
INTERNATIONAL PROGRAMME ON CHEMICAL SAFETY
ENVIRONMENTAL HEALTH CRITERIA 157
HYDROQUINONE
This report contains the collective views of an international group of
experts and does not necessarily represent the decisions or the stated
policy of the United Nations Environment Programme, the International
Labour Organisation, or the World Health Organization.
First draft prepared by Dr M. Gillner, Dr G.S. Moore, Dr H. Cederberg
and Dr K. Gustafsson, National Chemicals Inspectorate, Solna, Sweden
Published under the joint sponsorship of
the United Nations Environment Programme,
the International Labour Organisation,
and the World Health Organization
World Health Orgnization
Geneva, 1994
The International Programme on Chemical Safety (IPCS) is a joint
venture of the United Nations Environment Programme, the International
Labour Organisation, and the World Health Organization. The main
objective of the IPCS is to carry out and disseminate evaluations of
the effects of chemicals on human health and the quality of the
environment. Supporting activities include the development of
epidemiological, experimental laboratory, and risk-assessment methods
that could produce internationally comparable results, and the
development of manpower in the field of toxicology. Other activities
carried out by the IPCS include the development of know-how for coping
with chemical accidents, coordination of laboratory testing and
epidemiological studies, and promotion of research on the mechanisms
of the biological action of chemicals.
WHO Library Cataloguing in Publication Data
Hydroquinone.
Environmental health criteria: 157)
1. Environmental exposure 2. Hydroquinones - analysis
3. Hydroquinones - toxicity I.Series
ISBN 92 4 157127 8 (NLM Classification QD 341.P5)
ISSN 0250-863X
The World Health Organization welcomes requests for permission
to reproduce or translate its publications, in part or in full.
Applications and enquiries should be addressed to the Office of
Publications, World Health Organization, Geneva, Switzerland,
Which will be glad to provide the latest information on any changes
made to the text, plans for new editions, and reprints and
translations already available.
(c) World Health Organization 1994
Publications of the World Health Organization enjoy copyright
protection in accordance with the provisions of Protocol 2 of the
Universal Copyright Convention. All rights reserved.
The designations employed and the presentation of the material in
this publication do not imply the impression of any opinion whatsoever
on the part of the Secretariat of the World Health Organization
concerning the legal status of every country, territory, city, or area
or of its authorities, or concerning the delimitation of its frontiers
or boundaries.
The mention of specific companies or of certain manufacturers'
products does not imply that they are endorsed or recommended by the
World Health Organization in preference to others of a similar nature
that are not mentioned. Errors and omissions excepted, the names of
proprietary products are distinguished by initial capital letters.
CONTENTS
ENVIRONMENTAL HEALTH CRITERIA FOR HYDROQUINONE
1. SUMMARY
1.1. Identity, physical and chemical properties, analytical
methods
1.2. Sources of human and environmental exposure
1.3. Environmental transport, distribution and transformation
1.4. Environmental levels and human exposure
1.5. Kinetics and metabolism
1.6. Effects on laboratory mammals, and in vitro systems
1.7. Effects on humans
1.8. Effects on other organisms in the laboratory and field
2. IDENTITY, PHYSICAL AND CHEMICAL PROPERTIES, ANALYTICAL METHODS
2.1. Identity
2.2. Physical and chemical properties
2.2.1. Reduction-oxidation equilibria
2.2.2. Oxidation of hydroquinone
2.3. Conversion factors
2.4. Analytical methods
2.4.1. Sampling
2.4.2. Methods of analysis
3. SOURCES OF HUMAN AND ENVIRONMENTAL EXPOSURE
3.1. Natural occurrence
3.2. Anthropogenic sources
3.2.1. Production levels and processes
3.2.2. Uses
4. ENVIRONMENTAL TRANSPORT, DISTRIBUTION AND TRANSFORMATION
4.1. Transport and distribution between media
4.2. Transformation
4.2.1. Biodegradation
4.2.2. Abiotic degradation
4.2.3. Bioaccumulation
4.3. Interaction with other physical, chemical or biological
factors
4.4. Ultimate fate following use
5. ENVIRONMENTAL LEVELS AND HUMAN EXPOSURE
5.1. Environmental levels
5.1.1. Air, soil and water
5.1.2. Food
5.2. General population exposure
5.3. Occupational exposure
6. KINETICS AND METABOLISM
6.1. Absorption
6.2. Distribution
6.3. Metabolic transformation
6.4. Elimination and excretion
6.5. Reaction with body components
7. EFFECTS ON LABORATORY MAMMALS AND IN VITRO SYSTEMS
7.1. Single exposure
7.2. Skin and eye irritation; sensitization
7.2.1. Skin irritation
7.2.2. Eye irritation
7.2.3. Sensitization
7.3. Short-term exposure
7.4. Long-term exposure
7.5. Reproduction, embryotoxicity and teratogenicity
7.5.1. Effects on male reproduction
7.5.2. Effects on female reproduction
7.5.3. Embryotoxicity and teratogenicity
7.6. Mutagenicity and related end-points
7.7. Carcinogenicity
7.7.1. Long-term bioassays
7.7.2. Carcinogenicity-related studies
7.7.2.1 Skin
7.7.2.2 Bladder
7.7.2.3 Stomach
7.7.2.4 Liver
7.8. Special studies
7.8.1. Effects on spleen and bone marrow cells;
immunotoxicity
7.8.2. Effects on tumour cells
7.8.3. Neurotoxicity
7.8.4. Nephrotoxicity
7.8.5. Interaction with phenols
8. EFFECTS ON HUMANS
8.1. General population exposure
8.1.1. Acute toxicity - poisoning incidents
8.1.2. Short-term controlled human studies
8.1.3. Dermal effects; sensitization
8.2. Occupational exposure
8.2.1. Dermal effects
8.2.2. Ocular effects
8.2.3. Systemic effects
8.2.4. Epidemiological studies
8.2.4.1 Respiratory effects
8.2.4.2 Carcinogenicity studies
9. EFFECTS ON OTHER ORGANISMS IN THE LABORATORY AND FIELD
10. EVALUATION OF HUMAN HEALTH RISKS AND EFFECTS ON THE ENVIRONMENT
10.1. Toxicokinetics
10.2. Animal and in vitro studies
10.3. Evaluation of human health risks
10.3.1. Exposure
10.3.2. Human health effects
10.4. Evaluation of effects on the environment
11. RECOMMENDATIONS
12. PREVIOUS EVALUATIONS BY INTERNATIONAL BODIES
REFERENCES
APPENDIX
RESUME
RESUMEN
WHO TASK GROUP ON ENVIRONMENTAL HEALTH CRITERIA FOR HYDROQUINONE
Members
Dr L. Albert, Program of Health and Environment, Centre for Ecology
and Development, Xalapa, Veracruz, Mexico (Chairman)
Dr H. Cederberg, National Chemicals Inspectorate, Solna, Sweden
Dr J. Devillers, Centre de Traitement de l'Information Scientifique
(CTIS), Lyon, France
Dr D.A. Eastmond, Environmental Toxicology Graduate Program,
Department of Entomology, University of California, Riverside,
California, USA
Dr M. Gillner, Scientific Documentation and Research, National
Chemicals Inspectorate, Solna, Sweden (Rapporteur)
Dr S. Humphreys, Contaminants, Standards, and Monitoring Branch,
Center for Food Safety and Applied Nutrition, US Food and Drug
Administration, Washington, DC, USA
Dr G.A. Moore, Scientific Documentation and Research, National
Chemicals Inspectorate, Solna, Sweden
Professor H. Naito, Institute of Clinical Medicine, University of
Tsukuba, Tsukuba City, Ibaraki, Japan
Dr C.O. Nwokike, Medical Division, Lever Brothers (Nigeria) PLC,
Apapa, Lagos, Nigeria
Dr J. O'Donoghue, Corporate Health and Environment Laboratories,
Eastman Kodak Company, Rochester, New York, USA
Professor P.N. Viswanathan, Ecotoxicology Group, Industrial
Toxicology Research Centre, Lucknow, India
Observer
Mr P-G. Pontal, Rhône Poulenc Agro, Lyon, France
Secretariat
Dr M. Gilbert, International Programme on Chemical Safety, World
Health Organization, Geneva, Switzerland (Secretary)
Dr J. Wilbourn, Unit of Carcinogen Identification and Evaluation,
International Agency for Research on Cancer, Lyon, France
NOTE TO READERS OF THE CRITERIA MONOGRAPHS
Every effort has been made to present information in the
criteria monographs as accurately as possible without unduly
delaying their publication. In the interest of all users of the
Environmental Health Criteria monographs, readers are kindly
requested to communicate any errors that may have occurred to the
Director of the International Programme on Chemical Safety, World
Health Organization, Geneva, Switzerland, in order that they may be
included in corrigenda.
* * *
A detailed data profile and a legal file can be obtained from
the International Register of Potentially Toxic Chemicals, Case
postale 356, 1219 Châtelaine, Geneva, Switzerland (Telephone No.
9799111).
* * *
This publication was made possible by grant number 5 U01
ES02617-14 from the National Institute of Environmental Health
Sciences, National Institutes of Health, USA.
ENVIRONMENTAL HEALTH CRITERIA FOR HYDROQUINONE
A WHO Task Group meeting on Environmental Health Criteria for
Hydroquinone was held at the British Industrial Biological Research
Association (BIBRA), Carshalton, United Kingdom, from 24 to 28 May
1993. Dr D. Anderson welcomed the participants on behalf of the host
institution and Dr M. Gilbert opened the meeting on behalf of the
three cooperating organizations of the IPCS (ILO/UNEP/WHO). The Task
Group reviewed and revised the draft criteria monograph and made an
evaluation of the risks for human health and the environment from
exposure to hydroquinone.
The first draft of this monograph was prepared by Dr M.
Gillner, Dr G.A. Moore, Dr H. Cederberg and Dr K. Gustafsson,
National Chemicals Inspectorate, Solna, Sweden. Dr M. Gilbert and
Dr P.G. Jenkins, both members of the IPCS Central Unit, were
responsible for the overall scientific content and editing,
respectively.
The efforts of all who helped in the preparation and
finalization of the monograph are gratefully acknowledged.
ABBREVIATIONS
AUC area under the curve
BP benzo [a]pyrene
BHA butylated hydroxyanisole
cAMP adenosine 3',5'-phosphate
cGMP guanine 3',5'-phosphate
CLV ceiling value
HPLC high-performance liquid chromatography
HQ hydroquinone
IL-1 interleukin-1
IL-4 interleukin-4
i.p. intraperitoneal
i.v. intravenous
MDA malondialdehyde
MCL melanotic cell lines
NADPH reduced nicotinamide adenine dinucleotide
NMCL nonmelanolic cell lines
MNNG N-methyl- N'-nitro- N-nitrosoguanidine
NOAEL no-observable-adverse-effect level
NOEL no-observed-effect level
ODC ornithine decarboxylase
QSAR quantitative structure-activity relationship
s.c. subcutaneous
STEL short-term exposure limit
TLC thin-layer chromatography
TLV threshold limit value
TWA time-weighted average
1. SUMMARY
1.1 Identity, physical and chemical properties, analytical methods
Hydroquinone (1,4-benzenediol; C6H4(OH)2) is a white
crystalline substance when pure, with a melting point of 173-174 °C.
The specific gravity is 1.332 at 15 °C, and the vapour pressure is
2.4 x 10-3 Pa (1.8 x 10-5 mmHg) at 25 °C. It is highly soluble
in water (70 g/litre at 25 °C) and the log n-octanol/water
partition coefficient is 0.59. With respect to organic solvents, the
solubility varies from 57% in ethanol to less than 0.1% in benzene.
Hydroquinone is combustible when preheated. It is a reducing agent
which is reversibly oxidized to its semiquinone and quinone.
Hydroquinone in the air is sampled either by trapping in
solvent or on a mixed cellulose ester membrane filter.
Analysis of hydroquinone is carried out by titrimetric,
spectrophotometric or, most commonly, chromatographic techniques.
1.2 Sources of human and environmental exposure
Hydroquinone occurs both in free and conjugated forms in
bacteria, plants and some animals. Industrially, it is produced in
several countries. In 1979, the total world capacity for production
exceeded 40 000 tonnes, while in 1992 it was approximately 35 000
tonnes. It is extensively used as a reducing agent, as a
photographic developer, as an antioxidant or stabilizer for certain
materials that polymerize in the presence of free radicals, and as a
chemical intermediate for the production of antioxidants,
antiozonants, agrochemicals and polymers. Hydroquinone is also used
in cosmetics and medical preparations.
1.3 Environmental transport, distribution and transformation
Hydroquinone occurs in the environment as a result of man-made
processes as well as in natural products from plants and animals.
Due to its physicochemical properties, hydroquinone will be
distributed mainly to the water compartment when released into the
environment. It degrades both as a result of photochemical and
biological processes; consequently, it does not persist in the
environment. No bioaccumulation is observed.
1.4 Environmental levels and human exposure
No data on hydroquinone concentrations in air, soil or water
have been found. However, hydroquinone has been measured in
mainstream smoke from non-filter cigarettes in amounts varying from
110 to 300 µg per cigarette, and also in sidestream smoke.
Hydroquinone has been found in plant-derived food products (e.g.,
wheat germ), in brewed coffee, and in teas prepared from the leaves
of some berries where the concentration sometimes exceeds 1%.
Photohobbyists can be exposed to hydroquinone dermally or by
inhalation. However no data on exposure levels are available. Dermal
exposure may also result from the use of cosmetic and medical
products containing hydroquinone, such as skin lighteners. The
European Community (EC) countries have restricted its use in
cosmetics to 2% or less. In the USA, the Food and Drug
Administration has proposed concentrations between 1.5 and 2% in
skin lighteners. Concentrations up to 4% may be found in
prescription drugs. In some countries even higher concentrations may
be found in skin lighteners.
Few industrial hygiene monitoring data are available for
hydroquinone. Average concentrations in air during manufacturing and
processing of hydroquinone have been reported to be in the range of
0.13 to 0.79 mg/m3. Occupational air exposure limits
(time-weighted average) in different countries range from 0.5 to 2
mg/m3.
1.5 Kinetics and metabolism
Hydroquinone is rapidly and extensively absorbed from the gut
and trachea of animals. Absorption via the skin is slower but may be
more rapid with vehicles such as alcohols. Hydroquinone distributes
rapidly and widely among tissues. It is metabolized to
p-benzoquinone and other oxidized products, and is detoxified by
conjugation to monoglucuronide, monosulfate, and mercapturic
derivatives. The excretion of hydroquinone and its metabolites is
rapid, and occurs primarily via the urine.
Hydroquinone and/or its derivatives react with different
biological components such as macromolecules and low molecular
weight molecules, and they have effects on cellular metabolism.
1.6 Effects on laboratory mammals, and in vitro systems
Oral LD50 values for several animal species range between 300
and 1300 mg/kg body weight. However, for the cat LD50 values range
from 42 to 86 mg/kg body weight. Acute high-level exposure to
hydroquinone causes severe effects on the central nervous system
(CNS) including hyperexcitability, tremor, convulsions, coma and
death. At sublethal doses these effects are reversible. The dermal
LD50 value has been estimated to be > 3800 mg/kg in rodents.
Inhalation LC50 values are not available.
A formulation containing 2% hydroquinone in a single-insult
patch test in rabbits resulted in an irritation score of 1.22 (on a
scale of 0 to 4). Daily topical applications for three weeks of 2 or
5% hydroquinone in an oil-water emulsion on the depilated skin of
black guinea-pigs caused depigmentation, inflammatory changes and
thickening of the epidermis. The depigmentation was more marked at
higher concentrations, and female guinea-pigs were more sensitive
than males.
Sensitization tests in guinea-pigs have shown weak to strong
reactions depending on the methods or vehicles used. The strongest
reactions were obtained with the guinea-pig maximization test. A
cross-sensitization of almost 100% between hydroquinone and
p-methoxyphenol was also seen in guinea-pigs, but only restricted
evidence of cross-reactions to p-phenylenediamine, sulfanilic acid
and p-benzoquinone was obtained.
A 6-week oral toxicity study in male F-344 rats resulted in
nephropathy and renal cell proliferation. Thirteen-week oral gavage
studies in F-344 rats and in B6C3F1 mice resulted in
nephrotoxicity in rats at 100 and 200 mg/kg, and tremors and
convulsions in rats at 200 mg/kg; reduced body weight gain was seen
in both rats and mice. Dosing at 400 mg/kg was lethal in rats. In
mice dosed for 13 weeks at 400 mg/kg, tremors, convulsions and
lesions in the gastric epithelium were reported. Thirteen-week
hydroquinone exposure of Sprague Dawley rats resulted in decreased
body weight gain and CNS signs at 200 mg/kg. CNS signs were also
observed at a dose level of 64 mg/kg body weight but not at 20
mg/kg.
Hydroquinone injected subcutaneously reduced fertility in male
rats, and prolonged the estrus cycle in female rats. However, this
was not found in oral studies (a dominant lethality study and a
two-generation study). In a developmental study in rats, oral doses
of 300 mg/kg body weight caused slight maternal toxicity and reduced
fetal body weight. In rabbits, the no-observed-effect level (NOEL)
for maternal toxicity was 25 mg/kg per day, and it was 75 mg/kg per
day for developmental toxicity. In a two-generation reproduction
study in rats hydroquinone caused no reproductive effects at oral
doses of up to 150 mg/kg body weight per day. The no-observed-
adverse-effect level (NOAEL) for parental toxicity was determined to
be 15 mg/kg per day, and for reproductive effects through two
generations it was 150 mg/kg per day.
Hydroquinone induces micronuclei in vivo and in vitro.
Structural and numerical chromosome aberrations have been observed
in vitro and after intraperitoneal administration in vivo.
Furthermore, the induction of gene mutations, sister-chromatid
exchange and DNA damage has been demonstrated in vitro.
Hydroquinone caused chromosomal aberrations in male mouse germ cells
at the same order of magnitude as in mouse bone marrow cells after
intraperitoneal injection. Induction of germ-cell mutations could
not be established in a dominant lethal test in male rats dosed
orally.
In a two-year study, oral administration of hydroquinone caused
a dose-related incidence of renal tubular cell adenomas in male
F-344/N rats. The incidence was statistically significant in the
high-dose group. In the high-dose males, renal tubular cell
hyperplasia was also found. In female rats a dose-related increased
incidence of mononuclear cell leukaemia occurred. Female B6C3F1
mice developed a significantly increased incidence of hepatocellular
adenomas. In another study, hydroquinone (at a dietary level of
0.8%) produced a significantly increased incidence of epithelial
hyperplasia of the renal papilla and a significant increase of renal
tubular hyperplasia and adenomas in male rats. No increased
incidence of mononuclear cell leukaemia in female rats was observed.
In mice, the incidence of squamous cell hyperplasia of the
forestomach epithelium was significantly increased in both sexes. In
male mice, there was a significantly increased incidence of
hepatocellular adenomas and also of renal tubular hyperplasia. A few
renal cell adenomas were observed.
In vivo (intraperitoneal injection) and in vitro studies in
mice have demonstrated that hydroquinone has a cytotoxic effect by
reducing the bone marrow and spleen cellularity and also an
immunosuppressive potential by inhibiting the maturation of
B-lymphocytes and the natural killer cell activity. Results also
indicate that bone marrow macrophages may be the primary target for
hydroquinone myelotoxicity. Myelotoxic effects were not observed in
a long-term bioassay in rodents.
In a 90-day study in rats using a functional-observational
battery, dose levels of 64 and 200 mg hydroquinone/kg produced
tremors, and 200 mg/kg produced depression of general activity.
Neuropathological examinations were negative.
1.7 Effects on humans
Cases of intoxication have been reported after oral ingestion
of hydroquinone alone or of photographic developing agents
containing hydroquinone. The major signs of poisoning included dark
urine, vomiting, abdominal pain, tachycardia, tremors, convulsions
and coma. Deaths have been reported after ingestion of photographic
developing agents containing hydroquinone. In a controlled oral
study on human volunteers, ingestion of 300-500 mg hydroquinone
daily for 3-5 months did not produce any observable pathological
changes in the blood and urine.
Dermal applications of hydroquinone at concentration levels
below 3% in different bases caused negligible effects in male
volunteers from different human races. However, there are case
reports suggesting that skin lightening creams containing 2%
hydroquinone have produced leucoderma, as well as ochronosis.
Hydroquinone (1% aqueous solution or 5% cream) has caused irritation
(erythema or staining). Allergic contact dermatitis due to
hydroquinone has been diagnosed.
Combined exposure to hydroquinone and quinone airborne
concentrations causes eye irritation, sensitivity to light, injury
of the corneal epithelium, corneal ulcers and visual disturbances.
There have been cases of appreciable loss of vision. Irritation has
occurred at exposure levels of 2.25 mg/m3 or more. Long-term
exposure causes staining of the conjunctiva and cornea and also
opacity. Slowly developing inflammation and discoloration of the
cornea and conjunctiva have resulted after daily hydroquinone
exposure for at least two years of 0.05-14.4 mg/m3; serious cases
have not occurred until after five or more years. One report
described cases of corneal damage occurring several years after the
exposure to hydroquinone had stopped.
There are no adequate epidemiological data to assess the
carcinogenicity of hydroquinone in humans.
1.8 Effects on other organisms in the laboratory and field
The ecotoxicological behaviour of hydroquinone has to be
related to its physicochemical properties, which induce sensitivity
to light, pH and dissolved oxygen. Its ecotoxicity, which is
generally high (e.g., < 1 mg/litre for aquatic organisms), varies
from species to species.
Algae, yeasts, fungi and plants are less sensitive to
hydroquinone than the other organisms generally used for toxicity
testing. However, within the same taxonomic group, the sensitivity
of different species to hydroquinone may vary by a factor of 1000.
2. IDENTITY, PHYSICAL AND CHEMICAL PROPERTIES, ANALYTICAL METHODS
2.1 Identity
Primary constituent
Chemical formula: C6 H4 (OH)2
Chemical structure:
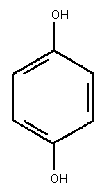 Relative molecular mass: 110.11
Common name: Hydroquinone
CAS registry number: 123-31-9
Synonyms: 1,4-benzenediol; p-benzenediol;
benzohydroquinone; benzoquinol; 1,4-
dihydroxybenzene; p-dihydroxybenzene;
p-dioxobenzene; p-dioxybenzene;
hydroquinol; hydroquinole; alpha-
hydroquinone; p-hydroquinone;
p-hydroxyphenol; quinol; ß-quinol
Technical product:
Trade name: Tecquinol
Impurities: none identified
Isomeric composition: None
Additives: None
2.2 Physical and chemical properties
Physical state: Long needles
Colour: White (analytical grade)
Odour: Odourless
Taste: Not documented
Melting point: 173-174 °C
Boiling point: 287 °C
Flash point: 165 °C (closed cup)
Flammability: Combustible when preheated
Explosion limits: Slight when exposed to heat.
Reactive at high temperature or pressure
Vapour pressure: 2.4 x 10-3 Pa (1.8 x 10-5 mmHg) at 25 °C
0.133 kPa (1 mmHg) at 132.4 °C
0.533 kPa (4 mmHg) at 150 °C
8.00 kPa (60 mmHg) at 203 °C
Specific gravity: 1.332 at 15 °C
Vapour density: 3.81
Log n-octanol/water
partition coefficient: 0.59
Solubility: Water: 59 g/litre at 15 °C
70 g/litre at 25 °C
94 g/litre at 28 °C
Organic solvents: Soluble in most polar organic solvents
ethyl alcohol 57 g/100 grams solvent at 25 °C
acetone 20 g/100 grams solvent at 25 °C
methyl isobutyl 27 g/100 grams solvent at 25 °C
ketone
2-ethylhexanol 12 g/100 grams solvent at 25 °C
ethyl acetate 22 g/100 grams solvent at 25 °C
Virtually insoluble (< 0.1%) in benzene, toluene and carbon
tetrachloride
Other properties: Reducing agent;
pK1 = 9.9, pK2 = 11.6;
Redox active (see below)
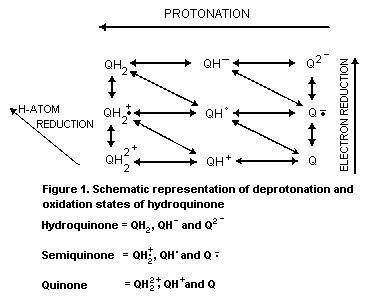
2.2.1 Reduction-oxidation equilibria
Hydroquinone undergoes reversible redox changes which can
involve a variety of pathways and redox couples (see Fig. 1). Each
redox couple has an electrochemical potential dependent upon the
degree of protonation and electron reduction.
Relative molecular mass: 110.11
Common name: Hydroquinone
CAS registry number: 123-31-9
Synonyms: 1,4-benzenediol; p-benzenediol;
benzohydroquinone; benzoquinol; 1,4-
dihydroxybenzene; p-dihydroxybenzene;
p-dioxobenzene; p-dioxybenzene;
hydroquinol; hydroquinole; alpha-
hydroquinone; p-hydroquinone;
p-hydroxyphenol; quinol; ß-quinol
Technical product:
Trade name: Tecquinol
Impurities: none identified
Isomeric composition: None
Additives: None
2.2 Physical and chemical properties
Physical state: Long needles
Colour: White (analytical grade)
Odour: Odourless
Taste: Not documented
Melting point: 173-174 °C
Boiling point: 287 °C
Flash point: 165 °C (closed cup)
Flammability: Combustible when preheated
Explosion limits: Slight when exposed to heat.
Reactive at high temperature or pressure
Vapour pressure: 2.4 x 10-3 Pa (1.8 x 10-5 mmHg) at 25 °C
0.133 kPa (1 mmHg) at 132.4 °C
0.533 kPa (4 mmHg) at 150 °C
8.00 kPa (60 mmHg) at 203 °C
Specific gravity: 1.332 at 15 °C
Vapour density: 3.81
Log n-octanol/water
partition coefficient: 0.59
Solubility: Water: 59 g/litre at 15 °C
70 g/litre at 25 °C
94 g/litre at 28 °C
Organic solvents: Soluble in most polar organic solvents
ethyl alcohol 57 g/100 grams solvent at 25 °C
acetone 20 g/100 grams solvent at 25 °C
methyl isobutyl 27 g/100 grams solvent at 25 °C
ketone
2-ethylhexanol 12 g/100 grams solvent at 25 °C
ethyl acetate 22 g/100 grams solvent at 25 °C
Virtually insoluble (< 0.1%) in benzene, toluene and carbon
tetrachloride
Other properties: Reducing agent;
pK1 = 9.9, pK2 = 11.6;
Redox active (see below)
2.2.1 Reduction-oxidation equilibria
Hydroquinone undergoes reversible redox changes which can
involve a variety of pathways and redox couples (see Fig. 1). Each
redox couple has an electrochemical potential dependent upon the
degree of protonation and electron reduction.
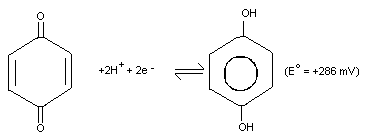
 Hydroquinone is a reducing agent with an electrochemical
potential (E°) of +286 mV for the benzoquinone/hydroquinone
(Q/H2Q) redox couple at 25 °C and pH 7.0, and under constant
conditions.
Hydroquinone is a reducing agent with an electrochemical
potential (E°) of +286 mV for the benzoquinone/hydroquinone
(Q/H2Q) redox couple at 25 °C and pH 7.0, and under constant
conditions.
 2.2.2 Oxidation of hydroquinone
Hydroquinone is oxidized by a variety of oxidants including
nitric acid, halogens, persulfates and metal salts (NIOSH, 1978). It
is also oxidized by molecular oxygen in alkaline solutions.
Hydroquinone reacts with molecular oxygen (autooxidation). In
an aqueous medium the rate of autooxidation is pH dependent,
occurring very rapidly at alkaline pH to produce a brown solution,
but very slowly in acidic medium. This reaction is strongly
catalysed by copper ions.
Some of the possible reactions during autooxidation of
hydroquinone in alkaline medium are outlined in Fig. 2. In alkaline
solution, p-benzoquinone can further react to form
2-hydroxyhydroquinone. In a similar manner to hydroquinone,
2-hydroxyhydroquinone can be oxidized to 2-hydroxy- p-benzoquinone
by electron transfer and disproportionation reactions (4a and b).
In addition, 2-hydroxy- p-benzoquinone (QI) is formed from
2-hydroxy-hydroquinone (HQI) by sequential mixed-redox reactions
with p-benzoquinone involving comproportionation [Eq. 1] and a
redox equilibrium reaction [Eq. 2].
2.2.2 Oxidation of hydroquinone
Hydroquinone is oxidized by a variety of oxidants including
nitric acid, halogens, persulfates and metal salts (NIOSH, 1978). It
is also oxidized by molecular oxygen in alkaline solutions.
Hydroquinone reacts with molecular oxygen (autooxidation). In
an aqueous medium the rate of autooxidation is pH dependent,
occurring very rapidly at alkaline pH to produce a brown solution,
but very slowly in acidic medium. This reaction is strongly
catalysed by copper ions.
Some of the possible reactions during autooxidation of
hydroquinone in alkaline medium are outlined in Fig. 2. In alkaline
solution, p-benzoquinone can further react to form
2-hydroxyhydroquinone. In a similar manner to hydroquinone,
2-hydroxyhydroquinone can be oxidized to 2-hydroxy- p-benzoquinone
by electron transfer and disproportionation reactions (4a and b).
In addition, 2-hydroxy- p-benzoquinone (QI) is formed from
2-hydroxy-hydroquinone (HQI) by sequential mixed-redox reactions
with p-benzoquinone involving comproportionation [Eq. 1] and a
redox equilibrium reaction [Eq. 2].
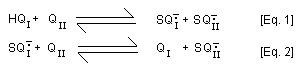 Formation of p-benzoquinone from hydroquinone also occurs in
a reverse manner by these mixed-redox reactions once
2-hydroxy- p-benzoquinone is formed. Hydrogen peroxide may be
generated by the reaction of hydroquinone and oxygen, and can then
react with p-benzoquinone forming 2,3-epoxy-hydroquinone. This
latter product, if reduced, forms 2-hydroxy-hydroquinone. Owing to
the large number of redox reactions possible between mono-benzo
products, the possible dimeric combinations, including formation of
charge transfer complexes between equal molar equivalents of
hydroquinones and benzoquinones (Q + HQ <-> Q ... HQ), oligomers
and polymers with various physical chemical properties are numerous
and, hence, their specific chemical formulae are not shown in Fig.
2.
Autooxidation of hydroquinone is not synonymous with
semiquinone autooxidation, which is also termed quinone redox
cycling. The latter phenomenon entails redox cycling between a
semiquinone and quinone in the presence of molecular oxygen,
generating the superoxide anion radical [Eq. 3]. With
p-benzosemiquinone and 2-hydroxy- p-benzoquinone, this reaction
is not marked because the equilibrium constant for the
disproportionation reaction (Ks) of p-benzosemiquinone to
hydroquinone and p-benzoquinone [Eq. 4] is around two orders of
magnitude higher than the equilibrium constant (Kc) for
autooxidation of benzosemiquinone [Eq. 3]. Thus autooxidation of the
semibenzoquinone does not significantly contribute to oxygen
depletion as for other hydroquinone/quinone couples. In contrast,
superoxide anion radical serves to reduce p-benzoquinone to
p-benzosemiquinone.
Formation of p-benzoquinone from hydroquinone also occurs in
a reverse manner by these mixed-redox reactions once
2-hydroxy- p-benzoquinone is formed. Hydrogen peroxide may be
generated by the reaction of hydroquinone and oxygen, and can then
react with p-benzoquinone forming 2,3-epoxy-hydroquinone. This
latter product, if reduced, forms 2-hydroxy-hydroquinone. Owing to
the large number of redox reactions possible between mono-benzo
products, the possible dimeric combinations, including formation of
charge transfer complexes between equal molar equivalents of
hydroquinones and benzoquinones (Q + HQ <-> Q ... HQ), oligomers
and polymers with various physical chemical properties are numerous
and, hence, their specific chemical formulae are not shown in Fig.
2.
Autooxidation of hydroquinone is not synonymous with
semiquinone autooxidation, which is also termed quinone redox
cycling. The latter phenomenon entails redox cycling between a
semiquinone and quinone in the presence of molecular oxygen,
generating the superoxide anion radical [Eq. 3]. With
p-benzosemiquinone and 2-hydroxy- p-benzoquinone, this reaction
is not marked because the equilibrium constant for the
disproportionation reaction (Ks) of p-benzosemiquinone to
hydroquinone and p-benzoquinone [Eq. 4] is around two orders of
magnitude higher than the equilibrium constant (Kc) for
autooxidation of benzosemiquinone [Eq. 3]. Thus autooxidation of the
semibenzoquinone does not significantly contribute to oxygen
depletion as for other hydroquinone/quinone couples. In contrast,
superoxide anion radical serves to reduce p-benzoquinone to
p-benzosemiquinone.
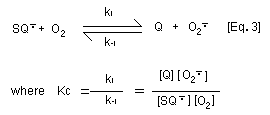
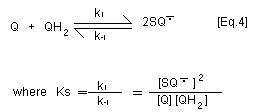 Confusion over the significance of redox cycling [Eq. 3] has
arisen from experiments performed in the presence of superoxide
dismutase (SOD) which catalyses the dismutation of superoxide anion
radical to H2O2 and O2 [Eq. 5]. Experiments in which addition
of SOD has been shown to modulate quinone toxicity have often been
interpreted as indicating that active oxygen species are involved in
hydroquinone/quinone mechanism of action (oxidative stress). In
fact, SOD "drives" the autooxidation of p-benzosemiquinone to
p-benzoquinone [Eq. 3] by removal of superoxide anion radical [Eq.
5] (Winterbourn, 1981; Rossi et al., 1986).
Confusion over the significance of redox cycling [Eq. 3] has
arisen from experiments performed in the presence of superoxide
dismutase (SOD) which catalyses the dismutation of superoxide anion
radical to H2O2 and O2 [Eq. 5]. Experiments in which addition
of SOD has been shown to modulate quinone toxicity have often been
interpreted as indicating that active oxygen species are involved in
hydroquinone/quinone mechanism of action (oxidative stress). In
fact, SOD "drives" the autooxidation of p-benzosemiquinone to
p-benzoquinone [Eq. 3] by removal of superoxide anion radical [Eq.
5] (Winterbourn, 1981; Rossi et al., 1986).
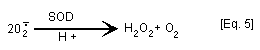 Dry pure hydroquinone is very stable to oxidation by oxygen,
darkening slowly upon prolonged exposure to air.
2.3 Conversion factors
1 ppm = 4.5 mg/m3 at 25 °C (1 atmosphere pressure)
1 mg/m3 = 0.222 ppm at 25 °C (1 atmosphere pressure)
2.4 Analytical methods
Information about analytical methods for hydroquinone are
contained in Devillers et al. (1990) and NIOSH (1978). The
procedures reported include colorimetry, column-, paper, thin-layer
and gas chromatography, and HPLC. It should be noted that
difficulties occur when hydroquinone is analysed by HPLC (Devillers
et al., 1990). Trace metal impurities, concentration of dissolved
oxygen in the mobile phase, pH of the solution, age of the water
sample, and age and history of the guard column may each influence
the analysis.
2.4.1 Sampling
Sampling techniques for air are outlined in Table 1.
2.4.2 Methods of analysis
Analytical methods are summarized in Table 2.
Dry pure hydroquinone is very stable to oxidation by oxygen,
darkening slowly upon prolonged exposure to air.
2.3 Conversion factors
1 ppm = 4.5 mg/m3 at 25 °C (1 atmosphere pressure)
1 mg/m3 = 0.222 ppm at 25 °C (1 atmosphere pressure)
2.4 Analytical methods
Information about analytical methods for hydroquinone are
contained in Devillers et al. (1990) and NIOSH (1978). The
procedures reported include colorimetry, column-, paper, thin-layer
and gas chromatography, and HPLC. It should be noted that
difficulties occur when hydroquinone is analysed by HPLC (Devillers
et al., 1990). Trace metal impurities, concentration of dissolved
oxygen in the mobile phase, pH of the solution, age of the water
sample, and age and history of the guard column may each influence
the analysis.
2.4.1 Sampling
Sampling techniques for air are outlined in Table 1.
2.4.2 Methods of analysis
Analytical methods are summarized in Table 2.
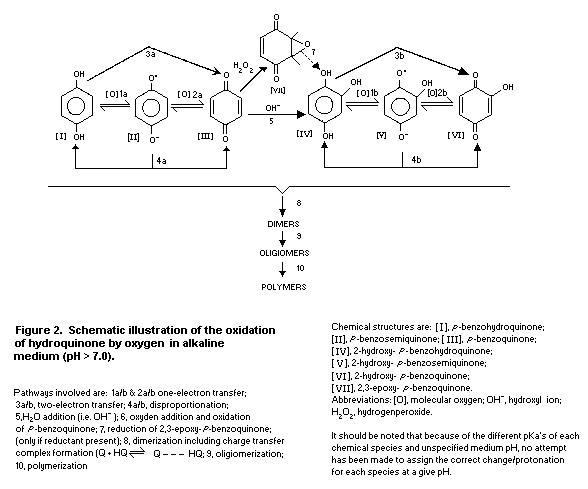 Table 1. Sampling techniques for hydroquinone in air in the occupational setting
Method Sample type Comments Technique Reference
Midget hydroquinone hydroquinone sample time = Oglesby
impinger dust absorbed in 5-10 min; sample et al. (1947)
isopropyl alcohol rate = 2.82 litres/min
in an all-glass
impinger
Midget hydroquinone hydroquinone air volume= 409-504 litres Chrostek (1975)
impinger mist collected in for about 430 min
distilled water;
disadvantage:
sample loss can
occur from spillage
Mixed cellulose hydroquinone filter with 0.8-µm sample time = NIOSH (1976)
ester aerosol pore size and 37-mm 60 min; sample
membrane diameter rate = 1.5 litres/min
filter recommended;
collection is >96%
Table 2. Analytical methods
Method Sample type Comments Detection limit Reference
Potentiometric aqueous hydroquinone extracted twice with ethyl- not stated Stott (1942),
titration acetate (<99.4% extraction) followed Levenson (1947),
by titration; requires little equipment Stevens (1945)
but is difficult and time consuming
Oxidiometric aqueous ceric sulfate with o-phenanthrolineferrous not stated; Kolthoff & Lee (1946),
titration sulfate complex (ferroin) used as indicator; accuracy <99.98% Brunner et al. (1949)
simple and fast with easily discernible
colour change
Iodometric aqueous single methyl acetate extraction involving not stated; Baumbach (1946),
titration potentiometric titration of metol (methyl- p- reproducibility Shaner & Sparks
amino-phenol sulfate) followed by oxidation (95.4-97.8%) (1946)
of both metol and hydroquinone with iodine
Iodometric urine urine hydrolysed at 100 °C for 2 h with conc. not stated Baernstein (1945)
titration H2SO4(pH 1.0); pH adjusted to 7.0 with sodium
sulfite followed by extraction of phenols for
4 h in a continuous liquid-liquid extractor;
hydroquinone precipitated with lead acetate
pH 6.5 plus pyridine-acetate buffer; filtrates
acidified, reacted with bromine, and excess
bromine back titrated with 0.2 mol/litre sodium
sulfite after addition of potassium iodide;
alternatively an iodine sensitive electrode can
be used as indicator; disadvantage: ketones
react in a similar manner to hydroquinone
Colorimetry aqueous hydroquinone reacted with phloroglucinol 1-35 mg/m3 Oglesby et al. (1947)
in NaOH; measured at 520 nm
Colorimetry aqueous hydroquinone in styrene reacted with sodium lower limit Whettem (1949)
tungstate and sodium carbonate; detected < 0.01 mg/ml
by visual comparison with standards
Table 2. (contd).
Method Sample type Comments Detection limit Reference
Colorimetry aqueous reaction with 4-aminoantipyrine; 0.05 ppm Jacquemain et al.
disadvantage; reacts with phenols (1975)
Spectrophotometry aqueous absorption wavelength not stated not stated Chrostek (1975)
Paper aqueous uses various solvent systems; separation qualitative Borecky (1963)
chromatography of mixtures with hydroquinone is indistinct
Paper aqueous three different solvent systems used; qualitative Stom (1975)
chromatography stable derivative formed by reaction with
benzene sulfinic acid
Paper aqueous developed with potassium meta periodate microgram quantities Clifford & Wight (1973)
chromatography
Chromatography cigarette methylether hydroquinone derivative formed qualitative Commins & Lindsey,
and smoke by reactions of dimethyl sulfate and (1956)
spectrophotometry hydroquinone
Gas aqueous phenols extracted into methyl isobutyl 0.1 mg/litre Cooper & Wheatstone,
chromatography ketone; trimethylsilyl ethers prepared, (1973)
separated on a Chromosorb W (AW-DCMS)
column coated with 5% tri-2,4-xylenyl
phosphate; detected by flame ionization
TLC aqueous reaction with feric chloride and qualitative Umpelev et al. (1974)
K3 [Fe (CN)6]
HPLC aqueous hydroquinone absorbed on mixed cellulose 0.84-4.05 mg/m3 NIOSH (1978)
ester filter membrane; filters are extracted
with 1% acetate; samples are injected onto a
Partisil TM 10-ODS column with 1% ethanoic acid
as mobile phase; detected at 290 nm
Table 2. (contd).
Method Sample type Comments Detection limit Reference
HPLC aqueous separated on Merckogel PGM 2000 column not stated Seki (1975)
with 0.05 mol/litre Pi (pH 6) followed by 0.05
mol/litre Pi plus 0.66 mol/litre borate pH 6;
detected at 280 nm
HPLC aqueous separated on µBondapak C18 column with > 2 µM Raghavan (1979)
0.01 mol/litre Pi (pH 7); detected at 280 nm
HPLC air hydroquinone oxidized to p-benzoquinone 0.005 mg/m3 Levin (1988)
by permanganate impregnated glassfibre in a 5-litre air
filter; p-benzoquinone formed is trapped on sample
XAD-2 adsorbent and desorbed with acetonitrile;
detection at 290 nm
3. SOURCES OF HUMAN AND ENVIRONMENTAL EXPOSURE
3.1 Natural occurrence
Hydroquinone occurs in a variety of forms as a natural product
from plants and animals. It has been found in non-volatile extracts
of coffee beans (Högl, 1958) and other foods (see section 5.1.2),
and as Arbutin (a glucoside of hydroquinone) in the leaves of
blueberry, cranberry, cowberry and bearberry plants (Varagnat,
1981). Hydroquinone formation from Arbutin in Pyrus spp. is
involved in fire blight resistance (Smale & Keil, 1966; Hildebrand
et al., 1969). Hydroquinone is considered to be the most important
component of the allelopathic interaction between the perennial weed
leafy spurge (Euphorbia esula) and the small everlasting
(Antennaria microphylla). A differential ability to detoxify
hydroquinone in the two species was observed in tissue cultures
(Hogan & Manners, 1990, 1991). Hydroquinones have been isolated from
marine sponges of Dysidea sp. (Iguchi et al., 1990) and from the
marine colonial tunicate Aplidium californicum (Howard et al.,
1979). Hydroquinone is also found in the bombardier beetle where it
is involved in defensive biochemistry: the beetle can shoot a hot
cloud of quinone, formed by the action of hydrogen peroxide,
hydroquinone and catalase-peroxidase in the explosion chamber of the
beetle, towards an oncoming enemy (Eisner et al., 1977).
The occurrence of hydroquinone in nature can originate from
metabolic processes. Direct hydroxylation of phenol to form
hydroquinone has been reported to occur when phenol was used as a
substrate by cytochrome P-450-enriched extracts of Streptomyces
griseus (Trower et al., 1988). Hydroquinone can also occur as a
metabolite in the biodegradation of substituted phenols (e.g. Spain
et al., 1979; Nyholm et al., 1984). Hydrolytic p-hydroxylation
initiates the degradation of many polychlorinated phenolic compounds
by Rhodococcus chlorophenolicus with the formation of substituted
hydroquinones (Häggblom et al., 1988).
3.2 Anthropogenic sources
3.2.1 Production levels and processes
In 1979, the world capacity for the production of hydroquinone
exceeded 40 000 tonnes (Varagnat, 1981). The annual production
volume of hydroquinone in the USA was estimated to be about 12 000
tonnes in 1985 (US EPA, 1985). Hydroquinone is manufactured in the
USA, Japan, France, Italy, and China (IARC, 1977; Varagnat, 1981).
In 1992, the world production was approximately 35 000 tonnes (USA:
16 000; Europe: 11 000; Japan: 6000; Central and South America and
Asian countries other than Japan: 2000) (personal communication from
H. Naito, University of Tsukuba, to the IPCS in 1993).
Hydroquinone can be manufactured commercially by several
processes. In the aniline oxidation process aniline is oxidized with
manganese dioxide and sulfuric acid to quinone; this is followed by
reduction of the latter to hydroquinone by an aqueous solution of
iron or by catalytic hydrogenation (Varagnat, 1981). Hydroquinone is
also manufactured by hydroxylation of phenol with hydrogen peroxide
as a hydroxylation agent. The reaction occurs with strong mineral
acids or ferrous or cobaltous salts as catalysts (Varagnat, 1981). A
third process to produce hydroquinone is hydroperoxidation of
diisopropylbenzene. The para isomer is isolated and oxidized with
oxygen to produce the corresponding dihydroperoxide, which is
treated with sulfuric acid to produce acetone and hydroquinone (NTP,
1989).
Hydroquinone can also be formed, based on Reppe's synthesis, by
carbonylation of acetylene under pressure. Finally, hydroquinone is
obtained from the reaction of p-isopropenylphenol and 30% aqueous
hydrogen peroxide in acidic conditions, but these syntheses are not
used for commercial production (Varagnat, 1981).
3.2.2 Uses
Hydroquinone has a multitude of used. It is used as a developer
in black-and-white photography and related graphic arts such as
lithography, rotogravure, and for medical and industrial X-ray films
(Varagnat, 1981). It is also widely used in the manufacture of
rubber antioxidants and antiozonants, monomer inhibitors, and food
antioxidants to prevent deterioration in many oxidizable products,
e.g., to stabilize vitamin A in fish oil, vitamins D and E,
ß-carotene, and antibiotics in feeds, and as a chemical intermediate
for the production of agrochemicals and performance polymers
(Varagnat, 1981). Hydroquinone and products containing hydroquinone
are used in cosmetics and medical skin preparations as a
depigmenting agent to lighten small areas of hyperpigmented skin. It
is also used in the treatment of melasma, freckles, senile
lentigines, and postinflammatory hyperpigmentation (Varagnat, 1981;
CIR, 1986). It is used as a coupler in oxidative hair dyeing (CIR,
1986).
In 1977, the use of hydroquinone in the USA was estimated to be
as follows: photographic developers, 45%; antioxidants and
polymerization inhibitors, 50%; other uses, 5%. Corresponding
figures in western Europe were, respectively, 70%, 15% and 15%
(Varagnat, 1981), and in Japan 30%, 50% and 20% for 1992 (personal
communication from H. Naito, University of Tsukuba, to the IPCS in
1993). In 1981, hydroquinone was an ingredient of 147 hair dyes and
colour preparations and 23 skin care products, including products
intended for medical use as skin lighteners in the USA (CIR, 1986).
Like hydroquinone, many of its derivatives are reducing agents
and have a wide variety of applications. Hydroquinone derivatives
that are used as rubber antioxidants and antiozonants include
dialkylated hydroquinone, N-alkyl-p-aminophenol and
diaryl- p-phenylenediamines. The main food antioxidants are
butylated hydroxyanisole (BHA) and tert-butylhydroquinone.
4. ENVIRONMENTAL TRANSPORT, DISTRIBUTION AND TRANSFORMATION
4.1 Transport and distribution between media
A calculation of fugacity, according to Mackay's model level I
(Mackay & Paterson, 1981), shows that hydroquinone will be
distributed mainly to the water compartment when released in
the environment. This was also concluded by Devillers et al.
(1990).
4.2 Transformation
4.2.1 Biodegradation
Biodegradation of hydroquinone is closely related to many
variables such as pH, temperature and whether conditions are aerobic
or anaerobic (Devillers et al., 1990). It also depends on the
acclimation level of the microorganisms involved (Tabak et al.,
1964; Harbison & Belly, 1982). Harbison & Belly (1982) investigated
various pure cultures of microorganisms for their ability to utilize
hydroquinone as sole carbon source. The pure cultures were isolated
from soil, photographic sludge and laboratory sludge. When incubated
with 750 mg/litre the isolates gave an average TOC (total organic
carbon) removal of 97.5% in 5 days. After various incubation
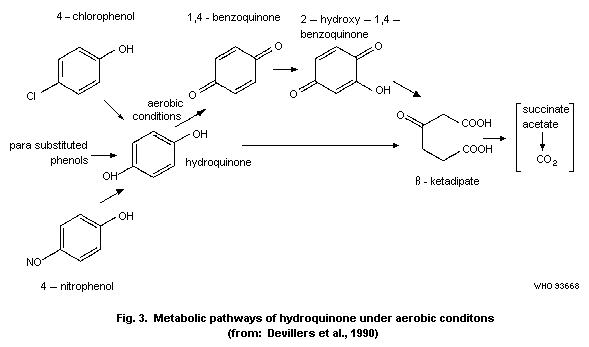
periods, the possible metabolites and end-products were analysed;
1,4-benzoquinone, 2-hydroxy-1,4-benzoquinone and ß-ketoadipic acid
were detected as metabolites. None of the compounds persisted in the
cultures. Neujahr & Varga (1970) proposed that the first step in the
degradation of hydroquinone by Trichosporon cutaneum should be a
hydroxylating step to hydroquinol. The ring fission should then
probably result in ß-hydroxymuconate.
The BOD5 (biological oxygen demand in 5 days)/COD (chemical
oxygen demand) ratio, which is an indicator of biodegradability, has
been reported to be 0.37 by Dore et al. (1975) and 0.53 by Young
et al. (1968). This indicates that under aerobic conditions
hydroquinone is readily biodegradable.
Devillers et al. (1990) have summarized various metabolic
pathways (Fig. 3).
Table 1. Sampling techniques for hydroquinone in air in the occupational setting
Method Sample type Comments Technique Reference
Midget hydroquinone hydroquinone sample time = Oglesby
impinger dust absorbed in 5-10 min; sample et al. (1947)
isopropyl alcohol rate = 2.82 litres/min
in an all-glass
impinger
Midget hydroquinone hydroquinone air volume= 409-504 litres Chrostek (1975)
impinger mist collected in for about 430 min
distilled water;
disadvantage:
sample loss can
occur from spillage
Mixed cellulose hydroquinone filter with 0.8-µm sample time = NIOSH (1976)
ester aerosol pore size and 37-mm 60 min; sample
membrane diameter rate = 1.5 litres/min
filter recommended;
collection is >96%
Table 2. Analytical methods
Method Sample type Comments Detection limit Reference
Potentiometric aqueous hydroquinone extracted twice with ethyl- not stated Stott (1942),
titration acetate (<99.4% extraction) followed Levenson (1947),
by titration; requires little equipment Stevens (1945)
but is difficult and time consuming
Oxidiometric aqueous ceric sulfate with o-phenanthrolineferrous not stated; Kolthoff & Lee (1946),
titration sulfate complex (ferroin) used as indicator; accuracy <99.98% Brunner et al. (1949)
simple and fast with easily discernible
colour change
Iodometric aqueous single methyl acetate extraction involving not stated; Baumbach (1946),
titration potentiometric titration of metol (methyl- p- reproducibility Shaner & Sparks
amino-phenol sulfate) followed by oxidation (95.4-97.8%) (1946)
of both metol and hydroquinone with iodine
Iodometric urine urine hydrolysed at 100 °C for 2 h with conc. not stated Baernstein (1945)
titration H2SO4(pH 1.0); pH adjusted to 7.0 with sodium
sulfite followed by extraction of phenols for
4 h in a continuous liquid-liquid extractor;
hydroquinone precipitated with lead acetate
pH 6.5 plus pyridine-acetate buffer; filtrates
acidified, reacted with bromine, and excess
bromine back titrated with 0.2 mol/litre sodium
sulfite after addition of potassium iodide;
alternatively an iodine sensitive electrode can
be used as indicator; disadvantage: ketones
react in a similar manner to hydroquinone
Colorimetry aqueous hydroquinone reacted with phloroglucinol 1-35 mg/m3 Oglesby et al. (1947)
in NaOH; measured at 520 nm
Colorimetry aqueous hydroquinone in styrene reacted with sodium lower limit Whettem (1949)
tungstate and sodium carbonate; detected < 0.01 mg/ml
by visual comparison with standards
Table 2. (contd).
Method Sample type Comments Detection limit Reference
Colorimetry aqueous reaction with 4-aminoantipyrine; 0.05 ppm Jacquemain et al.
disadvantage; reacts with phenols (1975)
Spectrophotometry aqueous absorption wavelength not stated not stated Chrostek (1975)
Paper aqueous uses various solvent systems; separation qualitative Borecky (1963)
chromatography of mixtures with hydroquinone is indistinct
Paper aqueous three different solvent systems used; qualitative Stom (1975)
chromatography stable derivative formed by reaction with
benzene sulfinic acid
Paper aqueous developed with potassium meta periodate microgram quantities Clifford & Wight (1973)
chromatography
Chromatography cigarette methylether hydroquinone derivative formed qualitative Commins & Lindsey,
and smoke by reactions of dimethyl sulfate and (1956)
spectrophotometry hydroquinone
Gas aqueous phenols extracted into methyl isobutyl 0.1 mg/litre Cooper & Wheatstone,
chromatography ketone; trimethylsilyl ethers prepared, (1973)
separated on a Chromosorb W (AW-DCMS)
column coated with 5% tri-2,4-xylenyl
phosphate; detected by flame ionization
TLC aqueous reaction with feric chloride and qualitative Umpelev et al. (1974)
K3 [Fe (CN)6]
HPLC aqueous hydroquinone absorbed on mixed cellulose 0.84-4.05 mg/m3 NIOSH (1978)
ester filter membrane; filters are extracted
with 1% acetate; samples are injected onto a
Partisil TM 10-ODS column with 1% ethanoic acid
as mobile phase; detected at 290 nm
Table 2. (contd).
Method Sample type Comments Detection limit Reference
HPLC aqueous separated on Merckogel PGM 2000 column not stated Seki (1975)
with 0.05 mol/litre Pi (pH 6) followed by 0.05
mol/litre Pi plus 0.66 mol/litre borate pH 6;
detected at 280 nm
HPLC aqueous separated on µBondapak C18 column with > 2 µM Raghavan (1979)
0.01 mol/litre Pi (pH 7); detected at 280 nm
HPLC air hydroquinone oxidized to p-benzoquinone 0.005 mg/m3 Levin (1988)
by permanganate impregnated glassfibre in a 5-litre air
filter; p-benzoquinone formed is trapped on sample
XAD-2 adsorbent and desorbed with acetonitrile;
detection at 290 nm
3. SOURCES OF HUMAN AND ENVIRONMENTAL EXPOSURE
3.1 Natural occurrence
Hydroquinone occurs in a variety of forms as a natural product
from plants and animals. It has been found in non-volatile extracts
of coffee beans (Högl, 1958) and other foods (see section 5.1.2),
and as Arbutin (a glucoside of hydroquinone) in the leaves of
blueberry, cranberry, cowberry and bearberry plants (Varagnat,
1981). Hydroquinone formation from Arbutin in Pyrus spp. is
involved in fire blight resistance (Smale & Keil, 1966; Hildebrand
et al., 1969). Hydroquinone is considered to be the most important
component of the allelopathic interaction between the perennial weed
leafy spurge (Euphorbia esula) and the small everlasting
(Antennaria microphylla). A differential ability to detoxify
hydroquinone in the two species was observed in tissue cultures
(Hogan & Manners, 1990, 1991). Hydroquinones have been isolated from
marine sponges of Dysidea sp. (Iguchi et al., 1990) and from the
marine colonial tunicate Aplidium californicum (Howard et al.,
1979). Hydroquinone is also found in the bombardier beetle where it
is involved in defensive biochemistry: the beetle can shoot a hot
cloud of quinone, formed by the action of hydrogen peroxide,
hydroquinone and catalase-peroxidase in the explosion chamber of the
beetle, towards an oncoming enemy (Eisner et al., 1977).
The occurrence of hydroquinone in nature can originate from
metabolic processes. Direct hydroxylation of phenol to form
hydroquinone has been reported to occur when phenol was used as a
substrate by cytochrome P-450-enriched extracts of Streptomyces
griseus (Trower et al., 1988). Hydroquinone can also occur as a
metabolite in the biodegradation of substituted phenols (e.g. Spain
et al., 1979; Nyholm et al., 1984). Hydrolytic p-hydroxylation
initiates the degradation of many polychlorinated phenolic compounds
by Rhodococcus chlorophenolicus with the formation of substituted
hydroquinones (Häggblom et al., 1988).
3.2 Anthropogenic sources
3.2.1 Production levels and processes
In 1979, the world capacity for the production of hydroquinone
exceeded 40 000 tonnes (Varagnat, 1981). The annual production
volume of hydroquinone in the USA was estimated to be about 12 000
tonnes in 1985 (US EPA, 1985). Hydroquinone is manufactured in the
USA, Japan, France, Italy, and China (IARC, 1977; Varagnat, 1981).
In 1992, the world production was approximately 35 000 tonnes (USA:
16 000; Europe: 11 000; Japan: 6000; Central and South America and
Asian countries other than Japan: 2000) (personal communication from
H. Naito, University of Tsukuba, to the IPCS in 1993).
Hydroquinone can be manufactured commercially by several
processes. In the aniline oxidation process aniline is oxidized with
manganese dioxide and sulfuric acid to quinone; this is followed by
reduction of the latter to hydroquinone by an aqueous solution of
iron or by catalytic hydrogenation (Varagnat, 1981). Hydroquinone is
also manufactured by hydroxylation of phenol with hydrogen peroxide
as a hydroxylation agent. The reaction occurs with strong mineral
acids or ferrous or cobaltous salts as catalysts (Varagnat, 1981). A
third process to produce hydroquinone is hydroperoxidation of
diisopropylbenzene. The para isomer is isolated and oxidized with
oxygen to produce the corresponding dihydroperoxide, which is
treated with sulfuric acid to produce acetone and hydroquinone (NTP,
1989).
Hydroquinone can also be formed, based on Reppe's synthesis, by
carbonylation of acetylene under pressure. Finally, hydroquinone is
obtained from the reaction of p-isopropenylphenol and 30% aqueous
hydrogen peroxide in acidic conditions, but these syntheses are not
used for commercial production (Varagnat, 1981).
3.2.2 Uses
Hydroquinone has a multitude of used. It is used as a developer
in black-and-white photography and related graphic arts such as
lithography, rotogravure, and for medical and industrial X-ray films
(Varagnat, 1981). It is also widely used in the manufacture of
rubber antioxidants and antiozonants, monomer inhibitors, and food
antioxidants to prevent deterioration in many oxidizable products,
e.g., to stabilize vitamin A in fish oil, vitamins D and E,
ß-carotene, and antibiotics in feeds, and as a chemical intermediate
for the production of agrochemicals and performance polymers
(Varagnat, 1981). Hydroquinone and products containing hydroquinone
are used in cosmetics and medical skin preparations as a
depigmenting agent to lighten small areas of hyperpigmented skin. It
is also used in the treatment of melasma, freckles, senile
lentigines, and postinflammatory hyperpigmentation (Varagnat, 1981;
CIR, 1986). It is used as a coupler in oxidative hair dyeing (CIR,
1986).
In 1977, the use of hydroquinone in the USA was estimated to be
as follows: photographic developers, 45%; antioxidants and
polymerization inhibitors, 50%; other uses, 5%. Corresponding
figures in western Europe were, respectively, 70%, 15% and 15%
(Varagnat, 1981), and in Japan 30%, 50% and 20% for 1992 (personal
communication from H. Naito, University of Tsukuba, to the IPCS in
1993). In 1981, hydroquinone was an ingredient of 147 hair dyes and
colour preparations and 23 skin care products, including products
intended for medical use as skin lighteners in the USA (CIR, 1986).
Like hydroquinone, many of its derivatives are reducing agents
and have a wide variety of applications. Hydroquinone derivatives
that are used as rubber antioxidants and antiozonants include
dialkylated hydroquinone, N-alkyl-p-aminophenol and
diaryl- p-phenylenediamines. The main food antioxidants are
butylated hydroxyanisole (BHA) and tert-butylhydroquinone.
4. ENVIRONMENTAL TRANSPORT, DISTRIBUTION AND TRANSFORMATION
4.1 Transport and distribution between media
A calculation of fugacity, according to Mackay's model level I
(Mackay & Paterson, 1981), shows that hydroquinone will be
distributed mainly to the water compartment when released in
the environment. This was also concluded by Devillers et al.
(1990).
4.2 Transformation
4.2.1 Biodegradation
Biodegradation of hydroquinone is closely related to many
variables such as pH, temperature and whether conditions are aerobic
or anaerobic (Devillers et al., 1990). It also depends on the
acclimation level of the microorganisms involved (Tabak et al.,
1964; Harbison & Belly, 1982). Harbison & Belly (1982) investigated
various pure cultures of microorganisms for their ability to utilize
hydroquinone as sole carbon source. The pure cultures were isolated
from soil, photographic sludge and laboratory sludge. When incubated
with 750 mg/litre the isolates gave an average TOC (total organic
carbon) removal of 97.5% in 5 days. After various incubation
periods, the possible metabolites and end-products were analysed;
1,4-benzoquinone, 2-hydroxy-1,4-benzoquinone and ß-ketoadipic acid
were detected as metabolites. None of the compounds persisted in the
cultures. Neujahr & Varga (1970) proposed that the first step in the
degradation of hydroquinone by Trichosporon cutaneum should be a
hydroxylating step to hydroquinol. The ring fission should then
probably result in ß-hydroxymuconate.
The BOD5 (biological oxygen demand in 5 days)/COD (chemical
oxygen demand) ratio, which is an indicator of biodegradability, has
been reported to be 0.37 by Dore et al. (1975) and 0.53 by Young
et al. (1968). This indicates that under aerobic conditions
hydroquinone is readily biodegradable.
Devillers et al. (1990) have summarized various metabolic
pathways (Fig. 3).
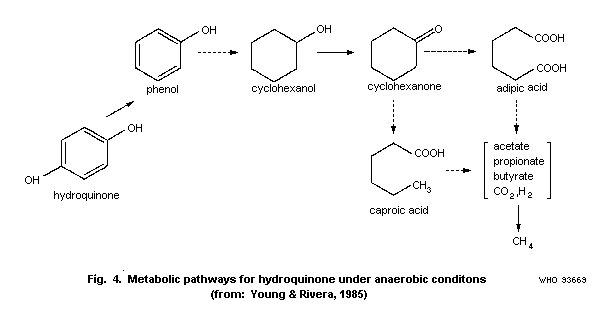
 Young & Rivera (1985) studied the methanogenic degradation of
hydroquinone. When the microbial community from a municipal sewage
treatment plant digester was acclimated to hydroquinone, the rate of
metabolism and gas formation increased. The rate of substrate
metabolism was 23.6 ± 2.0 (n=6) with acclimated microorganisms
compared to 5.7 ± 1.4 (n=6) mg/litre per day with non-acclimated
organisms. The rate of gas production (CO2 + CH4) was 9.33 ± 1.7
and 5.70 ± 1.1 ml/litre culture fluid per day for acclimated and non
acclimated organisms, respectively. Prior to mineralization
hydroquinone was metabolized to phenol. The authors have summarized
various anaerobic degradation steps and proposed the scheme in Fig.
4.
Stoichiometrically the anaerobic bioconversion of hydroquinone
is described as follows:
C6H6O2 + 3.5 H2O -> 2.75 CO2 + 3.25 CH4
4.2.2 Abiotic degradation
The photodegradation of hydroquinone has been discussed by
Devillers et al. (1990). Due to its intrinsic properties
hydroquinone is relatively readily degraded by means of
photodegradation. Phototransformation may occur from direct
excitation or from induced or photocatalytic reactions.
Freitag et al. (1985) reported that when 62 ng hydroquinone
adsorbed on silica gel was exposed to ultraviolet light (290 nm) for
17 h, 57.4% of the hydroquinone was mineralized.
Tissot et al. (1985) measured changed toxicity due to
phototransformation (Table 3). The phototransformation products were
p-benzoquinone after 0.5 h and hydroxy p-benzoquinone after 4
and 22 h.
Table 3. Photoirradiation of hydroquinone and toxicity to Daphnia magna measured as
inhibition of motility after 24 h (from: Tissot et al., 1985)
Initial Irradiation % EC50 (mg/litre HPLC analysis at the
concentration time degradation initial end of the irradiation
(h) concentration) period
67.1 mg/litre 0 0 0.15
(6.1 x 10-4 M) 0.5 15 0.2 p-benzoquinone
4 49 0.2 10-4 M hydroxy
p-benzoquinone
22 80 0.5 1.4 x 10-4 M hydroxy
p-benzoquinone
Young & Rivera (1985) studied the methanogenic degradation of
hydroquinone. When the microbial community from a municipal sewage
treatment plant digester was acclimated to hydroquinone, the rate of
metabolism and gas formation increased. The rate of substrate
metabolism was 23.6 ± 2.0 (n=6) with acclimated microorganisms
compared to 5.7 ± 1.4 (n=6) mg/litre per day with non-acclimated
organisms. The rate of gas production (CO2 + CH4) was 9.33 ± 1.7
and 5.70 ± 1.1 ml/litre culture fluid per day for acclimated and non
acclimated organisms, respectively. Prior to mineralization
hydroquinone was metabolized to phenol. The authors have summarized
various anaerobic degradation steps and proposed the scheme in Fig.
4.
Stoichiometrically the anaerobic bioconversion of hydroquinone
is described as follows:
C6H6O2 + 3.5 H2O -> 2.75 CO2 + 3.25 CH4
4.2.2 Abiotic degradation
The photodegradation of hydroquinone has been discussed by
Devillers et al. (1990). Due to its intrinsic properties
hydroquinone is relatively readily degraded by means of
photodegradation. Phototransformation may occur from direct
excitation or from induced or photocatalytic reactions.
Freitag et al. (1985) reported that when 62 ng hydroquinone
adsorbed on silica gel was exposed to ultraviolet light (290 nm) for
17 h, 57.4% of the hydroquinone was mineralized.
Tissot et al. (1985) measured changed toxicity due to
phototransformation (Table 3). The phototransformation products were
p-benzoquinone after 0.5 h and hydroxy p-benzoquinone after 4
and 22 h.
Table 3. Photoirradiation of hydroquinone and toxicity to Daphnia magna measured as
inhibition of motility after 24 h (from: Tissot et al., 1985)
Initial Irradiation % EC50 (mg/litre HPLC analysis at the
concentration time degradation initial end of the irradiation
(h) concentration) period
67.1 mg/litre 0 0 0.15
(6.1 x 10-4 M) 0.5 15 0.2 p-benzoquinone
4 49 0.2 10-4 M hydroxy
p-benzoquinone
22 80 0.5 1.4 x 10-4 M hydroxy
p-benzoquinone
 4.2.3 Bioaccumulation
With a log n-octanol/water partition coefficient of 0.59 it
can be considered that hydroquinone does not bioaccumulate. The
bioconcentration factors found in the literature for static tests
are listed in Table 4.
Table 4. Bioaccumulation factors (BCF)a
Species Test Hydroquinone BCF Comment
duration concentration
(days) (mg/litre)
Activated sludge 5 0.05 870 dry weight basis
Algae
Chlorella fusca 1 0.05 40 wet weight basis
Fish
Leuciscus idus
melanotus 3 0.05 40 wet weight basis
a From: Freitag et al. (1985)
4.3 Interaction with other physical, chemical or biological factors
Tratnyek & Macalady (1989) report on direct abiotic reductions
of nitro groups from nitro aromatic pesticides to amines by
hydroquinones. In homogeneous solutions of quinone-hydroquinone
redox couples, which were selected to model the redox-labile
functional groups in natural organic matter, rapid abiotic reduction
of nitro aromatic pesticides occurred. The authors proposed that
hydroquinones contribute to the reduction of pollutants in the
environment, but their role is likely to be complex.
The water hyacinth (Eichhornia crassipes), which is used for
water treatment, clears more than 98% hydroquinone (50 mg/litre)
after about 48 h (O'Keeffe et al., 1987). This property has been
attributed to enzymatic metabolism by polyphenol oxidases.
4.4 Ultimate fate following use
Hydroquinone occurs in photo-processing effluents (Dagon, 1973;
Harbison & Belly, 1982). However, it is not certain that it reaches
the water ecosystem, because reliable monitoring data are not
available.
5. ENVIRONMENTAL LEVELS AND HUMAN EXPOSURE
5.1 Environmental levels
5.1.1 Air, soil and water
No monitoring data have been found concerning ambient free
hydroquinone concentrations in air, soil or water. However,
hydroquinone has been identified in tobacco smoke and measured in
mainstream smoke from non-filtered cigarettes at amounts ranging
from 110 to 300 µg per cigarette, with a ratio of the sidestream to
mainstream concentration of 0.7-0.9 (IARC 1986).
5.1.2 Food
Free and conjugated (Arbutin) hydroquinone exist as natural
components of a variety of plant-derived beverages and food
products.
Högl (1958) identified hydroquinone in the non-volatile extract
of coffee beans. Hydroquinone concentrations in roasted coffee have
been reported to range between 25 and 40 mg/kg (Maier, 1981). Gold
et al. (1992) estimated that one cup of coffee would contain
approximately 100 µg hydroquinone. Teas prepared from leaves of
blueberry, cowberry, cranberry and bearberry have been reported to
contain hydroquinone at concentrations sometimes exceeding 1%
(Deichmann & Keplinger, 1981).
The concentrations of free and total (free hydroquinone and
Arbutin) hydroquinone have been measured in a variety of foods and
beverages by Hill et al. (1993); results indicate that significant
exposure to hydroquinone can occur through dietary sources. In most
of the samples (Table 5) derived from plant sources, the levels of
Arbutin are considerably higher than those of free hydroquinone.
However, Arbutin is hydrolysed readily by dilute acids yielding
hydroquinone and glucose. Therefore, both free hydroquinone and
Arbutin may contribute to hydroquinone exposure from natural sources
as well as to the daily intake of dietary antioxidants.
Adhesives containing trace amounts of hydroquinone are
permitted as a component of food packaging in the USA (FDA, 1981;
1991).
Table 5. Concentrations of free and total hydroquinone in various foods and
beverages
Food sample Concentrations (mg/kg ± SD)a
Free HQ Total HQb
Wheat germ, toasted 0.591 8.352
Drip-brewed coffee (pre-ground) 0.293 ± 0.003 0.385 ± 0.016
Whole wheat bread (100% whole wheat) 0.584 ± 0.202 0.893 ± 0.480
Whole wheat cereal (commercially available) 0.205 ± 0.019 0.992 ± 0.161
Processed corn cereal (commercially available) bkgb bkg
Pear skin (D'Anjou, fresh) bkg 38.057
Pear flesh (D'Anjou, fresh) bkg 1.301
Milkfat (2%) homogenized milk bkg bkg
Yogurt (black cherry) bkg bkg
Cantaloupe bkg bkg
Diet cola 0.0362 0.0287
a bkg = background levels comparable to that observed in control blanks
b Includes free hydroquinone and hydroquinone released following treatment
of the samples with ß-glucosidase
5.2 General population exposure
Photohobbyists, who develop their own black-and-white films (a
process which utilizes hydroquinone) may be exposed dermally.
Exposure to dust is also possible when preparing developer
solutions. In 1980, the number of photohobbyists was estimated to be
about 2.2 million in the USA (US EPA, 1985). There are no data on
exposure levels.
Dermal exposure to hydroquinone may also occur from products
intended for cosmetic and medical use. In the USA, hydroquinone has
been used in cosmetics, and in over-the-counter (OTC or
non-prescription) and prescription drugs. Both OTC and prescription
drugs are used to lighten areas of hyperpigmented skin. In
cosmetics, concentrations of < 0.1% to 5% have been reported
(CIR, 1986). OTC skin lighteners may contain up to 2% hydroquinone
and prescription drugs may contain higher concentrations. In the EC
countries, hydroquinone is restricted for use in cosmetics to 2% or
less (Boyle & Kennedy, 1985). The US Food and Drug Administration
has issued a Notice of Proposed Rule-making for the use of
hydroquinone as a skin lightener in OTC drugs at concentrations
below 1.5-2.0% (FDA, 1982).
Skin-lightening creams containing hydroquinone are frequently
inadequately labelled and the concentration often exceeds the limit
of 2%; it is likely to be much stronger than 2% (Brauer, 1985;
Godlee, 1992) and even up to 7% (Boyle & Kennedy, 1986).
