.
Patty FA (1963) Vinyl chloride. In: Patty FA ed. Industrial hygiene
and toxicology. New York, Interscience Publishers, pp 1303-1305.
Patty FA, Yant WP, & Waite CP (1930) Acute response of Guinea pigs to
vapors of some new commercial organic compounds: V. Vinyl chloride.
Public Health Rep, 45: 1963-1971.
Pau JC, Knoll JE, & Midgett MR (1988) Evaluation of a process gas
chromatograph as a continuos emission monitor for benzene and vinyl
chloride. Int J Air Pollut Control Waste Manage, 38: 1528-1529.
Pearson CR & McConnell G (1975) Chlorinated C1 and C2 hydrocarbons in
the marine environment. Proc R Soc (Lond), B189: 305-332.
Pellizzari ED, Erickson MD, & Zweidinger RA (1979) Formulation of a
preliminary assessment of halogenated organic compounds in man and
environmental media. Washington, DC, US Environmental Protection
Agency (EPA-560/13-79-006; prepared by the Research Triangle
Institute, Research Triangle Park, North Carolina.).
Penin H, Sargar G, Lange C-E & Veltman G (1975) [Neurological,
psychiatrical and electroencephalographical findings in patients with
vinyl chloride disease.] Verh Dtsch Ges Arbeitsmed, 15: 299-304 (in
German).
Peoples AS & Leake CD (1933) The anesthetic action of vinyl chloride.
J Pharmacol Exp Ther, 48: 284-285.
Perrard M-H (1985) Mutagenicity and toxicity of chloroethylene oxide
and chloroacetaldehyde. Experientia (Basel), 41: 676-677.
Perry RA, Atkinson R, & Pitts JN (1977) Rate constants for the
reaction of OH radicals with CH2=CHF, CH2=CHCl, and CH2=CHBr over the
temperature range 299-426°K. J Chem Phys, 67: 458-462.
Perticoni GF, Abbritti G, Cantisani TA, Bondi L, & Mauro L (1986)
Polineuropathy in workers with long exposure to vinyl chloride
electrophysiological study. Electromyogr Clin Neurophysiol, 26: 41-47.
Pessayre D, Wandscheer JC, Descatoire V, Artigou JY, & Benhamou JP
(1979) Formation and inactivation of a chemically reactive metabolite
of vinyl chloride. Toxicol Appl Pharmacol, 49: 505-515.
Peter S & Ungváry G (1980) Lack of mutagenic effect of vinyl chloride
monomer in the mammalian spot test. Mutat Res, 77: 193-196.
Pettit BR (1986) The analysis of thiodiglycollic acid by selected ion
monitoring. Clin Chim Acta, 156: 85-90.
Pfab W & Mücke G (1977) [Migration of chosen monomers in food
simulants.] Verpackungsrundschau, 6: 49-52 (in German).
Phelps TJ, Malachowsky K, Schram RM, & White DC (1991a) Aerobic
mineralization of vinyl chloride by a bacterium of the order
Actinomycetales. Appl Environ Microbiol, 57: 1252-1254.
Phelps TJ, Niedzielski JJ, Malachowsky KJ, Schram RM, Herbes SE, &
White DC (1991b) Biodegradation of mixed-organic wastes by microbial
consortia in continuous-recycle expanded-bed bioreactors. Environ Sci
Technol, 25: 1461-1465.
Picciano DJ, Flake RE, Gay PC, & Kilian DJ (1977) Vinyl chloride
cytogenetics. J Occup Med, 19: 527-530.
Pirastu R, Comba P, Reggiani A, Foa V, Masina A, & Maltoni C (1990)
Mortality from liver disease among Italian vinyl chloride
monomer/polyvinyl chloride manufacturers. Am J Ind Med, 17: 155-161.
Pirastu R, Bellis S, Bruno C, Maltoni C, Masina A, & Reggiani A (1991)
[The mortality among the workers of vinyl chloride in Italy.] Med Lav,
82: 388-423 (in Italian).
Pirastu R, Chellini E, Carnevale F, De Santis M, Bracci C, & Comba P
(1997) [Cohort mortality study of vinyl chloride exposed workers in
Terrara, Rosignano and Ravenna.] Rome, Istituto Superiore di Sanità
(in Italian).
Pirastu R, Bruno C, De Santis M, & Comba P (1998) Cohort mortality
study of vinyl chloride exposed workers in Ferrara, Rosignano and
Ravenna. Epidemiol Prevenz, 22: 226-236.
Pitts JN (1993) Atmospheric formation and fates of toxic ambient air
pollutants. Occup Med, 3: 621-662.
Platt UF, Winer AM, Biermann HW, Atkinson R, & Pitts JN (1984)
Measurement of nitrate radical concentrations in continental air.
Environ Sci Technol, 18: 365-369.
Pleil JD & Lindstrom AB (1997) Exhaled human breath measurement
methods for assessing exposure to halogenated volatile organic
compounds. Clin Chem, 43: 723-730.
Plugge H & Safe S (1977) Vinyl chloride metabolism -- a review.
Chemosphere, 6: 309-325.
Podoll K, Berg-Dammer E, & Noth J (1990) [Neurological and
psychiatrical disorders in vinyl chloride disease.] Fortschr Neurol
Psychiatr, 58: 439-443 (in German).
Popper H & Thomas LB (1975) Alterations of liver and spleen among
workers exposed to vinyl chloride. Ann NY Acad Sci, 246: 172-194.
Popper H, Maltoni C, & Selikoff IJ (1981) Vinyl chloride-induced
hepatic lesions in man and rodents: A comparison. Liver, 1: 7-20.
Portier R, Miller G, Hoover D, & Simar R (1992) Field applications of
biological reactors removing ethylene dichloride, trichloroethylene,
and vinyl chloride from contaminated groundwaters. Abstr Gen Meet Am
Soc Microbiol, 92: 382.
Portier RJ, Shane BS, Walsh MM, & Williams MB (1993) In situ
remediation of EDC contaminated vadose soil: A toxicological
assessment. Symposium on Emerging Technologies: Metals, Oxidation and
Separation, Belmont, Texas, 25-26 February. Waste Manage, 13(5-7):
532-533.
Poy F, Cobelli L, Banfi S, & Fossati F (1987) Determination of vinyl
chloride monomer residue in poly(vinyl chloride) at the
parts-per-billion level with an automatic purge- and - trap technique.
J Chromatogr, 395: 281-289.
Prodan L, Suciu I, Pîslaru V, Ilea E, & Pascu L (1975) Experimental
acute toxicity of vinyl chloride (monochloroethene). Ann NY Acad Sci,
246: 154-158.
Przygodzki RM, Finkelstein SD, Keohavong P, Zhu D, Bakker A, Swalsky
PA, Soini Y, Ishak KG, & Bennet WP (1997) Sporadic and
thorotrast-induced angiosarcomas of the liver manifest frequent and
multiple point mutations in K- ras-2. Lab Invest, 76: 153-159.
Pudill R (1993) [Comparative toxicological and ecotoxicological
evaluation of volatile emissions from waste deponies and
incinerators.] Umweltwiss Schadst -Forsch, 5: 223-227 (in German).
Purchase IFH, Richardson CR, Anderson D, Paddle GM, & Adams WGF (1978)
Chromosomal analyses in vinyl chloride-exposed workers. Mutat Res, 57:
325-334.
Purchase IFH, Stafford J, & Paddle GM (1987) Vinyl chloride: An
assessment of risk of occupational exposure. Food Chem Toxicol, 25:
187-202.
Puschmann J (1975) [Determination of vinyl chloride in PVC and food
simulants.] Angew Makromol Chem ,47: 29-41 (in German).
Radike MJ, Stemmer KL, & Bingham E (1981) Effect of ethanol on vinyl
chloride carcinogenesis. Environ Health Perspect, 41: 59-62.
Rakus J, Brantley T, & Szabo E (1991) Environmental concerns with
vinyl medical devices. J Vinyl Technol, 13: 195-198.
Ramsey JC & Andersen ME (1984) A physiologically based description of
the inhalation pharmacokinetics of styrene in rats and humans. Toxicol
Appl Pharmacol, 73: 159-175.
Randall PM (1994) Pollution prevention strategies for the minimization
of industrial wastes in the VCM-PVC industry. Environ Prog, 13:
269-277.
Rannug U, Johansson A, Ramel C, & Wachtmeister CA (1974) The
mutagenicity of vinyl chloride after metabolic activation. Ambio, 3:
194-197.
Rannug U, Göthe R, & Wachtmeister CA (1976) The mutagenicity of
chloroethylene oxide, chloroacetaldehyde, 2-chloroethanol and
chloroacetic acid, conceivable metabolites of vinyl chloride.
Chem-Biol Interact, 12: 251-263.
Rappoport Z & Gal A (1969) Vinylic cations from solvolysis: I. The
trianisylvinyl halide system. J Am Chem Soc, 91: 5246-5254.
Rasche ME, Hyman MR, & Arp DJ (1991) Factors limiting aliphatic
chlorocarbon degradation by Nitrosomonas europaea: Cometabolic
inactivation of ammonia monooxygenase and substrate specificity. Appl
Environ Microbiol, 57: 2986-2994
Rashad MM, El-Belbessy SF, Hussein NG, Helmey MH, & El-Toukhy MA
(1994) Effect on some enzyme activities of occupational exposure to
vinyl chloride monomer for five consecutive years. Med Sci Res, 22:
289-290.
Rehm T & Werner R (1996) [Polyvinyl chloride.] Kunststoffe, 86:
1474-1476 (in German).
Reitz RH, Gargas ML, Andersen ME, Provan WM, & Green TL (1996)
Predicting cancer risk from vinyl chloride exposure with a
physiologically based pharmacokinetic model. Toxicol Appl Pharmacol,
137: 253-267.
Reynolds ES, Jaeger RJ, & Murphy SD (1975a) Acute liver injury by
vinyl chloride: Involvement of endoplastic reticulum in
phenobarbital-pretreated rats. Environ Health Perspect, 11: 227-233.
Reynolds ES, Moslen MT, Szabo S, & Jaeger RJ (1975b) Vinyl
chloride-induced deactivation of cytochrome P-450 and other components
of the liver mixed function oxidase system: An in vivo study. Res
Commun Chem Pathol Pharmacol, 12: 685-694.
Reynolds ES, Moslen MT, Szabo S, Jaeger RJ, & Murphy SD (1975c)
Hepatotoxicity of vinyl chloride and 1,1-dichloroethylene. Am J
Pathol, 81: 219-231.
Richardson CR, Styles JA, & Bennett IP (1983) Activity of vinyl
chloride monomer in the mouse micronucleus assay. Mutat Res, 122:
139-142.
Riordan SM, Loo CK, Haber RW, & Thomas MC (1991) Vinyl chloride
related hepatic angiosarcoma in a polyvinyl chloride autoclave cleaner
in Australia. Med J Aust, 155: 125-128.
Rippen G (1995) Handbook of environmental chemicals. Landsberg,
Germany, Ecomed Verlag.
Roberts PV, Simprini L, Hopkins GD, Grbic-Galic D, McCarty PL, &
Reinhard M (1989) In-situ aquifer restoration of the chlorinated
aliphatics by methanotrophic bacteria. Washington, DC, US
Environmental Protection Agency, pp 1-232 (EPA/600/2-89/033; prepared
by Stanford University, Department of Civil Engineering, Stanford,
California.).
Rodics K, Barcza G, & Ungváry G (1981) Mutagenicity of VC exposition:
Studies of the dose-response relationship in the micronucleus test.
Egeszsegtudomany, 25: 313-416.
Rohrschneider L, Jaeschke A, & Kubik W (1971) [Investigation into air
pollution at heights of up to 500 m above an industrial site.] Chem
Ing Tech, 43: 1010-1017 (in German).
Rosenman KD, Rizzo JE, Conomos MG, & Halpin GJ (1989) Central nervous
system malformations in relation to two polyvinyl chloride production
facilities. Arch Environ Health, 44: 279-282.
Rösli M, Zimmerli B, & Marek B (1975) Residues of vinyl chloride
monomer in edible oils. Mitt Geb Lebensm.unters Hyg, 66: 507-511.
Rossberg M, Lendle W, Tögel A, Dreher EL, Langer E, Rassaerts H,
Kleinschmidt P, Strack H, Beck U, Lipper KA, Torkelson TR, Löser E, &
Beutel KK (1986) Chlorinated hydrocarbons. In: Gerhartz W, Yamamoto
YS, Campbell FT, Pfefferkorn R, & Rounsaville JF ed. Ullmann's
encyclopedia of industrial chemistry, 5th completely revis ed.
-- Volume A6: Ceramics to chlorohydrins. Weinheim, Germany, VCH
Verlagsgesellschaft mbH, pp 283-297.
Rössner P, Srám RJ, Nováková J, & Lambl V (1980) Cytogenetic analysis
in workers occupationally exposed to vinyl chloride. Mutat Res, 73:
425-427.
Rowe VK (1975) Experience in industrial exposure control. Ann NY Acad
Sci, 246: 306-310.
Ryan JA, Bell RM, Davidson JM, & O'Connor GA (1988) Plant uptake of
non-ionic organic chemicals from soils. Chemosphere, 17: 2299-2323.
Sabadie N, Malaveille C, Camus AM, & Bartsch H (1980) Comparison of
the hydroxylation of benzo(a)pyrene with the metabolism of vinyl
chloride, N-nitrosomorpholine, and N-nitroso-N'-methylpiperazine to
mutagens by human and rat liver microsomal fractions. Cancer Res, 40:
119-126.
Sabel GV & Clark TP (1984) Volatile organic compounds as indicators of
municipal solid waste leachate contamination. Waste Manage Res, 32:
119-130.
Sabljic A (1984) Prediction of the nature and strength of soil
sorption of organic pollutants by molecular topology. J Agric Food
Chem, 32: 243-246.
Saito T, Barbin A, Omori Y, & Yamasaki H (1997) Connexion 37 mutation
in rat hepatic angiosarcomas induced by vinyl chloride. Cancer Res,
57: 375-377.
Salkinoja-Salonen M, Uotila J, Jokela J, Laine M, & Saski E (1995)
Organic halogens in the environment: Studies of environmental
biodegradability and human exposure. Environ Health Perspect, 103:
63-69.
Samoiloff MR, Schulz S, Jordan Y, Denich K, & Arnott E (1980) A rapid
simple long-term toxicity assay for aquatic contaminants using the
nematode Panagrellus redivivus. Can J Fish Aquat Sci, 37: 1167-1174
Sanchez JH & Recio L (1991) Mutagenicity of chloroacetaldehyde in
human TK6 lymphoblasts. Environ Mol Mutagen, 17(suppl 19): 64.
Sanhueza E & Heicklen J (1975) Oxidation of chloroethylene. J Phys
Chem, 79: 677-681.
Sanhueza E, Hisatsune IC, & Heicklen J (1976) Oxidation of
haloethylenes. Chem Rev, 76: 801-826.
Sanotsky IV, Davtian RM, & Glushchenko VJ (1980) [Study of the
reproductive function in men exposed to chemicals.] Gig Tr Prof Zabol,
6: 28-32 (in Russian)
Saparbaev M & Laval J (1998) 3,N4-ethenocytosine, a highly mutagenic
adduct, is a primary substrate for Escherichia coli double-stranded
uracil-DNA glycosylase and human mismatch-specific thymine-DNA
glycosylas. Proc Natl Acad Sci (USA), 95: 8508-8513.
Saparbaev M, Kleibl K, & Laval J (1995) Eschericia coli,
Saccharomyces cerevisiae, rat and human 3-methyladenine DNA
glycosylases repair 1,N6-ethenoademine when present in DNA. Nucleic
Acids Res, 23: 3750-3755.
Saurin JC, Taniere P, Mion F, Jacob P, Partensky C, Paliard P, &
Berger F (1997) Primary hepatocellular carcinoma in workers exposed to
vinyl chloride: a report of two cases. Cancer, 79: 1671-1677.
Sauvant MP, Pépin D, & Bohatier J, & Groliere CA (1995) Comparison of
six bioassays for assessing in vitro acute toxicity and
structure-activity relationships for vinyl chloride monomer, its main
metabolites and derivates. Sci Total Environ, 172: 79-92.
Schaffner F (1979) Effect of long-term vinyl chloride exposure in
mouse liver structure. In: Remmer H, Bolt HM, Bannasch P, & Popper H
ed. Proceedings of the Symposium on Primary Liver Tumors, Titisee,
Germany, 3-5 October 1977. New York, MTP Press Limited, International
Medical Publishers, pp 189-199.
Scherb K (1978) [Investigations into the evaporation of low molecular
chlorinated hydrocarbons from running water.] Münch Beitr
Abwasser- Fisch- Flussbiol, 30: 235-248 (in German).
Scherer E, Van der Laken CJ, Gwinner LM, Laib RJ, & Emmelot P (1981)
Modification of deoxyguanosine by chloroethylene oxide.
Carcinogenesis, 2: 671-677.
Schippert E (1987) [Purification of water contaminated with
chlorinated of hydrocarbons - comparison of direct absorption to
activated charcoal and stripping with air by purification of the air
by activated charcoal.] In: Thome-Kozmiensky KJ ed. [Existing
chemicals.] Berlin, Publishers for Energy and Environmental Technics,
pp 674-680 (in German).
Schlett C & Pfeifer B (1993) [Gas chromatographic determination of
vinyl chloride after purge-and trap concentration and mass
spectrometric detection.] Vom Wasser, 81: 1-6 (in German).
Schleyer R, Arneth J-D, Kerndorff H, & Milde G (1988) [Main priority
contaminants in waste dump sites: Selection criteria for evaluation
with respect to ground water path.] In: Wolf K, Van den Brink WJ, &
Colon FJ ed. [Cleaning of contaminated sites '88: Second TNO/BMFT
International Congress on Sanitation of Contaminated Sites, Hamburg,
11-15 April 1988.] Berlin, Federal Environmental Agency, vol 1, pp
249-253 (in German).