A 2% upper limit on hydroquinone concentration, set by the
South African government in 1980 and followed by the United Kingdom
and USA, was based on tests of cutaneous irritancy (Arndt &
Fitzpatrick, 1965) and contact dermatitis (Bentley-Philips & Bayles,
1975).
5.3 Occupational exposure
Hydroquinone can be encountered in solid form or in solution
during its production and use (NIOSH, 1978). It has a very low
vapour pressure, but can be oxidized in the presence of moisture to
form quinone, which is more volatile. The saturated concentration in
air for hydroquinone vapour under standard conditions is estimated
to be 0.108 mg/m3 (approximately 0.024 ppm at 25 °C) (NIOSH,
1978).
There are some industrial hygiene monitoring data available for
hydroquinone. Oglesby et al. (1947) reported 20-35 mg
hydroquinone/m3 in a packaging area without exhaust cabinet and
1-4 mg/m3 in a packaging area with exhaust cabinet in a plant
manufacturing hydroquinone. However, the analytical methods did not
distinguish between hydroquinone and quinone. Industrial data,
provided to the US EPA (1985), indicated worker inhalation exposure
due to closed production processes within one manufacturing facility
at an arithmetical average concentration of 0.79 mg/m3 (± 0.52
standard deviation) and a highest average concentration of 0.2
mg/m3 in another facility. In the unloading area of a production
facility the arithmetic average air concentration was reported to be
0.13 mg/m3 (± 0.15 standard deviation). The concentration of
hydroquinone was measured in the workroom air in 12 Finnish plants
(altogether 36 samples) during the period 1950-89 (Rantanen et
al., personal communication to the IPCS, 1992). Most samples were
collected in the printing industry (23 samples from five plants).
The occupational exposure limit of 2 mg/m3 was exceeded in only
one measurement: 9.5 mg/m3 during charging of hydroquinone in a
gas plant in 1962, a short operation carried out once every three
weeks. Approximately 470 000 workers in the USA are potentially
exposed to hydroquinone in about 137 occupations (US EPA, 1985).
Certain occupations in which hydroquinone exposure may occur are
listed in Table 6. Some of the national occupational air exposure
limits used in various countries are compiled in Table 7.
Table 6. Occupations with potential exposure to hydroquinonea
Antioxidant makers
Drug makers
Hair dressers and cosmetologists
Hydroquinone manufacturing workers
Paint makers
Photo processors
Organic chemical synthesizers
Photographic developer makers
Plastic stabilizer workers
Rubber coating workers
a From: Key et al. (1977); NIOSH (1978); NIOSH (1990)
Table 7. National occupational air exposure limits used in various
countries (from: IRPTC, 1987; ILO, 1991)
Country TWAa STELb
CLVc
(mg/m3) (mg/m3) (mg/m3)
Australia 2
Belgium 2
Denmark 2
Finland 2 4
France 2
Germany 2d
The Netherlands 2
Poland 2 2
Romania 1 2
Sweden 0.5 1.5
Switzerland 2 4
United Kingdom 2 4
USA: ACGIH 2
NIOSH/OSHA 2
Yugoslavia 2
a TWA = time-weighted average; a maximum mean exposure limit based
generally over the period of a working day
b STEL = short-term exposure limit
c CLV = ceiling level value
d inhalable dust
6. KINETICS AND METABOLISM
The main results pertinent to this chapter, with the exception
of section 6.5, have been summarized in Table 8 and will be expanded
upon only where necessary. Table 8 shows that the majority of
studies have been performed in Fischer-344 rats.
6.1 Absorption
Absorption of orally administered or intratracheally instilled
hydroquinone is rapid and extensive (Garton & Williams, 1949;
Divincenzo et al., 1984; English et al., 1988). However, the
rate of hydroquinone absorption through the skin is low. Marty et
al. (1981) reported that the in vitro permeability constants for
rat and human skin were 28 x 10-6 and 4 x 10-6 cm/h,
respectively. Based on the data of Bucks et al. (1988), an in
vivo human dermal absorption rate of 3 µg/cm2.h and a
permeability constant of 2.25 x 10-6 cm/h can be calculated. The
actual amount of hydroquinone absorbed following dermal exposure
depends on the exposure concentrations, length of exposure and
vehicle, as well as other factors. When Bucks et al. (1988)
applied 14C-labelled hydroquinone in an alcoholic vehicle to the
foreheads of human volunteers for 24 h, 57% of the total 14C label
was excreted in the urine after 5 days. Addition of a sun screen to
the hydroquinone solution reduced total excretion to 26%.
6.2 Distribution
Following the oral administration of radiolabelled hydroquinone
to F-344 rats, radioactivity was widely distributed throughout the
animal tissues. The highest activity was localized in the kidney and
liver (Divincenzo et al., 1984). However, on a quantitative basis,
the amount retained within the animal was low, representing < 2%
of the total dose 48 h after exposure (Divincenzo et al., 1984;
English et al., 1988). Widespread distribution and extensive
elimination was also observed following intratracheal administration
of hydroquinone to F-344 rats (Lockhart & Fox, 1985b). However,
following the intravenous injection of radiolabelled hydroquinone to
F-344 rats, radioactivity was shown, using whole body
autoradiographic techniques, to concentrate in the bone marrow,
thymus and white pulp of the spleen (Greenlee et al., 1981a).
Subsequent experiments indicated that significant acid soluble and
covalently bound radioactivity could be recovered in the thymus,
bone marrow and white blood cells 24 h after intravenous
administration (Greenlee et al., 1981b). These results indicate
that the route of administration may influence the profile of
distribution and elimination observed following hydroquinone
administration.
Table 8. Summary of toxicokinetic data for hydroquinone (HQ)
Species and Absorption Distribution Metabolic Elimination and Reference
treatment transformation excretion
Oral administration
Species: rabbits less than 1% of the dose Garton &
Treatment: 3-6 rabbits was excreted unchanged; Williams
received 100 or 200 mg/kg about 80% of the dose was (1949)
HQ as a single dose; urine recovered as glucuronide
metabolites analysed after and monosulfate conjugates
24 h in the urine
Species: Sprague-Dawley T1 & T2: rapid and T1 & T2: for 200 mg/kg the major radio- mainly in the urine; Divincenzo
rats (m) extensive based 0.28-1.25% and labelled species in elimination for T1 and et al.
Treatment: (T1) 2-4 rats upon urinary 0.26-0.56% of the urine were: HQ T2 was similar; after (1984)
per group received 5, 30 excretion administered radioactivity monoglucuronide, HQ 48 h around 95% of
or 200 mg/kg [14C]-HQ as a recovered in carcass monosulfate and HQ dose had been excreted
single dose; rats were and tissues after 48 h (T1: 56%, 42% and in urine (90%), faeces (4%)
observed for 48 and 96 h and 96 h, respectively; 1%; T2: 72%, 23% and CO2 (0.4%); no
before sacrifice. widely distributedand 1%) difference in elimination
(T2) 4 rats pretreated with throughout tissues between single and
200 mg/kg unlabelled HQ with highest concentration repeated doses
once a day for 4 days in liver and
followed by 200 mg/kg kidney
[14C]-HQ on day 5; rats
observed for 48 h
Table 8. (contd).
Species and Absorption Distribution Metabolic Elimination and Reference
treatment transformation excretion
Species: Fischer-344 T1 & T2: rapid and T1 & T2: < 1% of T1 & T2: the major Excreta English
rats (m,f) extensive based administered radiolabelled species T1 & T2: mainly excreted et al.
Treatment: (T1) 8 (m,f) upon peak blood radioactivity recovered in the urine were in the urine; after 48 h (1988)
rats per group received 25 concentration in carcass and HQ monoglucuronide around 90% of the
or 350 mg/kg [14C]-HQ as within 0.8 h of tissues for each dose (44-54%), HQ mono- administered radioactivity
a single dose. dosing and urinary after 48 h; twice as sulfate (19-33%), was recovered as urine
(T2) 8 rats (m,f) excretion; much radioactivity HQ (0.25-7%), HQ (approx.78%), cage rinse
pretreated with 25 mg/kg no sex recovered in the mercapturate (approx.12%) and faeces
unlabelled HQ once a day differences liver and kidney (0.16-4.68%) and (approx.2.2%); dose-related
for 14 days followed by 25 of females compared p-benzoquinone differences were observed
mg/kg [14C]-HQ on day 15; with males (0.24-0.84%); no at 8 h, 54% (m) and 45% (f)
after oral administration sex difference of the dose was excreted
4 rats per dose and renally by the high-dose
sex were designated group compared with 81% (m)
for blood collection and 82% (f) for the low-
samples (up to 96 h) dose group
and for excreta and
radiodistribution Blood kinetics
(up to 7 days) AUC values were increased
by 17-fold (m) and 26-fold
(f) for a 13- and 14-fold
higher mean dose; most of
radioactivity was excreted
by 8 h and was associated
mainly with alpha-
elimination phase (T´ =
0.23-1.72 h); accurate ß T´
could not be determined
because of the appearance
of a second peak in the
blood concentration versus
Table 8. (contd).
Species and Absorption Distribution Metabolic Elimination and Reference
treatment transformation excretion
Species: Fischer-344 rapid and extensive low distribution, by 8 h, major time curve by 24 h, Lockhart
rats (m,f) absorption as i.e. less than 1%; metabolites found recovery was more than 92% et al.
Treatment: a single dose indicated by marked no significant in urinewere HQ in urine, approx.2% in (1984);
of 5, 25 or 50 mg [U-14C]- recovery of [14C] differences between glucuronide faeces and less than 0.2% Lockhart
HQ/kg body weight by in urine by 24 h sexes (approx.50%), in CO2 & Fox
gavage HQ sulfate (1985a)
(approx.30%) and
HQ (approx.2%);
neither dose- nor
sex-dependent
Species: Fischer-344 rapid and extensive low distribution: by 8 h, major by 24 h, recovery was Lockhart
rats (f) absorption as approx. 0.55% in liver and metabolites found in approx.92% in urine, & Fox
Treatment: 5 rats per dose indicated by marked 0.64-0.9% in carcass urine were HQ approx.2.6% in faeces and (1985a)
group 5, 25 or 50 mg recovery of [14C] glucuronide (approx. approx.0.3% in CO2
[U-14C]-HQ/kg body weight in urine by 24 h 46%), HQ sulfate
(single gavage dose) (approx.29-36%) and
HQ (approx.2.5%)
Dermal administration
In vitro: Rat or human overall absorption Marty et
skin biopsy; repeated and permeability al. (1981)
dosing with 40 mg/cm2 in constant were low
water; observed for 24 h but on average
7-fold greater for
rat than human skin
In vivo: Mouse or rat absorption by local cutaneous combined urine and faecal
mouse was low; distribution was high elimination was low;
1.6% after 6 h in rat approx.10% after 96 h in
the rat
Table 8. (contd).
Species and Absorption Distribution Metabolic Elimination and Reference
treatment transformation excretion
Species: human average percutaneous Bucks et
Treatment: 6 normal adult taneous absorption al. (1988)
male volunteers had 2% estimated from
(w/w) HQ in ethanol urinary elimination
(approx.70%) plus 0.2% data was 57% after
ascorbic acid applied to 120 days; sun-
their foreheads for 24 h; screens decreased
single dose = 125 µg/cm2; absorption but
observed for up to 120 h penetration
enhancers were
without effect
Species: Fischer-344 skin irritated but after 1 week, 15-18% HQ English
rats (m,f) poorly absorbed; was recovered in urine and et al.
Treatment: 8 (m,f) rats large interanimal cage rinsings, 1.7-3.7% in (1988)
per group were dermally variation in the faeces, 2.6 to 12.9%
exposed for 24 h to 25 disposition; removal in the body and 0.14 to
or 150 mg/kg [14C]-HQ of HQ after skin 2.2% in the excised
dissolved in distilled washing with soapy skin exposure site
water for 24 h water was close to
100% after 10 min of
exposure or around
65% after 24 h of
exposure
Table 8. (contd).
Species and Absorption Distribution Metabolic Elimination and Reference
treatment transformation excretion
Intravenous administration
Species: Fischer-344 rat (m) whole body Greenlee
Treatment: 1.3 mg/kg autoradiography showed et al.
[14C]-HQ in saline that [14C] concentrated (1981a)
administered as a single most in the white pulp
dose; one group of rats of the spleen, bone marrow
was pretreated with and thymus; Aroclor 1254
Aroclor 1254 (250 mg/kg pre-treatment decreased
i.p.) the tissue/blood optical
density by approx.60% for
the thymus and bone marrow
Species: Fischer-344 rat (m) acid-insoluble Greenlee
Treatment: rats received radioactivity associated et al.
14 mg/kg [14C]-HQ as a with protein increased (1981b)
single administration; one with time in the bone
group of rats was marrow > thymus > liver;
pretreated with Aroclor pretreatment with Aroclor
1254 resulted in a significant
decrease in the
radioactivity measured in
the bone marrow
Intratracheal instillation
Species: Fischer-344 rapid and extensive < 0.13% in lung, less by 8 h, major by 48 h recovery was Lockhart
rats (m) absorption as than 1% to other organs metabolites recovered more than 92% in urine, & Fox
Treatment: 5 rats per dose indicated by in the urine were approx.2% in faeces and (1985b)
group: 5, 25 or 50 mg recovery in urine HQ-glucuronide less than 0.2% in CO2
[U-14C]-HQ/kg; 2 rats per within 24 h (approx.50%),
control group HQ-sulfate
(approx.30%) and
HQ (approx.2%)
Table 8. (contd).
Species and Absorption Distribution Metabolic Elimination and Reference
treatment transformation excretion
Intraperitoneal administration
Species: Wistar rat (f) metabolites recovered elimination was rapid with Inoue et
Treatment: 9 rats received in urine were 1,2,4- 84% of the metabolites al.
a single 50 mg/kg dose benzenetriol (11%), recovered within 4 h after (1989a)
catechol (1%) and administration; by 24 h,
hydroquinone (87%) recovery of 1,2,4-
benzenetriol, catechol and
hydroquinone in the acid-
hydrolysed urine comprised
38% of the administered
dose
Species: Japanese white metabolites recovered by 24 h, recovery of Inoue et
rabbits in urine were 1,2,4- 1,2,4,-benzenetriol, al.
Treatment: 5 rabbits benzenehydrotriol catechol and quinone in (1989b)
received a single 50 mg/kg (12%), catechol (1%) the acid-hydrolysed urine
dose and hydroquinone comprised 40% of the
(86%) administered dose
6.3 Metabolic transformation
Hydroquinone is converted mainly by Phase II metabolism to
water-soluble conjugates, as shown by the recovery of only little
parent compound and p-benzoquinone (0.25-7%) but large amounts of
hydroquinone-monoglucuronide and hydroquinone-monosulfate (>90%) in
the urine (Divincenzo et al. 1984; English et al. 1988). A small
percentage of the dose was recovered as the mercapturic acid
conjugate of hydroquinone, suggesting the intermediate formation of
a glutathione conjugate of hydroquinone.
Divincenzo et al. (1984) demonstrated that repeated dosing
with 200 mg hydroquinone/kg did not alter the relative or absolute
rat liver weight or induce the hepatic mixed-function oxidase
system, nor did hydroquinone undergo Phase I oxidation to other
metabolites such as 1,2,4-trihydroxybenzene. In addition, the
formation of 1,2,4-trihydroxybenzene was not observed in the urine
after oral administration of hydroquinone to rabbits (Garton &
Williams, 1949). However, following intraperitoneal injection of
hydroquinone (50 mg/kg) in Wistar rats and Japanese white rabbits,
1,2,4-trihydroxybenzene represented a significant proportion
(approximately 12%) of the metabolites recovered in the urine (Inoue
et al., 1989a,b). This apparent difference in the metabolic
profile observed when hydroquinone is administered by the
intraperitoneal route rather than the oral route is probably related
to the efficient ability of the gastrointestinal system to conjugate
phenolic compounds absorbed in the intestine, thus reducing the
amount of free hydroquinone available for Phase I metabolism in the
liver (Powell et al., 1974; Cassidy & Houston, 1980a,b; Cassidy &
Houston, 1984). Fig. 5 shows proposed metabolic pathways for
hydroquinone biotransformation in Fischer-334 rats.
4.2.3 Bioaccumulation
With a log n-octanol/water partition coefficient of 0.59 it
can be considered that hydroquinone does not bioaccumulate. The
bioconcentration factors found in the literature for static tests
are listed in Table 4.
Table 4. Bioaccumulation factors (BCF)a
Species Test Hydroquinone BCF Comment
duration concentration
(days) (mg/litre)
Activated sludge 5 0.05 870 dry weight basis
Algae
Chlorella fusca 1 0.05 40 wet weight basis
Fish
Leuciscus idus
melanotus 3 0.05 40 wet weight basis
a From: Freitag et al. (1985)
4.3 Interaction with other physical, chemical or biological factors
Tratnyek & Macalady (1989) report on direct abiotic reductions
of nitro groups from nitro aromatic pesticides to amines by
hydroquinones. In homogeneous solutions of quinone-hydroquinone
redox couples, which were selected to model the redox-labile
functional groups in natural organic matter, rapid abiotic reduction
of nitro aromatic pesticides occurred. The authors proposed that
hydroquinones contribute to the reduction of pollutants in the
environment, but their role is likely to be complex.
The water hyacinth (Eichhornia crassipes), which is used for
water treatment, clears more than 98% hydroquinone (50 mg/litre)
after about 48 h (O'Keeffe et al., 1987). This property has been
attributed to enzymatic metabolism by polyphenol oxidases.
4.4 Ultimate fate following use
Hydroquinone occurs in photo-processing effluents (Dagon, 1973;
Harbison & Belly, 1982). However, it is not certain that it reaches
the water ecosystem, because reliable monitoring data are not
available.
5. ENVIRONMENTAL LEVELS AND HUMAN EXPOSURE
5.1 Environmental levels
5.1.1 Air, soil and water
No monitoring data have been found concerning ambient free
hydroquinone concentrations in air, soil or water. However,
hydroquinone has been identified in tobacco smoke and measured in
mainstream smoke from non-filtered cigarettes at amounts ranging
from 110 to 300 µg per cigarette, with a ratio of the sidestream to
mainstream concentration of 0.7-0.9 (IARC 1986).
5.1.2 Food
Free and conjugated (Arbutin) hydroquinone exist as natural
components of a variety of plant-derived beverages and food
products.
Högl (1958) identified hydroquinone in the non-volatile extract
of coffee beans. Hydroquinone concentrations in roasted coffee have
been reported to range between 25 and 40 mg/kg (Maier, 1981). Gold
et al. (1992) estimated that one cup of coffee would contain
approximately 100 µg hydroquinone. Teas prepared from leaves of
blueberry, cowberry, cranberry and bearberry have been reported to
contain hydroquinone at concentrations sometimes exceeding 1%
(Deichmann & Keplinger, 1981).
The concentrations of free and total (free hydroquinone and
Arbutin) hydroquinone have been measured in a variety of foods and
beverages by Hill et al. (1993); results indicate that significant
exposure to hydroquinone can occur through dietary sources. In most
of the samples (Table 5) derived from plant sources, the levels of
Arbutin are considerably higher than those of free hydroquinone.
However, Arbutin is hydrolysed readily by dilute acids yielding
hydroquinone and glucose. Therefore, both free hydroquinone and
Arbutin may contribute to hydroquinone exposure from natural sources
as well as to the daily intake of dietary antioxidants.
Adhesives containing trace amounts of hydroquinone are
permitted as a component of food packaging in the USA (FDA, 1981;
1991).
Table 5. Concentrations of free and total hydroquinone in various foods and
beverages
Food sample Concentrations (mg/kg ± SD)a
Free HQ Total HQb
Wheat germ, toasted 0.591 8.352
Drip-brewed coffee (pre-ground) 0.293 ± 0.003 0.385 ± 0.016
Whole wheat bread (100% whole wheat) 0.584 ± 0.202 0.893 ± 0.480
Whole wheat cereal (commercially available) 0.205 ± 0.019 0.992 ± 0.161
Processed corn cereal (commercially available) bkgb bkg
Pear skin (D'Anjou, fresh) bkg 38.057
Pear flesh (D'Anjou, fresh) bkg 1.301
Milkfat (2%) homogenized milk bkg bkg
Yogurt (black cherry) bkg bkg
Cantaloupe bkg bkg
Diet cola 0.0362 0.0287
a bkg = background levels comparable to that observed in control blanks
b Includes free hydroquinone and hydroquinone released following treatment
of the samples with ß-glucosidase
5.2 General population exposure
Photohobbyists, who develop their own black-and-white films (a
process which utilizes hydroquinone) may be exposed dermally.
Exposure to dust is also possible when preparing developer
solutions. In 1980, the number of photohobbyists was estimated to be
about 2.2 million in the USA (US EPA, 1985). There are no data on
exposure levels.
Dermal exposure to hydroquinone may also occur from products
intended for cosmetic and medical use. In the USA, hydroquinone has
been used in cosmetics, and in over-the-counter (OTC or
non-prescription) and prescription drugs. Both OTC and prescription
drugs are used to lighten areas of hyperpigmented skin. In
cosmetics, concentrations of < 0.1% to 5% have been reported
(CIR, 1986). OTC skin lighteners may contain up to 2% hydroquinone
and prescription drugs may contain higher concentrations. In the EC
countries, hydroquinone is restricted for use in cosmetics to 2% or
less (Boyle & Kennedy, 1985). The US Food and Drug Administration
has issued a Notice of Proposed Rule-making for the use of
hydroquinone as a skin lightener in OTC drugs at concentrations
below 1.5-2.0% (FDA, 1982).
Skin-lightening creams containing hydroquinone are frequently
inadequately labelled and the concentration often exceeds the limit
of 2%; it is likely to be much stronger than 2% (Brauer, 1985;
Godlee, 1992) and even up to 7% (Boyle & Kennedy, 1986).
A 2% upper limit on hydroquinone concentration, set by the
South African government in 1980 and followed by the United Kingdom
and USA, was based on tests of cutaneous irritancy (Arndt &
Fitzpatrick, 1965) and contact dermatitis (Bentley-Philips & Bayles,
1975).
5.3 Occupational exposure
Hydroquinone can be encountered in solid form or in solution
during its production and use (NIOSH, 1978). It has a very low
vapour pressure, but can be oxidized in the presence of moisture to
form quinone, which is more volatile. The saturated concentration in
air for hydroquinone vapour under standard conditions is estimated
to be 0.108 mg/m3 (approximately 0.024 ppm at 25 °C) (NIOSH,
1978).
There are some industrial hygiene monitoring data available for
hydroquinone. Oglesby et al. (1947) reported 20-35 mg
hydroquinone/m3 in a packaging area without exhaust cabinet and
1-4 mg/m3 in a packaging area with exhaust cabinet in a plant
manufacturing hydroquinone. However, the analytical methods did not
distinguish between hydroquinone and quinone. Industrial data,
provided to the US EPA (1985), indicated worker inhalation exposure
due to closed production processes within one manufacturing facility
at an arithmetical average concentration of 0.79 mg/m3 (± 0.52
standard deviation) and a highest average concentration of 0.2
mg/m3 in another facility. In the unloading area of a production
facility the arithmetic average air concentration was reported to be
0.13 mg/m3 (± 0.15 standard deviation). The concentration of
hydroquinone was measured in the workroom air in 12 Finnish plants
(altogether 36 samples) during the period 1950-89 (Rantanen et
al., personal communication to the IPCS, 1992). Most samples were
collected in the printing industry (23 samples from five plants).
The occupational exposure limit of 2 mg/m3 was exceeded in only
one measurement: 9.5 mg/m3 during charging of hydroquinone in a
gas plant in 1962, a short operation carried out once every three
weeks. Approximately 470 000 workers in the USA are potentially
exposed to hydroquinone in about 137 occupations (US EPA, 1985).
Certain occupations in which hydroquinone exposure may occur are
listed in Table 6. Some of the national occupational air exposure
limits used in various countries are compiled in Table 7.
Table 6. Occupations with potential exposure to hydroquinonea
Antioxidant makers
Drug makers
Hair dressers and cosmetologists
Hydroquinone manufacturing workers
Paint makers
Photo processors
Organic chemical synthesizers
Photographic developer makers
Plastic stabilizer workers
Rubber coating workers
a From: Key et al. (1977); NIOSH (1978); NIOSH (1990)
Table 7. National occupational air exposure limits used in various
countries (from: IRPTC, 1987; ILO, 1991)
Country TWAa STELb
CLVc
(mg/m3) (mg/m3) (mg/m3)
Australia 2
Belgium 2
Denmark 2
Finland 2 4
France 2
Germany 2d
The Netherlands 2
Poland 2 2
Romania 1 2
Sweden 0.5 1.5
Switzerland 2 4
United Kingdom 2 4
USA: ACGIH 2
NIOSH/OSHA 2
Yugoslavia 2
a TWA = time-weighted average; a maximum mean exposure limit based
generally over the period of a working day
b STEL = short-term exposure limit
c CLV = ceiling level value
d inhalable dust
6. KINETICS AND METABOLISM
The main results pertinent to this chapter, with the exception
of section 6.5, have been summarized in Table 8 and will be expanded
upon only where necessary. Table 8 shows that the majority of
studies have been performed in Fischer-344 rats.
6.1 Absorption
Absorption of orally administered or intratracheally instilled
hydroquinone is rapid and extensive (Garton & Williams, 1949;
Divincenzo et al., 1984; English et al., 1988). However, the
rate of hydroquinone absorption through the skin is low. Marty et
al. (1981) reported that the in vitro permeability constants for
rat and human skin were 28 x 10-6 and 4 x 10-6 cm/h,
respectively. Based on the data of Bucks et al. (1988), an in
vivo human dermal absorption rate of 3 µg/cm2.h and a
permeability constant of 2.25 x 10-6 cm/h can be calculated. The
actual amount of hydroquinone absorbed following dermal exposure
depends on the exposure concentrations, length of exposure and
vehicle, as well as other factors. When Bucks et al. (1988)
applied 14C-labelled hydroquinone in an alcoholic vehicle to the
foreheads of human volunteers for 24 h, 57% of the total 14C label
was excreted in the urine after 5 days. Addition of a sun screen to
the hydroquinone solution reduced total excretion to 26%.
6.2 Distribution
Following the oral administration of radiolabelled hydroquinone
to F-344 rats, radioactivity was widely distributed throughout the
animal tissues. The highest activity was localized in the kidney and
liver (Divincenzo et al., 1984). However, on a quantitative basis,
the amount retained within the animal was low, representing < 2%
of the total dose 48 h after exposure (Divincenzo et al., 1984;
English et al., 1988). Widespread distribution and extensive
elimination was also observed following intratracheal administration
of hydroquinone to F-344 rats (Lockhart & Fox, 1985b). However,
following the intravenous injection of radiolabelled hydroquinone to
F-344 rats, radioactivity was shown, using whole body
autoradiographic techniques, to concentrate in the bone marrow,
thymus and white pulp of the spleen (Greenlee et al., 1981a).
Subsequent experiments indicated that significant acid soluble and
covalently bound radioactivity could be recovered in the thymus,
bone marrow and white blood cells 24 h after intravenous
administration (Greenlee et al., 1981b). These results indicate
that the route of administration may influence the profile of
distribution and elimination observed following hydroquinone
administration.
Table 8. Summary of toxicokinetic data for hydroquinone (HQ)
Species and Absorption Distribution Metabolic Elimination and Reference
treatment transformation excretion
Oral administration
Species: rabbits less than 1% of the dose Garton &
Treatment: 3-6 rabbits was excreted unchanged; Williams
received 100 or 200 mg/kg about 80% of the dose was (1949)
HQ as a single dose; urine recovered as glucuronide
metabolites analysed after and monosulfate conjugates
24 h in the urine
Species: Sprague-Dawley T1 & T2: rapid and T1 & T2: for 200 mg/kg the major radio- mainly in the urine; Divincenzo
rats (m) extensive based 0.28-1.25% and labelled species in elimination for T1 and et al.
Treatment: (T1) 2-4 rats upon urinary 0.26-0.56% of the urine were: HQ T2 was similar; after (1984)
per group received 5, 30 excretion administered radioactivity monoglucuronide, HQ 48 h around 95% of
or 200 mg/kg [14C]-HQ as a recovered in carcass monosulfate and HQ dose had been excreted
single dose; rats were and tissues after 48 h (T1: 56%, 42% and in urine (90%), faeces (4%)
observed for 48 and 96 h and 96 h, respectively; 1%; T2: 72%, 23% and CO2 (0.4%); no
before sacrifice. widely distributedand 1%) difference in elimination
(T2) 4 rats pretreated with throughout tissues between single and
200 mg/kg unlabelled HQ with highest concentration repeated doses
once a day for 4 days in liver and
followed by 200 mg/kg kidney
[14C]-HQ on day 5; rats
observed for 48 h
Table 8. (contd).
Species and Absorption Distribution Metabolic Elimination and Reference
treatment transformation excretion
Species: Fischer-344 T1 & T2: rapid and T1 & T2: < 1% of T1 & T2: the major Excreta English
rats (m,f) extensive based administered radiolabelled species T1 & T2: mainly excreted et al.
Treatment: (T1) 8 (m,f) upon peak blood radioactivity recovered in the urine were in the urine; after 48 h (1988)
rats per group received 25 concentration in carcass and HQ monoglucuronide around 90% of the
or 350 mg/kg [14C]-HQ as within 0.8 h of tissues for each dose (44-54%), HQ mono- administered radioactivity
a single dose. dosing and urinary after 48 h; twice as sulfate (19-33%), was recovered as urine
(T2) 8 rats (m,f) excretion; much radioactivity HQ (0.25-7%), HQ (approx.78%), cage rinse
pretreated with 25 mg/kg no sex recovered in the mercapturate (approx.12%) and faeces
unlabelled HQ once a day differences liver and kidney (0.16-4.68%) and (approx.2.2%); dose-related
for 14 days followed by 25 of females compared p-benzoquinone differences were observed
mg/kg [14C]-HQ on day 15; with males (0.24-0.84%); no at 8 h, 54% (m) and 45% (f)
after oral administration sex difference of the dose was excreted
4 rats per dose and renally by the high-dose
sex were designated group compared with 81% (m)
for blood collection and 82% (f) for the low-
samples (up to 96 h) dose group
and for excreta and
radiodistribution Blood kinetics
(up to 7 days) AUC values were increased
by 17-fold (m) and 26-fold
(f) for a 13- and 14-fold
higher mean dose; most of
radioactivity was excreted
by 8 h and was associated
mainly with alpha-
elimination phase (T´ =
0.23-1.72 h); accurate ß T´
could not be determined
because of the appearance
of a second peak in the
blood concentration versus
Table 8. (contd).
Species and Absorption Distribution Metabolic Elimination and Reference
treatment transformation excretion
Species: Fischer-344 rapid and extensive low distribution, by 8 h, major time curve by 24 h, Lockhart
rats (m,f) absorption as i.e. less than 1%; metabolites found recovery was more than 92% et al.
Treatment: a single dose indicated by marked no significant in urinewere HQ in urine, approx.2% in (1984);
of 5, 25 or 50 mg [U-14C]- recovery of [14C] differences between glucuronide faeces and less than 0.2% Lockhart
HQ/kg body weight by in urine by 24 h sexes (approx.50%), in CO2 & Fox
gavage HQ sulfate (1985a)
(approx.30%) and
HQ (approx.2%);
neither dose- nor
sex-dependent
Species: Fischer-344 rapid and extensive low distribution: by 8 h, major by 24 h, recovery was Lockhart
rats (f) absorption as approx. 0.55% in liver and metabolites found in approx.92% in urine, & Fox
Treatment: 5 rats per dose indicated by marked 0.64-0.9% in carcass urine were HQ approx.2.6% in faeces and (1985a)
group 5, 25 or 50 mg recovery of [14C] glucuronide (approx. approx.0.3% in CO2
[U-14C]-HQ/kg body weight in urine by 24 h 46%), HQ sulfate
(single gavage dose) (approx.29-36%) and
HQ (approx.2.5%)
Dermal administration
In vitro: Rat or human overall absorption Marty et
skin biopsy; repeated and permeability al. (1981)
dosing with 40 mg/cm2 in constant were low
water; observed for 24 h but on average
7-fold greater for
rat than human skin
In vivo: Mouse or rat absorption by local cutaneous combined urine and faecal
mouse was low; distribution was high elimination was low;
1.6% after 6 h in rat approx.10% after 96 h in
the rat
Table 8. (contd).
Species and Absorption Distribution Metabolic Elimination and Reference
treatment transformation excretion
Species: human average percutaneous Bucks et
Treatment: 6 normal adult taneous absorption al. (1988)
male volunteers had 2% estimated from
(w/w) HQ in ethanol urinary elimination
(approx.70%) plus 0.2% data was 57% after
ascorbic acid applied to 120 days; sun-
their foreheads for 24 h; screens decreased
single dose = 125 µg/cm2; absorption but
observed for up to 120 h penetration
enhancers were
without effect
Species: Fischer-344 skin irritated but after 1 week, 15-18% HQ English
rats (m,f) poorly absorbed; was recovered in urine and et al.
Treatment: 8 (m,f) rats large interanimal cage rinsings, 1.7-3.7% in (1988)
per group were dermally variation in the faeces, 2.6 to 12.9%
exposed for 24 h to 25 disposition; removal in the body and 0.14 to
or 150 mg/kg [14C]-HQ of HQ after skin 2.2% in the excised
dissolved in distilled washing with soapy skin exposure site
water for 24 h water was close to
100% after 10 min of
exposure or around
65% after 24 h of
exposure
Table 8. (contd).
Species and Absorption Distribution Metabolic Elimination and Reference
treatment transformation excretion
Intravenous administration
Species: Fischer-344 rat (m) whole body Greenlee
Treatment: 1.3 mg/kg autoradiography showed et al.
[14C]-HQ in saline that [14C] concentrated (1981a)
administered as a single most in the white pulp
dose; one group of rats of the spleen, bone marrow
was pretreated with and thymus; Aroclor 1254
Aroclor 1254 (250 mg/kg pre-treatment decreased
i.p.) the tissue/blood optical
density by approx.60% for
the thymus and bone marrow
Species: Fischer-344 rat (m) acid-insoluble Greenlee
Treatment: rats received radioactivity associated et al.
14 mg/kg [14C]-HQ as a with protein increased (1981b)
single administration; one with time in the bone
group of rats was marrow > thymus > liver;
pretreated with Aroclor pretreatment with Aroclor
1254 resulted in a significant
decrease in the
radioactivity measured in
the bone marrow
Intratracheal instillation
Species: Fischer-344 rapid and extensive < 0.13% in lung, less by 8 h, major by 48 h recovery was Lockhart
rats (m) absorption as than 1% to other organs metabolites recovered more than 92% in urine, & Fox
Treatment: 5 rats per dose indicated by in the urine were approx.2% in faeces and (1985b)
group: 5, 25 or 50 mg recovery in urine HQ-glucuronide less than 0.2% in CO2
[U-14C]-HQ/kg; 2 rats per within 24 h (approx.50%),
control group HQ-sulfate
(approx.30%) and
HQ (approx.2%)
Table 8. (contd).
Species and Absorption Distribution Metabolic Elimination and Reference
treatment transformation excretion
Intraperitoneal administration
Species: Wistar rat (f) metabolites recovered elimination was rapid with Inoue et
Treatment: 9 rats received in urine were 1,2,4- 84% of the metabolites al.
a single 50 mg/kg dose benzenetriol (11%), recovered within 4 h after (1989a)
catechol (1%) and administration; by 24 h,
hydroquinone (87%) recovery of 1,2,4-
benzenetriol, catechol and
hydroquinone in the acid-
hydrolysed urine comprised
38% of the administered
dose
Species: Japanese white metabolites recovered by 24 h, recovery of Inoue et
rabbits in urine were 1,2,4- 1,2,4,-benzenetriol, al.
Treatment: 5 rabbits benzenehydrotriol catechol and quinone in (1989b)
received a single 50 mg/kg (12%), catechol (1%) the acid-hydrolysed urine
dose and hydroquinone comprised 40% of the
(86%) administered dose
6.3 Metabolic transformation
Hydroquinone is converted mainly by Phase II metabolism to
water-soluble conjugates, as shown by the recovery of only little
parent compound and p-benzoquinone (0.25-7%) but large amounts of
hydroquinone-monoglucuronide and hydroquinone-monosulfate (>90%) in
the urine (Divincenzo et al. 1984; English et al. 1988). A small
percentage of the dose was recovered as the mercapturic acid
conjugate of hydroquinone, suggesting the intermediate formation of
a glutathione conjugate of hydroquinone.
Divincenzo et al. (1984) demonstrated that repeated dosing
with 200 mg hydroquinone/kg did not alter the relative or absolute
rat liver weight or induce the hepatic mixed-function oxidase
system, nor did hydroquinone undergo Phase I oxidation to other
metabolites such as 1,2,4-trihydroxybenzene. In addition, the
formation of 1,2,4-trihydroxybenzene was not observed in the urine
after oral administration of hydroquinone to rabbits (Garton &
Williams, 1949). However, following intraperitoneal injection of
hydroquinone (50 mg/kg) in Wistar rats and Japanese white rabbits,
1,2,4-trihydroxybenzene represented a significant proportion
(approximately 12%) of the metabolites recovered in the urine (Inoue
et al., 1989a,b). This apparent difference in the metabolic
profile observed when hydroquinone is administered by the
intraperitoneal route rather than the oral route is probably related
to the efficient ability of the gastrointestinal system to conjugate
phenolic compounds absorbed in the intestine, thus reducing the
amount of free hydroquinone available for Phase I metabolism in the
liver (Powell et al., 1974; Cassidy & Houston, 1980a,b; Cassidy &
Houston, 1984). Fig. 5 shows proposed metabolic pathways for
hydroquinone biotransformation in Fischer-334 rats.
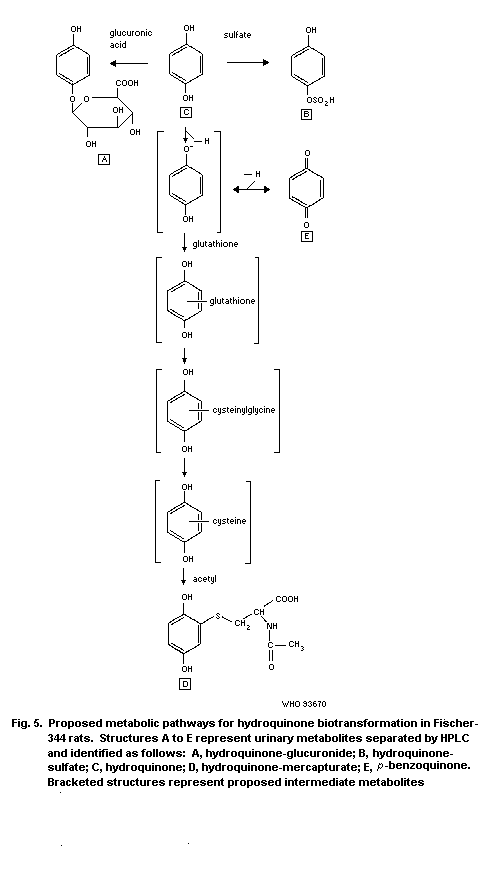 6.4 Elimination and excretion
Hydroquinone is excreted mainly in the form of water soluble
metabolites via the urine (about 90%). Dose-related differences have
been observed for rats receiving 25 or 350 mg/kg, which suggests
that elimination processes are saturated at high-dose levels
(English et al., 1988). The area under the curve (AUC) values for
plasma concentration, which provide an index of bioavailability,
also showed that saturation of elimination had occurred at high-dose
levels, particularly for females. The fact that most of the
radioactivity excreted is associated with the alpha-elimination
phase suggests that this may be due to conjugation of hydroquinone
to readily excreted metabolites. The appearance of a double peak in
the blood concentration versus time curve indicates that
enterohepatic recycling of hydroquinone may have occurred.
6.5 Reaction with body components
The available studies suggest that hydroquinone derivatives are
responsible for many of the toxicological effects associated with
in vivo and in vitro hydroquinone exposure. Hydroquinone itself
may be responsible for the acute CNS signs (tremors and convulsions)
that are seen within the first hour following hydroquinone exposure
(see section 7.8.3), since the signs appear soon after exposure when
significant metabolism has probably not occurred. However, it is
possible that derivatives even have a role in inducing CNS effects.
The derivatives formed from hydroquinone may differ between in
vivo and in vitro studies. Even when the in vivo situation
alone is considered, the derivatives may vary qualitatively and
quantitatively, and the concentrations of derivatives in the various
body compartments may be different depending on the route of
exposure. When hydroquinone is administered by expected routes of
exposure, the primary derivatives should be largely glucuronide and
sulfate conjugates, which are quickly exported, as well as
glutathione conjugates, which may represent activated metabolites.
When hydroquinone is given by intraperitoneal or intravenous routes,
the primary metabolites are expected to be 1,4-benzoquinone and
1,2,4-trihydroxybenzene. In most in vitro systems the primary
metabolite is expected to be 1,4-benzoquinone. Hydroquinone-and
oxygen-derived radical species are also likely to be formed both in
vivo and in vitro. The hydroquinone-derived metabolites and
radical species formed in vitro will depend on the oxygen content,
the pH, the ionic strength, the autooxidant and the protein content
of the culture or reaction medium used in the study, as well as
other factors including the metabolic capacity of the test system.
The differences in the potential derivatives and the
concentrations of the derivatives occurring in the different in
vivo and in vitro exposure systems studied indicate that
extrapolations from in vitro to in vivo systems and between
routes of exposure need to be made with a great deal of care.
The main results pertinent to this section have been summarized
in Table 9, which shows that many of the interactions of
hydroquinone have been identified in vitro but not all have been
demonstrated in vivo. Hydroquinone reacts with many different
biological components, including macromolecules such as protein,
DNA, tubulin, lipids, and low molecular weight molecules such as
sulfydryls and nucleotides, is toxic to different cell types, has
affects on cellular metabolism, and modulates enzyme activities.
Covalent binding and oxidative stress are mechanisms postulated
to be associated with hydroquinone-induced toxicity. Both oxidized
hydroquinone species ( p-benzosemiquinone radical and
p-benzoquinone) and thiol-hydroquinone/quinone conjugates are
believed to contribute to hydroquinone toxicity.
Oxidized hydroquinone derivatives can covalently bind cellular
macromolecules or alkylate low molecular weight nucleophiles, e.g.,
glutathione (GSH), resulting in enzyme inhibition, alterations in
nucleic acids and oxidative stress; however, redox cycling is not
likely to contribute significantly to oxidative stress in contrast
with other hydroquinones and quinones (see section 2.2; Rossi et
al., 1986; O'Brien, 1991). The reaction of benzoquinone with GSH
results in the formation of GSyl conjugates which can be processed
to cysteine conjugates. These latter thiol conjugates have been
speculated to mediate cellular toxicity in the kidney by alkylation
and/or oxidative stress, possibly involving redox cycling (Lau et
al., 1988).
Table 9. Summary of the reactions of hydroquinone (HQ) with biological componentsa
Index studied Method Result Reference
Reactions with macromolecules
Covalent binding to 14 mg/kg [14C]-HQ was administered acid-insoluble radioactivity associated with Greenlee et al.
cell protein i.v. as a single dose to Fischer-344 rats protein increased with time in the bone (1981b)
(in vivo) (m); after 2 and 24 h, acid-insoluble marrow > thymus > liver; pretreatment with
radioactivity associated with proteins Aroclor resulted in a significant decrease in
was determined; one group of rats was the radioactivity measured in the bone marrow
pretreated with Aroclor 1254
Covalent binding to 25-75 mg/kg [14C]-HQ was incubated radioactivity associated with protein; the Eastmond et al.
boiled rat liver in the absence or presence of phenol presence of PhOH enhanced this association (1987)
protein (in vitro) (PhOH) (75 mg/kg) with H2O2-horseradish
peroxidase or freshly isolated human
polymorphonuclear leucocytes in the
presence of boiled rat liver protein
Covalent binding to 75 mg/kg [14C]-HQ alone or coadministered after 18 h of administration, acid-insoluble Subrahmanyam et
cells (in vivo) with phenol (PhOH) (75 mg/kg) radioactivity was found associated with al. (1990)
was administered i.p. (probably as a kidney > blood > bone marrow; coadministration
single dose) to pathogen-free male with PhOH significantly (statistically)
B6C3F1 mice (5-12); after 4 and 18 h, increased binding to blood and bone marrow
acid-insoluble radioactivity associated but not kidney or liver
with macromolecules was isolated and
analysed for covalent binding
Covalent binding to isolated liver microsomes from male radioactivity associated with microsomal Wallin et al.
microsomal proteins S-D rats, either treated or untreated with proteins; binding was more extensive than (1985)
(in vitro) phenobarbital or 3-methyl cholanthrene, that of phenol and independent of electron
was incorporated with [14C]-HQ, both with donors
and without NADPH
Table 9. (contd).
Index studied Method Result Reference
Covalent binding to peritoneum macrophages isolated from [14C]-HQ was activated by macrophages to Schlosser et
cells (in vitro) male C57BL/6 mice were incorporated metabolites that bind irreversibly to protein; al. (1989);
with [14C]-HQ activation was inhibited by peroxidase inhibitor Schlosser &
aminotriazine and the nucleophile cysteine and Kalf (1989)
enhanced by arachidonic-acid-mediated
prostagladin synthesis catalysed reaction
Covalent binding to bone marrow macrophages and a fibroblastoid radioactivity associated with macrophages was Thomas et al.
cells (in vitro) stromal cell (LTF) line obtained 16-fold higher than for LTF cells; DT- (1989); Ross et
from male B6C3F1 mice were incorporated diaphorase activity [Q -> HQ] was 4 times al. (1990)
with [14C]-HQ higher on LTF cells than in macrophages;
slightly decreased (approx.16%) by addition of
dicoumarol, an inhibitor of DT-diaphorase,
for LTs but not macrophages
Chromosomal aberration example: bone marrow cells were isolated micronuclei induced in polychromatic Tunek et al.
(in vivo) (see also from male NMRI mice (4 per group) erythrocytes (1982)
section 7.6) administered between 20 and 100 mg HQ/kg
by subcutaneous injection once a day for
6 days
Mitochondrial DNA mitoplasts isolated from rabbit bone covalent adduct formed with guanine Rushmore et
(in vitro) marrow cells were prelabelled with al. (1984)
[3H]-dGTP incorporated with [14C]-HQ and
assayed for guanosine adduct formation
DNA damage (in vitro) example: calf thymus DNA was incubated two adducts identified Jowa et al.
(see also section 7.6) with [14C]-HQ in the presence of Fe3+ at (1990)
pH 7.2
Microtubulin binding T1: isolated brain microtubulin from male T1: HQ inhibited microtubulin polymerization Irons & Neptun
(in vitro) Fischer-344 rats was incubated with between and bound to high molecular weight tubulin; (1980)
1 and 1.5 x 10-4 mol/litre [14C]-HQ anaerobic conditions inhibited polymerization
Table 9. (contd).
Index studied Method Result Reference
T2: isolated spleen lymphocytes from rat T2: HQ suppressed lectin-induced blastogenesis Pfeifer & Irons
were incubated with HQ (10-6-10-4 mol/litre) and concomitant inhibition of cell (1983)
agglutination
Lipids (in vivo) SD rats received a single oral dose of 100 urinary MDA increased in HQ-treated rats Ekström et
or 200 mg HQ/kg; malondialdehyde (MDA), al. (1988)
a lipid peroxidation product, was analysed
in the excreted urine for up to 18 h
Cytochrome c3+ stop- and continuous-flow experiments HQ reduces cytochrome c3+ via Yamazaki & Ohnishi
reduction (in vitro) p-benzosemiquinone; reaction accelerated by (1969); Ohnishi
p-benzoquinone et al. (1969)
Reactions with low molecular weight molecules
Thiol conjugation glutathione thiol conjugates formed by reductive addition; Tunek et al.
(in vitro) monothiol HQ conjugate formed by the reaction (1980)
of p-benzoquinone with thiol after oxidation of
HQ to the semiquinone or quinone
glutathione di, tri and tetra (GSyl)-HQ conjugates are Eckert et al.
formed by reductive addition of the oxidized (1990)
(GSyl)-HQ conjugate, i.e. quinone conjugate with
GSH
monocysteine-HQ conjugate HQ oxidized by prostaglandin H systhetase Schlosser et
al. (1990)
Nucleotide adduct [3H]-deoxyguanosine and [14C]-HQ two doubly labelled products isolated; adduct Jowa et al.
(in vitro) incubated in the presence of Fe3+ at pH 7.2 2: 3-OH benzethano (1, N2) deoxyguanosine (1990)
2-Thiobarbituric glutamate or deoxyribonucleic acid 2-thiobarbituric acid produced; hydroxyl radical Rao & Pandya
acid (in vitro) incorporated with HQ plus Cu2+ at pH 7.4 (OH) formation thought to be involved (1989)
Table 9. (contd).
Index studied Method Result Reference
Toxicity to cells
Erythrocytes (in vivo) polychromatic erythrocytes isolated from 20 mg/kg: no haemotoxic effect; 100 mg/kg: Tunek et al.
male NMRI mice (4 per group) haemotoxic effects (suppressed bone marrow (1982)
administered between 20 and 100 mg cellularity)
HQ/kg s.c. once a day for 6 days
Bone marrow (in vivo) i.p. administration of HQ (100 mg/kg, twice transient, mild suppression in bone marrow Eastmond et
daily for 12 days to six male B6C3F1 mice) cellularity al. (1987)
i.p. co-administration of HQ (25-75 mg/kg) significant decrease in bone marrow cellularity; Eastmond et
and phenol (75 mg/kg) twice daily for phenol enhanced HQ-induced myelotoxicity al. (1987)
12 days to groups of six male B6C3F1 mice
Isolated rat spleen responses of spleen cells from F-344 rats low concentrations (10-7-10-6) enhanced Irons & Pfeifer
and lymphocytes were assayed after addition of mitogen and mitogenesis, higher concentrations (10-5) (1982)
(in vitro) phytohaemagglutinin A suppressed mitogen response
Pigment cells (in vitro) toxic effects of HQ on melanotic cell lines toxic effects occurred between 0.625 and HU (1966)
(MCL) and non-melanotic cell lines (NMCL) 2.5 µg/ml for MCL and NMCL
was studied
Cell line (in vitro) lymphoma-derived cell line Raji, erythro- percentage survival decreased for all cells Picardo et al.
leukaemia cell line K 562 and human (approx. 65%, low dose) (approx. 20%, high dose) (1987)
melanotic cell lines IRE 1 and IRE 2 were
incubated with 0.01 and 0.1 mmol HQ/litre
Bone marrow cells bone marrow cells isolated from the femurs HQ decreased the number of mature surface King et al.
(in vitro) and tibias of male B6C3F1 (C57BL/6J x IgM+ B cells and adherent cells; HQ may block (1987)
C3h/HeJ) mice were incubated with final maturation stages of B cell
between 10-7 and 10-5 mol HQ/litre differentiation
Table 9. (contd).
Index studied Method Result Reference
Cell line (in vitro) bone marrow macrophage and a fibroblastoid HQ (10-4 mol/litre) decreased viability and Thomas et al.
stromal cell line isolated from male colonies for macrophages (60% and 70%, (1989b)
B6C3F1 mice were incubated with respectively) and stromal cells (30% and
between 10-8 and 10-4 mol HQ/litre 0%, respectively)
Cell line (in vitro) bone marrow stromal cells isolated from HQ cytotoxicity was greater in stromal cells Twerdok & Trush
male DBA/2 mice and C57BL/6 mice derived from DBA/2 than C57BL/6 mice; tert- (1990); Twerdok
were incubated with HQ butylhydroquinone (tBHQ) or 1,2-dithiole-3- et al. (1992)
thione (DTT) preincubation protected against
HQ-induced toxicity; dicoumarol-sensitive
quinone reductase activity was increased by
tBHQ and DTT, and levels of GSH increased with
DTT
Cell line (in vitro) human promyelocytic leukaemia cell line, HQ dose-dependently inhibited TPA- and 1,25- Oliveira & Kalf
which can be induced to differentiate to dihydroxy vitamin D3-induced (but not (1992)
both monocyte and myeloid cells, was interleukin (IL)-1) acquisition of
incubated with HQ (0.01 µmol/litre to differentiation characteristics of monocytes
10 µmol/litre) (adherence, nonspecific esterase activity and
phagocytosis), but had no effect on cell
proliferation; retinoic-acid- or DMSO-induced
differentiation to granuloctyes was not
inhibited at the same doses
Hepatocytes (in vitro) freshly isolated hepatocytes (106/ml) time-dependent cell death; complete by 120 min O'Brien (1991)
incubated with 850 µM HQ; % cell viability
measured by Trypan blue inclusion
Effects on cellular metabolism
Haemoglobin (Hb) five cats were treated every second day up 15-30% Hb oxidized to ferric form Jung & Witt
(in vivo) to 12 times with HQ (40-160 mg/kg) (1947)
Table 9. (contd).
Index studied Method Result Reference
Reduction of haemoglobin HQ incubated with Hb in presence of O2 very slow formation of ferrihaemoglobin Oettel (1936)
(Hb) (in vitro)
Iron utilization female Swiss albino mice administered inhibition (70%) of erythroid 59Fe utilization Guy et al.
(in vivo) between 25 and 100 mg HQ/kg 3 times at occurred only at the highest dose; (1990, 1991)
64, 48 and 40 h prior to administration coadministration of PhOH or muconaldehyde
of 59Fe; coadministration with phenol enhanced the inhibitory effects of HQ
(PhOH) (50 mg/kg)
Cellular RNA and DNA the effects of HQ on nucleoside HQ selectively inhibited the metabolism of MCL Pennay et al.
synthesis (in vitro) incorporation in two melanotic cell lines cf. NMCL; [3H]-uridine incorporation was de- (1984)
(MCL) and three non-melanotic cell lines creased approx. 30-fold in MCL cf NMCL; [3H]-
(NMCL) was observed uridine incorporation was more sensitive to HQ
than [3H]-thymidine; DNA and RNA syntheses were
decreased by 80 and 50%, respectively, in MCL;
HQ may exert depigmenting effect by selective
action on MCL metabolism rather than specific
effect on melanin synthesis
Mitochondrial synthesis mitoplasts isolated from rabbit bone RNA synthesis inhibited (IC50 = 5.0 x 10-5 mol Rushmore
marrow cells were incorporated with HQ HQ/litre) et al. (1984)
and assayed for RNA synthesis
Cyclic nucleotides three different melanomas were treated cAMP and cGMP were elevated in 3/3 and 2/3 Abramowitz &
(in vitro) with HQ and assayed for cAMP and cGMP tumours, respectively Chavin (1980)
by radioimmuno assay
Cytokine synthesis murine P388D1 macrophages or bone HQ caused a concentration-dependent inhibition Renz et al.
marrow stromal macrophages were of the processing of 34-Kd pre-interleukin-1 (1991)
incubated with HQ (0.5-10 µmol/litre) alpha (IL-1alpha) to 71-Kd mature cytokine in
both types of macrophages; lipopolysaccharide-
induced production of the pre-IL-1alpha
precursor or cell viability or DNA and protein
synthesis were not inhibited
Table 9. (contd).
Index studied Method Result Reference
Effects on enzymes
Tyrosine-tyrosinase radiometric assay tyrosinase inhibited by HQ (9 x 10-4 mol/litre); Usmani et al.
(tyrosine --> dopa) suggested that HQ is a competitive inhibitor (1980); Palumbo
(required for skin et al. (1991)
pigmentation)
Catalase (in vivo) male Wistar rats received 5 mg HQ/kg per 5 mg HQ/kg per day inhibits catalase activity in Vladescu &
day p.o. for 10 days and H 18R tumour- the liver, spleen, blood and H 18R tumour Apetroae (1983)
bearing rats received 5 mg HQ/kg per day
p.o. for 7 days; [14C]-HQ was
administered i.p.
Ornithine decarboxylase SD rats received a single oral dose of 100 ODC activity was increased in a dose- Ekström et al.
(ODC) or 200 mg HQ/kg; liver ODC activity was dependent manner (1988)
measured after 18 h of exposure
Horseradish peroxidase [14C]-phenol incubated with H2O2-HRP in radioactivity associated with protein Eastmond et
(HRP) the presence of boiled rat liver protein; decreased when coincubated with HQ; al. (1987)
coincubated with HQ competitive inhibition presumed
Table 9. (contd).
Index studied Method Result Reference
Reaction of HQ metabolites
Benzoquinone (BQ)/ numerous: BQ/SQ formed by autooxidation rate of enzyme-mediated formation of BQ is Yamazaki et al.
benzosemiquinone (SQ) and 1e-mediated enzymic oxidation (e.g., much faster than autooxidation; formation of (1960); Sawada
horseradish peroxidase (HRP), BQ is enhanced by coincubation of HRP and et al. (1975);
myeloperoxidase (MPO) and prostaglandine MPO with phenol and other compounds; O'Brien (1991);
H synthase (PHS)) of HQ indomethacin partially inhibits H2O2-dependent Smith et al.
HQ oxidation with HRP or MPO or PHS, but (1989); Hsuanyu
substantially inhibits arachidonate-dependent & Dunford (1992);
oxidation mediated by PHS; this latter finding Eastmond et al.
indicates the involvement of cyclooxygenase; (1987); Thomas
both BQ and SQ are electrophiles which readily et al. (1989);
react with low and high molecular weight Subrahmanyam et
nucleophiles; BQ is believed to be the al. (1991);
proximate toxicant responsible for benzene-/ Schlosser
hydroquinone-induced myelotoxicity et al. (1990)
GSH conjugate (in vivo) male Sprague-Dawley rats were administered nephrotoxicity occurred with tris-(GSyl)-HQ > Lau et al.
various (GSyl)-HQ conjugates i.v.; di-(GSyl)-HQ conjugate; mono and tetra (1988)
nephro- and hepatotoxicity were measured conjugates were not toxic; toxicity of the tris
as increased plasma blood urea nitrogen conjugate was depressed by AT-125, a gamma
(BUN) and serum glutamate pyruvate glutamyl trans-peptidase (gamma-GT) inhibitor;
transaminase (SGPT), respectively it is suggested that gamma-GT is probably
required for the transport of the latent quinone
into proximal tubular cells as the corresponding
cysteine conjugate; alkylation and/or oxidation
are suggested mechanisms of molecular toxicity
Table 9. (contd).
Index studied Method Result Reference
GSH conjugate (in vitro) O2 consumption measurement of in situ autooxidation was stimulated by SOD Brunmark &
formed glutathionyl-p-benzoquinone HQ Cadenas (1988); cf.
conjugate in potassium phosphate buffer Eckert et al.
at pH 7.65 in the absence and presence of (1990)
superoxide dismutase (SOD)
a NADPH = reduced nicotinamide adenine dinucleotide
cAMP = cyclic adenosine monophosphate
cGMP = cyclic guanosine monophosphate
7. EFFECTS ON LABORATORY MAMMALS AND IN VITRO SYSTEMS
7.1 Single exposure
The acute toxicity of hydroquinone has been studied in several
animal species (Tables 10 and 11). Oral LD50 values for different
strains of rats range from 720 to 1300 mg/kg body weight (Carlson &
Brewer, 1953; Mozhaev et al., 1966). Fasting the animals for 18 h
previous to the administration of hydroquinone produced a two- to
three-fold increase in the observed toxicity (Carlson & Brewer,
1953). The LD50 of unfed rats was 310 mg/kg, in contrast to an
average of 1064 mg/kg observed in non-fasted animals. Oral LD50
values range from 340 mg/kg to 400 mg/kg body weights for mice, and
are 550 mg/kg body weight for guinea-pigs, 540 mg/kg body weight for
rabbits and 299 mg/kg body weight for dogs. Cats have a greater
sensitivity (LD50 values of 42-86 mg/kg body weight).
Signs of hydroquinone intoxication were developed 30-90 min
after single oral doses and consisted of hyperexcitability, tremors,
convulsions, dyspnoea and cyanosis. They also included salivation in
dogs and cats, emesis in dogs and pigeons, swelling of the tissues
around the eyes, and incoordination of the hind limbs of dogs
(Woodard 1951; Christian et al., 1976; Deichmann & Keplinger,
1981). These signs were followed by complete exhaustion,
hypothermia, paralysis, loss of reflexes, coma, respiratory failure
and death. When the dose was sublethal, recovery was complete within
three days (Christian et al., 1976).
Single-dose acute dermal toxicity studies have not been
reported. However, the acute dermal LD50 can be estimated to be >
3840 mg/kg for mice and 74 800 mg/kg for rats based on effects
observed in two-week dermal studies (NTP, 1989). Hydroquinone as a
2% solution in dimethyl phthalate caused no adverse local or
systemic effects in rabbits (Draize et al., 1944).
In the rat, the LD50 values for intraperitoneal
administration varied between 160 and 194 mg/kg body weight and the
LD50 for intravenous administration was 115 mg/kg body weight
(Woodard, 1951). In rabbits, intravenous injections of hydroquinone
caused death at doses of 100-150 mg/kg body weight (Delcambre et
al., 1962).
Table 10. Acute oral toxicity of hydroquinone in experimental animals
Species Material tested LD50 (mg/kg Comments Reference
body weight)
Rat 2% aqueous solution 320 rapid onset of symptoms: twitchings, Woodard (1951)
Osborne-Mendel tremors, convulsions, respiratory
failure and death within a few hours
Rat
Priestly glycerine 1005-1295 unfasted rats; the mean LD50 value Carlson & Brewer (1953)
Sprague-Dawley propylene-glycol 1090 was 1050 mg/kg
Sprague-Dawley distilled water 1182
Sprague-Dawley glycerine 1081
Wistar propylene-glycol 731
Sprague-Dawley propylene-glycol 323 fasted rats; the mean LD50 value Carlson & Brewer (1953)
Wistar 298 was 310 mg/kg
Rat water 743 (m) when the dose was sufficiently large, Christian et al. (1976)
627 (f) death occurred during a severe tonic
spasm within 2 h; when the dose was
sublethal, recovery was complete
within 3 days
Mouse 2% aqueous solution 400 the symptoms are similar to those Woodard (1951)
Swiss in rats
Guinea-pig 2% aqueous solution 550 the symptoms are similar to those Woodard (1951)
in rats
Cat 2% aqueous solution 70 the symptoms are similar to those in Woodard (1951)
rats except for salivation and swelling
of the area around the eye noted in cats
Cat sugar-coated tablets 42-86 Carlson & Brewer (1953)
Table 10. (contd).
Species Material tested LD50 (mg/kg Comments Reference
body weight)
Dog sugar-coated tablets 299 similar symptoms to those of cats Carlson & Brewer (1953)
Dog 2% aqueous solution 200 hyperexcitability, tremors, convulsions, Woodard (1951)
salivation, emesis, incoordination of the
hind legs, respiratory failure, and death;
100 mg/kg caused mild to severe swelling
of the area around the eye, of the nictating
membrane and of the upper lip
Rabbit 2% aqueous solution 540 the symptoms are similar to those in rats Woodard (1951)
Pigeon 2% aqueous solution 300 the symptoms are similar to those in Woodard (1951)
rats, except for emesis noted in pigeons
Table 11. Acute parenteral toxicity of hydroquinone in experimental animals
Species Material Administration LD50 (mg/kg body Comments Reference
tested route weight) (except
where otherwise
stated)
Rat (Osborne-Mendel) 2 or 3% aqueous intraperitoneal 160 Woodard (1951)
solution
Rat (Wistar) intraperitoneal 194 Delcambre et al. (1962)
Rat (Osborne-Mendel) 2 or 3% aqueous intravenous 115 Woodard (1951)
solution
Rabbit intravenous 150a death Delcambre et al. (1962)
100 tremor, hypotonia, death
10-20 hypertension, hyperkalaemia
Mouse subcutaneous 160-170 (LLD)b Busatto (1940)
Mouse 1% solution subcutaneous 182 Marquardt et al. (1947)
Mouse subcutaneous 190 most animals died within 24 h Nomiyama et al. (1967)
a Single dose
b LLD = lowest lethal dose
Single subcutaneous injections of up to 500 mg of
hydroquinone/kg body weight in white mice caused symptoms in the
central nervous system: markedly increased motor activity,
hyperactive reflexes, sensitivity to light and sound, laboured
breathing and cyanosis, followed by clonic convulsions, complete
motor exhaustion, paralysis, a nearly complete loss of sensitivity
and reflexes, semicoma and death (Busatto, 1940). The lowest lethal
dose was 160-170 mg/kg. The subcutaneous LD50 for hydroquinone has
been found to be 182-190 mg/kg in mice (Marquardt et al., 1947;
Nomiyama et al., 1967).
No experimental data on inhalation exposure have been located.
7.2 Skin and eye irritation; sensitization
7.2.1 Skin irritation
Hydroquinone was applied to both epilated and unepilated skin
of eight black guinea-pigs at concentrations of 1, 3, 5, 7 and 10%
in three vanishing creams (Bleehen et al., 1968). The number of
animals per dose group was not reported. Six animals served as
controls. The material was applied once daily, five times a week,
for one month. Hydroquinone was irritating only at concentrations of
5% or more. Weak to moderate depigmentation occurred in all areas of
skin to which creams containing 1-10% hydroquinone were applied.
Hydroquinone (0.001, 0.01 and 0.1% in 0.1 ml water) was not
found to be a primary irritant when administered intracutaneously in
18 female guinea-pigs during a period of 10 days (Rajka & Blohm,
1970).
Jimbow et al. (1974) reported depigmentation in the epilated
skin of 24 black guinea-pigs (both males and females) after topical
applications of hydroquinone. Creams containing 2 or 5% hydroquinone
in an oil-water emulsion were applied daily, 6 days a week, for 3
weeks. The depigmentation was first seen within 8-10 days and was
greatest between 14 and 20 days. It was more marked at the higher
concentration. Inflammatory changes and thickening of the epidermis
were also reported. When hydroquinone was applied topically for
three weeks, biopsy specimens showed that it had caused a marked
reduction both in the number of melanized melanosomes in the cells
and the number of actively functioning melanocytes.
In a preliminary screening study with eight guinea-pigs, an
aqueous solution of hydroquinone was slightly irritating at 10% but
not at 0.5, 1.0 or 5.0% (Springborn Institute for Bioresearch,
1984).
The potential of hydroquinone to produce skin depigmentation
and irritation has also recently been studied in male and female
black guinea-pigs (Maibach & Patrick, 1989). The study indicated
that females may be more sensitive than males. Groups of five male
and five female animals were administered hydroquinone (0.1 ml in a
hydrophilic ointment) at concentrations of 0.1, 1.0, and 5.0% on an
epilated area of the back five days a week for 13 weeks. The lowest
concentration caused marginal irritation without depigmentation,
while the medium concentration resulted in a slight to marginal
irritation in 30% of the animals (mainly females) and to weak
depigmentation in females. Moderate to severe irritation and severe
ulcerated inflammatory responses occurred with the highest
concentration. Moderate depigmen-tation was observed in
approximately 40% of the animals dosed (only females).
Hyperpigmentation was noticed in 80-100% of the animals in all dose
groups, but this was not considered to be attributable to the
treatment.
7.2.2 Eye irritation
Powdered hydroquinone (2-5 mg) instilled twice daily (5 days
per week for 9 weeks) into the eyes of dogs caused immediate but
transient irritation and lacrimation (Dreyer, 1940). Opacity of the
cornea, lacrimation and redness of the conjunctiva were produced
within 4 days, but no ulcers were seen. The eye returned to normal
within two days after cessation of treatment. In guinea-pigs
powdered hydroquinone (1-3 mg, twice daily for 9 weeks) also caused
immediate but transient irritation. During the second day of
application a slight corneal opacity was observed in some animals
and on the third day opacity of varying degrees occurred in most of
the animals. Ulcers appeared in two animals. The eyes had fully
recovered 3 days after cessation of treatment.
Following an injection of 0.1 ml of a solution (vehicle not
specified) of hydroquinone (0.012-0.05 mol/litre) into the cornea of
rabbits, the resultant reaction was graded 5 out of the possible
maximum of 100 (Hughes, 1948).
Finely powdered hydroquinone (amounts not specified) was
applied daily, from 2-4 months, to the eyes of rabbits in 6 groups,
which were, respectively, kept in the dark, in sunlight, in normal
light, irradiated with UV light, or pre-sensitized with
haematoporphyrin and then kept under either reduced light or
sunlight. Most rabbits developed pigmentation, first in the
conjunctiva and then in the cornea. Degenerative alterations of the
corneal parenchyma were also observed. Pigment formation appeared
earlier in animals exposed to light. Older animals seemed more prone
to develop pigment than younger ones. Pigment was deposited in
albino rabbit eyes as well as in those of rabbits with normal
pigmentation (Ferraris de Gaspare, 1949).
Hydroquinone in aqueous solution, e.g., in tears, is oxidized
by air, forming a brown colour partly due to conversion to
1,4-benzoquinone (Grant, 1986). No disturbance of the inner parts of
the eye is known to have been produced by exposure to hydroquinone
(Grant, 1986).
7.2.3 Sensitization
Several sensitization studies have been carried out with
hydroquinone; methods and results are summarized in Table 12.
The skin sensitizing potential of hydroquinone for female
guinea-pigs was investigated by Rajka & Blohm (1970). Hydroquinone
elicited "weak" sensitivity after sensitization with a 0.001%
solution injected intracutaneously and challenge with an equipotent
solution of hydroquinone.
The ability of guinea-pigs to detect known human contact
sensitizers was explored by Goodwin et al. (1981). Sensitization
induced by hydroquinone was "strong" when assayed by the Magnusson &
Kligman maximization test, "moderate" by the single injection
adjuvant test and "weak" by the modified Draize procedure.
Hydroquinone was found to be a "moderate" sensitizer in female
guinea-pigs in both the guinea-pig maximization test and Freund's
complete adjuvant test as performed by Van der Walle et al.
(1982a,b). Hydroquinone produced identical sensitization potentials
in the Freund's complete adjuvant test using induction
concentrations of 0.5 mol/litre and 0.45 µmol/litre. The study also
showed almost 100% cross reactivity of hydroquinone and
p-methoxyphenol. Both substances are used as inhibitors in acrylic
monomers to prevent unwanted polymerization.
More recently, Basketter & Goodwin (1988) used three
sensitization test methods representing both topical and intradermal
routes of application. Groups of 10 guinea-pigs were sensitized by
using the guinea-pig maximization test, a modified single injection
adjuvant test, and a cumulative contact enhancement test. The
sensitization potential of hydroquinone was assessed as "strong",
"weak", and "moderate", respectively, in these three tests.
Subsequent cross-challenges with p-phenylenediamine, sulfanilic
acid, and p-benzoquinone gave only "restricted evidence" of
cross-reactions.
Table 12. Contact allergy predictive tests with hydroquinone in guinea-pigs
Test Induction dose Challenge dose Sensitization Reference
(number positive/number
tested or percentage)
Intracutaneous sensitization 0.001% (0.1 ml, injection) 0.001% (injection) 4/18 Rajka & Blohm (1970)
Guinea-pig 2.0% (0.1 ml, injection) 5.0% (patch) 70%, "strong" sensitizer Goodwin et al. (1981)
maximization test 10.0% (patch) 5.0% (patch)
Guinea-pig 0.5 mol/litre (day 0) (patch) 0.125 mol/litre 5/10 (day 21); Van der Walle
maximization test 1 mol/litre (day 7) (patch) 0.250 mol/litre 5/10 (day 35) et al. (1982a,b)
Guinea-pig 2.0% (0.1 ml, injection) 0.5% (patch) "strong" sensitizer Basketter &
maximization test 1.0% (patch) 0.5% (patch) Goodwin (1988)
Single injection 2.0% (injection) 5.0% (patch) 40%, "moderate" sensitizer Goodwin et al. (1981)
adjuvant test
Modified single injection 2.0% (0.1 ml, injection) 10% (patch) "weak" sensitizer Basketter &
adjuvant test Goodwin (1988)
Modified Draize test 2.5% (injection) 1.0% (injection); 0%a, 30%b, "weak" Goodwin et al. (1981)
20% (application) sensitizer
Freund's complete 5 x 0.45 µmol/litre 0.115 µmol/litre 3/8 (day 21) Van der Walle
adjuvant test (0.1 ml, injection) (patch) 4/8 (day 35); et al. (1982a,b)
5 x 0.5 mol/litre 0.125 mol/litre 4/8 (day 21)
(0.1 ml, injection) (patch) 4/8 (day 35)
Cumulative contact 1.0% (patch) 20% (patch) "moderate" sensitizer Basketter &
enhancement test Goodwin (1988)
a the proportion of guinea-pigs sensitized after one induction treatment
b the proportion of guinea-pigs sensitized after two induction treatments
7.3 Short-term exposure
The short-term toxicity of hydroquinone has been studied in
rats and mice. The effects are summarized in Table 13.
Two groups of rats (14 animals/group, sex and strain not
reported) were fed a diet containing 0 or 5% HQ for nine weeks
(Carlson & Brewer, 1953). Findings at the end of the study consisted
of atrophy of the liver cord cells, adipose tissue, striated muscle
and lymphoid tissue of the spleen, as well as an average decrease of
66% in cellularity of the bone marrow (considered as aplastic
anaemia by the authors). No information was reported on mortality
but the fact that animal lost 46% of their body weight during the
course of the study makes the findings difficult to interpret.