Schulz-Berendt V (1993) Soil bio remediation in practice: Problems and
results. In: Eijsackers HJP & Hamers T ed. Integrated soil and
sediment research: A basis for proper protection. Dordrecht, The
Netherlands, Kluwer Academic Publishers, pp 559-575.
Seiber JN (1996) Toxic air contaminants in urban atmospheres:
Experience in California. Atmos Environ, 30: 751-756.
Semenza JC & Weasel LH (1997) Molecular epidemiology in environmental
health: The potential of tumor suppressor gene p53 as a biomarker.
Environ Health Perspect, 105: 155-163.
Semprini L (1995) In situ bioremediation of chlorinated solvents.
Environ Health Perspect, 103: 101-105.
Semprini L, Roberts PV, Hopkins GD, & McCarty PL (1990) A field
evaluation of in-situ biodegradation of chlorinated ethenes -- Part 2:
Results of biostimulation and biotransformation experiments. Ground
Water, 28: 715-727.
Semprini L, Hopkins GD, Roberts PV, Grbic-Galic D, & McCarty PL (1991)
A field evaluation of in situ biodegradation of chlorinated ethenes
-- Part 3: Studies of competitive inhibition. Ground Water, 29:
239-250.
Semprini L, Kitanidis PK, Kampbell DH, & Wilson JT (1995) Anaerobic
transformation of chlorinated aliphatic hydrocarbons in a sand aquifer
based on spatial chemical distributions. Water Resour Res, 31:
1051-1062.
Shah HC (1998) Vinyl chloride combined inhalation two-generation
reproduction and developmental toxicity study in CD rats: Reproduction
study -- Final report (Study No. 96-4080). East Millstone, Huntingdon
Life Sciences Inc., vol I, pp 1-1726 (Unpublished report).
Shah JJ & Singh HB (1988) Distribution of volatile organic chemicals
in outdoor and indoor air. Environ Sci Technol, 22: 1381-1388.
Shahin MM (1976) The non-mutagenicity and -- recombinogenicity of
vinyl chloride in the absence of metabolic activation. Mutat Res, 40:
269-272.
Sharma RP & Gehring PJ (1979) Immunologic effects of vinyl chloride in
mice. Ann NY Acad Sci, 320: 551-563.
Sharma PK & McCarty PL (1996) Isolation and characterization of a
facultatively aerobic bacterium that reductively dehalogenates
tetrachloroethene to cis-1,2-dichloroethene. Appl Environ Microbiol,
62: 761-765.
Shechter M (1985) An anatomy of a groundwater contamination episode. J
Environ Econ Manage, 12: 72-88.
Sheldon LS & Hites RA (1978) Organic compounds in the Delaware river.
Environ Sci Technol, 12: 1188-1194.
Shimada T, Swanson AF, Leber P, & Williams GM (1985) Activities of
chlorinated ethane and ethylene compounds in the Salmonella/rat
microsome mutagenesis and rat hepatocyte/DNA repair assays under vapor
phase exposure conditions. Cell Biol Toxicol, 1: 159-179.
Shimp JF, Tracy JC, Davis LC, Lee E, Huang W, & Erickson LE (1993)
Beneficial effects of plants in the remediation of soil and
groundwater contaminated with organic materials. Crit Rev Environ Sci
Technol, 23: 41-77.
Shin MS, Carpenter JT, & Ho K-J (1991) Epithelioid
hemangioendothelioma: CT manifestations and possible linkage to vinyl
chloride exposure. J Comput Assist Tomogr, 15: 505-507.
Shirey RE (1995) Rapid analysis of environmental samples using
solid-phase microextraction (SPME) and narrow bore capillary columns.
J High Resol Chromatogr, 18: 495-499.
Short RD, Minor JL, Winston JM, & Lee C-C (1977) A dominant lethal
study in male rats after repeated exposures to vinyl chloride or
vinylidene chloride. J Toxicol Environ Health, 3: 965-968.
Simha D, Palejwala VA, & Humayun MZ (1991) Mechanisms of mutagenesis
by exocyclic DNA adducts: Construction and in vitro template
characteristics of an oligonucleotide bearing a single site-specific
ethenocytosine. Biochemistry, 30: 8727-8735.
Simonato L, L'Abbé KA, Andersen A, Belli S, Comba P, Engholm G, Ferro
G, Hagmar L, Langard S, Lundberg I, Pirastu R, Thomas P, Winkelmann R,
& Saracci R (1991) A collaborative study of cancer incidence and
mortality among vinyl chloride workers. Scand J Work Environ Health,
17: 159-169.
Simpson G, Klasmeier M, Hill H, Atkinson D, Radolovich G, Lopez-Avila
V, & Jones TL (1996) Evaluation of gas chromatography coupled with ion
mobility spectrometry for monitoring Vinyl Chloride and other
chlorinated and aromatic compounds in air samples. J High Resol
Chromatogr, 19: 301-312.
Singer B (1996) DNA damage: Chemistry, repair, and mutagenic
potential. Regul Toxicol Pharmacol, 23: 2-13.
Singer B, Kusmierek JT, Folkman W, Chavez F, & Dosanjh MK (1991)
Evidence for the mutagenic potential of the vinyl chloride induced
adduct, N2,3-etheno-deoxyguanosine, using a site-directed kinetic
assay. Carcinogenesis, 12: 745-747.
Singh HB, Ludwig FL, & Johnson WB (1978) Tropospheric ozone:
Concentrations and variabilities in clean remote atmospheres. Atmos
Environ, 12: 2185-2196.
Singh HB, Jaber HM, & Davenport JE (1984) Reactivity/volatility
classification of selected organic chemicals: existing data. Research
Triangle Park, North Carolina, US Environmental Protection Agency, pp
1-190 (EPA-600/3-84-082; prepared by SRI International, Menlo Park,
California).
Sinués B, Sanz A, Bernal ML, Tres A, Alcala A, Lanuza J, Ceballos C, &
Sáenz MA (1991) Sister chromatid exchanges, proliferating rate index,
and micronuclei in biomonitoring of internal exposure to vinyl
chloride monomer in plastic industry workers. Toxicol Appl Pharmacol,
108: 37-45.
Sittig M (1985) Vinylchloride. In: Sittig M ed. Handbook of toxic and
hazardous chemicals and carcinogens, 2nd ed. Park Ridge, New Jersey,
Noyes Publications, pp 918-921.
Skeen RS, Gao J, & Hooker BS (1995) Kinetics of chlorinated ethylene
dehalogenation under methanogenic conditions. Biotechnol Bioeng, 48:
659-666.
Smerasta P, Srivburuang P, Tongtan N, Ishiwata H, & Yoshihira K (1991)
Migration and material tests of some food-contact plastic wares made
in Thailand. Bull Natl Inst Hyg Sci, 109: 105-106.
Smith LR & Dragun J (1984) Degradation of volatile chlorinated
aliphatic priority pollutants in groundwater. Environ Int, 10:
291-298.
Smith SJ, Li Y, Whitley R, Marion MJ, Partilo S, Carney WP, &
Brandt-Rauf PW (1998) Molecular epidemiology of p53 protein mutations
in workers exposed to vinyl chloride. Am J Epidemiol, 147: 302-308.
Smulevich VB, Fedotova IV, & Filatova VS (1988) Increasing evidence of
the risk of cancer in workers exposed to vinyl chloride. Br J Ind Med,
45: 93-97.
Soini Y, Welsh JA, Ishak KG, & Bennett WP (1995) p53 mutations in
primary hepatic angiosarcomas not associated with vinyl chloride
exposure. Carcinogenesis, 16: 2879-2881.
Sokal JA, Baranski B, Majka J, Rolecki R, Stetkiewicz J,
Ivanova-Chemishanska L, Vergieva T, Antonov G, Mirkova E, Kolakowski
A, Szendzikowski S, & Wróblewska K (1980) Experimental studies on the
chronic toxic effects of vinyl chloride in rats. J Hyg Epidemiol
Microbiol Immunol, 24: 285-294.
Solionova LG, Smulevich VB, Turbin EV, Krivosheyeva LV, & Plotnikov JV
(1992) Carcinogens in rubber production in the Soviet Union. Scand J
Work Environ Health, 18: 120-123.
Sood C & O'Brien PJ (1993) Molecular mechanisms of
chloroacetaldehyde-induced cytotoxicity in isolated rat hepatocytes.
Biochem Pharmacol, 46: 1621-1626.
Soule R, Symonik D, Jones D, Turgeon D, & Gerbec B (1996) Vinyl
Chloride loss during laboratory holding time. Regul Toxicol Pharmacol,
23: 209-212.
Spit BJ, Feron VJ, & Hendriksen CFM (1981) Ultrastructure of hepatic
angiosarcoma in rats induced by vinyl chloride. Exp Mol Pathol, 35:
277-284.
Staples CA, Werner AF, & Hoogheem TJ (1985) Assessment of priority
pollutant concentrations in the United States using STORET database.
Environ Toxicol Chem, 4: 131-142.
Stareczek T (1988) [Differences in content of free monomer during
storage and processing of mono- and copolymer vinyl chloride.] Polim
Tworz Wielk 33: 145-147 (in Polish).
Stephens RD, Ball NB, & Mar DM (1986) A multimedia study of hazardous
waste landfill gas migration. In: Cohen Y ed. Pollutants in a
multimedia environment. New York, London, Plenum Press, pp 265-287.
Storm JE & Rozman KK (1997) Evaluation of alternative methods for
establishing safe levels of occupational exposure to vinyl halides.
Regul Toxicol Pharmacol, 25: 240-255.
Strandberg GW, Donaldson TL, & Farr LL (1989) Degradation of
trichloroethylene and trans-1,2-dichloroethylene by a methanotrophic
consortium in a fixed-film, packed-bed bioreactor. Environ Sci
Technol, 23: 1422-1425.
Stringer RL, Costner P, & Johnston PA (1995) PVC manufacture as a
source of PCDD/Fs. Organohalog Compd, 24: 119-123.
Stuart JD (1983) Organics transported thru selected geologic media:
Assessment of organics transported away from industrial waste disposal
sites. Springfield, Virginia, US Department of the Interior, pp 1-10
(Document No. A-089 CONN, prepared by the University's Institute of
Water Resources, Storrs, Connecticut, USA).
Stuckey DC, Owen WF, & McCarty PL (1980) Anaerobic toxicity evaluation
by batch and semi-continuous assays. J Water Pollut Control Fed, 52:
720-729.
Studniarek M, Durski K, Liniecki J, Brykalski D, Poznanska A, &
Gluszcz M (1989) Effects of vinyl chloride on liver function of
exposed workers, evaluated by measurements of plasma clearance of the
99mTc-N-2,4-dimethylacetanilido-iminoiacetate complex. J Appl Toxicol,
9: 213-218.
Stblová V, Lambl V, Chumcal O, Kellerová V, Pasková V, Vítovcocá J, &
Zlab L (1981) Neurological changes in vinyl chloride-exposed workers.
J Hyg Epidemiol Microbiol Immunol, 25: 233-243.
Styles JA (1980) Studies on the detection of carcinogens using a
mammalian cell transformation assay with liver homogenate activation.
In: Norpoth KH & Garner RC ed. Short-term test systems for detecting
carcinogens. Berlin, Heidelberg, New York, Springer Verlag, pp
226-238.
Suciu I, Prodan L, Ilea E, Paduraru A, & Pascu L (1975) Clinical
manifestations in vinyl chloride poisoning. Ann NY Acad Sci, 246:
53-69.
Sugita M, Masuda Y, & Tschuiya K (1986) Early detection and signs of
hepatoangiosarcoma among vinyl chloride workers. Am J Ind Med, 10:
411-417.
Suter GW (1996) Toxicological benchmarks for screening contaminants of
potential concern for effects on freshwater biota. Environ Toxicol
Chem, 15: 1232-1241.
Suzuki Y (1978) Pulmonary tumors induced in mice by vinyl chloride
monomer. Environ Res, 16: 285-301.
Suzuki Y (1980) Nonneoplastic effects of vinyl chloride in mouse lung.
Environ Res, 21: 235-253.
Suzuki Y (1981) Neoplastic and nonneoplastic effects of vinylchloride
in mouse lung. Environ Health Perspect, 41:31-52.
Suzuki Y (1983) Neoplastic effect of vinyl chloride in mouse lung --
lower doses and short-term exposure. Environ Res, 32: 91-103.
Svensson K (1994) Legislation, control and research in the Nordic
countries on plastics for packaging food. Food Addit Contam, 11:
241-248.
Swenberg JA, Fedtke N, Ciroussel F, Barbin A, & Bartsch H (1992)
Etheno adducts formed in DNA of vinyl chloride-exposed rats are highly
persistent in liver. Carcinogenesis, 13: 727-729.
Swenberg JA, La KD, Scheller NA, & Kuen-Yuh W (1995) Dose-response
relationships for carcinogens. Toxicol Lett, 82/83: 751-756.
Szadkowski D & Lehnert G (1982) [Vinyl chloride as a cause of disease
- a bibliography.] In: Szadkowski D & Lehnert G ed. [On the subject of
VC/PVC.] Frankfurt am Main, Germany, Henssler KG, pp 1-20 (in German).
Szentesi I, Hornyák É, Ungváry G, Czeizel A, Bognár Z, & Timar M
(1976) High rate of chromosomal aberration in PVC workers. Mutat Res,
37: 313-316.
Tabershaw IR & Gaffey WR (1974) Mortality study of workers in the
manufacture of vinyl chloride and its polymers. J Occup Med, 16:
509-518.
Tamburro CH, Makk L, & Popper H (1984) Early hepatic histologic
alterations among chemical(vinyl monomer) workers. Hepatology, 4:
413-418.
Tandoi V, DiStefano TD, Bowser PA, Gossett JM, & Zinder SH (1994)
Reductive dehalogenation of chlorinated ethenes and halogenated
ethanes by a high-rate anaerobic enrichment culture. Environ Sci
Technol, 28: 973-979.
Tátrai E & Ungváry G (1981) On the acute hepatotoxicity of inhaled
vinyl chloride. Acta Morphol Acad Sci Hung, 29: 221-226.
Terwiesch B (1982) Vinyl chloride peroxide explosion in a vinyl
chloride recovery plant. J Macromol Sci Chem, A17: 1081-1092.
Tester DA (1976) The extraction of vinyl chloride from PVC containers.
J Soc Cosmet Chem, 49: 459-466.
Theisen J, Funcke W, Balfanz E, & König J (1989) Determination of
PCDFs and PCDDs in fire accidents and laboratory combustion tests
involving PVC-containing materials. Chemosphere, 19: 423-428.
Theisen J, Funcke W, & Hamm S (1991) [Investigation of possible risks
to the environment from burning plastics.] In: [Investigation of
possible risks to the environment from burning plastics.] Münster,
Germany, Society for Occupational and Environmental Analytic (GFA), pp
91-131 (in German).
Thériault G & Allard P (1981) Cancer mortality of a group of Canadian
workers exposed to vinyl chloride monomer. J Occup Med, 23: 671-676.
Thériault G, Iturra H, & Gingras S (1983) Evaluation of the
association between birth defects and exposures to ambient vinyl
chloride. Teratology, 27: 359-370.
Thiess AM & Versen P (1974) [Occupational medical thoughts on the
so-called 'vinyl chloride disease'.] Arbeitsmed Sozialmed
Präventivmed, 7: 146-148 (in German).
Thomas VN & Ramstad T (1992) A dynamic headspace GC method for the
determination of vinyl chloride monomer in stored solutions of
cefmetazole sodium in PVC bags. Acta Pharm Nord, 4: 97-104.
Tie X, Kao C-Y, Mroz EJ, Cicerone RJ, Alyea FN, & Cunnold DM (1992)
Three-dimensional simulations of atmospheric methyl chloroform: Effect
of an ocean sink. J Geophys Res Atmos, D97: 20.751-20.769.
Til HP, Feron VJ, & Immel HR (1991) Lifetime (149-week) oral
carcinogenicity study of vinyl chloride in rats. Food Chem Toxicol,
29: 713-718.
Til HP, Immel HP, & Feron FJ (1983) Lifespan oral carcinogenicity
study of vinyl chloride in rats. Zeist, The Netherlands, Organization
for Applied Scientific Research (TNO), pp 1-29 (TNO Report No.
V-83.285/291099).
Topudurti K (1992) A UV/oxidation technology demonstration to treat
groundwater contaminated with VOCs. Water Sci Technol, 25: 347-354.
Torkelson TR (1994) Halogenated aliphatic hydrocarbons containing
chlorine, bromine, and iodine.. In: Clayton GD & Clayton FE ed.
Patty's industrial hygiene and toxicology, 4th ed. New York, John
Wiley & Sons, Inc., vol 2, part E, pp 4007-4251.
Torkelson TR, Oyen F, & Rowe VK (1961) The toxicity of vinyl chloride
as determined by repeated exposure of laboratory animals. Am Ind Hyg
Assoc J, 22: 354-361.
Tribukh SL, Tikhomirova NP, Levin SV, & Kozlov LA (1949) Working
conditions and measures for their sanitation in the production and
utilization of vinyl chloride plastics. Gig i Sanit, 10: 38-45.
Trivers GE, Cawley HL, DeBenedetti VMG, Hollstein M, Marion MJ, Benett
WP, Hoover ML, Prives CC, Tamburro CC, & Harris CC (1995) Anti-p53
antibodies in sera of workers occupationally exposed to vinyl
chloride. J Natl Cancer Inst, 87: 1400-1407.
Tsien H-C, Brusseau GA, Hanson RS, & Wackett LP (1989) Biodegradation
of trichloroethylene by Methylosinus trichosporium OB3b. Appl
Environ Microbiol, 55: 3155-3161.
Tsivoglou EC (1967) Tracer measurement of stream reaeration.
Washington, DC, Federal Water Pollution Control Administration (PB-229
923).
Tu AS, Murray TA, Hatch KM, Sivak A, & Milman HA (1985) In vitro
transformation of BALB/c-3T3 cells by chlorinated ethanes and
ethylenes. Cancer Lett, 28: 85-92.
Tuazon EC, Atkinson R, Aschmann SM, Goodman MA, & Winer AM (1988)
Atmospheric reactions of chloroethenes with the OH radical. Int J Chem
Kinet, 20: 241-265.
Turner AC, Payne AP, & Bushby BR (1984) Determination of ambient
levels of vinyl chloride monomer (VCM) around manufacturers in the UK,
Part 7. Warren Spring Laboratory, pp 1-26 (Report No. LR 514[AP]).
Uchiyama H, Nakajima T, Yagi O, & Tabuchi T (1989) Aerobic degradation
of trichloroethylene at high concentration by a methane-utilizing
mixed culture. Agric Biol Chem, 53: 1019-1024.
UK MAFF (Ministry of Agriculture Fisheries and Food) (1978) Working
Party on Vinyl Chloride -- Survey of vinyl chloride content of
polyvinyl chloride for food contact and of foods: Second report of the
Steering Group on Food Surveillance. London, Her Majesty's Stationary
Office, pp 1-16 (Food Surveillance Paper No. 2).
UK MAFF (Ministry of Agriculture Fisheries and Food) (1984) Steering
Group on Food Surveillance -- Progress report 1984: Fourteenth report
of the Steering Group on Food Surveillance. London, Her Majesty's
Stationery Office, pp 11-15, 46 (Food Surveillance Paper No. 14).
Ungváry G, Hudák A, Tátrai E, Lörincz M, & Folly G (1978) Effects of
vinyl chloride exposure alone and in combination with trypan blue --
applied systematically during all thirds of pregnancy on the fetuses
of CFY rats. Toxicology, 11: 45-54.
US EPA (1975) Scientific and technical assessment report on vinyl
chloride and polyvinyl chloride. Research Triangle Park, North
Carolina, US Environmental Protection Agency, Office of Research and
Development, Environmental Research Center (EPA 600/6-75-004).
US EPA (1982) Vinyl chloride -- a review of national emission
standards. Research Triangle Park, North Carolina, US Environmental
Protection Agency, pp 1-37 (EPA 450/3-82-003).
US EPA (1985a) Health and environmental effects profile for
chloroethene. Cincinnati, Ohio, US Environmental Protection Agency, pp
1-176 (EPA 600/x-85/374; PB88-174529).
US EPA (1985b) Drinking water criteria document for vinyl chloride.
Washington, DC, US Environmental Protection Agency, pp 1-18 (Report
TR-540-162; prepared by ICAIR Life Systems, Inc., Cleveland, Ohio).
US EPA (1987) Incorporation of biological information in cancer risk
assessment: Example -- vinyl chloride. Washington, DC, US
Environmental Protection Agency, pp 1-51 (EPA 600/21).
US EPA (1992) Superfund record of decision, Hagen farm, WI: Second
remedial action- final. Washington, DC, US Environmental Protection
Agency, pp 1-40 (EPA/ROD/RO5-92/216; prepared by the Hagen Farm Site,
Groundwater Control Operable Unit, Dane Country, Wisconsin).
US EPA (1995a) Health effects assessment summary tables (HEAST).
Washington, DC, US Environmental Protection Agency, Office of Solid
Waste and Emergency Response (EPA 540 R-95/036).
US EPA (1995b) Drinking water standards and health advisories table.
San Francisco, California, US Environmental Protection Agency, Region
IX, 26 pp.
US EPA (1996) Proposed guidelines cancer risk assessment. Fed Reg,
61(79): 177960-180011.
US FDA (Food and Drug Administration) (1973) Prior-sanctioned
polyvinyl chloride resin. Fed Reg, 38: 12931.
US FDA (Food and Drug Administration) (1986) Vinyl chloride polymers:
Withdrawal of proposal. Fed Reg, 51: 4173-4188.
Uzych L (1988) Human male exposure to vinyl chloride and possible
teratogenic and mutagenic risks: A review. Hum Toxicol, 7: 517-527.
Van der Meer JR, Bosma TNP, De Bruin WP, Harms H, Holliger C,
Rijnaarts HHM, Tros ME, Schraa G, & Zehnder AJB (1992) Versatility of
soil column experiments to study biodegradation of halogenated
compounds under environmental conditions. Biodegradation, 3: 265-284.
Van Duuren BL (1975) On the possible mechanism of carcinogenic action
of vinyl chloride. Ann NY Acad Sci, 246: 258-267.
Van Lierop JBH (1979) Determination of potentially carcinogenic
compounds in food. In: Van Lierop JBH ed. Determination of potentially
carcinogenic compounds in food:Trace analysis of vinylchloride,
vinylidenechloride, acrylonitrile, epichlorohydrin and
diethylpyrocarbonate. Utrecht, The Netherlands, University of Utrecht,
pp 1-187.
van Sittert NJ & De Jong G (1985) Biomonitoring of exposure to
potential mutagens and carcinogens in industrial populations. Food
Chem Toxicol, 23: 23-31.
Van't Hof J & Schairer LA (1982) Tradescantia assay system for gaseous
mutagens. A report of the US Environmental Protection Agency Gene-Tox
Program. Mutat Res, 99: 303-315.
Vannelli T, Logan M, Arciero DM, & Hooper AB (1990) Degradation of
halogenated aliphatic compounds by the ammonia-oxidizing bacterium
Nitrosomonas europaea. Appl Environ Microbiol, 56: 1169-1171.
Veltman G, Lange CE, Jühe S, Stein G, & Bachner U (1975) Clinical
manifestations and course of vinyl chloride disease. Ann NY Acad Sci,
246: 6-17.
Verburgt FG & Vogel E (1977) Vinyl chloride mutagenesis in Drosophila
melanogaster. Mutat Res, 48: 327-337.
Victorin K & Ståhlberg M (1988) A method for studying the mutagenicity
of some gaseous compounds in Salmonella typhimurium. Environ Mol
Mutagen, 11: 65-77.
Victorin K & Ståhlberg M (1991) Mutagenic activity of photoreaction
products formed from chlorinated ethenes and nitrogen dioxide. Environ
Res, 55: 178-187.
Viinanen R (1993) Monitoring results of VC in a PVC plant in
1981-1993. Porvoo, Finland, Neste Environmental and Industrial Hygiene
Institute, pp 1-13 (Research Report No. 93025T).
Viola PL (1970) Pathology of vinyl chloride. Med Lav, 61: 174-180.
Viola PL, Bigotti A, & Caputo A (1971) Oncogenic response of rat skin,
lungs and bones to vinyl chloride. Cancer Res, 31: 516-522.
Vogel TM & McCarty PL (1985) Biotransformation of tetrachloroethylene
to trichloroethylene, dichloroethylene, vinyl chloride, and carbon
dioxide under methanogenic conditions. Appl Environ Microbiol, 49:
1080-1083.
Vogel EW & Nivard MJM (1993) Performance of 181 chemicals in a
Drosophila assay predominantly monitoring interchromosomal mitotic
recombination. Mutagenesis, 8: 57-81.
Vogel TM, Criddle CS, & McCarty PL (1987) Transformations of
halogenated aliphatic compounds. Environ Sci Technol, 21: 722-736.
Vom Bruck CG, Rudolph FB, Figge K, & Eckert WR (1979) Application of
the diffusion theory to migration of plastics components into packed
goods: Survey of recent migration studies. Food Cosmet Toxicol, 17:
153-157.
Wackett LP, Brusseau GA, Householder SR, & Hanson RS (1989) Survey of
microbial oxygenases: Trichloroethylene degradation by
propane-oxidizing bacteria. Appl Environ Microbiol, 55: 2960-2964.
Wagenaar H, Langeland K, Hardman R, Sergeant Y, Brenner K, Sandra P,
Rappe C, Fernandes A, & Tiernan T (1998) Analysis of PCDDs and PCDFs
in virgin suspension PVC resin. Chemosphere, 36: 1-12.
Wakeman IB & Johnson HR (1978) Vinyl chloride formation from the
thermal degradation of poly(vinyl chloride). Polymer, 18: 404-407.
Walker AE (1976) Clinical aspects of vinyl chloride disease: Skin.
Proc R Soc Med, 69: 286-289.
Walles SA & Holmberg B (1984) Induction of single-strand breaks in DNA
of mice after inhalation of vinyl chloride. Cancer Lett, 25: 13-18.
Walles SA, Holmberg B, Svensson K, Osterman-Golkar S, & Sigvardsson K
(1988) Induction of single-strand breaks in liver DNA of mice after
inhalation of vinylchloride. In: Bartsch H, Hemminki K, & O-Neill IK
ed. Methods for detecting DNA damaging agents in humans: Applications
in cancer epidemiology and prevention. Lyon, International Agency for
Research on Cancer, pp 227-231 (IARC Scientific Publications No. 89).
Wang T, Lenahan R, & Kanik M (1985) Impact of trichlorethylene
contaminated groundwater discharged to the main canal and Indian River
Lagoon, Vero Beach, Florida. Bull Environ Contam Toxicol, 34: 578-586.
Wang YL & Zhao XH (1987) Occupational health of working women in
China. Asia-Pac J Public Health, 1(4): 66-71.
Ward AM, Udnoon S, Watkins J, Walker AE, & Darke CS (1976)
Immunological mechanisms in the pathogenesis of vinyl chloride
disease. Br Med J, 1: 936-938.
Ward RS, Williams GM, & Hills CC (1996) Changes in major and trace
components of landfill gas during subsurface migration. Waste Manage
Res, 14: 243-261.
Watanabe PG & Gehring PJ (1976) Dose-dependent fate of vinyl chloride
and its possible relationship to oncogenicity in rats. Environ Health
Perspect, 17: 145-152.
Watanabe PG, McGowan GR, & Gehring PJ (1976a) Fate of [14C]vinyl
chloride after single oral administration in rats. Toxicol Appl
Pharmacol, 36: 339-352.
Watanabe PG, McGowan RG, Madrid EO, & Gehring PJ (1976b) Fate of
[14C]vinyl chloride following inhalation exposure in rats. Toxicol
Appl Pharmacol, 37: 49-59.
Watanabe PG, Hefner RE, & Gehring PJ (1976c) Vinyl chloride-induced
depression of hepatic non-protein sulfhydryl content and effects on
bromosulphalein (BSP) clearance in rats. Toxicology, 6: 1-8.
Watson JR, Lawrence RC, & Lovering EG (1977) A GLC method for
monitoring vinyl chloride in drugs and in polyvinyl chloride drug
containers. Can J Pharmacol Sci, 12: 94-97.
Watanabe PG, Zempel JA, Pegg DG, & Gehring PJ (1978a) Hepatic
macromolecular binding following exposure to vinyl chloride. Toxicol
Appl Pharmacol, 44: 571-579.
Watanabe PG, Zempel JA, & Gehring PJ (1978b) Comparison of the fate of
vinyl chloride following single and repeated exposure in rats. Toxicol
Appl Pharmacol, 44: 391-399.
Watson JR, Lawrence RC, & Lovering EG (1979) A GC headspace method for
monitoring vinyl chloride in liquid drug and cosmetic products. Can J
Pharmacol Sci, 14: 57-60.
Waxweiler RJ, Stringer W, Wagoner JK, Jones J, Falk H, & Carter C
(1976) Neoplastic risk among workers exposed to vinyl chloride. Ann NY
Acad Sci 271: 40-48.
Waxweiler J, Falk H, McMichael A, Mallov JS, Grivas AS, & Stringer WT
(1977) A cross-sectional epidemiologic survey of vinyl chloride
workers. Cincinnati, Ohio, National Institute of Occupational Safety
and Health, pp 1-43 (DHEW (NIOSH) Publication No.77-177).
Weaver JW, Wilson JT, & Kampbell DH (1996) Extraction of degradation
rate constants from the St Joseph, Michigan, trichloroethene site. In:
Proceedings of the Symposium on Natural Attenuation of Chlorinated
Organics in Ground Water, 11-13 September 1996. Washington, DC, US
Environmental Protection Agency, pp 69-73 (EPA 540/R-96/509).
Weber H, Reinl W, & Greiser E (1981) German investigations on
morbidity and mortality of workers exposed to vinyl chloride. Environ
Health Perspect, 41: 95-99.
Weisman WH (1992) Toxicity, mutagenicity, and mutational spectra of
vinyl chloride, 2-chloroethylene oxide, and chloroacetaldehyde in a
human lymphoblastoid line expressing cytochrome P 450IIE1. Chapel
Hill, North Carolina, University of North Carolina, pp 1-85
(AFIT/CI/CIA-92-030).
Westrick JJ, Mello JW, & Thomas RF (1984) The ground water supply
survey. J Am Water Works Assoc, 76: 52-59.
Wheeler RN (1981) Poly(vinyl Chloride) processes and products. Environ
Health Perspect, 41: 123-128.
WHO (1987) Vinyl chloride. In: Air quality guidelines for Europe.
Copenhagen, World Health Organization, Regional Office for Europe, pp
158-169 (WHO Regional Publications, European Series No. 23).
WHO (1996) Vinyl chloride. In: Guidelines for drinking-water quality
-- Volume 2: Health criteria and other supporting information. Geneva,
World Health Organization, pp 424-431.
Wild SR & Jones KC (1992) Organic chemicals entering agricultural
soils in sewage sludges: screening for their potential to transfer to
crop plants and livestock. Sci Total Environ, 119: 85-119.
Williams DT (1976a) Gas-liquid chromatographic headspace method for
vinyl chloride in vinegars and alcoholic beverages. J Assoc Off Anal
Chem, 59: 30-31.
Williams DT (1976b) Confirmation of vinyl chloride in food by
conversion to 1-chloro-1,2-dibromoethane. J Assoc Off Anal Chem, 59:
32-34.
Williams DT & Miles WF (1975) Gas-liquid chromatographic determination
of vinyl chloride in alcoholic beverages, vegetable oils, and
vinegars. J Am Optom Assoc, 58: 272-275.
Williamson J & Kavanagh B (1987) Vinyl chloride monomer and other
contaminants in PVC welding fumes. Am Ind Hyg Assoc J, 48: 432-436.
Wilson RH, McCormick WE, Tatum CF, & Creech JL (1967) Occupational
acroosteolysis: Report of 31 cases. J Am Med Assoc, 201: 577-580.
Wisniewska-Knypl JM, Klimczak J, & Kolakowski J (1980) Monooxygenase
activity and ultrastructural changes of liver in the course of chronic
exposure of rats to vinyl chloride. Int Arch Occup Environ Health, 46:
241-249.
Withey JR (1976) Pharmacodynamics and uptake of vinyl chloride monomer
administered by various routes to rats. J Toxicol Environ Health, 1:
381-394.
Wittsiepe J (1990) Occurrence of vinyl chloride and other halogenated
C1- and C2- hydrocarbons in German surface water. Organohalog Compd,
4: 425-434.
Wittsiepe J, Selenka F, & Jackwerth E (1990) Gas-chromatographic
determination of trace amounts of vinyl chloride in water and air
after derivatization to 1,2-dibromochloroethane. Fresenius J Anal
Chem, 336: 322-327.
Wittsiepe J, Wallschläger D, Selenka F, & Jackwerth E (1993)
Gas-chromatographic determination of trace amounts of vinyl chloride
and dichloroethenes in water after purge-and-trap preconcentration and
conversion into their dibrominated derivatives. Fresenius J Anal Chem,
346: 1028-1034.
Wittsiepe J, Selenka F, & Jackwerth E (1996) Gas chromatographic
determination of trace amounts of vinyl chloride and dichloroethenes
in landfill-gas. Fresenius J Anal Chem, 354: 910-914.
Woldbaek T & Klaboe P (1978) The photochemical reaction of
vinylchloride with oxygen studied by ir spectroscopy. Spectrochim
Acta, A34: 481-487.
Wolf K, Holland R, & Rjaratnam A (1987) Vinyl chloride contamination:
The hidden threat. J Hazard Mater, 15: 163-184.
Wong O, Whorton MD, Foliart DE, & Ragland D (1991) An industry-wide
epidemiologic study of vinyl chloride workers, 1942-1982. Am J Ind
Med, 20: 317-334.
Wood JA & Porter ML (1987) Hazardous pollutants in class II landfills.
Int J Air Pollut Control Hazard Waste Manage, 37: 609-615.
Wrede F (1995) [Polyvinyl chloride (PVC).] Kunststoffe, 85: 1515-1521
(in German).
Wright DK, Mahler KO, & Davis J (1992) The application of
multidimensional GC techniques to the analysis of sub-PPB levels of
vinyl chloride monomer in polyvinylchloride. J Chromatogr Sci, 30:
291-295.
Wu W, Steenland K, Brown D, Wells V, Jones J, Schulte P, & Halperin W
(1989) Cohort and case-control analyses of workers exposed to vinyl
chloride: An update. J Occup Med, 31: 518-523.
Yamamoto K, Fukushima M, Kakutani N, & Kuroda K (1997) Volatile
organic compounds in urban rivers and their estuaries in Osaka, Japan.
Environ Pollut, 95: 135-143.
Zajdela F, Croisy A, Barbin A, Malaveille C, Tomatis L, & Bartsch H
(1980) Carcinogenicity of chloroethylene oxide, an ultimate reactive
metabolite of vinyl chloride, and bis(chloromethyl)ether after
subcutaneous administration and in initiation-promotion experiments in
mice. Cancer Res, 40: 352-356.