Aqueous solutions of hydroquinone (0, 7.5 or 15 mg/kg, 6 days
per week) administered by oral gavage to male Wistar rats for 40
days resulted in haematological changes, including anisocytosis,
polychromatophilia and acidophilic erythroblasts, at the highest
dose level (Delcambre et al., 1962). Administration of 0, 5 or 10
mg/kg for four months to groups of 15 male Wistar rats resulted in
mortality in the highest dose group; 5 mg/kg was well tolerated.
"Mild hyperplastic and hyperkeratotic areas near the
oesophageal entry" occurred in groups of male and female Wistar rats
fed powdered diets containing 2% hydroquinone. However, there were
some deficiencies in the reporting of experimental design (Altmann
et al., 1985). No other treatment-related changes were found, nor
were there any sex-related changes concerning forestomach lesions.
A two-week oral study on Fischer-344 rats and B6C3F1 mice was
carried out by the National Toxicology Program (NTP, 1989; Kari et
al., 1992). The animals were given hydroquinone in corn oil by
gavage 5 days per week (12 doses over 14 days). Five rats per sex
and dose group were administered 0, 63, 125, 250, 500 or 1000 mg/kg
body weight and five mice per sex and dose group 0, 31, 63, 125, 250
or 500 mg/kg. All rats given 1000 mg/kg died during the dosing
period; deaths were also reported at the 500 mg/kg dose level.
Clinical signs of treatment-related toxicity included tremors and
convulsions at the 500 and 1000 mg/kg dose levels. Tremors,
convulsions and deaths also occurred in mice during the study period
at the 250 and 500 mg/kg dose levels.
Table 13. Effects of short-term exposure to hydroquinone via the oral route
Species Concentration Means of Duration Observation Reference
administration
Rat 5% diet 9 weeks reduced weight; aplastic anaemia; decreased Carlson & Brewer
bone marrow cellularity; atrophy of liver,(1953)
spleen, adipose tissue and striated muscle;
ulceration and haemorrhage of the stomach
mucosa
Rat 500, 750, 1000, stomach tube 12 days increased mortality Carlson & Brewer
1250, 1500, (1953)
1750 mg/kg
Rat 7.5, 15 mg/kg intubation 40 days 15 mg/kg: anisocytosis, polychromatophilia, Delcambre et al.
acidophilic erythroblasts (1962)
Rat 5, 10 mg/kg intubation 4 months 10 mg/kg: deaths (7/15) Delcambre et al.
(1962)
Rat 2% diet 4 weeks; mild hyperplasia and hyperkeratosis Altmann et al.
8 weeks of forestomach (1985)
Rat 20, 64, 200 mg/kg gavage 90 days 64 mg/kg: tremors, reduced activity; Eastman Kodak
200 mg/kg: tremors, reduced activity, reduced Company (1988)
body weight and feed consumption (males)
Rat 63, 125, 250, gavage 14 days 1000 mg/kg: tremors, convulsions andNTP (1989); Kari
500, 1000 mg/kg death (10/10); et al. (1992)
500 mg/kg: tremors, convulsions and
death (1/5 male and 4/5 female)
Table 13. (contd).
Species Concentration Means of Duration Observation Reference
administration
Rat 25, 50, 100, 200, gavage 13 weeks 25 mg/kg: decreased relative liver weight NTP (1989)
400 mg/kg (males); 50 and 100 mg/kg: decreased (males)
and increased (females) relative liver weight;
200 mg/kg: lethargy, decreased body weight gain
and increased relative liver weight; tremors,
convulsions and deaths (females); nephropathy;
inflammation and/or epithelial hyperplasia of the
forestomach; 400 mg/kg: tremors, convulsions
and deaths
Rat 2.5, 25, 50 mg/kg gavage 1,3,6 weeks 50 mg/kg: increased urinary enzyme excretion; English et al. (1992)
renal tubular degeneration/regeneration; and
increased renal tubular cell proliferation in
male F-344 rats; female F-344 rats and male
SD rats: no effects
Mouse 25, 50, 100, 200, gavage 13 weeks 25 and 50 mg/kg: lethargy (males); increased NTP (1989)
400 mg/kg relative liver weight (males); 100 mg/kg: lethargy;
increased relative liver weight (males); 200 mg/kg:
lethargy; increased relative liver weight (males);
tremors (males); lesions in the forestomach (one
female); deaths (males); 400 mg/kg: lethargy,
tremors, convulsions, lesions in the
forestomach, deaths
Mouse 31, 63, 125, 250, gavage 14 days 500 mg/kg: tremors, convulsions and death NTP (1989)
500 mg/kg (4/5 male and 5/5 female);
250 mg/kg: tremors, convulsions and death
(3/5 male)
Hydroquinone in 95% ethanol was dermally applied (12 doses over
14 days) to Fischer-344 rats and B6C3F1 mice (NTP, 1989). Rats
(five per sex and dose group) received 0, 240, 480, 1920 or 3840
mg/kg and mice (five per sex and dose group) 0, 300, 600, 1200, 2400
or 4800 mg/kg. The survival rate was not affected. The only findings
were a 6% lower body weight in male rats administered 3840 mg/kg and
crystals on the skin and fur of animals at 3840 mg/kg. No
histopathological examination of the tissues was carried out.
In a 13-week oral toxicity study, 10 male and 10 female
CD(SD)BR rats (initially, approximately 7 weeks old) in each of four
groups were administered hydroquinone (0, 20, 64 or 200 mg/kg per
day) by gavage on 5 days/week (Eastman Kodak Company, 1988). Brown
urine was seen in rats of both sexes from all dose groups. Males
also showed lower feed consumption; however, this was only
significantly (P < 0.05) reduced during the first week of the
study. Female body weight gain and food consumption were not
significantly altered in any dose group during the study. Signs of
behavioural effects were observed at both the 64 and 200 mg/kg dose
levels (see section 7.8.3). The animals were sacrificed after the
exposure period, and six males and six females from each group were
perfused for neuropathological examination (see section 7.8.3). No
treatment-related changes were observed at gross necropsy.
Administration of 200 mg hydroquinone/kg reduced body weight gain in
male rats so that the final body weight of the treated rats was 7%
less than the mean weight for the controls.
Thirteen-week studies in rodents have also been presented by
the NTP (1989). Groups of 10 males and 10 females of each species
(F-344/N rats, initially 4 to 5 weeks old, and B6C3F1 mice,
initially 5 to 6 weeks old) were administered hydroquinone (0, 25,
50, 100, 200, or 400 mg/kg) in corn oil by gavage, five days per
week. A dose level of 400 mg/kg was lethal to all rats. Tremors and
convulsions were observed in most rats at this dose level and in
several female rats receiving 200 mg/kg. Doses of 100 mg/kg or less
did not cause signs of central nervous system (CNS) stimulation.
Rats receiving 200 mg/kg also showed reduced body weight gain,
nephropathy, and inflammation and/or epithelial hyperplasia of the
forestomach. The kidney lesions in male rats were judged to be more
severe than in females.
In mice, a dose level of 400 mg/kg caused mortality in both
males (8/l0) and females (8/10). In the 200 mg/kg male group two
animals died (one due to gavage error). Lethargy was seen in all
dosed males and in females in the three highest-dose groups of each
sex. Tremors, often followed by convulsions, were noted in the
highest-dose group. Liver-to-body weight ratios for all dosed males
were significantly (P < 0.01) greater than for vehicle controls.
Ulceration, inflammation or epithelial hyperplasia of the
forestomach occurred in mice receiving 400 mg/kg (3/10 males and
2/10 females) and in one female mouse receiving 200 mg/kg.
Male and female F-344 rats were given hydroquinone (0, 2.5, 25
or 50 mg/kg) 5 days/week in water by oral gavage for 1, 3 or 6 weeks
(English et al., 1992). At each time point, 5 rats per sex and
dose group were examined for urinalysis changes, renal tubular cell
proliferation and histopathology. Body and kidney weights were not
altered by hydroquinone exposure. Increased excretion of urinary
enzymes was observed in male F-344 rats given 50 mg/kg as early as
one week into the exposure. The incidence of tubular degeneration
and regeneration was mild in the 50 mg/kg male group, and cell
proliferation was increased by 87%, 50% and 34% in the P1, P2 and P3
tubular segments, respectively. Female rats and males in the lower
dose level groups were not affected by hydroquinone exposure. Male
Sprague-Dawley rats given 50 mg/kg for 6 weeks were also not
affected by hydroquinone exposure.
7.4 Long-term exposure
The effects of long-term exposure to hydroquinone in
experimental animals are shown in Table 14. In addition to the acute
(see section 7.1) and subacute (see section 7.3) toxicity tests,
Carlson & Brewer (1953) carried out a "cumulative" toxicity study
and a series of long-term experiments with rats and dogs. In the
cumulative toxicity study a group of 16 rats (strain and sex not
reported) received 500 mg hydroquinone/kg by stomach tube 101 times
in 151 days. More than 50% of the rats died during the study period.
However, the survivors grew at the same rate as the controls and
were autopsied at the end of the experiment. However, no information
on autopsy findings was reported.
In the first long-term study, four groups of male and four
groups of female Sprague-Dawley rats (23 to 24 days old, 10 rats in
each group) were given a basic diet containing 0, 0.1, 0.5 or 1.0%
hydroquinone (Carlson & Brewer, 1953). In the second experiment the
hydroquinone was heated together with the lard in the feed. Eight
groups of rats (16 to 23 in each group) were fed the basic diet
containing 0, 0.1, 0.25 or 0.5% hydroquinone. In the third
experiment eight groups of rats (20 rats in each group) were given a
basic diet containing 0, 0.1, 0.5 or 1.0% hydroquinone with 0.1%
citric acid added. The experiments lasted for 103 weeks. During the
first month of the study the animals given 0.5 or 1.0% hydroquinone
in their diets gained weight at a slower rate than did control
animals. A similar reduction was not found in the rats given
hydroquinone previously heated with lard before feeding. However,
the final body weights of the dose groups were not significantly
different from those of the controls. Haematological analysis (red
blood cell count, % haemoglobin and differential white blood cell
count) showed no statistically significant deviations from the
control values. The microscopic examinations (liver, omentum,
kidney, spleen, heart, lung, bone marrow, stomach wall, pancreas,
adrenal, subperitoneal and intramuscular abdominal fat) also failed
to reveal compound-related changes. Some of the males and females
(number and dose groups not specified) in each experiment were mated
after six months of dosing to produce two successive litters. The
average numbers of offspring for the two successive litters showed
no difference between experimental and control groups. The offspring
given diets containing 0.1, 0.25 or 0.5% hydroquinone previously
heated with lard grew at the same rate as the controls.
Carlson & Brewer (1953) also studied the long-term effects of
hydroquinone in male and female dogs (four months of age at the
beginning of the study). One dog was maintained on a diet containing
16 mg of hydroquinone/kg per day for 80 weeks, while two dogs
received 1.6 mg of hydroquinone/kg per day for 31 weeks and 40 mg/kg
per day for the succeeding 49 weeks. The compound was administered
in sugar-coated tablets mixed with the food. Two dogs served as
controls. The sex distribution in the different groups was not
reported. In addition, five adult male dogs were fed 100 mg of
hydroquinone/kg per day for 26 weeks. Routine blood and urine
analyses (not specified) were made periodically. After the
experiment, the dogs were killed and autopsied. The dogs given
hydroquinone in their diet from four months of age grew at the same
rate as controls. The adults maintained their body weights.
Haematological analyses and urinalyses showed no differences between
exposed rats and controls. No effects on gross pathology or
histopathology were observed.
Fifteen-month oral toxicity studies on rats and mice were also
included in two-year studies presented by the National Toxicology
Program (NTP, 1989) (see also section 7.). Groups of F-44/N rats and
B6C3F1 mice (64 or 65 males and 65 females of each species) were
administered 0, 25 or 50 mg hydroquinone/kg and 0, 50 or 100 mg/kg,
respectively, in deionized water by gavage 5 days per week. No
compound-related clinical signs were observed during the study
period. At 15 months, ten animals from each group were selected for
haematological and clinical chemical analyses, killed and
necropsied. In male rats significantly (P < 0.01) higher mean
relative kidney and liver weights were observed in the high-dose
group and there was also a compound-related increase in the severity
of nephropathy. For high-dose female rats, the haematocrit value,
haemoglobin concentration and erythrocyte counts were decreased. In
mice the relative liver weights were significantly (P < 0.01)
higher for high-dose males and females than for vehicle controls. A
significantly (P < 0.05) higher relative brain weight was noted for
high-dose female mice and kidney weights were significantly (P <
0.01) increased for dosed females. In dosed males, but not in
females, compound-related lesions in the liver were seen, including
centrilobular fatty changes, cytomegaly and syncytial cells.
Table 14. Effects of long-term oral administration of hydroquinone in experimental animals
Species Concentration Duration Observation Reference
Rat 500 mg/kg body weight 151 days > 50% died Carlson &
(by stomach tube) (101 dosings) Brewer (1953)
Rat 0, 0.1, 0.5, 1.0% 103 weeks decreased weight gain for the first months at 0.5 Carlson &
(in the diet) and 1.0%; final body weights did not differ Brewer (1953)
Rat 0, 0.1, 0.25, 0.5% 103 weeks no adverse effects Carlson &
(heated together with Brewer (1953)
lard in the diet)
Rat 0, 0.1, 0.5, 1.0% + 103 weeks no adverse effects Carlson &
0.1% citric acid (in the diet) Brewer (1953)
Rat 0, 25 or 50 mg/kg in 15 months significantly higher relative kidney weight in high-dose NTP (1989)
deionized water (by gavage) males and increased severity of nephropathy in dosed
males; decreased haematocrit value, haemoglobin
concentration and erythrocyte count in females
Mouse 0, 50 or 100 mg/kg in 15 months significantly higher relative liver weights for high- NTP (1989)
deionized water (by gavage) dose males and females; liver lesions in males
Dog 16 mg/kg per day 80 weeks no adverse effects Carlson &
(in the diet) Brewer (1953)
Dog 1.6 mg/kg per day; 31 weeks no adverse effects during the total experimental Carlson &
40 mg/kg per day 49 succeeding period Brewer (1953)
(sugar coated tablets) weeks
7.5 Reproduction, embryotoxicity and teratogenicity
7.5.1 Effects on male reproduction
In a study by Skalka (1964), hydroquinone was injected
subcutaneously into 16 male rats (100 mg/kg body weight per day for
51 days), while 17 male rats served as controls. The average weights
of the testes, epididymides, seminal vesicles and adrenals were
decreased after the treatment period. The fertility was reduced by
33% in the males and the number of pregnancies in mated females was
reduced by 19% compared with the corresponding results for the
control animals. Histological examinations indicated a disruption in
sperm production. Diminished content of DNA in sperm heads was also
noted.
In 13-week and two-year oral studies in rats and mice, no
effects on testicular or epididymal weights or on the histopathology
of these organs were observed (NTP, 1989). Hydroquinone in corn oil
was given by gavage to groups of males F-344/N rats and to male
B6C3F1 mice for 13 weeks. The dose ranged from 0 to 400 mg/kg body
weight for both animal species. In the two-year studies hydroquinone
in water was given in doses of 0-50 mg/kg body weight to rats and in
doses of 0-100 mg/kg to mice.
In a dominant lethal assay in male rats (CRL:COBS CD (SD) BR)
(see also section 7.6), the males (25 per dose group) were given
doses of 0, 30, 100 or 300 mg hydroquinone/kg by gavage 5 days per
week for ten weeks (Krasavage, 1984b). The controls consisted of two
groups, one positive and one negative. During the 2 weeks
immediately following the final treatment, the males were mated
(1:1) with untreated females. All females were killed on day 14 of
gestation. In the high-dose group (300 mg/kg) the mean body weight
and the feed intake of the males were significantly reduced compared
to the negative controls (P < 0.05). The high-dose males also
exhibited clinical signs of toxicity such as brown urine,
sialorrhoea, swollen eyelids, tremors, convulsions and death. No
compound-related effects were seen on male fertility and no dominant
lethality was observed. There were no compound-related effects on
the reproductive parameters studied in the mated females
(insemination rate, pregnancy rate, mean number of corpora lutea,
implantation sites, viable implants, early and late deaths, and pre-
and postimplantation losses).
In a two-generation study oral administration of hydroquinone
did not appear to affect the reproduction of Fo and F1 parental
rats at dose levels up to 150 mg/kg per day (see also section 7.5.3)
(Bio/dynamics Inc., 1989b). Male fertility indices, mating indices
and pregnancy rates did not differ significantly between the control
and the hydroquinone-treated groups.
7.5.2 Effects on female reproduction
Hydroquinone was shown to affect the rat estrus cycle when
given parenterally (Rosen & Millman, 1955). Three rats were given 10
mg hydroquinone/day subcutaneously for 11 days, and vaginal smears
were used to indicate estrus or diestrus. Following an induction
period of about three days, the estrus cycle was interrupted for 5
days, after which normal cycling was observed.
Similar results were obtained in a study performed by Racz et
al. (1958). One group of rats was given 200 mg hydroquinone/kg
body weight per day by gavage for 14 days and one group was given
100 mg hydroquinone plus 100 mg phloridzin/kg per day for 14 days.
Ten animals were used per group. Some of the rats treated with
hydroquinone remained in diestrus. In the group treated with both
hydroquinone and phloridzin no effects on the estrus cycle were
shown. As the compound caused effects on the central nervous system
and mortalities occurred after 4-5 days in the 200 mg/kg dose group,
the dose of hydroquinone was reduced to 50 and 100 mg/kg per day. At
autopsy no mature Graafian follicles were seen, but some were
growing.
Hydroquinone in stock diets (0.003 and 0.3%) fed to female rats
(10/group) for 10 days prior to insemination had no effect on
gestation length, maternal mortality or on other reproductive
parameters studied (fertility index, litter efficiency, mean litter
size, fetal viability or lactation index (Ames et al., 1956).
However, it is not clearly stated if the dosing also included the
gestation period.
Hydroquinone did not produce adverse affects on female
reproduction in a two-generation study in rats (see section 7.5.3)
after daily oral administration of doses up to 150 mg/kg per day
(Bio/dynamics Inc., 1989b).
7.5.3 Embryotoxicity and teratogenicity
The earliest experimental study of the developmental toxicity
of hydroquinone and other antioxidants was performed by Telford et
al. (1962). They reported that hydroquinone added to the diet
caused increased resorption rates. Ten Walter Reed-Carworth Farm
strain rats (first gestation animals), weighing about 200 g at the
time of breeding, were given 0.5 g hydroquinone in their diet (about
100 mg/kg body weight per day); however, the treatment period was
not documented. Based on the number of implantation sites, the
resorption rate was 26.8% compared to 10.6% for controls (126
untreated, pregnant rats). The report made no mention of the numbers
of corpora lutea. Hydroquinone caused no maternal toxicity.
Burnett et al. (1976) reported the findings of a teratology
study on twelve hair dye composite formulations. The study groups
were tested along with one positive and three negative control
groups. Mated Charles River CD female rats (20 per group) were
treated by topical application with 2 ml/kg (0.2% hydroquinone) on
pregnancy days 1, 4, 7, 10, 13, 16, and 19. No signs of toxicity
were seen throughout the study. There were no differences between
untreated controls and the hydroquinone-treated group in any
reported parameter (maternal toxicity, body weight and food
consumption, mean number of corpora lutea, implantation sites,
resorption sites, mean resorptions per pregnancy, live fetuses and
sex ratio) and no significant soft tissue or skeletal changes.
Hydroquinone has been found to induce micronuclei
transplacentally in fetal liver cells (Ciranni et al., 1988a) (see
also section 7.6). The compound was given by gastric intubation at a
dose level of 80 mg/kg to four pregnant Swiss CD-1 mice on the 13th
day of gestation. Micronuclei were detected from 9 h after the
administration.
In a pilot study on developmental toxicity in rats,
hydroquinone (5% in distilled water) was administered daily by
gavage to groups of ten pregnant COBSCD(SD)BR rats on gestation days
6 to 15 at dose levels of 0, 50, 100 or 200 mg/kg (Krasavage,
1984a). The animals were sacrificed and autopsied on gestation day
16. No significant maternal toxicity or embryo-toxicity was produced
in any dose group during the treatment period with the exception of
slightly reduced weight gain and feed consumption in the
highest-dose. A dose-dependent brownish discolouration of the urine
was observed in all treated groups. No other treatment-related
changes were found.
In the subsequent developmental toxicity study, hydroquinone
(5% in distilled water) was administered daily by gavage to groups
of 30 plug-positive female (COBSCD(SD)BR rats (initially 6 weeks
old) on gestation days 6 to 15 (0, 30, 100 or 300 mg/kg) (Krasavage
et al., 1985; Krasavage et al., 1992). Maternal toxicity,
manifested as a slight but statistically significant (P < 0.05)
reduction in body weight gain and feed intake, was observed in those
dams given 300 mg/kg. A reduction in the fetal body weight was seen
at 300 mg/kg (statistically significant (P < 0.05) only for the
females). No compound-related teratogenic effects were recorded.
Dilated renal pelvis, hydronephrosis and hydroureter were seen more
frequently in the treated groups than in the controls; however, the
changes did not appear to be dose-related and were not significantly
different from the controls. Skeletal variations were seen in
fetuses from all dose groups and from the control group. The total
number of fetuses with a vertebral variation was statistically
increased in the highest dose group compared with the controls, but
analyses of individual skeletal variations and statistical analyses
of total number of fetuses with a skeletal variation indicated no
significant effects. The authors concluded that the
no-observed-effect level (NOEL) for maternal and developmental
toxicity in rats was 100 mg/kg body weight, and the
no-observed-adverse-effect level (NOAEL) for fetal development was
set at 300 mg/kg (Krasavage et al., 1992).
In a pilot study for developmental toxicity in rabbits,
hydroquinone (0, 50, 100, 200, 300, 400 or 500 mg/kg per day) was
administered by gavage to mated New Zealand White rabbits (five
rabbits/group) from days 6 to 18 of gestation (Bio/dynamics Inc.,
1988). Dose levels of 300, 400 and 500 mg/kg per day caused maternal
death. At dose levels of 50, 100 and 200 mg/kg per day, dose-related
reductions in body weight and food consumption were recorded. In the
200 mg/kg per day dose group, an increased number of resorptions
were observed, suggesting an embryotoxic effect. Mean fetal weights
were unaffected at 50 and 100 mg/kg per day, but at 200 mg/kg per
day they were lower than those of the controls (by 20.7%). No
fetuses were recovered at the higher dose levels as no females
survived throughout the study. The fetal external examinations
showed no treatment-related adverse effects.
In the subsequent developmental toxicity study, New Zealand
White rabbits (18 mated females/group) were given hydroquinone by
gavage at dose levels of 0, 25, 75 or 150 mg/kg per day on gestation
days 6 to 18 (Bio/dynamics Inc., 1989a; Murphy et al., 1992). The
exposure of rabbits to 25 or 75 mg hydroquinone/kg per day had no
effect on any maternal or fetal parameter. However, there was a
reduction in mean food consumption during the 11-14 day gestational
interval at 75 mg/kg per day, but only on days 11 and 12 were these
differences statistically (P < 0.05) different from control data.
In the 150 mg/kg per day dose group there were statistically
significant (P < 0.01) reductions in the body weight and food
consumption during the dosing period, suggesting maternal toxicity.
No other parameter (clinical observations, uterine implantations,
liver or kidney weights or gross pathology) showed any
treatment-related adverse effect on the females. No embryotoxicity
was found from uterine implantation data. An increased incidence
(not statistically significant) of external, visceral and skeletal
malformations was noticed in the fetuses from the 150 mg/kg per day
group. The NOEL for maternal toxicity was 25 mg/kg per day and the
NOEL for developmental toxicity was 75 mg/kg per day.
The results of a two-generation reproduction study in rats
revealed no hydroquinone-related reproductive toxicity (Bio/dynamics
Inc., 1989b). Charles River CD rats (180 Fo and 180 F1) were
exposed via gavage to hydroquinone at concentrations of 15, 50 or
150 mg/kg per day (30 rats per sex and group; 120 rats served as
controls). The two lower dose levels did not affect mortality rates,
body weight, or feed consumption either in the Fo or the F1
parental animals. One Fo male given 50 mg/kg per day had tremors
during the post-dosing period. There were no adverse effects on mean
litter size, body weight, sex distribution or survival, or in
postmortem evaluations of pups from females given 15 or 50 mg/kg per
day. At the highest-dose level, tremors were observed in Fo and
F1 parental animals of both sexes following dosing. Reproductive
indices and pregnancy rates for the Fo and F1 parents were not
considered to have been adversely affected by hydroquinone
treatment, nor were there any adverse effects of treatment for pups
from Fo and F1 parental animals. The NOAEL for parental toxicity
was estimated to be 15 mg/kg per day and for reproductive effects
through two generations to be 150 mg/kg per day.
A quantitative approach to relate the physico-chemical
properties of a series of substituted phenols (including
hydroquinone) to maternal and developmental toxicity in rats was
conducted by Kavlock (1990) and by Kavlock et al. (1991). However,
due to theoretical and statistical mistakes, no formal conclusions
can be made on the possibility of modelling these particular
end-points quantitatively.
A review of the different reproduction studies is given in
Table 15.
7.6 Mutagenicity and related end-points
The results of genotoxicity studies, with or without S9
metabolic activation, are indicated in Table 16.
Hydroquinone has been found to be non-mutagenic in Salmonella
typhimurium tester strains TA97, TA98, TA100, TA1535, TA1537 and
TA1538, with and without metabolic activation, at doses up to 1000
µg/plate (Bulman & Wampler, 1979; Bulman & Van der Sluis, 1980;
Florin et al., 1980; Gocke et al., 1981; Serva & Bulman, 1981;
Bulman & Serva, 1982; Haworth et al., 1983; Sakai et al., 1985).
However, in one study hydroquinone was reported to be clearly
mutagenic without metabolic activation in strain 1535A (a strain
that the authors suggested might harbour an undefined genetic
alteration from TA1535 because of differences in length of storage)
with ZLM minimal medium (a modified minimal medium for Escherichia
coli) but not with the standard VB (Vogel-Bonner) minimal medium
(Gocke et al., 1981). The concentration of citrate was 3.5 times
higher in VB medium than in ZLM medium. In a fluctuation test using
the Salmonella typhimurium tester strain TA100, hydroquinone was
shown to be mutagenic at the concentrations 100 and 200 ng/well with
metabolic activation (Koike et al., 1988).
In Escherichia coli hydroquinone causes differential killing
of DNA-repair-deficient (Pol A-) and -proficient (Pol A+) strains
without a supplementary metabolic activation system, indicating
induction of repairable DNA damage (Bilimoria, 1975; Van der Sluis,
1980; Wampler, 1980). In Salmonella typhimurium no SOS-inducing
activity of hydroquinone (3300 mg/litre liquid medium) could be
demonstrated (Nakamura et al., 1987).
Table 15. Studies on reproductive effects in laboratory animals
Species Route of Number of Dosage Time of Result Reference
exposure animals treatment
Rat subcutaneous 16 males 100 mg/kg body 51 days 33% reduced fertility, diminished Skalka (1964)
weight content of DNA in sperm heads; 19%
reduced number of pregnancies in
mated females
Rat subcutaneous 3 10 mg/day 11 days interrupted estrus cycle Rosen & Millman
(1955)
Rat oral (gavage) 10/group 200 mg/kg body 14 days some rats remained in diestrus, Racz et al.
weight per daya CNS effects and death within 5 days (1958)
Rat oral (diet) 10 females/ 0.003% 10 days prior to no effects on gestation and Ames et al.
group 0.3% insemination maternal mortality (1956)
Rat oral (diet) 10 0.5 g not clearly stated increased resorption rate Telford et al.
(1962)
Rat oral (gavage) 10 females/ 0, 50, 100 or 200 gestation days reduced feed consumption and body Krasavage (1984a)
group mg/kg body weight 6-15 weight gain at 200 mg/kg
Rat oral (gavage) 25/group 0, 30, 100 or 300 5 days per week no treatment-related effects on Krasavage (1984b)
(male) mg/kg body weight for 10 weeks reproductive parameters studied
Rat oral (gavage) 30/group 0, 30, 100 or 300 gestation days reduced fetal body weight at Krasavage et al.
mg/kg 6-15 300 mg/kg (1985)
Rat oral (gastric 30/group 0, 15, 50 or 150 two-generation no effect on reproduction at Bio/dynamics
intubation) mg/kg body weight study 150 mg/kg Inc. (1989b)
Table 15. (contd).
Species Route of Number of Dosage Time of Result Reference
exposure animals treatment
Rat oral 16/group 0, 100, 333, 667 gestation day 11 reduced weight gain at 667 and 1000 Kavlock (1990)
(intubation) or 1000 mg/kg mg/kg; increased mortality at 1000
mg/kg; malformations of the limbs,
tail and urogenital system
Rat embryo < 0.5 mmol/litre from day 10 growth retardation, structural Kavlock et al.
culture (approx. 12 somite) abnormalities of hind limb and tail (1991)
system
Mouse oral (gastric 4 80 mg/kg body gestation day 13 micronuclei in fetal liver cells Ciranni et al.
intubation) weight (1988a)
Rabbit oral (gastric 5/group 0, 50, 100, 200, gestation days increased number of resorptions Bio/dynamics
intubation) 300, 400 or 500 6-18 and lower fetal weights at 200 mg/kg; Inc. (1988)
mg/kg body weight no females survived 300-500 mg/kg
Rabbit oral (gastric 18/group 0, 25, 75 or 150 gestation days an increased incidence (not Bio/dynamics Inc.
intubation) mg/kg body weight 6-18 significant) of malformations at (1989a); Murphy
150 mg/kg et al. (1992)
a Due to high mortality at 200 mg/kg, the dose was reduced for the rest of the 14-day period.
In Aspergillus nidulans hydroquinone was found to induce
mitotic segregation at 110-330 mg/litre (Crebelli et al., 1987)
and 264-396 mg/litre (Crebelli et al., 1991) and mitotic
crossing-over at 250-750 mg/litre (Kappas, 1989) and 264-396
mg/litre (Crebelli et al., 1991). The studies were performed
without a supplementary metabolic activation system.
Hydroquinone has been shown to induce chromosome aberrations or
karyotypic alterations in several plant species, e.g., Allium cepa
(Levan & Tjio, 1948a,b), Allium sativum (Sharma & Chatterjee,
1964; Valadaud & Izard, 1971), Callisia fragrans (Roy, 1973),
Chara zeylanica (Chatterjee & Sharma, 1972), Nigella sativa
(Sharma & Chatterjee, 1964), Trigonella foenum-graecum (Sharma &
Chatterjee, 1964), and Vicia faba (Sharma & Chatterjee, 1964;
Valadaud & Izard, 1971; Valadaud-Barrieu & Izard, 1973).
Hydroquinone produced an equivocal increase in recessive lethal
mutations in the X-chromosome of Drosophila melanogaster after
feeding adult males with a sucrose solution containing 26.4 or 30.0
mg hydroquinone/litre (NTP, 1989). No increase in the frequency of
recessive lethal mutations could be established in similar feeding
studies with hydroquinone at concentrations of 5500 to 11 000
mg/litre (Gocke et al., 1981) and 1000 mg/litre (Serva & Murphy,
1981), or in a study where males were injected with a dose of 1.5
mg/litre (NTP, 1989) (see also section 7.5.1).
At concentrations of 1.25 mg/litre (without a supplementary
metabolic activation system) and 2.5 mg/litre (with a supplementary
metabolic activation system), hydroquinone induced forward mutations
in vitro in the thymidine kinase locus of the mouse lymphoma cell
line L5178Y (McGregor et al., 1988a,b).
Gene mutations to 6-thioguanine resistance were induced in
vitro in V79 Chinese hamster cells exposed to hydroquinone at 350
µg/litre (Glatt et al., 1989). No supplementary metabolic
activation system was used.
Structural chromosome aberrations were induced in vitro in
Chinese hamster ovary cells treated with hydroquinone, but only in
the presence of a supplementary metabolic activation system
(Galloway et al., 1987).
Table 16. Studies on genotoxicity
Genetic end-point Resulta References
+S9 -S9
Gene mutations
Salmonella typhimurium - Bulman & Wampler (1979)
Salmonella typhimurium - - Bulman & Van der Sluis (1980)
Salmonella typhimurium - - Florin et al. (1980)
Salmonella typhimurium - - Gocke et al. (1981)
Salmonella typhimurium - - Serva & Bulman (1981)
Salmonella typhimurium - - Bulman & Serva (1982)
Salmonella typhimurium - - Haworth et al. (1983)
Salmonella typhimurium - - Sakai et al. (1985)
Salmonella typhimurium - + Gocke et al. (1981)
TA1535A, ZLM medium
Salmonella typhimurium + - Koike et al. (1988)
TA100, fluctuation test
Mouse lymphoma cells L5178Y + + McGregor et al. (1988a,b)
Chinese hamster cells V79 nd + Glatt et al. (1989)
Drosophila melanogaster, (+) NTP (1989)
feeding
Drosophila melanogaster, - Gocke et al. (1981)
feeding
Drosophila melanogaster, - Serva & Murphy (1981)
feeding
Drosophila melanogaster, - NTP (1989)
injection
Mouse, spot test - Gocke et al. (1983)
Rat, dominant lethals - Krasavage (1984b)
Chromosomal aberrations
Aspergillus nidulans nd + Crebelli et al. (1987, 1991)
Chinese hamster ovary cells + - Galloway et al. (1987)
Allium cepa + Levan & Tjio (1948a,b)
Allium sativum + Sharma & Chatterjee (1964)
Allium sativum + Valadaud & Izard (1971)
Callisia fragrans + Roy (1973)
Chara zeylanica + Chatterjee & Sharma (1972)
Nigella sativa + Sharma & Chatterjee (1964)
Trigonella foenum-graecum + Sharma & Chatterjee (1964)
Vicia faba + Sharma & Chatterjee (1964)
Vicia faba + Valadaud & Izard (1971)
Vicia faba + Valadaud-Barrieu & Izard (1973)
Mouse, bone marrow + Xu & Adler (1990)
Table 16. (contd).
Genetic end-point Resulta References
+S9 -S9
Mouse, bone marrow + Pacchierotti et al. (1991)
Mouse, spermatocytes and + Ciranni & Adler (1991)
differentiating spermatogonia
Rat, dominant lethals - Krasavage (1984b)
Micronuclei
Chinese hamster cells V79nd + Glatt et al. (1989, 1990)
Rat intestinal cells nd + Glatt et al. (1990)
Embryonal human liver cells nd + Glatt et al. (1990)
Human lymphocytes nd + Yager et al. (1990)
Human lymphocytes nd + Robertson et al. (1991)
Chinese hamster embryonicnd + Antoccia et al. (1991)
lung cells
Mouse, bone marrow + Gocke et al. (1981)
Mouse, bone marrow + Gad-El-Karim et al. (1985)
Mouse, bone marrow + Ciranni et al. (1988a,b)
Mouse, bone marrow + Adler & Kliesch (1990)
Mouse, bone marrow + Barale et al. (1990)
Mouse, bone marrow + Adler et al. (1991)
Mouse, bone marrow + Pachierotti et al. (1991)
Mouse, fetal liver + Ciranni et al. (1988a)
Chinese hamster cells V79 nd + Glatt et al. (1989)
Chinese hamster ovary cells + + Galloway et al. (1987)
Human lymphocytes nd + Erexson et al. (1985)
Human lymphocytes nd + Morimoto & Wolff (1980)
Human lymphocytes nd ± Knadle (1985)
Human lymphocytes + nd Morimoto et al. (1983)
Mouse, bone marrow - Pacchierotti et al. (1991)
Mitotic crossing-over
Aspergillus nidulans nd + Kappas (1989)
Aspergillus nidulans nd + Crebelli et al. (1991)
C-mitotic effects
Mouse, small intestine + Parmentier & Dustin (1948)
Mouse, bone marrow + Miller & Adler (1989)
DNA damage
Mouse lymphoma cells L5178YS, nd - Pellack-Walker & Blumer
breaks (1986)
Phi X-174 DNA, breaks + Lewis et al. (1988a)
Table 16. (contd).
Genetic end-point Resulta References
+S9 -S9
Rat, liver (hepatectomy), + Stenius et al. (1989)
single-strand breaks
DNA repair
Escherichia coli nd + Bilimoria (1975)
Escherichia coli nd + Van der Sluis (1980)
Escherichia coli nd + Wampler (1980)
Salmonella typhimurium nd + Nakamura et al. (1987)
HeLa cells + + Painter & Howard (1982)
Mouse lymphoma cells L5178YS nd + Pellack-Walker et al. (1985)
DNA adducts
In vitro + Rushmore et al. (1984)
In vitro + Jowa et al. (1986)
In vitro + Reddy et al. (1989)
In vitro + Jowa et al. (1990)
In vitro + Leanderson & Tagesson (1990)
In vitro + Reddy et al. (1990)
In vitro + Levay et al. (1991)
a + = positive; - = negative; (+) = equivocal; ± = induction in some
cases but not in others; nd = not determined
The induction of sister-chromatid exchanges in vitro after
exposure to hydroquinone without the use of a supplementary
metabolic activation system has been demonstrated in V79 Chinese
hamster cells at 2.2 mg/litre (Glatt et al., 1989) and human
lymphocytes at 0.55-11 mg/litre (Erexson et al., 1985), 4.4-22
mg/litre (Morimoto & Wolff, 1980), and 4.4 mg/litre in cells from
some individuals but not from others (Knadle, 1985), with a
supplementary metabolic activation system in human lymphocytes at
110 mg/litre (Morimoto et al., 1983), and both with and without a
supplementary metabolic activation system in Chinese hamster ovary
cells (Galloway et al., 1987).
Hydroquinone has been found to induce micronuclei in vitro in
V79 Chinese hamster cells (Glatt et al., 1989, 1990), rat
intestinal cells IEC-17 and IEC-18, and embryonal human liver cells
HuFoe-15 (Glatt et al., 1990). In cultured human lymphocytes an
11-fold and 3-fold increase in the frequency of micronuclei, was
reported after exposure to hydroquinone at 13.75 mg/litre (Yager et
al., 1990) and 8.25 mg/litre (Robertson et al., 1991),
respectively. Hydroquinone induced micronuclei in vitro in Cl-1
Chinese hamster embryonic lung cells at concentrations of 1, 3, and
4.5 mg/litre (Antoccia et al., 1991). The use of an
antikinetochore antibody in the three latter studies indicated the
occurrence of numerical as well as structural chromosome
aberrations. All studies were performed without a supplementary
metabolic activation system.
With the in vitro porcine brain tubulin assembly assay,
hydroquinone had no effect with respect to lag-phase, polymerization
velocity or end absorption at doses up to 2750 mg/litre. The
disassembly process was stimulated at doses higher than 1100
mg/litre (Brunner et al., 1991). In another study, hydroquinone
exhibited a weak inhibition of microtubule assembly and resulted in
abnormal microtubule formation at a concentration of 110 mg/litre
(Wallin & Hartley-Asp, 1993). Although hydroquinone itself appears
to be a poor inhibitor of microtubule assembly, its oxidation
products have been shown to bind to both alpha- and beta-tubulin and
to inhibit microtubule assembly at low concentrations (5 µmol/litre
or less) (Epe et al., 1990). In another study it was demonstrated
that polymerization of tubulin was inhibited by hydroquinone in a
concentration-dependent manner (Irons et al., 1981).
An effect on DNA synthesis, indicating DNA damage, following
in vitro exposure to hydroquinone has been demonstrated both with
and without a supplementary metabolic activation system in HeLa
cells (Painter & Howard, 1982) and without the use of a metabolic
activation system in the mouse lymphoma cell line L5178YS
(Pellack-Walker et al., 1985).
Female mice (C57BL/6JHan), mated with T-stock males, were
injected intraperitoneally with hydroquinone (110 mg/kg) on the 10th
day of pregnancy and subsequently analysed in accordance with the
mammalian spot test, which detects somatic gene mutations in mouse
embryos. No substance-related effect on the mutation frequency was
detected (Gocke et al., 1983).
In a study by Xu & Adler (1990), mice [(101/E1 x C3H/E1)F1],
10-14 weeks old and weighing 25-28 g, were injected
intraperito-neally with hydroquinone (40, 75 and 100 mg/kg). Bone
marrow cells, sampled 6 and 24 h later in the case of the two lower
doses and 6, 12, 18, 24 and 36 h later in the case of the highest
dose, were analysed for the presence of structural chromosome
aber-rations. At the highest dose level hydroquinone significantly
increased the frequency of aberrations 6-24 h after the treatment,
while at 75 mg/kg such an effect was detectable only 24 h after
treatment.
Male mice [(101/E1 x C3H/E1)F1], 10-14 weeks old and weighing
25-28 g, were injected intraperitoneally with hydroquinone (80, 100
and 150 mg/kg), and bone marrow cells, sampled 2 h later, were
analysed for the induction of c-mitotic effects. At dose levels of
100 and 150 mg/kg hydroquinone significantly increased the frequency
of metaphases with spread chromosomes (Miller & Adler, 1989).
Colchicine-like accumulation of metaphases and unusual "three-group
metaphases" in the small intestine were described in mice injected
with hydroquinone (125 mg/kg) (Parmentier & Dustin, 1948, 1951).
Similar results were observed in the intestine and bone marrow cells
of rats and hamsters following hydroquinone administration
(Parmentier, 1952, 1953).
In a study by Pacchierotti et al. (1991), male mice
[(C57Bl/Cne x C3H/Cne)F1], 12 weeks old, were injected with
hydroquinone (40, 80 and 120 mg/kg). Bone marrow cells, sampled 18
and 24 h later were analysed for the presence of numerical
chromosome changes, micronuclei and sister-chromatid exchanges. At a
dose level of 80 mg/kg, hydroquinone significantly increased the
frequency of hyperploid cells with 41-42 chromosomes after 18 h. At
all concentrations tested the frequency of micronuclei was
significantly increased after 24 h, while after 18 h such an effect
was only detectable at the concentration of 80 mg/kg. An increase in
sister-chromatid exchange frequencies over control values was not
detected in any treated animal.
Mice [(101/E1 x C3H/E1)F1], 10-14 weeks old and weighing
25-28 g, were injected intraperitoneally with hydroquinone (30, 50,
75 and 100 mg/kg), and bone marrow cells, sampled 18, 24 and 30 h
later (75 mg/kg), 6 and 24 h later (50 mg/kg) and 24 h later (30 and
100 mg/kg), were analysed for the presence of micronuclei. At a dose
level of 75 mg/kg, hydroquinone significantly increased the
frequency of micronuclei in cells at all sampling intervals.
Treatment with hydroquinone at 50 and 100 mg/kg significantly
increased the frequency of micronuclei after 24 h (Adler & Kliesch,
1990). The authors also reported results from daily treatments (up
to 3 days) of male mice of the same strain with hydroquinone (15 or
75 mg/kg) by the intraperitoneal route of administration. Bone
marrow cells were sampled 24 h after the first, second and third
injections, respectively. The frequency of micronuclei increased in
the case of the lower dose and decreased in the case of the higher
dose with increasing number of treatments.
The induction of micronuclei in mouse bone marrow cells after
intraperitoneal injection of hydroquinone has also been demonstrated
by Gocke et al. (1981), Ciranni et al. (1988a,b), Barale et al.
(1990) and Adler et al. (1991). Oral administration of
hydroquinone (200 mg/kg) induced an increase in the frequency of
micronuclei in mice (Gad-El-Karim et al., 1985). Ciranni et al.
(1988b) demonstrated that oral administration of hydroquinone (80
mg/kg) produced a weak increase in the frequency of micronuclei
compared to the effect found following intraperitoneal
administration. In male mice (outbred NMRI) daily subcutaneous
injections of hydroquinone on six consecutive days induced
micronuclei in the bone marrow at dose levels of 25 to 100 mg/kg
(Tunek et al., 1982).
When pregnant mice (Swiss CD-1, three months old) were given
hydroquinone (80 mg/kg) by gastric intubation on the 13th day of
gestation, micronuclei were induced in fetal liver cells (Ciranni
et al., 1988a).
In male rats (Sprague-Dawley), weighing 200 g and subjected to
70% partial hepatectomy, single-strand breaks were induced in
hepatic DNA after the rats had received daily hydroquinone doses of
200 mg/kg, given in a liquid casein-based diet, for 7 weeks (Stenius
et al., 1989).
Male mice [(102/ElxC3H/El)F1], 12-14 weeks old and weighing
25-28 g, were injected intraperitoneally with hydroquinone (40, 80
and 120 mg/kg). In spermatocytes sampled 12 days after treatment
(representing cells treated at preleptotene), the frequency of
chromosomal aberrations excluding gaps was significantly increased
at 40 and 80 mg/kg but not at 120 mg/kg. In differentiating
spermatogonia sampled 24 h after treatment, the frequency of
chromosomal aberrations excluding gaps was significantly increased
at all dose levels (Ciranni & Adler, 1991).
No increased frequency of dominant lethal mutations was
detected in male rats [CRL:COBS"CD"(SD)BR] given repeated doses of
hydroquinone (30, 100 or 300 mg/kg, 5 days/week for 10 weeks) by
gavage (Krasavage, 1984b). The induction of sperm-head abnormalities
could not be demonstrated in male mice after intraperitoneal
injection of hydroquinone (55-220 mg/kg) (Wild et al., 1981).
The ability of hydroquinone to produce adducts with DNA or its
nucleotides in vitro has been shown by Rushmore et al. (1984),
Jowa et al. (1986), Reddy et al. (1989), Jowa et al. (1990),
Leanderson & Tagesson (1990), Reddy et al. (1990) and Levay et
al. (1991). There is less evidence for the hydroquinone-induced
formation of DNA adducts in vivo. Using the 32P postlabelling
assay, no treatment-related adducts were detected in the kidneys of
either male or female F-344 rats following the oral administration
of hydroquinone at dose levels of up to 50 mg/kg for 6 weeks
(English et al., 1992). In additional studies by Reddy et al.
(1990), also using the postlabelling assay, no treatment-related DNA
adducts were detected in the bone marrow, zymbal gland or liver of
female F-344 rats following the oral co-administration of phenol and
hydroquinone at either 75 or 150 mg/kg for 4 days (Reddy et al.,
1990).
Hydroquinone has been shown to be capable of producing breaks
in phi X-174 DNA (Lewis et al., 1988a). No increase in the
frequency of breaks was detected in DNA from hydroquinone-treated
mouse lymphoma cells (L5178YS) at concentrations of up to 11
mg/litre (Pellack-Walker & Blumer, 1986).
7.7 Carcinogenicity
The available studies on the carcinogenicity of hydroquinone
are summarized in Table 17.
7.7.1 Long-term bioassays
In an NTP study (NTP, 1989; Kari et al., 1992), groups of 65
F-344/N rats of each sex were given hydroquinone (0, 25 or 50 mg/kg
body weight) in deionized water by gavage 5 days/week for up to 103
weeks, and groups of 65 B6C3F1 mice of each sex were administered
0, 50 or 100 mg/kg body weight according to the same schedule. A
15-month interim kill of ten animals from each group showed that the
kidney of male rats was a target organ forthe toxicity (see also
section 7.4), since there was a compound-related increased severity
of nephropathy. The lesions were less severe in female rats, in
which a mild regenerative anaemia was also found (slightly decreased
haematocrit, haemoglobin and erythrocyte count). After termination
of the experiment, a dose-related increase in the incidence of renal
tubular cell adenomas was observed in male rats (controls 0/55, low
dose 4/55, high dose 8/55; P = 0.003). The incidence of adenomas was
closely associated with the severity of chronic nephropathy. No
renal adenomas were observed in animals examined at 15 months, when
the severity of nephropathy was less severe, or in female rats,
which developed nephropathy to a lesser degree. In the male rats,
9/12 adenomas were seen in kidneys with marked nephropathy, two were
seen in animals with moderate nephropathy, and only one was seen in
an animal with mild nephropathy. In the high-dose group single
tubules exhibited tubular cell hyperplasia. No renal tumours were
seen in females. A dose-related increase in the incidence of
mononuclear cell leukaemia was found in female rats (controls 9/55,
low dose 15/55, high dose 22/55) (P < 0.01 in the high-dose group
versus controls). However, this was not observed in the animals
killed at 15 months. The incidence in controls was lower than the
historical control mean incidence but was within the historical
control group range.
Table 17. Carcinogenicity studies in animals
Species Route of Number of Dosage Time of Result Remarks Reference
exposure animals treatment
Long-term bioassays
Mouse oral 64 or 65 of 50 or 100 103 weeks liver lesions (males), some evidence of NTP (1989);
each sex mg/kg hepatocellular adenomas carcinogenic activity Kari et al.
per group 5 days/week (females) for female mice (1992)
Mouse oral 30 m, 30 f 0.8% in 96 weeks squamous cell hyperplasia of potential of Shibata et al.
the diet the forestomach epithelium; hepatocarcinogenicity (1991)
renal tubular hyperplasia and in male mice
adenomas (males); increased
incidence of liver foci and
hepatocellular adenomas
(males)
Rat oral 65 of each 25 or 50 103 weeks nephropathy (more severe in some evidence of NTP (1989);
sex per mg/kg males), renal tubular cell carcinogenic activity Kari et al.
group 5 days/week hyperplasia and adenomas for male and female (1992)
(males), leukaemia (females) rats
Rat oral 30 m, 30 f 0.8% in 104 weeks renal tubular hyperplasia, potential of renal Shibata et al.
the diet adenomas and epithelial carcinogencity in (1991)
hyperplasia of the renal male rats
papilla (males); decreased
incidence of liver foci
Table 17. (contd).
Species Route of Number of Dosage Time of Result Remarks Reference
exposure animals treatment
Carcinogenicity-related studies
Mouse skin 24 m 0.3 ml of 6.7% one skin papilloma (1/24) no initiating Roe & Salaman
application solution; application; activity (1955)
0.3 ml of then three
0.5% croton weeks later,
oil 18 weekly
applications
Mouse skin 50 f 5 mg three 368 days papilloma (7/50), squamous no co-carcinogenic van Duuren &
application times carcinoma (3/50) or tumour-promoting Goldschmidt
weeklya activity; partial (1976)
inhibition of BP
carcinogenicity
Mouse implantation not stated 2 mg 25 weeks carcinomas (6/19) Boyland et al.
in urinary (1964)
bladder
Rat oral 20 f 0.8% in 32 weeks no preneoplastic lesions Kurata et al.
basal dietb or papillomas of the (1990)
urinary bladder
Rat oral 15-16 m 0.8% in 51 weeks no increase in forestomach or Hirose et al.
dietc glandular stomach neoplasms (1989)
Rat oral 5 m 8 weeks no proliferative changes Shibata et al.
in forestomach or glandular (1990)
stomach
Table 17. (contd).
Species Route of Number of Dosage Time of Result Remarks Reference
exposure animals treatment
Rat oral 7-10 m per 100 mg/kg 7 weeks increased number of liver foci relatively weak Stenius et al.
group diet per dayd decreased number of liver foci inducer of enzyme- (1989)
200 mg/kg compared to the 100 mg/kg altered liver foci
diet per dayd dose
Hamster oral 15 m 0.5% in basal 20 weeks no proliferative changes in Hirose et al.
diet forestomach (1986)
a after initiating dose of benzo[ a] pyrene (BP)
b after initiating with N-butyl-2 N-(4-hydroxybutyl) nitrosamine for four weeks
c one week after 150 mg/kg body weight
d after partial hepatectomy
In male mice centrilobular fatty changes and cytomegaly were
found in the animals killed at 15 months, but these findings were
not seen in mice killed at 2 years. The authors reported that
hydroquinone dosing stopped two weeks before necropsy and that the
microscopic lesions were likely to be reversible after cessation of
treatment. There was a significantly (P=0.0005) increased incidence
of hepatocellular adenomas in female mice given hydroquinone for 2
years (controls 2/55, low dose 15/55, high dose 12/55) and the
incidences of hepatocellular carcinomas were 1/55, 2/55 and 2/55,
respectively. In males the incidence of adenomas was increased in
treated mice but the incidence of hepatocellular carcinomas was
decreased. Preneoplastic changes (anisokaryosis, multinucleated
hepatocytes, and basophilic foci) were increased in high-dose male
mice. Treatment-related, but not statistically significant,
follicular cell hyperplasia of the thyroid gland was observed in
both male and female mice (NTP, 1989; Kari et al., 1992).
The NTP concluded that there was "some evidence of carcinogenic
activity" of hydroquinone for male F-344/N rats (tubular cell
adenomas of the kidney) and also for female F-344/N rats
(mononuclear cell leukaemia). There was "no evidence of carcinogenic
activity" for male B6C3F1 mice and "some evidence of carcinogenic
activity" for female B6C3F1 mice (hepatocellular adenomas and
carcinomas combined).
Shibata et al. (1991) administered hydroquinone at dietary
levels of 0.% or 8 g/kg to groups of 30 Fischer-344 rats and
B6C3F1 mice of each sex. The rats were dosed for 104 weeks and the
mice for 96 weeks. Average daily intakes were reported to be 351 and
368 mg/kg body weight per day in male and female rats, respectively,
and 1046 and 1486 mg/kg per day in male and female mice,
respectively. No treatment-related clinical signs and no significant
differences in mortality were found between treated and control
animals of either species. The final body weight was significantly
(P < 0.05) lower in treated female rats than in corresponding
controls. In male rats the absolute and relative liver and kidney
weights were significantly (P < 0.01) increased, but in females
this applied only to the relative kidney weights (P < 0.05).
Histologically, chronic nephropathy was seen in both control and
treated groups of male rats. However, treated males were more
severely affected than the controls, while treated females showed
only slight nephropathy. The incidence of epithelial hyperplasia of
the renal papilla was significantly (P < 0.05) increased in treated
male rats as was the incidence of renal tubular hyperplasia (30/30)
and renal tubular adenomas (14/30).
The authors found that renal cell tumour development in male
rats under the long-term influence of hydroquinone was not
associated with alpha2u-globulin nephropathy. The incidence of liver
foci showed a tendency to decrease in treated males. A quantitative
analysis showed a statistically significant (P < 0.05 in males, P<
0.01 in females) reduction in both sexes given hydroquinone. The
authors did not find an increased incidence of mononuclear cell
leukaemia in female rats (personal communication).
In mice, the final body weight was significantly (P < 0.05)
lower in females given hydroquinone; the relative liver and kidney
weights were significantly (P < 0.05) increased. Histologically,
the incidence of squamous cell hyperplasia of the forestomach
epithelium was significantly (P < 0.01) increased in both sexes. A
significant increase in the incidence of renal tubular hyperplasia
(P < 0.01) and three renal cell adenomas were seen in 30 males
given hydroquinone. In treated males the incidence of liver foci and
hepatocellular adenomas (14/30) was also significantly (P < 0.05)
increased.
7.7.2 Carcinogenicity-related studies
7.7.2.1 Skin
In a study by Roe & Salaman (1955), stock albino mice (24
males, "S" strain) were given a single skin application of 0.3 ml of
a 6.7% solution of hydroquinone in acetone (total dose 20.0 mg).
Three weeks later the mice received 18 weekly applications of 0.3 ml
of 0.5% croton oil in acetone as a promoter on the same area of the
skin. Of the 24 treated animals, two died during the experiment and
one mouse developed a skin papilloma.
In a two-stage carcinogenesis test on mouse skin using
benzo[ a]pyrene (BP) as the initiating agent, no tumour-promoting
activity was shown (Van Duuren & Goldschmidt, 1976). Hydroquinone (5
mg) was applied to mouse skin (50 female ICR/Ha Swiss mice/group;
both positive and negative controls) three times weekly for 368
days, together with 5 µg BP. Hydroquinone showed no potential as a
co-carcinogen when applied simultaneously with BP; in fact, it
partially inhibited BP carcinogenicity.
7.7.2.2 Bladder
Implantation of cholesterol pellets containing hydroquinone
into the urinary bladder of mice (strain and sex unspecified) has
been studied by Boyland et al. (1964). The amount of hydroquinone
was 20% in 10 mg cholesterol pellets (2 mg hydroquinone per mouse).
Bladder carcinomas were produced in 6 out of 19 mice (32%) surviving
25 weeks. The incidence of urinary bladder carcinomas in survivors
of the dosed group was significantly (P=0.03) higher than in
controls (11.7%) given cholesterol pellets only. However, the number
of animals surviving the study was low, and the original number of
animals and their sex distribution were not specified.
In a study by Kurata et al. (1990), groups of 20 male
Fischer-344 rats received 0.05% N-butyl- N-(4-hydroxybutyl)
nitrosamine in the drinking-water for four weeks (as initiation)
followed by 8 g hydroquinone/kg in the basal diet for 32 weeks. No
increase in the incidence of preneoplastic lesions or
papillomas/carcinomas of the urinary bladder was observed when
compared to the incidences in rats given nitrosamine alone.
7.7.2.3 Stomach
Hirose et al. (1989) examined the promotion activity and the
carcinogenic potential of some dihydroxybenzenes, such as
hydroquinone, in the glandular stomach and forestomach of F-344
rats. Groups of 15-16 male rats were given a single intragastric
dose of 150 mg/kg body weight N-methyl- N'-nitro- N-
nitrosoguanidine (MNNG), followed one week later by powdered diet
containing hydroquinone (8g/kg) or basal diet alone for 51 weeks.
Further groups of 10 and 15 animals, respectively, were administered
the basal diet alone or a diet containing hydroquinone (8 g/kg) for
51 weeks without pretreatment with MNNG. Hydroquinone did not cause
an increased incidence of forestomach or glandular stomach lesions,
either with or without pretreatment with MNNG, in comparison with
the control groups.
In studies performed by Hirose et al. (1986), hydroquinone
did not produce proliferative lesions in the stomach of hamsters.
Male Syrian golden hamsters (15/group, seven weeks old at the
beginning of the study) were given basal diet with hydroquinone (5
g/kg) added or basal diet alone for 20 weeks. The dose was chosen as
approximately a quarter of the LD50. Tissues from forestomach and
glandular stomach showed mild to moderate hyperplasia in the group
given hydroquinone, but at the same incidence as in the controls.
Similar results were obtained by Shibata et al. (1990) in an
8-week oral study using five male F-344 rats. Hydroquinone did not
induce any proliferative changes in the forestomach or the glandular
stomach epithelium.
7.7.2.4 Liver
Hydroquinone has been shown to be a relatively weak inducer of
enzyme-altered foci in rat liver when tested for tumour-promoting
activity in a liver focus test (Stenius et al., 1989). Male
Sprague-Dawley rats (7-10/group) given diethylnitrosamine (30 mg/kg
intraperitoneally) after partial hepatectomy were treated with
hydroquinone (0, 100 and 200 mg/kg per day) in their diet for 7
weeks. At 100 mg/kg there was a significantly (P < 0.01) increased
number of liver foci and an increased focus volume. The 200-mg dose
caused less foci (0.34 ± 0.16 per cm2) than the 100-mg dose (0.65
± 0.25 per cm2), but the incidence was higher than in the control
group (0.08 ± 0.08 per cm2).
A study by Kurata et al. (1990) yielded similar results
concerning the tumour-promoting potential of hydroquinone in rats.
Dietary administration of hydroquinone (8g/kg in basal diet) for 32
weeks, after initiation for four weeks with N-butyl- N-
(4-hydroxybutyl) nitrosamine, caused no preneoplastic lesions or
papillomas of the urinary bladder.
7.8 Special studies
7.8.1 Effects on spleen and bone marrow cells; immunotoxicity
The bone marrow is the target in benzene toxicity; among the
many metabolites of benzene, hydroquinone has received increased
scrutiny as one of the possible contributing factors. Intravenous or
intraperitoneal administration of hydroquinone (100 mg/kg) for three
consecutive days to male C57BL/6 CRIBR mice significantly (P <
0.05) reduced the spleen and bone marrow cellularity, with bone
marrow demonstrating the greatest sensitivity (Wierda & Irons,
1982). Laskin et al. (1989) found that after injection in Balb/c
mice hydroquinone (50 mg/kg) caused a 30-40% decrease in bone marrow
cellularity.
In vitro studies have demonstrated direct myelotoxic effects
of hydroquinone toward mouse bone marrow stromal cells (Gaido &
Wierda, 1984; Gaido & Wierda 1987). Hydroquinone inhibited stromal
cell colony growth along with the ability of these cells to support
granulocyte/monocyte colony formation in co-culture. The bone marrow
stroma predominantly consists of macrophages and fibroblastoid
stromal cells which interact to regulate myelopoiesis. Treatment
with hydroquinone thus results in reduced capacity of the stroma to
support myelopoiesis.
In addition to this cytotoxic effect, Wierda & Irons (1982)
found in in vivo studies that hydroquinone also affected the
immune function by reducing the number of progenitor B-lymphocytes
in the spleen and bone marrow in mice, thus demonstrating an
immunosuppressive potential. The rapid generation and maturation of
progenitor B cells renders them highly susceptible to toxic agents
that affect dividing cells. Evidence has accumulated concerning the
effect of hydroquinone on the cellular activity of the immune system
in vitro. Exposure of lymphocytes in vitro to hydroquinone has
been shown to result in a dose-dependent inhibition of RNA synthesis
in the lymphocytes (Post et al., 1985). A hydroquinone
concentration of 1-2 x 10-5 mol/litre inhibited the RNA synthesis
by 50%.
In vitro exposure (one hour) of mouse bone marrow cells to
hydroquinone (10-7-10-5 mol/litre) inhibited the maturation of
B-lymphocytes from pre B-cells after 24 and 48 h in culture (King
et al., 1987). More recent data have demonstrated that
hydroquinone-induced inhibition of pre-B cell maturation results
from toxicity to adherent stromal cells, and that bone marrow
macrophages may be the primary target for hydroquinone
myelotoxicity, rather than fibroblastic stromal cells or pre-B cells
(King et al., 1989; Thomas et al., 1989a). Results also indicate
a dose-related reduction of macrophage interleukin-1 (IL-1)
secretion in cultures of bone marrow macrophages exposed to
hydroquinone (King et al., 1989; Thomas et al., 1989b). IL-1 is
necessary for the induction of interleukin-4 (IL-4), which is
produced by fibroblastic stromal cells and is required for
maturation of pre-B cells to B cells (King et al., 1989).
Fan et al. (1989) demonstrated that hydroquinone can inhibit
the natural killer activity of mouse spleen cells in vitro at low
concentrations. Concentrations of 1 x 10-5 mol/litre and 1 x
10-6 mol/litre inhibited 29 and 22% of the activity, respectively.
Lewis et al. (1988b) found that hydroquinone had a selective
effect on macrophage functions important in host defense. At
concentrations of 3-100 µmol/litre, hydroquinone significantly
(P < 0.05) inhibited the release of hydrogen peroxide and at 100
µmol/litre it significantly (P < 0.05) inhibited priming by
interferon for tumour cell cytolysis. Cheung et al. (1989) have
shown a concentration-dependent inhibition of interferon-alpha/ß
production following exposure to hydroquinone in murine L-929 cell
cultures.
7.8.2 Effects on tumour cells
The cytotoxic activity of hydroquinone has been tested on
different tumour cells. Chavin et al. (1980) studied the effect on
melanoma transplants in female BALB/c mice. The incidence of
melanoma transplants was reduced and the survival significantly (P
< 0.0005) increased in mice that received hydroquinone treatment
(80 mg/kg).
Vladescu & Apetroae (1983) studied the molecular mechanisms of
antitumour action and the possibilities of using hydroquinone as a
toxic agent against cancer cells. In H 18R tumour-bearing male
Wistar rats treated with hydroquinone (5 mg/kg per day) for seven
days, the catalase activity was markedly depressed in liver, spleen,
blood and H 18R tumour. In vitro studies on tumour and liver
homogenates from normal and tumour-bearing rats showed a marked
inhibition of catalase activity in the tumour, which was less
evident in the liver. The activity was less reduced in normal liver
homogenates. It was suggested that the mechanism of action of
hydroquinone as an antitumour agent is achieved mainly via peroxide
production.
When tested on cultured rat hepatoma cells hydroquinone showed
a dose-dependent cytotoxic activity (Assaf et al. 1987). A dose of
33 mg/litre (300 µmol/litre) caused cellular mortality of 40% after
24 h of incubation and 66 mg/litre (600 µmol/litre) resulted in 100%
cellular mortality.
7.8.3 Neurotoxicity
Hydroquinone, given as single oral or subcutaneous lethal
doses, causes nonspecific effects on the nervous system such as
hyperexcitability, tremor and convulsions in several experimental
animal species (see section 7.1). Animals given sublethal oral doses
recover within a few days.
These central nervous system stimulation effects were confirmed
in a 90-day oral study on rats (Eastman Kodak Company, 1988) (see
also section 7.3). Male and female weanling rats (CD(SD)BR),
initially seven weeks old, were treated with hydroquinone (20, 64 or
200 mg/kg per day) dissolved in water at a concentration of 5%.
Doses were given by gavage 5 days per week. Functional-observational
battery examinations were performed throughout the study. The
battery included observations of body position, activity level,
coordination of movement and gait, behaviour, presence of
convulsions, tremors, lacrimation, salivation, piloerection,
pupillary dilatation or constriction, respiration, diarrhoea,
urination, vocalization, forelimb/hindlimb grip strength and sensory
function. Tremors and depression of general activity were observed
in both sexes shortly after dosing with 64 or 200 mg
hydroquinone/kg. Functional-observational battery examinations did
not result in any evidence of neurotoxicity as assessed by
quantitative grip strength measurement, brain weight or
neuropathological examination. The NOEL was considered to be 20 mg
hydroquinone/kg body weight.
Otsuka & Nonomura (1963) reported that hydroquinone reversed
curare blockage at neuromuscular junctions in frog sciatic nerve -
sartorius muscle preparations. The authors suggested that this
effect was due to an increased release of transmitter at the
neuromuscular junction induced by hydroquinone.
7.8.4 Nephrotoxicity
Until recently, exposure to hydroquinone has not been
associated with nephrotoxicity. Nephrotoxicity has not been reported
following either occupational exposure to hydroquinone or acute
exposures in humans. Carlson & Brewer (1953) gave human volunteers
daily hydroquinone doses of 300 or 500 mg/day for periods of up to
20 weeks without effects on urinalysis parameters. Exposure of five
male mixed-breed dogs to 100 mg hydroquinone/kg per day for 26 weeks
had no effect on urinalysis parameters or renal histopathology
(Carlson & Brewer, 1953). Christian et al. (1976) reported that
exposure of Carworth rats to hydroquinone in the drinking-water at
concentrations of up to 10 g/litre (6 rats of each sex per group for
8 weeks) or up to 4 g/litre (20 rats of each sex per group for 15
weeks) resulted in slight changes in kidney weight but no
histopathological changes. Carlson & Brewer (1953) also reported no
evidence of renal histopathological changes in Sprague-Dawley rats
fed diets containing 10g hydroquinone/kg for 104 weeks.
NTP (1989) reported that oral gavage of hydroquinone (0, 25,
50, 100, 200 or 400 mg/kg) in corn oil for 13 weeks resulted in
toxic nephropathy in F-344 rats at the two highest dose levels (200
mg/kg: 7/10 males, 6/10 females; 100 mg/kg: 1/10 females). Oral
gavage of 0, 25 or 50 mg/kg in water for 15 months resulted in an
increased incidence of chronic nephropathy in male F-344 rats (25
mg/kg: 5/5 males; 50 mg/kg: 6/10 males). When male F-344 rats were
dosed at 0, 25 or 50 mg/kg for two years, there was an increased
severity of chronic progressive nephropathy in 20/55 animals given
50 mg/kg. At a dosage level of 50 mg/kg for either 15 months or 2
years, male rats had heavier relative kidney weights.
Shibata et al. (1991) also reported that F-344 rats developed
chronic nephropathy when fed 8g hydroquinone/kg diet for 2 years.
Male rats showed increased relative and absolute kidney weight, as
well as an increased severity of chronic nephropathy (14/30
animals). Female rats showed an increased relative kidney weight,
but only a minimal increase in severity of chronic nephropathy in
7/30 animals.
Boatman et al. (1992) reported on the urinalysis changes
observed in male and female F-344 rats and Sprague-Dawley rats given
single doses of 0, 200 or 400 mg hydroquinone/kg in water by oral
gavage. B6C3F1 mice were examined after receiving doses of 0 or
350 mg/kg in a similar fashion. The placement of venous catheters in
F-344 rats increased their response to hydroquinone. At 400 mg/kg,
male and female F-344 rats, but not Sprague-Dawley rats, displayed
pronounced enzymuria and glucosuria, which resolved in 72-96 h. At
200 mg/kg, enzymuria and glucosuria were present in female F-344
rats but not males. Epithelial cell counts in the urine were
statistically significantly increased (P < 0.05) at 400 mg/kg
(male and female F-344 rats only) and 200 mg/kg (female F-344 rats
only). Statistically significant (P < 0.05) decreases in
osmolality were reported at 400 mg/kg for F-344 (both sexes) and
female Sprague-Dawley rats. Diuresis (ml urine/h) was statistically
significant (P < 0.005) only for female F-344 rats at 200 mg/kg
and 400 mg/kg. Although differences were observed in some of the
urinary parameters measured, mice were generally not responsive to
hydroquinone.
To characterize the early development of renal toxicity in
rats, cell proliferation was quantified within the proximal (P1, P2
and P3) and distal tubular segments of the kidney in rats given 0,
2.5, 25 or 50 mg hydroquinone/kg by oral gavage. Male and female
F-344 rats were treated for 1, 3 or 6 weeks, and male Sprague-Dawley
rats were treated for 6 weeks. At 6 weeks, an 87% increase in cell
proliferation was measured in the P1 segment, a 50% increase in the
P2 segment, and a 34% increase in the P3 segment from kidneys of
male F-344 rats dosed with 50 mg/kg. Urinalysis indicated increased
enzymuria in this same dose group, and mild histological changes
were present in the kidneys. Animals examined at other time points
or from other dose groups were not affected by hydroquinone.
The increased incidence of renal adenomas only in male F-344
rats (NTP, 1989) has led to speculation that the tumours observed
may be related to alpha2u-globulin-induced nephropathy. This
mechanism of action for induction of kidney tumours does not appear
to be relevant for hydroquinone as none of the studies cited above
has reported finding evidence of hyalin droplet nephropathy
following subacute, subchronic or chronic hydroquinone exposure.
Glutathione metabolites, which are at least partially formed in
the liver and transported to the kidney, are reported to be involved
in the nephrotoxicity observed. Some of the potential glutathione
conjugates of hydroquinone have been shown to be more nephrotoxic
when injected parenterally than the parent chemical (Boatman et
al., 1992; Hill et al., 1992a,b; Kleiner et al., 1992).
Administration of hydroquinone by parenteral injection, a route
which is likely to increase hydroquinone glutathione conjugates,
induces nephrotoxicity in otherwise non-responsive male
Sprague-Dawley rats (Hill et al., 1992a,b; Kleiner et al.,
1992). The formation of glutathione metabolites and an increased
susceptibility of the male F-344 rat to the conjugates appear to be
mechanistically linked to the nephrotoxicity observed in these rats.
7.8.5 Interaction with phenols
Recently there have been a number of studies reporting
interactive effects between hydroquinone and other phenolic
compounds. Initially, Eastmond et al. (1987) showed that the
co-administration of hydroquinone and phenol (75 mg/kg), when given
by intraperitoneal injection twice per day, produced a synergistic
decrease in bone marrow cellularity in B6C3F1 mice that was
similar to that induced by benzene. This combined treatment was
significantly more myelotoxic than that observed when either
hydroquinone or phenol was administered separately. Associated in
vitro studies suggested that this interactive effect was due to a
phenol-induced stimulation of the myeloperoxidase-mediated
conversion of hydroquinone to 1,4-benzoquinone in the bone marrow
(Eastmond et al., 1987; Smith et al., 1989; Subrahmanyam et
al., 1991).