Zeff JD & Barich JT (1992) UV/Oxidation of organic contaminants in
ground, waste and leachate waters. Water Pollut Res J Can, 27:139-150.
Zhang J, Hatakeyama S, & Akimoto H (1983) Rate constants of the
reaction of ozone with trans-1,2-dichloroethene and vinyl chloride
in air. Int J Chem Kinet, 15: 655-668.
Zhang W, Johnson F, Grollman AP, & Shibutani S (1995) Miscoding by the
exocyclic and related DNA adducts 3,N4-etheno-2'deoxycytidine, 3,
N4-ethano-2'deoxycytidine, and 3-(2-hydroxyethyl)-2'deoxyuridine.
Chem Res Toxicol, 8: 157-163.
Zhao M-Y, Ying C-J, Shao N, Yang Y, Yang C-F, Shi L, & Liu W-Q (1994)
The study of health effects of vinyl chloride air pollution on
population. Biomed Environ Sci, 7: 136-143.
Zuccato E, Marcucci F, Fanelli R ,& Mussini E (1979) Head-space
gas-chromatographic analysis of vinyl chloride monomer in rat blood
and tissues. Xenobiotica, 9: 27-31.
ANNEX 1. REGULATIONS CONCERNING VINYL CHLORIDE
1. Regulations regarding VC levels in PVC materials and
food and drink
According to EC regulations PVC materials intended to come in
contact with foodstuffs must not contain VC at levels > 1 mg/kg; the
limit in the packed food is 10 µg/kg (CEC, 1978). The corresponding
German regulation additionally includes commodities, toys and tricks,
and articles which are for toiletry use (German Federal Health Office,
1998). The US FDA proposed to restrict the VC content of polymers used
for food contact to 5-50 µg/kg (US FDA, 1986).
A limit value of 2 µg/litre was issued by the US EPA for
drinking-water (US EPA, 1995b) and by the US FDA for bottled water
(Anon, 1993). The Maximum Contaminant Level of the Californian
Department of Health Services (DOHS) is 0.5 µg/litre (US EPA, 1995b).
In Germany, there are recommendations to tolerate 2 µg/litre in
drinking-water (German Federal Health Office, 1992).
2. Occupational exposure limit values for vinyl
chloride
Some examples of present exposure limit values are given in
Table 55.
Table 55. Occupational exposure limit values for vinyl chloride
Country/organization Exposure limit Value Value Reference
description (ppm) (mg/m3)
Europe; United Kingdom; personal 8-h TWA 7 18.2 CEC (1978); HSC (1995);
Germany; Finland BIA (1997)
5 12.8 Finnish Government
(1992)
Europe; United Kingdom; working area 3 8 CEC (1978); Finnish
Germany; Finland (annual) Government (1992);
HSC (1995); BIA (1997)
Czech Republic, Slovakia, MACKa 10 Bencko & Ungváry
Hungary and Poland (1994)
USA 15 min 5 12.8 OSHA (1998)
8 h 1 2.6 OSHA (1998)
TLV 8-h TWA 1 2.6 ACGIH (1999)
0b NIOSH (1997)
a Maximal allowable concentration (K indicates the carcinogenic properties of VC)
b No detectable exposure
TLV = threshold limit value; TWA = time weighted average;
OSHA = US Occupational Safety and Health Administration;
ACGIH = US American Conference of Governmental and Industrial
Hygienists Inc.; HSC = UK Health and Safety Commission;
BIA = German Professional Associations
Institute for Occupational Safety; Finnish Government = Government
Resolution 919/92, Finland
ANNEX 2. PHYSIOLOGICAL MODELLING AND RECENT RISK ASSESSMENTS
1. Physiological modelling of toxicokinetic data for vinyl chloride
Physiologically based toxicokinetic (PBTK) models intend to
predict the dose of active metabolites reaching target tissues in
different species, including humans, and thereby to improve the
toxicokinetic extrapolation in cancer risk assessments. PBTK models
have also been used as a tool to examine the behaviour of VC in
mammalian systems.
The model of Chen & Blancato (1989), also cited as a US EPA
(1987) model, proposed a PBTK model based on the styrene model of
Ramsey & Andersen (1984) with four compartments, representing the
liver, a fat compartment that includes all of the fatty tissues, a
richly-perfused tissue compartment that includes all organs except the
liver, and a slowly perfused compartment that includes muscular and
skin tissues. Liver was assumed to be the sole metabolizing organ, and
the metabolism of VC was assumed to occur via one pathway following
Michaelis-Menten-kinetics and saturable at high concentration levels.
Physiological parameters (e.g., body weight, lung weight, ventilation
rate, tissue volume and blood flows to various tissue groups) were
reference values from US EPA. This model was validated by Clement
International Corporation (1990) with gas uptake experiments in rats
and with data sets available in the literature (Gehring et al., 1978).
At concentrations < 65 mg/m3 the model fitted observed data, but
at higher concentrations the model predicted a greater amount of VC
metabolism than observed.
A second model (Gargas et al., 1990) was a generic model of
volatile chemical kinetics in a recirculated closed chamber, which was
used to identify global metabolic parameters in the rat for a number
of chemicals including VC. It incorporated a second, linear metabolic
pathway (presumed to be glutathione conjugation) in parallel with the
saturable (oxidative) pathway.
Continuing on from these above models, Clement International
Corporation (1990) re-fitted the one- and two-pathway descriptions to
gas uptake data and then compared their predictions to measurements of
total metabolism by Gehring et al. (1978) and Watanabe et al. (1976b).
Two refinements to the model were investigated: 1) reaction of VC with
glutathione (GSH), or 2) the products of both the saturable and the
linear pathways were assumed to react with GSH. Neither description
fitted both total metabolism and GSH depletion data and the authors
suggested a new PBTK model featuring two saturable oxidative pathways,
both producing reactive metabolites. Clewell et al. (1995a) developed
a more elaborate model of VC based on the suggestions from Clement
International Corporation (1990). The initial metabolism of VC was
hypothesized to occur via two saturable pathways (one representing
high affinity, low capacity oxidation by CYP2E1 and one representing
low affinity, high capacity oxidation by other P-450 isozymes),
producing in both cases chloroethylene oxide (CEO) as an intermediate
product. CAA (from CEO) was modelled as the major substrate in GSH
conjugation, with a lesser amount of CEO as the glutathione
transferase substrate. The model was similar to that proposed by Chen
& Blancato (1989) with regard to number and type of compartments,
physiological parameters and the assumption that metabolism of VC
takes place solely in the liver. Partition coefficients taken from
literature data (Gargas et al., 1989) were used. The metabolic
parameters for the two oxidative pathways were estimated from gas
uptake experiments conducted by Clement International Corporation
(1990). In this model it is assumed that the reactive VC metabolites
were further degraded to carbon dioxide, or reacted with GSH or with
cellular material. Parameters for subsequent metabolism were taken
from the PBTK model for vinylidene chloride (D'Souza & Andersen, 1988)
and were also used for species other than the rat after appropriate
allometric scaling, i.e., metabolic capacity scales approximately
according to body weight raised to 3/4 power (Andersen et al.,
1987). Support for the use of this principle for VC comes from data on
the metabolism of VC in non-human primates (Buchter et al., 1980). In
humans there is no evidence of a low-affinity high-capacity oxidation
by other P-450 isozymes so this parameter was set at zero for humans.
There is a wide variability of human CYP2E1 activity of about an order
of magnitude, whereas in inbred strains typically used in animal
studies the variability in CYP2E1 activity is small. The Clewell model
was then used to calculate dose metrics for ASL in animal bioassays
(Maltoni et al., 1981, 1984; Feron et al., 1981) as well as for human
inhalation exposure (see Table 56).
Reitz et al. (1996) developed a PBTK model for VC similar to that
proposed by Clewell et al. (1995a) with a single saturable pathway
based on a model for methylene chloride (Andersen et al., 1987) with
modifications on the size of liver compartment and allometric
constants. Partition coefficients were obtained from Gargas et al.
(1989). Metabolic parameters were calculated from gas uptake
experiments with male and female Sprague-Dawley rats. Metabolic
parameters for mice and humans were estimated from these experimental
data on rats and interspecies ratios obtained from literature
(Andersen et al., 1987). The model has been validated against data on
total metabolism in the rat (Watanabe et al., 1976b), gas uptake data
in the mouse, and inhalation data in the human on exhaled
concentrations of VC following VC exposure (Baretta et al., 1969).
2. Recent risk assessments of VC
2.1 Simonato et al. (1991)
Simonato et al. (1991) performed a regression analysis from
epidemiological data (see sections 8.2 and 8.3) to assess the relative
risk of liver cancer and ASL in occupational exposure to VC using the
variables cumulative exposure (ppm-years) and years since first
employment (Tables 48 and 49). On the basis of this regression
analysis and including cumulative exposure and years since first
exposure in the model, the absolute risk of ASL per 100 000 was
calculated (Table 50).
2.2 Clewell et al. (1995b)
A PBTK model was used to predict the total production of reactive
metabolites from VC in the animal bioassays and in human exposure
scenarios. These measures of internal exposure were then used in the
linearized multistage model (LMS) (Crump, 1984) to predict the risk
associated with lifetime exposure to VC in air or drinking-water.
The most appropriate toxicokinetic dose metric for a reactive
metabolite was considered to be the total amount of the metabolite
generated divided by the volume of the tissue in which it is produced
(Andersen et al., 1987), which, in the case of VC and ASL, would be
the total amount of metabolism divided by the volume of the liver.
Specifically, the average amount generated in a single day is used,
averaged over the lifetime (i.e., the lifetime average daily dose, or
LADD). The use of a dose rate such as LADD, rather than total lifetime
dose, has been reported to provide a better cross-species
extrapolation of chemical carcinogenic potency (D'Souza & Andersen,
1988). According to the authors, a body surface area adjustment is no
longer necessary when using a LADD.
The animal bioassays used are shown in Table 56. The 95% upper
confidence limits on the human risk estimates for lifetime exposure to
2.6 µg VC/m3 (1 ppb) were then calculated on the basis of each of the
sets of bioassay data, using the LMS model. The risk estimates on
inhalation studies in rats agreed with those in mice and were also
comparable with those from studies on rat diet. The estimates on oral
gavage were much higher (× 6), which the authors suggested might be
due to the use of corn oil.
Table 56. Human risk estimates (per million) for lifetime exposure to
2.6 µg VC/m3 (1 ppb) air based on the incidence
of ASL in animal bioassay a
Animal bioassay 95% UCL risk/million Reference
study per ppb
Males Females
Mouse inhalation 1.52 3.27 Maltoni et al. (1981, 1984)
Rat inhalation 5.17 2.24 Maltoni et al. (1981, 1984)
Rat diet 3.05 1.10 Feron et al. (1981)
Rat gavage 8.68 15.70 Maltoni et al. (1981, 1984)
a From: Clewell et al. (1995b); UCL = the 95% upper confidence limit of the
human risk estimates for lifetime exposure to 2.6 µg VC/m3 (1 ppb)
Risk calculations were also carried out on the basis of
epidemiological data using a linear relative risk dose-response model
(see Table 57). A PBTK model was run for the exposure scenario
appropriate to each of the selected subcohorts from each of the
studies.
Table 57. Human risk estimates (per million) for lifetime inhalation
of 2.6 µg VC/m3 (1 ppb) air based on the incidence of
ASL in human epidemiological studies a
95% UCL risk/million per ppb b Epidemiological study
0.71-4.22 Fox & Collier (1977)
0.97-3.60 Jones et al. (1988)
0.40-0.79 Simonato et al. (1991)
a From: Clewell et al. (1995b)
b The range of risk estimates reflects uncertainty in
the appropriate value for P0, the background
probability of death from liver cancer. The lower
risk estimate was calculated using the value
of P0 used in the study of Fox & Collier (1977),
while the higher risk estimate was calculated using
an estimate of the lifetime liver cancer mortality
in the USA population (Chen & Blancato, 1989).
Therefore there is not a true range but a
reflection of two assumptions about the background
rate of liver cancer in humans.
To obtain an estimate of the carcinogenic potency, the resulting
internal dose metrics were multiplied by the appropriate durations to
obtain the cumulative internal doses, which were then input into the
relative risk model together with observed and expected liver cancer
deaths for each subcohort. A 95% upper confidence limit on the
lifetime risk per ppb of VC was estimated to compare results with the
animal-based results obtained with the LMS model. The estimates from
the three epidemiological studies compared well with those from the
toxicokinetic animal-based studies. From the Simonato data, a 0.4 to
0.8 × 10-6 risk per ppm VC was estimated, which Clewell et al.
(1995b) noted is about 30 below the published inhalation unit risk of
8.4 × 10-6 (µg/m3)-1. A carcinogenic risk estimate of ASL from a
lifetime exposure to 1 µg VC/litre in human drinking-water was
1.14 × 10-6 (µg/litre)-1 and was derived from Feron's study on male
rats after dietary administration of VC. This value was compared with
the published unit risk of 5.4 × 10-5 (µg/litre)-1 (US EPA, 1995a).
2.3 Reitz et al. (1996)
Reitz et al. (1996) used a PBTK model to calculate LADDs and
these were fit to a LMS model. The data generated in rats were used to
predict ASL incidence in mice and were found to fit with the data from
Maltoni's experiment BT 4 and those of Lee et al. (1978).
The PBTK model was used to calculate LADDs for humans: mg VC
metabolites formed/day per litre of liver tissue for conditions
thought likely to have been present in the workplace in past years
(i.e., VC concentrations 50-2000 ppm; 8 h/day; 5 days/week and 10 or
20 years/70 years). From these estimates of dose the LMS model was
used to estimate the likelihood that tumours would be produced based
on the rat model. These results were then compared with the actual
data from Simonato et al. (1991). Predictions using this PBTK model
were substantially higher (10-35 fold) than actually observed in
humans (see Table 58). But the PBTK-based value for the 95% UCL for
excess lifetime risk associated with continuous inhalation of 1 µg/m3
was 5.7 × 10-7, compared with the much higher risk, 8.4 × 10-5, using
conventional risk estimates (US EPA, 1995a). Reitz et al. (1996)
concluded that even using the PBTK model, the risk of ASL is
overestimated and suggested that the livers of humans are less
sensitive to the carcinogenic effect of reactive VC metabolites than
the livers of the commonly used inbred laboratory rodents.
2.4 Storm & Rozman (1997)
Storm & Rozman (1997) use a no-threshold (LMS model and benchmark
dose approach with linear extrapolation) and threshold (NOEL/LOEL and
benchmark dose uncertainty factor approaches) models and compare them
with the present occupational exposure limits in the 0.5 to 5 ppm
range (Table 59). Although VC is a genotoxic carcinogen, the authors
argued that for VC a threshold exists because DNA adducts formed by
reaction with vinyl chloride metabolites are repaired by DNA
glycosylase in both rat and human liver in vitro (Dosanjh et al.,
1994; Saparbaev et al., 1995), and only when repair capability is
exceeded does cancer develop (Swenberg et al., 1995).
Storm & Rozman (1997) used Maltoni's inhalation study with
Sprague-Dawley rats (Maltoni et al., 1981) and, after adjusting all
exposures to reflect equivalent continuous human exposures and then to
LADD in mg/kg-day, used these dose measures in the low-dose
extrapolation models. (Occupational exposures were derived by
converting appropriately derived LADDs to equivalent occupational
exposures, assuming an inhalation rate of 10 m3/working day and that
exposures occurred 8 h/day, 50 weeks/year for 45 years).
No-threshold models evaluated were the LMS model for
extrapolation of genetic carcinogen dose-response curves to low doses,
and the benchmark dose approach (BMD) with linear extrapolation (US
EPA, 1996). Maximum likelihood estimates (MLE) and 95% UCL (qi*) on
Table 58. Predicted versus observed ASL incidence for humans
assuming occupational exposure to VC a
Ppm Years Ppm-years Linearized multistage Human observed
exposed prediction based on (incidence/100 000)
animal studies
(incidence/100 000)
100 10 1000 374 b 6.2 e
50 20 1000 376 b 6.2 e
200 20 4000 1465 b 42.2 f
1 45 45 21 c 0 g
6-11 d
a From: Storm & Rozman (1997)
b Derived by Reitz et al. (1996) assuming an occupational exposure of
5 days/week, 50 weeks/year
c Calculated from unit risk derived by Reitz et al. (1996), assuming
occupational rather than continuous exposures
d Calculated from human risk/million per ppb based on Simonato et al. (1991)
derived by Clewell et al. (1995b), assuming occupational rather than
continuous exposures
e Derived by Simonato et al. (1991) for exposure < 2000 ppm-years
and > 25 years since first employment
f Derived by Simonato et al. (1991) for exposure 2000-5999 ppm-years
and > 25 years since first employment
g No ASL reported among workers first exposed in or after 1974 when the
OSHA permissible exposure limit (PEL) of 1 ppm was promulgated.
the linear coefficient (q i) were derived using a LMS model. The
resultant linear coefficients from these and the results from Reitz et
al. (1996) described above using the PBTK model were then used to
derive safe levels of occupational exposure. For the BMD approach, a
computer programme was used to derive MLEs as well as 95% lower bound
(LB) estimates for specified risk levels along with X2
goodness-of-fit statistics, in this case P > 0.01. The MLE and LB
of both the exposure associated with 10% incidence (ED10 and LED10)
and the exposure associated with 1% incidence (ED01 and LED01),
respectively, were used for linear extrapolation to zero (US EPA,
1996). "Safe" levels of occupational exposure were derived from the
slopes of these lines.
Threshold models evaluated included a) the LOEL/NOEL
uncertainty factor approach traditionally used for non-carcinogenic
effects and b) the BMD and uncertainty factor approach being developed
as a replacement for the former. A NOEL of 13 mg/m3 (5 ppm) was
derived from the Maltoni et al. (1981) bioassay in rats. An
uncertainty factor of 2 was applied to account for the half-lifetime
exposure duration of the VC study, a standard uncertainty factor of 10
to account for potential intraspecies variability in susceptibility,
an interspecies uncertainty factor of 3 or 0.3 to cover the assumption
that humans were more or less sensitive than rats in this study giving
total uncertainty factors of 60 or 6, respectively. The same factors
were applied to MLE and LB estimates of dose using the BMD
curve-fitting approach.
The X2 goodness-of-fit P value for the BMD curve was only
acceptable when the apparent outliers at 100 and 250 ppm and high-dose
responses at 500, 2500 and 6000 ppm were eliminated, and these values
were therefore also eliminated in the LMS model for purpose of
comparison.
Both no-threshold models based on administered dose provide
estimates of occupational safe levels of about 0.004 ppm, at least two
orders of magnitude lower than the present accepted "safety" limit
value of 0.5-5 ppm when using an acceptable level of risk of 1 in
100 000 (1 × 10-5). With the PBTK model, estimates were about
0.04 ppm. For threshold models, using the BMD curve-fitting approach,
with an ED01 and LED01 point of departure, occupational levels of 0.1
to 0.7 ppm, respectively, were estimated for humans less or more
sensitive than rats. Estimations based on the rat NOEL approach gave
values of 0.4 to 4 ppm, respectively, for humans more and less
sensitive than rats.
Table 59. Comparison of estimated safe levels for occupational
exposure to VC (from Storm & Rozman, 1997)
No-threshold Slope Risk 1/100 000
models (mg/kg-day)-1 assuming
occupational
exposure a (ppb)
Linearized Administered dose b MLE 1.4 × 10-2 4.3
multistage UCL 1.8 × 10-2 3.4
PBTK dose c MLE 1.6 × 10-3 39.4
UCL 2.0 × 10-3 31.2
US EPA d UCL 2.9 × 10-1 0.2
Benchmark ED01-Point of MLE 1.4 × 10-2 4.3
dose departure LB 1.8 × 10-2 3.4
Threshold BMD b Uncertainty Risk 1/100 000
models (mg/kg- factor assuming
day) occupational
exposure a (ppm)
Benchmark Humans less MLE 0.7 6 e 0.7
dose-ED01 sensitive LB 0.5 6 e 0.7
Human = rat MLE 0.7 20 f 0.2
sensitivity LB 0.5 20 f 0.2
Humans more MLE 0.7 60 g 0.1
sensitive LB 0.5 60 g 0.1
Table 59. (cont'd)
LADD Uncertainty Risk 1/100 000
(mg/kg- factor assuming
day) occupational
exposure a (ppm)
Rat NOEL Humans less 3.6 6 e 3.8
(5 ppm) sensitive
Human = rat 3.6 20 f 1.1
sensitivity
Humans more 3.6 60 g 0.4
sensitive
a Occupational exposures assume 10 m3/day inhalation rate, 5 days/week, 50 weeks/year,
for 45 years averaged over a lifetime of 70 years (Note: Levels are ppb for
no-threshold models, ppm for threshold models)
b Derived using a computer programme
c Slope from Reitz et al. (1996)
d Slope from US EPA Health Effects Assessment Summary Tables (US EPA, 1995)
e Safety factors of 10 for intraspecies, 0.3 for interspecies extrapolation and
2 for half-lifetime exposure
f Safety factors of 10 for intraspecies extrapolation and 2 for
half-lifetime exposure
g Safety factors of 10 for intraspecies, 3 for interspecies extrapolation, and
2 for half-lifetime exposure
MLE = maximum likelihood estimate;
UCL = 95% upper confidence limit;
LB = 95% lower bound on dose
ANNEX 3. EXECUTIVE SUMMARY OF VINYL CHLORIDE PANEL REPORT
Executive summary of
Mundt KA, Dell LD, Austin RA, Luippold RS, Noess R & Bigelow C.
Epidemiological study of men employed in the vinyl chloride industry
between 1942 and 1972: I. Re-analysis of mortality through December
31, 1982; and II. Update of mortality through December 31, 1995.
Final Report. Arlington, Virginia: The Vinyl Chloride Panel, Chemical
Manufacturers Association, January 8, 1999. 178 pagesa
A cohort of 10 109 men who were employed for at least one year in
vinyl chloride exposed jobs between 1942 and 1972 at any one of
37 facilities in the USA or Canada was followed for mortality. Through
December 31, 1995, a total of 3191 deaths had occurred among cohort
members. A re-analysis of mortality through December 1982, at which
time 1569 deaths had occurred, was conducted to establish baseline
results against which the updated mortality results could be compared.
The re-analysis of mortality through December 1982 was necessary for
four reasons: (1) the cohort was reduced from 10 173 to 10 109 after
the removal of study subjects who were duplicates or were not eligible
for cohort inclusion; (2) a substantial proportion of cohort members
previously lost to follow-up were identified and restored to the
cohort; (3) an appropriate referent population, based on regional
mortality rates, was constructed and used for the mortality update
through 1995; and (4) the number of categories of death that could be
evaluated in the analysis increased from 61 to 92 causes of death.
Through 1995, mortality from all causes of death was 17% lower
than state mortality weighted according to person-years accumulated
among employees in the state where the plant was located. This result
was similar to the 16% deficit seen for mortality from all causes of
death through 1982. Mortality from all cancers, however, showed a
deficit of 4% through 1995, in contrast with the 2% excess seen as of
1982. Specific cancers showing meaningful excesses both through 1982
and through 1995 included cancers of the liver and biliary tract,
brain, and connective and soft tissues. However, several causes of
death previously believed to be related to vinyl chloride exposure
were not seen in excess, including lung cancer, cancers of lymphatic
and haematopoietic tissue, emphysema and pneumoconioses and other lung
diseases, including chronic obstructive pulmonary disease (COPD).
a This summary is reprinted by permission of the Vinyl Chloride
Health Committee. This study, which is an update of the Wong et
al. (1991) study, was not available at the Task Group meeting and
thus did not affect the evaluation of the health risks of vinyl
chloride. It is printed here as additional information to the
reader.
Through 1995, mortality from liver and biliary tract cancer,
including angiosarcoma of the liver, was significantly increased
(SMR=359; 95% CI: 284-446) although the excess was smaller than that
observed through 1982 (SMR=428; 95% CI: 304-585). The SMR for liver
and biliary tract cancer increased with duration of exposure from 83
(95% CI: 33-171) to 215 (95% CI: 103-396) to 679 (95% CI: 483-929) to
688 (95% CI: 440-1023) for those exposed from 1 to 4 years, 5 to 9
years, 10 to 19 years and 20 years or more, respectively. The SMR for
liver and biliary tract cancer also increased with time since first
exposure from 0 (no deaths observed) to 287 (95% CI: 131-544) to 323
(95% CI: 200-493) to 434 (95% CI: 322-572) for those 1 to 9 years, 10
to 19 years, 20 to 29 years, and 30 or more years since first
exposure, respectively. Results of a Cox proportional hazards model
that fitted age at first exposure, duration of exposure, and year of
first exposure indicated that duration of exposure was most strongly
associated with increased risk of mortality from liver and biliary
tract cancer. The adjusted hazards ratio for 1 to 4 years, 5 to 9
years, 10 to 19 years and 20 years or more duration of exposure were
1.0 (referent group), 2.8 (95% CI: 1.0-7.3), 9.0 (95% CI: 4.0-20.7)
and 6.0 (95% CI: 2.5-14.4), respectively. This confirms the recognized
association between vinyl chloride exposure and liver and biliary
tract cancer, mainly due to a large excess of angiosarcoma of the
liver.
Mortality from brain cancer showed an excess (SMR=142; 95% CI:
100-197) that was slightly smaller than that seen in the re-analysis
through 1982 (SMR=169; 95% CI: 105-255). Since 1982, an additional 14
brain cancer deaths were observed (12.2 expected). The SMRs for brain
cancer were elevated among those exposed for 5 to 9 years (SMR=193;
95% CI: 96-346) and those exposed for 20 years or more (SMR=290; 95%
CI: 132-551). Results of a Cox proportional hazards model adjusted for
duration of employment showed that age at employment of 35 years or
older and longer duration of employment were both associated with
increased risk of mortality from brain cancer, and these associations
were not confounded. The association of brain cancer deaths with vinyl
chloride exposure is uncertain, as the older age at first employment
in the vinyl chloride industry suggests that these cohort members
might have sustained exposure to some other carcinogen prior to
employment in vinyl chloride.
Mortality from cancers of the connective and soft tissues also
showed an excess (SMR=270; 95% CI: 139-472), based on 12 deaths.
Through 1982, mortality from connective and soft tissue cancers was
also elevated (SMR=367; 95% CI: 147-755), based on seven observed
deaths. This result was not reported in earlier investigations of this
cohort because mortality for this cause of death category had not been
previously evaluated. Although the total number of deaths due to this
rare cause was small, the observed excess is potentially important for
two reasons. First, very little is known about the risk factors for
connective and soft tissue cancers, a category including numerous
histological types of cancer. Second, some of these cancers may have
been mis-diagnosed or mis-reported angiosarcomas of the liver, one
form of soft tissue sarcoma known to be related to vinyl chloride
exposure.
No excesses in mortality were seen for lung cancer or cancers of
the lymphatic and haematopoietic system. In contrast to the previous
report which showed an excess of mortality from emphysema and chronic
obstructive disease, mortality from emphysema and mortality from
pneumoconioses and other respiratory diseases (PORD), the category
which included chronic obstructive pulmonary disease, each showed
deficits (SMR=61; 95% CI: 40-89 for emphysema and SMR=73; 95% CI:
60-88 for PORD). The previously published findings with respect to
mortality due to these causes of death may have resulted from
methodological errors.
In conclusion, this update of the industry-wide cohort confirmed
a strong association between duration of employment in the vinyl
chloride industry prior to 1974 and cancers of the liver and biliary
tract, mostly resulting from a large excess of deaths due to
angiosarcoma of the liver. This association has been observed in all
similar studies and represents one of the most consistent effects
observed in the occupational health literature. Cancers of the
connective and soft tissues, though occurring in relatively small
numbers, also appear to be related to employment in the industry. The
elucidation of the actual mechanism for this observed association will
require further investigation. The brain cancer mortality excess
observed in the analyses through 1982 is less clearly associated with
vinyl chloride, and may reflect risk factors other than vinyl chloride
present in the early years of the industry or reflecting exposures
sustained prior to employment in this industry. Since 1982, no such
excess has occurred.
This study represents one of the largest and longest-followed
cohorts potentially exposed to substantial levels of vinyl chloride.
The brain cancer and emphysema excesses previously reported are not
sustained, however, the brain cancer excess remains unexplained.
Except for deaths due [to] cancers of the liver and biliary tract, and
to a lesser extent cancers of connective and soft tissues, the
mortality patterns for this cohort remain at or below that expected in
the general population.
RESUME
La présente monographie traite du chlorure de vinyle monomère
lui-même, à l'exclusion de son polymère, le chlorure de polyvinyle
(PVC). Le problème de l'exposition à des mélanges contenant du
chlorure de vinyle n'est pas abordé.
1. Identité, propriétés physiques et chimiques et méthodes d'analyse
Dans les conditions ambiantes, le chlorure de vinyle se présente
sous la forme d'un gaz incolore inflammable à l'odeur douceâtre. Sa
tension de vapeur est élevée, de même que la valeur de sa constante
pour la loi d'Henry; sa solubilité dans l'eau est relativement faible.
Il est plus lourd que l'air et il est soluble dans presque tous les
solvants organiques. On le transporte à l'état liquide sous pression.
A la température ambiante et en l'absence d'air, le chlorure de
vinyle pur et sec est très stable et non corrosif. Toutefois,
au-dessus de 450°C ou en présence d'hydroxyde de sodium ou de
potassium, il peut subir une décomposition partielle. Sa combustion
dans l'air donne naissance à du dioxyde de carbone et à du chlorure
d'hydrogène. En présence d'air et d'oxygène, des peroxydes très
explosifs peuvent se former, ce qui exige une surveillance permanente
et la limitation de la teneur en oxygène, en particulier dans les
installations de récupération du chlorure de vinyle. En présence
d'eau, il se forme de l'acide chlorhydrique.
Du point de vue industriel, ce sont les réactions de
polymérisation conduisant au chlorure de polyvinyle qui sont
techniquement les plus importantes, mais les réactions d'addition des
halogènes sur la double liaison, qui conduisent au
1,1,2-trichloréthane ou au 1,1-dichloréthane, ont aussi leur
importance.
On peut surveiller la concentration de chlorure de vinyle dans
l'atmosphère en piégeant le composé sur un support adsorbant et en
procédant à une analyse par chromatographie en phase gazeuse après
désorption liquide ou thermique. Pour le dosage dans l'air ambiant, il
peut être nécessaire d'utiliser plusieurs supports adsorbants ou
pièges froids disposés en série pour mieux capter le composé. Les pics
de concentration sur les lieux de travail peuvent se mesurer au moyen
d'instruments à lecture directe utilisant notamment des détecteurs à
ionisation de flamme ou à photo-ionisation. Pour le contrôle en
continu, ont utilise des analyseurs basés sur la spectrophotométrie
infrarouge ou sur la chromatographie en phase gazeuse avec détection
par ionisation de flamme et qui sont munis d'un système
d'enregistrement et de traitement des données. Pour doser le chlorure
de vinyle dans les liquides et les solides, on a recours à l'injection
directe, à l'extraction et de plus en plus, à la technique de l'espace
de tête ou à celle de purgeage-piégeage. Pour l'analyse de ces
échantillons, on utilise aussi la chromatographie en phase gazeuse
avec détection par ionisation de flamme ou spectrométrie de masse.
2. Sources d'exposition humaine dans l'environnement
Le chlorure de vinyle n'existe pas à l'état naturel, même si on
en a décelé la présence dans les gaz qui s'échappent des décharges et
dans les eaux souterraines, par suite de la décomposition de rejets de
solvants constitués d'hydrocarbures chlorés, ou encore dans
l'environnement des lieux de travail où on fait usage de ce genre de
solvants. Il y a également du chlorure de vinyle dans la fumée de
cigarette.
La production industrielle du chlorure de vinyle utilise deux
réactions principales: a) l'hydrochloration de l'acétylène et b) le
craquage thermique (à environ 500°C) du 1,2-dichloréthane produit par
chloration ou oxychloration directes de l'éthylène par le chlore ou
par un mélange HCl + air/O2. Ce procédé est actuellement le plus
couramment utilisé.
La production mondiale de PVC et par conséquent celle de chlorure
de vinyle monomère a été d'environ 27 millions de tonnes en 1998.
Vingt pour cent des matières plastiques utilisées dans le monde sont à
base de PVC et ce polymère se retrouve dans presque tous les secteurs
de l'industrie. Environ 95% de la production mondiale de chlorure de
vinyle sert à la fabrication de PVC. Le reste est utilisé pour la
production de solvants chlorés, essentiellement du
1,1,1-trichloréthane (10 000 tonnes par an).
Trois procédés principaux sont utilisés pour la production
industrielle du PVC: la polymérisation en suspension (80% de la
production mondiale), la polymérisation en émulsion (12%) et la
polymérisation en masse (8%). La plupart des études de cas faisant
état des effets nocifs du chlorure de vinyle concernent des usines
produisant du PVC par polymérisation en suspension (on dit aussi
polymérisation en perles).
On a signalé des émissions de chlorure de vinyle lors d'accidents
survenus dans des usines produisant du PVC ou pendant le transport.
Dans de nombreux pays, on récupère le chlorure de vinyle résiduel non
polymérisé lors de la production de PVC ou qui se trouve dans les
rejets gazeux ou les eaux usées. Faute de certaines précautions, il
est possible de retrouver du chlorure de vinyle dans les résines et
autres produits à base de PVC.
Le taux de chlorure de vinyle résiduel présent dans le PVC est
réglementé dans de nombreux pays depuis la fin des années 1970. Depuis
lors, les émissions de chlorure de vinyle dues à la décomposition
thermique du PVC sont soit indétectables, soit très faibles.
Des dioxines peuvent se former lors de la production de chlorure
de vinyle. Les quantités qui sont répandues dans l'environnement sont
controversées.
3. Transport, distribution et transformation dans l'environnement
En raison de sa forte tension de vapeur, le chlorure de vinyle
libéré dans l'environnement devrait exister presque uniquement en
phase gazeuse. Il existe des indices de dépôts sous forme liquide.
Le chlorure de vinyle est relativement peu soluble dans l'eau et
sa capacité d'adsorption aux particules et aux solides en suspension
est faible. C'est par volatilisation que le composé s'élimine le plus
rapidement des eaux de surface. La demi-vie de volatilisation à partir
des eaux de surface varie d'environ 1h à 40 h.
A partir du sol, la demi-vie de volatilisation est, selon les
calculs, égale à 0,2-0,5 jours. On estime que les pertes de chlorure
de vinyle (au bout d'un an sous un mètre de terre) vont de 0,1 à 45%,
selon le type de sol. Le coefficient de sorption pédologique tiré des
données physico-chimiques indique que le potentiel de sorption du
composé est faible et que par conséquent il est très mobile dans le
sol. Il existe une autre voie importante de distribution dans
l'environnement, à savoir le lessivage qui peut entraîner le chlorure
de vinyle jusqu'aux nappes d'eau souterraines où il est susceptible de
persister pendant des années.
Les études de laboratoire portant sur des organismes aquatiques
révèlent une certaine tendance à la bioaccumulation, mais pas de
bioamplification le long de la chaîne alimentaire.