Subsequent studies have indicated that interactions between
hydroquinone and other phenolic compounds can result in a variety of
cytotoxic, immunotoxic and genotoxic effects. Some of the adverse
interactive effects that have been reported are outlined below:
a) Decreased uptake of 59Fe, an indicator of toxicity to the
bone marrow, has been reported with the combined administration
of hydroquinone and various phenolic metabolites (Guy et al.,
1990, 1991).
b) The combined administration of phenol and radiolabelled
hydroquinone results in increased binding of hydroquinone
equivalents in the bone marrow (Subrahmanyam et al., 1990).
c) Decreased bone marrow cellularity and increased production of
reactive oxygen species in phagocytes when stimulated with a
phorbol ester tumour promoter have been observed following
hydroquinone and phenol co-administration (Laskin et al.,
1989).
d) Synergistic increases in the formation of micronuclei have been
observed in mice and human lymphocytes obtained from one
individual following exposure to hydroquinone and other
phenolic metabolites (Barale et al., 1990; Robertson et
al., 1991).
e) Three- to six-fold increases in DNA adduct formation (over that
observed using the sum of the individual metabolites) were
observed in HL-60 cells treated with the combination of
hydroquinone and either catechol or 1,2,4-trihydroxybenzene. In
addition, the combined treatment of hydroquinone and
1,2,4-trihydroxybenzene produced DNA adducts not detected after
treatment with either metabolite alone (Levay & Bodell, 1992).
f) Co-treatment of phenol and hydroquinone was reported to shift
the optimal concentration of hydroquinone inducing the maximal
recombinant granulocyte/macrophage colony-stimulating factor
response from 1 µmol/litre to 100 pmol/litre (Irons et al.,
1992).
g) The co-administration of hydroquinone with either phenol or
catechol in vivo to B6C3F1 mice increased the formation of
oxidative DNA damage as measured by the formation of
8-hydroxy-2'-deoxyguanosine which occurred in the bone marrow
of B6C3F1 mice (Kolachana et al., in press).
8. EFFECTS ON HUMANS
8.1 General population exposure
8.1.1 Acute toxicity - poisoning incidents
There have been reports of poisoning due to accidental or
suicidal ingestion of hydroquinone alone (Mitchell & Webster, 1919;
Rémond & Colombies, 1927) or of photographic developers containing
hydroquinone (Busatto, 1939; Zeidman & Deutl, 1945; Grudzinski,
1969; Larcan et al., 1974). Deaths have been reported after
ingestion of photographic developers containing hydroquinone in
amounts of 3-12 g (80-200 mg/kg body weight). The main symptoms of
intoxication by hydroquinone include tremors, vomiting, abdominal
pain, headache, tachycardia, convulsions, loss of reflexes, dark
urine, dyspnoea, cyanosis and coma. No adverse systemic effects have
been reported after acute inhalation of hydroquinone dust (Anderson,
1947; Oglesby et al., 1947; Sterner et al., 1947).
8.1.2 Short-term controlled human studies
In a controlled oral study, ingestion of 500 mg hydroquinone
daily for 5 months (two males) or 300 mg/day for 3-5 months (17
volunteers, both males and females) produced no observable
pathological changes in blood and urine (Carlson & Brewer, 1953). No
further data were given.
8.1.3 Dermal effects; sensitization
Skin lighteners often contain hydroquinone (1.5 to 2%) as the
bleaching agent, which inhibits the production of melanin. Prolonged
use (about three years) of strong (>5%) hydroquinone bleaching
creams has been reported to cause ochronosis and pigmented colloid
milium in South African black women (Findlay et al., 1975; Findlay
& de Beer, 1980). Sporadic skin reactions have also occurred among
amateurs who develop their own films manually (Fisher, 1986).
In a study to assess the safety of hydroquinone in cosmetic
skin-lightening products, 840 male volunteers from different human
races such as Blacks (Zulu), Asians (Indians), and Coloureds (mixed
ethnic origins) were treated with various concentrations of
hydroquinone in different bases (Bentley-Philips & Bayles, 1975).
They were subjected to open-patch tests, normal usage tests, and
standard 48 h closed-patch tests. The results of the study showed
that concentrations of hydroquinone below 3% produced negligible
adverse effects, irrespective of the base used or the colour of the
user's skin. In earlier, less extensive studies, Fitzpatrick et al.
(1966) found that a 5% cream of hydroquinone caused a high incidence
of primary irritant reactions such as erythema and tingling at the
site of application.
Fisher (1982) reported four cases of leucoderma following the
use of bleaching creams containing 2% hydroquinone. The leucoderma
was not preceded by inflammation and the patients had no positive
patch-test reaction to 1% hydroquinone in petrolatum after 72 h.
Hypopigmentation produced by 1% hydroquinone occurred in one patient
(Fisher, 1986). Spencer (1965) found that higher concentrations (5%)
of hydroquinone might cause sensitization.
Van Ketel (1984) reported a case of probable sensitization to
hydroquinone with cross-sensitization to hydroquinone monobenzyl
ether. Two days after the first application of a cream containing 5%
hydroquinone monobenzyl ether (after a treatment period of three
months with 2% hydroquinone in a cream base), an acute dermatitis
developed. Patch testing was positive for the two substances.
Sensitization to hydroquinone monobenzyl ether occurs fairly
frequently (Fisher, 1986), while hydroquinone is regarded as a weak
sensitizer.
There have been several cases in Europe and the USA of
ochronosis in people who have used creams containing 2% hydroquinone
or less (Connor & Braunstein, 1987; Lawrence et al., 1988).
Hardwick et al. (1989) established a causal link between
hydroquinone and exogenous ochronosis.
Hydroquinone, like its monobenzyl ether or monoethyl ether, has
been reported to cause severe patchy depigmentation disorders in a
confetti-like pattern in a single black man (Markey et al., 1989).
These authors also noted that four other cases had been reported.
Several cases of brown discoloration of the finger-nails due to
hydroquinone-containing skin-lightening creams have been reviewed by
Mann & Harman (1983). The colour change is considered to be due to
hydroquinone oxidation products resulting from exposure to sunlight.
8.2 Occupational exposure
8.2.1 Dermal effects
There have been case reports of occupational depigmentation of
the skin where a causal relationship between photographic developers
containing hydroquinone and depigmentation (leucoderma or vitiligo)
has been suggested (Frenk & Loi-Zedda, 1980; Kersey & Stevenson,
1981). In one case a vitiligo-like depigmentation was induced in a
black man by contact with a dilute hydroquinone solution (0.06%)
after 8-9 months (Frenk & Loi-Zedda, 1980). The depigmentation
occurred without any inflammatory skin changes and without itching.
Biopsy revealed lack of epidermal melanin pigment and melanocytes.
However, in the upper dermis there were numerous melanin-laden
macrophages. Developer containing 7% hydroquinone has also caused
vitiligo in a man wearing protective gloves (Kersey & Stevenson,
1981). The gloves were considered to have functioned as an occlusive
bandage.
Lidén (1989) carried out dermatological examination and patch
testing on 78 employees exposed to film chemicals at a film
laboratory. Hydroquinone (1% aqueous solution) was found to cause
irritation (erythema or staining) in previously non-exposed, healthy
volunteers. Contact allergy to hydroquinone (1% in water and
petrolatum) was diagnosed in four out of seven tested employees.
8.2.2 Ocular effects
Ocular lesions of various degrees have been observed in workers
exposed to quinone vapour and hydroquinone dust in the manufacture
of hydroquinone, and the clinical characteristics are well described
(Sterner et al., 1947; Oglesby et al., 1947; Anderson & Oglesby,
1958). Airborne concentrations of hydroquinone dust were
occasionally 20-35 mg/m3 (Oglesby et al., 1947). In the presence
of air and moisture hydroquinone is rapidly converted into the more
volatile quinone. Acute exposure to high (not specified) vapour
concentrations resulted in irritation, sensitivity to light,
lacrimation, injury of the corneal epithelium, and corneal
ulceration. Acute exposure to hydroquinone dust caused eye
irritation. Chronic exposure to hydroquinone dust led to corneal
staining (greenish-brown), corneal opacity, and conjunctival
staining (brownish to brownish-black), with a distribution
corresponding to the palpebral fissure (Anderson, 1947; Sterner et
al., 1947). In some cases, an appreciable loss of vision due to
permanent fine opacities, astigmatism and irregularity occurred in
the cornea (Anderson & Oglesby, 1958). Eye irritation occurred
following exposure to 2.25 mg/m3 (0.5 ppm) and became marked at
13.5 mg/m3 (3 ppm). Slowly developing inflammation and
discoloration of the cornea and conjunctiva followed daily exposures
of 0.05 to 14.4 mg hydroquinone/m3 (0.01 to 3.2 ppm) for two or
more years (Oglesby et al. 1947). The degree of eye injury showed
a positive correlation only with length of employment (Anderson,
1947; Sterner et al., 1947; Anderson & Oglesby, 1958). The ocular
lesions developed gradually over a period of several years of
exposure; no serious cases were seen until after five or more years
of exposure. Removal from exposure resulted in considerable
improvement of the staining, but improvement of the corneal
opacities was questionable. The relative contribution of quinone
vapour and hydroquinone dust to eye injury was not assessed.
Three cases of corneal pigmentation were described among men
working in a hydroquinone factory (Naumann, 1966). At a later date,
corneal damage became apparent even though exposure had been
discontinued, and progressive deterioration of vision was reported.
Histopathological examinations revealed pigmentary and degenerative
changes. Two distinct forms of pigment were distinguished. One was
intraepithelial and contained iron, the other was confined to a
band-like zone between normal and altered stroma and was considered
to be quinone.
An operation nurse repeatedly developed a corneal ulcer while
mixing bone cement containing methyl methacrylate and hydroquinone
(Norrelykke Nissen & Corydon, 1985). It was suggested that the eye
symptoms developed because of a composite effect of methyl
methacrylate and hydroquinone vapours. However, the presence of
hydroquinone or quinone vapour in the air was not determined.
8.2.3 Systemic effects
Clinical and laboratory evaluation of 88 workers in 1943 and
101 workers in 1945, who had been exposed to high airborne
concentrations of both quinone and hydroquinone for periods up to 15
years, showed no evidence of any systemic toxicity (Sterner et
al., 1947).
8.2.4 Epidemiological studies
8.2.4.1 Respiratory effects
In a study on airway responses to hydroquinone (Choudat et
al., 1988), 33 workers exposed to hydroquinone, trimethyl-
hydroquinone, and retinene-hydroquinone were compared to a reference
group of 55 matched, non-exposed workers regarding the potential
allergic effect of the exposure. No further information on exposure
conditions (nature, extent and duration) and possible exposure to
other chemicals was reported. The prevalence of respiratory symptoms
was increased in workers exposed to hydroquinone and its
derivatives. The exposed workers had a significantly (P < 0.01)
higher prevalence of cough induced by a smoky atmosphere or by cold
air. The prevalences of eczema and coughing at work were also
higher. Pulmonary function values were significantly (P < 0.01)
lower in the exposed than in the non-exposed group. According to the
results from a bronchodilator test, hydroquinone or its derivatives
also seemed to induce intermittent dyspnoea and reversible
obstruction. The exposed workers had higher levels of IgG (P <
0.002) and IgE (not significant) than the non-exposed workers.
However, because of the higher chemical reactivity of trimethyl-
hydroquinone compared with hydroquinone, it is difficult to
determine the effects of hydroquinone alone (O'Brien, 1991).
8.2.4.2 Carcinogenicity studies
Greenwald et al. (1981) performed a nested case-referent
study of brain cancer in a group of employees from the Eastman Kodak
Company. This study was prompted by preliminary data suggesting a
possible increase of brain cancer incidence in New York state, USA,
and included a case group of 56 employees who had died with primary
brain tumours during the period 1956-1975. An elevated relative risk
was found among photographic processing workers exposed to colour
developers, but the results were not statistically significant. The
authors stated that the excess of brain neoplasms noted may have
resulted from a diagnostic sensitivity bias arising from more
complete medical evaluation of Kodak employees than of other plant
employees.
A cohort study of 478 photographic processors in nine Eastman
Kodak Color Print and Processing laboratories was undertaken as a
follow-up study. The primary objectives of the study were to
evaluate mortality, cancer incidence, and absence due to sickness of
employees exposed to photographic chemicals. Both industrial and
general population references were used. There was no significant
increase in the incidence of the parameters investigated. Two cases
classified as malignant neoplasms on the brain and CNS were observed
versus 0.4 expected (Friedlander et al., 1982), but this
difference was not statistically significant. Individual exposures
were not examined, but hydroquinone was identified among the many
possible exposures. One air sample analysed for hydroquinone
contained < 0.01 mg/m3.
Work-related mortality among male employees at Tennessee
Eastman Company was studied by Pifer et al. (1986). The exposure
included hydroquinone among other chemical agents. Cancer mortality
was lower than that of the general population; the standard
mortality ratio was 56. No information on levels of hydroquinone
exposure or total number of workers exposed to hydroquinone was
reported. However, work is underway to explore the feasibility of
conducting a mortality and, possibly, a morbidity study of
hydroquinone workers at this facility (personal communication to the
IPCS, 1992).
9. EFFECTS ON OTHER ORGANISMS IN THE LABORATORY AND FIELD
Hydroquinone has been shown to be an outlier in numerous QSAR
(Quantitative Structure-Activity Relationship) studies (e.g.,
Devillers et al., 1986, 1987; Hodson et al., 1988; Nendza &
Seydel, 1988a,b,c). Due to this fact Devillers et al. (1990)
reviewed the environmental and health risks of hydroquinone.
Ecotoxicity data for hydroquinone are listed in Table 18. From these
data, it appears that hydroquinone is highly toxic for most of the
organisms studied. However, a difference in sensitivity exists among
the taxons. It should be noted that the results need to be analysed
in relation to the experimental conditions under which they were
obtained (e.g., pH, light). Thus, numerous studies have been
performed under static condition. In only a few experiments has the
concentration of hydroquinone been monitored.
Table 18. Ecotoxicity data for hydroquinone
Species Effect Test Hydroquinone Comments Reference
duration concentration
(mg/litre)
Bacteria
Beneckea harveyi 50% inhibition of luminescence 10 sec 82.6 Stom et al.
100% inhibition of luminescence 10 sec 550.6 (1986)
50% inhibition of dehydrogenase 1 h 110
activity
Escherichia coli toxicity threshold concentration 6 h 50 Bringmann &
for inhibition of acid production Kühn (1959a)
from glucose
50% inhibition of cell 6-8 h 34.0 Devillers et
multiplication al. (1990)
Photobacterium EC10, luminescence 30 min 0.022 Devillers et
phosphoreum EC50 0.072 al. (1990)
EC90 0.210
EC50, luminescence 5 min 0.042 Ribo & Kaiser
10 min 0.038 (1983)
30 min 0.038
Pseudomonas zone of growth inhibition on 24 h 200 the zone of inhibition Trevors &
fluorescens agar was 14 mm; after exposure Basaraba
the % survival of resting (1980)
cells was 0.11
Pseudomonas putida toxicity threshold concentration 16 h 58 Bringmann &
for inhibition of cell Kühn (1977a)
multiplication
Table 18. (contd).
Species Effect Test Hydroquinone Comments Reference
duration concentration
(mg/litre)
Dinoflagellates
Crypthecodinium 50% mortality 40 h 50.0 Devillers et
cohnii al. (1990)
Prorocentrum micans 50% immobilization 1 h 0.750 Devillers et
50% immobilization 2 h 0.300 al. (1990)
100% immobilization 18 h 5.00
Cyanobacteria
(Blue-green algae)
Anabaena flos-aque lowest concentration for no 14 days 39.8 irradiance of 2 W/m2 Wängberg &
detectable growth Blanck (1988)
Anabaena sp. lowest concentration for no 14 days 10.0 irradiance of 2 W/m2 Wängberg &
detectable growth Blanck (1988)
"LPP sp" 1 PCC6402 lowest concentration for no 14 days 5.01 irradiance of 10 W/m2 Wängberg &
detectable growth 20.0 irradiance of 2 W/m2 Blanck (1988)
"LPP sp" 2 PCC73110 lowest concentration for no 14 days 5.01 irradiance of 2 W/m2 Wängberg &
detectable growth Blanck (1988)
Microcystis toxicity threshold concentration 8 days 1.1 Bringmann &
aeruginosa for inhibition of cell Kühn (1978)
multiplication
EC100 24 h 0.5 Fitzgerald et
al. (1952)
Synechococcus lowest concentration for no 14 days 10.0 irradiance of 10 W/m2 Wängberg &
leopoliensis detectable growth 10.0 irradiance of 2 W/m2 Blanck (1988)
Table 18. (contd).
Species Effect Test Hydroquinone Comments Reference
duration concentration
(mg/litre)
Algae
Bumilleriopsis lowest concentration for no 14 days 10.0 irradiance of 10 W/m2 Wängberg &
filiformis detectable growth Blanck (1988)
Chlamydomonas lowest concentration for no 14 days 79.4 irradiance of 10 W/m2 Wängberg &
dysosmos detectable growth Blanck (1988)
Chlamydomonas 100% inhibition of motility 15 min 55.1 Stom & Roth
reinhardii (1981)
Chlorella emersonii lowest concentration for no 14 days 79.4 irradiance of 10 W/m2 Wängberg &
detectable growth Blanck (1988)
slight activation of growth 24 h 10.0 Devillers et
al. (1990)
Dunaliella marina activation of growth 24 h 10.0 Devillers et
al. (1990)
Dunaliella salina 100% inhibition of motility 15 min 330.3 Stom & Roth
(1981)
Euglena gracilis 100% inhibition of motility 15 min 7708 Stom & Roth
(1981)
Kirchneriella contorta lowest concentration for no 14 days 79.4 irradiance of 10 W/m2 Wängberg &
detectable growth Blanck (1988)
Klebsormidium lowest concentration for no 14 days 20.0 irradiance of 10 W/m2 Wängberg &
marinum detectable growth Blanck (1988)
Table 18. (contd).
Species Effect Test Hydroquinone Comments Reference
duration concentration
(mg/litre)
Monodus lowest concentration for no 14 days 20.0 irradiance of 10 W/m2 Wängberg &
subterraneus detectable growth Blanck (1988)
Monoraphidium lowest concentration for no 14 days 20.0 irradiance of 10 W/m2 Wängberg &
pusillum detectable growth 79.4 irradiance of 2 W/m2 Blanck (1988)
Nitella sp. 100% inhibition of cytoplasmic 15 min 2753 Stom & Roth
streaming (1981)
Raphidonema lowest concentration for no 14 days 0.316 irradiance of 10 W/m2 Wängberg &
longiseta detectable growth Blanck (1988)
Scenedesmus lowest concentration for no 14 days 39.8 irradiance of 10 W/m2 Wängberg &
obtusiusculus detectable growth Blanck (1988)
Scenedesmus toxicity threshold concentration for 96 h 4 Bringmann &
quadricauda inhibition of cell multiplication Kühn (1959a)
toxicity threshold concentration for 7 days 0.93 Bringmann &
inhibition of cell multiplication Kühn (1978)
Selenastrum EC50, growth 3 days 0.335 irradiance of 17 W/m2 Devillers et
capricornutum al. (1990)
lowest concentration for no 14 days 79.4 irradiance of 10 W/m2 Wängberg &
detectable growth Blanck (1988)
Tribonema aequale lowest concentration for no 14 days 2.51 irradiance of 10 W/m2 Wängberg &
detectable growth Blanck (1988)
Table 18. (contd).
Species Effect Test Hydroquinone Comments Reference
duration concentration
(mg/litre)
Yeasts
Candida albicans activation of growth 24 h 500 Devillers et
50% inhibition of growth 24 h 3750 al. (1990)
Candida tropicalis R2 100% inhibition of growth 24 h 1000 Devillers et
al. (1990)
Saccharomyces 50% inhibition of growth 24 h 2750 Devillers et
cerevisiae al. (1990)
Torulopsis glabrata 50% inhibition of growth 24 h 1000 Devillers et
100% inhibition of growth 3000 al. (1990)
Fungi
Fusarium oxysporum no significant inhibition of spore 1000 Ismail et al.
f sp lycopersici germination (1987)
no significant inhibition of length 1000
of germ tube
Plants
Elodea canadensis 50% inhibition of growth 9 days 42.9 Stom & Roth
(1981)
Lemna minor 50% inhibition of plant 12 days 7.71 Stom & Roth
multiplication (1981)
Vallisneria spiralis 100% inhibition of cytoplasmic 15 min 2753 in leaves Stom & Roth
streaming 275.3 in roots (1981)
Protozoa
Chilomonas toxicity threshold concentration for 48 h 22 Bringmann &
paramaecium inhibition of cell multiplication Kühn (1981)
Table 18. (contd).
Species Effect Test Hydroquinone Comments Reference
duration concentration
(mg/litre)
Colpidium campylum EC50, growth 24 h 73.3 Devillers et
al. (1990)
Entosiphon sulcatum toxicity threshold concentration for 72 h 11 Bringmann
inhibition of cell multiplication (1978)
Microregma toxicity threshold concentration for 28 h 2 Bringmann &
nutrient uptake Kühn (1959b)
Tetrahymena EC50, growth 60 h 95.0 Schultz et al.
pyriformis (1987)
Uronema parduczi toxicity threshold concentration for 20 h 21 Bringmann &
inhibition of cell multiplication Kühn (1980)
Mollusc
Deroceras reticulatum 0% mortality 4 days 0.020a Briggs &
20% mortality 4 days 0.200a Henderson
(1987)
Crustacea
Artemia salina LC50 2 h 321 Devillers et
4 h 67.5 al. (1990)
6 h 57.5
24 h 20.7
Crangon LT50 84 h 0.83 time to 50% mortality McLeese et al.
septemspinosa (1979)
Daphnia magna toxicity threshold concentration for 48 h 0.60 Bringmann &
inhibition of mobility Kühn (1959a)
Table 18. (contd).
Species Effect Test Hydroquinone Comments Reference
duration concentration
(mg/litre)
Daphnia magna LC0 24 h 0.04 Bringmann &
LC50 0.09 Kühn (1977b)
LC100 0.31
EC0, inhibition of mobility 24 h 0.05 Bringmann &
EC50 0.12 Kühn (1982)
EC100 0.19
EC50, inhibition of mobility 24 h 0.137 Devillers et
al. (1987)
EC0, inhibition of mobility 24 h 0.13 Kühn et al.
EC50 0.32 (1989)
EC100 0.71
EC0 48 h 0.13 Kühn et al.
EC50 0.29 (1989)
EC100 0.71
EC50, inhibition of mobility 24 h 0.15 Tissot et al.
(1985)
Daphnia pulicaria LC50 48 h 0.162 DeGraeve et
al. (1980)
Daphnia pulex LC100 6 min 8809 Stom et al.
(1986)
Gammarus toxicity threshold concentration 1.5 Bandt (1955)
Insect
Apis mellifera LD50 24 h 0.200 concentration in mg per bee Devillers et
al. (1990)
Table 18. (contd).
Species Effect Test Hydroquinone Comments Reference
duration concentration
(mg/litre)
Fish
Brachydanio rerio LC50 24 h 0.265 Devillers et
al. (1988)
LC0 96 h 0.12 Wellens (1982)
LC50 0.17
LC100 0.25
Carassius auratus 100% mortality 22 h 5 US EPA (1987)
LC100 48 h 0.287 Sollmann
(1949)
Lepomis macrochirus 100% mortality 22 h 5 US EPA (1987)
Leuciscus idus LC0 48 h 0.1 Juhnke &
melanotus LC50 0.15 Lüdemann
LC100 0.2 (1978)
LC0 0.1
LC50 0.16
LC100 0.25
Oncorhynchus mykiss 100% mortality 2 h 7.69 Devillers et
4 h 4.50 al. (1990)
LC50 96 h 0.097 DeGraeve et
al. (1980)
LC50 96 h 0.639 Hodson et al.
LD50, intraperitoneal injection 24.2 concentration in mg per kg (1984)
Table 18. (contd).
Species Effect Test Hydroquinone Comments Reference
duration concentration
(mg/litre)
Pimephales promelas LC50 96 h 0.044 DeGraeve et
al. (1980)
LT50 11 h 0.2 time to 50% mortality Terhaar et al.
(1972)
Salmo trutta 100% mortality 22 h 5 US EPA (1987)
a Slugs injected with 2 µl of glycerol formal containing 20 or 200 µg of hydroquinone.
10. EVALUATION OF HUMAN HEALTH RISKS AND ON THE ENVIRONMENT
10.1 Toxicokinetics
The property of hydroquinone most pertinent to its toxicity is
its ability to undergo reversible redox reactions. Autooxidation of
hydroquinone leads to p-benzoquinone and/or p-benzosemiquinone.
These two strong electrophiles do not redox-cycle under
physiological conditions to form active oxygen species, but readily
arylate nucleophiles. They are probably the two major toxic
metabolites of hydroquinone.
Toxicokinetic studies with hydroquinone show that although it
is readily absorbed from the gut of animals it has a low potential
for bioaccumulation < 2% distributed out of total administered
dose). Extensive conjugation and rapid excretion, primarily via the
urine, suggests that hydroquinone is effectively detoxified.
However, because hydroquinone is oxidized to p-benzosemiquinone
and/or p-benzoquinone, which are able to readily react with
nucleophilic body components, it represents a potentially harmful
toxicant. Indeed, hydroquinone and/or its metabolites covalently
bind to cellular components in vitro.
It is, therefore, possible that although the bioaccumulation
potential of hydroquinone is low critical body components may still
be adversely affected.
10.2 Animal and in vitro studies
Hydroquinone exhibits moderately high acute oral toxicity for
animals, with LD50 values generally being in the range of 300 to
1300 mg/kg. However, cats are more sensitive, LD50 values of 40-85
mg/kg having been reported. The principle toxic effects of a single
oral lethal dose of hydroquinone are increased motor activity,
dyspnoea and cyanosis followed by convulsions, paralysis, coma and
death. Repeated oral dosing has caused tremors and reduced activity
(> 64 mg/kg), reduced body weight gain (> 200 mg/kg),
convulsions (> 400 mg/kg) and adverse effects on the liver and
kidneys (> 200 mg/kg).
Dermal exposure of black guinea-pigs to hydroquinone (2 or 5%)
has been found to cause depigmentation, inflammatory changes and
thickening of the epidermis. Slight skin irritation has been
recorded following topical application of hydrophillic ointment
containing 1% hydroquinone. The results also showed that female
guinea-pigs were more sensitive than males.
Limited data suggest that powdered hydroquinone causes
transient eye irritation and corneal opacity in dogs and
guinea-pigs; in rabbits powdered hydroquinone induced brownish
pigmentation of conjunctiva and cornea, but only after a period of
at least 2-4 months.
Hydroquinone is a skin sensitizer in rabbits. The ability to
induce sensitization has been found to vary from "weak" to "strong"
depending on the test procedure and vehicle used. The cross-
reactivity of hydroquinone and p-methoxyphenol has been reported
to be almost 100%.
Early reproductive studies indicated reduced fertility in male
rats and a disturbed sexual cycle in female rats when hydro-quinone
was administered parentally. However, this was not confirmed in more
recent studies in rats, i.e. a dominant lethality study and a
two-generation study with oral doses of hydroquinone up to 300 mg/kg
per day and 150 mg/kg per day, respectively.
Oral dosing of 100 or 300 mg hydroquinone/kg to pregnant rats
on days 6-15 of gestation caused maternal toxicity at the higher
dose level (a statistically significant reduction in body weight
gain and feed consumption). A reduction in mean fetal body weight
was correlated with the reduced maternal body weight. No
compound-related teratogenic effects were produced at this dose
level; thus, 100 mg/kg was considered the NOEL for maternal and
developmental toxicity in rats. Findings of increased resorption
rates in rats given hydroquinone orally at about 100 mg/kg per day
were not confirmed in this study, and, consequently, the NOAEL for
maternal reproductive effects and teratogenicity was 300 mg/kg. In
rabbits, 150 mg/kg caused reductions in body weight and feed intake
and an increased (but not statistically significant) increase of
malformations in the fetuses. The malformations may have been
associated with maternal toxicity. The dose level of 200 mg/kg
produced an increased number of resorptions, indicating
embryotoxicity. The NOEL for developmental toxicity in rabbits was
25 mg/kg per day.
In a two-generation reproduction study in rats the NOAEL for
reproductive effects through two generations was 150 mg/kg per day
(the highest tested dose). In mice, 80 mg hydroquinone/kg given
orally on the 13th day of gestation transplacentally induced
micronuclei in fetal liver cells. A single oral dose of 1000 mg/kg
on gestation day 11 caused maternal toxicity (decreased weight gain)
and an increased incidence of mortality. Reduced litter size and
perinatal loss occurred in the treated groups (333, 667 and 1000
mg/kg), together with dose-related malformations of limbs, tail and
urogenital system. In a rat embryo culture system, hydroquinone
(effective concentration < 55 mg/litre, < 0.5 mmol/litre)
caused growth retardation and an increased incidence of structural
abnormalities involving tail and hindlimb buds.
Genotoxicity data indicates that hydroquinone induces
micronuclei, structural chromosome aberrations, and c-mitotic
effects in vivo in mouse bone-marrow cells. in vitro studies
with various cell lines showed that hydroquinone was capable of
inducing gene mutations, structural chromosome aberrations,
sister-chromatid exchange and DNA damage. Hydroquinone induces
chromosome aberrations or karyotypic alterations in plant species
and mitotic crossing-over in fungi. Hydroquinone is not mutagenic in
the Salmonella/microsome test, but induces repairable DNA damage
in Escherichia coli. It produces adducts with DNA.
Hydroquinone induces chromosome aberrations in germ cells of
male mice.
Cholesterol pellets containing 20% hydroquinone implanted into
the bladders of mice produced bladder tumours in 6 out of 19 mice
surviving 25 weeks. However, the study was incompletely reported and
the method is not generally recognized as a valid measure of
carcinogenic potential as small pellets of cholesterol are known to
induce transitional carcinoma of the bladder in both rats and mice.
There is no support for hydroquinone being a stomach carcinogen in
experimental animals after oral dosing. Hydroquinone produced an
increase in the number of liver foci in rats at a dose level of 100
mg/kg per day for 7 weeks. However, increased dose levels (200 mg/kg
per day for 7 weeks or 8 g/kg diet for about two years) caused a
reduction in the number of foci of cellular alteration of the liver.
In mice, the incidence of liver foci was increased when hydroquinone
was added to the diet at 8 g/kg.
Hydroquinone did not show any potential as an initiator or a
co-carcinogen when dermally applied to mice before application of a
tumour promoter (croton oil or BP) or following co-exposure with BP.
Orally administered hydroquinone showed no promotion activity after
MNNG initiation or carcinogenic potential in the forestomach and
glandular stomach of rats.
Most of the earlier carcinogenicity studies on hydroquinone
lasted for less than one year, which might be considered too short
for the assessment of carcinogenicity.
Two-year studies performed recently give support for
hydroquinone being a carcinogen in F-344 rats and B6C3F1 mice. In
an NTP study, renal tubular cell adenomas occurred in male rats and
leukaemia in females, and hepatocellular neoplasms, mainly adenomas,
in female mice. The NTP concluded that these data indicated "some
evidence of carcinogenic activity" in male and female rats and in
female mice. In another study, renal tubular cell adenomas were
again noted in male rats; hepatocellular adenomas occurred in male
mice along with a biologically significant increase in the incidence
of renal tubular cell adenomas.
Both in vivo and in vitro studies have shown that
hydroquinone causes direct myelotoxic effects in mouse bone marrow
stromal cells by reducing bone marrow cellularity. In reducing the
number of progenitor B-lymphocytes in mouse spleen and bone marrow,
hydroquinone also demonstrates an immunosuppressive potential.
Moreover, hydroquinone may inhibit the natural killer activity of
mouse spleen cells in vitro, and have a selective effect on
macrophage functions important in host defence.
Hydroquinone has also demonstrated cytotoxic activity on
various tumour cells such as cells from melanoma transplants and rat
hepatoma cells.
Central nervous system stimulatory effects have been produced
in animal studies. However, functional-observational battery and
neuropathological examinations failed to give any evidence of
neurotoxicity after repeated dosing for 90 days. The NOEL was 20 mg
hydroquinone/kg per day.
10.3 Evaluation of human health risks
10.3.1 Exposure
Potential exposure of the general population to hydroquinone
may occur through the consumption of foods that contain hydroquinone
as a natural component, through smoking or exposure to cigarette
smoke, or from the use of cosmetics and skin-lightening creams.
People who use skin lighteners with concentrations of hydroquinone
exceeding 2%, apply creams over large areas of the body or use
creams for long time periods represent one group with significant
and sometimes excessive exposure. Heavy cigarette smokers and those
living and working in environments contaminated by cigarette smoke
represent another group that experiences significant exposure to
hydroquinone. Photohobbyists who develop their film manually may
also be exposed to hydroquinone solutions through skin contact and
inhalation.
Exposure to hydroquinone may occur in a variety of occupations,
particularly among those involved in its manufacture.
10.3.2 Human health effects
Ingestion of large quantities of hydroquinone may produce
tremors, vomiting, convulsions, dyspnoea, cyanosis and coma. Deaths
have been reported to occur after ingestion estimated at 3-12 g of
hydroquinone in developing agents. In studies with human volunteers,
ingestion of up to 500 mg hydroquinone per day over a 20-week period
resulted in no observable pathological changes in the blood and
urine.
Dermal exposure to hydroquinone causes skin depigmentation;
cases of ochronosis, patchy depigmentation and brown staining of the
nails after repeated usage of skin-lightening products have been
reported. Hydroquinone has shown a sensitizing potential in both
animals and humans.
Eye irritation, sensitivity to light, staining of the cornea
and conjunctiva, corneal opacities and visual disturbances are
associated with long-term occupational exposure to airborne
hydroquinone. Isolated cases of corneal ulceration have also been
described.
Effects on the central nervous system have been seen in cases
of acute human poisoning. Similar symptoms have been observed in
animal studies; these effects were reversible when exposure was
discontinued.
Nephrotoxicity has been seen in F-344 rats dosed repeatedly
with hydroquinone. Male rats are more susceptible to these effects
than females. Nephrotoxic effects due to hydroquinone have not been
observed in humans.
Myelotoxic and immunotoxic effects have been observed in
animals exposed to hydroquinone. However, the routes of exposure
differed from those via which humans are normally exposed.
Studies conducted by routes of exposure similar to those by
which humans are exposed have not revealed specific reproductive and
developmental effects.
Numerous studies using cell culture systems and in vivo
rodent experiments have shown that co-exposure to hydroquinone and
various phenolic compounds can result in toxic effects that are
substantially greater than the sum of the effects of the individual
compounds. The relevance of these interactive effects in
understanding and predicting the toxic effects of human exposure to
hydroquinone is uncertain. However, since many of the human
exposures to hydroquinone occur under conditions in which other
phenolic compounds are present, the possibility that significant
interactive effects may occur should be considered.
Several experiments with hydroquinone in vivo and in vitro
have shown mutagenic effects; the relevance of these results to
human risk is uncertain. The evidence for carcinogenicity in animals
is limited. Adequate epidemiological studies are lacking, and, at
present, the experimental data are insufficient to allow a thorough
assessment of the carcinogenic potential for humans.
10.4 Evaluation of effects on the environment
When hydroquinone is released into the environment it is
distributed mainly to the water compartment due to its
physicochemical properties. However, hydroquinone can be degraded
both photochemically and biologically and will therefore not persist
in the environment. The ecotoxicity of hydroquinone, which can be
also related to its physicochemical properties, is generally high
but varies from species to species. Therefore, a battery of tests
using organisms occupying different trophic levels in the ecosystems
is required to assess thoroughly the adverse effects of hydroquinone
in the environment.
11. RECOMMENDATIONS
a) In view of the widespread inappropriate use of skin-lightening
creams, it is recommended that over-the-counter sales of creams
containing hydroquinone be restricted. Health Education
Programmes should be developed to discourage the use of
hydroquinone-containing creams for whole body skin lightening.
b) Further investigations into the safety of long-term use of
creams containing 1-2% hydroquinone is needed.
c) Hydroquinone in waste water effluent should be allowed
sufficient time for degradation before reaching recipient
water.
d) Multispecies toxicity testing performed under controlled
experimental conditions is required to make a thorough
assessment of the environmental effects of hydroquinone and its
derivatives.
e) Epidemiological studies, including precise exposure data, would
assist in an assessment of the occupational hazards from
hydroquinone. Epidemiological information, including
reproductive effects, is also required for users of skin
lighteners, particularly women. More data on human exposure
from different sources are required, particularly with respect
to dietary exposure.
12. PREVIOUS EVALUATIONS BY INTERNATIONAL BODIES
In 1977 the International Agency for Research on Cancer (IARC)
Working Group concluded that the available data on hydroquinone did
not allow an evaluation of its carcinogenicity.
Hydroquinone was evaluated by a Nordic Expert Group for
Documentation of Occupational Exposure Limits in 1989. It was
recommended that its genotoxic effects should be given attention and
also its possible effects on the immune system, bone marrow, skin
and mucous membranes.
REFERENCES
Abramowitz J & Chavin W (1980) Acute effects of two melanocytolytic
agents, hydroquinone and ß-mercaptoethanolamine, upon tyrosinase
activity and cyclic nucleotide levels in murine melanomas. Chem-Biol
Interact, 32: 195-208.
Adler I-D & Kliesch U (1990) Comparison of single and multiple
treatment regimens in the mouse bone marrow micronucleus assay for
hydroquinone (HQ) and cyclophosphamide (CP). Mutat Res, 234:
115-123.
Adler I-D, Kliesch U, van Hummelen P & Kirsch-Volders M (1991) Mouse
micronucleus tests with known and suspect spindle poisons: results
from two laboratories. Mutagenesis, 6(1): 47-53.
Altmann H-J, Grunow W, Wester PW, & Mohr U (1985) Induction of
forestomach lesions by butylhydroxyanisole and structurally related
substances. Arch Toxicol, 8(Suppl): 114-116.
Ames SR, Ludwig MJ, Swanson WJ, & Harris PL (1956) Effect of DPPD,
methylene blue, BHT, and hydroquinone on reproductive process in the
rat. Proc Soc Exp Biol Med, 93: 39-42.
Anderson B (1947) Corneal and conjunctival pigmentation among
workers engaged in manufacture of hydroquinone. Arch Ophthalmol, 38:
812-826.
Anderson B & Oglesby F (1958) Corneal changes from
quinone-hydroquinone exposure. Am Med Assoc Arch Opthalmol, 59:
495-501.
Antoccia A, Degrassi F, Battistoni A, Ciliutti P, & Tanzarella C
(1991) In vitro micronucleus test with kinetochore staining:
evaluation of test performance. Mutagenesis, 6(4): 319-324.
Arndt KA & Fitzpatrick TB (1965) Topical use of hydroquinone as a
depigment agent. J Am Med Assoc, 194: 965-967.
Assaf MH, Ali AA, Makboul MA, Beck JP, & Anton R (1987) Preliminary
study of phenolic glycosides from Origanum majorana; quantitative
estimation of arbutin; cytotoxic activity of hydroquinone. Planta
Med, 53: 343-345.
Baernstein HD (1945) The determination of catechol, phenol, and
hydroquinone in urine. J Biol Chem, 161: 685-692.
Bandt HJ (1955) [Damage to fishing grounds caused by waste water
containing phenols.] Wasserwirtsch-Wassertech, 5: 290-294 (in
German).
Barale R, Marazzini A, Betti C, Vangelisti V, Loprieno N, & Barrai I
(1990) Genotoxicity of two metabolites of benzene: phenol and
hydroquinone show strong synergistic effects in vivo. Mutat Res,
244: 15-20.
Basketter DA & Goodwin BFJ (1988) Investigation of the prohapten
concept. Contact Dermatitis, 19: 248-253.
Baumbach HL (1946) An improved method for the determination of
hydroquinone and metol in photographic developers. J Soc Motion Pict
Eng, 47: 403-408.
Bentley-Phillips B & Bayles MAH (1975) Cutaneous reactions to
topical application of hydroquinone. Results of a 6-year
investigation. S Afr Med J, 49: 1391-1395.
Bilimoria MH (1975) The detection of mutagenic activity of chemicals
and tobacco smoke in a bacterial system (Abstract). Mutat Res, 31:
328.
Bio/dynamics Inc. (1988) A range-finding study to evaluate toxicity
of hydroquinone in the pregnant rabbit (Project No. 87-3218): Final
report. East Millstone, New Jersey, Bio/dynamics Inc. (Prepared for
the Chemical Manufacturers Association, Washington).
Bio/dynamics Inc. (1989a) A developmental toxicity study in rabbits
with hydroquinone (Project No. 87-3220): Final report. East
Millstone, New Jersey, Bio/dynamics Inc. (Prepared for the Chemical
Manufacturers Association, Washington).
Bio/dynamics Inc. (1989b) A two-generation reproduction study in
rats with hydroquinone (Project No. 87-32l9): Final report. East
Millstone, New Jersey, Bio/dynamics Inc. (Prepared for the Chemical
Manufacturers Association, Washington).
Bleehen SS, Pathak MA, Hori Y, & Fitzpatrick TB (1968)
Depigmentation of skin with 4-isopropylcatechol, mercaptoamines, and
other compounds. J Invest Dermatol, 50: 103-117.
Boatman RS, English JC, Perry LG, & Bialecki VE (1992) Indications
of nephrotoxicity for hydroquinone and for 2-(glutathion-S-YL)
hydroquinone in Fischer-344 and Sprague-Dawley rats and in B6C3F1
mice. Rochester, New York, Eastman Kodak Company, Health and
Environment Laboratories (Unpublished data).
Borecky J (1963) [Identification of organic compounds-Report LIV.
Separation and identification of photographic developers by paper
chromatography.] J Chromatogr, 12: 385-393 (in German).
Boyland E, Busby ER, Dukes CE, Grover PL, & Manson D (1964) Further
experiments on implantation of materials into the urinary bladder of
mice. Br J Cancer, 18: 575-581.
Boyle J & Kennedy CTC (1985) Leukoderma from hydroquinone. Contact
Dermatitis, 13: 287-288.
Boyle J & Kennedy CTC (1986) Hydroquinone concentrations in skin
lightening creams. Br J Dermatol, 114: 501-504.
Brauer EW (1985) Safety of over-the-counter hydroquinone bleaching
creams. Arch Dermatol, 121: 1239.
Briggs GG & Henderson IF (1987) Some factors affecting the toxicity
of poisons to the slug Deroceras reticulatum (Müller) (Pulmonata:
Limacidae). Crop Prot, 6: 341-346.
Bringmann G (1978) [Determination of the biological harmful effect
of water pollutants on protozoa. I. Bacteriophagic flagellates
(model organism: Entosiphon sulcatum Stein).] Z Wasser
Abwasser-Forsch, 11: 210-215 (in German).
Bringmann G & Kühn R (1959a) [Comparative water toxicology studies
on bacteria, algae and small crustaceans.] Gesundheits-Ingenieur, 4:
115-120 (in German).
Bringmann G & Kühn R (1959b) [Water toxicology studies using
protozoa as test organisms.] Gesundheits-Ingenieur, 8: 239-240 (in
German).
Bringmann G & Kühn R (1977a) [Threshold values for the harmful
effect of water pollutants on bacterio (Pseudomonas putida) and
green algae (Scenedesmus quadricauda) in the cell reproduction
inhibition test.] Z Wasser Abwasser-Forsch, 10: 87-98 (in German).
Bringmann G & Kühn R (1977b) [Findings concerning the harmful effect
of water pollutants on Daphnia magna.] Z Wasser Abwasser Forsch,
10: 161-166 (in German).
Bringmann G & Kühn R (1978) [Threshold values for the harmful effect
of water pollutants on blue algae (Microcystis aeruginosa) and
green algae (Scenedesmus quadricauda) in the cell reproduction
inhibition test.] Vom Wasser, 50: 45-60 (in Grman).
Bringmann G & Kühn R (1980) [Determination of the biological harmful
effect of water pollutants on protozoa. II. Bacteriophagic
ciliates.] Z Wasser Abwasser Forsch, 13: 26-31 (in German).
Bringmann G & Kühn R (1981) [Comparison of the effect of harmful
substances on flagellates and ciliates with the effect of holozoic
bacteriophagic and saprozoic protozoa.] GWF-Wasser/Abwasser, 122:
308-313 (in German).
Bringmann G & Kühn R (1982) [Findings concerning the harmful effect
of water pollutants on Daphnia magna in an advanced standardized
test procedure.] Z Wasser Abwasser Forsch, 15: 1-6 (in German).
Brunmark A & Cadenas E (1988) Reductive addition of glutathione to
p-benzoquinone, 2-hydroxy- p-benzoquinone, and p-benzoquinone
epoxides. Effect of the hydroxy- and glutathionyl substituents on
p-benzohydroquinone autoxidation. Chem-Biol Interact, 68: 273-298.
Brunner AH Jr, Means PB Jr, & Zappert RH (1949) Analysis of
developers and bleach for Ansco color film. J Soc Motion Pict Eng,
53: 25-35.
Brunner M, Albertini S, & Würgler FE (1991) Effects of 10 known or
suspected spindle poisons in the in vitro porcine brain tubulin
assembly assay. Mutagenesis, 6(1): 65-70.
Bucks DAW, McMaster JR, Guy RH, & Maibach HI (1988) Percutaneous
absorption of hydroquinone in humans: effect of
1-dodecylazacycloheptan-2-one (azone) and the 2-ethylhexyl ester of
4-(dimethylamino)benzoic acid (escalol 507). J Toxicol Environ
Health, 24: 279-289.
Bulman C & Serva RJ (1982) Mutagenicity evaluation of Eastman
hydroquinone. Salmonella typhimurium/microsome bioassay. Akron,
Ohio, The Goodyear Tyre & Rubber Company (Laboratory report No
82-5-1).
Bulman C & Van der Sluis D (1980) Mutagenicity evaluation of 302
bottoms (Bayport Plant Stream). Salmonella typhimurium/microsome
bioassay. Akron, Ohio, The Goodyear Tire & Rubber Company
(Laboratory report No. 80-8-1).
Bulman C & Wampler DA (1979) Mutagenicity evaluation of
hydroquinone. Akron, Ohio, The Goodyear Tire & Rubber Company
(Laboratory report No. 79-63).
Burnett C, Goldenthal EI, Harris SB, Wazeter FX, Strausburg J, Kapp
R, & Voelker R (1976) Teratology and percutaneous toxicity studies
on hair dyes. J Toxicol Environ Health, 1: 1027-1040.
Busatto S (1939) [Fatal poisoning with a photographic developer
containing hydroquinone.] Dtsch Z Gesamte Gerichtl Med, 31: 285-297
(in German).
Busatto S (1940) [The biological diagnosis of hydroquinone
poisoning.] Arch Antropol Crim Psichiatr Med Leg, 60: 620-625 (in
Italian).
Carlson AJ & Brewer NR (1953) Toxicity studies on hydroquinone. Proc
Soc Exp Biol Med, 84: 684-688.
Cassidy MK & Houston JB (1980) In vivo assessment of extrahepatic
conjugative metabolism in first pass effects using the model
compound phenol. J Pharm Pharmacol, 32: 57-59.
Cassidy MK & Houston JB (1984) In vivo capacity of hepatic and
extrahepatic enzymes to conjugate phenol. Drug Metab Dispos, 12(5):
619-624.
Chatterjee P & Sharma AK (1972) Effect of phenols on nuclear
division in Chara zeylanica. Nucleus, 15: 214-218.
Chavin W, Jelonek EJ Jr, Reed AH, & Binder LR (1980) Survival of
mice receiving melanoma transplants is promoted by hydroquinone.
Science, 208: 408-410.
Cheung SC, Nerland DE, & Sonnenfeld G (1989) Inhibition of
interferon-alpha/beta induction in L-929 cells by benzene and
benzene metabolites. Oncology, 46: 335-338.
Choudat D, Neukirch F, Brochard P, Barrat G, Marsac J, Conso F, &
Philbert M (1988) Allergy and occupational exposure to hydroquinone
and to methionine. Br J Ind Med, 45: 376-380.
Christian RT, Clark CS, Cody TE, Witherup S, Gartside PS, Elia VJ,
Eller PM, Lingg R, & Cooper GP (1976) The development of a test for
the potability of water treated by a direct refuse system.
Cincinnati, Ohio, University of Cincinnati, College of Medicine,
Department of Environmental Health, pp 126-146 (Report No 8:83-87).
Chrostek WJ (1975) Health hazard evaluation/Toxicity determination
report H.H.E. 75-84-235. Springfield, Virginia, US Department of
Commerce, National Technical Information Service, 7 pp
(NTIS/PB-249.415).
CIR (1986) CIR expert panel - Cosmetic ingredient review. Final
report on the safety assessment of hydroquinone and pyrocatechol. J
Am Coll Toxicol, 5(3): 123-165.
Ciranni R & Adler I-D (1991) Clastogenic effects of hydroquinone:
induction of chromosomal aberrations in mouse germ cells. Mutat Res,
263: 223-229.
Ciranni R, Barale R, Marrazzini A, & Loprieno N (1988a) Benzene and
the genotoxicity of its metabolites I. Transplancental activity in
mouse fetuses and in their dams. Mutat Res, 208: 61-67.
Ciranni R, Barale R, Ghelardini G, & Loprieno N (1988b) Benzene and
the genotoxicity of its metabolites. II. The effect of the route of
administration on the micronuclei and bone marrow depression in
mouse bone marrow cells. Mutat Res, 209: 23-28.
Clifford MN & Wight J (1973) Metaperiodate - A new structure-
specific locating reagent for phenolic compounds. J Chromatogr, 86:
222-224.
Commins BT & Lindsey AJ (1956) The determination of phenols by
chromatography and spectrophotometry of their methyl ethers. IV. The
determination of phenols in cigarette smoke. Anal Chim Acta, 15:
557-558.
Connor T & Braunstein B (1987) Hyper-pigmentation following the use
of bleaching creams. Arch Dermatol, 123: 105, 110.
Cooper RL & Wheatstone KC (1973) The determination of phenols in
aqueous effluents. Water Res, 7: 1375-1384.
Crebelli R, Conti G, & Carere A (1987) On the mechanism of mitotic
segregation induction in Aspergillus nidulans by benzene hydroxy
metabolites. Mutagenesis, 2: 235-238.
Crebelli R, Conti G, & Carere A (1991) In vitro studies with nine
known or suspected spindle poisons: results in tests for chromosome
malsegregation in Aspergillus nidulans. Mutagenesis, 6(2):
131-136.
Dagon TJ (1973) Biological treatment of photo processing effluents.
J Water Pollut Control Fed, 45: 2123-2135.
DeGraeve GM, Geiger DL, Meyer JS, & Bergman HL (1980) Acute and
embryo-larval toxicity of phenolic compounds to aquatic biota. Arch
Environ Contam Toxicol, 9: 557-568.
Deichmann WB & Keplinger ML (1981) Hydroquinone. In: Clayton GD &
Crlayton FE ed. Patty's industrial hygiene and toxicology - Volume
2A: Toxicology. New York, John Wiley & Sons, pp 2589-2592.
Delcambre JP, Weber B, & Baron C (1962) Toxicité de l'hydroquinone.
Agressologie, 3: 311-315.
Devillers J, Chambon P, Zakarya D, & Chastrette M (1986) A new
approach in ecotoxicological QSAR studies. Chemosphere, 15:
993-1002.
Devillers J, Chambon P, Zakarya D, Chastrette M, & Chambon R (1987)
A predictive structure-toxicity model with Daphnia magna.
Chemosphere, 16: 1149-1163.
Devillers J, Zakarya D, & Chastrette M (1988) Structure-activity
relationships for the toxicity of organic pollutants to Brachydanio
rerio. In: Turner JE, England MW, Schultz TW, & Kwaak NJ ed.
Proceedings of the Third International Workshop on Quantitative
Structure-Activity Relationships in Environmental Toxicology.
Dordrecht, Reidel Publishing Company, pp 91-90.
Devillers J, Boule P, Vasseur P, Prevot P, Steiman R, Seigle-Murandi
F, Benoit-Guyod JL, Nendza M, Grioni C, Dive D, & Chambon P (1990)
Environmental and health risks of hydroquinone. Ecotoxicol Environ
Saf, 19: 327-354.
Divincenzo GD, Hamilton ML, Reynolds RC, & Ziegler DA (1984)
Metabolic fate and disposition of [14C]hydroquinone given orally
to Sprague-Dawley rats. Toxicology, 33: 9-18.
Dore M, Brunet N, & Legube B (1975) Participation de différents
composés organiques à la valeur des critères globaux de pollution.
Trib CEBEDEAU, 374: 3-11.
Draize JH, Woodard G, & Calvery HO (1944) Methods for the study of
irritation and toxicity of substances applied topically to the skin
and mucous membranes. J Pharmacol Exp Ther, 82: 377-390.
Dreyer NB (1940) Toxicity of hydroquinone (Medical Research Project
No. MR-78). Wilmington, Delaware, Haskell Laboratory of Industrial
Toxicology.
Eastman Kodak Company (1988) Subchronic oral toxicity study of
hydroquinone in rats utilizing a functional observational battery
and neuropathology to detect neurotoxicity. Rochester, New York,
Eastman Kodak Company (Report No. TX-88-78, prepared for the
Chemical Manufacturers Association, Washington).
Eastmond DA, Smityh MT, & Irons DA (1987) An interaction of benzene
metabolites reproduces the myelotoxicity observed with benzene
exposure. Toxicol Appl Pharmacol, 91: 85-95.
Eckert K-G, Eyer P, Sonnenbichler J, & Zetl I (1990) Activation and
detoxication of aminophenols. III. Synthesis and structural
elucidation of various glutathione addition products to
1,4-benzoquinone. Xenobiotica, 20: 351-361.
Eisner T, Jones TH, Aneshansley DJ, Tschinkel WR, Silberglied RH, &
Meinwald J (1977) Chemistry of defensive secretions of bombardier
beetles (Brachinini, Metriini, Ozaenini, Paussini). J Insect
Physiol, 23: 1383-1386.
Ekström T, Warholm M, Kronevi T, & Högberg J (1988) Recovery of
malondialdehyde in urine as a 2,4-dinitrophenylhydrazine derivative
after exposure to chloroform or hydroquinone. Chem-Biol Interact,
67: 25-31.
English JC, Desinger PJ, Perry LG, Schum DB, & Guest D (1988)
Toxicokinetics studies with hydroquinone in male and female Fischer
344 rats. Rochester, New York, Eastman Kodak Company (Report No.
TX-88-84, prepared for the Chemical Manufactures Association,
Washington).
English JC, Perry LG, Vlaovic M, Moyer C, & O'Donoghue JL (1992)
Measurement of cell proliferation in the kidneys of Fischer 344 and
Sprague-Dawley rats after gavage administration of hydroquinone.
Rochester, New York, Eastman Kodak Company (Unpublished report).
Epe B, Harttig U, Stopper H, & Metzler M (1990) Covalent binding of
reactive estrogen metabolites to microtubular protein as a possible
mechanism of aneuploidy induction and neoplastic cell
transformation. Environ Health Perspect, 88: 123-127.
Erexson GL, Wilmer JL, & Kligerman AD (1985) Sister chromatid
exchange induction in human lymphocytes exposed to benzene and its
metabolites in vitro. Cancer Res, 45: 2471-2477.
Fan X-H, Hirata Y, & Minami M (1989) Effects of benzene and its
metabolites on natural killer activity of mouse spleen cells in
vitro. Jpn J Ind Health, 31: 330-334.
FDA (1981) Indirect food additives: Adhesives coatings and
components. Code Fed Regul, 21(175): 114.
FDA (1982) Skin bleaching drug products for over-the-counter human
use; tentative final monograph. Fed Reg, 47:(172) 39108-39117.
FDA (1991) Indirect food additives: Adhesives and components of
coatings. Code Fed Regul, 21(175): 129, 135, 171, 176.
Ferraris de Gaspare PF (1949) [Experimental studies on the
keratoconjunctivitis from hydroquinone.] Boll Ocul, 28: 361-367 (in
Italian).
Findlay GH & De Beer HA (1980) Chronic hydroquinone poisoning of the
skin from skin-lightening cosmetics. A South African epidemic of
ochronosis of the face in dark-skinned individuals. S Afr Med J, 57:
187-190.
Findlay GH, Morrison JGL, & Simson IW (1975) Exogenous ochronosis
and pigmented colloid milium from hydroquinone bleaching creams. Br
J Dermatol, 93: 613-622.
Fisher AA (1982) Can bleaching creams containing 2% hydroquinone
produce leukoderma. J Am Acad Dermatol, 7: 134.
Fisher AA (1986) Contact dermatitis, 3rd ed. Philadelphia,
Pennsylvania, Lea & Febiger.
Fitzgerald GP, Gerloff GC, & Skoog F (1952) Studies on chemicals
with selective toxicity to blue-green algae. Sew Ind Wastes, 24:
888-896.
Fitzpatrick TB, Arndt KA, Mofty AM, & Pathak MA (1966) Hydroquinone
and psoralens in the therapy of hypermelanosis and vitiligo. Arch
Dermatol, 93: 589.
Florin I, Rutberg L, Curvall M, & Enzell CR (1980) Screening of
tobacco smoke constituents for mutagenicity using the Ames' test.
Toxicology, 18: 219-232.
Freitag D, Ballhorn L, Geyer H, & Korte F (1985) Environmental
hazard profile of organic chemicals. Chemosphere, 14: 1589-1616.
Frenk E & Loi-Zedda P (1980) Occupational depigmentation due to a
hydroquinone-containing photographic developer. Contact Dermatitis,
6: 238-239.
Friedlander BR, Hearne FT, & Newman BJ (1982) Mortality, cancer
incidence, and sickness-absence in photographic processors: an
epidemiologic study. J Occup Med, 24(8): 605-613.
Gad-El-Karim MM, Ramanujam VMS, Ahmed AE, & Legator MS (1985)
Benzene myeloclastogenicity: A function of its metabolism. Am J Ind
Med, 7: 475-484.
Gaido K & Wierda D (1984) In vitro effects of benzene metabolites
on mouse bone marrow stromal cells. Toxicol Appl Pharmacol, 76:
45-55.
Gaido KW & Wierda D (1987) Suppression of bone marrow stromal cell
function by benzene and hydroquinone is ameliorated by indomethacin.
Toxicol Appl Pharmacol, 89: 378-390.
Galloway SM, Armstrong MJ, Reuben C, Colman S, Brown B, Cannon C,
Bloom AD, Nakamura F, Ahmed M, Duk S, Rimpo J, Margolin BH, Resnick
MA, Anderson B, & Zeiger E (1987) Chromosome aberrations and sister
chromatid exchanges in Chinese hamster ovary cells: Evaluations of
108 chemicals. Environ Mol Mutagen, 10(Suppl 10): 1-175.
Garton GA & Williams RT (1949) Studies in detoxification: 21. The
fates of quinol and resorcinol in the rabbit in relation to the
metabolism of benzene. Biochem J, 44: 234-238.
Glatt H, Padykula R, Berchtold GA, Ludewig G, Platt KL, Klein J, &
Oesch F (1989) Multiple activation pathways of benzene leading to
products with varying genotoxic characteristics. Environ Health
Perspect, 82: 81-89.
Glatt H, Gemperlein I, Setiabudi F, Platt KL, & Oesch F (1990)
Expression of xenobiotic-metabolizing enzymes in propagatable cell
cultures and induction of micronuclei by 13 compounds. Mutagenesis,
5(3): 241-249.
Gocke E, King M-T, Eckhardt K, & Wild D (1981) Mutagenicity of
cosmetics ingredients licenced by the European Communities. Mutat
Res, 90: 91-109.
Gocke E, Wild D, Eckhardt K, & King M-T (1983) Mutagenicity studies
with the mouse spot test. Mutat Res, 117: 201-212.
Godlee F (1992) Skin lighteners cause permanent damage. Br Med J,
305: 332-333.
Gold LS, Slone TS, Stern BR, Manley NB, & Ames BN (1992) Rodent
carcinogens: setting priorities. Science, 258: 261-265.
Goodwin BTJ, Crevel RWR, & Johnson AW (1981) A comparison of three
guinea-pig sensitization procedures for the detection of 19 reported
human contact sensitizers. Contact Dermatitis, 7: 248-258.
Grant W (1986) Toxicology of the eye, 3rd ed. Springfield, Illinois,
Charles C. Thomas, pp 97-500.
Greenlee WF, Gross EA, & Irons RD (1981a) Relationship between
benzene toxicity and the disposition of 14C-labelled benzene
metabolites in the rat. Chem-Biol Interact, 33: 285-299.
Greenlee WF, Sun JD, & Bus JS (1981b) A proposed mechanism of
benzene toxicity: Formation of reactive intermediates from
polyphenol metabolites. Toxicol Appl Pharmacol, 59: 187-195.
Greenwald P, Friedlander BR, Lawrence CE, Hearne T, & Earle K (1981)
Diagnostic sensitivity bias - an epidemiologic explanation for an
apparent brain tumor excess. J Occup Med, 23: 690-694.
Grudzinski W (1969) [A case of lethal intoxication with
methol-hydroquinone photographic developer.] Pol Tyg Lek, 24:
1460-1462 (in Polish).
Guy RL, Dimitriadis EA, Hu P, Cooper KR, & Snyder R (1990)
Interactive inhibition of erythroid 59Fe utilization by benzene
metabolites in female mice. Chem-Biol Interact, 74: 55-62.
Guy RL, Hu P, Witz G, Goldstein BD & Snyder R (1991) Depression of
iron uptake into erythrocytes in mice by treatment with the combined
benzene metabolites p-benzoquinone, muconaldehyde and hydroquinone.
J Appl Toxicol, 11: 443-446.
Häggblom MM, Apajalahti JHA, & Salkinoja-Salonen MS (1988)
Degradation of chlorinated phenolic compounds occurring in pulp mill
effluents. Water Sci Technol, 20: 205-208.
Harbison KG & Belly RT (1982) The biodegradation of hydroquinone.
Environ Toxicol Chem, 1: 9-15.
Hardwick N, Van Gelder JW, Van der Merwe CA, & Van der Merwe MP
(1989) Exogenous ochronosis: an epidemiological study, 120: 229-238.
Haworth S, Lawlor T, Mortelmans K, Speck W, & Zeiger E (1983)
Salmonella mutagenicity test results for 250 chemicals. Environ
Mutagen, 5(Suppl 1): 3-142.
Hildebrand DC, Powell CC Jr, & Schroth MN (1969) Fire blight
resistance in Pyrus: Localization of arbutin and ß-glucosidase.
Phytopathology, 59: 1534-1539.
Hill BA, Monks TJ, & Lau SS (1992a) Metabolism and toxicity of
2-(glutathion-S-YL) hydroquinone and 2,3,5-(triglutathion-S-YL)
hydroquinone in the in situ perfused rat kidney. Toxicologist, 12:
345 (Abstract).
Hill BA, Monks TJ, & Lau SS (1992b) The effects of
2,3,5-(triglutathion-S-YL) hydroquinone on renal mitochondrial
respiratory function in vivo and in vitro: Possible role in
cytotoxicity. Toxicol Appl Pharmacol, 117: 165-171.
Hill T, English JC, & Deisinger P (1993) Human exposure to naturally
occurring hydroquinone. Rochester, New York, Eastman Kodak Company
(Unpublished data).
Hirose M, Inoue T, Asamoto M, Tagawa Y, & Ito N (1986) Comparison of
the effects of 13 phenolic compounds in induction of proliferative
lesions of the forestomach and increase in the labelling indices of
the glandular stomach and urinary bladder epithelium of Syrian
golden hamsters. Carcinogenesis, 7(8): 1285-1289.
Hirose M, Yamaguchi S, Fukushima S, Hasegawa R, Takahashi S, &
Nobuyuki I (1989) Promotion by dihydroxybenzene derivatives of
N-methyl-N1-vitro-N-nitrosoguanidine-induced F344 rat forestomach
and glandular stomach carcinogenesis. Cancer Res, 49: 5143-5147.
Hodson PV, Dixon DG, & Kaiser KLE (1984) Measurement of median
lethal dose as a rapid indication of contaminant toxicity to fish.
Environ Toxicol Chem, 3: 243-254.
Hodson PV, Dixon DG, & Kaiser KLE (1988) Estimating the acute
toxicity of waterborne chemicals in trout from measurements of
median lethal dose and the octanol-water partition coefficient.
Environ Toxicol Chem, 7: 443-454.
Hogan ME & Manners GD (1990) Allelopathy of small everlasting
(Antennaria microphylla). Phytotoxicity to leafy spurge
(Euphorbia esula) in tissue culture. J Chem Ecol, 16(3): 931-939.
Hogan ME & Manners GD (1991) Differential allelochemical
detoxification mechanism in tissue cultures of Antennaria
microphylla and Euphorbia esula. J Chem Ecol, 17: 167-174.
Högl O (1958) [Some non-volatile extracts of coffee.] Mitt Geb
Lebensm Hyg, 49: 433-441 (in German).
Howard BM, Clarkson K, & Bernstein RL (1979) Simple prenylated
hydroquinone derivatives from the marine urochordate Aplidium
californicum. Natural anticancer and antimutagenic agents.
Tetrahedron Lett, 46: 4449-4452.
Hsuanyu Y & Dunford HB (1992) Reduction of prostaglandin H synthase
compound II by phenol and hydroquinone, and the effect of
indomethacin. Arch Biochem Biophys, 292: 213-220.
Hu F (1966) The influence of certain hormones and chemicals on
mammalian pigment cells. J Invest Dermatol, 46: 117-124.
Hughes W (1948) The tolerance of rabbit cornea for various chemical
substances. Bull John Hopkins Hosp, 82: 338-349.
IARC (1977) Dihydroxybenzenes. In: Some fumigants, the herbicides
2,4-D and 2,4,5-T, chlorinated dibenzodioxins and miscellaneous
industrial chemicals. Lyon, International Agency for Research on
Cancer, pp 155-174 (IARC Monographs on the Evaluation of the
Carcinogenic Risk of Chemicals to Man, Volume 15).
IARC (1986) Tobacco smoking. Lyon, International Agency for Research
on Cancer, pp 86, 105, 122 (IARC Monographs on the Evaluation of the
Carcinogenic Risk of Chemicals to Humans, Volume 38).
IARC (1987) Overall evaluations of carcinogenicity: An updating of
IARC monographs, volumes 1 to 42. Lyon, International Agency for
Research on Cancer, p 64 (IARC Monographs on the Evaluation of
Carcinogenic Risks to Humans, Supplement 7).
Iguchi K, Sahashi A, Kohno J, & Yamada Y (1990) New sesquiterpenoid
hydroquinone and quinones from the Okinawan marine sponge (Dysidea
sp.). Chem Pharm Bull (Tokyo), 38(5): 1121-1123.
ILO (1991) Occupational exposure limits for airborne toxic
substances. Geneva, International Labour Office (Occupational Safety
and Health Series No. 37).
Inoue O, Seiji K, Nakatsuka H, Watanabe T, Yin S-N, Li G-L, Cai S-X,
Jin C, & Ikeda M (1989a) Excretion of 1,2,4-benzenetriol in the
urine of workers exposed to benzene. Br J Ind Med, 46: 559-565.
Inoue O, Seiji K, & Ikeda M (1989b) Pathways for formation of
catechol and 1,2,4-benzenetriol in rabbits. Bull Environ Contam
Toxicol, 43: 220-224.
Irons RD & Neptun DA (1980) Effects of the principal
hydroxy-metabolites of benzene on microtubule polymerization. Arch
Toxicol, 45: 297-305.
Irons RD & Pfeifer RW (1982) Benzene metabolites: evidence for an
epigenetic mechanism of toxicity. Environ Sci Res, 25: 241-256.
Irons RD, Neptun DA, & Pfeifer RW (1981) Inhibition of lymphocyte
transformation and microtubule assembly by quinone metabolites of
benzene: Evidence for a common mechanism. J Reticuloendothel Soc,
30(5): 359-372.
Irons RD, Stillman WS, Colagiovanni DB, & Henry VA (1992)
Synergistic action of the benzene metabolite hydroquinone on
myelopoietic stimulating activity of granulocyte/macrophage
colony-stimulating factor in vitro. Proc Natl Acad Sci (USA), 89:
3691-3695.
IRPTC (1987) IRPTC data file on hydroquinone. Geneva, United Nations
Environment Programme, International Register of Potentially Toxic
Chemicals.
Ismail IMK, Salama AMA, Ali MIA, & Ouf SAE (1987) Effect of some
phenolic compounds on spore germination and germ-tube length of
Aspergillus fumigatus and Fusarium oxysporium f sp lycopersici.
Cryptogam Mycol, 8: 51-60.
Jacquemain R, Remy F, & Guinchard C (1975) Etudes et comparaisons
des déterminations des phénols dans les eaux; application à l'examen
d'un rejet de papeterie. J Fr Hydrol, 16: 25-31.
Jimbow K, Obata H, Pathak MA, & Fitzpatrick TB (1974) Mechanisms of
depigmentation by hydroquinone. J Invest Dermatol, 62: 436-449.
Jowa L, Winkle S, Kalf G, Witz G, & Snyder R (1986) Deoxyguanosine
adducts formed from benzoquinone and hydroquinone. Adv Exp Med Biol,
197: 825-832.
Jowa L, Witz G, Snyder R, Winkle S, & Kalf GF (1990) Synthesis and
characterization of deoxyguanosine-benzoquinone adducts. J Appl
Toxicol, 10: 47-54.
Juhnke I & Lüdemann D (1978) [Results of the testing of 200 chemical
compounds for acute toxicity for fish by means of the golden orfe
test.] Z Wasser Abwasser Forsch, 11: 161-164 (in German).
Jung F & Witt P (1947) [Studies on methaemoglobin formation. XXX.
Tests with polyphenols.] Naunyn-Schmiedebergs Arch Exp Pathol
Pharmakol, 204: 246-436 (in German).
Kappas A (1989) On the mechanism of induced aneuploidy in
Aspergillus nidulans and validation of tests for genomic
mutations. In: Resnick MA & Vig BK ed. Mechanisms of chromosome
distribution and aneuploidi. New York, Alan R. Liss, Inc., pp
377-384.
Kari FW, Bucher J, Eustis SL, Haseman JK, & Huff JE (1992) Toxicity
and carcinogenicity of hydroquinone in F344/N rats and B6C3F1
mice. Food Chem Toxicol, 30: 737-747.
Kavlock RJ (1990) Structure-activity relationships in the
developmental toxicity of substituted phenols: in vivo effects.
Teratology, 41: 43-59.
Kavlock RJ, Oglesby LA, Hall LL, Fisher HL, Copeland F, Logsdon T, &
Ebron-McCoy M (1991) In vivo and in vitro structure-dosimetry-
activity relationships of substituted phenols in developmental
toxicity assays. Reprod Toxicol, 5: 255-258.
Kersey P & Stevenson CJ (1981) Vitiligo and occupational exposure to
hydroquinone from servicing self-photographing machines. Contact
Dermatitis, 7: 285-287.
Key MM, Henschel AF, Butler J, Ligo RN, & Tabershaw IR ed. (1977)
Hydroquinone. In: Occupational diseases. A guide to their
recognition. Washington, DC, US Department of Health, Education and
Welfare, pp 249-250.
King AG, Landreth KS, & Wierda D (1987) Hydroquinone inhibits bone
marrow pre-B cell maturation in vitro. Mol Pharmacol, 32: 807-812.
King AG, Landreth KS, & Wierda D (1989) Bone marrow stromal cell
regulation of B-lymphopoiesis. II. Mechanisms of hydroquinone
inhibition of pre-B cell maturation. J Pharmacol Exp Ther, 250(2):
582-590.
Kleiner HE, Hill BA, Monks TJ, & Lau SS (1992) In vivo and in
vitro formation of several S-conjugates of hydroquinone.
Toxicologist, 12: 345 (Abstract).
Knadle S (1985) Synergistic interaction between hydroquinone and
acetaldehyde in the induction of sister chromatid exchange in human
lymphocytes in vitro. Cancer Res, 45: 4853-4857.
Koike N, Haga S, Ubukata N, Sakurai M, Shimizu H, & Sato A (1988)
Mutagenicity of benzene metabolites by fluctuation test. Jpn J Ind
Health, 30: 475-480.
Kolachana P, Subrahmanyam V, Meyer KB, Zhang L, & Smith MT (in
press) Benzene and its phenolic metabolites produce oxidative DNA
damage in HL60 cells in vitro and in the bone marrow in vivo.
Can Res.
Kolthoff IM & Lee TS (1946) Assay of hydroquinone. Ind Eng Chem,
18(7): 452.
Krasavage WJ (1984a) Hydroquinone: Teratology probe study in rats.
Rochester, New York, Eastman Kodak Company, Health and Environment
Laboratories (Report No. TX-84-01).
Krasavage WJ (1984b) Hydroquinone: A dominant lethal assay in male
rats. Rochester, New York, Eastman Kodak Company, Health and
Environment Laboratories (Report No. TX-84-23).
Krasavage WJ, Terhaar CJ, Ely TS, Vlaovic MS, & Katz GV (1985)
Hydroquinone: A developmental toxicity study in rats. Rochester, New
York, Eastman Kodak Company, Health and Environment Laboratories
(Report No. TX-85-50, prepared for the Chemical Manufacturers
Association, Washington).
Krasavage WJ, Blacker AM, English JC, & Murphy SJ (1992)
Hydroquinone: A developmental toxicity study in rats. Fundam Appl
Toxicol, 18: 370-375.
Kühn R, Pattard M, Pernak K-D, & Winter A (1989) Results of the
harmful effects of selected water pollutants (anilines, phenols,
aliphatic compounds) to Daphnia magna. Water Res, 23: 495-499.
Kurata Y, Fukushima S, Hasegawa R, Hirose M, Shibata M-A, Shirai T,
& Ito N (1990) Structure-activity relations in promotion of rat
urinary bladder carcinogenesis by phenolic antioxidants. Jpn J
Cancer Res, 81: 754-759.
Larcan A, Lambert H, Laprevote-Heully M-C, Bertrand D, & Bertrand D
(1974) Les intoxications par les produits utilisés en photographie
(bains, fixateurs, rélévateurs). J Eur Toxicol, 7: 17-21.
Laskin DL, MacEachern L, & Snyder R (1989) Activation of bone marrow
phagocytes following benzene treatment of mice. Environ Health
Perspect, 82: 75-79.
Lau SS, Hill BA, Highet RJ, & Monks TJ (1988) Sequential oxidation
and glutathione addition to 1,4-benzoquinone: Correlation of
toxicity with increased glutathione substitution. Mol Pharmacol, 34:
829-836.
Lawrence N, Bligard CA, Reed R, & Perret WJ (1988) Exogenous
ochronosis in the United States. J Am Acad Dermatol, 18: 1207-1211.
Leanderson P & Tagesson C (1990) Cigarette smoke-induced DNA-damage:
Role of hydroquinone and catechol in the formation of the oxidative
DNA-adduct, 8-hydroxyde-oxyguanosine. Chem-Biol Interact, 75: 71-81.
Leopardi P, Zijno A, Bassani B, & Pacchierotti F (1993) In vivo
studies on chemically induced aneuploidy in mouse somatic and
germinal cells. Mutat Res, 287: 119-130.
Levan A & Tjio JH (1948a) Chromosome fragmentation induced by
phenols. Hereditas, 34: 250-252.
Levan A & Tjio JH (1948b) Induction of chromosome fragmentation by
phenols. Hereditas, 34: 453-484.
Levay G & Bodell WJ (1992) Potentiation of DNA adduct formation in
HL-60 cells by combinations of benzene metabolites. Proc Natl Acad
Sci (USA), 89(15): 7105-7109.
Levay G, Pongracz K, & Bodell WJ (1991) Detection of DNA adducts in
HL-60 cells treated with hydroquinone and p-benzoquinone by
32P-postlabeling. Carcinogenesis, 12(7): 1181-1186.
Levenson GIP (1947) Determination of Elon and hydroquinone in
developer--an examination of Stott's method. Photogr J, B87: 18-24.
Levin J-O (1988) High performance liquid chromatographic
determination of hydroquinone in air as benzoquinone, using combined
oxidizing filter and XAD-2 adsorbent preconcentration. Chemosphere,
17: 671-679.
Lewis JG, Stewart W, & Adams DO (1988a) Role of oxygen radicals in
induction of DNA damage by metabolites of benzene. Cancer Res, 48:
4762-4765.
Lewis JG, Odom B, & Adams DO (1988b) Toxic effects of benzene and
benzene metabolites of mononuclear phagocytes. Toxicol Appl
Pharmacol, 92: 246-254.
Lidén C (1989) Occupational dermatoses at a film laboratory.
Follow-up after modernization. Contact Dermatitis, 20: 191-200.
Lockhart HB & Fox JA (1985a) Metabolic fate of [U-14C]
hydroquinone administered by gavage to female Fischer 344 rats.
Rochester, New York, Eastman Kodak Company, Health and Environment
Laboratories (Report No. TX-85-55).
Lockhart HB & Fox JA (1985b) The metabolic fate CF [14C]
hydroquinone administered by intratracheal instillation to male
Fischer 344 rats. Rochester, New York Eastman Kodak Company, Health
and Environment Laboratories (Report No. TX-85-76).
Lockhart HB, Fox JA, & Divincenzo GD (1984) The metabolic fate of
[U-14C] hydroquinone administered by gavage to male Fischer 344
rats. Rochester, New York, Eastman Kodak Company, Health and
Environment Laboratories (Report No. BC-84-14, prepared for the
Chemical Manufacturers Association, Washington).
McGregor DB, Brown A, Cattanach P, Edwards I, McBride D, & Caspary
WJ (1988a) Responses of the L5178Y tk+/tk- mouse lymphoma cell
forward mutation assay. II. 18 coded chemicals. Environ Mol Mutagen,
11: 91-118.
McGregor DB, Riach CG, Brown A, Edwards I, Reynolds D, West K, &
Willington S (1988b) Reactivity of catecholamines and related
substances in the mouse lymphoma L5178Y cell assay for mutagens.
Environ Mol Mutagen, 11: 523-544.
Mackay D & Paterson S (1981) Calculating fugacity. Environ Sci
Technol, 15: 1006-1014.
McLeese DW, Zitko V, & Peterson MR (1979) Structure-lethality
relationships for phenols, anilines and other aromatic compounds in
shrimp and clams. Chemosphere, 2: 53-57.
Maibach HJ & Patrick E (1989) A study to evaluate the potential of
mono-T-butyl hydroquinone to produce skin depigmentation. Kingsport,
Tennessee, Eastman Kodak Company (Report No. HIM 88-KOD-DEPIG-01).
Maier HG (1981) [Coffee.] In: Mayer HG ed. [Advances in food
research and food technology]. Berlin, Paul Parey Publisher, vol 18,
p 57 (in German).
Mann RJ & Harman RRM (1983) Nail staining due to hydroquinone
skin-lightening creams. Br J Dermatol, 108: 363-365.
Markey AC, Black AK, & Rycroft RJG (1989) Confetti-like
depigmentation from hydroquinone. Contact Dermatitis, 20: 148-149.
Marquardt P, Koch R, & Aubert J-P (1947) [The toxicity of the
monovalent, divalent and trivalent phenols.] J Gesamte Inn Med, 2:
333-346 (in German).
Marty JP, Trouvin JH, Jacquot C, & Wepierre J (1981)
Pharmacocinétique percutanée de l'hydroquinone 14C. C R Congrès
Eur Biopharm Pharmacocinet, 2: 221-228.
Miller BM & Adler I-D (1989) Suspect spindle poisons: analysis of
c-mitotic effects in mouse bone marrow cells. Mutagenesis, 4:
208-215.
Mitchell A & Webster J (1919) Notes on a case of poisoning by
hydroquinone. Br Med J, 21: 465.
Moore GA, Rossi L, Orrenius S, & O'Brien PJ (1987) Ca2+ release
from rat liver mitochondria by benzoquinone derivatives. Arch
Biochem Biophys, 259: 283-295.
Moore GA, Weis M, Orrenius S, & O'Brien PJ (1988) Role of sulfhydryl
group(s) in benzoquinone induced Ca2+ release by rat liver
mitochondria. Arch Biochem Biophys, 267: 539-550.
Morimoto K & Wolff S (1980) Increase of sister chromatid exchanges
and perturbations of cell division kinetics in human lymphocytes by
benzene metabolites. Cancer Res, 40: 1189-1193.
Morimoto K, Wolff S, & Koizumi A (1983) Induction of
sister-chromatid exchanges in human lymphocytes by microsomal
activation of benzene metabolites. Mutat Res, 119: 355-360.
Mozhaev EA, Osintseva VP, & Arzamastsev EV (1966) [Hydroquinone
toxicity in chronic poisoning.] Farmakol Toksikol, 29: 238-240 (in
Russian).
Murphy SJ, Schroeder RE, Blacker AM, Krasavage WJ, & English JC
(1992) A study of developmental toxicity of hydroquinone in the
rabbit. Fundam Appl Toxicol, 19: 214-221.
Nakamura S, Oda Y, Shimada T, Oki I, & Sugimoto K (1987)
SOS-inducing activity of chemical carcinogens and mutagens in
Salmonella typhimurium TA1535/pSK1002: examination with 151
chemicals. Mutat Res, 192: 239-246.
Naumann G (1966) Corneal damage in hydroquinone workers. Arch
Ophthalmol, 76: 189-194.
Nendza M & Seydel JK (1988a) Multivariate data analysis of various
biological test systems used for the quantification of ecotoxic
compounds. Quant Struct-Act Relat, 7: 165-174.
Nendza M & Seydel JK (1988b) Quantitative structure-toxicity
relationships and multivariate data analysis for ecotoxic chemicals
in different biotest systems. Chemosphere, 7: 1575-1584.
Nendza M & Seydel JK (1988c) Quantitative structure-toxicity
relationships for ecotoxicologically relevant biotest systems and
chemicals. Chemosphere, 17: 1585-1602.
Neujahr H & Varga JM (1970) Degradation of phenols by intact cells
and cell-free preparations of Trichosporon cutaneum. Eur J
Biochem, 13: 37-44.
NIOSH (1976) Hydroquinone: Method No. S57 - Standards Completion
Program. Cincinnati, Ohio, National Institute for Occupational
Safety and Health, Division of Laboratories and Criteria
Development, p 66.
NIOSH (1978) Criteria for a recommended standard ... Occupational
exposure to hydroquinone, Cincinnati, Ohio, US National Institute
for Occupational Safety and Health, 182 pp (Publication DHEW (NIOSH)
No. 78-155).
NIOSH (1987) Registry of toxic effects of chemical substances
(RTECS). 1985-1986 edition, Cincinnati, Ohio, National Institute for
Occupational Safety and Health (NIOSH Publication No. 87-114).
Nomiyama K, Minai M, Suzuki T, & Kita H (1967) Studies on poisoning
by benzene and its homologues. (10). Median lethal doses of benzene
metabolites. Ind Health, 5: 143-148.
Norrelykke Nissen J & Corydon L (1985) Corneal ulcer after exposure
to vapours from bone cement (methyl metacrylate and hydroquinone).
Int Arch Ocup Environ Health, 56: 161-165.
NTP (1989) Technical report on the toxicology and carcinogenesis
studies of hydroquinone (CAS No. 123-31-9) in F344/N rats and
B6C3F1 mice (gavage studies). Research Triangle Park, North
Carolina, National Toxicology Program (NTP TR 366).
Nyholm N, Jorgensen-Lindgaard P, & Hansen N (1984) Biodegradation of
4-nitrophenol in standardized aquatic degradation tests. Ecotoxicol
Environ Saf, 8: 451-470.
O'Brien PJ (1991) Molecular mechanisms of quinone cytotoxicity.
Chem-Biol Interact, 80: 1-41.
OECD (1981) OECD guidelines for testing of chemicals. Paris,
Organisation for Economic Co-operation and Development.
Oettel H (1936) [Hydroquinone poisoning.] Naunyn-Schmiedebergs Arch
Exp Pathol Pharmakol, 183: 319-362 (in German).
Oglesby FL, Sterner JH, & Anderson B (1947) Quinone vapors and their
harmful effects. II. Plant exposures associated with eye injuries. J
Ind Hyg Toxicol, 29: 74-84.
Oglesby LA, Ebron-McCoy MT, Logsdon TR, Copeland F, Beyer PE, &
Kavlock RJ (1992) In vitro embryotoxicity of a series of
para-substituted phenols: structure, activity, and correlation with
in vitro data. Teratology, 45: 11-33.
Ohnishi T, Yamazaki H, Iyanage T, Nakamura T, & Yamazaki I (1969)
One-electron-transfer reactions in biochemical systems. II. The
reaction of free radicals formed in the enzymic oxidation. Biochim
Biophys Acta, 172: 357-369.
O'Keeffe DH, Wiese TE, Brummet SR, & Miller TW (1987) Uptake and
metabolism of phenolic compounds by the water hyacinth (Eichhornia
crassipes). Recent Adv Phytochem, 21(87): 101-129.
Oliveira NL & Kalf GF (1992) Induced differentiation of HL-60
promyelocytic leukemia cells to monocyte/macrophages is inhibited by
hydroquinone, a hematotoxic metabolite. Blood, 79: 627-633.
Otsuka M & Nonomura Y (1963) The action of phenolic substances on
nerve endings. J Pharmacol Exp Ther, 140: 41-45.
Pacchierotti F, Bassani B, Leopardi P, & Zijno A (1991) Origin of
aneuploidi in relation to disturbances of cell-cycle progression.
II: Cytogenetic analysis of various parameters in mouse bone marrow
cells after colchicine or hydroquinone treatment. Mutagenesis, 6(4):
307-311.
Painter RB & Howard R (1982) The HeLa DNA-synthesis inhibition test
as a rapid screen for mutagenic carcinogens. Mutat Res, 92: 427-437.
Palumbo A, d'Ischia M, Misuraca G, & Prota G (1991) Mechanism of
inhibition of melanogenesis by hydroquinone. Biochim Biophys Acta,
1073: 85-90.
Parmentier R (1952) Etude des lésions cellulaires provoquées par
divers phénols et amines aromatiques. Revue Belge Pathol Méd Exp,
22: 1-54.
Parmentier R (1953) Production of `three-group metaphases' in the
bone-marrow of the golden hamster. Nature (Lond), 171: 1029-1030.
Parmentier R & Dustin P Jr (1948) Early effects of hydroquinone on
mitosis. Nature (Lond), 161: 527-528.
Parmentier R & Dustin P Jr (1951) Reproduction expérimentale d'une
anomalie particulière de la métaphase des cellules malignes
(métaphase "à trois groupes"). Caryologia, IV(1): 98-109.
Pellack-Walker P & Blumer JL (1986) DNA damage in L5178YS cells
following exposure to benzene metabolites. Mol Pharmacol, 30: 42-47.
Pellack-Walker P, Walker JK, Evans HH, & Blumer JL (1985)
Relationship between the oxidation potential of benzene metabolites
and their inhibitory effect on DNA synthesis in L5178YS cells. Mol
Pharmacol, 28: 560-566.
Penney BK, Smith JC, & Allen CJ (1984) Depigmenting action of
hydroquinone depends on disruption of fundamental cell processes. J
Invest Dermatol, 82: 308-310.
Pfeifer PW & Irons RD (1983) Alteration of lymphocyte function by
quinones through a sulfhydryl-dependent disruption of microtubule
assembly. Int J Immunopharmacol, 5: 463-470.
Picardo M, Nazzaro-Porro MN, Breathnach A, Zompetta C, Faggioni A, &
Riley P (1987) Mechanism of antitumoral activity of catechols in
culture. Biochem Pharmacol, 36(4): 417-425.
Pifer JW, Hearne FT, Friedlander BR, & McDonough JR (1986) Mortality
study of men employed at a large chemical plant, 1972 through 1982.
J Occup Med, 28: 438-444.
Post GB, Snyder R, & Kalf GF (1985) Inhibition of RNA synthesis and
interleukin-2 production in lymphocytes in vitro by benzene and
its metabolites, hydroquinone and p-benzoquinone. Toxicol Lett,
29: 161-167.
Rácz G, Füzi J, Kemény G, & Kisgyörgy Z (1958) [The effect of
hydroquinone and Phloridzin on the sexual cycle of white rats.] Orv
Sz, 5: 65-67 (in Hungarian).
Raghavan NV (1979) Separation and quantification of trace isomeric
hydroxyphenols in aqueous solution by high-performance liquid
chromatography. J Chromatogr, 168: 523-525.
Rajka G & Blohm SG (1970) The allergenicity of paraphenylendiamine
II. Acta Dermatovenerol (Stockholm), 50: 51-54.
Rao GS & Pandya KP (1989) Release of 2-thiobarbituric acid reactive
products from glutamate or deoxyribonucleic acid by
1,2,4-benzenetriol or hydroquinone in the presence of copper ions.
Toxicology, 59: 59-65.
Reddy MV, Blackburn GR, Irwin SE, Kommineni C, Mackerer CR, &
Mehlman MA (1989) A method for in vitro culture of rat zymbal
gland: Use in mechanistic studies of benzene carcinogenesis in
combination with 32P-postlabeling. Environ Health Perspect, 82:
239-247.
Reddy MV, Bleicher T, Blackburn GR, & Mackerer CR (1990) DNA
adduction by phenol, hydroquinone, or benzoquinone in vitro but
not in vivo: nuclease P1-enhanced 32P-postlabeling of adducts as
labeled nucleoside biphosphates, dinucleotides and nucleoside
monophosphates. Carcinogenesis, 11: 1349-1357.
Rémond A & Colombies H (1927) Intoxication par l'hydroquinone. Ann
Méd Lég, 7: 79-81.
Renz JF, Georg F, & Kalf GF (1991) Role for interleukin-1 (IL-1) in
benzene-induced hematotoxicity: Inhibition of conversion of
Pre-IL-1alpha to mature cytokine in murine macrophages by
hydroquinone and prevention of benzene-induced hematotoxicity in
mice by IL-1alpha. Blood, 78(4): 938-944.
Ribo JM & Kaiser KLE (1983) Effects of selected chemicals to
photoluminescent bacteria and their correlations with acute and
sublethal effects on other organisms. Chemosphere, 12: 1421-1442.
Robertson ML, Eastmond DA, & Smith MT (1991) Two benzene
metabolites, catechol and hydroquinone, produced a synergistic
induction of micronuclei and toxicity in cultured human lymphocytes.
Mutat Res, 249: 201-209.
Roe FJC & Salaman MH (1955) Further studies on incomplete
carcinogenesis: triethylene melamine (T.E.M.), 1,2-benzanthracene
and ß-propiolactone, as initiators of skin tumour formation in the
mouse. Br J Cancer, 9: 177-203.
Rosen F & Millman N (1955) Anti-gonadotrophic activities of quinones
and related compounds. Endocrinology, 57: 466-471.
Ross D, Siegel D, Gibson NW, Pacheco D, Thomas DJ, Reasor M, &
Wierda D (1990) Activation and deactivation of quinones catalyzed by
DT-diaphorase. Evidence for bioreductive activation of diaziquone
(AZQ) in human tumor cells and detoxification of benzene metabolites
in bone marrow stroma. Free Radic Res Commun, 8: 4-6, 373-381.
Rossi L, Moore GA, Orrenius S, & O'Brien PJ (1986) Quinone toxicity
in hepatocytes without oxidative stress. Arch Biochem Biophys, 251:
1, 25-35.
Rott B, Viswanathan R, Freitag D, & Korte F (1982) [Comparative
investigation of the applicability of various tests for evaluating
the degradability of environmental chemicals.] Chemosphere, 11:
531-538 (in German).
Roy SC (1973) Comparative effects of colchicine, caffeine and
hydroquinone on nodal roots of Callisia fragrans. Biol Plant, 15:
383-390.
Rushmore T, Snyder R, & Kalf G (1984) Covalent binding of benzene
and its metabolites to DNA in rabbit bone marrow mitochondria in
vitro. Chem-Biol Interact, 49: 133-154.
Sakai M, Yoshida D, & Mizusaki S (1985) Mutagenicity of polycyclic
aromatic hydrocarbons and quinones on Salmonella typhimurium TA97.
Mutat Res, 156: 61-67.
Sawada Y, Iyanagi T, & Yamazaki I (1975) Relation between redox
potentials and rate constants in reactions coupled with the system
oxygen-superoxide. Biochemistry, 14: 3761-3764.
Sawahata T & Neal RA (1983) Biotransformation of phenol to
hydroquinone and catechol by rat liver microsomes. Mol Pharmacol,
23: 453-460.
Schlosser MJ & Kalf GF (1989) Metabolic activation of hydroquinone
by macrophage peroxidase. Chem-Biol Interact, 72: 191-207.
Schlosser MJ, Shurina RD, & Kalf GF (1989) Metabolism of phenol and
hydroquinone to reactive products by macrophage peroxidase or
purified prostaglandin H synthase. Environ Health Perspect, 82:
229-237.
Schlosser MJ, Shurina RD, & Galf GF (1990) Prostaglandin H synthase
catalyzed oxidation of hydroquinone to a sulfhydryl-binding and
DNA-damaging metabolite. Chem Res Toxicol, 3: 333-339.
Schultz TW, Riggin GW, & Wesley SK (1987) Structure-activity
relationships for para-substituted phenols. In: Kaiser KLE ed. QSAR
in environmental toxicology - II. Dordrecht, D. Reidel Publishing
Company, pp 333-345.
Seki T (1975) Chromatographic separation of dihydroxybenzenes on a
column of Merckogel PGM 2000. J Chromatogr, 115: 262-263.
Serva RJ & Bulman C (1981) Mutagenicity evaluation of hydroquinone
(MIBK process). Akron, Ohio, The Goodyear Tire & Rubber Company
(Report No 81-4-3).
Serva RJ & Murphy SJ (1981) Evaluation of hydroquinone using the
Drosophila melanogaster/sex-linked recessive lethal test. Akron,
Ohio, The Goodyear Tire & Rubber Company (Report No. 56).
Shaner VC & Sparks MR (1946) Application of methyl ethyl ketone to
the analysis of developers for Elon and hydroquinone. J Soc Motion
Pict Eng, 47: 409-417.
Sharma AK & Chatterjee T (1964) Effect of oxygen on chromosomal
aberrations induced by hydroquinone. Nucleus, 7: 113-124.
Shibata M-A, Yamada M, Hirose M, Asakawa E, Tatematsu M, & Ito N
(1990) Early proliferative responses of forestomach and glandular
stomach of rats treated with five different phenolic antioxidants.
Carcinogenesis, 11(3): 425-429.
Shibata M-A, Hirose M, Tanaka H, Asakawa E, Shirai T, & Ito N (1991)
Induction of renal cell tumors in rats and mice, and enhancement of
hepatocellular tumor development in mice after long-term
hydroquinone treatment. Jpn J Cancer Res, 82: 1211-1219.
Skalka P (1964) [The influence of hydroquinone on the fertility of
male rats.] Sb Vys Sk Zemed (Brne), BXII: 491-494 (in Czech).
Smale BC & Keil HL (1966) A biochemical study of the intervarietal
resistance of Pyrus communis to fire blight. Phytochemistry, 5:
1113-1120.
Smith MT, Yager JW, Steinmetz KL, & Eastmond DA (1989)
Peroxidase-dependent metabolism of benzene's phenolic metabolites
and its potential role in benzene toxicity and carcinogenicity.
Environ Health Perspect, 82: 23-29.
Sollmann T (1949) Correlation of the aquarium goldfish toxicities of
some phenols, quinones and other benzene derivatives with their
inhibition of autooxidative reactions. J Gen Physiol, 32: 671-679.
Spain JC, Wyss O, & Gibson DT (1979) Enzymatic oxidation of
p-nitrophenol. Biochem Biophys Res Commun, 88(2): 634-641.
Spencer MC (1965) Topical use of hydroquinone for depigmentation. J
Am Med Assoc, 194(9): 114-116.
Springborn Institute for Bioresearch (1984) Photoallergic contact
dermatitis in guinea-pigs (Armstrong method): Final report
(Unpublished data from Springborn Institute for Bioresearch,
submitted to WHO by CFTA).
Stenius U (1989) [Nordic Expert Group for Documentation of
Occupational Exposure Limits. 84. Hydroquinone.] Solna, Sweden,
National Institute of Occupational Health (Work and Health -
Scientific Publication Series No. 1989:15) (in Swedish with English
summary).
Stenius U, Warholm M, Rannug A, Walles S, Lundberg J, & Högberg J
(1989) The role of GSH depletion and toxicity in
hydroquinone-induced development of enzyme-altered foci.
Carcinogenesis, 10(3): 593-599.
Sterner JH, Oglesby FL, & Anderson B (1947) Quinone vapors and their
harmful effects. I. Corneal and conjunctival injury. J Ind Hyg
Toxicol, 26: 60-73.
Stevens HP (1945) Rubber photogelling agents - Part II. J Soc Chem
Ind, 64: 312-315.
Stom DJ (1975) Use of thin-layer and paper chromatography for
detection of ortho- and para-quinones formed in the course of phenol
oxidation. Acta Hydrochim Hydrobiol, 3: 39-45.
Stom DJ & Roth R (1981) Some effects of polyphenols on aquatic
plants. I. Toxicity of phenols in aquatic plants. Bull Environ
Contam Toxicol, 27: 332-337.
Stom DJ, Geel TA, & Balayan AE (1986) Effect of individual phenolic
compounds and their mixtures on luminous bacteria. Part I: Use of
bacterial luminescence extinguishing for biotesting of phenols. Acta
Hydrochim Hydrobiol, 14: 283-292.
Stott JG (1942) The application of potentiometric methods to
developer analysis. J Soc Motion Pict Eng, 39: 37-54.
Stubberfield CR & Cohen GM (1989) Interconversion of NAD(H) to
NADP(H) a cellular response to quinone-induced oxidative stress in
isolated hepatocytes. Biochem Pharmacol, 38: 2631-2637.
Subrahmanyam VV, Kolachana P, & Smith MT (1991) Metabolism of
hydroquinone by human myeloperoxidase: Mechanisms of stimulation by
other phenolic compounds. Arch Biochem Biophys, 286: 76-84.
Subrahmanyam VV, Doane-Setzer P, Steinmetz KL, Ross D, & Smith MT
(1990) Phenol-induced stimulation of hydroquinone bioactivation in
mouse bone marrow in vivo: possible implications in benzene
myelotoxicity. Toxicology, 62: 107-116.
Tabak HH, Chambers CW, & Kabler PW (1964) Microbial metabolism of
aromatic compounds. I Decomposition of phenolic compounds and
aromatic hydrocarbons by phenol-adapted bacteria. J Bacteriol, 87:
910-919.
Telford IR, Woodruff CS, & Linford RH (1962) Fetal resorption in the
rat as influenced by certain antioxidants. Am J Anat, 110: 29-36.
Terhaar CJ, Ewell WS, Dziuba SP, & Fassett DW (1972) Toxicity of
photographic processing chemicals to fish. Photogr Sci Eng, 16:
370-377.
Thomas DJ, Sadler A, Subrahmanyam VV, Siegel D, Reasor MJ, Wierda D,
& Ross D (1989a) Bone marrow stromal cell bioactivation and
detoxification of the benzene metabolite hydroquinone: Comparison of
macrophages and fibroblastoid cells. Mol Pharmacol, 37: 255-262.
Thomas DJ, Reasor MJ, & Wierda D (1989b) Macrophage regulation of
myelopoiesis is altered by exposure to the benzene metabolite
hydroquinone. Toxicol Appl Pharmacol, 97: 440-453.
Tissot A, Boule P, Lemaire J, Lambert S, & Palla JC (1985)
Photochimie et environnement. X. Evaluation de la toxicité des
produits de phototransformation de l'hydroquinone et des
chlorophénols en milieu aqueux. Chemosphere, 14: 1221-1230.
Tratnyek PG & Macalady DL (1989) Abiotic reduction of nitro aromatic
pesticides in anaerobic laboratory systems. J Agric Food Chem, 37:
248-254.
Trevors JT & Basaraba J (1980) Toxicity of benzoquinone and
hydroquinone in short-term bacterial bioassays. Bull Environ Contam
Toxicol, 25: 672-675.
Trower MK, Sariaslani FS, & Kitson FG (1988) Xenobiotic oxidation by
cytochrome P-450 enriched extracts of Streptomyces griseus.
Biochem Biophys Res Commun, 157(3): 1417-1422.
Tunek A, Platt KL, Przybylski M, & Oesch F (1980) Multi-step
metabolic activation of benzene: effect of superoxide dismutase on
covalent binding to microsomal macromolecules, and identification of
glutathione conjugates using high pressure liquid chromatography and
field desorption mass spectrometry. Chem-Biol Interact, 33: 1-17.
Tunek A, Högstedt B, & Olofsson T (1982) Mechanism of benzene
toxicity. Effects of benzene and benzene metabolites on bone marrow
cellularity, number of granulopoietic stem cells and frequency of
micronuclei in mice. Chem-Biol Interact, 39: 129-138.
Twerdok LE & Trush MA (1990) Differences in quinone reductase
activity in primary bone marrow stromal cells derived from C57BL/6
and DBA/2 mice. Res Commun Chem Pathol Pharmacol, 67(3): 375-386.
Twerdok LE, Rembish SJ, & Trush MA (1992) Induction of quinone
reductase and glutathione in bone marrow cells by
1,2-dithiole-3-thione: Effect on hydroquinone-induced cytotoxicity.
Toxicol Appl Pharmacol, 112: 273-281.
Umpelev VL, Kogan LA, & Gagarinova LM (1974)
[Thin-layer-chromatographic separation of polyhydric phenols in
effluents.] Zh Anal Khim, 29: 179-180 (in Russian).
US EPA (1985) Hydroquinone: Testing requirements. Fed Reg, 50(250):
53145-53156.
US EPA (1987) Part 1: The toxicity of 3400 chemicals to fish. Part
2: The toxicity of 1085 chemicals to fish. Washington, DC, US
Environmental Protection Agency, Office of Toxic Substances (EPA
560/6-87-002.
Usami Y, Landau AB, Fukuyama K, & Gellin GA (1980) Inhibition of
tyrosinase activity by 4-tert-butylcatechol and other depigmenting
agents. J Toxicol Environ Health, 6: 559-567.
Valadaud D & Izard C (1971) Contribution à l'étude des effets
biologiques de l'hydroqui-none. Action sur la division cellulaire. C
R Acad Sci Paris, D273: 2247-2248.
Valadaud-Barrieu D & Izard C (1973) Modifications du cycle
cellulaire sous l'influence de l'hydroquinone dans les méristèmes
radiculaires de Vicia faba. C R Acad Sci Paris, D276: 33-35.
Van der Sluis D (1980) DNA damage by hydroquinone (sublimed) #
8349-34 in the E. coli Pol A1- assay. Akron, Ohio, The
Goodyear Tire & Rubber Company (Report No 80-6-7).
Van der Walle HB, Klecak G, Geleick H, & Bensink T (1982b)
Sensitizing potential of 14 mono(meth)acrylates in the guinea-pig.
Contact Dermatitis, 8: 223-235.
Van der Walle HB, Delbressine LPC, & Seutter E (1982a) Concomitant
sensitization to hydroquinone and P-methoxyphenol in the guinea-pig;
inhibitors in acrylic monomers. Contact Dermatitis, 8: 147-154.
Van Duuren BL & Goldschmidt BM (1976) Cocarcinogenic and
tumor-promoting agents in tobacco carcinogenesis. J Natl Cancer
Inst, 56: 1237-1242.
Van Ketel WG (1984) Sensitization to hydroquinone and the monobenzyl
ether of hydroquinone. Contact Dermatitis, 10: 253.
Varagnat J (1981) Hydroquinone, resorcinol, and catechol. In:
Grayson M ed. Kirk-Othmer encyclopedia of chemical technology, 3rd
ed. New York, John Wiley & Sons, pp 39-69.
Vladescu C & Apetroae M (1983) Biophysical radiosensitization.
Radiat Environ Biophys, 22: 141-148.
Wallin M & Hartley-Asp B (1993) Effects of potential aneuploidy
inducing agents on microtubule assembly in vitro. Mutat Res, 287:
17-22.
Wallin H, Melin P, Schelin C, & Jergil B (1985) Evidence that
covalent binding of metabolically activated phenol to microsomal
proteins is caused by oxidised products of hydroquinone and
catechol. Chem-Biol Interact, 55: 335-346.
Wampler DA (1980) DNA damage by hydroquinone lot 07319A in the E.
coli Pol A1- assay. Akron, Ohio, The Goodyear Tire & Rubber
Company (Report No 80-3-7).
Wängberg SA & Blanck H (1988) Multivariate patterns of algal
sensitivity to chemicals in relation to phylogeny. Ecotoxicol
Environ Saf, 16: 72-82.
Wellens H (1982) [Comparison of the sensitivity of Brachydanio
rerio and Leuciscus idus by testing the fish toxicity of
chemicals and wastewaters.] Z Wasser Abwasser Forsch, 15: 49-52 (in
German).
Whettem SMA (1949) The determination of small amounts of
hydroquinone in styrene. Analyst, 74: 185-188.
Wierda D & Irons RD (1982) Hydroquinone and catechol reduce the
frequency of progenitor B lymphocytes in mouse spleen and bone
marrow. Immunopharmacology, 4: 41-54.
Wild D, King M-T, Eckhardt K, & Gocke E (1981) Mutagenic activity of
aminophenols and diphenols, and relations with chemical structure.
Mutat Res, 85: 456.
Winterbourn CC (1981) Cytochrome c reduction by semiquinone radicals
can be indirectly inhibited by superoxide dismutase. Arch Biochem
Biophys, 209(1): 159-167.
Woodard GDL (1951) The toxicity, mechanism of action, and metabolism
of hydroquinone. Washington, DC, George Washington University, 81 pp
(Dissertation).
Wynder EL & Hoffman D (1967) Tobacco and tobacco smoke. Studies in
experimental carcinogenesis. New York, Academic Press, pp 388-389.
Xu W & Adler I-D (1990) Clastogenic effects of known and suspect
spindle poisons studied by chromosome analysis in mouse bone marrow
cells. Mutagenesis, 5: 371-374.
Yager JW, Eastmond DA, Robertson ML, Paradisin WM, & Smith MT (1990)
Characterization of micronuclei induced in human lymphocytes by
benzene metabolites. Cancer Res, 50: 393-399.
Yamazaki I & Ohnishi T (1969) One-electron-transfer reactions in
biochemical systems. I. Kinetic analysis of the oxidation-reduction
equilibrium between quinol-quinone and ferro-ferricytochrome c.
Biochim Biophys Acta, 112: 469-481.
Yamazaki I, Mason HS, & Piette L (1960) Identification, by electron
paramagnetic resonance spectroscopy, of free radicals generated from
substrates by peroxidase. J Biol Chem, 235: 2444-2449.
Young LY & Rivera MD (1985) Methanogenic degradation of four
phenolic compounds. Water Res, 19(10): 1325-1332.
Young RHF, Ryckman DW, & Buzzell JC Jr (1968) An improved tool for
measuring biodegradability. J Water Pollut Control Fed, 40: 354-370.
Zeidman I & Deutl R (1945) Poisoning by hydroquinone and
mono-methyl-paraaminophenol sulfate. Am J Med Sci, 210: 328-333.
APPENDIX 1.
Bibliographic data bases consulted
Agricola
Aqualine
Aquatic Science and Fisheries Abstracts
BIBLINE
BIBRA
Biosis Previews
CA Search
CAB Abstracts
Cancerlit
CHEMLIST
CHRIS
ECDIN
Enviroline
EXICHEM
FEDREG
Food Science and Technical Abstracts
HSDB
Medline
Oceanic Abstracts
OHMTADS
RISKLINE
RTECS
ROADMAPS
SERIX
Toxline
TOXLIT
TSCATS
RESUME
1. Identité, propriétés physiques et chimiques et méthodes
d'analyse
L'hydroquinone (1,4-benzènediol; C6H4(OH)2) se présente à
l'état pur sous la forme d'un solide cristallin blanc dont le point
de fusion est égal à 173-174°C. Sa densité est de 1,332 à 15°C et sa
tension de vapeur de 2,4 x 10-3 Pa (1,8 x 10-5 mmHg) à 25°C.
Elle est extrêmement soluble dans l'eau (70g/litre à 25°C) et le
logarithme de son coefficient de partage n-octanol/eau est de
0,59. Sa solubilité dans les solvants organiques varie de 57% dans
l'éthanol à moins de 0,1% dans le benzène. L'hydroquinone est
combustible à condition de subir un chauffage préalable. C'est un
réducteur qui est oxydé réversiblement en semiquinone correspondante
et en quinone.
L'échantillonnage de l'hydroquinone dans l'air s'effectue soit
par piégeage dans un solvant soit par filtration sur membrane
d'ester cellulosique mixte.
Le dosage de l'hydroquinone s'effectue soit par titrimétrie
soit par spectrophotométrie ou plus couramment par chromatographie.
2. Sources d'exposition humaine et environnementale
L'hydroquinone existe sous forme libre ou conjuguée dans les
bactéries, les plantes et certains animaux. Plusieurs pays la
produisent en quantités industrielles. En 1979, la capacité mondiale
totale de production dépassait 40 000 tonnes et était retombée à
environ 35 000 tonnes en 1992. L'hydroquinone est très largement
utilisée comme réducteur, comme développateur en photographie ainsi
que antioxydant ou comme stabilisant pour certaines substances qui
se polymérisent en présence de radicaux libres; elle sert encore
d'intermédiaire dans la production des antioxydants, antiozonants,
produits agrochimiques et polymères. L'hydroquinone est également
utilisée pour la fabrication de cosmétiques et de préparations
médicinales.
3. Transport, distribution et transformation dans l'environnement
L'hydroquinone qui est présente dans l'environnement est le
produit de l'activité humaine mais elle se trouve également dans des
substances naturelles d'origine végétale ou animale.
En raison de ses propriétés physico-chimiques, l'hydroquinone
se répartit essentiellement dans le compartiment aquatique
lorsqu'elle est libérée dans l'environnement. Sa décomposition
résulte principalement de processus photochimiques et biologiques;
elle n'est donc pas persistante et ne manifeste aucune tendance à la
bioaccumulation.
4. Concentrations dans l'environnement et exposition humaine
On ne dispose d'aucune données sur la concentration de
l'hydroquinone dans l'air, le sol ou l'eau. Toutefois le dosage de
l'hydroquinone dans la fumée de cigarettes (courant principal) sans
bout-filtre a donné des résultats allant de 110 à 300 ug/cigarette,
résultats qui valent également pour la fumée du courant latéral. On
trouve également de l'hydroquinone dans des denrées alimentaires
d'origine végétale (par exemple le germe de blé), dans le café
infusé, dans les thés préparés à partir des feuilles de certaines
baies, avec des concentrations pouvant dépasser quelquefois 1%.
Les photographes amateurs peuvent être exposés à l'hydroquinone
par voie percutanée ou respiratoire. Toutefois on ne dispose
d'aucune donnée sur l'importance de cette exposition. L'exposition
percutanée peut également résulter de l'utilisation de produits
cosmétiques ou médicinaux contenant de l'hydroquinone, tels que les
éclaircissants. Dans les pays de la Communauté européenne, la teneur
des cosmétiques en hydroquinone est limitée à 2% au maximum. Aux
Etats-Unis d'Amérique, la Food and Drug Administration a proposé une
concentration comprise entre 1,5 et 2% pour les éclaircissants
cutanés. La concentration peut atteindre 4% dans certains
médicaments délivrés sur ordonnance. Dans certains pays, les
éclaircissants cutanés peuvent en contenir des quantités encore plus
importantes.
Du point de vue hygiène et sécurité, on ne dispose que de peu
de données concernant la surveillance de l'hydroquinone. On indique
des concentrations moyennes dans l'air au cours de la fabrication de
l'hydroquinone et des divers traitements subis par cette substance,
qui se situeraient dans la gamme de 0,13 à 0,79 mg/m3. Les limites
d'exposition professionnelles dans l'air (moyenne pondérée par
rapport au temps) dans les différents pays vont de 0,5 à 2 mg/m3.
5. Cinétique et métabolisme
L'hydroquinone est rapidement et largement résorbée chez
l'animal au niveau de l'intestin et de la trachée. L'absorption
percutanée est plus lente mais elle peut s'accélérer en présence de
véhicules tels que les alcools. L'hydroquinone se répartit
rapidement et largement dans les différents tissus. Elle est
métabolisée en p-benzoquinone et autres produits d'oxydation et sa
détoxification s'effectue par conjugaison sous forme de
monoglucuronide, monosulfate et mercapturates. L'excrétion de
l'hydroquinone et de ses métabolites est rapide et s'effectue
principalement par la voie urinaire.
L'hydroquinone et/ou ses dérivés réagissent avec différents
constituants biologiques tels que les macromolécules et les
molécules de faible masse moléculaire et ils exercent des effets sur
la métabolisme cellulaire.
6. Effets sur les mammifères de laboratoire et les systèmes
d'épreuve in vitro
Les valeurs de la DL50 par voie orale varient de 300 à 1300
mg/kg de poids corporel pour un certain nombre d'espèces animales.
Toutefois pour le chat, elle se situe entre 42 et 86 mg/kg de poids
corporel. Une intoxication aiguë par de fortes concentrations
d'hydroquinone entraîne de graves effets sur le système nerveux
central et notamment une hyperexcitabilité, des tremblements, des
convulsions, le coma et la mort. A des doses sublétales, ces effets
sont réversibles. Pour les rongeurs, on estime la DL50 par voie
percutanée à > 3800 mg/kg. On ne dispose d'aucun renseignement sur
les valeurs de la CL50.
L'épidermo-réaction effectuée en une seule fois au moyen d'une
préparation à 2% d'hydroquinone a provoqué une irritation chez le
lapin notée à 1,22 sur une échelle allant de 0 à 4. Des applications
topiques quotidiennes effectuées pendant trois semaines avec de
l'hydroquinone à 2 ou 5% dans une émulsion huile/eau, sur la peau
rasée de cobayes noirs, a entraîné une dépigmentation, des
altérations inflammatoires et un épaississement de l'épiderme. La
dépigmentation était plus marquée à fortes concentrations et les
femelles se sont révélées plus sensibles que les mâles.
Les tests de sensibilisation effectués sur des cobayes
suscitent des réactions faibles à fortes selon la méthode ou le
véhicule utilisé. Les réactions les plus fortes ont été obtenues
avec le test de sensibilisation maximale sur le cobaye. On a
également observé une sensibilisation croisée de presque 100 pour
cent entre l'hydroquinone et le p-méthoxyphénol chez le cobaye
mais les éléments qui pourraient militer en faveur de l'existence de
réactions analogues avec la p-phénylénediamine, l'acide
sulfanilique et la p-benzoquinone restent limités.
Une étude de toxicité par voie orale de six semaines chez des
rats mâles F-344 a permis de mettre en évidence des néphropathies et
une prolifération des cellules rénales. Après 13 semaines de gavage,
on a mis en observation des rats F-344 et des souris B6C3F1. Des
signes de néphrotoxicité se sont manifestés chez les rats aux doses
respectives de 100 et 200 mg/kg, avec des tremblements et des
convulsions à cette dernière dose; chez les deux espèces, on a
observé une diminution du gain de poids. L'administration d'une dose
de 400 mg/kg a entraîné la mort des rats. Chez les souris qui
avaient reçu cette même dose pendant 13 semaines, on a relevé des
tremblements, des convulsions et des lésions affectant l'épithélium
gastrique. Des rats Sprague Dawley exposés pendant 13 semaines à de
l'hydroquinone ont présenté une réduction du gain de poids et des
signes neurologiques témoignant d'une atteinte centrale à 200 mg/kg.
Ces signes ont également été observés à la dose de 64 mg/kg de poids
corporel mais ils étaient absents à 20 mg/kg.
Après injection sous-cutanée d'hydroquinone à des rats, on a
observé une diminution de la fécondité chez les mâles et un
allongement du cycle oestral chez les femelles. Toutefois ces effets
n'ont pas été observés dans les études portant sur une
administration par voie orale (étude de létalité dominante et étude
sur deux générations). Une étude portant sur le développement de
rats ayant reçu par voie orale des doses de 300 mg/kg de poids
corporel a révélé une légère toxicité pour les femelles gravides et
une réduction du poids du foetus. Chez le lapin, la dose sans effets
toxiques observables sur la mère était de 25 mg/kg/jour; la dose
sans effets toxiques sur le développement était de 75 mg/kg/jour.
Lors d'une étude de deux générations portant sur la reproduction de
rats, l'administration d'hydroquinone n'a entraîné aucun effet nocif
sur la reproduction à des doses orales quotidiennes allant jusqu'à
150 mg/kg de poids corporel. La dose sans effets toxiques
observables pour les géniteurs a été fixée par 15 mg/kg/jour; en ce
qui concerne les effets sur la reproduction observés en l'espace de
deux générations, on a obtenu une valeur de 150 mg/kg/jour.
L'hydroquinone provoque la formation de micronoyaux in vivo
et in vitro. Des aberrations portant sur la structure et le nombre
des chromosomes ont été observées in vitro ainsi qu'après
administration intrapéritonéale in vivo. En outre, on a pu mettre
en évidence in vitro des mutations géniques, des échanges entre
chromatides soeurs et des lésions de l'ADN. Après injection
intrapéritonéale d'hydroquinone à des souris, on a observé, dans les
cellules germinales des mâles, de aberrations chromosomiques d'une
ampleur comparable à celles que l'on observait dans les cellules de
la moelle osseuse. Une épreuve de létalité dominante effectuée sur
des rats mâles recevant de l'hydroquinone par voie orale n'a pas
permis d'établir l'existence de mutations au niveau des cellules
germinales.
Lors d'une étude de deux ans, au cours de laquelle on a
administré à des rats F-344/N de l'hydroquinone par voie orale, on a
observé, chez les mâles, des adénomes affectant les tubules rénaux
dont la fréquence était liée à la dose. L'incidence de ces adénomes
était statistiquement significative dans le groupe qui recevait une
forte dose. Dans ce même groupe, on a également observé une
hyperplasie des cellules tubulaires rénales. Chez les femelles, on a
observé un accroissement de l'incidence, lié à la dose, des
leucémies à monocytes. Chez des souris B6C3F1, on a observé une
incidence sensiblement accrue des adénomes hépatocellulaires. Dans
une autre étude, l'hydroquinone administrée dans la proportion de
0,8% de la nourriture, a entraîné une augmentation significative de
l'incidence de l'hyperplasie épithéliale des papilles rénales et une
augmentation également significative des hyperplasies et des
adénomes au niveau des tubules rénaux chez les rats mâles. En
revanche, on n'a pas observé d'augmentation dans l'incidence des
leucémies à monocytes chez les femelles. Chez les souris,
l'incidence de l'hyperplasie spinocellulaire affectant l'épithélium
de la portion cardiaque de l'estomac présentait une augmentation
significative dans les deux sexes. Chez les mâles, on notait
également une incidence sensiblement accrue des adénomes
hépatocellulaires et des hyperplasies tubulaires rénales. Quelques
adénomes rénaux ont été observés.
Des études sur des souris, effectuées in vivo (injection
intrapéritonéale) et in vitro montrent que l'hydroquinone a un
effet cytotoxique, à savoir qu'elle diminue la cellularité
médullaire et splénique et qu'elle possède également un pouvoir
immunodépresseur puisqu'elle inhibe la maturation des lymphocytes-B
et bloque l'activité des cellules tueuses naturelles. Les résultats
obtenus indiquent également que les macrophages de la moelle osseuse
pourraient être les principales cibles des effets myélotoxiques
exercés par l'hydroquinone. Cependant, une étude biologique au long
cours sur des rongeurs n'a pas révélé l'existence d'effets
myélotoxiques.
Une étude de 90 jours sur des rats au cours de laquelle on a
utilisé une batterie de tests fonctionnels et observationnels, a
montré qu'à des doses respectives de 64 et 200 mg d'hydroquinone/kg,
les animaux étaient pris de tremblements et qu'à la dose de 200
mg/kg, il y avait réduction de l'activité générale. L'examen
anatomopathologique du système nerveux n'a rien donné.
7. Effets sur l'homme
On a fait état de cas d'intoxication consécutifs à l'ingestion
d'hydroquinone seule ou de développateurs photographiques contenant
de l'hydroquinone. Les principaux signes de ces intoxications
étaient les suivants: urines foncées, vomissements, douleurs
abdominales, tachycardie, tremblements, convulsions et coma. On a
également signalé des décès après l'ingestion de développateurs
photographiques à base d'hydroquinone. Lors d'une étude contrôlée au
cours de laquelle des volontaires humains ont ingéré quotidiennement
pendant 3 à 5 mois 300 à 500 mg d'hydroquinone, on n'a pas relevé le
moindre signe pathologique dans le sang et les urines.
L'application cutanée d'hydroquinone dans divers excipients à
des concentrations inférieures à 3% n'a causé que des effets
négligeables sur des volontaires humains appartenant à diverses
races. Cependant, on dispose de rapports selon lesquels des crèmes
destinées à éclaircir la peau et contenant 2% d'hydroquinone ont
produit une leucodermie ainsi qu'une ochronose. L'hydroquinone
(solution aqueuse à 1% ou crème à 5%) peut provoquer des irritations
(érythème ou taches épidermiques). On a également observé des
dermatites de contact d'origine allergique dues à l'hydroquinone.
Une double exposition à de l'air chargé d'hydroquinone et de
quinone entraîne une irritation oculaire, une photophobie, des
lésions de l'épithélium cornéen, voire des ulcères cornéens et des
troubles visuels. On connaît des cas où l'acuité visuelle a
sensiblement baissé. Une irritation peut se manifester à partir de
2,25 mg/m3. Une exposition de longue durée peut faire apparaître
des taches sur la conjonctive et la cornée et provoquer
l'opacification de cette dernière. Après exposition quotidienne
pendant au moins deux ans à 0,05-14,4 mg d'hydroquinone/m3, on a
observé une inflammation et une dyschromie de la cornée et de la
conjonctive; on n'a pas observé de cas graves avant cinq ans au
moins d'exposition. On dispose d'un rapport qui décrit des cas de
lésions cornéennes apparues plusieurs années après cessation de
l'exposition à l'hydroquinone.
On ne dispose pas de données épidémiologiques suffisantes pour
évaluer la cancérogénicité de l'hydroquinone chez l'homme.
8. Effets sur les autres êtres vivants au laboratoire et dans leur
milieu naturel
Pour expliquer le comportement écotoxicologique de
l'hydroquinone il faut se rapporter à ses propriétés
physico-chimiques, et notamment à sa sensibilité à la lumière, au pH
et à l'oxygène dissous. L'écotoxicité de l'hydroquinone, qui est
généralement élevée (par exemple < 1 mg/litre pour les organismes
aquatiques), varie d'une espèce à l'autre.
Les algues, les levures, les champignons et les plantes en
général sont moins sensibles à l'hydroquinone que les autres
organismes habituellement utilisés pour les épreuves toxicologiques.
Toutefois,au sein d'un même groupe toxonomique, la sensibilité des
différentes espèces à l'hydroquinone peut varier d'un facteur 1000.
RESUMEN
1. Identidad, propiedades físicas y químicas y métodos analíticos
La hidroquinona (1,4-bencenodiol; C6H4(OH)2) es una
sustancia cristalina blanca en estado puro, cuyo punto de fusión es
de 173-174 °C. Su peso específico es de 1,332 a 15 °C, y su presión
de vapor, de 2,4 x 10-3Pa (1,8 x 10-5 mmHg) a 25 °C. Es muy
hidrosoluble (70 g/litro a 25 °C), y el logaritmo de su coeficiente
de reparto n-octanol/agua es de 0,59. Por lo que se refiere a los
disolventes orgánicos, su solubilidad varía entre el 57% para el
etanol, y menos del 0,1% para el benceno. La hidroquinona es
combustible si se calienta previamente. Es un agente reductor, que
se oxida reversiblemente transformándose en semiquinona y quinona.
Se pueden obtener muestras de la hidroquinona presente en el
aire capturándola, bien sea en un disolvente o sobre una membrana-
filtro de éster de celulosa mixto.
Para analizar los niveles de hidroquinona se utilizan técnicas
de valorimetría, espectrofotometría o, muy a menudo, cromatografía.
2. Fuentes de exposición humana y ambiental
La hidroquinona está presente en forma tanto libre como
conjugada en bacterias, plantas y algunos animales. Varios países la
producen industrialmente. En 1979, la capacidad mundial total de
producción de esta sustancia superaba las 40 000 toneladas, mientras
que en 1992 fue de aproximadamente 35 000 toneladas. Es ampliamente
utilizada como agente reductor, como medio de revelado fotográfico,
como antioxidante o estabilizador de ciertos materiales que se
polimerizan en presencia de radicales libres, y como producto
químico intermedio en la producción de antioxidantes, agentes
antiozono, productos agroquímicos y polímeros. La hidroquinona se
emplea también en la elaboración de productos cosméticos y
preparados médicos.
3. Transporte, distribución y transformación en el medio ambiente
La hidroquinona presente en el medio ambiente procede de la
actividad del hombre o forma parte de productos naturales de las
plantas y animales.
Debido a sus propiedades fisicoquímicas, la hidroquinona
liberada en el medio penetra sobre todo en los compartimientos de
agua. Se degrada como resultado de procesos tanto fotoquímicos como
biológicos, por lo que no persiste en el medio. No se produce
bioacumulación.
4. Niveles ambientales y exposición humana
No se han hallado datos sobre las concentraciones de
hidroquinona en la atmósfera, el suelo o el agua. Sin embargo, se ha
analizado la hidroquinona presente en la corriente principal de humo
emitida por cigarrillos sin filtro, en la que se han hallado entre
110 y 300 µg por cigarrillo, así como en el humo lateral. Se ha
hallado hidroquinona en alimentos derivados de las plantas (por
ejemplo, el germen de trigo), en el café listo para beber y en los
tes preparados a partir de las hojas de algunas bayas en que la
concentración supera a veces el 1%.
Los aficionados a la fotografía pueden verse expuestos a la
hidroquinona por vía cutánea o por inhalación; sin embargo, no se
dispone de datos sobre niveles de exposición. Otra causa de
exposición cutánea es el uso de productos cosméticos o médicos con
hidroquinona, como los utilizados para aclarar el color de la piel.
Los países de la Comunidad Europea (CE) han restringido su empleo en
los productos cosméticos a un máximo del 2%. En los Estados Unidos,
la Administración de Alimentos y Medicamentos ha propuesto
concentraciones de entre 1,5% y 2% para los productos de maquillaje
decolorante. Algunos medicamentos de venta con receta contienen
concentraciones de hasta un 4%, y en ciertos países se venden
cosméticos decolorantes que contienen concentraciones incluso
mayores.
Son pocos los datos disponibles sobre la hidroquinona por lo
que se refiere a la vigilancia de la higiene industrial. Según se ha
señalado, el valor promedio de las concentraciones que este producto
alcanza en el aire durante su fabricación y transformación oscila
entre 0,13 y 0,79 mg/m3. El límite de la exposición atmosférica
ocupacional (promedio ponderado por el tiempo) oscila entre 0,5 y 2
mg/m3, según los países.
5. Cinética y metabolismo
La hidroquinona es rápida y ampliamente absorbida en el tubo
digestivo y la tráquea de los animales. La absorción cutánea es más
lenta, pero se ve acelerada por vehículos como los alcoholes. La
hidroquinona se distribuye de forma rápida y generalizada por los
tejidos. Es metabolizada en p-benzoquinona y otros productos de
oxidación, y la detoxificación se produce por conjugación,
formándose los derivados monoglucurónido, monosulfato y
mercaptúrico. La excreción de la hidroquinona y de sus metabolitos
es rápida y se produce principalmente a través de la orina.
La hidroquinona y/o sus derivados reaccionan con distintos
componentes biológicos, como macromoléculas o moléculas de bajo peso
molecular, y tienen efectos sobre el metabolismo celular.
6. Efectos en mamíferos de laboratorio y en los sistemas in vitro
Los valores de la DL50 por vía oral en varias especies
animales oscilan entre 300 y 1300 mg/kg de peso corporal. En el
gato, sin embargo, la DL50 varía entre 42 y 86 mg/kg de peso
corporal. La exposición aguda a altos niveles de hidroquinona tiene
efectos graves sobre el sistema nervioso central (SNC), que abarcan
desde la hiperexcitabilidad, pasando por temblores, convulsiones y
coma, hasta la muerte. A dosis subletales esos efectos son
reversibles. Se ha calculado que la DL50 por vía cutánea es de >
3800 mg/kg en roedores. No se dispone de los valores de la CL50
por inhalación.
Un preparado de hidroquinona al 2% aplicado a conejos mediante
una única prueba del parche tuvo unos efectos irritantes de 1,22 (en
una escala de 0 a 4). La aplicación tópica diaria durante tres
semanas de hidroquinona al 2% o 5% en una emulsión de aceite/agua
sobre la piel depilada de cobayos negros provocó despigmentación,
cambios inflamatorios y espesamiento de la epidermis. La
despigmentación fue más marcada con las concentraciones mayores, y
los cobayos hembra se revelaron más sensibles que los machos.
Las pruebas de sensibilización realizadas en cobayos han
provocado respuestas de carácter entre débil y fuerte, según los
métodos o vehículos empleados. Las reacciones más intensas fueron
las observadas en la prueba de maximización aplicada a cobayos. Se
observó también en estos animales una sensibilización cruzada de
casi el 100% entre la hidroquinona y el p-metoxifenol, pero los
indicios de reacción cruzada con la p-fenilendiamina, el ácido
sulfanílico y la p-benzoquinona fueron sólo limitados.
Un estudio de toxicidad oral realizado durante seis semanas con
ratas macho F-344 reveló la aparición de nefropatía y proliferación
de las células renales. En estudios de sobrealimentación forzada
mediante sonda esofágica llevados a cabo durante 13 semanas con
ratas F-344 y ratones B6C3F1, se observaron en las ratas signos de
nefrotoxicidad a dosis de 100 y 200 mg/kg, temblores y convulsiones
a dosis de 200 mg/kg, así como una disminución del aumento del peso
corporal tanto en las ratas como en los ratones. Las dosis de 400
mg/kg resultaron letales para las ratas. En los ratones sometidos
durante 13 semanas a dosis de 400 mg/kg se observaron temblores,
convulsiones y lesiones del epitelio gástrico. La exposición de
ratas Sprague Dawley a hidroquinona durante 13 semanas dio lugar a
una atenuación del aumento del peso corporal y la aparición de
signos de afección del SNC a la dosis de 200 mg/kg. Estos signos
neurológicos se observaron también a la dosis de 64 mg/kg de peso
corporal, pero no así a la de 20 mg/kg.
La hidroquinona inyectada por vía subcutánea redujo la
fecundidad de las ratas macho y prolongó el ciclo menstrual de las
ratas hembra. Esa observación no se reprodujo en cambio en los
estudios de administración oral (un estudio de dominancia letal y
otro efectuado con dos generaciones). En un estudio sobre el
desarrollo de la rata, dosis orales de 300 mg/kg de peso corporal
tuvieron un ligero efecto tóxico en las madres y provocaron una
disminución del peso corporal del feto. En el conejo, el nivel sin
efectos observados (NOEL) por lo que se refiere a la toxicidad
materna fue de 25 mg/kg al día, y de 75 mg/kg al día en lo que
respecta a la toxicidad ontogénica. En un estudio de los efectos
sobre la reproducción realizado con dos generaciones de ratas, la
hidroquinona no tuvo efecto alguno a dosis orales de hasta 150 mg/kg
de peso corporal al día. El nivel sin efectos adversos observados
(NOAEL) fue de 15 mg/kg al día por lo que hace a la toxicidad
materna, y de 150 mg/kg al día en lo referente a los efectos sobre
la reproducción observados a lo largo de dos generaciones.
La hidroquinona tiene un efecto inductor sobre los micronúcleos
in vivo e in vitro. Se han observado aberraciones cromosómicas
estructurales y numéricas in vitro y tras la administración
intraperitoneal in vivo. Se ha demostrado además la inducción de
mutaciones genéticas, intercambio de cromátides hermanas y lesiones
del ADN in vitro. La hidroquinona causó aberraciones cromosómicas
en las células germinales de ratones macho, del mismo orden de
magnitud que en las células de médula ósea de esa misma especie tras
inyección intraperitoneal. En una prueba de dominancia letal llevada
a cabo con ratas macho tratadas por vía oral no se observó ningún
efecto de inducción de mutaciones de las células germinales.
En un estudio de dos años de duración, la administración oral
de hidroquinona provocó una incidencia dosisdependiente de adenomas
de las células tubulares renales en ratas macho F-344/N. La
incidencia fue estadísticamente significativa en el grupo tratado
con la dosis más alta; en los machos sometidos a esa dosis se halló
también hiperplasia de las células tubulares renales. En las ratas
hembra se observó un aumento dosisdependiente de la incidencia de
leucemia de células mononucleares. En los ratones hembra B6C3F1 se
observó una incidencia significativamente mayor de adenomas
hepatocelulares. En otro estudio, la hidroquinona (presente a un
nivel del 0,8% en la ingesta alimentaria) provocó un aumento
significativo de la incidencia de hiperplasia epitelial de la papila
renal y un aumento significativo de la incidencia de adenomas e
hiperplasia de los túbulos renales en las ratas macho. En las ratas
hembra no se observó ningún incremento de la incidencia de leucemia
de células mononucleares. En los ratones, la incidencia de
hiperplasia de las células escamosas del epitelio del antro cardiaco
aumentó significativamente en los dos sexos. En los ratones macho se
observó un aumento significativo de la incidencia de adenomas
hepatocelulares, así como de hiperplasia tubular renal. Se observó
también un reducido número de adenomas de células renales.
Los estudios realizados in vivo (inyección intraperitoneal) e
in vitro con ratones han demostrado que el efecto citotóxico de la
hidroquinona se debe a que reduce la celularidad de la médula ósea y
del bazo, y a su posible efecto inmunosupresor, mediado por la
inhibición de la maduración de los linfocitos B y de la actividad
natural de las células asesinas. Los resultados indican además que
las células más afectadas por la mielotoxicidad de la hidroquinona
son quizá los macrófagos de la médula ósea. En una biovaloración
prolongada realizada con roedores no se observaron esos efectos
mielotóxicos.
En un estudio realizado durante 90 días con ratas mediante una
batería de pruebas funcionales y de observación, se advirtieron
temblores a dosis de 64 y 200 mg de hidroquinona/kg, dosis esta
última que además provocó una disminución de la actividad general.
El resultado de los análisis neuropatológicos fue negativo.
7. Efectos en el hombre
Se han notificado casos de intoxicación tras la ingestión oral
de hidroquinona sola o de productos de revelado fotográfico que
contenían dicho producto. Los principales signos de intoxicación
fueron el oscurecimiento de la orina, vómitos, dolor abdominal,
taquicardia, temblores, convulsiones y coma. Se han notificado
muertes provocadas por la ingestión de productos de revelado
fotográfico que contenían hidroquinona. En un estudio controlado
realizado con voluntarios, la ingestión de 300-500 mg de
hidroquinona diarios durante 3-5 meses no produjo cambios
patológicos observables en la sangre y la orina.
La aplicación cutánea de distintas bases que contenían
concentraciones de hidroquinona inferiores al 3% tuvo efectos
insignificantes en hombres voluntarios de distintas razas. Sin
embargo, se han notificado algunos casos que llevan a pensar que hay
cremas cutáneas decolorantes con hidroquinona al 2% que han
provocado la aparición de leucoderma, así como de ocronosis. Se han
producido casos de irritación (eritema o coloración) por
hidroquinona (en solución acuosa al 1% o en forma de crema al 5%).
También se han diagnosticado casos de dermatitis alérgica de
contacto por hidroquinona.
La exposición simultánea a hidroquinona y quinona presentes en
el aire causa irritación ocular, sensibilidad a la luz, lesiones del
epitelio corneal, úlceras corneales y trastornos visuales. En
algunos casos se han producido pérdidas considerables de visión. Se
han observado efectos irritantes a niveles de exposición de 2,25
mg/m3 o más. La exposición prolongada provoca coloración de la
conjuntiva y la córnea, así como opacidad. La exposición diaria
durante al menos dos años a concentraciones de hidroquinona de 0,05-
14,4 mg/m3 ha dado lugar al lento desarrollo de inflamación y
decoloración de la córnea y la conjuntiva; pero no se han observado
casos graves hasta transcurridos al menos 5 años. En un estudio se
describieron casos de aparición de lesiones de la córnea al cabo de
varios años de interrumpida la exposición a la hidroquinona.
No se dispone de datos epidemiológicos adecuados para evaluar
la carcinogenicidad de la hidroquinona en el hombre.
8. Efectos en otros organismos en el laboratorio y sobre el terreno
Los efectos ecotoxicológicos de la hidroquinona guardan
relación con sus propiedades fisicoquímicas, entre ellas su
sensibilidad a la luz, al pH y al oxígeno disuelto. Su ecotoxicidad,
que por lo general es alta (por ejemplo, < 1 mg/litro para los
organismos acuáticos), varía de una especie a otra.
Las algas, levaduras, hongos y plantas son menos sensibles
a la hidroquinona que los otros organismos empleados habitualmente
para evaluar la toxicidad. No obstante, dentro de un mismo grupo
taxonómico, la sensibilidad de las distintas especies a la
hidroquinona puede variar de 1 a 1000.
6.4 Elimination and excretion
Hydroquinone is excreted mainly in the form of water soluble
metabolites via the urine (about 90%). Dose-related differences have
been observed for rats receiving 25 or 350 mg/kg, which suggests
that elimination processes are saturated at high-dose levels
(English et al., 1988). The area under the curve (AUC) values for
plasma concentration, which provide an index of bioavailability,
also showed that saturation of elimination had occurred at high-dose
levels, particularly for females. The fact that most of the
radioactivity excreted is associated with the alpha-elimination
phase suggests that this may be due to conjugation of hydroquinone
to readily excreted metabolites. The appearance of a double peak in
the blood concentration versus time curve indicates that
enterohepatic recycling of hydroquinone may have occurred.
6.5 Reaction with body components
The available studies suggest that hydroquinone derivatives are
responsible for many of the toxicological effects associated with
in vivo and in vitro hydroquinone exposure. Hydroquinone itself
may be responsible for the acute CNS signs (tremors and convulsions)
that are seen within the first hour following hydroquinone exposure
(see section 7.8.3), since the signs appear soon after exposure when
significant metabolism has probably not occurred. However, it is
possible that derivatives even have a role in inducing CNS effects.
The derivatives formed from hydroquinone may differ between in
vivo and in vitro studies. Even when the in vivo situation
alone is considered, the derivatives may vary qualitatively and
quantitatively, and the concentrations of derivatives in the various
body compartments may be different depending on the route of
exposure. When hydroquinone is administered by expected routes of
exposure, the primary derivatives should be largely glucuronide and
sulfate conjugates, which are quickly exported, as well as
glutathione conjugates, which may represent activated metabolites.
When hydroquinone is given by intraperitoneal or intravenous routes,
the primary metabolites are expected to be 1,4-benzoquinone and
1,2,4-trihydroxybenzene. In most in vitro systems the primary
metabolite is expected to be 1,4-benzoquinone. Hydroquinone-and
oxygen-derived radical species are also likely to be formed both in
vivo and in vitro. The hydroquinone-derived metabolites and
radical species formed in vitro will depend on the oxygen content,
the pH, the ionic strength, the autooxidant and the protein content
of the culture or reaction medium used in the study, as well as
other factors including the metabolic capacity of the test system.
The differences in the potential derivatives and the
concentrations of the derivatives occurring in the different in
vivo and in vitro exposure systems studied indicate that
extrapolations from in vitro to in vivo systems and between
routes of exposure need to be made with a great deal of care.
The main results pertinent to this section have been summarized
in Table 9, which shows that many of the interactions of
hydroquinone have been identified in vitro but not all have been
demonstrated in vivo. Hydroquinone reacts with many different
biological components, including macromolecules such as protein,
DNA, tubulin, lipids, and low molecular weight molecules such as
sulfydryls and nucleotides, is toxic to different cell types, has
affects on cellular metabolism, and modulates enzyme activities.
Covalent binding and oxidative stress are mechanisms postulated
to be associated with hydroquinone-induced toxicity. Both oxidized
hydroquinone species ( p-benzosemiquinone radical and
p-benzoquinone) and thiol-hydroquinone/quinone conjugates are
believed to contribute to hydroquinone toxicity.
Oxidized hydroquinone derivatives can covalently bind cellular
macromolecules or alkylate low molecular weight nucleophiles, e.g.,
glutathione (GSH), resulting in enzyme inhibition, alterations in
nucleic acids and oxidative stress; however, redox cycling is not
likely to contribute significantly to oxidative stress in contrast
with other hydroquinones and quinones (see section 2.2; Rossi et
al., 1986; O'Brien, 1991). The reaction of benzoquinone with GSH
results in the formation of GSyl conjugates which can be processed
to cysteine conjugates. These latter thiol conjugates have been
speculated to mediate cellular toxicity in the kidney by alkylation
and/or oxidative stress, possibly involving redox cycling (Lau et
al., 1988).
Table 9. Summary of the reactions of hydroquinone (HQ) with biological componentsa
Index studied Method Result Reference
Reactions with macromolecules
Covalent binding to 14 mg/kg [14C]-HQ was administered acid-insoluble radioactivity associated with Greenlee et al.
cell protein i.v. as a single dose to Fischer-344 rats protein increased with time in the bone (1981b)
(in vivo) (m); after 2 and 24 h, acid-insoluble marrow > thymus > liver; pretreatment with
radioactivity associated with proteins Aroclor resulted in a significant decrease in
was determined; one group of rats was the radioactivity measured in the bone marrow
pretreated with Aroclor 1254
Covalent binding to 25-75 mg/kg [14C]-HQ was incubated radioactivity associated with protein; the Eastmond et al.
boiled rat liver in the absence or presence of phenol presence of PhOH enhanced this association (1987)
protein (in vitro) (PhOH) (75 mg/kg) with H2O2-horseradish
peroxidase or freshly isolated human
polymorphonuclear leucocytes in the
presence of boiled rat liver protein
Covalent binding to 75 mg/kg [14C]-HQ alone or coadministered after 18 h of administration, acid-insoluble Subrahmanyam et
cells (in vivo) with phenol (PhOH) (75 mg/kg) radioactivity was found associated with al. (1990)
was administered i.p. (probably as a kidney > blood > bone marrow; coadministration
single dose) to pathogen-free male with PhOH significantly (statistically)
B6C3F1 mice (5-12); after 4 and 18 h, increased binding to blood and bone marrow
acid-insoluble radioactivity associated but not kidney or liver
with macromolecules was isolated and
analysed for covalent binding
Covalent binding to isolated liver microsomes from male radioactivity associated with microsomal Wallin et al.
microsomal proteins S-D rats, either treated or untreated with proteins; binding was more extensive than (1985)
(in vitro) phenobarbital or 3-methyl cholanthrene, that of phenol and independent of electron
was incorporated with [14C]-HQ, both with donors
and without NADPH
Table 9. (contd).
Index studied Method Result Reference
Covalent binding to peritoneum macrophages isolated from [14C]-HQ was activated by macrophages to Schlosser et
cells (in vitro) male C57BL/6 mice were incorporated metabolites that bind irreversibly to protein; al. (1989);
with [14C]-HQ activation was inhibited by peroxidase inhibitor Schlosser &
aminotriazine and the nucleophile cysteine and Kalf (1989)
enhanced by arachidonic-acid-mediated
prostagladin synthesis catalysed reaction
Covalent binding to bone marrow macrophages and a fibroblastoid radioactivity associated with macrophages was Thomas et al.
cells (in vitro) stromal cell (LTF) line obtained 16-fold higher than for LTF cells; DT- (1989); Ross et
from male B6C3F1 mice were incorporated diaphorase activity [Q -> HQ] was 4 times al. (1990)
with [14C]-HQ higher on LTF cells than in macrophages;
slightly decreased (approx.16%) by addition of
dicoumarol, an inhibitor of DT-diaphorase,
for LTs but not macrophages
Chromosomal aberration example: bone marrow cells were isolated micronuclei induced in polychromatic Tunek et al.
(in vivo) (see also from male NMRI mice (4 per group) erythrocytes (1982)
section 7.6) administered between 20 and 100 mg HQ/kg
by subcutaneous injection once a day for
6 days
Mitochondrial DNA mitoplasts isolated from rabbit bone covalent adduct formed with guanine Rushmore et
(in vitro) marrow cells were prelabelled with al. (1984)
[3H]-dGTP incorporated with [14C]-HQ and
assayed for guanosine adduct formation
DNA damage (in vitro) example: calf thymus DNA was incubated two adducts identified Jowa et al.
(see also section 7.6) with [14C]-HQ in the presence of Fe3+ at (1990)
pH 7.2
Microtubulin binding T1: isolated brain microtubulin from male T1: HQ inhibited microtubulin polymerization Irons & Neptun
(in vitro) Fischer-344 rats was incubated with between and bound to high molecular weight tubulin; (1980)
1 and 1.5 x 10-4 mol/litre [14C]-HQ anaerobic conditions inhibited polymerization
Table 9. (contd).
Index studied Method Result Reference
T2: isolated spleen lymphocytes from rat T2: HQ suppressed lectin-induced blastogenesis Pfeifer & Irons
were incubated with HQ (10-6-10-4 mol/litre) and concomitant inhibition of cell (1983)
agglutination
Lipids (in vivo) SD rats received a single oral dose of 100 urinary MDA increased in HQ-treated rats Ekström et
or 200 mg HQ/kg; malondialdehyde (MDA), al. (1988)
a lipid peroxidation product, was analysed
in the excreted urine for up to 18 h
Cytochrome c3+ stop- and continuous-flow experiments HQ reduces cytochrome c3+ via Yamazaki & Ohnishi
reduction (in vitro) p-benzosemiquinone; reaction accelerated by (1969); Ohnishi
p-benzoquinone et al. (1969)
Reactions with low molecular weight molecules
Thiol conjugation glutathione thiol conjugates formed by reductive addition; Tunek et al.
(in vitro) monothiol HQ conjugate formed by the reaction (1980)
of p-benzoquinone with thiol after oxidation of
HQ to the semiquinone or quinone
glutathione di, tri and tetra (GSyl)-HQ conjugates are Eckert et al.
formed by reductive addition of the oxidized (1990)
(GSyl)-HQ conjugate, i.e. quinone conjugate with
GSH
monocysteine-HQ conjugate HQ oxidized by prostaglandin H systhetase Schlosser et
al. (1990)
Nucleotide adduct [3H]-deoxyguanosine and [14C]-HQ two doubly labelled products isolated; adduct Jowa et al.