A quelques exceptions près, le chlorure de vinyle ne se laisse
pas facilement dégrader par les groupements de microorganismes
inadaptés dans les conditions ambiantes. On estime que la demi-vie de
biodégradation par des micro-organismes non acclimatés est de l'ordre
de plusieurs mois ou années. Toutefois, certaines cultures
microbiennes enrichies ou pures (par exemple Mycobacterium sp.) sont
capables de décomposer le chlorure de vinyle lorsque les conditions
culturales sont optimales. Les principaux produits de dégradation sont
l'acide glycolique ou le dioxyde de carbone après conversion aérobie
et l'éthane, l'éthylène, le méthane ou le chlorométhane après
transformation anaérobie. Souvent, les microorganismes aérobies
décomposent plus rapidement le chlorure de vinyle que les
microorganismes anaérobies.
Le principal processus atmosphérique est la réaction avec les
radicaux OH produits par voie photochimique; la demi-vie
troposphérique résultant de ce processus est de 1 à 4 jours. Les
réactions de photolyse réalisées dans des conditions expérimentales
donnent naissance à plusieurs composés importants, comme le
chloracétaldéhyde, le formaldéhyde et le chlorure de formyle.
On pense que les réactions de photolyse, de même que l'hydrolyse
chimique, sont de peu d'importance en milieu aqueux. Toutefois, la
présence de photosensibilisateurs peut faciliter la transformation du
chlorure de vinyle.
On a des raisons de penser que le chlorure de vinyle réagit avec
le chlore ou les chlorures utilisés pour la désinfection de l'eau pour
donner du chloracétaldéhyde et autres composés indésirables. Il existe
d'autres interactions possibles, notamment avec les sels, dont un
grand nombre sont susceptibles de former des complexes avec le
chlorure de vinyle, ce qui peut en accroître la solubilité.
Parmi les méthodes utilisées (avec des succès divers) pour
éliminer le chlorure de vinyle présent dans l'eau, on peut citer le
lavage, l'extraction, l'adsorption et l'oxydation. Certaines méthodes
de biopurification (applicables aux eaux souterraines et aux sols)
associent l'évaporation ou d'autres techniques à un traitement
microbien. Le chlorure de vinyle présent dans les rejets gazeux peut
être recyclé, incinéré ou décomposé par voie microbienne. La majeure
partie du chlorure de vinyle produit industriellement se retrouve à
l'état lié dans des articles en PVC. L'incinération de ces produits
comporte un risque de formation de PCDD, de PCDF et d'autres dérivés
chlorés indésirables.
4. Concentrations dans l'environnement et exposition humaine
La population dans son ensemble est très peu exposée au chlorure
de vinyle.
La concentration de chlorure de vinyle dans l'air ambiant est
faible, généralement inférieure à 3 µg/m3. La population peut être
davantage exposée lorsque de grandes quantités de chlorure de vinyle
sont libérées accidentellement dans l'environnement, par exemple en
cas de déversement pendant le transport. Il s'agit cependant d'une
exposition qui a toutes chances d'être passagère. A proximité de sites
industriels où l'on produit du chlorure de vinyle et du PVC ou près de
décharges, on a enregistré des concentrations beaucoup plus élevées
(pouvant aller respectivement jusqu'à 8000 µg/m3 et 100 µg/m3).
La principale voie d'exposition professionnelle est la voie
respiratoire et alle intervient principalement dans les usines qui
produisent du chlorure de vinyle et du PVC. Dans les années 1940 et
1950, l'exposition professionnelle au chlorure de vinyle était de
plusieurs milliers de mg/m3; elle atteignait encore plusieurs
centaines de mg/m3 dans les années 1960 et au début des années
1970.Une fois reconnu le risque cancérogène inhérent au chlorure de
vinyle, la norme d'exposition à ce composé a été fixée au cours des
années 1970 à environ 13-26 mg/m3 (5-10 ppm) dans la plupart des
pays. Le respect de ces directives a fait fortement chuter la
concentration du chlorure de vinyle sur les lieux de travail, mais au
cours des années 1990 on a tout de même fait état de concentrations
plus élevées et ce pourrait encore être le cas actuellement dans
quelques pays.
Il est arrivé que l'on trouve du chlorure de vinyle dans les eaux
de surface, dans les sédiments et dans les boues d'égout, avec des
concentrations maximales respectivement égales à 570 µg/litre,
580 µg/kg et 62 000 µg/litre. Des échantillons de sol prélevés près
d'une teinturerie abandonnée se sont révélés présenter une forte
teneur en chlorure de vinyle (jusqu'à 900 mg/kg). On a mis en évidence
des concentrations de l'ordre de 60 000 µg/litre ou davantage dans des
eaux souterraines ou dans des eaux de lessivage provenant de zones
contaminées par des hydrocarbures chlorés. Des teneurs élevées
(jusqu'à 200 mg/litre) ont également été mises en évidence, 10 ans
après le repérage des premières fuites, dans l'eau de puits situés à
proximité d'une usine de PVC.
Les quelques données disponibles montrent que les tissus des
petits vertébrés aquatiques et des poissons peuvent contenir du
chlorure de vinyle.
Dans la majorité des échantillons d'eau de boisson analysés, le
chlorure de vinyle n'était pas présent à des concentrations
décelables. La concentration maximale dont il ait été fait état était
de 10 µg/litre dans de l'eau prête à être consommée. On manque de
données récentes sur la concentration du composé dans l'eau de
boisson, mais elle devrait être inférieure à 10 µg/litre. Si la source
utilisée pour la boisson est contaminée, l'exposition peut être plus
importante. Des études récentes ont permis de déceler la présence de
chlorure de vinyle dans de l'eau minérale en bouteilles de PVC à une
concentration inférieure à 1 µg/litre. On peut penser que la présence
de chlorure de vinyle est plus fréquente dans ces eaux en bouteilles
que dans l'eau du robinet.
L'utilisation de PVC pour l'emballage peut entraîner la présence
de chlorure de vinyle dans les denrées alimentaires, les produits
pharmaceutiques et les cosmétiques. On en a décelé jusqu'à 20 mg/kg
dans des liqueurs, jusqu'à 18 mg/kg dans des huiles végétales et
jusqu'à 9,8 mg/kg dans du vinaigre. Depuis le début des années 1970,
le nombre d'échantillons contrôlés positifs pour le chlorure de vinyle
est moindre et la concentration est plus faible qu'auparavant, grâce
aux mesures législatives prises par un certain nombre de pays.
Plusieurs organismes ont calculé l'exposition au chlorure de
vinyle due à la contamination des denrées alimentaires par leur
emballage de PVC. En se basant sur valeur estimative moyenne de la
dose ingérée au Royaume-Uni et aux Etats-Unis, on a calculé qu'à la
fin des années 1970 et au début des années 1980, l'exposition devait
être inférieure à 0,0004 µg/kg par jour. Une étude ancienne a mis
évidence du chlorure de vinyle dans la fumée de tabac à des
concentrations de quelques ng par cigarette.
5. Cinétique et métabolisme chez les animaux de laboratoire et
l'Homme
Après inhalation ou ingestion, le chlorure de vinyle est
facilement et rapidement absorbé. Des études effectuées sur des
animaux et des sujets humains, dans des conditions d'équilibre
dynamique, ont montré qu'environ 40% de la dose inhalée étaient
absorbés après exposition par la voie respiratoire. L'expérimentation
animale montre que l'absorption peut dépasser 95% après ingestion.
L'absorption percutanée de chlorure de vinyle à l'état gazeux est peu
importante.
Les données fournies par les études sur l'inhalation et
l'ingestion de chlorure de vinyle par des rats montrent que le composé
se répartit rapidement et largement dans l'organisme. Par suite d'une
métabolisation et d'une excrétion rapides, son accumulation est
limitée. Chez le rat, le passage transplacentaire se produit
rapidement. On n'a pas connaissance d'études consacrées à la
répartition du composé dans l'organisme après exposition par voie
percutanée.
Après inhalation ou ingestion, la principale voie métabolique
consiste en une oxydation par le cytochrome P-450 (CYP2E1) qui conduit
à la formation d'oxyde de chloréthylène, un époxyde à courte vie
extrêmement réactif, qui se transpose très vite en chloracétaldéhyde.
La principale réaction de détoxication de ces deux métabolites
réactifs ainsi que de l'acide chloracétique, produit de
déshydrogénation du chloracétaldéhyde, consiste en une conjugaison
avec le glutathion catalysée par la glutathion- S-transférase. Les
conjugués sont ensuite transformés en dérivés de la cystéine
[ S-(2-hydroxyéthyl)cystéine, N-acétyl- S-
(2-hydroxyéthyl)cystéine, S-carboxyméthylcystéine et acide
thiodiglycolique] et ils sont excrétés dans l'urine. Un autre
métabolite, le dioxyde de carbone, est excrété dans l'air exhalé.
Les isozymes de la CYP2E1 et de la glutathion- S-transférase
présentent d'importantes variations d'activité interspécifiques et
interindividuelles.
Après exposition par inhalation ou ingestion de faibles doses de
chlorure de vinyle, le composé est éliminé par métabolisation et ses
métabolites non volatils sont principalement excrétés par la voie
urinaire. L'étude comparée de l'absorption après inhalation montre que
relativement au poids corporel, le chlorure de vinyle est métabolisé
moins rapidement par l'organisme humain que par l'organisme animal.
Toutefois, si on tient compte de la superficie du corps, on constate
que la clairance métabolique est comparable chez l'Homme et chez les
autres mammifères. Lorsque l'exposition - toujours par inhalation ou
ingestion - est plus importante, la principale voie d'excrétion chez
l'animal consiste dans l'élimination du composé initial dans l'air
expiré, ce qui témoigne de la saturation des voies métaboliques.
Quelle que soit la dose, l'excrétion par la voie fécale est
secondaire. On n'a pas trouvé d'étude qui soit spécialement consacrée
à l'excrétion par la voie biliaire.
On estime que l'oxyde de chloréthylène est le métabolite le plus
important in vivo, pour ce qui est des effets cancérogènes et
mutagènes du chlorure de vinyle. Cet époxyde réagit sur d'ADN pour
former un certain nombre d'adduits, principalement de la
7-(2'-oxoéthyl)guanine (7-OEG) et, en moindre quantité, des
éthénoadduits exocycliques comme la 1, N 6-éthénoadénine (Epsilon
A), la 3, N 4-éthénocytosine (Epsilon C) et la N2,3-éthénoguanine
(Epsilon G). Contrairement à l'adduit principal, la 7-OEG, ces
éthénoadduits avec l'ADN ont des propriétés promutagènes. On a pu
doser la 7-OEG, la Epsilon A, la Epsilon C et la Epsilon G dans les
divers tissus de rongeurs exposés à du chlorure de vinyle. On a mis au
point des modèles toxicocinétiques à base physiologique pour rendre
compte de la relation entre la dose aux tissus cibles et les points
d'aboutissement toxicologique du chlorure de vinyle.
6. Effets sur les mammifères de laboratoire et les systèmes d'épreuve
in vitro
Administré par inhalation à des animaux de plusieurs espèces, le
chlorure de vinyle s'est révélé d'une faible toxicité aiguë. La CL50
à 2 h pour le rat, la souris, le cobaye et le lapin a été trouvée
respectivement égale à 390 000, 293 000, 595 000 et 295 000 mg/m3. On
ne possède aucune donnée sur la toxicité aiguë du composé par voie
orale ou percutanée. L'inhalation brutale de chlorure de vinyle a un
effet stuporeux. Chez des rats, des souris et des hamsters, la mort a
été précédée d'un accroissement de l'activité motrice, d'ataxie et de
convulsions suivies d'une défaillance respiratoire. Chez des chiens
plongés dans un état stuporeux après exposition brutale à une
concentration de 260 000 mg/m3, on a noté de graves arythmies
cardiaques. Des rats, exposés par la voie respiratoire à du chlorure
de vinyle, ont présenté un certain nombre d'anomalies
anatomopathologiques telles que congestion des organes internes,
notamment des poumons, du foie et des reins, et oedème pulmonaire.
On ne dispose pas d'études ni de données appropriées qui
permettent d'évaluer les effets de l'exposition par voie percutanée ou
encore le pouvoir irritant ou sensibilisateur au niveau cutané.
Une exposition de courte durée (13 semaines) au chlorure de
vinyle par la voie orale a permis d'obtenir une valeur de 30 mg/kg
pour la NOEL, c'est-à-dire la dose sans effet observable, le critère
retenu étant l'augmentation du poids du foie.
Chez plusieurs espèces, le principal organe cible après
exposition de courte durée (jusqu'à 6 mois) par voie respiratoire se
révèle être le foie. Chez des rats soumis à une dose de 26 mg/m3 (la
dose la plus faible utilisée) on a constaté une augmentation du poids
relatif du foie et des modification hépatocellulaires; à dose plus
élevée (> 260 mg/m3), les anomalies hépatiques étaient plus
prononcées et dépendaient de la dose. Les autres organes cibles
étaient le rein, le poumon et le testicule. Les rats, les souris et
les lapins se sont révélés plus sensibles que les cobayes et les
chiens.
Une exposition de longue durée par la voie respiratoire a
entraîné une augmentation statistiquement significative de la
mortalité chez certaines souches de rats à des doses ne dépassant pas
260 mg/m3, chez des souris à la dose de 130 mg/m3 et chez des
hamsters à la dose de 520 mg/m3 sur des durées d'exposition
variables. Des rats exposés à une dose de 130 mg/m3 ont présenté une
diminution du poids corporel et une augmentation du poids relatif de
la rate ainsi qu'une dégénérescence hépatocellulaire et une
prolifération des cellules pariétales des capillaires sinusoïdes. A
dose plus élevée, on a noté une dégénérescence testiculaire, une
néphrose tubulaire et des foyers de dégénérescence au niveau du
myocarde. Chez des rats et des souris exposés par la voie respiratoire
la dose sans effet nocif observable (NOAEL) est inférieure à 130
mg/m3 pour ce qui est des effets cancérogènes.
Des études d'alimentation de longue durée ont fait ressortir une
augmentation de la mortalité, un accroissement du poids du foie et une
modification de la morphologie de cet organe.
Après exposition par la voie orale, on a observé un polymorphisme
hépatocellulaire (variation de la taille et de la forme des
hépatocytes et de leur noyau) chez des rats soumis à des doses de
chlorure de vinyle ne dépassant pas 1,3 mg/kg de poids corporel. La
NOAEL était égale à 0,13 mg/kg de poids corporel.
Après administration prolongée à des rats de chlorure de vinyle
dans des granulés de PVC par la voie alimentaire, on a observé un
accroissement significatif de l'incidence des tumeurs au niveau du
foie. Il s'agissait d'angiosarcomes à la dose quotidienne de 5,0 mg/kg
p.c., et de nodules néoplasiques (femelles) ou de carcinomes
hépatocellulaires (mâles) à la dose de 1,3 mg/kg p.c.
Des études au cours desquelles on a fait inhaler du chlorure de
vinyle à des rats Sprague-Dawley ont fait ressortir une relation
dose-réponse dans le cas des angiosarcomes du foie et, à forte
concentration, pour les carcinomes de la glande de Zymbal. En
revanche, on n'a pas observé de relation dose-réponse bien nette dans
le cas des hépatomes ou des angiosarcomes extrahépatiques, des
néphroblastomes, des neuroblastomes ou des tumeurs des glandes
mammaires. Chez la souris, le spectre tumoral induit par une
exposition respiratoire de longue durée est analogue à celui que l'on
observe chez le rat, mais on a aussi noté une augmentation des tumeurs
pulmonaires propre à la souris. Chez des hamsters, on a relevé une
augmentation de l'incidence des angiosarcomes hépatiques, des tumeurs
des glandes mammaires et du conduit auditif, des mélanomes et des
épithéliomas gastriques et cutanés.
Un certain nombre de systèmes d'épreuve in vitro ont permis de
mettre en évidence les effets mutagènes et génotoxiques du chlorure de
vinyle, surtout après activation métabolique. Le composé s'est révélé
mutagène dans le test d'Ames sur les souches TA100, TA1530 et TA1535
de S. typhimurium, à l'exclusion des souches TA98, TA1537 et TA1538,
ce qui dénote des mutations par substitution de paires de bases
(transversion et transition) plutôt que des mutations par décalage du
cadre de lecture. Ces résultats concordent avec une autre observation,
à savoir que les éthénoadduits qui se forment lors de l'attaque de
l'ADN par l'oxyde de chloréthylène et par le chloracétaldéhyde
aboutissent effectivement à des mutations par substitution de paires
de bases.
D'autres tests de mutation génique effectués sur des bactéries,
des levures et des cellules mammaliennes ont donné des résultats
positifs, mais seulement en présence d'activation métabolique. Des
effets mutagènes ont également été observés dans des lignées
cellulaires humaines contenant le cytochrome P-450IIE1 obtenu par
clonage, qui est capable de métaboliser le chlorure de vinyle. On a
aussi décelé des mutations dans des fragments d'un végétal
(Tradescantia) mis en présence de chlorure de vinyle. Des tests de
conversion génique ont donné des résultats positifs dans le cas de
Saccharomyces cerevisiae en présence d'un système d'activation
métabolique. En présence de chlorure de vinyle, des hépatocytes de rat
ont été le siège d'une synthèse non programmée de l'ADN et un
accroissement des échanges entre chromatides-soeurs a été observé dans
des lymphocytes humains après addition d'un système activateur
exogène. Chez des bactéries dont le système de réparation de l'ADN
était défectueux, on n'a pas décelé d'inhibition de la croissance en
l'absence de système activateur. Les tests de transformation
cellulaire ont donné des résultats positifs avec ou sans activation
métabolique.