(in vitro) incubated in the presence of Fe3+ at pH 7.2 2: 3-OH benzethano (1, N2) deoxyguanosine (1990)
2-Thiobarbituric glutamate or deoxyribonucleic acid 2-thiobarbituric acid produced; hydroxyl radical Rao & Pandya
acid (in vitro) incorporated with HQ plus Cu2+ at pH 7.4 (OH) formation thought to be involved (1989)
Table 9. (contd).
Index studied Method Result Reference
Toxicity to cells
Erythrocytes (in vivo) polychromatic erythrocytes isolated from 20 mg/kg: no haemotoxic effect; 100 mg/kg: Tunek et al.
male NMRI mice (4 per group) haemotoxic effects (suppressed bone marrow (1982)
administered between 20 and 100 mg cellularity)
HQ/kg s.c. once a day for 6 days
Bone marrow (in vivo) i.p. administration of HQ (100 mg/kg, twice transient, mild suppression in bone marrow Eastmond et
daily for 12 days to six male B6C3F1 mice) cellularity al. (1987)
i.p. co-administration of HQ (25-75 mg/kg) significant decrease in bone marrow cellularity; Eastmond et
and phenol (75 mg/kg) twice daily for phenol enhanced HQ-induced myelotoxicity al. (1987)
12 days to groups of six male B6C3F1 mice
Isolated rat spleen responses of spleen cells from F-344 rats low concentrations (10-7-10-6) enhanced Irons & Pfeifer
and lymphocytes were assayed after addition of mitogen and mitogenesis, higher concentrations (10-5) (1982)
(in vitro) phytohaemagglutinin A suppressed mitogen response
Pigment cells (in vitro) toxic effects of HQ on melanotic cell lines toxic effects occurred between 0.625 and HU (1966)
(MCL) and non-melanotic cell lines (NMCL) 2.5 µg/ml for MCL and NMCL
was studied
Cell line (in vitro) lymphoma-derived cell line Raji, erythro- percentage survival decreased for all cells Picardo et al.
leukaemia cell line K 562 and human (approx. 65%, low dose) (approx. 20%, high dose) (1987)
melanotic cell lines IRE 1 and IRE 2 were
incubated with 0.01 and 0.1 mmol HQ/litre
Bone marrow cells bone marrow cells isolated from the femurs HQ decreased the number of mature surface King et al.
(in vitro) and tibias of male B6C3F1 (C57BL/6J x IgM+ B cells and adherent cells; HQ may block (1987)
C3h/HeJ) mice were incubated with final maturation stages of B cell
between 10-7 and 10-5 mol HQ/litre differentiation
Table 9. (contd).
Index studied Method Result Reference
Cell line (in vitro) bone marrow macrophage and a fibroblastoid HQ (10-4 mol/litre) decreased viability and Thomas et al.
stromal cell line isolated from male colonies for macrophages (60% and 70%, (1989b)
B6C3F1 mice were incubated with respectively) and stromal cells (30% and
between 10-8 and 10-4 mol HQ/litre 0%, respectively)
Cell line (in vitro) bone marrow stromal cells isolated from HQ cytotoxicity was greater in stromal cells Twerdok & Trush
male DBA/2 mice and C57BL/6 mice derived from DBA/2 than C57BL/6 mice; tert- (1990); Twerdok
were incubated with HQ butylhydroquinone (tBHQ) or 1,2-dithiole-3- et al. (1992)
thione (DTT) preincubation protected against
HQ-induced toxicity; dicoumarol-sensitive
quinone reductase activity was increased by
tBHQ and DTT, and levels of GSH increased with
DTT
Cell line (in vitro) human promyelocytic leukaemia cell line, HQ dose-dependently inhibited TPA- and 1,25- Oliveira & Kalf
which can be induced to differentiate to dihydroxy vitamin D3-induced (but not (1992)
both monocyte and myeloid cells, was interleukin (IL)-1) acquisition of
incubated with HQ (0.01 µmol/litre to differentiation characteristics of monocytes
10 µmol/litre) (adherence, nonspecific esterase activity and
phagocytosis), but had no effect on cell
proliferation; retinoic-acid- or DMSO-induced
differentiation to granuloctyes was not
inhibited at the same doses
Hepatocytes (in vitro) freshly isolated hepatocytes (106/ml) time-dependent cell death; complete by 120 min O'Brien (1991)
incubated with 850 µM HQ; % cell viability
measured by Trypan blue inclusion
Effects on cellular metabolism
Haemoglobin (Hb) five cats were treated every second day up 15-30% Hb oxidized to ferric form Jung & Witt
(in vivo) to 12 times with HQ (40-160 mg/kg) (1947)
Table 9. (contd).
Index studied Method Result Reference
Reduction of haemoglobin HQ incubated with Hb in presence of O2 very slow formation of ferrihaemoglobin Oettel (1936)
(Hb) (in vitro)
Iron utilization female Swiss albino mice administered inhibition (70%) of erythroid 59Fe utilization Guy et al.
(in vivo) between 25 and 100 mg HQ/kg 3 times at occurred only at the highest dose; (1990, 1991)
64, 48 and 40 h prior to administration coadministration of PhOH or muconaldehyde
of 59Fe; coadministration with phenol enhanced the inhibitory effects of HQ
(PhOH) (50 mg/kg)
Cellular RNA and DNA the effects of HQ on nucleoside HQ selectively inhibited the metabolism of MCL Pennay et al.
synthesis (in vitro) incorporation in two melanotic cell lines cf. NMCL; [3H]-uridine incorporation was de- (1984)
(MCL) and three non-melanotic cell lines creased approx. 30-fold in MCL cf NMCL; [3H]-
(NMCL) was observed uridine incorporation was more sensitive to HQ
than [3H]-thymidine; DNA and RNA syntheses were
decreased by 80 and 50%, respectively, in MCL;
HQ may exert depigmenting effect by selective
action on MCL metabolism rather than specific
effect on melanin synthesis
Mitochondrial synthesis mitoplasts isolated from rabbit bone RNA synthesis inhibited (IC50 = 5.0 x 10-5 mol Rushmore
marrow cells were incorporated with HQ HQ/litre) et al. (1984)
and assayed for RNA synthesis
Cyclic nucleotides three different melanomas were treated cAMP and cGMP were elevated in 3/3 and 2/3 Abramowitz &
(in vitro) with HQ and assayed for cAMP and cGMP tumours, respectively Chavin (1980)
by radioimmuno assay
Cytokine synthesis murine P388D1 macrophages or bone HQ caused a concentration-dependent inhibition Renz et al.
marrow stromal macrophages were of the processing of 34-Kd pre-interleukin-1 (1991)
incubated with HQ (0.5-10 µmol/litre) alpha (IL-1alpha) to 71-Kd mature cytokine in
both types of macrophages; lipopolysaccharide-
induced production of the pre-IL-1alpha
precursor or cell viability or DNA and protein
synthesis were not inhibited
Table 9. (contd).
Index studied Method Result Reference
Effects on enzymes
Tyrosine-tyrosinase radiometric assay tyrosinase inhibited by HQ (9 x 10-4 mol/litre); Usmani et al.
(tyrosine --> dopa) suggested that HQ is a competitive inhibitor (1980); Palumbo
(required for skin et al. (1991)
pigmentation)
Catalase (in vivo) male Wistar rats received 5 mg HQ/kg per 5 mg HQ/kg per day inhibits catalase activity in Vladescu &
day p.o. for 10 days and H 18R tumour- the liver, spleen, blood and H 18R tumour Apetroae (1983)
bearing rats received 5 mg HQ/kg per day
p.o. for 7 days; [14C]-HQ was
administered i.p.
Ornithine decarboxylase SD rats received a single oral dose of 100 ODC activity was increased in a dose- Ekström et al.
(ODC) or 200 mg HQ/kg; liver ODC activity was dependent manner (1988)
measured after 18 h of exposure
Horseradish peroxidase [14C]-phenol incubated with H2O2-HRP in radioactivity associated with protein Eastmond et
(HRP) the presence of boiled rat liver protein; decreased when coincubated with HQ; al. (1987)
coincubated with HQ competitive inhibition presumed
Table 9. (contd).
Index studied Method Result Reference
Reaction of HQ metabolites
Benzoquinone (BQ)/ numerous: BQ/SQ formed by autooxidation rate of enzyme-mediated formation of BQ is Yamazaki et al.
benzosemiquinone (SQ) and 1e-mediated enzymic oxidation (e.g., much faster than autooxidation; formation of (1960); Sawada
horseradish peroxidase (HRP), BQ is enhanced by coincubation of HRP and et al. (1975);
myeloperoxidase (MPO) and prostaglandine MPO with phenol and other compounds; O'Brien (1991);
H synthase (PHS)) of HQ indomethacin partially inhibits H2O2-dependent Smith et al.
HQ oxidation with HRP or MPO or PHS, but (1989); Hsuanyu
substantially inhibits arachidonate-dependent & Dunford (1992);
oxidation mediated by PHS; this latter finding Eastmond et al.
indicates the involvement of cyclooxygenase; (1987); Thomas
both BQ and SQ are electrophiles which readily et al. (1989);
react with low and high molecular weight Subrahmanyam et
nucleophiles; BQ is believed to be the al. (1991);
proximate toxicant responsible for benzene-/ Schlosser
hydroquinone-induced myelotoxicity et al. (1990)
GSH conjugate (in vivo) male Sprague-Dawley rats were administered nephrotoxicity occurred with tris-(GSyl)-HQ > Lau et al.
various (GSyl)-HQ conjugates i.v.; di-(GSyl)-HQ conjugate; mono and tetra (1988)
nephro- and hepatotoxicity were measured conjugates were not toxic; toxicity of the tris
as increased plasma blood urea nitrogen conjugate was depressed by AT-125, a gamma
(BUN) and serum glutamate pyruvate glutamyl trans-peptidase (gamma-GT) inhibitor;
transaminase (SGPT), respectively it is suggested that gamma-GT is probably
required for the transport of the latent quinone
into proximal tubular cells as the corresponding
cysteine conjugate; alkylation and/or oxidation
are suggested mechanisms of molecular toxicity
Table 9. (contd).
Index studied Method Result Reference
GSH conjugate (in vitro) O2 consumption measurement of in situ autooxidation was stimulated by SOD Brunmark &
formed glutathionyl-p-benzoquinone HQ Cadenas (1988); cf.
conjugate in potassium phosphate buffer Eckert et al.
at pH 7.65 in the absence and presence of (1990)
superoxide dismutase (SOD)
a NADPH = reduced nicotinamide adenine dinucleotide
cAMP = cyclic adenosine monophosphate
cGMP = cyclic guanosine monophosphate
7. EFFECTS ON LABORATORY MAMMALS AND IN VITRO SYSTEMS
7.1 Single exposure
The acute toxicity of hydroquinone has been studied in several
animal species (Tables 10 and 11). Oral LD50 values for different
strains of rats range from 720 to 1300 mg/kg body weight (Carlson &
Brewer, 1953; Mozhaev et al., 1966). Fasting the animals for 18 h
previous to the administration of hydroquinone produced a two- to
three-fold increase in the observed toxicity (Carlson & Brewer,
1953). The LD50 of unfed rats was 310 mg/kg, in contrast to an
average of 1064 mg/kg observed in non-fasted animals. Oral LD50
values range from 340 mg/kg to 400 mg/kg body weights for mice, and
are 550 mg/kg body weight for guinea-pigs, 540 mg/kg body weight for
rabbits and 299 mg/kg body weight for dogs. Cats have a greater
sensitivity (LD50 values of 42-86 mg/kg body weight).
Signs of hydroquinone intoxication were developed 30-90 min
after single oral doses and consisted of hyperexcitability, tremors,
convulsions, dyspnoea and cyanosis. They also included salivation in
dogs and cats, emesis in dogs and pigeons, swelling of the tissues
around the eyes, and incoordination of the hind limbs of dogs
(Woodard 1951; Christian et al., 1976; Deichmann & Keplinger,
1981). These signs were followed by complete exhaustion,
hypothermia, paralysis, loss of reflexes, coma, respiratory failure
and death. When the dose was sublethal, recovery was complete within
three days (Christian et al., 1976).
Single-dose acute dermal toxicity studies have not been
reported. However, the acute dermal LD50 can be estimated to be >
3840 mg/kg for mice and 74 800 mg/kg for rats based on effects
observed in two-week dermal studies (NTP, 1989). Hydroquinone as a
2% solution in dimethyl phthalate caused no adverse local or
systemic effects in rabbits (Draize et al., 1944).
In the rat, the LD50 values for intraperitoneal
administration varied between 160 and 194 mg/kg body weight and the
LD50 for intravenous administration was 115 mg/kg body weight
(Woodard, 1951). In rabbits, intravenous injections of hydroquinone
caused death at doses of 100-150 mg/kg body weight (Delcambre et
al., 1962).
Table 10. Acute oral toxicity of hydroquinone in experimental animals
Species Material tested LD50 (mg/kg Comments Reference
body weight)
Rat 2% aqueous solution 320 rapid onset of symptoms: twitchings, Woodard (1951)
Osborne-Mendel tremors, convulsions, respiratory
failure and death within a few hours
Rat
Priestly glycerine 1005-1295 unfasted rats; the mean LD50 value Carlson & Brewer (1953)
Sprague-Dawley propylene-glycol 1090 was 1050 mg/kg
Sprague-Dawley distilled water 1182
Sprague-Dawley glycerine 1081
Wistar propylene-glycol 731
Sprague-Dawley propylene-glycol 323 fasted rats; the mean LD50 value Carlson & Brewer (1953)
Wistar 298 was 310 mg/kg
Rat water 743 (m) when the dose was sufficiently large, Christian et al. (1976)
627 (f) death occurred during a severe tonic
spasm within 2 h; when the dose was
sublethal, recovery was complete
within 3 days
Mouse 2% aqueous solution 400 the symptoms are similar to those Woodard (1951)
Swiss in rats
Guinea-pig 2% aqueous solution 550 the symptoms are similar to those Woodard (1951)
in rats
Cat 2% aqueous solution 70 the symptoms are similar to those in Woodard (1951)
rats except for salivation and swelling
of the area around the eye noted in cats
Cat sugar-coated tablets 42-86 Carlson & Brewer (1953)
Table 10. (contd).
Species Material tested LD50 (mg/kg Comments Reference
body weight)
Dog sugar-coated tablets 299 similar symptoms to those of cats Carlson & Brewer (1953)
Dog 2% aqueous solution 200 hyperexcitability, tremors, convulsions, Woodard (1951)
salivation, emesis, incoordination of the
hind legs, respiratory failure, and death;
100 mg/kg caused mild to severe swelling
of the area around the eye, of the nictating
membrane and of the upper lip
Rabbit 2% aqueous solution 540 the symptoms are similar to those in rats Woodard (1951)
Pigeon 2% aqueous solution 300 the symptoms are similar to those in Woodard (1951)
rats, except for emesis noted in pigeons
Table 11. Acute parenteral toxicity of hydroquinone in experimental animals
Species Material Administration LD50 (mg/kg body Comments Reference
tested route weight) (except
where otherwise
stated)
Rat (Osborne-Mendel) 2 or 3% aqueous intraperitoneal 160 Woodard (1951)
solution
Rat (Wistar) intraperitoneal 194 Delcambre et al. (1962)
Rat (Osborne-Mendel) 2 or 3% aqueous intravenous 115 Woodard (1951)
solution
Rabbit intravenous 150a death Delcambre et al. (1962)
100 tremor, hypotonia, death
10-20 hypertension, hyperkalaemia
Mouse subcutaneous 160-170 (LLD)b Busatto (1940)
Mouse 1% solution subcutaneous 182 Marquardt et al. (1947)
Mouse subcutaneous 190 most animals died within 24 h Nomiyama et al. (1967)
a Single dose
b LLD = lowest lethal dose
Single subcutaneous injections of up to 500 mg of
hydroquinone/kg body weight in white mice caused symptoms in the
central nervous system: markedly increased motor activity,
hyperactive reflexes, sensitivity to light and sound, laboured
breathing and cyanosis, followed by clonic convulsions, complete
motor exhaustion, paralysis, a nearly complete loss of sensitivity
and reflexes, semicoma and death (Busatto, 1940). The lowest lethal
dose was 160-170 mg/kg. The subcutaneous LD50 for hydroquinone has
been found to be 182-190 mg/kg in mice (Marquardt et al., 1947;
Nomiyama et al., 1967).
No experimental data on inhalation exposure have been located.
7.2 Skin and eye irritation; sensitization
7.2.1 Skin irritation
Hydroquinone was applied to both epilated and unepilated skin
of eight black guinea-pigs at concentrations of 1, 3, 5, 7 and 10%
in three vanishing creams (Bleehen et al., 1968). The number of
animals per dose group was not reported. Six animals served as
controls. The material was applied once daily, five times a week,
for one month. Hydroquinone was irritating only at concentrations of
5% or more. Weak to moderate depigmentation occurred in all areas of
skin to which creams containing 1-10% hydroquinone were applied.
Hydroquinone (0.001, 0.01 and 0.1% in 0.1 ml water) was not
found to be a primary irritant when administered intracutaneously in
18 female guinea-pigs during a period of 10 days (Rajka & Blohm,
1970).
Jimbow et al. (1974) reported depigmentation in the epilated
skin of 24 black guinea-pigs (both males and females) after topical
applications of hydroquinone. Creams containing 2 or 5% hydroquinone
in an oil-water emulsion were applied daily, 6 days a week, for 3
weeks. The depigmentation was first seen within 8-10 days and was
greatest between 14 and 20 days. It was more marked at the higher
concentration. Inflammatory changes and thickening of the epidermis
were also reported. When hydroquinone was applied topically for
three weeks, biopsy specimens showed that it had caused a marked
reduction both in the number of melanized melanosomes in the cells
and the number of actively functioning melanocytes.
In a preliminary screening study with eight guinea-pigs, an
aqueous solution of hydroquinone was slightly irritating at 10% but
not at 0.5, 1.0 or 5.0% (Springborn Institute for Bioresearch,
1984).
The potential of hydroquinone to produce skin depigmentation
and irritation has also recently been studied in male and female
black guinea-pigs (Maibach & Patrick, 1989). The study indicated
that females may be more sensitive than males. Groups of five male
and five female animals were administered hydroquinone (0.1 ml in a
hydrophilic ointment) at concentrations of 0.1, 1.0, and 5.0% on an
epilated area of the back five days a week for 13 weeks. The lowest
concentration caused marginal irritation without depigmentation,
while the medium concentration resulted in a slight to marginal
irritation in 30% of the animals (mainly females) and to weak
depigmentation in females. Moderate to severe irritation and severe
ulcerated inflammatory responses occurred with the highest
concentration. Moderate depigmen-tation was observed in
approximately 40% of the animals dosed (only females).
Hyperpigmentation was noticed in 80-100% of the animals in all dose
groups, but this was not considered to be attributable to the
treatment.
7.2.2 Eye irritation
Powdered hydroquinone (2-5 mg) instilled twice daily (5 days
per week for 9 weeks) into the eyes of dogs caused immediate but
transient irritation and lacrimation (Dreyer, 1940). Opacity of the
cornea, lacrimation and redness of the conjunctiva were produced
within 4 days, but no ulcers were seen. The eye returned to normal
within two days after cessation of treatment. In guinea-pigs
powdered hydroquinone (1-3 mg, twice daily for 9 weeks) also caused
immediate but transient irritation. During the second day of
application a slight corneal opacity was observed in some animals
and on the third day opacity of varying degrees occurred in most of
the animals. Ulcers appeared in two animals. The eyes had fully
recovered 3 days after cessation of treatment.
Following an injection of 0.1 ml of a solution (vehicle not
specified) of hydroquinone (0.012-0.05 mol/litre) into the cornea of
rabbits, the resultant reaction was graded 5 out of the possible
maximum of 100 (Hughes, 1948).
Finely powdered hydroquinone (amounts not specified) was
applied daily, from 2-4 months, to the eyes of rabbits in 6 groups,
which were, respectively, kept in the dark, in sunlight, in normal
light, irradiated with UV light, or pre-sensitized with
haematoporphyrin and then kept under either reduced light or
sunlight. Most rabbits developed pigmentation, first in the
conjunctiva and then in the cornea. Degenerative alterations of the
corneal parenchyma were also observed. Pigment formation appeared
earlier in animals exposed to light. Older animals seemed more prone
to develop pigment than younger ones. Pigment was deposited in
albino rabbit eyes as well as in those of rabbits with normal
pigmentation (Ferraris de Gaspare, 1949).
Hydroquinone in aqueous solution, e.g., in tears, is oxidized
by air, forming a brown colour partly due to conversion to
1,4-benzoquinone (Grant, 1986). No disturbance of the inner parts of
the eye is known to have been produced by exposure to hydroquinone
(Grant, 1986).
7.2.3 Sensitization
Several sensitization studies have been carried out with
hydroquinone; methods and results are summarized in Table 12.
The skin sensitizing potential of hydroquinone for female
guinea-pigs was investigated by Rajka & Blohm (1970). Hydroquinone
elicited "weak" sensitivity after sensitization with a 0.001%
solution injected intracutaneously and challenge with an equipotent
solution of hydroquinone.
The ability of guinea-pigs to detect known human contact
sensitizers was explored by Goodwin et al. (1981). Sensitization
induced by hydroquinone was "strong" when assayed by the Magnusson &
Kligman maximization test, "moderate" by the single injection
adjuvant test and "weak" by the modified Draize procedure.
Hydroquinone was found to be a "moderate" sensitizer in female
guinea-pigs in both the guinea-pig maximization test and Freund's
complete adjuvant test as performed by Van der Walle et al.
(1982a,b). Hydroquinone produced identical sensitization potentials
in the Freund's complete adjuvant test using induction
concentrations of 0.5 mol/litre and 0.45 µmol/litre. The study also
showed almost 100% cross reactivity of hydroquinone and
p-methoxyphenol. Both substances are used as inhibitors in acrylic
monomers to prevent unwanted polymerization.
More recently, Basketter & Goodwin (1988) used three
sensitization test methods representing both topical and intradermal
routes of application. Groups of 10 guinea-pigs were sensitized by
using the guinea-pig maximization test, a modified single injection
adjuvant test, and a cumulative contact enhancement test. The
sensitization potential of hydroquinone was assessed as "strong",
"weak", and "moderate", respectively, in these three tests.
Subsequent cross-challenges with p-phenylenediamine, sulfanilic
acid, and p-benzoquinone gave only "restricted evidence" of
cross-reactions.
Table 12. Contact allergy predictive tests with hydroquinone in guinea-pigs
Test Induction dose Challenge dose Sensitization Reference
(number positive/number
tested or percentage)
Intracutaneous sensitization 0.001% (0.1 ml, injection) 0.001% (injection) 4/18 Rajka & Blohm (1970)
Guinea-pig 2.0% (0.1 ml, injection) 5.0% (patch) 70%, "strong" sensitizer Goodwin et al. (1981)
maximization test 10.0% (patch) 5.0% (patch)
Guinea-pig 0.5 mol/litre (day 0) (patch) 0.125 mol/litre 5/10 (day 21); Van der Walle
maximization test 1 mol/litre (day 7) (patch) 0.250 mol/litre 5/10 (day 35) et al. (1982a,b)
Guinea-pig 2.0% (0.1 ml, injection) 0.5% (patch) "strong" sensitizer Basketter &
maximization test 1.0% (patch) 0.5% (patch) Goodwin (1988)
Single injection 2.0% (injection) 5.0% (patch) 40%, "moderate" sensitizer Goodwin et al. (1981)
adjuvant test
Modified single injection 2.0% (0.1 ml, injection) 10% (patch) "weak" sensitizer Basketter &
adjuvant test Goodwin (1988)
Modified Draize test 2.5% (injection) 1.0% (injection); 0%a, 30%b, "weak" Goodwin et al. (1981)
20% (application) sensitizer
Freund's complete 5 x 0.45 µmol/litre 0.115 µmol/litre 3/8 (day 21) Van der Walle
adjuvant test (0.1 ml, injection) (patch) 4/8 (day 35); et al. (1982a,b)
5 x 0.5 mol/litre 0.125 mol/litre 4/8 (day 21)
(0.1 ml, injection) (patch) 4/8 (day 35)
Cumulative contact 1.0% (patch) 20% (patch) "moderate" sensitizer Basketter &
enhancement test Goodwin (1988)
a the proportion of guinea-pigs sensitized after one induction treatment
b the proportion of guinea-pigs sensitized after two induction treatments
7.3 Short-term exposure
The short-term toxicity of hydroquinone has been studied in
rats and mice. The effects are summarized in Table 13.
Two groups of rats (14 animals/group, sex and strain not
reported) were fed a diet containing 0 or 5% HQ for nine weeks
(Carlson & Brewer, 1953). Findings at the end of the study consisted
of atrophy of the liver cord cells, adipose tissue, striated muscle
and lymphoid tissue of the spleen, as well as an average decrease of
66% in cellularity of the bone marrow (considered as aplastic
anaemia by the authors). No information was reported on mortality
but the fact that animal lost 46% of their body weight during the
course of the study makes the findings difficult to interpret.
Aqueous solutions of hydroquinone (0, 7.5 or 15 mg/kg, 6 days
per week) administered by oral gavage to male Wistar rats for 40
days resulted in haematological changes, including anisocytosis,
polychromatophilia and acidophilic erythroblasts, at the highest
dose level (Delcambre et al., 1962). Administration of 0, 5 or 10
mg/kg for four months to groups of 15 male Wistar rats resulted in
mortality in the highest dose group; 5 mg/kg was well tolerated.
"Mild hyperplastic and hyperkeratotic areas near the
oesophageal entry" occurred in groups of male and female Wistar rats
fed powdered diets containing 2% hydroquinone. However, there were
some deficiencies in the reporting of experimental design (Altmann
et al., 1985). No other treatment-related changes were found, nor
were there any sex-related changes concerning forestomach lesions.
A two-week oral study on Fischer-344 rats and B6C3F1 mice was
carried out by the National Toxicology Program (NTP, 1989; Kari et
al., 1992). The animals were given hydroquinone in corn oil by
gavage 5 days per week (12 doses over 14 days). Five rats per sex
and dose group were administered 0, 63, 125, 250, 500 or 1000 mg/kg
body weight and five mice per sex and dose group 0, 31, 63, 125, 250
or 500 mg/kg. All rats given 1000 mg/kg died during the dosing
period; deaths were also reported at the 500 mg/kg dose level.
Clinical signs of treatment-related toxicity included tremors and
convulsions at the 500 and 1000 mg/kg dose levels. Tremors,
convulsions and deaths also occurred in mice during the study period
at the 250 and 500 mg/kg dose levels.
Table 13. Effects of short-term exposure to hydroquinone via the oral route
Species Concentration Means of Duration Observation Reference
administration
Rat 5% diet 9 weeks reduced weight; aplastic anaemia; decreased Carlson & Brewer
bone marrow cellularity; atrophy of liver,(1953)
spleen, adipose tissue and striated muscle;
ulceration and haemorrhage of the stomach
mucosa
Rat 500, 750, 1000, stomach tube 12 days increased mortality Carlson & Brewer
1250, 1500, (1953)
1750 mg/kg
Rat 7.5, 15 mg/kg intubation 40 days 15 mg/kg: anisocytosis, polychromatophilia, Delcambre et al.
acidophilic erythroblasts (1962)
Rat 5, 10 mg/kg intubation 4 months 10 mg/kg: deaths (7/15) Delcambre et al.
(1962)
Rat 2% diet 4 weeks; mild hyperplasia and hyperkeratosis Altmann et al.
8 weeks of forestomach (1985)
Rat 20, 64, 200 mg/kg gavage 90 days 64 mg/kg: tremors, reduced activity; Eastman Kodak
200 mg/kg: tremors, reduced activity, reduced Company (1988)
body weight and feed consumption (males)
Rat 63, 125, 250, gavage 14 days 1000 mg/kg: tremors, convulsions andNTP (1989); Kari
500, 1000 mg/kg death (10/10); et al. (1992)
500 mg/kg: tremors, convulsions and
death (1/5 male and 4/5 female)
Table 13. (contd).
Species Concentration Means of Duration Observation Reference
administration
Rat 25, 50, 100, 200, gavage 13 weeks 25 mg/kg: decreased relative liver weight NTP (1989)
400 mg/kg (males); 50 and 100 mg/kg: decreased (males)
and increased (females) relative liver weight;
200 mg/kg: lethargy, decreased body weight gain
and increased relative liver weight; tremors,
convulsions and deaths (females); nephropathy;
inflammation and/or epithelial hyperplasia of the
forestomach; 400 mg/kg: tremors, convulsions
and deaths
Rat 2.5, 25, 50 mg/kg gavage 1,3,6 weeks 50 mg/kg: increased urinary enzyme excretion; English et al. (1992)
renal tubular degeneration/regeneration; and
increased renal tubular cell proliferation in
male F-344 rats; female F-344 rats and male
SD rats: no effects
Mouse 25, 50, 100, 200, gavage 13 weeks 25 and 50 mg/kg: lethargy (males); increased NTP (1989)
400 mg/kg relative liver weight (males); 100 mg/kg: lethargy;
increased relative liver weight (males); 200 mg/kg:
lethargy; increased relative liver weight (males);
tremors (males); lesions in the forestomach (one
female); deaths (males); 400 mg/kg: lethargy,
tremors, convulsions, lesions in the
forestomach, deaths
Mouse 31, 63, 125, 250, gavage 14 days 500 mg/kg: tremors, convulsions and death NTP (1989)
500 mg/kg (4/5 male and 5/5 female);
250 mg/kg: tremors, convulsions and death
(3/5 male)
Hydroquinone in 95% ethanol was dermally applied (12 doses over
14 days) to Fischer-344 rats and B6C3F1 mice (NTP, 1989). Rats
(five per sex and dose group) received 0, 240, 480, 1920 or 3840
mg/kg and mice (five per sex and dose group) 0, 300, 600, 1200, 2400
or 4800 mg/kg. The survival rate was not affected. The only findings
were a 6% lower body weight in male rats administered 3840 mg/kg and
crystals on the skin and fur of animals at 3840 mg/kg. No
histopathological examination of the tissues was carried out.
In a 13-week oral toxicity study, 10 male and 10 female
CD(SD)BR rats (initially, approximately 7 weeks old) in each of four
groups were administered hydroquinone (0, 20, 64 or 200 mg/kg per
day) by gavage on 5 days/week (Eastman Kodak Company, 1988). Brown
urine was seen in rats of both sexes from all dose groups. Males
also showed lower feed consumption; however, this was only
significantly (P < 0.05) reduced during the first week of the
study. Female body weight gain and food consumption were not
significantly altered in any dose group during the study. Signs of
behavioural effects were observed at both the 64 and 200 mg/kg dose
levels (see section 7.8.3). The animals were sacrificed after the
exposure period, and six males and six females from each group were
perfused for neuropathological examination (see section 7.8.3). No
treatment-related changes were observed at gross necropsy.
Administration of 200 mg hydroquinone/kg reduced body weight gain in
male rats so that the final body weight of the treated rats was 7%
less than the mean weight for the controls.
Thirteen-week studies in rodents have also been presented by
the NTP (1989). Groups of 10 males and 10 females of each species
(F-344/N rats, initially 4 to 5 weeks old, and B6C3F1 mice,
initially 5 to 6 weeks old) were administered hydroquinone (0, 25,
50, 100, 200, or 400 mg/kg) in corn oil by gavage, five days per
week. A dose level of 400 mg/kg was lethal to all rats. Tremors and
convulsions were observed in most rats at this dose level and in
several female rats receiving 200 mg/kg. Doses of 100 mg/kg or less
did not cause signs of central nervous system (CNS) stimulation.
Rats receiving 200 mg/kg also showed reduced body weight gain,
nephropathy, and inflammation and/or epithelial hyperplasia of the
forestomach. The kidney lesions in male rats were judged to be more
severe than in females.
In mice, a dose level of 400 mg/kg caused mortality in both
males (8/l0) and females (8/10). In the 200 mg/kg male group two
animals died (one due to gavage error). Lethargy was seen in all
dosed males and in females in the three highest-dose groups of each
sex. Tremors, often followed by convulsions, were noted in the
highest-dose group. Liver-to-body weight ratios for all dosed males
were significantly (P < 0.01) greater than for vehicle controls.
Ulceration, inflammation or epithelial hyperplasia of the
forestomach occurred in mice receiving 400 mg/kg (3/10 males and
2/10 females) and in one female mouse receiving 200 mg/kg.
Male and female F-344 rats were given hydroquinone (0, 2.5, 25
or 50 mg/kg) 5 days/week in water by oral gavage for 1, 3 or 6 weeks
(English et al., 1992). At each time point, 5 rats per sex and
dose group were examined for urinalysis changes, renal tubular cell
proliferation and histopathology. Body and kidney weights were not
altered by hydroquinone exposure. Increased excretion of urinary
enzymes was observed in male F-344 rats given 50 mg/kg as early as
one week into the exposure. The incidence of tubular degeneration
and regeneration was mild in the 50 mg/kg male group, and cell
proliferation was increased by 87%, 50% and 34% in the P1, P2 and P3
tubular segments, respectively. Female rats and males in the lower
dose level groups were not affected by hydroquinone exposure. Male
Sprague-Dawley rats given 50 mg/kg for 6 weeks were also not
affected by hydroquinone exposure.
7.4 Long-term exposure
The effects of long-term exposure to hydroquinone in
experimental animals are shown in Table 14. In addition to the acute
(see section 7.1) and subacute (see section 7.3) toxicity tests,
Carlson & Brewer (1953) carried out a "cumulative" toxicity study
and a series of long-term experiments with rats and dogs. In the
cumulative toxicity study a group of 16 rats (strain and sex not
reported) received 500 mg hydroquinone/kg by stomach tube 101 times
in 151 days. More than 50% of the rats died during the study period.
However, the survivors grew at the same rate as the controls and
were autopsied at the end of the experiment. However, no information
on autopsy findings was reported.
In the first long-term study, four groups of male and four
groups of female Sprague-Dawley rats (23 to 24 days old, 10 rats in
each group) were given a basic diet containing 0, 0.1, 0.5 or 1.0%
hydroquinone (Carlson & Brewer, 1953). In the second experiment the
hydroquinone was heated together with the lard in the feed. Eight
groups of rats (16 to 23 in each group) were fed the basic diet
containing 0, 0.1, 0.25 or 0.5% hydroquinone. In the third
experiment eight groups of rats (20 rats in each group) were given a
basic diet containing 0, 0.1, 0.5 or 1.0% hydroquinone with 0.1%
citric acid added. The experiments lasted for 103 weeks. During the
first month of the study the animals given 0.5 or 1.0% hydroquinone
in their diets gained weight at a slower rate than did control
animals. A similar reduction was not found in the rats given
hydroquinone previously heated with lard before feeding. However,
the final body weights of the dose groups were not significantly
different from those of the controls. Haematological analysis (red
blood cell count, % haemoglobin and differential white blood cell
count) showed no statistically significant deviations from the
control values. The microscopic examinations (liver, omentum,
kidney, spleen, heart, lung, bone marrow, stomach wall, pancreas,
adrenal, subperitoneal and intramuscular abdominal fat) also failed
to reveal compound-related changes. Some of the males and females
(number and dose groups not specified) in each experiment were mated
after six months of dosing to produce two successive litters. The
average numbers of offspring for the two successive litters showed
no difference between experimental and control groups. The offspring
given diets containing 0.1, 0.25 or 0.5% hydroquinone previously
heated with lard grew at the same rate as the controls.
Carlson & Brewer (1953) also studied the long-term effects of
hydroquinone in male and female dogs (four months of age at the
beginning of the study). One dog was maintained on a diet containing
16 mg of hydroquinone/kg per day for 80 weeks, while two dogs
received 1.6 mg of hydroquinone/kg per day for 31 weeks and 40 mg/kg
per day for the succeeding 49 weeks. The compound was administered
in sugar-coated tablets mixed with the food. Two dogs served as
controls. The sex distribution in the different groups was not
reported. In addition, five adult male dogs were fed 100 mg of
hydroquinone/kg per day for 26 weeks. Routine blood and urine
analyses (not specified) were made periodically. After the
experiment, the dogs were killed and autopsied. The dogs given
hydroquinone in their diet from four months of age grew at the same
rate as controls. The adults maintained their body weights.
Haematological analyses and urinalyses showed no differences between
exposed rats and controls. No effects on gross pathology or
histopathology were observed.
Fifteen-month oral toxicity studies on rats and mice were also
included in two-year studies presented by the National Toxicology
Program (NTP, 1989) (see also section 7.). Groups of F-44/N rats and
B6C3F1 mice (64 or 65 males and 65 females of each species) were
administered 0, 25 or 50 mg hydroquinone/kg and 0, 50 or 100 mg/kg,
respectively, in deionized water by gavage 5 days per week. No
compound-related clinical signs were observed during the study
period. At 15 months, ten animals from each group were selected for
haematological and clinical chemical analyses, killed and
necropsied. In male rats significantly (P < 0.01) higher mean
relative kidney and liver weights were observed in the high-dose
group and there was also a compound-related increase in the severity
of nephropathy. For high-dose female rats, the haematocrit value,
haemoglobin concentration and erythrocyte counts were decreased. In
mice the relative liver weights were significantly (P < 0.01)
higher for high-dose males and females than for vehicle controls. A
significantly (P < 0.05) higher relative brain weight was noted for
high-dose female mice and kidney weights were significantly (P <
0.01) increased for dosed females. In dosed males, but not in
females, compound-related lesions in the liver were seen, including
centrilobular fatty changes, cytomegaly and syncytial cells.
Table 14. Effects of long-term oral administration of hydroquinone in experimental animals
Species Concentration Duration Observation Reference
Rat 500 mg/kg body weight 151 days > 50% died Carlson &
(by stomach tube) (101 dosings) Brewer (1953)
Rat 0, 0.1, 0.5, 1.0% 103 weeks decreased weight gain for the first months at 0.5 Carlson &
(in the diet) and 1.0%; final body weights did not differ Brewer (1953)
Rat 0, 0.1, 0.25, 0.5% 103 weeks no adverse effects Carlson &
(heated together with Brewer (1953)
lard in the diet)
Rat 0, 0.1, 0.5, 1.0% + 103 weeks no adverse effects Carlson &
0.1% citric acid (in the diet) Brewer (1953)
Rat 0, 25 or 50 mg/kg in 15 months significantly higher relative kidney weight in high-dose NTP (1989)
deionized water (by gavage) males and increased severity of nephropathy in dosed
males; decreased haematocrit value, haemoglobin
concentration and erythrocyte count in females
Mouse 0, 50 or 100 mg/kg in 15 months significantly higher relative liver weights for high- NTP (1989)
deionized water (by gavage) dose males and females; liver lesions in males
Dog 16 mg/kg per day 80 weeks no adverse effects Carlson &
(in the diet) Brewer (1953)
Dog 1.6 mg/kg per day; 31 weeks no adverse effects during the total experimental Carlson &
40 mg/kg per day 49 succeeding period Brewer (1953)
(sugar coated tablets) weeks
7.5 Reproduction, embryotoxicity and teratogenicity
7.5.1 Effects on male reproduction
In a study by Skalka (1964), hydroquinone was injected
subcutaneously into 16 male rats (100 mg/kg body weight per day for
51 days), while 17 male rats served as controls. The average weights
of the testes, epididymides, seminal vesicles and adrenals were
decreased after the treatment period. The fertility was reduced by
33% in the males and the number of pregnancies in mated females was
reduced by 19% compared with the corresponding results for the
control animals. Histological examinations indicated a disruption in
sperm production. Diminished content of DNA in sperm heads was also
noted.
In 13-week and two-year oral studies in rats and mice, no
effects on testicular or epididymal weights or on the histopathology
of these organs were observed (NTP, 1989). Hydroquinone in corn oil
was given by gavage to groups of males F-344/N rats and to male
B6C3F1 mice for 13 weeks. The dose ranged from 0 to 400 mg/kg body
weight for both animal species. In the two-year studies hydroquinone
in water was given in doses of 0-50 mg/kg body weight to rats and in
doses of 0-100 mg/kg to mice.
In a dominant lethal assay in male rats (CRL:COBS CD (SD) BR)
(see also section 7.6), the males (25 per dose group) were given
doses of 0, 30, 100 or 300 mg hydroquinone/kg by gavage 5 days per
week for ten weeks (Krasavage, 1984b). The controls consisted of two
groups, one positive and one negative. During the 2 weeks
immediately following the final treatment, the males were mated
(1:1) with untreated females. All females were killed on day 14 of
gestation. In the high-dose group (300 mg/kg) the mean body weight
and the feed intake of the males were significantly reduced compared
to the negative controls (P < 0.05). The high-dose males also
exhibited clinical signs of toxicity such as brown urine,
sialorrhoea, swollen eyelids, tremors, convulsions and death. No
compound-related effects were seen on male fertility and no dominant
lethality was observed. There were no compound-related effects on
the reproductive parameters studied in the mated females
(insemination rate, pregnancy rate, mean number of corpora lutea,
implantation sites, viable implants, early and late deaths, and pre-
and postimplantation losses).
In a two-generation study oral administration of hydroquinone
did not appear to affect the reproduction of Fo and F1 parental
rats at dose levels up to 150 mg/kg per day (see also section 7.5.3)
(Bio/dynamics Inc., 1989b). Male fertility indices, mating indices
and pregnancy rates did not differ significantly between the control
and the hydroquinone-treated groups.
7.5.2 Effects on female reproduction
Hydroquinone was shown to affect the rat estrus cycle when
given parenterally (Rosen & Millman, 1955). Three rats were given 10
mg hydroquinone/day subcutaneously for 11 days, and vaginal smears
were used to indicate estrus or diestrus. Following an induction
period of about three days, the estrus cycle was interrupted for 5
days, after which normal cycling was observed.
Similar results were obtained in a study performed by Racz et
al. (1958). One group of rats was given 200 mg hydroquinone/kg
body weight per day by gavage for 14 days and one group was given
100 mg hydroquinone plus 100 mg phloridzin/kg per day for 14 days.
Ten animals were used per group. Some of the rats treated with
hydroquinone remained in diestrus. In the group treated with both
hydroquinone and phloridzin no effects on the estrus cycle were
shown. As the compound caused effects on the central nervous system
and mortalities occurred after 4-5 days in the 200 mg/kg dose group,
the dose of hydroquinone was reduced to 50 and 100 mg/kg per day. At
autopsy no mature Graafian follicles were seen, but some were
growing.
Hydroquinone in stock diets (0.003 and 0.3%) fed to female rats
(10/group) for 10 days prior to insemination had no effect on
gestation length, maternal mortality or on other reproductive
parameters studied (fertility index, litter efficiency, mean litter
size, fetal viability or lactation index (Ames et al., 1956).
However, it is not clearly stated if the dosing also included the
gestation period.
Hydroquinone did not produce adverse affects on female
reproduction in a two-generation study in rats (see section 7.5.3)
after daily oral administration of doses up to 150 mg/kg per day
(Bio/dynamics Inc., 1989b).
7.5.3 Embryotoxicity and teratogenicity
The earliest experimental study of the developmental toxicity
of hydroquinone and other antioxidants was performed by Telford et
al. (1962). They reported that hydroquinone added to the diet
caused increased resorption rates. Ten Walter Reed-Carworth Farm
strain rats (first gestation animals), weighing about 200 g at the
time of breeding, were given 0.5 g hydroquinone in their diet (about
100 mg/kg body weight per day); however, the treatment period was
not documented. Based on the number of implantation sites, the
resorption rate was 26.8% compared to 10.6% for controls (126
untreated, pregnant rats). The report made no mention of the numbers
of corpora lutea. Hydroquinone caused no maternal toxicity.
Burnett et al. (1976) reported the findings of a teratology
study on twelve hair dye composite formulations. The study groups
were tested along with one positive and three negative control
groups. Mated Charles River CD female rats (20 per group) were
treated by topical application with 2 ml/kg (0.2% hydroquinone) on
pregnancy days 1, 4, 7, 10, 13, 16, and 19. No signs of toxicity
were seen throughout the study. There were no differences between
untreated controls and the hydroquinone-treated group in any
reported parameter (maternal toxicity, body weight and food
consumption, mean number of corpora lutea, implantation sites,
resorption sites, mean resorptions per pregnancy, live fetuses and
sex ratio) and no significant soft tissue or skeletal changes.
Hydroquinone has been found to induce micronuclei
transplacentally in fetal liver cells (Ciranni et al., 1988a) (see
also section 7.6). The compound was given by gastric intubation at a
dose level of 80 mg/kg to four pregnant Swiss CD-1 mice on the 13th
day of gestation. Micronuclei were detected from 9 h after the
administration.
In a pilot study on developmental toxicity in rats,
hydroquinone (5% in distilled water) was administered daily by
gavage to groups of ten pregnant COBSCD(SD)BR rats on gestation days
6 to 15 at dose levels of 0, 50, 100 or 200 mg/kg (Krasavage,
1984a). The animals were sacrificed and autopsied on gestation day
16. No significant maternal toxicity or embryo-toxicity was produced
in any dose group during the treatment period with the exception of
slightly reduced weight gain and feed consumption in the
highest-dose. A dose-dependent brownish discolouration of the urine
was observed in all treated groups. No other treatment-related
changes were found.
In the subsequent developmental toxicity study, hydroquinone
(5% in distilled water) was administered daily by gavage to groups
of 30 plug-positive female (COBSCD(SD)BR rats (initially 6 weeks
old) on gestation days 6 to 15 (0, 30, 100 or 300 mg/kg) (Krasavage
et al., 1985; Krasavage et al., 1992). Maternal toxicity,
manifested as a slight but statistically significant (P < 0.05)
reduction in body weight gain and feed intake, was observed in those
dams given 300 mg/kg. A reduction in the fetal body weight was seen
at 300 mg/kg (statistically significant (P < 0.05) only for the
females). No compound-related teratogenic effects were recorded.
Dilated renal pelvis, hydronephrosis and hydroureter were seen more
frequently in the treated groups than in the controls; however, the
changes did not appear to be dose-related and were not significantly
different from the controls. Skeletal variations were seen in
fetuses from all dose groups and from the control group. The total
number of fetuses with a vertebral variation was statistically
increased in the highest dose group compared with the controls, but
analyses of individual skeletal variations and statistical analyses
of total number of fetuses with a skeletal variation indicated no
significant effects. The authors concluded that the
no-observed-effect level (NOEL) for maternal and developmental
toxicity in rats was 100 mg/kg body weight, and the
no-observed-adverse-effect level (NOAEL) for fetal development was
set at 300 mg/kg (Krasavage et al., 1992).
In a pilot study for developmental toxicity in rabbits,
hydroquinone (0, 50, 100, 200, 300, 400 or 500 mg/kg per day) was
administered by gavage to mated New Zealand White rabbits (five
rabbits/group) from days 6 to 18 of gestation (Bio/dynamics Inc.,
1988). Dose levels of 300, 400 and 500 mg/kg per day caused maternal
death. At dose levels of 50, 100 and 200 mg/kg per day, dose-related
reductions in body weight and food consumption were recorded. In the
200 mg/kg per day dose group, an increased number of resorptions
were observed, suggesting an embryotoxic effect. Mean fetal weights
were unaffected at 50 and 100 mg/kg per day, but at 200 mg/kg per
day they were lower than those of the controls (by 20.7%). No
fetuses were recovered at the higher dose levels as no females
survived throughout the study. The fetal external examinations
showed no treatment-related adverse effects.
In the subsequent developmental toxicity study, New Zealand
White rabbits (18 mated females/group) were given hydroquinone by
gavage at dose levels of 0, 25, 75 or 150 mg/kg per day on gestation
days 6 to 18 (Bio/dynamics Inc., 1989a; Murphy et al., 1992). The
exposure of rabbits to 25 or 75 mg hydroquinone/kg per day had no
effect on any maternal or fetal parameter. However, there was a
reduction in mean food consumption during the 11-14 day gestational
interval at 75 mg/kg per day, but only on days 11 and 12 were these
differences statistically (P < 0.05) different from control data.
In the 150 mg/kg per day dose group there were statistically
significant (P < 0.01) reductions in the body weight and food
consumption during the dosing period, suggesting maternal toxicity.
No other parameter (clinical observations, uterine implantations,
liver or kidney weights or gross pathology) showed any
treatment-related adverse effect on the females. No embryotoxicity
was found from uterine implantation data. An increased incidence
(not statistically significant) of external, visceral and skeletal
malformations was noticed in the fetuses from the 150 mg/kg per day
group. The NOEL for maternal toxicity was 25 mg/kg per day and the
NOEL for developmental toxicity was 75 mg/kg per day.
The results of a two-generation reproduction study in rats
revealed no hydroquinone-related reproductive toxicity (Bio/dynamics
Inc., 1989b). Charles River CD rats (180 Fo and 180 F1) were
exposed via gavage to hydroquinone at concentrations of 15, 50 or
150 mg/kg per day (30 rats per sex and group; 120 rats served as
controls). The two lower dose levels did not affect mortality rates,
body weight, or feed consumption either in the Fo or the F1
parental animals. One Fo male given 50 mg/kg per day had tremors
during the post-dosing period. There were no adverse effects on mean
litter size, body weight, sex distribution or survival, or in
postmortem evaluations of pups from females given 15 or 50 mg/kg per
day. At the highest-dose level, tremors were observed in Fo and
F1 parental animals of both sexes following dosing. Reproductive
indices and pregnancy rates for the Fo and F1 parents were not
considered to have been adversely affected by hydroquinone
treatment, nor were there any adverse effects of treatment for pups
from Fo and F1 parental animals. The NOAEL for parental toxicity
was estimated to be 15 mg/kg per day and for reproductive effects
through two generations to be 150 mg/kg per day.
A quantitative approach to relate the physico-chemical
properties of a series of substituted phenols (including
hydroquinone) to maternal and developmental toxicity in rats was
conducted by Kavlock (1990) and by Kavlock et al. (1991). However,
due to theoretical and statistical mistakes, no formal conclusions
can be made on the possibility of modelling these particular
end-points quantitatively.
A review of the different reproduction studies is given in
Table 15.
7.6 Mutagenicity and related end-points
The results of genotoxicity studies, with or without S9
metabolic activation, are indicated in Table 16.
Hydroquinone has been found to be non-mutagenic in Salmonella
typhimurium tester strains TA97, TA98, TA100, TA1535, TA1537 and
TA1538, with and without metabolic activation, at doses up to 1000
µg/plate (Bulman & Wampler, 1979; Bulman & Van der Sluis, 1980;
Florin et al., 1980; Gocke et al., 1981; Serva & Bulman, 1981;
Bulman & Serva, 1982; Haworth et al., 1983; Sakai et al., 1985).
However, in one study hydroquinone was reported to be clearly
mutagenic without metabolic activation in strain 1535A (a strain
that the authors suggested might harbour an undefined genetic
alteration from TA1535 because of differences in length of storage)
with ZLM minimal medium (a modified minimal medium for Escherichia
coli) but not with the standard VB (Vogel-Bonner) minimal medium
(Gocke et al., 1981). The concentration of citrate was 3.5 times
higher in VB medium than in ZLM medium. In a fluctuation test using
the Salmonella typhimurium tester strain TA100, hydroquinone was
shown to be mutagenic at the concentrations 100 and 200 ng/well with
metabolic activation (Koike et al., 1988).
In Escherichia coli hydroquinone causes differential killing
of DNA-repair-deficient (Pol A-) and -proficient (Pol A+) strains
without a supplementary metabolic activation system, indicating
induction of repairable DNA damage (Bilimoria, 1975; Van der Sluis,
1980; Wampler, 1980). In Salmonella typhimurium no SOS-inducing
activity of hydroquinone (3300 mg/litre liquid medium) could be
demonstrated (Nakamura et al., 1987).
Table 15. Studies on reproductive effects in laboratory animals
Species Route of Number of Dosage Time of Result Reference
exposure animals treatment
Rat subcutaneous 16 males 100 mg/kg body 51 days 33% reduced fertility, diminished Skalka (1964)
weight content of DNA in sperm heads; 19%
reduced number of pregnancies in
mated females
Rat subcutaneous 3 10 mg/day 11 days interrupted estrus cycle Rosen & Millman
(1955)
Rat oral (gavage) 10/group 200 mg/kg body 14 days some rats remained in diestrus, Racz et al.
weight per daya CNS effects and death within 5 days (1958)
Rat oral (diet) 10 females/ 0.003% 10 days prior to no effects on gestation and Ames et al.
group 0.3% insemination maternal mortality (1956)
Rat oral (diet) 10 0.5 g not clearly stated increased resorption rate Telford et al.
(1962)
Rat oral (gavage) 10 females/ 0, 50, 100 or 200 gestation days reduced feed consumption and body Krasavage (1984a)
group mg/kg body weight 6-15 weight gain at 200 mg/kg
Rat oral (gavage) 25/group 0, 30, 100 or 300 5 days per week no treatment-related effects on Krasavage (1984b)
(male) mg/kg body weight for 10 weeks reproductive parameters studied
Rat oral (gavage) 30/group 0, 30, 100 or 300 gestation days reduced fetal body weight at Krasavage et al.
mg/kg 6-15 300 mg/kg (1985)
Rat oral (gastric 30/group 0, 15, 50 or 150 two-generation no effect on reproduction at Bio/dynamics
intubation) mg/kg body weight study 150 mg/kg Inc. (1989b)
Table 15. (contd).
Species Route of Number of Dosage Time of Result Reference
exposure animals treatment
Rat oral 16/group 0, 100, 333, 667 gestation day 11 reduced weight gain at 667 and 1000 Kavlock (1990)
(intubation) or 1000 mg/kg mg/kg; increased mortality at 1000
mg/kg; malformations of the limbs,
tail and urogenital system
Rat embryo < 0.5 mmol/litre from day 10 growth retardation, structural Kavlock et al.
culture (approx. 12 somite) abnormalities of hind limb and tail (1991)
system
Mouse oral (gastric 4 80 mg/kg body gestation day 13 micronuclei in fetal liver cells Ciranni et al.
intubation) weight (1988a)
Rabbit oral (gastric 5/group 0, 50, 100, 200, gestation days increased number of resorptions Bio/dynamics
intubation) 300, 400 or 500 6-18 and lower fetal weights at 200 mg/kg; Inc. (1988)
mg/kg body weight no females survived 300-500 mg/kg
Rabbit oral (gastric 18/group 0, 25, 75 or 150 gestation days an increased incidence (not Bio/dynamics Inc.
intubation) mg/kg body weight 6-18 significant) of malformations at (1989a); Murphy
150 mg/kg et al. (1992)
a Due to high mortality at 200 mg/kg, the dose was reduced for the rest of the 14-day period.
In Aspergillus nidulans hydroquinone was found to induce
mitotic segregation at 110-330 mg/litre (Crebelli et al., 1987)
and 264-396 mg/litre (Crebelli et al., 1991) and mitotic
crossing-over at 250-750 mg/litre (Kappas, 1989) and 264-396
mg/litre (Crebelli et al., 1991). The studies were performed
without a supplementary metabolic activation system.
Hydroquinone has been shown to induce chromosome aberrations or
karyotypic alterations in several plant species, e.g., Allium cepa
(Levan & Tjio, 1948a,b), Allium sativum (Sharma & Chatterjee,
1964; Valadaud & Izard, 1971), Callisia fragrans (Roy, 1973),
Chara zeylanica (Chatterjee & Sharma, 1972), Nigella sativa
(Sharma & Chatterjee, 1964), Trigonella foenum-graecum (Sharma &
Chatterjee, 1964), and Vicia faba (Sharma & Chatterjee, 1964;
Valadaud & Izard, 1971; Valadaud-Barrieu & Izard, 1973).
Hydroquinone produced an equivocal increase in recessive lethal
mutations in the X-chromosome of Drosophila melanogaster after
feeding adult males with a sucrose solution containing 26.4 or 30.0
mg hydroquinone/litre (NTP, 1989). No increase in the frequency of
recessive lethal mutations could be established in similar feeding
studies with hydroquinone at concentrations of 5500 to 11 000
mg/litre (Gocke et al., 1981) and 1000 mg/litre (Serva & Murphy,
1981), or in a study where males were injected with a dose of 1.5
mg/litre (NTP, 1989) (see also section 7.5.1).
At concentrations of 1.25 mg/litre (without a supplementary
metabolic activation system) and 2.5 mg/litre (with a supplementary
metabolic activation system), hydroquinone induced forward mutations
in vitro in the thymidine kinase locus of the mouse lymphoma cell
line L5178Y (McGregor et al., 1988a,b).
Gene mutations to 6-thioguanine resistance were induced in
vitro in V79 Chinese hamster cells exposed to hydroquinone at 350
µg/litre (Glatt et al., 1989). No supplementary metabolic
activation system was used.
Structural chromosome aberrations were induced in vitro in
Chinese hamster ovary cells treated with hydroquinone, but only in
the presence of a supplementary metabolic activation system
(Galloway et al., 1987).
Table 16. Studies on genotoxicity
Genetic end-point Resulta References
+S9 -S9
Gene mutations
Salmonella typhimurium - Bulman & Wampler (1979)
Salmonella typhimurium - - Bulman & Van der Sluis (1980)
Salmonella typhimurium - - Florin et al. (1980)
Salmonella typhimurium - - Gocke et al. (1981)
Salmonella typhimurium - - Serva & Bulman (1981)
Salmonella typhimurium - - Bulman & Serva (1982)
Salmonella typhimurium - - Haworth et al. (1983)
Salmonella typhimurium - - Sakai et al. (1985)
Salmonella typhimurium - + Gocke et al. (1981)
TA1535A, ZLM medium
Salmonella typhimurium + - Koike et al. (1988)
TA100, fluctuation test
Mouse lymphoma cells L5178Y + + McGregor et al. (1988a,b)
Chinese hamster cells V79 nd + Glatt et al. (1989)
Drosophila melanogaster, (+) NTP (1989)
feeding
Drosophila melanogaster, - Gocke et al. (1981)
feeding
Drosophila melanogaster, - Serva & Murphy (1981)
feeding
Drosophila melanogaster, - NTP (1989)
injection
Mouse, spot test - Gocke et al. (1983)
Rat, dominant lethals - Krasavage (1984b)
Chromosomal aberrations
Aspergillus nidulans nd + Crebelli et al. (1987, 1991)
Chinese hamster ovary cells + - Galloway et al. (1987)
Allium cepa + Levan & Tjio (1948a,b)
Allium sativum + Sharma & Chatterjee (1964)
Allium sativum + Valadaud & Izard (1971)
Callisia fragrans + Roy (1973)
Chara zeylanica + Chatterjee & Sharma (1972)
Nigella sativa + Sharma & Chatterjee (1964)
Trigonella foenum-graecum + Sharma & Chatterjee (1964)
Vicia faba + Sharma & Chatterjee (1964)
Vicia faba + Valadaud & Izard (1971)
Vicia faba + Valadaud-Barrieu & Izard (1973)
Mouse, bone marrow + Xu & Adler (1990)
Table 16. (contd).
Genetic end-point Resulta References
+S9 -S9
Mouse, bone marrow + Pacchierotti et al. (1991)
Mouse, spermatocytes and + Ciranni & Adler (1991)
differentiating spermatogonia
Rat, dominant lethals - Krasavage (1984b)
Micronuclei
Chinese hamster cells V79nd + Glatt et al. (1989, 1990)
Rat intestinal cells nd + Glatt et al. (1990)
Embryonal human liver cells nd + Glatt et al. (1990)
Human lymphocytes nd + Yager et al. (1990)
Human lymphocytes nd + Robertson et al. (1991)
Chinese hamster embryonicnd + Antoccia et al. (1991)
lung cells
Mouse, bone marrow + Gocke et al. (1981)
Mouse, bone marrow + Gad-El-Karim et al. (1985)
Mouse, bone marrow + Ciranni et al. (1988a,b)
Mouse, bone marrow + Adler & Kliesch (1990)
Mouse, bone marrow + Barale et al. (1990)
Mouse, bone marrow + Adler et al. (1991)
Mouse, bone marrow + Pachierotti et al. (1991)
Mouse, fetal liver + Ciranni et al. (1988a)
Chinese hamster cells V79 nd + Glatt et al. (1989)
Chinese hamster ovary cells + + Galloway et al. (1987)
Human lymphocytes nd + Erexson et al. (1985)
Human lymphocytes nd + Morimoto & Wolff (1980)
Human lymphocytes nd ± Knadle (1985)
Human lymphocytes + nd Morimoto et al. (1983)
Mouse, bone marrow - Pacchierotti et al. (1991)
Mitotic crossing-over
Aspergillus nidulans nd + Kappas (1989)
Aspergillus nidulans nd + Crebelli et al. (1991)
C-mitotic effects
Mouse, small intestine + Parmentier & Dustin (1948)
Mouse, bone marrow + Miller & Adler (1989)
DNA damage
Mouse lymphoma cells L5178YS, nd - Pellack-Walker & Blumer
breaks (1986)
Phi X-174 DNA, breaks + Lewis et al. (1988a)
Table 16. (contd).
Genetic end-point Resulta References
+S9 -S9
Rat, liver (hepatectomy), + Stenius et al. (1989)
single-strand breaks
DNA repair
Escherichia coli nd + Bilimoria (1975)
Escherichia coli nd + Van der Sluis (1980)
Escherichia coli nd + Wampler (1980)
Salmonella typhimurium nd + Nakamura et al. (1987)
HeLa cells + + Painter & Howard (1982)
Mouse lymphoma cells L5178YS nd + Pellack-Walker et al. (1985)
DNA adducts
In vitro + Rushmore et al. (1984)
In vitro + Jowa et al. (1986)
In vitro + Reddy et al. (1989)
In vitro + Jowa et al. (1990)
In vitro + Leanderson & Tagesson (1990)
In vitro + Reddy et al. (1990)
In vitro + Levay et al. (1991)
a + = positive; - = negative; (+) = equivocal; ± = induction in some
cases but not in others; nd = not determined
The induction of sister-chromatid exchanges in vitro after
exposure to hydroquinone without the use of a supplementary
metabolic activation system has been demonstrated in V79 Chinese
hamster cells at 2.2 mg/litre (Glatt et al., 1989) and human
lymphocytes at 0.55-11 mg/litre (Erexson et al., 1985), 4.4-22
mg/litre (Morimoto & Wolff, 1980), and 4.4 mg/litre in cells from
some individuals but not from others (Knadle, 1985), with a
supplementary metabolic activation system in human lymphocytes at
110 mg/litre (Morimoto et al., 1983), and both with and without a
supplementary metabolic activation system in Chinese hamster ovary
cells (Galloway et al., 1987).
Hydroquinone has been found to induce micronuclei in vitro in
V79 Chinese hamster cells (Glatt et al., 1989, 1990), rat
intestinal cells IEC-17 and IEC-18, and embryonal human liver cells
HuFoe-15 (Glatt et al., 1990). In cultured human lymphocytes an
11-fold and 3-fold increase in the frequency of micronuclei, was
reported after exposure to hydroquinone at 13.75 mg/litre (Yager et
al., 1990) and 8.25 mg/litre (Robertson et al., 1991),
respectively. Hydroquinone induced micronuclei in vitro in Cl-1
Chinese hamster embryonic lung cells at concentrations of 1, 3, and
4.5 mg/litre (Antoccia et al., 1991). The use of an
antikinetochore antibody in the three latter studies indicated the
occurrence of numerical as well as structural chromosome
aberrations. All studies were performed without a supplementary
metabolic activation system.
With the in vitro porcine brain tubulin assembly assay,
hydroquinone had no effect with respect to lag-phase, polymerization
velocity or end absorption at doses up to 2750 mg/litre. The
disassembly process was stimulated at doses higher than 1100
mg/litre (Brunner et al., 1991). In another study, hydroquinone
exhibited a weak inhibition of microtubule assembly and resulted in
abnormal microtubule formation at a concentration of 110 mg/litre
(Wallin & Hartley-Asp, 1993). Although hydroquinone itself appears
to be a poor inhibitor of microtubule assembly, its oxidation
products have been shown to bind to both alpha- and beta-tubulin and
to inhibit microtubule assembly at low concentrations (5 µmol/litre
or less) (Epe et al., 1990). In another study it was demonstrated
that polymerization of tubulin was inhibited by hydroquinone in a
concentration-dependent manner (Irons et al., 1981).
An effect on DNA synthesis, indicating DNA damage, following
in vitro exposure to hydroquinone has been demonstrated both with
and without a supplementary metabolic activation system in HeLa
cells (Painter & Howard, 1982) and without the use of a metabolic
activation system in the mouse lymphoma cell line L5178YS
(Pellack-Walker et al., 1985).
Female mice (C57BL/6JHan), mated with T-stock males, were
injected intraperitoneally with hydroquinone (110 mg/kg) on the 10th
day of pregnancy and subsequently analysed in accordance with the
mammalian spot test, which detects somatic gene mutations in mouse
embryos. No substance-related effect on the mutation frequency was
detected (Gocke et al., 1983).
In a study by Xu & Adler (1990), mice [(101/E1 x C3H/E1)F1],
10-14 weeks old and weighing 25-28 g, were injected
intraperito-neally with hydroquinone (40, 75 and 100 mg/kg). Bone
marrow cells, sampled 6 and 24 h later in the case of the two lower
doses and 6, 12, 18, 24 and 36 h later in the case of the highest
dose, were analysed for the presence of structural chromosome
aber-rations. At the highest dose level hydroquinone significantly
increased the frequency of aberrations 6-24 h after the treatment,
while at 75 mg/kg such an effect was detectable only 24 h after
treatment.
Male mice [(101/E1 x C3H/E1)F1], 10-14 weeks old and weighing
25-28 g, were injected intraperitoneally with hydroquinone (80, 100
and 150 mg/kg), and bone marrow cells, sampled 2 h later, were
analysed for the induction of c-mitotic effects. At dose levels of
100 and 150 mg/kg hydroquinone significantly increased the frequency
of metaphases with spread chromosomes (Miller & Adler, 1989).
Colchicine-like accumulation of metaphases and unusual "three-group
metaphases" in the small intestine were described in mice injected
with hydroquinone (125 mg/kg) (Parmentier & Dustin, 1948, 1951).
Similar results were observed in the intestine and bone marrow cells
of rats and hamsters following hydroquinone administration
(Parmentier, 1952, 1953).
In a study by Pacchierotti et al. (1991), male mice
[(C57Bl/Cne x C3H/Cne)F1], 12 weeks old, were injected with
hydroquinone (40, 80 and 120 mg/kg). Bone marrow cells, sampled 18
and 24 h later were analysed for the presence of numerical
chromosome changes, micronuclei and sister-chromatid exchanges. At a
dose level of 80 mg/kg, hydroquinone significantly increased the
frequency of hyperploid cells with 41-42 chromosomes after 18 h. At
all concentrations tested the frequency of micronuclei was
significantly increased after 24 h, while after 18 h such an effect
was only detectable at the concentration of 80 mg/kg. An increase in
sister-chromatid exchange frequencies over control values was not
detected in any treated animal.
Mice [(101/E1 x C3H/E1)F1], 10-14 weeks old and weighing
25-28 g, were injected intraperitoneally with hydroquinone (30, 50,
75 and 100 mg/kg), and bone marrow cells, sampled 18, 24 and 30 h
later (75 mg/kg), 6 and 24 h later (50 mg/kg) and 24 h later (30 and
100 mg/kg), were analysed for the presence of micronuclei. At a dose
level of 75 mg/kg, hydroquinone significantly increased the
frequency of micronuclei in cells at all sampling intervals.
Treatment with hydroquinone at 50 and 100 mg/kg significantly
increased the frequency of micronuclei after 24 h (Adler & Kliesch,
1990). The authors also reported results from daily treatments (up
to 3 days) of male mice of the same strain with hydroquinone (15 or
75 mg/kg) by the intraperitoneal route of administration. Bone
marrow cells were sampled 24 h after the first, second and third
injections, respectively. The frequency of micronuclei increased in
the case of the lower dose and decreased in the case of the higher
dose with increasing number of treatments.
The induction of micronuclei in mouse bone marrow cells after
intraperitoneal injection of hydroquinone has also been demonstrated
by Gocke et al. (1981), Ciranni et al. (1988a,b), Barale et al.
(1990) and Adler et al. (1991). Oral administration of
hydroquinone (200 mg/kg) induced an increase in the frequency of
micronuclei in mice (Gad-El-Karim et al., 1985). Ciranni et al.
(1988b) demonstrated that oral administration of hydroquinone (80
mg/kg) produced a weak increase in the frequency of micronuclei
compared to the effect found following intraperitoneal
administration. In male mice (outbred NMRI) daily subcutaneous
injections of hydroquinone on six consecutive days induced
micronuclei in the bone marrow at dose levels of 25 to 100 mg/kg
(Tunek et al., 1982).
When pregnant mice (Swiss CD-1, three months old) were given
hydroquinone (80 mg/kg) by gastric intubation on the 13th day of
gestation, micronuclei were induced in fetal liver cells (Ciranni
et al., 1988a).
In male rats (Sprague-Dawley), weighing 200 g and subjected to
70% partial hepatectomy, single-strand breaks were induced in
hepatic DNA after the rats had received daily hydroquinone doses of
200 mg/kg, given in a liquid casein-based diet, for 7 weeks (Stenius
et al., 1989).
Male mice [(102/ElxC3H/El)F1], 12-14 weeks old and weighing
25-28 g, were injected intraperitoneally with hydroquinone (40, 80
and 120 mg/kg). In spermatocytes sampled 12 days after treatment
(representing cells treated at preleptotene), the frequency of
chromosomal aberrations excluding gaps was significantly increased
at 40 and 80 mg/kg but not at 120 mg/kg. In differentiating
spermatogonia sampled 24 h after treatment, the frequency of
chromosomal aberrations excluding gaps was significantly increased
at all dose levels (Ciranni & Adler, 1991).
No increased frequency of dominant lethal mutations was
detected in male rats [CRL:COBS"CD"(SD)BR] given repeated doses of
hydroquinone (30, 100 or 300 mg/kg, 5 days/week for 10 weeks) by
gavage (Krasavage, 1984b). The induction of sperm-head abnormalities
could not be demonstrated in male mice after intraperitoneal
injection of hydroquinone (55-220 mg/kg) (Wild et al., 1981).
The ability of hydroquinone to produce adducts with DNA or its
nucleotides in vitro has been shown by Rushmore et al. (1984),
Jowa et al. (1986), Reddy et al. (1989), Jowa et al. (1990),
Leanderson & Tagesson (1990), Reddy et al. (1990) and Levay et
al. (1991). There is less evidence for the hydroquinone-induced
formation of DNA adducts in vivo. Using the 32P postlabelling
assay, no treatment-related adducts were detected in the kidneys of
either male or female F-344 rats following the oral administration
of hydroquinone at dose levels of up to 50 mg/kg for 6 weeks
(English et al., 1992). In additional studies by Reddy et al.
(1990), also using the postlabelling assay, no treatment-related DNA
adducts were detected in the bone marrow, zymbal gland or liver of
female F-344 rats following the oral co-administration of phenol and
hydroquinone at either 75 or 150 mg/kg for 4 days (Reddy et al.,
1990).
Hydroquinone has been shown to be capable of producing breaks
in phi X-174 DNA (Lewis et al., 1988a). No increase in the
frequency of breaks was detected in DNA from hydroquinone-treated
mouse lymphoma cells (L5178YS) at concentrations of up to 11
mg/litre (Pellack-Walker & Blumer, 1986).
7.7 Carcinogenicity
The available studies on the carcinogenicity of hydroquinone
are summarized in Table 17.
7.7.1 Long-term bioassays
In an NTP study (NTP, 1989; Kari et al., 1992), groups of 65
F-344/N rats of each sex were given hydroquinone (0, 25 or 50 mg/kg
body weight) in deionized water by gavage 5 days/week for up to 103
weeks, and groups of 65 B6C3F1 mice of each sex were administered
0, 50 or 100 mg/kg body weight according to the same schedule. A
15-month interim kill of ten animals from each group showed that the
kidney of male rats was a target organ forthe toxicity (see also
section 7.4), since there was a compound-related increased severity
of nephropathy. The lesions were less severe in female rats, in
which a mild regenerative anaemia was also found (slightly decreased
haematocrit, haemoglobin and erythrocyte count). After termination
of the experiment, a dose-related increase in the incidence of renal
tubular cell adenomas was observed in male rats (controls 0/55, low
dose 4/55, high dose 8/55; P = 0.003). The incidence of adenomas was
closely associated with the severity of chronic nephropathy. No
renal adenomas were observed in animals examined at 15 months, when
the severity of nephropathy was less severe, or in female rats,
which developed nephropathy to a lesser degree. In the male rats,
9/12 adenomas were seen in kidneys with marked nephropathy, two were
seen in animals with moderate nephropathy, and only one was seen in
an animal with mild nephropathy. In the high-dose group single
tubules exhibited tubular cell hyperplasia. No renal tumours were
seen in females. A dose-related increase in the incidence of
mononuclear cell leukaemia was found in female rats (controls 9/55,
low dose 15/55, high dose 22/55) (P < 0.01 in the high-dose group
versus controls). However, this was not observed in the animals
killed at 15 months. The incidence in controls was lower than the
historical control mean incidence but was within the historical
control group range.