Chez Drosophila melanogaster, l'exposition au chlorure de
vinyle a provoqué des mutations géniques et des recombinaisons
mitotiques, mais ces effets n'ont pas été observés sur des cellules
germinales de mammifères. Le composé a des effets clastogènes chez des
rongeurs, il augmente les échanges entre chromatides-soeurs chez le
hamster et provoque la rupture des brins de l'ADN chez la souris. Des
tests par passage sur hôte (rat) ont montré que le chlorure de vinyle
provoquait une conversion génique et les mutations directes chez des
levures.
L'oxyde de chloréthylène et le chloracétaldéhyde se sont révélés
mutagènes dans un certain nombre de systèmes. L'oxyde de chloréthylène
est un mutagène puissant, alors que le chloracétaldéhyde est fortement
toxique. Ces deux composés sont cancérogènes pour la souris, l'oxyde
de chloréthylène étant de loin le plus actif.
Les mutations observées au niveau des gènes ras et p53 ont
été analysées sur des tumeurs hépatiques induites chez des rats
Sprague-Dawley par du chlorure de vinyle: dans les carcinomes, on a
constaté la présence de substitutions de paires de bases au niveau du
gène Ha- ras; dans les angiosarcomes, ces substitutions intéressaient
le gène p53. La présence de ces mutations concorde avec la
formation, observée après exposition au chlorure de vinyle,
d'éthénoadduits persistants dans l'ADN des hépatocytes, éthénoadduits
dont on connaît le caractère promutagène.
L'étude du mécanisme par lequel s'exerce l'effet cancérogène du
chlorure de vinyle donne à penser que l'intermédiaire réactif que
constitue l'oxyde de chloréthylène attaque l'ADN pour former des
éthénoadduits, ce qui conduit à la substitution de paires de bases et
à la transformation néoplasique.
7. Effets sur l'Homme
L'exposition à des concentrations de l'ordre de 2590 mg/m3
(1000 ppm), qui n'étaient pas rares avant 1974, pendant des périodes
de 1 mois à plusieurs années, seraient à l'origine d'un syndrome
pathologique particulier observé chez des ouvriers travaillant sur le
chlorure de vinyle et appelé "maladie du chlorure de vinyle". Les
symptômes évoqués consistaient en douleurs auriculaires et céphalées,
étourdissements, troubles visuels, fatigue et perte d'appétit,
nausées, insomnies, essoufflement, douleurs au niveau de l'estomac et
dans la région du foie et de la rate, douleurs et picotements dans les
membres, sensation de froid aux extrémités, diminution de la libido et
perte de poids. Sur le plan clinique, on a relevé au niveau des doigts
des modifications à type de sclérodermie évoluant vers des anomalies
osseuses qualifiées d'acro-ostéolyse, avec des anomalies de la
circulation périphérique identiques à celles qui sont caractéristiques
de la maladie de Raynaud, une hypertrophie du foie et de la rate
d'histologie particulière et des manifestations respiratoires.
Les études sur l'Homme ne sont pas suffisantes pour pouvoir
confirmer la présence d'effets sur la fonction de reproduction. Un
certain nombre d'études font état d'une augmentation de l'incidence
des affections de l'appareil circulatoire chez des ouvriers
travaillant sur le chlorure de vinyle. Toutefois, les études
effectuées sur des cohortes d'effectif plus important ont mis en
évidence une diminution de la mortalité due aux maladies
cardiovasculaires.
Les études épidémiologiques fournissent une argumentation solide
et cohérente tendant à prouver que l'exposition au chlorure de vinyle
provoque une forme rare de cancer, l'angiosarcome du foie. On a
également établi un lien entre certaines tumeurs cérébrales ou
carcinomes hépatocellulaires et le chlorure de vinyle, sans qu'on
puisse considérer les données obtenues à cet égard comme définitives.
Les autres localisations où l'on a observé une augmentation des
cancers sont le poumon, les tissus lymphatiques et hématopoïétiques et
la peau.
Le chlorure de vinyle a des effets mutagènes et clastogènes chez
l'Homme. On a ainsi constaté que chez des ouvriers exposés à de fortes
concentrations de chlorure de vinyle, la fréquence des aberrations
chromosomiques, des micronoyaux et des échanges entre
chromatides-soeurs dans les lymphocytes du sang périphériques, était
plus élevée que chez les témoins. Dans de nombreuses études,
l'intensité et la durée de l'exposition ne sont qu'estimatives, mais
on peut néanmoins observer l'existence d'une relation dose-réponse et
une "normalisation" des effets génotoxiques avec le temps lorsque
l'exposition diminue.
Des mutations ponctuelles ont été décelées au niveau des gènes
p53 et ras dans des tumeurs (angiosarcomes du foie) prélevées sur
des ouvriers très exposés travaillant sur des autoclaves (avant 1974)
ainsi que dans un carcinome hépatocellulaire dont était porteur un
autre ouvrier également exposé au chlorure de vinyle.
Parmi les marqueurs biologiques dont on a étudié la possibilité
d'utilisation comme indicateurs d'une exposition au chlorure de
vinyle, on peut citer a) l'excrétion de métabolites (par exemple
l'acide thiodiglycolique), b) des marqueurs génétiques comme la
présence d'anomalies chromosomiques ou de micronoyaux, c) le taux de
certaines enzymes (par exemple celles qui sont mesurées dans les tests
de la fonction hépatique), d) les oncoprotéines sériques et leurs
anticorps en tant que biomarqueurs des effets induits par le chlorure
de vinyle.
Les enfants qui vivent à proximité de décharges et autres sources
ponctuelles d'exposition au chlorure de vinyle pourraient courir un
risque accru, compte tenu de la sensibilité plus forte constatée chez
les jeunes animaux. On n'a toutefois pas de preuves directes d'une
telle sensibilité chez l'Homme.
C'est seulement dans le cas de l'angiosarcome du foie-seul ou en
association avec d'autres tumeurs hépatiques - qu'une relation
dose-réponse claire ressort des études épidémiologiques. Il n'existe
qu'une seule étude épidémiologique qui ait produit des données
suffisantes pour une estimation quantitative de la relation
dose-réponse.
8. Effets sur les autres êtres vivants au laboratoire et dans leur
milieu naturel
On manque des données toxicologiques habituelles au sujet de la
survie et de la reproduction des organismes aquatiques exposés au
chlorure de vinyle. Il faut interpréter avec prudence les données
disponibles car pour la plupart, elles proviennent de tests au cours
desquels on n'a pas mesuré les concentrations auxquelles ces
organismes étaient exposés et il n'a donc pas été tenu compte des
pertes par volatilisation.
La concentration la plus faible de chlorure de vinyle qui
produise un effet sur des microorganismes a été trouvée égale à 40
mg/litre. Il s'agit d'une valeur de la CE50 obtenue lors d'un test
statique de 3,5 jours sur des microorganismes anaérobies pour lequel
le critère retenu était l'inhibition de la respiration.
La concentration la plus faible qui produise un effet sur des
organismes supérieurs a été trouvée égale à 210 mg/litre (CL50 à 48 h
pour un poisson d'eau douce); avec une concentration sans effet nocif
observable (NOAEC) de 128 mg/litre. Chez d'autres espèces, on a
constaté des effets à plus faible concentration, mais la portée
écologique de ces effets n'a pas été vérifiée.
On peut prévoir que la concentration de chlorure de vinyle sans
effet nocif pour les poissons d'eau douce se situe entre 0,088 et
29 mg/litre.
On manque de données concernant des effets du chlorure de vinyle
sur les organismes terrestres.
RESUMEN
Esta monografía se ocupa exclusivamente del cloruro de vinilo
(VC) como monómero y no es una evaluación del cloruro de polivinilo
(PVC), polímero del VC. No se tratan las exposiciones a mezclas con
VC.
1. Identidad, propiedades físicas y químicas y métodos analíticos
En condiciones normales, el VC es un gas incoloro, inflamable,
con un olor ligeramente dulce. Tiene una presión de vapor alta, un
valor elevado para la constante de la Ley de Henry y una solubilidad
en agua relativamente baja. Es más pesado que el aire y soluble en
casi todos los disolventes orgánicos. Se transporta en forma líquida
bajo presión.
A temperatura ambiente en ausencia de aire, el VC purificado seco
es muy estable y no corrosivo, pero por encima de 450°C, o en
presencia de hidróxido sódico o potásico, se puede producir una
descomposición parcial. La combustión del VC en el aire produce
anhídrido carbónico y cloruro de hidrógeno. En presencia de aire y de
oxígeno, se pueden formar peróxidos muy explosivos, por lo que hay que
mantener una vigilancia constante y limitar el contenido de oxígeno,
en particular en las plantas de recuperación de VC. En presencia de
agua se forma ácido clorhídrico.
Desde el punto de vista industrial, las reacciones de
polimerización para obtener PVC son técnicamente las más importantes,
aunque también lo son las reacciones de adición con otros halógenos en
el doble enlace, por ejemplo, para obtener 1,1,2-tricloroetano o
1,1-dicloroetano.
La concentración de VC en el aire se puede vigilar mediante su
retención en adsorbentes y, tras la desorción líquida o térmica, el
análisis por cromatografía de gases. En las mediciones del aire
ambiente, se pueden necesitar varios adsorbentes en serie o colectores
refrigerados para aumentar la eficacia de la retención. Las
concentraciones máximas en los lugares de trabajo se pueden medir con
instrumentos de lectura directa, por ejemplo basados en la detección
por FID o la PID. En la vigilancia continua se han utilizado
analizadores de rayos infrarrojos y de cromatografía de
gases/detección de ionización de llama combinados con el registro y
procesamiento de datos. En el análisis del VC en líquidos y sólidos se
utilizan la inyección directa, la extracción y cada vez más las
técnicas del espacio libre superior y de purga y retención. En estas
muestras también se analiza el VC mediante cromatografía de gases,
combinada, por ejemplo, con detectores de ionización de llama o de
espectrometría de masas.
2. Fuentes de exposición humana y ambiental
No se tiene conocimiento de que el VC se produzca de forma
natural, aunque se ha encontrado en los gases de vertedero y en el
agua freática como producto de la degradación de hidrocarburos
clorados depositados como residuos de disolventes en los vertederos o
en el entorno de lugares de trabajo en los que se utilizan dichos
disolventes. El VC también está presente en el humo de los
cigarrillos.
La producción industrial de VC se lleva a cabo mediante dos
reacciones principales: a) hidrocloración del acetileno; y b)
descomposición térmica (a unos 500°C) del 1,2-dicloroetano producido
mediante cloración directa (etileno y cloro) o la oxicloración
(etileno, ClH y aire/O2) de etileno en el "proceso equilibrado". En
la actualidad se utiliza más el segundo proceso.
La producción mundial de PVC (y por consiguiente de VC) en 1998
fue de unos 27 millones de toneladas. El PVC representa el 20% del
material plástico utilizado y se emplea en la mayoría de los sectores
industriales. Alrededor del 95% de la producción mundial de VC se
utiliza para la fabricación de PVC. El resto se destina a la
producción de disolventes clorados, fundamentalmente de
1,1,1-tricloroetano (10 000 toneladas/año).
Son tres los procesos principales que se utilizan en la
fabricación comercial de PVC: suspensión (equivalente al 80% de la
producción mundial), emulsión (12%) y en masa o a granel (8%). La
mayoría de los estudios monográficos en los que se describen efectos
adversos del VC se refieren a instalaciones que utilizan el proceso de
suspensión (llamado también de dispersión).
Hay varios informes de liberación de VC a causa de accidentes en
instalaciones de fabricación de PVC o durante el transporte. En
numerosos países se ha introducido la recuperación del VC no
convertido residual procedente de la polimerización y de otras fuentes
del proceso, como por ejemplo los efluentes de gases residuales y de
agua. Cuando no se toman precauciones especiales, se puede detectar VC
en resinas y productos de PVC.
La concentración residual de VC en el PVC está reglamentada en
muchos países desde finales de los años setenta. Desde entonces, la
liberación de VC a partir de la degradación térmica del PVC no es
detectable o se produce a niveles muy bajos.
En la producción de VC se pueden formar dioxinas como
contaminantes. Las concentraciones de dioxinas liberadas al medio
ambiente son un tema polémico.
3. Transporte, distribución y transformación en el medio ambiente
Debido a su alta presión de vapor, cabe esperar que el VC que se
libera a la atmósfera se mantenga casi totalmente en la fase de vapor.
Hay indicios de deposición húmeda.
La solubilidad del VC en agua es relativamente baja y su
capacidad de adsorción a la materia particulada y los sedimentos
escasa. La volatilización es el proceso más rápido de eliminación del
VC que se incorpora al agua superficial. Se han notificado semividas
para la volatilización del agua superficial que oscilan entre
alrededor de una y 40 horas.
Las semividas de volatilización a partir del suelo se calcularon
en 0,2-0,5 días. Las pérdidas estimadas de VC (tras un año bajo una
cubierta de suelo de 1 m) oscilaron entre el 0,1% y el 45%, en función
del tipo de suelo. Los coeficientes de sorción en el suelo estimados a
partir de datos fisicoquímicos indican un potencial escaso y, por
consiguiente, una movilidad alta en el suelo. Otra vía importante de
distribución es la lixiviación a través del suelo hacia el agua
freática, donde el VC puede persistir durante años.
En experimentos de laboratorio con organismos acuáticos se
observó una cierta bioacumulación, pero no hubo bioamplificación en la
cadena alimentaria.
Salvo en un pequeño número de excepciones, las agrupaciones
microbianas no adaptadas no degradan fácilmente el VC en condiciones
normales. Se estimó que las semividas de biodegradación máxima sin
adaptación del VC eran del orden de varios meses o años. Sin embargo,
los cultivos enriquecidos especiales o puros (por ejemplo,
Mycobacterium spp.) son capaces de degradar el CV en condiciones de
cultivo óptimas. Los productos principales de degradación fueron el
ácido glicólico o el anhídrido carbónico tras la conversión aerobia y
etano, eteno, metano o clorometano mediante transformación anaerobia.
Con frecuencia, la reacción de degradación del CV es más rápida por
vía aerobia que anaerobia.
El proceso predominante de transformación en la atmósfera es la
reacción con radicales OH producidos por vía fotoquímica, dando lugar
a semividas en la troposfera estimadas en 1-4 días. Durante las
reacciones experimentales de fotolisis se generan varios compuestos
críticos, como cloroacetaldehído, formaldehído y cloruro de formilo.
Se considera que las relaciones fotolíticas, así como la
hidrólisis química, tienen escasa importancia en los medios acuosos.
Sin embargo, la presencia de fotosensibilizadores puede potenciar la
transformación del VC.
Hay indicios de que el VC reacciona con el cloro o el cloruro
utilizado en la desinfección del agua, produciendo de esta manera
cloroacetaldehído y otros compuestos no deseados. Otra posibilidad de
interacción es con las sales, muchas de las cuales tienen la capacidad
de formar complejos con el VC, aumentando tal vez su solubilidad.
Los métodos utilizados (con diferente éxito) para la eliminación
del VC de las aguas contaminadas son la separación, la extracción, la
adsorción y la oxidación. Algunas técnicas de biocorrección in situ
(para el agua freática o el suelo) combinan la evaporación y otros
métodos con el tratamiento microbiano. El VC de los gases de desecho
se puede reciclar, incinerar o degradar por medios microbiológicos. La
mayor parte del CV de producción industrial se encuentra en los
artículos de PVC. Con su incineración se corre el riesgo de que se
formen dibenzodioxinas policloradas/dibenzofuranos policlorados y
otros compuestos orgánicos clorados perjudiciales.
4. Niveles medioambientales y exposición humana
La exposición de la población general al VC es muy pequeña.
Las concentraciones atmosféricas de VC en el aire ambiente son
bajas, normalmente inferiores a 3 µg/m3. La exposición de la
población general puede ser mayor en situaciones en las cuales se haya
producido una liberación accidental de grandes cantidades de VC a la
atmósfera, por ejemplo por un escape durante el transporte. Sin
embargo, esta exposición probablemente es transitoria. Cerca de zonas
industriales y de eliminación de desechos de VC/PVC se han registrado
concentraciones mucho más altas (hasta 8000 µg/m3 y 100 µg/m3,
respectivamente).
Las concentraciones en el aire de los espacios cerrados en casas
adyacentes a vertederos alcanzaron concentraciones máximas de
1000 µg/m3.
La vía más importante de exposición ocupacional es la inhalación
y se produce fundamentalmente en instalaciones de VC/PVC. La
exposición ocupacional al VC ascendió a varios miles de mg/m3 en los
años cuarenta y cincuenta y fue de varios cientos de mg/m3 en los
sesenta y comienzo de los setenta. Tras el reconocimiento del peligro
carcinogénico del VC, en los años setenta se establecieron en la
mayoría de los países normas de exposición ocupacional de alrededor de
13-26 mg/m3 (5-10 ppm). El cumplimiento de estas directrices ha
reducido considerablemente las concentraciones de VC en los lugares de
trabajo, pero incluso en los años noventa se han notificado
concentraciones más altas, que se pueden encontrar todavía en algunos
países.
Ocasionalmente se ha detectado VC en aguas superficiales,
sedimento y fangos cloacales, con máximos de 570 µg/litro, 580 µg/kg y
62 000 µg/litro, respectivamente. Las muestras de suelo recogidas
cerca de una tienda de productos químicos de limpieza abandonada
contenían concentraciones muy elevadas de VC (hasta 900 mg/kg). Las
concentraciones máximas de VC en el agua freática o lixiviada de zonas
contaminadas por hidrocarburos clorados ascendieron a 60 000 µg/litro
(o más). Se detectaron concentraciones altas (hasta 200 mg/litro) en
el agua de un pozo cercano a una instalación de PVC 10 años después de
las filtraciones.
Los escasos datos disponibles ponen de manifiesto que el VC puede
estar presente en los tejidos de pequeños invertebrados acuáticos y de
peces.