Table 17. Carcinogenicity studies in animals
Species Route of Number of Dosage Time of Result Remarks Reference
exposure animals treatment
Long-term bioassays
Mouse oral 64 or 65 of 50 or 100 103 weeks liver lesions (males), some evidence of NTP (1989);
each sex mg/kg hepatocellular adenomas carcinogenic activity Kari et al.
per group 5 days/week (females) for female mice (1992)
Mouse oral 30 m, 30 f 0.8% in 96 weeks squamous cell hyperplasia of potential of Shibata et al.
the diet the forestomach epithelium; hepatocarcinogenicity (1991)
renal tubular hyperplasia and in male mice
adenomas (males); increased
incidence of liver foci and
hepatocellular adenomas
(males)
Rat oral 65 of each 25 or 50 103 weeks nephropathy (more severe in some evidence of NTP (1989);
sex per mg/kg males), renal tubular cell carcinogenic activity Kari et al.
group 5 days/week hyperplasia and adenomas for male and female (1992)
(males), leukaemia (females) rats
Rat oral 30 m, 30 f 0.8% in 104 weeks renal tubular hyperplasia, potential of renal Shibata et al.
the diet adenomas and epithelial carcinogencity in (1991)
hyperplasia of the renal male rats
papilla (males); decreased
incidence of liver foci
Table 17. (contd).
Species Route of Number of Dosage Time of Result Remarks Reference
exposure animals treatment
Carcinogenicity-related studies
Mouse skin 24 m 0.3 ml of 6.7% one skin papilloma (1/24) no initiating Roe & Salaman
application solution; application; activity (1955)
0.3 ml of then three
0.5% croton weeks later,
oil 18 weekly
applications
Mouse skin 50 f 5 mg three 368 days papilloma (7/50), squamous no co-carcinogenic van Duuren &
application times carcinoma (3/50) or tumour-promoting Goldschmidt
weeklya activity; partial (1976)
inhibition of BP
carcinogenicity
Mouse implantation not stated 2 mg 25 weeks carcinomas (6/19) Boyland et al.
in urinary (1964)
bladder
Rat oral 20 f 0.8% in 32 weeks no preneoplastic lesions Kurata et al.
basal dietb or papillomas of the (1990)
urinary bladder
Rat oral 15-16 m 0.8% in 51 weeks no increase in forestomach or Hirose et al.
dietc glandular stomach neoplasms (1989)
Rat oral 5 m 8 weeks no proliferative changes Shibata et al.
in forestomach or glandular (1990)
stomach
Table 17. (contd).
Species Route of Number of Dosage Time of Result Remarks Reference
exposure animals treatment
Rat oral 7-10 m per 100 mg/kg 7 weeks increased number of liver foci relatively weak Stenius et al.
group diet per dayd decreased number of liver foci inducer of enzyme- (1989)
200 mg/kg compared to the 100 mg/kg altered liver foci
diet per dayd dose
Hamster oral 15 m 0.5% in basal 20 weeks no proliferative changes in Hirose et al.
diet forestomach (1986)
a after initiating dose of benzo[ a] pyrene (BP)
b after initiating with N-butyl-2 N-(4-hydroxybutyl) nitrosamine for four weeks
c one week after 150 mg/kg body weight
d after partial hepatectomy
In male mice centrilobular fatty changes and cytomegaly were
found in the animals killed at 15 months, but these findings were
not seen in mice killed at 2 years. The authors reported that
hydroquinone dosing stopped two weeks before necropsy and that the
microscopic lesions were likely to be reversible after cessation of
treatment. There was a significantly (P=0.0005) increased incidence
of hepatocellular adenomas in female mice given hydroquinone for 2
years (controls 2/55, low dose 15/55, high dose 12/55) and the
incidences of hepatocellular carcinomas were 1/55, 2/55 and 2/55,
respectively. In males the incidence of adenomas was increased in
treated mice but the incidence of hepatocellular carcinomas was
decreased. Preneoplastic changes (anisokaryosis, multinucleated
hepatocytes, and basophilic foci) were increased in high-dose male
mice. Treatment-related, but not statistically significant,
follicular cell hyperplasia of the thyroid gland was observed in
both male and female mice (NTP, 1989; Kari et al., 1992).
The NTP concluded that there was "some evidence of carcinogenic
activity" of hydroquinone for male F-344/N rats (tubular cell
adenomas of the kidney) and also for female F-344/N rats
(mononuclear cell leukaemia). There was "no evidence of carcinogenic
activity" for male B6C3F1 mice and "some evidence of carcinogenic
activity" for female B6C3F1 mice (hepatocellular adenomas and
carcinomas combined).
Shibata et al. (1991) administered hydroquinone at dietary
levels of 0.% or 8 g/kg to groups of 30 Fischer-344 rats and
B6C3F1 mice of each sex. The rats were dosed for 104 weeks and the
mice for 96 weeks. Average daily intakes were reported to be 351 and
368 mg/kg body weight per day in male and female rats, respectively,
and 1046 and 1486 mg/kg per day in male and female mice,
respectively. No treatment-related clinical signs and no significant
differences in mortality were found between treated and control
animals of either species. The final body weight was significantly
(P < 0.05) lower in treated female rats than in corresponding
controls. In male rats the absolute and relative liver and kidney
weights were significantly (P < 0.01) increased, but in females
this applied only to the relative kidney weights (P < 0.05).
Histologically, chronic nephropathy was seen in both control and
treated groups of male rats. However, treated males were more
severely affected than the controls, while treated females showed
only slight nephropathy. The incidence of epithelial hyperplasia of
the renal papilla was significantly (P < 0.05) increased in treated
male rats as was the incidence of renal tubular hyperplasia (30/30)
and renal tubular adenomas (14/30).
The authors found that renal cell tumour development in male
rats under the long-term influence of hydroquinone was not
associated with alpha2u-globulin nephropathy. The incidence of liver
foci showed a tendency to decrease in treated males. A quantitative
analysis showed a statistically significant (P < 0.05 in males, P<
0.01 in females) reduction in both sexes given hydroquinone. The
authors did not find an increased incidence of mononuclear cell
leukaemia in female rats (personal communication).
In mice, the final body weight was significantly (P < 0.05)
lower in females given hydroquinone; the relative liver and kidney
weights were significantly (P < 0.05) increased. Histologically,
the incidence of squamous cell hyperplasia of the forestomach
epithelium was significantly (P < 0.01) increased in both sexes. A
significant increase in the incidence of renal tubular hyperplasia
(P < 0.01) and three renal cell adenomas were seen in 30 males
given hydroquinone. In treated males the incidence of liver foci and
hepatocellular adenomas (14/30) was also significantly (P < 0.05)
increased.
7.7.2 Carcinogenicity-related studies
7.7.2.1 Skin
In a study by Roe & Salaman (1955), stock albino mice (24
males, "S" strain) were given a single skin application of 0.3 ml of
a 6.7% solution of hydroquinone in acetone (total dose 20.0 mg).
Three weeks later the mice received 18 weekly applications of 0.3 ml
of 0.5% croton oil in acetone as a promoter on the same area of the
skin. Of the 24 treated animals, two died during the experiment and
one mouse developed a skin papilloma.
In a two-stage carcinogenesis test on mouse skin using
benzo[ a]pyrene (BP) as the initiating agent, no tumour-promoting
activity was shown (Van Duuren & Goldschmidt, 1976). Hydroquinone (5
mg) was applied to mouse skin (50 female ICR/Ha Swiss mice/group;
both positive and negative controls) three times weekly for 368
days, together with 5 µg BP. Hydroquinone showed no potential as a
co-carcinogen when applied simultaneously with BP; in fact, it
partially inhibited BP carcinogenicity.
7.7.2.2 Bladder
Implantation of cholesterol pellets containing hydroquinone
into the urinary bladder of mice (strain and sex unspecified) has
been studied by Boyland et al. (1964). The amount of hydroquinone
was 20% in 10 mg cholesterol pellets (2 mg hydroquinone per mouse).
Bladder carcinomas were produced in 6 out of 19 mice (32%) surviving
25 weeks. The incidence of urinary bladder carcinomas in survivors
of the dosed group was significantly (P=0.03) higher than in
controls (11.7%) given cholesterol pellets only. However, the number
of animals surviving the study was low, and the original number of
animals and their sex distribution were not specified.
In a study by Kurata et al. (1990), groups of 20 male
Fischer-344 rats received 0.05% N-butyl- N-(4-hydroxybutyl)
nitrosamine in the drinking-water for four weeks (as initiation)
followed by 8 g hydroquinone/kg in the basal diet for 32 weeks. No
increase in the incidence of preneoplastic lesions or
papillomas/carcinomas of the urinary bladder was observed when
compared to the incidences in rats given nitrosamine alone.
7.7.2.3 Stomach
Hirose et al. (1989) examined the promotion activity and the
carcinogenic potential of some dihydroxybenzenes, such as
hydroquinone, in the glandular stomach and forestomach of F-344
rats. Groups of 15-16 male rats were given a single intragastric
dose of 150 mg/kg body weight N-methyl- N'-nitro- N-
nitrosoguanidine (MNNG), followed one week later by powdered diet
containing hydroquinone (8g/kg) or basal diet alone for 51 weeks.
Further groups of 10 and 15 animals, respectively, were administered
the basal diet alone or a diet containing hydroquinone (8 g/kg) for
51 weeks without pretreatment with MNNG. Hydroquinone did not cause
an increased incidence of forestomach or glandular stomach lesions,
either with or without pretreatment with MNNG, in comparison with
the control groups.
In studies performed by Hirose et al. (1986), hydroquinone
did not produce proliferative lesions in the stomach of hamsters.
Male Syrian golden hamsters (15/group, seven weeks old at the
beginning of the study) were given basal diet with hydroquinone (5
g/kg) added or basal diet alone for 20 weeks. The dose was chosen as
approximately a quarter of the LD50. Tissues from forestomach and
glandular stomach showed mild to moderate hyperplasia in the group
given hydroquinone, but at the same incidence as in the controls.
Similar results were obtained by Shibata et al. (1990) in an
8-week oral study using five male F-344 rats. Hydroquinone did not
induce any proliferative changes in the forestomach or the glandular
stomach epithelium.
7.7.2.4 Liver
Hydroquinone has been shown to be a relatively weak inducer of
enzyme-altered foci in rat liver when tested for tumour-promoting
activity in a liver focus test (Stenius et al., 1989). Male
Sprague-Dawley rats (7-10/group) given diethylnitrosamine (30 mg/kg
intraperitoneally) after partial hepatectomy were treated with
hydroquinone (0, 100 and 200 mg/kg per day) in their diet for 7
weeks. At 100 mg/kg there was a significantly (P < 0.01) increased
number of liver foci and an increased focus volume. The 200-mg dose
caused less foci (0.34 ± 0.16 per cm2) than the 100-mg dose (0.65
± 0.25 per cm2), but the incidence was higher than in the control
group (0.08 ± 0.08 per cm2).
A study by Kurata et al. (1990) yielded similar results
concerning the tumour-promoting potential of hydroquinone in rats.
Dietary administration of hydroquinone (8g/kg in basal diet) for 32
weeks, after initiation for four weeks with N-butyl- N-
(4-hydroxybutyl) nitrosamine, caused no preneoplastic lesions or
papillomas of the urinary bladder.
7.8 Special studies
7.8.1 Effects on spleen and bone marrow cells; immunotoxicity
The bone marrow is the target in benzene toxicity; among the
many metabolites of benzene, hydroquinone has received increased
scrutiny as one of the possible contributing factors. Intravenous or
intraperitoneal administration of hydroquinone (100 mg/kg) for three
consecutive days to male C57BL/6 CRIBR mice significantly (P <
0.05) reduced the spleen and bone marrow cellularity, with bone
marrow demonstrating the greatest sensitivity (Wierda & Irons,
1982). Laskin et al. (1989) found that after injection in Balb/c
mice hydroquinone (50 mg/kg) caused a 30-40% decrease in bone marrow
cellularity.
In vitro studies have demonstrated direct myelotoxic effects
of hydroquinone toward mouse bone marrow stromal cells (Gaido &
Wierda, 1984; Gaido & Wierda 1987). Hydroquinone inhibited stromal
cell colony growth along with the ability of these cells to support
granulocyte/monocyte colony formation in co-culture. The bone marrow
stroma predominantly consists of macrophages and fibroblastoid
stromal cells which interact to regulate myelopoiesis. Treatment
with hydroquinone thus results in reduced capacity of the stroma to
support myelopoiesis.
In addition to this cytotoxic effect, Wierda & Irons (1982)
found in in vivo studies that hydroquinone also affected the
immune function by reducing the number of progenitor B-lymphocytes
in the spleen and bone marrow in mice, thus demonstrating an
immunosuppressive potential. The rapid generation and maturation of
progenitor B cells renders them highly susceptible to toxic agents
that affect dividing cells. Evidence has accumulated concerning the
effect of hydroquinone on the cellular activity of the immune system
in vitro. Exposure of lymphocytes in vitro to hydroquinone has
been shown to result in a dose-dependent inhibition of RNA synthesis
in the lymphocytes (Post et al., 1985). A hydroquinone
concentration of 1-2 x 10-5 mol/litre inhibited the RNA synthesis
by 50%.
In vitro exposure (one hour) of mouse bone marrow cells to
hydroquinone (10-7-10-5 mol/litre) inhibited the maturation of
B-lymphocytes from pre B-cells after 24 and 48 h in culture (King
et al., 1987). More recent data have demonstrated that
hydroquinone-induced inhibition of pre-B cell maturation results
from toxicity to adherent stromal cells, and that bone marrow
macrophages may be the primary target for hydroquinone
myelotoxicity, rather than fibroblastic stromal cells or pre-B cells
(King et al., 1989; Thomas et al., 1989a). Results also indicate
a dose-related reduction of macrophage interleukin-1 (IL-1)
secretion in cultures of bone marrow macrophages exposed to
hydroquinone (King et al., 1989; Thomas et al., 1989b). IL-1 is
necessary for the induction of interleukin-4 (IL-4), which is
produced by fibroblastic stromal cells and is required for
maturation of pre-B cells to B cells (King et al., 1989).
Fan et al. (1989) demonstrated that hydroquinone can inhibit
the natural killer activity of mouse spleen cells in vitro at low
concentrations. Concentrations of 1 x 10-5 mol/litre and 1 x
10-6 mol/litre inhibited 29 and 22% of the activity, respectively.
Lewis et al. (1988b) found that hydroquinone had a selective
effect on macrophage functions important in host defense. At
concentrations of 3-100 µmol/litre, hydroquinone significantly
(P < 0.05) inhibited the release of hydrogen peroxide and at 100
µmol/litre it significantly (P < 0.05) inhibited priming by
interferon for tumour cell cytolysis. Cheung et al. (1989) have
shown a concentration-dependent inhibition of interferon-alpha/ß
production following exposure to hydroquinone in murine L-929 cell
cultures.
7.8.2 Effects on tumour cells
The cytotoxic activity of hydroquinone has been tested on
different tumour cells. Chavin et al. (1980) studied the effect on
melanoma transplants in female BALB/c mice. The incidence of
melanoma transplants was reduced and the survival significantly (P
< 0.0005) increased in mice that received hydroquinone treatment
(80 mg/kg).
Vladescu & Apetroae (1983) studied the molecular mechanisms of
antitumour action and the possibilities of using hydroquinone as a
toxic agent against cancer cells. In H 18R tumour-bearing male
Wistar rats treated with hydroquinone (5 mg/kg per day) for seven
days, the catalase activity was markedly depressed in liver, spleen,
blood and H 18R tumour. In vitro studies on tumour and liver
homogenates from normal and tumour-bearing rats showed a marked
inhibition of catalase activity in the tumour, which was less
evident in the liver. The activity was less reduced in normal liver
homogenates. It was suggested that the mechanism of action of
hydroquinone as an antitumour agent is achieved mainly via peroxide
production.
When tested on cultured rat hepatoma cells hydroquinone showed
a dose-dependent cytotoxic activity (Assaf et al. 1987). A dose of
33 mg/litre (300 µmol/litre) caused cellular mortality of 40% after
24 h of incubation and 66 mg/litre (600 µmol/litre) resulted in 100%
cellular mortality.
7.8.3 Neurotoxicity
Hydroquinone, given as single oral or subcutaneous lethal
doses, causes nonspecific effects on the nervous system such as
hyperexcitability, tremor and convulsions in several experimental
animal species (see section 7.1). Animals given sublethal oral doses
recover within a few days.
These central nervous system stimulation effects were confirmed
in a 90-day oral study on rats (Eastman Kodak Company, 1988) (see
also section 7.3). Male and female weanling rats (CD(SD)BR),
initially seven weeks old, were treated with hydroquinone (20, 64 or
200 mg/kg per day) dissolved in water at a concentration of 5%.
Doses were given by gavage 5 days per week. Functional-observational
battery examinations were performed throughout the study. The
battery included observations of body position, activity level,
coordination of movement and gait, behaviour, presence of
convulsions, tremors, lacrimation, salivation, piloerection,
pupillary dilatation or constriction, respiration, diarrhoea,
urination, vocalization, forelimb/hindlimb grip strength and sensory
function. Tremors and depression of general activity were observed
in both sexes shortly after dosing with 64 or 200 mg
hydroquinone/kg. Functional-observational battery examinations did
not result in any evidence of neurotoxicity as assessed by
quantitative grip strength measurement, brain weight or
neuropathological examination. The NOEL was considered to be 20 mg
hydroquinone/kg body weight.
Otsuka & Nonomura (1963) reported that hydroquinone reversed
curare blockage at neuromuscular junctions in frog sciatic nerve -
sartorius muscle preparations. The authors suggested that this
effect was due to an increased release of transmitter at the
neuromuscular junction induced by hydroquinone.
7.8.4 Nephrotoxicity
Until recently, exposure to hydroquinone has not been
associated with nephrotoxicity. Nephrotoxicity has not been reported
following either occupational exposure to hydroquinone or acute
exposures in humans. Carlson & Brewer (1953) gave human volunteers
daily hydroquinone doses of 300 or 500 mg/day for periods of up to
20 weeks without effects on urinalysis parameters. Exposure of five
male mixed-breed dogs to 100 mg hydroquinone/kg per day for 26 weeks
had no effect on urinalysis parameters or renal histopathology
(Carlson & Brewer, 1953). Christian et al. (1976) reported that
exposure of Carworth rats to hydroquinone in the drinking-water at
concentrations of up to 10 g/litre (6 rats of each sex per group for
8 weeks) or up to 4 g/litre (20 rats of each sex per group for 15
weeks) resulted in slight changes in kidney weight but no
histopathological changes. Carlson & Brewer (1953) also reported no
evidence of renal histopathological changes in Sprague-Dawley rats
fed diets containing 10g hydroquinone/kg for 104 weeks.
NTP (1989) reported that oral gavage of hydroquinone (0, 25,
50, 100, 200 or 400 mg/kg) in corn oil for 13 weeks resulted in
toxic nephropathy in F-344 rats at the two highest dose levels (200
mg/kg: 7/10 males, 6/10 females; 100 mg/kg: 1/10 females). Oral
gavage of 0, 25 or 50 mg/kg in water for 15 months resulted in an
increased incidence of chronic nephropathy in male F-344 rats (25
mg/kg: 5/5 males; 50 mg/kg: 6/10 males). When male F-344 rats were
dosed at 0, 25 or 50 mg/kg for two years, there was an increased
severity of chronic progressive nephropathy in 20/55 animals given
50 mg/kg. At a dosage level of 50 mg/kg for either 15 months or 2
years, male rats had heavier relative kidney weights.
Shibata et al. (1991) also reported that F-344 rats developed
chronic nephropathy when fed 8g hydroquinone/kg diet for 2 years.
Male rats showed increased relative and absolute kidney weight, as
well as an increased severity of chronic nephropathy (14/30
animals). Female rats showed an increased relative kidney weight,
but only a minimal increase in severity of chronic nephropathy in
7/30 animals.
Boatman et al. (1992) reported on the urinalysis changes
observed in male and female F-344 rats and Sprague-Dawley rats given
single doses of 0, 200 or 400 mg hydroquinone/kg in water by oral
gavage. B6C3F1 mice were examined after receiving doses of 0 or
350 mg/kg in a similar fashion. The placement of venous catheters in
F-344 rats increased their response to hydroquinone. At 400 mg/kg,
male and female F-344 rats, but not Sprague-Dawley rats, displayed
pronounced enzymuria and glucosuria, which resolved in 72-96 h. At
200 mg/kg, enzymuria and glucosuria were present in female F-344
rats but not males. Epithelial cell counts in the urine were
statistically significantly increased (P < 0.05) at 400 mg/kg
(male and female F-344 rats only) and 200 mg/kg (female F-344 rats
only). Statistically significant (P < 0.05) decreases in
osmolality were reported at 400 mg/kg for F-344 (both sexes) and
female Sprague-Dawley rats. Diuresis (ml urine/h) was statistically
significant (P < 0.005) only for female F-344 rats at 200 mg/kg
and 400 mg/kg. Although differences were observed in some of the
urinary parameters measured, mice were generally not responsive to
hydroquinone.
To characterize the early development of renal toxicity in
rats, cell proliferation was quantified within the proximal (P1, P2
and P3) and distal tubular segments of the kidney in rats given 0,
2.5, 25 or 50 mg hydroquinone/kg by oral gavage. Male and female
F-344 rats were treated for 1, 3 or 6 weeks, and male Sprague-Dawley
rats were treated for 6 weeks. At 6 weeks, an 87% increase in cell
proliferation was measured in the P1 segment, a 50% increase in the
P2 segment, and a 34% increase in the P3 segment from kidneys of
male F-344 rats dosed with 50 mg/kg. Urinalysis indicated increased
enzymuria in this same dose group, and mild histological changes
were present in the kidneys. Animals examined at other time points
or from other dose groups were not affected by hydroquinone.
The increased incidence of renal adenomas only in male F-344
rats (NTP, 1989) has led to speculation that the tumours observed
may be related to alpha2u-globulin-induced nephropathy. This
mechanism of action for induction of kidney tumours does not appear
to be relevant for hydroquinone as none of the studies cited above
has reported finding evidence of hyalin droplet nephropathy
following subacute, subchronic or chronic hydroquinone exposure.
Glutathione metabolites, which are at least partially formed in
the liver and transported to the kidney, are reported to be involved
in the nephrotoxicity observed. Some of the potential glutathione
conjugates of hydroquinone have been shown to be more nephrotoxic
when injected parenterally than the parent chemical (Boatman et
al., 1992; Hill et al., 1992a,b; Kleiner et al., 1992).
Administration of hydroquinone by parenteral injection, a route
which is likely to increase hydroquinone glutathione conjugates,
induces nephrotoxicity in otherwise non-responsive male
Sprague-Dawley rats (Hill et al., 1992a,b; Kleiner et al.,
1992). The formation of glutathione metabolites and an increased
susceptibility of the male F-344 rat to the conjugates appear to be
mechanistically linked to the nephrotoxicity observed in these rats.
7.8.5 Interaction with phenols
Recently there have been a number of studies reporting
interactive effects between hydroquinone and other phenolic
compounds. Initially, Eastmond et al. (1987) showed that the
co-administration of hydroquinone and phenol (75 mg/kg), when given
by intraperitoneal injection twice per day, produced a synergistic
decrease in bone marrow cellularity in B6C3F1 mice that was
similar to that induced by benzene. This combined treatment was
significantly more myelotoxic than that observed when either
hydroquinone or phenol was administered separately. Associated in
vitro studies suggested that this interactive effect was due to a
phenol-induced stimulation of the myeloperoxidase-mediated
conversion of hydroquinone to 1,4-benzoquinone in the bone marrow
(Eastmond et al., 1987; Smith et al., 1989; Subrahmanyam et
al., 1991).
Subsequent studies have indicated that interactions between
hydroquinone and other phenolic compounds can result in a variety of
cytotoxic, immunotoxic and genotoxic effects. Some of the adverse
interactive effects that have been reported are outlined below:
a) Decreased uptake of 59Fe, an indicator of toxicity to the
bone marrow, has been reported with the combined administration
of hydroquinone and various phenolic metabolites (Guy et al.,
1990, 1991).
b) The combined administration of phenol and radiolabelled
hydroquinone results in increased binding of hydroquinone
equivalents in the bone marrow (Subrahmanyam et al., 1990).
c) Decreased bone marrow cellularity and increased production of
reactive oxygen species in phagocytes when stimulated with a
phorbol ester tumour promoter have been observed following
hydroquinone and phenol co-administration (Laskin et al.,
1989).
d) Synergistic increases in the formation of micronuclei have been
observed in mice and human lymphocytes obtained from one
individual following exposure to hydroquinone and other
phenolic metabolites (Barale et al., 1990; Robertson et
al., 1991).
e) Three- to six-fold increases in DNA adduct formation (over that
observed using the sum of the individual metabolites) were
observed in HL-60 cells treated with the combination of
hydroquinone and either catechol or 1,2,4-trihydroxybenzene. In
addition, the combined treatment of hydroquinone and
1,2,4-trihydroxybenzene produced DNA adducts not detected after
treatment with either metabolite alone (Levay & Bodell, 1992).
f) Co-treatment of phenol and hydroquinone was reported to shift
the optimal concentration of hydroquinone inducing the maximal
recombinant granulocyte/macrophage colony-stimulating factor
response from 1 µmol/litre to 100 pmol/litre (Irons et al.,
1992).
g) The co-administration of hydroquinone with either phenol or
catechol in vivo to B6C3F1 mice increased the formation of
oxidative DNA damage as measured by the formation of
8-hydroxy-2'-deoxyguanosine which occurred in the bone marrow
of B6C3F1 mice (Kolachana et al., in press).
8. EFFECTS ON HUMANS
8.1 General population exposure
8.1.1 Acute toxicity - poisoning incidents
There have been reports of poisoning due to accidental or
suicidal ingestion of hydroquinone alone (Mitchell & Webster, 1919;
Rémond & Colombies, 1927) or of photographic developers containing
hydroquinone (Busatto, 1939; Zeidman & Deutl, 1945; Grudzinski,
1969; Larcan et al., 1974). Deaths have been reported after
ingestion of photographic developers containing hydroquinone in
amounts of 3-12 g (80-200 mg/kg body weight). The main symptoms of
intoxication by hydroquinone include tremors, vomiting, abdominal
pain, headache, tachycardia, convulsions, loss of reflexes, dark
urine, dyspnoea, cyanosis and coma. No adverse systemic effects have
been reported after acute inhalation of hydroquinone dust (Anderson,
1947; Oglesby et al., 1947; Sterner et al., 1947).
8.1.2 Short-term controlled human studies
In a controlled oral study, ingestion of 500 mg hydroquinone
daily for 5 months (two males) or 300 mg/day for 3-5 months (17
volunteers, both males and females) produced no observable
pathological changes in blood and urine (Carlson & Brewer, 1953). No
further data were given.
8.1.3 Dermal effects; sensitization
Skin lighteners often contain hydroquinone (1.5 to 2%) as the
bleaching agent, which inhibits the production of melanin. Prolonged
use (about three years) of strong (>5%) hydroquinone bleaching
creams has been reported to cause ochronosis and pigmented colloid
milium in South African black women (Findlay et al., 1975; Findlay
& de Beer, 1980). Sporadic skin reactions have also occurred among
amateurs who develop their own films manually (Fisher, 1986).
In a study to assess the safety of hydroquinone in cosmetic
skin-lightening products, 840 male volunteers from different human
races such as Blacks (Zulu), Asians (Indians), and Coloureds (mixed
ethnic origins) were treated with various concentrations of
hydroquinone in different bases (Bentley-Philips & Bayles, 1975).
They were subjected to open-patch tests, normal usage tests, and
standard 48 h closed-patch tests. The results of the study showed
that concentrations of hydroquinone below 3% produced negligible
adverse effects, irrespective of the base used or the colour of the
user's skin. In earlier, less extensive studies, Fitzpatrick et al.
(1966) found that a 5% cream of hydroquinone caused a high incidence
of primary irritant reactions such as erythema and tingling at the
site of application.
Fisher (1982) reported four cases of leucoderma following the
use of bleaching creams containing 2% hydroquinone. The leucoderma
was not preceded by inflammation and the patients had no positive
patch-test reaction to 1% hydroquinone in petrolatum after 72 h.
Hypopigmentation produced by 1% hydroquinone occurred in one patient
(Fisher, 1986). Spencer (1965) found that higher concentrations (5%)
of hydroquinone might cause sensitization.
Van Ketel (1984) reported a case of probable sensitization to
hydroquinone with cross-sensitization to hydroquinone monobenzyl
ether. Two days after the first application of a cream containing 5%
hydroquinone monobenzyl ether (after a treatment period of three
months with 2% hydroquinone in a cream base), an acute dermatitis
developed. Patch testing was positive for the two substances.
Sensitization to hydroquinone monobenzyl ether occurs fairly
frequently (Fisher, 1986), while hydroquinone is regarded as a weak
sensitizer.
There have been several cases in Europe and the USA of
ochronosis in people who have used creams containing 2% hydroquinone
or less (Connor & Braunstein, 1987; Lawrence et al., 1988).
Hardwick et al. (1989) established a causal link between
hydroquinone and exogenous ochronosis.
Hydroquinone, like its monobenzyl ether or monoethyl ether, has
been reported to cause severe patchy depigmentation disorders in a
confetti-like pattern in a single black man (Markey et al., 1989).
These authors also noted that four other cases had been reported.
Several cases of brown discoloration of the finger-nails due to
hydroquinone-containing skin-lightening creams have been reviewed by
Mann & Harman (1983). The colour change is considered to be due to
hydroquinone oxidation products resulting from exposure to sunlight.
8.2 Occupational exposure
8.2.1 Dermal effects
There have been case reports of occupational depigmentation of
the skin where a causal relationship between photographic developers
containing hydroquinone and depigmentation (leucoderma or vitiligo)
has been suggested (Frenk & Loi-Zedda, 1980; Kersey & Stevenson,
1981). In one case a vitiligo-like depigmentation was induced in a
black man by contact with a dilute hydroquinone solution (0.06%)
after 8-9 months (Frenk & Loi-Zedda, 1980). The depigmentation
occurred without any inflammatory skin changes and without itching.
Biopsy revealed lack of epidermal melanin pigment and melanocytes.
However, in the upper dermis there were numerous melanin-laden
macrophages. Developer containing 7% hydroquinone has also caused
vitiligo in a man wearing protective gloves (Kersey & Stevenson,
1981). The gloves were considered to have functioned as an occlusive
bandage.
Lidén (1989) carried out dermatological examination and patch
testing on 78 employees exposed to film chemicals at a film
laboratory. Hydroquinone (1% aqueous solution) was found to cause
irritation (erythema or staining) in previously non-exposed, healthy
volunteers. Contact allergy to hydroquinone (1% in water and
petrolatum) was diagnosed in four out of seven tested employees.
8.2.2 Ocular effects
Ocular lesions of various degrees have been observed in workers
exposed to quinone vapour and hydroquinone dust in the manufacture
of hydroquinone, and the clinical characteristics are well described
(Sterner et al., 1947; Oglesby et al., 1947; Anderson & Oglesby,
1958). Airborne concentrations of hydroquinone dust were
occasionally 20-35 mg/m3 (Oglesby et al., 1947). In the presence
of air and moisture hydroquinone is rapidly converted into the more
volatile quinone. Acute exposure to high (not specified) vapour
concentrations resulted in irritation, sensitivity to light,
lacrimation, injury of the corneal epithelium, and corneal
ulceration. Acute exposure to hydroquinone dust caused eye
irritation. Chronic exposure to hydroquinone dust led to corneal
staining (greenish-brown), corneal opacity, and conjunctival
staining (brownish to brownish-black), with a distribution
corresponding to the palpebral fissure (Anderson, 1947; Sterner et
al., 1947). In some cases, an appreciable loss of vision due to
permanent fine opacities, astigmatism and irregularity occurred in
the cornea (Anderson & Oglesby, 1958). Eye irritation occurred
following exposure to 2.25 mg/m3 (0.5 ppm) and became marked at
13.5 mg/m3 (3 ppm). Slowly developing inflammation and
discoloration of the cornea and conjunctiva followed daily exposures
of 0.05 to 14.4 mg hydroquinone/m3 (0.01 to 3.2 ppm) for two or
more years (Oglesby et al. 1947). The degree of eye injury showed
a positive correlation only with length of employment (Anderson,
1947; Sterner et al., 1947; Anderson & Oglesby, 1958). The ocular
lesions developed gradually over a period of several years of
exposure; no serious cases were seen until after five or more years
of exposure. Removal from exposure resulted in considerable
improvement of the staining, but improvement of the corneal
opacities was questionable. The relative contribution of quinone
vapour and hydroquinone dust to eye injury was not assessed.
Three cases of corneal pigmentation were described among men
working in a hydroquinone factory (Naumann, 1966). At a later date,
corneal damage became apparent even though exposure had been
discontinued, and progressive deterioration of vision was reported.
Histopathological examinations revealed pigmentary and degenerative
changes. Two distinct forms of pigment were distinguished. One was
intraepithelial and contained iron, the other was confined to a
band-like zone between normal and altered stroma and was considered
to be quinone.
An operation nurse repeatedly developed a corneal ulcer while
mixing bone cement containing methyl methacrylate and hydroquinone
(Norrelykke Nissen & Corydon, 1985). It was suggested that the eye
symptoms developed because of a composite effect of methyl
methacrylate and hydroquinone vapours. However, the presence of
hydroquinone or quinone vapour in the air was not determined.
8.2.3 Systemic effects
Clinical and laboratory evaluation of 88 workers in 1943 and
101 workers in 1945, who had been exposed to high airborne
concentrations of both quinone and hydroquinone for periods up to 15
years, showed no evidence of any systemic toxicity (Sterner et
al., 1947).
8.2.4 Epidemiological studies
8.2.4.1 Respiratory effects
In a study on airway responses to hydroquinone (Choudat et
al., 1988), 33 workers exposed to hydroquinone, trimethyl-
hydroquinone, and retinene-hydroquinone were compared to a reference
group of 55 matched, non-exposed workers regarding the potential
allergic effect of the exposure. No further information on exposure
conditions (nature, extent and duration) and possible exposure to
other chemicals was reported. The prevalence of respiratory symptoms
was increased in workers exposed to hydroquinone and its
derivatives. The exposed workers had a significantly (P < 0.01)
higher prevalence of cough induced by a smoky atmosphere or by cold
air. The prevalences of eczema and coughing at work were also
higher. Pulmonary function values were significantly (P < 0.01)
lower in the exposed than in the non-exposed group. According to the
results from a bronchodilator test, hydroquinone or its derivatives
also seemed to induce intermittent dyspnoea and reversible
obstruction. The exposed workers had higher levels of IgG (P <
0.002) and IgE (not significant) than the non-exposed workers.
However, because of the higher chemical reactivity of trimethyl-
hydroquinone compared with hydroquinone, it is difficult to
determine the effects of hydroquinone alone (O'Brien, 1991).
8.2.4.2 Carcinogenicity studies
Greenwald et al. (1981) performed a nested case-referent
study of brain cancer in a group of employees from the Eastman Kodak
Company. This study was prompted by preliminary data suggesting a
possible increase of brain cancer incidence in New York state, USA,
and included a case group of 56 employees who had died with primary
brain tumours during the period 1956-1975. An elevated relative risk
was found among photographic processing workers exposed to colour
developers, but the results were not statistically significant. The
authors stated that the excess of brain neoplasms noted may have
resulted from a diagnostic sensitivity bias arising from more
complete medical evaluation of Kodak employees than of other plant
employees.
A cohort study of 478 photographic processors in nine Eastman
Kodak Color Print and Processing laboratories was undertaken as a
follow-up study. The primary objectives of the study were to
evaluate mortality, cancer incidence, and absence due to sickness of
employees exposed to photographic chemicals. Both industrial and
general population references were used. There was no significant
increase in the incidence of the parameters investigated. Two cases
classified as malignant neoplasms on the brain and CNS were observed
versus 0.4 expected (Friedlander et al., 1982), but this
difference was not statistically significant. Individual exposures
were not examined, but hydroquinone was identified among the many
possible exposures. One air sample analysed for hydroquinone
contained < 0.01 mg/m3.
Work-related mortality among male employees at Tennessee
Eastman Company was studied by Pifer et al. (1986). The exposure
included hydroquinone among other chemical agents. Cancer mortality
was lower than that of the general population; the standard
mortality ratio was 56. No information on levels of hydroquinone
exposure or total number of workers exposed to hydroquinone was
reported. However, work is underway to explore the feasibility of
conducting a mortality and, possibly, a morbidity study of
hydroquinone workers at this facility (personal communication to the
IPCS, 1992).
9. EFFECTS ON OTHER ORGANISMS IN THE LABORATORY AND FIELD
Hydroquinone has been shown to be an outlier in numerous QSAR
(Quantitative Structure-Activity Relationship) studies (e.g.,
Devillers et al., 1986, 1987; Hodson et al., 1988; Nendza &
Seydel, 1988a,b,c). Due to this fact Devillers et al. (1990)
reviewed the environmental and health risks of hydroquinone.
Ecotoxicity data for hydroquinone are listed in Table 18. From these
data, it appears that hydroquinone is highly toxic for most of the
organisms studied. However, a difference in sensitivity exists among
the taxons. It should be noted that the results need to be analysed
in relation to the experimental conditions under which they were
obtained (e.g., pH, light). Thus, numerous studies have been
performed under static condition. In only a few experiments has the
concentration of hydroquinone been monitored.
Table 18. Ecotoxicity data for hydroquinone
Species Effect Test Hydroquinone Comments Reference
duration concentration
(mg/litre)
Bacteria
Beneckea harveyi 50% inhibition of luminescence 10 sec 82.6 Stom et al.
100% inhibition of luminescence 10 sec 550.6 (1986)
50% inhibition of dehydrogenase 1 h 110
activity
Escherichia coli toxicity threshold concentration 6 h 50 Bringmann &
for inhibition of acid production Kühn (1959a)
from glucose
50% inhibition of cell 6-8 h 34.0 Devillers et
multiplication al. (1990)
Photobacterium EC10, luminescence 30 min 0.022 Devillers et
phosphoreum EC50 0.072 al. (1990)
EC90 0.210
EC50, luminescence 5 min 0.042 Ribo & Kaiser
10 min 0.038 (1983)
30 min 0.038
Pseudomonas zone of growth inhibition on 24 h 200 the zone of inhibition Trevors &
fluorescens agar was 14 mm; after exposure Basaraba
the % survival of resting (1980)
cells was 0.11
Pseudomonas putida toxicity threshold concentration 16 h 58 Bringmann &
for inhibition of cell Kühn (1977a)
multiplication
Table 18. (contd).
Species Effect Test Hydroquinone Comments Reference
duration concentration
(mg/litre)
Dinoflagellates
Crypthecodinium 50% mortality 40 h 50.0 Devillers et
cohnii al. (1990)
Prorocentrum micans 50% immobilization 1 h 0.750 Devillers et
50% immobilization 2 h 0.300 al. (1990)
100% immobilization 18 h 5.00
Cyanobacteria
(Blue-green algae)
Anabaena flos-aque lowest concentration for no 14 days 39.8 irradiance of 2 W/m2 Wängberg &
detectable growth Blanck (1988)
Anabaena sp. lowest concentration for no 14 days 10.0 irradiance of 2 W/m2 Wängberg &
detectable growth Blanck (1988)
"LPP sp" 1 PCC6402 lowest concentration for no 14 days 5.01 irradiance of 10 W/m2 Wängberg &
detectable growth 20.0 irradiance of 2 W/m2 Blanck (1988)
"LPP sp" 2 PCC73110 lowest concentration for no 14 days 5.01 irradiance of 2 W/m2 Wängberg &
detectable growth Blanck (1988)
Microcystis toxicity threshold concentration 8 days 1.1 Bringmann &
aeruginosa for inhibition of cell Kühn (1978)
multiplication
EC100 24 h 0.5 Fitzgerald et
al. (1952)
Synechococcus lowest concentration for no 14 days 10.0 irradiance of 10 W/m2 Wängberg &
leopoliensis detectable growth 10.0 irradiance of 2 W/m2 Blanck (1988)
Table 18. (contd).
Species Effect Test Hydroquinone Comments Reference
duration concentration
(mg/litre)
Algae
Bumilleriopsis lowest concentration for no 14 days 10.0 irradiance of 10 W/m2 Wängberg &
filiformis detectable growth Blanck (1988)
Chlamydomonas lowest concentration for no 14 days 79.4 irradiance of 10 W/m2 Wängberg &
dysosmos detectable growth Blanck (1988)
Chlamydomonas 100% inhibition of motility 15 min 55.1 Stom & Roth
reinhardii (1981)
Chlorella emersonii lowest concentration for no 14 days 79.4 irradiance of 10 W/m2 Wängberg &
detectable growth Blanck (1988)
slight activation of growth 24 h 10.0 Devillers et
al. (1990)
Dunaliella marina activation of growth 24 h 10.0 Devillers et
al. (1990)
Dunaliella salina 100% inhibition of motility 15 min 330.3 Stom & Roth
(1981)
Euglena gracilis 100% inhibition of motility 15 min 7708 Stom & Roth
(1981)
Kirchneriella contorta lowest concentration for no 14 days 79.4 irradiance of 10 W/m2 Wängberg &
detectable growth Blanck (1988)
Klebsormidium lowest concentration for no 14 days 20.0 irradiance of 10 W/m2 Wängberg &
marinum detectable growth Blanck (1988)
Table 18. (contd).
Species Effect Test Hydroquinone Comments Reference
duration concentration
(mg/litre)
Monodus lowest concentration for no 14 days 20.0 irradiance of 10 W/m2 Wängberg &
subterraneus detectable growth Blanck (1988)
Monoraphidium lowest concentration for no 14 days 20.0 irradiance of 10 W/m2 Wängberg &
pusillum detectable growth 79.4 irradiance of 2 W/m2 Blanck (1988)
Nitella sp. 100% inhibition of cytoplasmic 15 min 2753 Stom & Roth
streaming (1981)
Raphidonema lowest concentration for no 14 days 0.316 irradiance of 10 W/m2 Wängberg &
longiseta detectable growth Blanck (1988)
Scenedesmus lowest concentration for no 14 days 39.8 irradiance of 10 W/m2 Wängberg &
obtusiusculus detectable growth Blanck (1988)
Scenedesmus toxicity threshold concentration for 96 h 4 Bringmann &
quadricauda inhibition of cell multiplication Kühn (1959a)
toxicity threshold concentration for 7 days 0.93 Bringmann &
inhibition of cell multiplication Kühn (1978)
Selenastrum EC50, growth 3 days 0.335 irradiance of 17 W/m2 Devillers et
capricornutum al. (1990)
lowest concentration for no 14 days 79.4 irradiance of 10 W/m2 Wängberg &
detectable growth Blanck (1988)
Tribonema aequale lowest concentration for no 14 days 2.51 irradiance of 10 W/m2 Wängberg &
detectable growth Blanck (1988)
Table 18. (contd).
Species Effect Test Hydroquinone Comments Reference
duration concentration
(mg/litre)
Yeasts
Candida albicans activation of growth 24 h 500 Devillers et
50% inhibition of growth 24 h 3750 al. (1990)
Candida tropicalis R2 100% inhibition of growth 24 h 1000 Devillers et
al. (1990)
Saccharomyces 50% inhibition of growth 24 h 2750 Devillers et
cerevisiae al. (1990)
Torulopsis glabrata 50% inhibition of growth 24 h 1000 Devillers et
100% inhibition of growth 3000 al. (1990)
Fungi
Fusarium oxysporum no significant inhibition of spore 1000 Ismail et al.
f sp lycopersici germination (1987)
no significant inhibition of length 1000
of germ tube
Plants
Elodea canadensis 50% inhibition of growth 9 days 42.9 Stom & Roth
(1981)
Lemna minor 50% inhibition of plant 12 days 7.71 Stom & Roth
multiplication (1981)
Vallisneria spiralis 100% inhibition of cytoplasmic 15 min 2753 in leaves Stom & Roth
streaming 275.3 in roots (1981)
Protozoa
Chilomonas toxicity threshold concentration for 48 h 22 Bringmann &
paramaecium inhibition of cell multiplication Kühn (1981)
Table 18. (contd).
Species Effect Test Hydroquinone Comments Reference
duration concentration
(mg/litre)
Colpidium campylum EC50, growth 24 h 73.3 Devillers et
al. (1990)
Entosiphon sulcatum toxicity threshold concentration for 72 h 11 Bringmann
inhibition of cell multiplication (1978)
Microregma toxicity threshold concentration for 28 h 2 Bringmann &
nutrient uptake Kühn (1959b)
Tetrahymena EC50, growth 60 h 95.0 Schultz et al.
pyriformis (1987)
Uronema parduczi toxicity threshold concentration for 20 h 21 Bringmann &
inhibition of cell multiplication Kühn (1980)
Mollusc
Deroceras reticulatum 0% mortality 4 days 0.020a Briggs &
20% mortality 4 days 0.200a Henderson
(1987)
Crustacea
Artemia salina LC50 2 h 321 Devillers et
4 h 67.5 al. (1990)
6 h 57.5
24 h 20.7
Crangon LT50 84 h 0.83 time to 50% mortality McLeese et al.
septemspinosa (1979)
Daphnia magna toxicity threshold concentration for 48 h 0.60 Bringmann &
inhibition of mobility Kühn (1959a)
Table 18. (contd).
Species Effect Test Hydroquinone Comments Reference
duration concentration
(mg/litre)
Daphnia magna LC0 24 h 0.04 Bringmann &
LC50 0.09 Kühn (1977b)
LC100 0.31
EC0, inhibition of mobility 24 h 0.05 Bringmann &
EC50 0.12 Kühn (1982)
EC100 0.19
EC50, inhibition of mobility 24 h 0.137 Devillers et
al. (1987)
EC0, inhibition of mobility 24 h 0.13 Kühn et al.
EC50 0.32 (1989)
EC100 0.71
EC0 48 h 0.13 Kühn et al.
EC50 0.29 (1989)
EC100 0.71
EC50, inhibition of mobility 24 h 0.15 Tissot et al.
(1985)
Daphnia pulicaria LC50 48 h 0.162 DeGraeve et
al. (1980)
Daphnia pulex LC100 6 min 8809 Stom et al.
(1986)
Gammarus toxicity threshold concentration 1.5 Bandt (1955)
Insect
Apis mellifera LD50 24 h 0.200 concentration in mg per bee Devillers et
al. (1990)
Table 18. (contd).
Species Effect Test Hydroquinone Comments Reference
duration concentration
(mg/litre)
Fish
Brachydanio rerio LC50 24 h 0.265 Devillers et
al. (1988)
LC0 96 h 0.12 Wellens (1982)
LC50 0.17
LC100 0.25
Carassius auratus 100% mortality 22 h 5 US EPA (1987)
LC100 48 h 0.287 Sollmann
(1949)
Lepomis macrochirus 100% mortality 22 h 5 US EPA (1987)
Leuciscus idus LC0 48 h 0.1 Juhnke &
melanotus LC50 0.15 Lüdemann
LC100 0.2 (1978)
LC0 0.1
LC50 0.16
LC100 0.25
Oncorhynchus mykiss 100% mortality 2 h 7.69 Devillers et
4 h 4.50 al. (1990)
LC50 96 h 0.097 DeGraeve et
al. (1980)
LC50 96 h 0.639 Hodson et al.
LD50, intraperitoneal injection 24.2 concentration in mg per kg (1984)
Table 18. (contd).
Species Effect Test Hydroquinone Comments Reference
duration concentration
(mg/litre)
Pimephales promelas LC50 96 h 0.044 DeGraeve et
al. (1980)
LT50 11 h 0.2 time to 50% mortality Terhaar et al.
(1972)
Salmo trutta 100% mortality 22 h 5 US EPA (1987)
a Slugs injected with 2 µl of glycerol formal containing 20 or 200 µg of hydroquinone.
10. EVALUATION OF HUMAN HEALTH RISKS AND ON THE ENVIRONMENT
10.1 Toxicokinetics
The property of hydroquinone most pertinent to its toxicity is
its ability to undergo reversible redox reactions. Autooxidation of
hydroquinone leads to p-benzoquinone and/or p-benzosemiquinone.
These two strong electrophiles do not redox-cycle under
physiological conditions to form active oxygen species, but readily
arylate nucleophiles. They are probably the two major toxic
metabolites of hydroquinone.
Toxicokinetic studies with hydroquinone show that although it
is readily absorbed from the gut of animals it has a low potential
for bioaccumulation < 2% distributed out of total administered
dose). Extensive conjugation and rapid excretion, primarily via the
urine, suggests that hydroquinone is effectively detoxified.
However, because hydroquinone is oxidized to p-benzosemiquinone
and/or p-benzoquinone, which are able to readily react with
nucleophilic body components, it represents a potentially harmful
toxicant. Indeed, hydroquinone and/or its metabolites covalently
bind to cellular components in vitro.
It is, therefore, possible that although the bioaccumulation
potential of hydroquinone is low critical body components may still
be adversely affected.
10.2 Animal and in vitro studies
Hydroquinone exhibits moderately high acute oral toxicity for
animals, with LD50 values generally being in the range of 300 to
1300 mg/kg. However, cats are more sensitive, LD50 values of 40-85
mg/kg having been reported. The principle toxic effects of a single
oral lethal dose of hydroquinone are increased motor activity,
dyspnoea and cyanosis followed by convulsions, paralysis, coma and
death. Repeated oral dosing has caused tremors and reduced activity
(> 64 mg/kg), reduced body weight gain (> 200 mg/kg),
convulsions (> 400 mg/kg) and adverse effects on the liver and
kidneys (> 200 mg/kg).
Dermal exposure of black guinea-pigs to hydroquinone (2 or 5%)
has been found to cause depigmentation, inflammatory changes and
thickening of the epidermis. Slight skin irritation has been
recorded following topical application of hydrophillic ointment
containing 1% hydroquinone. The results also showed that female
guinea-pigs were more sensitive than males.
Limited data suggest that powdered hydroquinone causes
transient eye irritation and corneal opacity in dogs and
guinea-pigs; in rabbits powdered hydroquinone induced brownish
pigmentation of conjunctiva and cornea, but only after a period of
at least 2-4 months.
Hydroquinone is a skin sensitizer in rabbits. The ability to
induce sensitization has been found to vary from "weak" to "strong"
depending on the test procedure and vehicle used. The cross-
reactivity of hydroquinone and p-methoxyphenol has been reported
to be almost 100%.
Early reproductive studies indicated reduced fertility in male
rats and a disturbed sexual cycle in female rats when hydro-quinone
was administered parentally. However, this was not confirmed in more
recent studies in rats, i.e. a dominant lethality study and a
two-generation study with oral doses of hydroquinone up to 300 mg/kg
per day and 150 mg/kg per day, respectively.
Oral dosing of 100 or 300 mg hydroquinone/kg to pregnant rats
on days 6-15 of gestation caused maternal toxicity at the higher
dose level (a statistically significant reduction in body weight
gain and feed consumption). A reduction in mean fetal body weight
was correlated with the reduced maternal body weight. No
compound-related teratogenic effects were produced at this dose
level; thus, 100 mg/kg was considered the NOEL for maternal and
developmental toxicity in rats. Findings of increased resorption
rates in rats given hydroquinone orally at about 100 mg/kg per day
were not confirmed in this study, and, consequently, the NOAEL for
maternal reproductive effects and teratogenicity was 300 mg/kg. In
rabbits, 150 mg/kg caused reductions in body weight and feed intake
and an increased (but not statistically significant) increase of
malformations in the fetuses. The malformations may have been
associated with maternal toxicity. The dose level of 200 mg/kg
produced an increased number of resorptions, indicating
embryotoxicity. The NOEL for developmental toxicity in rabbits was
25 mg/kg per day.
In a two-generation reproduction study in rats the NOAEL for
reproductive effects through two generations was 150 mg/kg per day
(the highest tested dose). In mice, 80 mg hydroquinone/kg given
orally on the 13th day of gestation transplacentally induced
micronuclei in fetal liver cells. A single oral dose of 1000 mg/kg
on gestation day 11 caused maternal toxicity (decreased weight gain)
and an increased incidence of mortality. Reduced litter size and
perinatal loss occurred in the treated groups (333, 667 and 1000
mg/kg), together with dose-related malformations of limbs, tail and
urogenital system. In a rat embryo culture system, hydroquinone
(effective concentration < 55 mg/litre, < 0.5 mmol/litre)
caused growth retardation and an increased incidence of structural
abnormalities involving tail and hindlimb buds.
Genotoxicity data indicates that hydroquinone induces
micronuclei, structural chromosome aberrations, and c-mitotic
effects in vivo in mouse bone-marrow cells. in vitro studies
with various cell lines showed that hydroquinone was capable of
inducing gene mutations, structural chromosome aberrations,
sister-chromatid exchange and DNA damage. Hydroquinone induces
chromosome aberrations or karyotypic alterations in plant species
and mitotic crossing-over in fungi. Hydroquinone is not mutagenic in
the Salmonella/microsome test, but induces repairable DNA damage
in Escherichia coli. It produces adducts with DNA.
Hydroquinone induces chromosome aberrations in germ cells of
male mice.
Cholesterol pellets containing 20% hydroquinone implanted into
the bladders of mice produced bladder tumours in 6 out of 19 mice
surviving 25 weeks. However, the study was incompletely reported and
the method is not generally recognized as a valid measure of
carcinogenic potential as small pellets of cholesterol are known to
induce transitional carcinoma of the bladder in both rats and mice.
There is no support for hydroquinone being a stomach carcinogen in
experimental animals after oral dosing. Hydroquinone produced an
increase in the number of liver foci in rats at a dose level of 100
mg/kg per day for 7 weeks. However, increased dose levels (200 mg/kg
per day for 7 weeks or 8 g/kg diet for about two years) caused a
reduction in the number of foci of cellular alteration of the liver.
In mice, the incidence of liver foci was increased when hydroquinone
was added to the diet at 8 g/kg.
Hydroquinone did not show any potential as an initiator or a
co-carcinogen when dermally applied to mice before application of a
tumour promoter (croton oil or BP) or following co-exposure with BP.
Orally administered hydroquinone showed no promotion activity after
MNNG initiation or carcinogenic potential in the forestomach and
glandular stomach of rats.
Most of the earlier carcinogenicity studies on hydroquinone
lasted for less than one year, which might be considered too short
for the assessment of carcinogenicity.
Two-year studies performed recently give support for
hydroquinone being a carcinogen in F-344 rats and B6C3F1 mice. In
an NTP study, renal tubular cell adenomas occurred in male rats and
leukaemia in females, and hepatocellular neoplasms, mainly adenomas,
in female mice. The NTP concluded that these data indicated "some
evidence of carcinogenic activity" in male and female rats and in
female mice. In another study, renal tubular cell adenomas were
again noted in male rats; hepatocellular adenomas occurred in male
mice along with a biologically significant increase in the incidence
of renal tubular cell adenomas.
Both in vivo and in vitro studies have shown that
hydroquinone causes direct myelotoxic effects in mouse bone marrow
stromal cells by reducing bone marrow cellularity. In reducing the
number of progenitor B-lymphocytes in mouse spleen and bone marrow,
hydroquinone also demonstrates an immunosuppressive potential.
Moreover, hydroquinone may inhibit the natural killer activity of
mouse spleen cells in vitro, and have a selective effect on
macrophage functions important in host defence.
Hydroquinone has also demonstrated cytotoxic activity on
various tumour cells such as cells from melanoma transplants and rat
hepatoma cells.
Central nervous system stimulatory effects have been produced
in animal studies. However, functional-observational battery and
neuropathological examinations failed to give any evidence of
neurotoxicity after repeated dosing for 90 days. The NOEL was 20 mg
hydroquinone/kg per day.
10.3 Evaluation of human health risks
10.3.1 Exposure
Potential exposure of the general population to hydroquinone
may occur through the consumption of foods that contain hydroquinone
as a natural component, through smoking or exposure to cigarette
smoke, or from the use of cosmetics and skin-lightening creams.
People who use skin lighteners with concentrations of hydroquinone
exceeding 2%, apply creams over large areas of the body or use
creams for long time periods represent one group with significant
and sometimes excessive exposure. Heavy cigarette smokers and those
living and working in environments contaminated by cigarette smoke
represent another group that experiences significant exposure to
hydroquinone. Photohobbyists who develop their film manually may
also be exposed to hydroquinone solutions through skin contact and
inhalation.
Exposure to hydroquinone may occur in a variety of occupations,
particularly among those involved in its manufacture.
10.3.2 Human health effects
Ingestion of large quantities of hydroquinone may produce
tremors, vomiting, convulsions, dyspnoea, cyanosis and coma. Deaths
have been reported to occur after ingestion estimated at 3-12 g of
hydroquinone in developing agents. In studies with human volunteers,
ingestion of up to 500 mg hydroquinone per day over a 20-week period
resulted in no observable pathological changes in the blood and
urine.
Dermal exposure to hydroquinone causes skin depigmentation;
cases of ochronosis, patchy depigmentation and brown staining of the
nails after repeated usage of skin-lightening products have been
reported. Hydroquinone has shown a sensitizing potential in both
animals and humans.
Eye irritation, sensitivity to light, staining of the cornea
and conjunctiva, corneal opacities and visual disturbances are
associated with long-term occupational exposure to airborne
hydroquinone. Isolated cases of corneal ulceration have also been
described.
Effects on the central nervous system have been seen in cases
of acute human poisoning. Similar symptoms have been observed in
animal studies; these effects were reversible when exposure was
discontinued.
Nephrotoxicity has been seen in F-344 rats dosed repeatedly
with hydroquinone. Male rats are more susceptible to these effects
than females. Nephrotoxic effects due to hydroquinone have not been
observed in humans.
Myelotoxic and immunotoxic effects have been observed in
animals exposed to hydroquinone. However, the routes of exposure
differed from those via which humans are normally exposed.
Studies conducted by routes of exposure similar to those by
which humans are exposed have not revealed specific reproductive and
developmental effects.
Numerous studies using cell culture systems and in vivo
rodent experiments have shown that co-exposure to hydroquinone and
various phenolic compounds can result in toxic effects that are
substantially greater than the sum of the effects of the individual
compounds. The relevance of these interactive effects in
understanding and predicting the toxic effects of human exposure to
hydroquinone is uncertain. However, since many of the human
exposures to hydroquinone occur under conditions in which other
phenolic compounds are present, the possibility that significant
interactive effects may occur should be considered.
Several experiments with hydroquinone in vivo and in vitro
have shown mutagenic effects; the relevance of these results to
human risk is uncertain. The evidence for carcinogenicity in animals
is limited. Adequate epidemiological studies are lacking, and, at
present, the experimental data are insufficient to allow a thorough
assessment of the carcinogenic potential for humans.
10.4 Evaluation of effects on the environment
When hydroquinone is released into the environment it is
distributed mainly to the water compartment due to its
physicochemical properties. However, hydroquinone can be degraded
both photochemically and biologically and will therefore not persist
in the environment. The ecotoxicity of hydroquinone, which can be
also related to its physicochemical properties, is generally high
but varies from species to species. Therefore, a battery of tests
using organisms occupying different trophic levels in the ecosystems
is required to assess thoroughly the adverse effects of hydroquinone
in the environment.
11. RECOMMENDATIONS
a) In view of the widespread inappropriate use of skin-lightening
creams, it is recommended that over-the-counter sales of creams
containing hydroquinone be restricted. Health Education
Programmes should be developed to discourage the use of
hydroquinone-containing creams for whole body skin lightening.
b) Further investigations into the safety of long-term use of
creams containing 1-2% hydroquinone is needed.
c) Hydroquinone in waste water effluent should be allowed
sufficient time for degradation before reaching recipient
water.
d) Multispecies toxicity testing performed under controlled
experimental conditions is required to make a thorough
assessment of the environmental effects of hydroquinone and its
derivatives.
e) Epidemiological studies, including precise exposure data, would
assist in an assessment of the occupational hazards from
hydroquinone. Epidemiological information, including
reproductive effects, is also required for users of skin
lighteners, particularly women. More data on human exposure
from different sources are required, particularly with respect
to dietary exposure.
12. PREVIOUS EVALUATIONS BY INTERNATIONAL BODIES
In 1977 the International Agency for Research on Cancer (IARC)
Working Group concluded that the available data on hydroquinone did
not allow an evaluation of its carcinogenicity.
Hydroquinone was evaluated by a Nordic Expert Group for
Documentation of Occupational Exposure Limits in 1989. It was
recommended that its genotoxic effects should be given attention and
also its possible effects on the immune system, bone marrow, skin
and mucous membranes.
REFERENCES
Abramowitz J & Chavin W (1980) Acute effects of two melanocytolytic
agents, hydroquinone and ß-mercaptoethanolamine, upon tyrosinase
activity and cyclic nucleotide levels in murine melanomas. Chem-Biol
Interact, 32: 195-208.
Adler I-D & Kliesch U (1990) Comparison of single and multiple
treatment regimens in the mouse bone marrow micronucleus assay for
hydroquinone (HQ) and cyclophosphamide (CP). Mutat Res, 234:
115-123.
Adler I-D, Kliesch U, van Hummelen P & Kirsch-Volders M (1991) Mouse
micronucleus tests with known and suspect spindle poisons: results
from two laboratories. Mutagenesis, 6(1): 47-53.
Altmann H-J, Grunow W, Wester PW, & Mohr U (1985) Induction of
forestomach lesions by butylhydroxyanisole and structurally related
substances. Arch Toxicol, 8(Suppl): 114-116.
Ames SR, Ludwig MJ, Swanson WJ, & Harris PL (1956) Effect of DPPD,
methylene blue, BHT, and hydroquinone on reproductive process in the
rat. Proc Soc Exp Biol Med, 93: 39-42.
Anderson B (1947) Corneal and conjunctival pigmentation among
workers engaged in manufacture of hydroquinone. Arch Ophthalmol, 38:
812-826.
Anderson B & Oglesby F (1958) Corneal changes from
quinone-hydroquinone exposure. Am Med Assoc Arch Opthalmol, 59:
495-501.
Antoccia A, Degrassi F, Battistoni A, Ciliutti P, & Tanzarella C
(1991) In vitro micronucleus test with kinetochore staining:
evaluation of test performance. Mutagenesis, 6(4): 319-324.
Arndt KA & Fitzpatrick TB (1965) Topical use of hydroquinone as a
depigment agent. J Am Med Assoc, 194: 965-967.
Assaf MH, Ali AA, Makboul MA, Beck JP, & Anton R (1987) Preliminary
study of phenolic glycosides from Origanum majorana; quantitative
estimation of arbutin; cytotoxic activity of hydroquinone. Planta
Med, 53: 343-345.
Baernstein HD (1945) The determination of catechol, phenol, and
hydroquinone in urine. J Biol Chem, 161: 685-692.
Bandt HJ (1955) [Damage to fishing grounds caused by waste water
containing phenols.] Wasserwirtsch-Wassertech, 5: 290-294 (in
German).
Barale R, Marazzini A, Betti C, Vangelisti V, Loprieno N, & Barrai I
(1990) Genotoxicity of two metabolites of benzene: phenol and
hydroquinone show strong synergistic effects in vivo. Mutat Res,
244: 15-20.
Basketter DA & Goodwin BFJ (1988) Investigation of the prohapten
concept. Contact Dermatitis, 19: 248-253.
Baumbach HL (1946) An improved method for the determination of
hydroquinone and metol in photographic developers. J Soc Motion Pict
Eng, 47: 403-408.
Bentley-Phillips B & Bayles MAH (1975) Cutaneous reactions to
topical application of hydroquinone. Results of a 6-year
investigation. S Afr Med J, 49: 1391-1395.
Bilimoria MH (1975) The detection of mutagenic activity of chemicals
and tobacco smoke in a bacterial system (Abstract). Mutat Res, 31:
328.
Bio/dynamics Inc. (1988) A range-finding study to evaluate toxicity
of hydroquinone in the pregnant rabbit (Project No. 87-3218): Final
report. East Millstone, New Jersey, Bio/dynamics Inc. (Prepared for
the Chemical Manufacturers Association, Washington).
Bio/dynamics Inc. (1989a) A developmental toxicity study in rabbits
with hydroquinone (Project No. 87-3220): Final report. East
Millstone, New Jersey, Bio/dynamics Inc. (Prepared for the Chemical
Manufacturers Association, Washington).
Bio/dynamics Inc. (1989b) A two-generation reproduction study in
rats with hydroquinone (Project No. 87-32l9): Final report. East
Millstone, New Jersey, Bio/dynamics Inc. (Prepared for the Chemical
Manufacturers Association, Washington).
Bleehen SS, Pathak MA, Hori Y, & Fitzpatrick TB (1968)
Depigmentation of skin with 4-isopropylcatechol, mercaptoamines, and
other compounds. J Invest Dermatol, 50: 103-117.
Boatman RS, English JC, Perry LG, & Bialecki VE (1992) Indications
of nephrotoxicity for hydroquinone and for 2-(glutathion-S-YL)
hydroquinone in Fischer-344 and Sprague-Dawley rats and in B6C3F1
mice. Rochester, New York, Eastman Kodak Company, Health and
Environment Laboratories (Unpublished data).
Borecky J (1963) [Identification of organic compounds-Report LIV.
Separation and identification of photographic developers by paper
chromatography.] J Chromatogr, 12: 385-393 (in German).
Boyland E, Busby ER, Dukes CE, Grover PL, & Manson D (1964) Further
experiments on implantation of materials into the urinary bladder of
mice. Br J Cancer, 18: 575-581.
Boyle J & Kennedy CTC (1985) Leukoderma from hydroquinone. Contact
Dermatitis, 13: 287-288.
Boyle J & Kennedy CTC (1986) Hydroquinone concentrations in skin
lightening creams. Br J Dermatol, 114: 501-504.
Brauer EW (1985) Safety of over-the-counter hydroquinone bleaching
creams. Arch Dermatol, 121: 1239.
Briggs GG & Henderson IF (1987) Some factors affecting the toxicity
of poisons to the slug Deroceras reticulatum (Müller) (Pulmonata:
Limacidae). Crop Prot, 6: 341-346.
Bringmann G (1978) [Determination of the biological harmful effect
of water pollutants on protozoa. I. Bacteriophagic flagellates
(model organism: Entosiphon sulcatum Stein).] Z Wasser
Abwasser-Forsch, 11: 210-215 (in German).
Bringmann G & Kühn R (1959a) [Comparative water toxicology studies
on bacteria, algae and small crustaceans.] Gesundheits-Ingenieur, 4:
115-120 (in German).
Bringmann G & Kühn R (1959b) [Water toxicology studies using
protozoa as test organisms.] Gesundheits-Ingenieur, 8: 239-240 (in
German).
Bringmann G & Kühn R (1977a) [Threshold values for the harmful
effect of water pollutants on bacterio (Pseudomonas putida) and
green algae (Scenedesmus quadricauda) in the cell reproduction
inhibition test.] Z Wasser Abwasser-Forsch, 10: 87-98 (in German).
Bringmann G & Kühn R (1977b) [Findings concerning the harmful effect
of water pollutants on Daphnia magna.] Z Wasser Abwasser Forsch,
10: 161-166 (in German).
Bringmann G & Kühn R (1978) [Threshold values for the harmful effect
of water pollutants on blue algae (Microcystis aeruginosa) and
green algae (Scenedesmus quadricauda) in the cell reproduction
inhibition test.] Vom Wasser, 50: 45-60 (in Grman).
Bringmann G & Kühn R (1980) [Determination of the biological harmful
effect of water pollutants on protozoa. II. Bacteriophagic
ciliates.] Z Wasser Abwasser Forsch, 13: 26-31 (in German).
Bringmann G & Kühn R (1981) [Comparison of the effect of harmful
substances on flagellates and ciliates with the effect of holozoic
bacteriophagic and saprozoic protozoa.] GWF-Wasser/Abwasser, 122:
308-313 (in German).
Bringmann G & Kühn R (1982) [Findings concerning the harmful effect
of water pollutants on Daphnia magna in an advanced standardized
test procedure.] Z Wasser Abwasser Forsch, 15: 1-6 (in German).
Brunmark A & Cadenas E (1988) Reductive addition of glutathione to
p-benzoquinone, 2-hydroxy- p-benzoquinone, and p-benzoquinone
epoxides. Effect of the hydroxy- and glutathionyl substituents on
p-benzohydroquinone autoxidation. Chem-Biol Interact, 68: 273-298.
Brunner AH Jr, Means PB Jr, & Zappert RH (1949) Analysis of
developers and bleach for Ansco color film. J Soc Motion Pict Eng,
53: 25-35.
Brunner M, Albertini S, & Würgler FE (1991) Effects of 10 known or
suspected spindle poisons in the in vitro porcine brain tubulin
assembly assay. Mutagenesis, 6(1): 65-70.
Bucks DAW, McMaster JR, Guy RH, & Maibach HI (1988) Percutaneous
absorption of hydroquinone in humans: effect of
1-dodecylazacycloheptan-2-one (azone) and the 2-ethylhexyl ester of
4-(dimethylamino)benzoic acid (escalol 507). J Toxicol Environ
Health, 24: 279-289.
Bulman C & Serva RJ (1982) Mutagenicity evaluation of Eastman
hydroquinone. Salmonella typhimurium/microsome bioassay. Akron,
Ohio, The Goodyear Tyre & Rubber Company (Laboratory report No
82-5-1).
Bulman C & Van der Sluis D (1980) Mutagenicity evaluation of 302
bottoms (Bayport Plant Stream). Salmonella typhimurium/microsome
bioassay. Akron, Ohio, The Goodyear Tire & Rubber Company
(Laboratory report No. 80-8-1).
Bulman C & Wampler DA (1979) Mutagenicity evaluation of
hydroquinone. Akron, Ohio, The Goodyear Tire & Rubber Company
(Laboratory report No. 79-63).
Burnett C, Goldenthal EI, Harris SB, Wazeter FX, Strausburg J, Kapp
R, & Voelker R (1976) Teratology and percutaneous toxicity studies
on hair dyes. J Toxicol Environ Health, 1: 1027-1040.
Busatto S (1939) [Fatal poisoning with a photographic developer
containing hydroquinone.] Dtsch Z Gesamte Gerichtl Med, 31: 285-297
(in German).
Busatto S (1940) [The biological diagnosis of hydroquinone
poisoning.] Arch Antropol Crim Psichiatr Med Leg, 60: 620-625 (in
Italian).
Carlson AJ & Brewer NR (1953) Toxicity studies on hydroquinone. Proc
Soc Exp Biol Med, 84: 684-688.
Cassidy MK & Houston JB (1980) In vivo assessment of extrahepatic
conjugative metabolism in first pass effects using the model
compound phenol. J Pharm Pharmacol, 32: 57-59.
Cassidy MK & Houston JB (1984) In vivo capacity of hepatic and
extrahepatic enzymes to conjugate phenol. Drug Metab Dispos, 12(5):
619-624.
Chatterjee P & Sharma AK (1972) Effect of phenols on nuclear
division in Chara zeylanica. Nucleus, 15: 214-218.
Chavin W, Jelonek EJ Jr, Reed AH, & Binder LR (1980) Survival of
mice receiving melanoma transplants is promoted by hydroquinone.
Science, 208: 408-410.
Cheung SC, Nerland DE, & Sonnenfeld G (1989) Inhibition of
interferon-alpha/beta induction in L-929 cells by benzene and
benzene metabolites. Oncology, 46: 335-338.
Choudat D, Neukirch F, Brochard P, Barrat G, Marsac J, Conso F, &
Philbert M (1988) Allergy and occupational exposure to hydroquinone
and to methionine. Br J Ind Med, 45: 376-380.
Christian RT, Clark CS, Cody TE, Witherup S, Gartside PS, Elia VJ,
Eller PM, Lingg R, & Cooper GP (1976) The development of a test for
the potability of water treated by a direct refuse system.
Cincinnati, Ohio, University of Cincinnati, College of Medicine,
Department of Environmental Health, pp 126-146 (Report No 8:83-87).
Chrostek WJ (1975) Health hazard evaluation/Toxicity determination
report H.H.E. 75-84-235. Springfield, Virginia, US Department of
Commerce, National Technical Information Service, 7 pp
(NTIS/PB-249.415).
CIR (1986) CIR expert panel - Cosmetic ingredient review. Final
report on the safety assessment of hydroquinone and pyrocatechol. J
Am Coll Toxicol, 5(3): 123-165.
Ciranni R & Adler I-D (1991) Clastogenic effects of hydroquinone:
induction of chromosomal aberrations in mouse germ cells. Mutat Res,
263: 223-229.
Ciranni R, Barale R, Marrazzini A, & Loprieno N (1988a) Benzene and
the genotoxicity of its metabolites I. Transplancental activity in
mouse fetuses and in their dams. Mutat Res, 208: 61-67.