En la mayoría de las muestras de agua de bebida analizadas, no
había VC presente en concentraciones detectables. La concentración
máxima de VC notificada en agua de bebida tratada fue de 10 µg/litro.
No se dispone de datos recientes sobre las concentraciones de VC en el
agua de bebida, pero cabe prever que sean inferiores a 10 µg/litro. Si
se utiliza agua contaminada como fuente de agua de bebida, se podrían
producir exposiciones más elevadas. En algunos estudios recientes se
ha identificado VC en agua de bebida embotellada en envases de PVC en
concentraciones inferiores a 1 µg/litro. Probablemente sea más
frecuente la presencia de VC en este tipo de agua que en la de grifo.
El envasado con ciertos materiales de PVC puede producir la
contaminación por CV de productos alimenticios, farmacéuticos o
cosméticos, incluso licores (hasta 20 mg/kg), aceites vegetales (hasta
18 mg/kg), vinagres (hasta 9,8 mg/kg) y colutorios (hasta 7,9 mg/kg).
Gracias a las medidas legislativas adoptadas por numerosos países,
desde comienzos de los años setenta se ha logrado una reducción
significativa de las concentraciones de VC y/o en el número de
muestras positivas.
Varios organismos han calculado la exposición al VC a través de
los envases de PVC utilizados para productos alimenticios y, teniendo
cuenta el promedio de ingesta estimado en el Reino Unido y en los
Estados Unidos, se calculó una exposición < 0,0004 µg/kg para finales
de los años setenta y comienzos de los ochenta. En un estudio inicial
se identificó VC en el humo del tabaco en concentraciones del orden de
ng/cigarrillo.
5. Cinética y metabolismo en animales de laboratorio y en el ser
humano
Tras las exposición por inhalación o por vía oral, el VC se
absorbe con rapidez y facilidad. La vía primaria de exposición al VC
es la inhalación. En estudios realizados con animales y con personas
en condiciones estables, después de la exposición por inhalación se
absorbe aproximadamente el 40% del VC inspirado. En estudios de
exposición oral con animales se observó una absorción de más del 95%.
La absorción cutánea al VC en estado gaseoso no es significativa.
Los datos obtenidos en estudios de administración oral y por
inhalación en ratas indican una distribución rápida y generalizada del
VC. La rapidez del metabolismo y la excreción limita la acumulación de
VC en el organismo. En las ratas se produce un desplazamiento rápido
del VC a través de la placenta. No se ha informado de estudios sobre
distribución tras la exposición cutánea.
La principal ruta de metabolismo del CV después de la inhalación
o la ingestión oral consiste en la oxidación por el citocromo P-450
(CYP2E1) para formar óxido de cloroetileno (CEO), epóxido muy reactivo
de vida breve que reacciona de nuevo rápidamente para formar
cloroacetaldehído (CAA). La reacción primaria de desintoxicación de
estos dos metabolitos reactivos, así como del ácido cloroacético,
producto de la deshidrogenación del CAA, es la conjugación con el
glutatión en una reacción catalizada por la glutatión- S-transferasa.
Los productos de la conjugación sufren ulteriores modificaciones para
formar derivados de la cisteína con sustituciones
(S-(2-hidroxietil)-cisteína, N-acetil- S-(2-hidroxietil)
cisteína, S-carboximetil cisteína y ácido tiodiglicólico) y se
excretan en la orina. Otro metabolito, el anhídrido carbónico, se
exhala en el aire.
Se sabe que el CYP2E1 y las isoenzimas de la
glutatión-S-transferasa tienen variaciones individuales importantes de
actividad interespecíficas e intraespecíficas.
Tras la exposición por inhalación o por vía oral a dosis bajas,
el CV se elimina metabólicamente y los metabolitos no volátiles se
excretan fundamentalmente en la orina. En investigaciones comparativas
de la absorción de VC por inhalación se puso de manifiesto una
velocidad de eliminación metabólica en el ser humano menor que en los
animales de laboratorio, en función del peso corporal. Sin embargo,
una vez corregida con arreglo a la superficie corporal, la eliminación
metabólica del VC en el ser humano es comparable a la observada en
otras especies de mamíferos. Al aumentar la exposición oral o por
inhalación, la vía más importante de excreción en los animales es la
exhalación de VC inalterado, lo cual indica que hay una saturación de
las rutas metabólicas. Con independencia de la dosis aplicada, la
excreción de metabolitos por las heces es sólo una vía secundaria. No
se encontraron estudios en los que se investigase específicamente la
excreción por la bilis.
Se considera que el CEO es el metabolito más importante in vivo
con respecto a los efectos mutagénicos y carcinogénicos del VC. El
CEO reacciona con el ADN para producir el aducto principal
7-(2'-oxoetil)guanina (7-OEG) y, a niveles más bajos, los aductos
exocíclicos de eteno, 1, N 6-etenoadenina (Epsilon A),
3, N4-etenocitosina (Epsilon C) y N 2,3-etenoguanina (Epsilon G).
Los aductos de eteno del ADN tienen propiedades promutagénicas, a
diferencia del aducto principal 7-OEG. Se han medido las
concentraciones de 7-OEG, Epsilon A, Epsilon C y Epsilon G en diversos
tejidos de roedores expuestos al VC. Se han creado modelos
toxicocinéticos con una base fisiológica para describir la relación
entre la concentración en el tejido destinatario y los efectos finales
tóxicos del VC.
6. Efectos en mamíferos de laboratorio y en sistemas de prueba
in vitro
La toxicidad aguda del VC administrado por inhalación a diversas
especies parece ser baja. Se notificaron CL50 a las 2 horas para
ratas, ratones, cobayas y conejos de 390 000, 293 000, 595 000 y
295 000 mg/m3, respectivamente. No hay datos disponibles sobre la
toxicidad aguda tras la administración oral o la aplicación cutánea.
El VC tiene un efecto estupefaciente después de la administración
aguda por inhalación. En ratas, ratones y hámsteres, la muerte estuvo
precedida por un aumento de la actividad motora, ataxia y
convulsiones, y a continuación colapso respiratorio. En perros se
produjo una arritmia cardíaca grave en estado de narcosis tras la
exposición por inhalación a 260 000 mg/m3. Después de la exposición
aguda de ratas al VC por inhalación se observaron diversos efectos
patológicos, entre ellos congestión de los órganos internos, sobre
todo los pulmones, el hígado y los riñones, así como edema pulmonar.
No hay estudios o datos importantes disponibles para la
evaluación de los efectos de la exposición cutánea, la irritación de
la piel o la propiedad de sensibilización del VC.
De la exposición oral de ratas al VC durante un período breve de
13 semanas se obtuvo una concentración sin efectos observados (NOEL),
basada en el aumento de peso del hígado, de 30 mg/kg.
En diversas especies, el principal órgano destinatario en la
exposición breve (hasta 6 meses) al VC por inhalación fue el hígado.
Con una concentración de 26 mg/m3 (la dosis más baja utilizada) se
observaron en ratas un aumento del peso relativo del hígado y cambios
hepatocelulares; a concentraciones superiores (> 260 mg/m3) se
produjeron también cambios hepáticos más acentuados dependientes de la
dosis. Otros órganos destinatarios fueron los riñones, los pulmones y
los testículos. Las ratas, los ratones y los conejos parecen ser más
sensibles que los cobayas y los perros.
La exposición prolongada al VC por inhalación produjo un aumento
estadísticamente significativo de la mortalidad en algunas estirpes de
ratas con concentraciones de sólo 260 mg/m3, en ratones con 130
mg/m3 y en hámsteres con 520 mg/m3 para diversos períodos de
exposición. Las ratas expuestas a 130 mg/m3 mostraron una reducción
del peso corporal y un aumento del peso relativo del bazo,
degeneración hepatocelular y proliferación de las células de
revestimiento de los sinusoides del hígado. La exposición de ratas a
concentraciones más elevadas produjo una alteración degenerativa de
los testículos, nefrosis tubular y degeneración focal del miocardio.
En ratas y ratones expuestos por inhalación, la concentración sin
efectos adversos observados (NOAEL) para los efectos no neoplásicos es
inferior a 130 mg/m3.
En estudios de alimentación crónica se puso de manifiesto un
aumento de la mortalidad, un peso mayor del hígado y una alteración
morfológica del hígado.
Tras la exposición oral de ratas a concentraciones de sólo
1,3 mg/kg de peso corporal se pudo observar polimorfismo de las
células hepáticas (variación del tamaño y la forma de los hepatocitos
y sus núcleos). La NOAEL fue de 0,13 mg/kg de peso corporal.
En estudios prolongados de alimentación realizados en ratas con
gránulos de VC y PVC se produjo un aumento significativo de la
incidencia del angiosarcoma hepático (ASH) con 5,0 mg/kg de peso
corporal al día y nódulos neoplásicos en el hígado (hembras) y
carcinoma hepatocelular (CHC) (machos) con 1,3 mg/kg de peso corporal
al día.
En estudios de inhalación de VC en ratas Sprague-Dawley se
observó una relación dosis-respuesta en el caso del ASH y, a
concentraciones más altas, carcinoma de las glándulas de Zymbal. No se
observó una dependencia clara de la dosis para el hepatoma o el
angiosarcoma extrahepático, los nefroblastomas, los neuroblastomas o
los tumores mamarios malignos. En ratones, el espectro de tumores
inducidos por la exposición prolongada mediante inhalación es
semejante al observado en ratas, pero se detectó sólo en ratones un
aumento de los tumores pulmonares. En hámsteres se notificó un aumento
de la incidencia de tumores de ASH, tumores de las glándulas mamarias
y el conducto acústico, melanomas, tumores de estómago y del epitelio
cutáneo.
Se han detectado efectos mutagénicos y genotóxicos del VC en
varios sistemas de prueba in vitro, fundamentalmente después de la
activación metabólica. El VC tiene un efecto mutagénico en la prueba
de Ames con las cepas TA100, TA1530 y TA1535 de S. typhimurium, pero
no con las cepas TA98, TA1537 y TA1538, lo cual indica que las
mutaciones son el resultado de la sustitución de pares de bases
(transversión y transición) más que de mutaciones por desfase. Esto
está en consonancia con el resultado de que los aductos de eteno del
ADN formados por los metabolitos reactivos CEO y CAA se convierten en
mutaciones reales mediante las sustituciones de pares de bases.
Otras valoraciones de mutaciones genéticas en bacterias,
levaduras y células de mamíferos han puesto de manifiesto resultados
positivos exclusivamente en presencia de una activación metabólica. Se
notificaron asimismo efectos mutagénicos en una línea de células
humanas con citocromo P-450IIE1 clonado, que es capaz de metabolizar
el VC. También se detectó una mutación genética en esquejes de plantas
(Tradescantia) expuestos al VC. En las valoraciones de la conversión
genética, se notificaron resultados positivos con Saccharomyces
cerevisiae en presencia de un sistema de activación metabólica. La
exposición al VC indujo síntesis no programada de ADN en hepatocitos
de rata y un aumento del intercambio de crómatidas hermanas en
linfocitos humanos tras la adición del sistema de activación exógeno.
No se detectó inhibición del crecimiento en bacterias deficientes en
enzimas de reparación del ADN sin activación metabólica. En
valoraciones de la transformación celular se obtuvieron resultados
positivos con activación metabólica y sin ella.
La exposición al VC indujo mutaciones genéticas y recombinación
mitótica en Drosophila melanogaster, pero no produjo ninguna
mutación genética en células germinales de mamíferos. El VC mostró
efectos clastogénicos en roedores, aumentó el intercambio de
cromátidas hermanas de hámster e indujo el fraccionamiento del ADN en
ratones. En valoraciones mediadas por huéspedes (ratas), el VC indujo
una conversión genética y mutaciones adaptativas en levaduras.
Se observó un efecto mutagénico del CEO y el CAA en diferentes
sistemas de prueba. El CEO es un mutágeno potente, mientras que el CAA
es muy tóxico. Ambos mostraron efectos carcinogénicos en ratones,
siendo el CEO mucho más activo que el CAA.
Se analizaron las mutaciones de los genes ras y p53 en
tumores de hígado inducidos por el VC en ratas Sprague-Dawley: se
encontraron sustiticiones de pares de bases en el gen Ha- ras en el
carcinoma hepatocelular y en el gen p-53 en el ASH. Estas mutaciones
coinciden con la formación observada y la persistencia de aductos de
eteno en el ADN del hígado, tras la exposición de ratas a VC, y con
las propiedades promutagénicas conocidas de los aductos de eteno.
Los estudios sobre los mecanismos de la carcinogenicidad del VC
parecen indicar que el epóxido intermedio reactivo CEO tiene una
interacción con el ADN para formar aductos de eteno, que dan lugar a
una sustitución de pares de bases que lleva a una transformación
neoplásica.
7. Efectos en el ser humano
Se ha notificado que concentraciones de VC del orden de
2590 mg/m3 (1000 ppm), que no eran raras antes de 1974, durante
periodos comprendidos entre un mes y varios años provocaban un
síndrome patológico específico observado en los trabajadores del VC,
llamado "enfermedad del cloruro de vinilo". Los síntomas descritos
fueron dolor de oídos y de cabeza, vértigo, visión borrosa, cansancio
y falta de apetito, náuseas, insomnio, dificultad respiratoria, dolor
de estómago, dolor en la zona del hígado/bazo, dolor y sensación de
hormigueo en los brazos y las piernas, sensación de frío en las
extremidades, pérdida de la libido y disminución del peso. Entre los
resultados clínicos figuraron cambios en los dedos del tipo del
escleroderma, con modificaciones óseas posteriores en la punta de los
dedos descritas como acroosteólisis, cambios en la circulación
periférica idénticos a los clásicos de la enfermedad de Raynaud y
agrandamiento del hígado y del bazo con una aspecto histológico
específico, así como manifestaciones respiratorias.
Los estudios en seres humanos no han sido adecuados para
confirmar los efectos en el sistema reproductor. En un pequeño número
de estudios de morbilidad se ha notificado una elevada incidencia de
enfermedades circulatorias entre los trabajadores relacionados con el
cloruro de vinilo. Sin embargo, en estudios de cohortes amplios se ha
observado una mortalidad más baja que la debida a enfermedades
cardiovasculares.
Hay pruebas manifiestas y convincentes obtenidas de estudios
epidemiológicos de que la exposición al VC produce un tumor raro, el
angiosarcoma hepático. También pueden asociarse con el VC casos de
tumores cerebrales y carcinoma hepatocelular, aunque las pruebas no
pueden considerarse definitivas. Otros puntos notificados como de una
mayor incidencia de cáncer, pero de manera menos convincente, son el
pulmón, los tejidos linfático y hematopoyético y la piel.
El VC es mutagénico y clastogénico en el ser humano. Se ha
observado un aumento de la frecuencia de aberraciones cromosómicas,
micronúcleos e intercambio de cromátidas hermanas en los linfocitos de
la sangre periférica de los trabajadores expuestos a concentraciones
elevadas de VC en comparación con los testigos. Aunque en muchos
estudios solamente se estimaron las concentraciones y la duración de
la exposición, se puede observar una relación dosis-respuesta y la
"normalización" de los efectos genotóxicos con el paso del tiempo
después de la reducción de la exposición.
Se han detectado mutaciones puntuales en los genes p 53 y ras
en tumores de personas que trabajaban con autoclaves y que estaban
muy expuestas (antes de 1974) afectadas de angiosarcoma hepático y en
otro trabajador relacionado con el CV con carcinoma hepatocelular.
Los marcadores biológicos investigados como indicadores de la
exposición al VC o de los efectos inducidos por el VC son los
siguientes: a) excreción de metabolitos del VC (por ejemplo, ácido
tiodiglicólico), b) valoraciones genéticas (por ejemplo, anomalías
cromosómicas o valoración de micronúcleos), c) concentraciones de
enzimas (por ejemplo, en pruebas de la función hepática), d)
oncoproteínas séricas (p21 y p53) y/o sus anticuerpos como
biomarcadores de los efectos inducidos por el VC.
Los niños que viven en lugares cercanos a vertederos y otras
fuentes puntuales pueden correr un riesgo mayor, tomando como base las
pruebas que parecen derivarse de la sensibilidad en las primeras fases
de la vida en estudios realizados con animales. Sin embargo, no hay
pruebas directas en el ser humano.
En los estudios epidemiológicos solamente hay una relación
dosis-respuesta clara para el angiocarcinoma hepático solo o en
combinación con otros tumores del hígado. Sólo en un estudio
epidemiológico hay datos suficientes para una estimación cuantitativa
de la relación dosis-respuesta.
8. Efectos en otros organismos en el laboratorio y en el medio
ambiente
Se carece de datos normalizados de toxicidad relativos a la
supervivencia y la reproducción de los organismos acuáticos expuestos
al VC. Hay que interpretar con cautela los datos disponibles, porque
la mayoría de ellos se obtuvieron en pruebas en las cuales no se midió
la concentración de la exposición, por lo que no se tuvieron en cuenta
las pérdidas debidas a la volatilización.
La concentración más baja de VC que produjo un efecto en los
microorganismos fue de 40 mg/litro. Fue un valor de la CE50 basado en
la inhibición de la respiración de microorganismos anaerobios en la
valoración de un lote durante 3,5 días.
La concentración más baja que produjo un efecto en organismos
superiores fue de 210 mg/litro (CL50 a las 48 horas para un pez de
agua dulce), con una concentración sin efectos adversos observados
(NOAEC) correspondiente de 128 mg/litro. Se han notificado efectos en
otras especies debidos a concentraciones más bajas de VC, pero no se
comprobó la importancia ecológica de dichos efectos.
Las concentraciones de VC estimadas como no peligrosas para los
peces de agua dulce se calculó que oscilaban entre 0,088 y 29
mg/litro.
Hay pocos datos sobre los efectos del VC en los organismos
terrestres.