Ciranni R, Barale R, Ghelardini G, & Loprieno N (1988b) Benzene and
the genotoxicity of its metabolites. II. The effect of the route of
administration on the micronuclei and bone marrow depression in
mouse bone marrow cells. Mutat Res, 209: 23-28.
Clifford MN & Wight J (1973) Metaperiodate - A new structure-
specific locating reagent for phenolic compounds. J Chromatogr, 86:
222-224.
Commins BT & Lindsey AJ (1956) The determination of phenols by
chromatography and spectrophotometry of their methyl ethers. IV. The
determination of phenols in cigarette smoke. Anal Chim Acta, 15:
557-558.
Connor T & Braunstein B (1987) Hyper-pigmentation following the use
of bleaching creams. Arch Dermatol, 123: 105, 110.
Cooper RL & Wheatstone KC (1973) The determination of phenols in
aqueous effluents. Water Res, 7: 1375-1384.
Crebelli R, Conti G, & Carere A (1987) On the mechanism of mitotic
segregation induction in Aspergillus nidulans by benzene hydroxy
metabolites. Mutagenesis, 2: 235-238.
Crebelli R, Conti G, & Carere A (1991) In vitro studies with nine
known or suspected spindle poisons: results in tests for chromosome
malsegregation in Aspergillus nidulans. Mutagenesis, 6(2):
131-136.
Dagon TJ (1973) Biological treatment of photo processing effluents.
J Water Pollut Control Fed, 45: 2123-2135.
DeGraeve GM, Geiger DL, Meyer JS, & Bergman HL (1980) Acute and
embryo-larval toxicity of phenolic compounds to aquatic biota. Arch
Environ Contam Toxicol, 9: 557-568.
Deichmann WB & Keplinger ML (1981) Hydroquinone. In: Clayton GD &
Crlayton FE ed. Patty's industrial hygiene and toxicology - Volume
2A: Toxicology. New York, John Wiley & Sons, pp 2589-2592.
Delcambre JP, Weber B, & Baron C (1962) Toxicité de l'hydroquinone.
Agressologie, 3: 311-315.
Devillers J, Chambon P, Zakarya D, & Chastrette M (1986) A new
approach in ecotoxicological QSAR studies. Chemosphere, 15:
993-1002.
Devillers J, Chambon P, Zakarya D, Chastrette M, & Chambon R (1987)
A predictive structure-toxicity model with Daphnia magna.
Chemosphere, 16: 1149-1163.
Devillers J, Zakarya D, & Chastrette M (1988) Structure-activity
relationships for the toxicity of organic pollutants to Brachydanio
rerio. In: Turner JE, England MW, Schultz TW, & Kwaak NJ ed.
Proceedings of the Third International Workshop on Quantitative
Structure-Activity Relationships in Environmental Toxicology.
Dordrecht, Reidel Publishing Company, pp 91-90.
Devillers J, Boule P, Vasseur P, Prevot P, Steiman R, Seigle-Murandi
F, Benoit-Guyod JL, Nendza M, Grioni C, Dive D, & Chambon P (1990)
Environmental and health risks of hydroquinone. Ecotoxicol Environ
Saf, 19: 327-354.
Divincenzo GD, Hamilton ML, Reynolds RC, & Ziegler DA (1984)
Metabolic fate and disposition of [14C]hydroquinone given orally
to Sprague-Dawley rats. Toxicology, 33: 9-18.
Dore M, Brunet N, & Legube B (1975) Participation de différents
composés organiques à la valeur des critères globaux de pollution.
Trib CEBEDEAU, 374: 3-11.
Draize JH, Woodard G, & Calvery HO (1944) Methods for the study of
irritation and toxicity of substances applied topically to the skin
and mucous membranes. J Pharmacol Exp Ther, 82: 377-390.
Dreyer NB (1940) Toxicity of hydroquinone (Medical Research Project
No. MR-78). Wilmington, Delaware, Haskell Laboratory of Industrial
Toxicology.
Eastman Kodak Company (1988) Subchronic oral toxicity study of
hydroquinone in rats utilizing a functional observational battery
and neuropathology to detect neurotoxicity. Rochester, New York,
Eastman Kodak Company (Report No. TX-88-78, prepared for the
Chemical Manufacturers Association, Washington).
Eastmond DA, Smityh MT, & Irons DA (1987) An interaction of benzene
metabolites reproduces the myelotoxicity observed with benzene
exposure. Toxicol Appl Pharmacol, 91: 85-95.
Eckert K-G, Eyer P, Sonnenbichler J, & Zetl I (1990) Activation and
detoxication of aminophenols. III. Synthesis and structural
elucidation of various glutathione addition products to
1,4-benzoquinone. Xenobiotica, 20: 351-361.
Eisner T, Jones TH, Aneshansley DJ, Tschinkel WR, Silberglied RH, &
Meinwald J (1977) Chemistry of defensive secretions of bombardier
beetles (Brachinini, Metriini, Ozaenini, Paussini). J Insect
Physiol, 23: 1383-1386.
Ekström T, Warholm M, Kronevi T, & Högberg J (1988) Recovery of
malondialdehyde in urine as a 2,4-dinitrophenylhydrazine derivative
after exposure to chloroform or hydroquinone. Chem-Biol Interact,
67: 25-31.
English JC, Desinger PJ, Perry LG, Schum DB, & Guest D (1988)
Toxicokinetics studies with hydroquinone in male and female Fischer
344 rats. Rochester, New York, Eastman Kodak Company (Report No.
TX-88-84, prepared for the Chemical Manufactures Association,
Washington).
English JC, Perry LG, Vlaovic M, Moyer C, & O'Donoghue JL (1992)
Measurement of cell proliferation in the kidneys of Fischer 344 and
Sprague-Dawley rats after gavage administration of hydroquinone.
Rochester, New York, Eastman Kodak Company (Unpublished report).
Epe B, Harttig U, Stopper H, & Metzler M (1990) Covalent binding of
reactive estrogen metabolites to microtubular protein as a possible
mechanism of aneuploidy induction and neoplastic cell
transformation. Environ Health Perspect, 88: 123-127.
Erexson GL, Wilmer JL, & Kligerman AD (1985) Sister chromatid
exchange induction in human lymphocytes exposed to benzene and its
metabolites in vitro. Cancer Res, 45: 2471-2477.
Fan X-H, Hirata Y, & Minami M (1989) Effects of benzene and its
metabolites on natural killer activity of mouse spleen cells in
vitro. Jpn J Ind Health, 31: 330-334.
FDA (1981) Indirect food additives: Adhesives coatings and
components. Code Fed Regul, 21(175): 114.
FDA (1982) Skin bleaching drug products for over-the-counter human
use; tentative final monograph. Fed Reg, 47:(172) 39108-39117.
FDA (1991) Indirect food additives: Adhesives and components of
coatings. Code Fed Regul, 21(175): 129, 135, 171, 176.
Ferraris de Gaspare PF (1949) [Experimental studies on the
keratoconjunctivitis from hydroquinone.] Boll Ocul, 28: 361-367 (in
Italian).
Findlay GH & De Beer HA (1980) Chronic hydroquinone poisoning of the
skin from skin-lightening cosmetics. A South African epidemic of
ochronosis of the face in dark-skinned individuals. S Afr Med J, 57:
187-190.
Findlay GH, Morrison JGL, & Simson IW (1975) Exogenous ochronosis
and pigmented colloid milium from hydroquinone bleaching creams. Br
J Dermatol, 93: 613-622.
Fisher AA (1982) Can bleaching creams containing 2% hydroquinone
produce leukoderma. J Am Acad Dermatol, 7: 134.
Fisher AA (1986) Contact dermatitis, 3rd ed. Philadelphia,
Pennsylvania, Lea & Febiger.
Fitzgerald GP, Gerloff GC, & Skoog F (1952) Studies on chemicals
with selective toxicity to blue-green algae. Sew Ind Wastes, 24:
888-896.
Fitzpatrick TB, Arndt KA, Mofty AM, & Pathak MA (1966) Hydroquinone
and psoralens in the therapy of hypermelanosis and vitiligo. Arch
Dermatol, 93: 589.
Florin I, Rutberg L, Curvall M, & Enzell CR (1980) Screening of
tobacco smoke constituents for mutagenicity using the Ames' test.
Toxicology, 18: 219-232.
Freitag D, Ballhorn L, Geyer H, & Korte F (1985) Environmental
hazard profile of organic chemicals. Chemosphere, 14: 1589-1616.
Frenk E & Loi-Zedda P (1980) Occupational depigmentation due to a
hydroquinone-containing photographic developer. Contact Dermatitis,
6: 238-239.
Friedlander BR, Hearne FT, & Newman BJ (1982) Mortality, cancer
incidence, and sickness-absence in photographic processors: an
epidemiologic study. J Occup Med, 24(8): 605-613.
Gad-El-Karim MM, Ramanujam VMS, Ahmed AE, & Legator MS (1985)
Benzene myeloclastogenicity: A function of its metabolism. Am J Ind
Med, 7: 475-484.
Gaido K & Wierda D (1984) In vitro effects of benzene metabolites
on mouse bone marrow stromal cells. Toxicol Appl Pharmacol, 76:
45-55.
Gaido KW & Wierda D (1987) Suppression of bone marrow stromal cell
function by benzene and hydroquinone is ameliorated by indomethacin.
Toxicol Appl Pharmacol, 89: 378-390.
Galloway SM, Armstrong MJ, Reuben C, Colman S, Brown B, Cannon C,
Bloom AD, Nakamura F, Ahmed M, Duk S, Rimpo J, Margolin BH, Resnick
MA, Anderson B, & Zeiger E (1987) Chromosome aberrations and sister
chromatid exchanges in Chinese hamster ovary cells: Evaluations of
108 chemicals. Environ Mol Mutagen, 10(Suppl 10): 1-175.
Garton GA & Williams RT (1949) Studies in detoxification: 21. The
fates of quinol and resorcinol in the rabbit in relation to the
metabolism of benzene. Biochem J, 44: 234-238.
Glatt H, Padykula R, Berchtold GA, Ludewig G, Platt KL, Klein J, &
Oesch F (1989) Multiple activation pathways of benzene leading to
products with varying genotoxic characteristics. Environ Health
Perspect, 82: 81-89.
Glatt H, Gemperlein I, Setiabudi F, Platt KL, & Oesch F (1990)
Expression of xenobiotic-metabolizing enzymes in propagatable cell
cultures and induction of micronuclei by 13 compounds. Mutagenesis,
5(3): 241-249.
Gocke E, King M-T, Eckhardt K, & Wild D (1981) Mutagenicity of
cosmetics ingredients licenced by the European Communities. Mutat
Res, 90: 91-109.
Gocke E, Wild D, Eckhardt K, & King M-T (1983) Mutagenicity studies
with the mouse spot test. Mutat Res, 117: 201-212.
Godlee F (1992) Skin lighteners cause permanent damage. Br Med J,
305: 332-333.
Gold LS, Slone TS, Stern BR, Manley NB, & Ames BN (1992) Rodent
carcinogens: setting priorities. Science, 258: 261-265.
Goodwin BTJ, Crevel RWR, & Johnson AW (1981) A comparison of three
guinea-pig sensitization procedures for the detection of 19 reported
human contact sensitizers. Contact Dermatitis, 7: 248-258.
Grant W (1986) Toxicology of the eye, 3rd ed. Springfield, Illinois,
Charles C. Thomas, pp 97-500.
Greenlee WF, Gross EA, & Irons RD (1981a) Relationship between
benzene toxicity and the disposition of 14C-labelled benzene
metabolites in the rat. Chem-Biol Interact, 33: 285-299.
Greenlee WF, Sun JD, & Bus JS (1981b) A proposed mechanism of
benzene toxicity: Formation of reactive intermediates from
polyphenol metabolites. Toxicol Appl Pharmacol, 59: 187-195.
Greenwald P, Friedlander BR, Lawrence CE, Hearne T, & Earle K (1981)
Diagnostic sensitivity bias - an epidemiologic explanation for an
apparent brain tumor excess. J Occup Med, 23: 690-694.
Grudzinski W (1969) [A case of lethal intoxication with
methol-hydroquinone photographic developer.] Pol Tyg Lek, 24:
1460-1462 (in Polish).
Guy RL, Dimitriadis EA, Hu P, Cooper KR, & Snyder R (1990)
Interactive inhibition of erythroid 59Fe utilization by benzene
metabolites in female mice. Chem-Biol Interact, 74: 55-62.
Guy RL, Hu P, Witz G, Goldstein BD & Snyder R (1991) Depression of
iron uptake into erythrocytes in mice by treatment with the combined
benzene metabolites p-benzoquinone, muconaldehyde and hydroquinone.
J Appl Toxicol, 11: 443-446.
Häggblom MM, Apajalahti JHA, & Salkinoja-Salonen MS (1988)
Degradation of chlorinated phenolic compounds occurring in pulp mill
effluents. Water Sci Technol, 20: 205-208.
Harbison KG & Belly RT (1982) The biodegradation of hydroquinone.
Environ Toxicol Chem, 1: 9-15.
Hardwick N, Van Gelder JW, Van der Merwe CA, & Van der Merwe MP
(1989) Exogenous ochronosis: an epidemiological study, 120: 229-238.
Haworth S, Lawlor T, Mortelmans K, Speck W, & Zeiger E (1983)
Salmonella mutagenicity test results for 250 chemicals. Environ
Mutagen, 5(Suppl 1): 3-142.
Hildebrand DC, Powell CC Jr, & Schroth MN (1969) Fire blight
resistance in Pyrus: Localization of arbutin and ß-glucosidase.
Phytopathology, 59: 1534-1539.
Hill BA, Monks TJ, & Lau SS (1992a) Metabolism and toxicity of
2-(glutathion-S-YL) hydroquinone and 2,3,5-(triglutathion-S-YL)
hydroquinone in the in situ perfused rat kidney. Toxicologist, 12:
345 (Abstract).
Hill BA, Monks TJ, & Lau SS (1992b) The effects of
2,3,5-(triglutathion-S-YL) hydroquinone on renal mitochondrial
respiratory function in vivo and in vitro: Possible role in
cytotoxicity. Toxicol Appl Pharmacol, 117: 165-171.
Hill T, English JC, & Deisinger P (1993) Human exposure to naturally
occurring hydroquinone. Rochester, New York, Eastman Kodak Company
(Unpublished data).
Hirose M, Inoue T, Asamoto M, Tagawa Y, & Ito N (1986) Comparison of
the effects of 13 phenolic compounds in induction of proliferative
lesions of the forestomach and increase in the labelling indices of
the glandular stomach and urinary bladder epithelium of Syrian
golden hamsters. Carcinogenesis, 7(8): 1285-1289.
Hirose M, Yamaguchi S, Fukushima S, Hasegawa R, Takahashi S, &
Nobuyuki I (1989) Promotion by dihydroxybenzene derivatives of
N-methyl-N1-vitro-N-nitrosoguanidine-induced F344 rat forestomach
and glandular stomach carcinogenesis. Cancer Res, 49: 5143-5147.
Hodson PV, Dixon DG, & Kaiser KLE (1984) Measurement of median
lethal dose as a rapid indication of contaminant toxicity to fish.
Environ Toxicol Chem, 3: 243-254.
Hodson PV, Dixon DG, & Kaiser KLE (1988) Estimating the acute
toxicity of waterborne chemicals in trout from measurements of
median lethal dose and the octanol-water partition coefficient.
Environ Toxicol Chem, 7: 443-454.
Hogan ME & Manners GD (1990) Allelopathy of small everlasting
(Antennaria microphylla). Phytotoxicity to leafy spurge
(Euphorbia esula) in tissue culture. J Chem Ecol, 16(3): 931-939.
Hogan ME & Manners GD (1991) Differential allelochemical
detoxification mechanism in tissue cultures of Antennaria
microphylla and Euphorbia esula. J Chem Ecol, 17: 167-174.
Högl O (1958) [Some non-volatile extracts of coffee.] Mitt Geb
Lebensm Hyg, 49: 433-441 (in German).
Howard BM, Clarkson K, & Bernstein RL (1979) Simple prenylated
hydroquinone derivatives from the marine urochordate Aplidium
californicum. Natural anticancer and antimutagenic agents.
Tetrahedron Lett, 46: 4449-4452.
Hsuanyu Y & Dunford HB (1992) Reduction of prostaglandin H synthase
compound II by phenol and hydroquinone, and the effect of
indomethacin. Arch Biochem Biophys, 292: 213-220.
Hu F (1966) The influence of certain hormones and chemicals on
mammalian pigment cells. J Invest Dermatol, 46: 117-124.
Hughes W (1948) The tolerance of rabbit cornea for various chemical
substances. Bull John Hopkins Hosp, 82: 338-349.
IARC (1977) Dihydroxybenzenes. In: Some fumigants, the herbicides
2,4-D and 2,4,5-T, chlorinated dibenzodioxins and miscellaneous
industrial chemicals. Lyon, International Agency for Research on
Cancer, pp 155-174 (IARC Monographs on the Evaluation of the
Carcinogenic Risk of Chemicals to Man, Volume 15).
IARC (1986) Tobacco smoking. Lyon, International Agency for Research
on Cancer, pp 86, 105, 122 (IARC Monographs on the Evaluation of the
Carcinogenic Risk of Chemicals to Humans, Volume 38).
IARC (1987) Overall evaluations of carcinogenicity: An updating of
IARC monographs, volumes 1 to 42. Lyon, International Agency for
Research on Cancer, p 64 (IARC Monographs on the Evaluation of
Carcinogenic Risks to Humans, Supplement 7).
Iguchi K, Sahashi A, Kohno J, & Yamada Y (1990) New sesquiterpenoid
hydroquinone and quinones from the Okinawan marine sponge (Dysidea
sp.). Chem Pharm Bull (Tokyo), 38(5): 1121-1123.
ILO (1991) Occupational exposure limits for airborne toxic
substances. Geneva, International Labour Office (Occupational Safety
and Health Series No. 37).
Inoue O, Seiji K, Nakatsuka H, Watanabe T, Yin S-N, Li G-L, Cai S-X,
Jin C, & Ikeda M (1989a) Excretion of 1,2,4-benzenetriol in the
urine of workers exposed to benzene. Br J Ind Med, 46: 559-565.
Inoue O, Seiji K, & Ikeda M (1989b) Pathways for formation of
catechol and 1,2,4-benzenetriol in rabbits. Bull Environ Contam
Toxicol, 43: 220-224.
Irons RD & Neptun DA (1980) Effects of the principal
hydroxy-metabolites of benzene on microtubule polymerization. Arch
Toxicol, 45: 297-305.
Irons RD & Pfeifer RW (1982) Benzene metabolites: evidence for an
epigenetic mechanism of toxicity. Environ Sci Res, 25: 241-256.
Irons RD, Neptun DA, & Pfeifer RW (1981) Inhibition of lymphocyte
transformation and microtubule assembly by quinone metabolites of
benzene: Evidence for a common mechanism. J Reticuloendothel Soc,
30(5): 359-372.
Irons RD, Stillman WS, Colagiovanni DB, & Henry VA (1992)
Synergistic action of the benzene metabolite hydroquinone on
myelopoietic stimulating activity of granulocyte/macrophage
colony-stimulating factor in vitro. Proc Natl Acad Sci (USA), 89:
3691-3695.
IRPTC (1987) IRPTC data file on hydroquinone. Geneva, United Nations
Environment Programme, International Register of Potentially Toxic
Chemicals.
Ismail IMK, Salama AMA, Ali MIA, & Ouf SAE (1987) Effect of some
phenolic compounds on spore germination and germ-tube length of
Aspergillus fumigatus and Fusarium oxysporium f sp lycopersici.
Cryptogam Mycol, 8: 51-60.
Jacquemain R, Remy F, & Guinchard C (1975) Etudes et comparaisons
des déterminations des phénols dans les eaux; application à l'examen
d'un rejet de papeterie. J Fr Hydrol, 16: 25-31.
Jimbow K, Obata H, Pathak MA, & Fitzpatrick TB (1974) Mechanisms of
depigmentation by hydroquinone. J Invest Dermatol, 62: 436-449.
Jowa L, Winkle S, Kalf G, Witz G, & Snyder R (1986) Deoxyguanosine
adducts formed from benzoquinone and hydroquinone. Adv Exp Med Biol,
197: 825-832.
Jowa L, Witz G, Snyder R, Winkle S, & Kalf GF (1990) Synthesis and
characterization of deoxyguanosine-benzoquinone adducts. J Appl
Toxicol, 10: 47-54.
Juhnke I & Lüdemann D (1978) [Results of the testing of 200 chemical
compounds for acute toxicity for fish by means of the golden orfe
test.] Z Wasser Abwasser Forsch, 11: 161-164 (in German).
Jung F & Witt P (1947) [Studies on methaemoglobin formation. XXX.
Tests with polyphenols.] Naunyn-Schmiedebergs Arch Exp Pathol
Pharmakol, 204: 246-436 (in German).
Kappas A (1989) On the mechanism of induced aneuploidy in
Aspergillus nidulans and validation of tests for genomic
mutations. In: Resnick MA & Vig BK ed. Mechanisms of chromosome
distribution and aneuploidi. New York, Alan R. Liss, Inc., pp
377-384.
Kari FW, Bucher J, Eustis SL, Haseman JK, & Huff JE (1992) Toxicity
and carcinogenicity of hydroquinone in F344/N rats and B6C3F1
mice. Food Chem Toxicol, 30: 737-747.
Kavlock RJ (1990) Structure-activity relationships in the
developmental toxicity of substituted phenols: in vivo effects.
Teratology, 41: 43-59.
Kavlock RJ, Oglesby LA, Hall LL, Fisher HL, Copeland F, Logsdon T, &
Ebron-McCoy M (1991) In vivo and in vitro structure-dosimetry-
activity relationships of substituted phenols in developmental
toxicity assays. Reprod Toxicol, 5: 255-258.
Kersey P & Stevenson CJ (1981) Vitiligo and occupational exposure to
hydroquinone from servicing self-photographing machines. Contact
Dermatitis, 7: 285-287.
Key MM, Henschel AF, Butler J, Ligo RN, & Tabershaw IR ed. (1977)
Hydroquinone. In: Occupational diseases. A guide to their
recognition. Washington, DC, US Department of Health, Education and
Welfare, pp 249-250.
King AG, Landreth KS, & Wierda D (1987) Hydroquinone inhibits bone
marrow pre-B cell maturation in vitro. Mol Pharmacol, 32: 807-812.
King AG, Landreth KS, & Wierda D (1989) Bone marrow stromal cell
regulation of B-lymphopoiesis. II. Mechanisms of hydroquinone
inhibition of pre-B cell maturation. J Pharmacol Exp Ther, 250(2):
582-590.
Kleiner HE, Hill BA, Monks TJ, & Lau SS (1992) In vivo and in
vitro formation of several S-conjugates of hydroquinone.
Toxicologist, 12: 345 (Abstract).
Knadle S (1985) Synergistic interaction between hydroquinone and
acetaldehyde in the induction of sister chromatid exchange in human
lymphocytes in vitro. Cancer Res, 45: 4853-4857.
Koike N, Haga S, Ubukata N, Sakurai M, Shimizu H, & Sato A (1988)
Mutagenicity of benzene metabolites by fluctuation test. Jpn J Ind
Health, 30: 475-480.
Kolachana P, Subrahmanyam V, Meyer KB, Zhang L, & Smith MT (in
press) Benzene and its phenolic metabolites produce oxidative DNA
damage in HL60 cells in vitro and in the bone marrow in vivo.
Can Res.
Kolthoff IM & Lee TS (1946) Assay of hydroquinone. Ind Eng Chem,
18(7): 452.
Krasavage WJ (1984a) Hydroquinone: Teratology probe study in rats.
Rochester, New York, Eastman Kodak Company, Health and Environment
Laboratories (Report No. TX-84-01).
Krasavage WJ (1984b) Hydroquinone: A dominant lethal assay in male
rats. Rochester, New York, Eastman Kodak Company, Health and
Environment Laboratories (Report No. TX-84-23).
Krasavage WJ, Terhaar CJ, Ely TS, Vlaovic MS, & Katz GV (1985)
Hydroquinone: A developmental toxicity study in rats. Rochester, New
York, Eastman Kodak Company, Health and Environment Laboratories
(Report No. TX-85-50, prepared for the Chemical Manufacturers
Association, Washington).
Krasavage WJ, Blacker AM, English JC, & Murphy SJ (1992)
Hydroquinone: A developmental toxicity study in rats. Fundam Appl
Toxicol, 18: 370-375.
Kühn R, Pattard M, Pernak K-D, & Winter A (1989) Results of the
harmful effects of selected water pollutants (anilines, phenols,
aliphatic compounds) to Daphnia magna. Water Res, 23: 495-499.
Kurata Y, Fukushima S, Hasegawa R, Hirose M, Shibata M-A, Shirai T,
& Ito N (1990) Structure-activity relations in promotion of rat
urinary bladder carcinogenesis by phenolic antioxidants. Jpn J
Cancer Res, 81: 754-759.
Larcan A, Lambert H, Laprevote-Heully M-C, Bertrand D, & Bertrand D
(1974) Les intoxications par les produits utilisés en photographie
(bains, fixateurs, rélévateurs). J Eur Toxicol, 7: 17-21.
Laskin DL, MacEachern L, & Snyder R (1989) Activation of bone marrow
phagocytes following benzene treatment of mice. Environ Health
Perspect, 82: 75-79.
Lau SS, Hill BA, Highet RJ, & Monks TJ (1988) Sequential oxidation
and glutathione addition to 1,4-benzoquinone: Correlation of
toxicity with increased glutathione substitution. Mol Pharmacol, 34:
829-836.
Lawrence N, Bligard CA, Reed R, & Perret WJ (1988) Exogenous
ochronosis in the United States. J Am Acad Dermatol, 18: 1207-1211.
Leanderson P & Tagesson C (1990) Cigarette smoke-induced DNA-damage:
Role of hydroquinone and catechol in the formation of the oxidative
DNA-adduct, 8-hydroxyde-oxyguanosine. Chem-Biol Interact, 75: 71-81.
Leopardi P, Zijno A, Bassani B, & Pacchierotti F (1993) In vivo
studies on chemically induced aneuploidy in mouse somatic and
germinal cells. Mutat Res, 287: 119-130.
Levan A & Tjio JH (1948a) Chromosome fragmentation induced by
phenols. Hereditas, 34: 250-252.
Levan A & Tjio JH (1948b) Induction of chromosome fragmentation by
phenols. Hereditas, 34: 453-484.
Levay G & Bodell WJ (1992) Potentiation of DNA adduct formation in
HL-60 cells by combinations of benzene metabolites. Proc Natl Acad
Sci (USA), 89(15): 7105-7109.
Levay G, Pongracz K, & Bodell WJ (1991) Detection of DNA adducts in
HL-60 cells treated with hydroquinone and p-benzoquinone by
32P-postlabeling. Carcinogenesis, 12(7): 1181-1186.
Levenson GIP (1947) Determination of Elon and hydroquinone in
developer--an examination of Stott's method. Photogr J, B87: 18-24.
Levin J-O (1988) High performance liquid chromatographic
determination of hydroquinone in air as benzoquinone, using combined
oxidizing filter and XAD-2 adsorbent preconcentration. Chemosphere,
17: 671-679.
Lewis JG, Stewart W, & Adams DO (1988a) Role of oxygen radicals in
induction of DNA damage by metabolites of benzene. Cancer Res, 48:
4762-4765.
Lewis JG, Odom B, & Adams DO (1988b) Toxic effects of benzene and
benzene metabolites of mononuclear phagocytes. Toxicol Appl
Pharmacol, 92: 246-254.
Lidén C (1989) Occupational dermatoses at a film laboratory.
Follow-up after modernization. Contact Dermatitis, 20: 191-200.
Lockhart HB & Fox JA (1985a) Metabolic fate of [U-14C]
hydroquinone administered by gavage to female Fischer 344 rats.
Rochester, New York, Eastman Kodak Company, Health and Environment
Laboratories (Report No. TX-85-55).
Lockhart HB & Fox JA (1985b) The metabolic fate CF [14C]
hydroquinone administered by intratracheal instillation to male
Fischer 344 rats. Rochester, New York Eastman Kodak Company, Health
and Environment Laboratories (Report No. TX-85-76).
Lockhart HB, Fox JA, & Divincenzo GD (1984) The metabolic fate of
[U-14C] hydroquinone administered by gavage to male Fischer 344
rats. Rochester, New York, Eastman Kodak Company, Health and
Environment Laboratories (Report No. BC-84-14, prepared for the
Chemical Manufacturers Association, Washington).
McGregor DB, Brown A, Cattanach P, Edwards I, McBride D, & Caspary
WJ (1988a) Responses of the L5178Y tk+/tk- mouse lymphoma cell
forward mutation assay. II. 18 coded chemicals. Environ Mol Mutagen,
11: 91-118.
McGregor DB, Riach CG, Brown A, Edwards I, Reynolds D, West K, &
Willington S (1988b) Reactivity of catecholamines and related
substances in the mouse lymphoma L5178Y cell assay for mutagens.
Environ Mol Mutagen, 11: 523-544.
Mackay D & Paterson S (1981) Calculating fugacity. Environ Sci
Technol, 15: 1006-1014.
McLeese DW, Zitko V, & Peterson MR (1979) Structure-lethality
relationships for phenols, anilines and other aromatic compounds in
shrimp and clams. Chemosphere, 2: 53-57.
Maibach HJ & Patrick E (1989) A study to evaluate the potential of
mono-T-butyl hydroquinone to produce skin depigmentation. Kingsport,
Tennessee, Eastman Kodak Company (Report No. HIM 88-KOD-DEPIG-01).
Maier HG (1981) [Coffee.] In: Mayer HG ed. [Advances in food
research and food technology]. Berlin, Paul Parey Publisher, vol 18,
p 57 (in German).
Mann RJ & Harman RRM (1983) Nail staining due to hydroquinone
skin-lightening creams. Br J Dermatol, 108: 363-365.
Markey AC, Black AK, & Rycroft RJG (1989) Confetti-like
depigmentation from hydroquinone. Contact Dermatitis, 20: 148-149.
Marquardt P, Koch R, & Aubert J-P (1947) [The toxicity of the
monovalent, divalent and trivalent phenols.] J Gesamte Inn Med, 2:
333-346 (in German).
Marty JP, Trouvin JH, Jacquot C, & Wepierre J (1981)
Pharmacocinétique percutanée de l'hydroquinone 14C. C R Congrès
Eur Biopharm Pharmacocinet, 2: 221-228.
Miller BM & Adler I-D (1989) Suspect spindle poisons: analysis of
c-mitotic effects in mouse bone marrow cells. Mutagenesis, 4:
208-215.
Mitchell A & Webster J (1919) Notes on a case of poisoning by
hydroquinone. Br Med J, 21: 465.
Moore GA, Rossi L, Orrenius S, & O'Brien PJ (1987) Ca2+ release
from rat liver mitochondria by benzoquinone derivatives. Arch
Biochem Biophys, 259: 283-295.
Moore GA, Weis M, Orrenius S, & O'Brien PJ (1988) Role of sulfhydryl
group(s) in benzoquinone induced Ca2+ release by rat liver
mitochondria. Arch Biochem Biophys, 267: 539-550.
Morimoto K & Wolff S (1980) Increase of sister chromatid exchanges
and perturbations of cell division kinetics in human lymphocytes by
benzene metabolites. Cancer Res, 40: 1189-1193.
Morimoto K, Wolff S, & Koizumi A (1983) Induction of
sister-chromatid exchanges in human lymphocytes by microsomal
activation of benzene metabolites. Mutat Res, 119: 355-360.
Mozhaev EA, Osintseva VP, & Arzamastsev EV (1966) [Hydroquinone
toxicity in chronic poisoning.] Farmakol Toksikol, 29: 238-240 (in
Russian).
Murphy SJ, Schroeder RE, Blacker AM, Krasavage WJ, & English JC
(1992) A study of developmental toxicity of hydroquinone in the
rabbit. Fundam Appl Toxicol, 19: 214-221.
Nakamura S, Oda Y, Shimada T, Oki I, & Sugimoto K (1987)
SOS-inducing activity of chemical carcinogens and mutagens in
Salmonella typhimurium TA1535/pSK1002: examination with 151
chemicals. Mutat Res, 192: 239-246.
Naumann G (1966) Corneal damage in hydroquinone workers. Arch
Ophthalmol, 76: 189-194.
Nendza M & Seydel JK (1988a) Multivariate data analysis of various
biological test systems used for the quantification of ecotoxic
compounds. Quant Struct-Act Relat, 7: 165-174.
Nendza M & Seydel JK (1988b) Quantitative structure-toxicity
relationships and multivariate data analysis for ecotoxic chemicals
in different biotest systems. Chemosphere, 7: 1575-1584.
Nendza M & Seydel JK (1988c) Quantitative structure-toxicity
relationships for ecotoxicologically relevant biotest systems and
chemicals. Chemosphere, 17: 1585-1602.
Neujahr H & Varga JM (1970) Degradation of phenols by intact cells
and cell-free preparations of Trichosporon cutaneum. Eur J
Biochem, 13: 37-44.
NIOSH (1976) Hydroquinone: Method No. S57 - Standards Completion
Program. Cincinnati, Ohio, National Institute for Occupational
Safety and Health, Division of Laboratories and Criteria
Development, p 66.
NIOSH (1978) Criteria for a recommended standard ... Occupational
exposure to hydroquinone, Cincinnati, Ohio, US National Institute
for Occupational Safety and Health, 182 pp (Publication DHEW (NIOSH)
No. 78-155).
NIOSH (1987) Registry of toxic effects of chemical substances
(RTECS). 1985-1986 edition, Cincinnati, Ohio, National Institute for
Occupational Safety and Health (NIOSH Publication No. 87-114).
Nomiyama K, Minai M, Suzuki T, & Kita H (1967) Studies on poisoning
by benzene and its homologues. (10). Median lethal doses of benzene
metabolites. Ind Health, 5: 143-148.
Norrelykke Nissen J & Corydon L (1985) Corneal ulcer after exposure
to vapours from bone cement (methyl metacrylate and hydroquinone).
Int Arch Ocup Environ Health, 56: 161-165.
NTP (1989) Technical report on the toxicology and carcinogenesis
studies of hydroquinone (CAS No. 123-31-9) in F344/N rats and
B6C3F1 mice (gavage studies). Research Triangle Park, North
Carolina, National Toxicology Program (NTP TR 366).
Nyholm N, Jorgensen-Lindgaard P, & Hansen N (1984) Biodegradation of
4-nitrophenol in standardized aquatic degradation tests. Ecotoxicol
Environ Saf, 8: 451-470.
O'Brien PJ (1991) Molecular mechanisms of quinone cytotoxicity.
Chem-Biol Interact, 80: 1-41.
OECD (1981) OECD guidelines for testing of chemicals. Paris,
Organisation for Economic Co-operation and Development.
Oettel H (1936) [Hydroquinone poisoning.] Naunyn-Schmiedebergs Arch
Exp Pathol Pharmakol, 183: 319-362 (in German).
Oglesby FL, Sterner JH, & Anderson B (1947) Quinone vapors and their
harmful effects. II. Plant exposures associated with eye injuries. J
Ind Hyg Toxicol, 29: 74-84.
Oglesby LA, Ebron-McCoy MT, Logsdon TR, Copeland F, Beyer PE, &
Kavlock RJ (1992) In vitro embryotoxicity of a series of
para-substituted phenols: structure, activity, and correlation with
in vitro data. Teratology, 45: 11-33.
Ohnishi T, Yamazaki H, Iyanage T, Nakamura T, & Yamazaki I (1969)
One-electron-transfer reactions in biochemical systems. II. The
reaction of free radicals formed in the enzymic oxidation. Biochim
Biophys Acta, 172: 357-369.
O'Keeffe DH, Wiese TE, Brummet SR, & Miller TW (1987) Uptake and
metabolism of phenolic compounds by the water hyacinth (Eichhornia
crassipes). Recent Adv Phytochem, 21(87): 101-129.
Oliveira NL & Kalf GF (1992) Induced differentiation of HL-60
promyelocytic leukemia cells to monocyte/macrophages is inhibited by
hydroquinone, a hematotoxic metabolite. Blood, 79: 627-633.
Otsuka M & Nonomura Y (1963) The action of phenolic substances on
nerve endings. J Pharmacol Exp Ther, 140: 41-45.
Pacchierotti F, Bassani B, Leopardi P, & Zijno A (1991) Origin of
aneuploidi in relation to disturbances of cell-cycle progression.
II: Cytogenetic analysis of various parameters in mouse bone marrow
cells after colchicine or hydroquinone treatment. Mutagenesis, 6(4):
307-311.
Painter RB & Howard R (1982) The HeLa DNA-synthesis inhibition test
as a rapid screen for mutagenic carcinogens. Mutat Res, 92: 427-437.
Palumbo A, d'Ischia M, Misuraca G, & Prota G (1991) Mechanism of
inhibition of melanogenesis by hydroquinone. Biochim Biophys Acta,
1073: 85-90.
Parmentier R (1952) Etude des lésions cellulaires provoquées par
divers phénols et amines aromatiques. Revue Belge Pathol Méd Exp,
22: 1-54.
Parmentier R (1953) Production of `three-group metaphases' in the
bone-marrow of the golden hamster. Nature (Lond), 171: 1029-1030.
Parmentier R & Dustin P Jr (1948) Early effects of hydroquinone on
mitosis. Nature (Lond), 161: 527-528.
Parmentier R & Dustin P Jr (1951) Reproduction expérimentale d'une
anomalie particulière de la métaphase des cellules malignes
(métaphase "à trois groupes"). Caryologia, IV(1): 98-109.
Pellack-Walker P & Blumer JL (1986) DNA damage in L5178YS cells
following exposure to benzene metabolites. Mol Pharmacol, 30: 42-47.
Pellack-Walker P, Walker JK, Evans HH, & Blumer JL (1985)
Relationship between the oxidation potential of benzene metabolites
and their inhibitory effect on DNA synthesis in L5178YS cells. Mol
Pharmacol, 28: 560-566.
Penney BK, Smith JC, & Allen CJ (1984) Depigmenting action of
hydroquinone depends on disruption of fundamental cell processes. J
Invest Dermatol, 82: 308-310.
Pfeifer PW & Irons RD (1983) Alteration of lymphocyte function by
quinones through a sulfhydryl-dependent disruption of microtubule
assembly. Int J Immunopharmacol, 5: 463-470.
Picardo M, Nazzaro-Porro MN, Breathnach A, Zompetta C, Faggioni A, &
Riley P (1987) Mechanism of antitumoral activity of catechols in
culture. Biochem Pharmacol, 36(4): 417-425.
Pifer JW, Hearne FT, Friedlander BR, & McDonough JR (1986) Mortality
study of men employed at a large chemical plant, 1972 through 1982.
J Occup Med, 28: 438-444.
Post GB, Snyder R, & Kalf GF (1985) Inhibition of RNA synthesis and
interleukin-2 production in lymphocytes in vitro by benzene and
its metabolites, hydroquinone and p-benzoquinone. Toxicol Lett,
29: 161-167.
Rácz G, Füzi J, Kemény G, & Kisgyörgy Z (1958) [The effect of
hydroquinone and Phloridzin on the sexual cycle of white rats.] Orv
Sz, 5: 65-67 (in Hungarian).
Raghavan NV (1979) Separation and quantification of trace isomeric
hydroxyphenols in aqueous solution by high-performance liquid
chromatography. J Chromatogr, 168: 523-525.
Rajka G & Blohm SG (1970) The allergenicity of paraphenylendiamine
II. Acta Dermatovenerol (Stockholm), 50: 51-54.
Rao GS & Pandya KP (1989) Release of 2-thiobarbituric acid reactive
products from glutamate or deoxyribonucleic acid by
1,2,4-benzenetriol or hydroquinone in the presence of copper ions.
Toxicology, 59: 59-65.
Reddy MV, Blackburn GR, Irwin SE, Kommineni C, Mackerer CR, &
Mehlman MA (1989) A method for in vitro culture of rat zymbal
gland: Use in mechanistic studies of benzene carcinogenesis in
combination with 32P-postlabeling. Environ Health Perspect, 82:
239-247.
Reddy MV, Bleicher T, Blackburn GR, & Mackerer CR (1990) DNA
adduction by phenol, hydroquinone, or benzoquinone in vitro but
not in vivo: nuclease P1-enhanced 32P-postlabeling of adducts as
labeled nucleoside biphosphates, dinucleotides and nucleoside
monophosphates. Carcinogenesis, 11: 1349-1357.
Rémond A & Colombies H (1927) Intoxication par l'hydroquinone. Ann
Méd Lég, 7: 79-81.
Renz JF, Georg F, & Kalf GF (1991) Role for interleukin-1 (IL-1) in
benzene-induced hematotoxicity: Inhibition of conversion of
Pre-IL-1alpha to mature cytokine in murine macrophages by
hydroquinone and prevention of benzene-induced hematotoxicity in
mice by IL-1alpha. Blood, 78(4): 938-944.
Ribo JM & Kaiser KLE (1983) Effects of selected chemicals to
photoluminescent bacteria and their correlations with acute and
sublethal effects on other organisms. Chemosphere, 12: 1421-1442.
Robertson ML, Eastmond DA, & Smith MT (1991) Two benzene
metabolites, catechol and hydroquinone, produced a synergistic
induction of micronuclei and toxicity in cultured human lymphocytes.
Mutat Res, 249: 201-209.
Roe FJC & Salaman MH (1955) Further studies on incomplete
carcinogenesis: triethylene melamine (T.E.M.), 1,2-benzanthracene
and ß-propiolactone, as initiators of skin tumour formation in the
mouse. Br J Cancer, 9: 177-203.
Rosen F & Millman N (1955) Anti-gonadotrophic activities of quinones
and related compounds. Endocrinology, 57: 466-471.
Ross D, Siegel D, Gibson NW, Pacheco D, Thomas DJ, Reasor M, &
Wierda D (1990) Activation and deactivation of quinones catalyzed by
DT-diaphorase. Evidence for bioreductive activation of diaziquone
(AZQ) in human tumor cells and detoxification of benzene metabolites
in bone marrow stroma. Free Radic Res Commun, 8: 4-6, 373-381.
Rossi L, Moore GA, Orrenius S, & O'Brien PJ (1986) Quinone toxicity
in hepatocytes without oxidative stress. Arch Biochem Biophys, 251:
1, 25-35.
Rott B, Viswanathan R, Freitag D, & Korte F (1982) [Comparative
investigation of the applicability of various tests for evaluating
the degradability of environmental chemicals.] Chemosphere, 11:
531-538 (in German).
Roy SC (1973) Comparative effects of colchicine, caffeine and
hydroquinone on nodal roots of Callisia fragrans. Biol Plant, 15:
383-390.
Rushmore T, Snyder R, & Kalf G (1984) Covalent binding of benzene
and its metabolites to DNA in rabbit bone marrow mitochondria in
vitro. Chem-Biol Interact, 49: 133-154.
Sakai M, Yoshida D, & Mizusaki S (1985) Mutagenicity of polycyclic
aromatic hydrocarbons and quinones on Salmonella typhimurium TA97.
Mutat Res, 156: 61-67.
Sawada Y, Iyanagi T, & Yamazaki I (1975) Relation between redox
potentials and rate constants in reactions coupled with the system
oxygen-superoxide. Biochemistry, 14: 3761-3764.
Sawahata T & Neal RA (1983) Biotransformation of phenol to
hydroquinone and catechol by rat liver microsomes. Mol Pharmacol,
23: 453-460.
Schlosser MJ & Kalf GF (1989) Metabolic activation of hydroquinone
by macrophage peroxidase. Chem-Biol Interact, 72: 191-207.
Schlosser MJ, Shurina RD, & Kalf GF (1989) Metabolism of phenol and
hydroquinone to reactive products by macrophage peroxidase or
purified prostaglandin H synthase. Environ Health Perspect, 82:
229-237.
Schlosser MJ, Shurina RD, & Galf GF (1990) Prostaglandin H synthase
catalyzed oxidation of hydroquinone to a sulfhydryl-binding and
DNA-damaging metabolite. Chem Res Toxicol, 3: 333-339.
Schultz TW, Riggin GW, & Wesley SK (1987) Structure-activity
relationships for para-substituted phenols. In: Kaiser KLE ed. QSAR
in environmental toxicology - II. Dordrecht, D. Reidel Publishing
Company, pp 333-345.
Seki T (1975) Chromatographic separation of dihydroxybenzenes on a
column of Merckogel PGM 2000. J Chromatogr, 115: 262-263.
Serva RJ & Bulman C (1981) Mutagenicity evaluation of hydroquinone
(MIBK process). Akron, Ohio, The Goodyear Tire & Rubber Company
(Report No 81-4-3).
Serva RJ & Murphy SJ (1981) Evaluation of hydroquinone using the
Drosophila melanogaster/sex-linked recessive lethal test. Akron,
Ohio, The Goodyear Tire & Rubber Company (Report No. 56).
Shaner VC & Sparks MR (1946) Application of methyl ethyl ketone to
the analysis of developers for Elon and hydroquinone. J Soc Motion
Pict Eng, 47: 409-417.
Sharma AK & Chatterjee T (1964) Effect of oxygen on chromosomal
aberrations induced by hydroquinone. Nucleus, 7: 113-124.
Shibata M-A, Yamada M, Hirose M, Asakawa E, Tatematsu M, & Ito N
(1990) Early proliferative responses of forestomach and glandular
stomach of rats treated with five different phenolic antioxidants.
Carcinogenesis, 11(3): 425-429.
Shibata M-A, Hirose M, Tanaka H, Asakawa E, Shirai T, & Ito N (1991)
Induction of renal cell tumors in rats and mice, and enhancement of
hepatocellular tumor development in mice after long-term
hydroquinone treatment. Jpn J Cancer Res, 82: 1211-1219.
Skalka P (1964) [The influence of hydroquinone on the fertility of
male rats.] Sb Vys Sk Zemed (Brne), BXII: 491-494 (in Czech).
Smale BC & Keil HL (1966) A biochemical study of the intervarietal
resistance of Pyrus communis to fire blight. Phytochemistry, 5:
1113-1120.
Smith MT, Yager JW, Steinmetz KL, & Eastmond DA (1989)
Peroxidase-dependent metabolism of benzene's phenolic metabolites
and its potential role in benzene toxicity and carcinogenicity.
Environ Health Perspect, 82: 23-29.
Sollmann T (1949) Correlation of the aquarium goldfish toxicities of
some phenols, quinones and other benzene derivatives with their
inhibition of autooxidative reactions. J Gen Physiol, 32: 671-679.
Spain JC, Wyss O, & Gibson DT (1979) Enzymatic oxidation of
p-nitrophenol. Biochem Biophys Res Commun, 88(2): 634-641.
Spencer MC (1965) Topical use of hydroquinone for depigmentation. J
Am Med Assoc, 194(9): 114-116.
Springborn Institute for Bioresearch (1984) Photoallergic contact
dermatitis in guinea-pigs (Armstrong method): Final report
(Unpublished data from Springborn Institute for Bioresearch,
submitted to WHO by CFTA).
Stenius U (1989) [Nordic Expert Group for Documentation of
Occupational Exposure Limits. 84. Hydroquinone.] Solna, Sweden,
National Institute of Occupational Health (Work and Health -
Scientific Publication Series No. 1989:15) (in Swedish with English
summary).
Stenius U, Warholm M, Rannug A, Walles S, Lundberg J, & Högberg J
(1989) The role of GSH depletion and toxicity in
hydroquinone-induced development of enzyme-altered foci.
Carcinogenesis, 10(3): 593-599.
Sterner JH, Oglesby FL, & Anderson B (1947) Quinone vapors and their
harmful effects. I. Corneal and conjunctival injury. J Ind Hyg
Toxicol, 26: 60-73.
Stevens HP (1945) Rubber photogelling agents - Part II. J Soc Chem
Ind, 64: 312-315.
Stom DJ (1975) Use of thin-layer and paper chromatography for
detection of ortho- and para-quinones formed in the course of phenol
oxidation. Acta Hydrochim Hydrobiol, 3: 39-45.
Stom DJ & Roth R (1981) Some effects of polyphenols on aquatic
plants. I. Toxicity of phenols in aquatic plants. Bull Environ
Contam Toxicol, 27: 332-337.
Stom DJ, Geel TA, & Balayan AE (1986) Effect of individual phenolic
compounds and their mixtures on luminous bacteria. Part I: Use of
bacterial luminescence extinguishing for biotesting of phenols. Acta
Hydrochim Hydrobiol, 14: 283-292.
Stott JG (1942) The application of potentiometric methods to
developer analysis. J Soc Motion Pict Eng, 39: 37-54.
Stubberfield CR & Cohen GM (1989) Interconversion of NAD(H) to
NADP(H) a cellular response to quinone-induced oxidative stress in
isolated hepatocytes. Biochem Pharmacol, 38: 2631-2637.
Subrahmanyam VV, Kolachana P, & Smith MT (1991) Metabolism of
hydroquinone by human myeloperoxidase: Mechanisms of stimulation by
other phenolic compounds. Arch Biochem Biophys, 286: 76-84.
Subrahmanyam VV, Doane-Setzer P, Steinmetz KL, Ross D, & Smith MT
(1990) Phenol-induced stimulation of hydroquinone bioactivation in
mouse bone marrow in vivo: possible implications in benzene
myelotoxicity. Toxicology, 62: 107-116.
Tabak HH, Chambers CW, & Kabler PW (1964) Microbial metabolism of
aromatic compounds. I Decomposition of phenolic compounds and
aromatic hydrocarbons by phenol-adapted bacteria. J Bacteriol, 87:
910-919.
Telford IR, Woodruff CS, & Linford RH (1962) Fetal resorption in the
rat as influenced by certain antioxidants. Am J Anat, 110: 29-36.
Terhaar CJ, Ewell WS, Dziuba SP, & Fassett DW (1972) Toxicity of
photographic processing chemicals to fish. Photogr Sci Eng, 16:
370-377.
Thomas DJ, Sadler A, Subrahmanyam VV, Siegel D, Reasor MJ, Wierda D,
& Ross D (1989a) Bone marrow stromal cell bioactivation and
detoxification of the benzene metabolite hydroquinone: Comparison of
macrophages and fibroblastoid cells. Mol Pharmacol, 37: 255-262.
Thomas DJ, Reasor MJ, & Wierda D (1989b) Macrophage regulation of
myelopoiesis is altered by exposure to the benzene metabolite
hydroquinone. Toxicol Appl Pharmacol, 97: 440-453.
Tissot A, Boule P, Lemaire J, Lambert S, & Palla JC (1985)
Photochimie et environnement. X. Evaluation de la toxicité des
produits de phototransformation de l'hydroquinone et des
chlorophénols en milieu aqueux. Chemosphere, 14: 1221-1230.
Tratnyek PG & Macalady DL (1989) Abiotic reduction of nitro aromatic
pesticides in anaerobic laboratory systems. J Agric Food Chem, 37:
248-254.
Trevors JT & Basaraba J (1980) Toxicity of benzoquinone and
hydroquinone in short-term bacterial bioassays. Bull Environ Contam
Toxicol, 25: 672-675.
Trower MK, Sariaslani FS, & Kitson FG (1988) Xenobiotic oxidation by
cytochrome P-450 enriched extracts of Streptomyces griseus.
Biochem Biophys Res Commun, 157(3): 1417-1422.
Tunek A, Platt KL, Przybylski M, & Oesch F (1980) Multi-step
metabolic activation of benzene: effect of superoxide dismutase on
covalent binding to microsomal macromolecules, and identification of
glutathione conjugates using high pressure liquid chromatography and
field desorption mass spectrometry. Chem-Biol Interact, 33: 1-17.
Tunek A, Högstedt B, & Olofsson T (1982) Mechanism of benzene
toxicity. Effects of benzene and benzene metabolites on bone marrow
cellularity, number of granulopoietic stem cells and frequency of
micronuclei in mice. Chem-Biol Interact, 39: 129-138.
Twerdok LE & Trush MA (1990) Differences in quinone reductase
activity in primary bone marrow stromal cells derived from C57BL/6
and DBA/2 mice. Res Commun Chem Pathol Pharmacol, 67(3): 375-386.
Twerdok LE, Rembish SJ, & Trush MA (1992) Induction of quinone
reductase and glutathione in bone marrow cells by
1,2-dithiole-3-thione: Effect on hydroquinone-induced cytotoxicity.
Toxicol Appl Pharmacol, 112: 273-281.
Umpelev VL, Kogan LA, & Gagarinova LM (1974)
[Thin-layer-chromatographic separation of polyhydric phenols in
effluents.] Zh Anal Khim, 29: 179-180 (in Russian).
US EPA (1985) Hydroquinone: Testing requirements. Fed Reg, 50(250):
53145-53156.
US EPA (1987) Part 1: The toxicity of 3400 chemicals to fish. Part
2: The toxicity of 1085 chemicals to fish. Washington, DC, US
Environmental Protection Agency, Office of Toxic Substances (EPA
560/6-87-002.
Usami Y, Landau AB, Fukuyama K, & Gellin GA (1980) Inhibition of
tyrosinase activity by 4-tert-butylcatechol and other depigmenting
agents. J Toxicol Environ Health, 6: 559-567.
Valadaud D & Izard C (1971) Contribution à l'étude des effets
biologiques de l'hydroqui-none. Action sur la division cellulaire. C
R Acad Sci Paris, D273: 2247-2248.
Valadaud-Barrieu D & Izard C (1973) Modifications du cycle
cellulaire sous l'influence de l'hydroquinone dans les méristèmes
radiculaires de Vicia faba. C R Acad Sci Paris, D276: 33-35.
Van der Sluis D (1980) DNA damage by hydroquinone (sublimed) #
8349-34 in the E. coli Pol A1- assay. Akron, Ohio, The
Goodyear Tire & Rubber Company (Report No 80-6-7).
Van der Walle HB, Klecak G, Geleick H, & Bensink T (1982b)
Sensitizing potential of 14 mono(meth)acrylates in the guinea-pig.
Contact Dermatitis, 8: 223-235.
Van der Walle HB, Delbressine LPC, & Seutter E (1982a) Concomitant
sensitization to hydroquinone and P-methoxyphenol in the guinea-pig;
inhibitors in acrylic monomers. Contact Dermatitis, 8: 147-154.
Van Duuren BL & Goldschmidt BM (1976) Cocarcinogenic and
tumor-promoting agents in tobacco carcinogenesis. J Natl Cancer
Inst, 56: 1237-1242.
Van Ketel WG (1984) Sensitization to hydroquinone and the monobenzyl
ether of hydroquinone. Contact Dermatitis, 10: 253.
Varagnat J (1981) Hydroquinone, resorcinol, and catechol. In:
Grayson M ed. Kirk-Othmer encyclopedia of chemical technology, 3rd
ed. New York, John Wiley & Sons, pp 39-69.
Vladescu C & Apetroae M (1983) Biophysical radiosensitization.
Radiat Environ Biophys, 22: 141-148.
Wallin M & Hartley-Asp B (1993) Effects of potential aneuploidy
inducing agents on microtubule assembly in vitro. Mutat Res, 287:
17-22.
Wallin H, Melin P, Schelin C, & Jergil B (1985) Evidence that
covalent binding of metabolically activated phenol to microsomal
proteins is caused by oxidised products of hydroquinone and
catechol. Chem-Biol Interact, 55: 335-346.
Wampler DA (1980) DNA damage by hydroquinone lot 07319A in the E.
coli Pol A1- assay. Akron, Ohio, The Goodyear Tire & Rubber
Company (Report No 80-3-7).
Wängberg SA & Blanck H (1988) Multivariate patterns of algal
sensitivity to chemicals in relation to phylogeny. Ecotoxicol
Environ Saf, 16: 72-82.
Wellens H (1982) [Comparison of the sensitivity of Brachydanio
rerio and Leuciscus idus by testing the fish toxicity of
chemicals and wastewaters.] Z Wasser Abwasser Forsch, 15: 49-52 (in
German).
Whettem SMA (1949) The determination of small amounts of
hydroquinone in styrene. Analyst, 74: 185-188.
Wierda D & Irons RD (1982) Hydroquinone and catechol reduce the
frequency of progenitor B lymphocytes in mouse spleen and bone
marrow. Immunopharmacology, 4: 41-54.
Wild D, King M-T, Eckhardt K, & Gocke E (1981) Mutagenic activity of
aminophenols and diphenols, and relations with chemical structure.
Mutat Res, 85: 456.
Winterbourn CC (1981) Cytochrome c reduction by semiquinone radicals
can be indirectly inhibited by superoxide dismutase. Arch Biochem
Biophys, 209(1): 159-167.
Woodard GDL (1951) The toxicity, mechanism of action, and metabolism
of hydroquinone. Washington, DC, George Washington University, 81 pp
(Dissertation).
Wynder EL & Hoffman D (1967) Tobacco and tobacco smoke. Studies in
experimental carcinogenesis. New York, Academic Press, pp 388-389.
Xu W & Adler I-D (1990) Clastogenic effects of known and suspect
spindle poisons studied by chromosome analysis in mouse bone marrow
cells. Mutagenesis, 5: 371-374.
Yager JW, Eastmond DA, Robertson ML, Paradisin WM, & Smith MT (1990)
Characterization of micronuclei induced in human lymphocytes by
benzene metabolites. Cancer Res, 50: 393-399.
Yamazaki I & Ohnishi T (1969) One-electron-transfer reactions in
biochemical systems. I. Kinetic analysis of the oxidation-reduction
equilibrium between quinol-quinone and ferro-ferricytochrome c.
Biochim Biophys Acta, 112: 469-481.
Yamazaki I, Mason HS, & Piette L (1960) Identification, by electron
paramagnetic resonance spectroscopy, of free radicals generated from
substrates by peroxidase. J Biol Chem, 235: 2444-2449.
Young LY & Rivera MD (1985) Methanogenic degradation of four
phenolic compounds. Water Res, 19(10): 1325-1332.
Young RHF, Ryckman DW, & Buzzell JC Jr (1968) An improved tool for
measuring biodegradability. J Water Pollut Control Fed, 40: 354-370.
Zeidman I & Deutl R (1945) Poisoning by hydroquinone and
mono-methyl-paraaminophenol sulfate. Am J Med Sci, 210: 328-333.
APPENDIX 1.
Bibliographic data bases consulted
Agricola
Aqualine
Aquatic Science and Fisheries Abstracts
BIBLINE
BIBRA
Biosis Previews
CA Search
CAB Abstracts
Cancerlit
CHEMLIST
CHRIS
ECDIN
Enviroline
EXICHEM
FEDREG
Food Science and Technical Abstracts
HSDB
Medline
Oceanic Abstracts
OHMTADS
RISKLINE
RTECS
ROADMAPS
SERIX
Toxline
TOXLIT
TSCATS
RESUME
1. Identité, propriétés physiques et chimiques et méthodes
d'analyse
L'hydroquinone (1,4-benzènediol; C6H4(OH)2) se présente à
l'état pur sous la forme d'un solide cristallin blanc dont le point
de fusion est égal à 173-174°C. Sa densité est de 1,332 à 15°C et sa
tension de vapeur de 2,4 x 10-3 Pa (1,8 x 10-5 mmHg) à 25°C.
Elle est extrêmement soluble dans l'eau (70g/litre à 25°C) et le
logarithme de son coefficient de partage n-octanol/eau est de
0,59. Sa solubilité dans les solvants organiques varie de 57% dans
l'éthanol à moins de 0,1% dans le benzène. L'hydroquinone est
combustible à condition de subir un chauffage préalable. C'est un
réducteur qui est oxydé réversiblement en semiquinone correspondante
et en quinone.
L'échantillonnage de l'hydroquinone dans l'air s'effectue soit
par piégeage dans un solvant soit par filtration sur membrane
d'ester cellulosique mixte.
Le dosage de l'hydroquinone s'effectue soit par titrimétrie
soit par spectrophotométrie ou plus couramment par chromatographie.
2. Sources d'exposition humaine et environnementale
L'hydroquinone existe sous forme libre ou conjuguée dans les
bactéries, les plantes et certains animaux. Plusieurs pays la
produisent en quantités industrielles. En 1979, la capacité mondiale
totale de production dépassait 40 000 tonnes et était retombée à
environ 35 000 tonnes en 1992. L'hydroquinone est très largement
utilisée comme réducteur, comme développateur en photographie ainsi
que antioxydant ou comme stabilisant pour certaines substances qui
se polymérisent en présence de radicaux libres; elle sert encore
d'intermédiaire dans la production des antioxydants, antiozonants,
produits agrochimiques et polymères. L'hydroquinone est également
utilisée pour la fabrication de cosmétiques et de préparations
médicinales.
3. Transport, distribution et transformation dans l'environnement
L'hydroquinone qui est présente dans l'environnement est le
produit de l'activité humaine mais elle se trouve également dans des
substances naturelles d'origine végétale ou animale.
En raison de ses propriétés physico-chimiques, l'hydroquinone
se répartit essentiellement dans le compartiment aquatique
lorsqu'elle est libérée dans l'environnement. Sa décomposition
résulte principalement de processus photochimiques et biologiques;
elle n'est donc pas persistante et ne manifeste aucune tendance à la
bioaccumulation.
4. Concentrations dans l'environnement et exposition humaine
On ne dispose d'aucune données sur la concentration de
l'hydroquinone dans l'air, le sol ou l'eau. Toutefois le dosage de
l'hydroquinone dans la fumée de cigarettes (courant principal) sans
bout-filtre a donné des résultats allant de 110 à 300 ug/cigarette,
résultats qui valent également pour la fumée du courant latéral. On
trouve également de l'hydroquinone dans des denrées alimentaires
d'origine végétale (par exemple le germe de blé), dans le café
infusé, dans les thés préparés à partir des feuilles de certaines
baies, avec des concentrations pouvant dépasser quelquefois 1%.
Les photographes amateurs peuvent être exposés à l'hydroquinone
par voie percutanée ou respiratoire. Toutefois on ne dispose
d'aucune donnée sur l'importance de cette exposition. L'exposition
percutanée peut également résulter de l'utilisation de produits
cosmétiques ou médicinaux contenant de l'hydroquinone, tels que les
éclaircissants. Dans les pays de la Communauté européenne, la teneur
des cosmétiques en hydroquinone est limitée à 2% au maximum. Aux
Etats-Unis d'Amérique, la Food and Drug Administration a proposé une
concentration comprise entre 1,5 et 2% pour les éclaircissants
cutanés. La concentration peut atteindre 4% dans certains
médicaments délivrés sur ordonnance. Dans certains pays, les
éclaircissants cutanés peuvent en contenir des quantités encore plus
importantes.
Du point de vue hygiène et sécurité, on ne dispose que de peu
de données concernant la surveillance de l'hydroquinone. On indique
des concentrations moyennes dans l'air au cours de la fabrication de
l'hydroquinone et des divers traitements subis par cette substance,
qui se situeraient dans la gamme de 0,13 à 0,79 mg/m3. Les limites
d'exposition professionnelles dans l'air (moyenne pondérée par
rapport au temps) dans les différents pays vont de 0,5 à 2 mg/m3.
5. Cinétique et métabolisme
L'hydroquinone est rapidement et largement résorbée chez
l'animal au niveau de l'intestin et de la trachée. L'absorption
percutanée est plus lente mais elle peut s'accélérer en présence de
véhicules tels que les alcools. L'hydroquinone se répartit
rapidement et largement dans les différents tissus. Elle est
métabolisée en p-benzoquinone et autres produits d'oxydation et sa
détoxification s'effectue par conjugaison sous forme de
monoglucuronide, monosulfate et mercapturates. L'excrétion de
l'hydroquinone et de ses métabolites est rapide et s'effectue
principalement par la voie urinaire.
L'hydroquinone et/ou ses dérivés réagissent avec différents
constituants biologiques tels que les macromolécules et les
molécules de faible masse moléculaire et ils exercent des effets sur
la métabolisme cellulaire.
6. Effets sur les mammifères de laboratoire et les systèmes
d'épreuve in vitro
Les valeurs de la DL50 par voie orale varient de 300 à 1300
mg/kg de poids corporel pour un certain nombre d'espèces animales.
Toutefois pour le chat, elle se situe entre 42 et 86 mg/kg de poids
corporel. Une intoxication aiguë par de fortes concentrations
d'hydroquinone entraîne de graves effets sur le système nerveux
central et notamment une hyperexcitabilité, des tremblements, des
convulsions, le coma et la mort. A des doses sublétales, ces effets
sont réversibles. Pour les rongeurs, on estime la DL50 par voie
percutanée à > 3800 mg/kg. On ne dispose d'aucun renseignement sur
les valeurs de la CL50.
L'épidermo-réaction effectuée en une seule fois au moyen d'une
préparation à 2% d'hydroquinone a provoqué une irritation chez le
lapin notée à 1,22 sur une échelle allant de 0 à 4. Des applications
topiques quotidiennes effectuées pendant trois semaines avec de
l'hydroquinone à 2 ou 5% dans une émulsion huile/eau, sur la peau
rasée de cobayes noirs, a entraîné une dépigmentation, des
altérations inflammatoires et un épaississement de l'épiderme. La
dépigmentation était plus marquée à fortes concentrations et les
femelles se sont révélées plus sensibles que les mâles.
Les tests de sensibilisation effectués sur des cobayes
suscitent des réactions faibles à fortes selon la méthode ou le
véhicule utilisé. Les réactions les plus fortes ont été obtenues
avec le test de sensibilisation maximale sur le cobaye. On a
également observé une sensibilisation croisée de presque 100 pour
cent entre l'hydroquinone et le p-méthoxyphénol chez le cobaye
mais les éléments qui pourraient militer en faveur de l'existence de
réactions analogues avec la p-phénylénediamine, l'acide
sulfanilique et la p-benzoquinone restent limités.
Une étude de toxicité par voie orale de six semaines chez des
rats mâles F-344 a permis de mettre en évidence des néphropathies et
une prolifération des cellules rénales. Après 13 semaines de gavage,
on a mis en observation des rats F-344 et des souris B6C3F1. Des
signes de néphrotoxicité se sont manifestés chez les rats aux doses
respectives de 100 et 200 mg/kg, avec des tremblements et des
convulsions à cette dernière dose; chez les deux espèces, on a
observé une diminution du gain de poids. L'administration d'une dose
de 400 mg/kg a entraîné la mort des rats. Chez les souris qui
avaient reçu cette même dose pendant 13 semaines, on a relevé des
tremblements, des convulsions et des lésions affectant l'épithélium
gastrique. Des rats Sprague Dawley exposés pendant 13 semaines à de
l'hydroquinone ont présenté une réduction du gain de poids et des
signes neurologiques témoignant d'une atteinte centrale à 200 mg/kg.
Ces signes ont également été observés à la dose de 64 mg/kg de poids
corporel mais ils étaient absents à 20 mg/kg.
Après injection sous-cutanée d'hydroquinone à des rats, on a
observé une diminution de la fécondité chez les mâles et un
allongement du cycle oestral chez les femelles. Toutefois ces effets
n'ont pas été observés dans les études portant sur une
administration par voie orale (étude de létalité dominante et étude
sur deux générations). Une étude portant sur le développement de
rats ayant reçu par voie orale des doses de 300 mg/kg de poids
corporel a révélé une légère toxicité pour les femelles gravides et
une réduction du poids du foetus. Chez le lapin, la dose sans effets
toxiques observables sur la mère était de 25 mg/kg/jour; la dose
sans effets toxiques sur le développement était de 75 mg/kg/jour.
Lors d'une étude de deux générations portant sur la reproduction de
rats, l'administration d'hydroquinone n'a entraîné aucun effet nocif
sur la reproduction à des doses orales quotidiennes allant jusqu'à
150 mg/kg de poids corporel. La dose sans effets toxiques
observables pour les géniteurs a été fixée par 15 mg/kg/jour; en ce
qui concerne les effets sur la reproduction observés en l'espace de
deux générations, on a obtenu une valeur de 150 mg/kg/jour.
L'hydroquinone provoque la formation de micronoyaux in vivo
et in vitro. Des aberrations portant sur la structure et le nombre
des chromosomes ont été observées in vitro ainsi qu'après
administration intrapéritonéale in vivo. En outre, on a pu mettre
en évidence in vitro des mutations géniques, des échanges entre
chromatides soeurs et des lésions de l'ADN. Après injection
intrapéritonéale d'hydroquinone à des souris, on a observé, dans les
cellules germinales des mâles, de aberrations chromosomiques d'une
ampleur comparable à celles que l'on observait dans les cellules de
la moelle osseuse. Une épreuve de létalité dominante effectuée sur
des rats mâles recevant de l'hydroquinone par voie orale n'a pas
permis d'établir l'existence de mutations au niveau des cellules
germinales.
Lors d'une étude de deux ans, au cours de laquelle on a
administré à des rats F-344/N de l'hydroquinone par voie orale, on a
observé, chez les mâles, des adénomes affectant les tubules rénaux
dont la fréquence était liée à la dose. L'incidence de ces adénomes
était statistiquement significative dans le groupe qui recevait une
forte dose. Dans ce même groupe, on a également observé une
hyperplasie des cellules tubulaires rénales. Chez les femelles, on a
observé un accroissement de l'incidence, lié à la dose, des
leucémies à monocytes. Chez des souris B6C3F1, on a observé une
incidence sensiblement accrue des adénomes hépatocellulaires. Dans
une autre étude, l'hydroquinone administrée dans la proportion de
0,8% de la nourriture, a entraîné une augmentation significative de
l'incidence de l'hyperplasie épithéliale des papilles rénales et une
augmentation également significative des hyperplasies et des
adénomes au niveau des tubules rénaux chez les rats mâles. En
revanche, on n'a pas observé d'augmentation dans l'incidence des
leucémies à monocytes chez les femelles. Chez les souris,
l'incidence de l'hyperplasie spinocellulaire affectant l'épithélium
de la portion cardiaque de l'estomac présentait une augmentation
significative dans les deux sexes. Chez les mâles, on notait
également une incidence sensiblement accrue des adénomes
hépatocellulaires et des hyperplasies tubulaires rénales. Quelques
adénomes rénaux ont été observés.
Des études sur des souris, effectuées in vivo (injection
intrapéritonéale) et in vitro montrent que l'hydroquinone a un
effet cytotoxique, à savoir qu'elle diminue la cellularité
médullaire et splénique et qu'elle possède également un pouvoir
immunodépresseur puisqu'elle inhibe la maturation des lymphocytes-B
et bloque l'activité des cellules tueuses naturelles. Les résultats
obtenus indiquent également que les macrophages de la moelle osseuse
pourraient être les principales cibles des effets myélotoxiques
exercés par l'hydroquinone. Cependant, une étude biologique au long
cours sur des rongeurs n'a pas révélé l'existence d'effets
myélotoxiques.
Une étude de 90 jours sur des rats au cours de laquelle on a
utilisé une batterie de tests fonctionnels et observationnels, a
montré qu'à des doses respectives de 64 et 200 mg d'hydroquinone/kg,
les animaux étaient pris de tremblements et qu'à la dose de 200
mg/kg, il y avait réduction de l'activité générale. L'examen
anatomopathologique du système nerveux n'a rien donné.
7. Effets sur l'homme
On a fait état de cas d'intoxication consécutifs à l'ingestion
d'hydroquinone seule ou de développateurs photographiques contenant
de l'hydroquinone. Les principaux signes de ces intoxications
étaient les suivants: urines foncées, vomissements, douleurs
abdominales, tachycardie, tremblements, convulsions et coma. On a
également signalé des décès après l'ingestion de développateurs
photographiques à base d'hydroquinone. Lors d'une étude contrôlée au
cours de laquelle des volontaires humains ont ingéré quotidiennement
pendant 3 à 5 mois 300 à 500 mg d'hydroquinone, on n'a pas relevé le
moindre signe pathologique dans le sang et les urines.
L'application cutanée d'hydroquinone dans divers excipients à
des concentrations inférieures à 3% n'a causé que des effets
négligeables sur des volontaires humains appartenant à diverses
races. Cependant, on dispose de rapports selon lesquels des crèmes
destinées à éclaircir la peau et contenant 2% d'hydroquinone ont
produit une leucodermie ainsi qu'une ochronose. L'hydroquinone
(solution aqueuse à 1% ou crème à 5%) peut provoquer des irritations
(érythème ou taches épidermiques). On a également observé des
dermatites de contact d'origine allergique dues à l'hydroquinone.
Une double exposition à de l'air chargé d'hydroquinone et de
quinone entraîne une irritation oculaire, une photophobie, des
lésions de l'épithélium cornéen, voire des ulcères cornéens et des
troubles visuels. On connaît des cas où l'acuité visuelle a
sensiblement baissé. Une irritation peut se manifester à partir de
2,25 mg/m3. Une exposition de longue durée peut faire apparaître
des taches sur la conjonctive et la cornée et provoquer
l'opacification de cette dernière. Après exposition quotidienne
pendant au moins deux ans à 0,05-14,4 mg d'hydroquinone/m3, on a
observé une inflammation et une dyschromie de la cornée et de la
conjonctive; on n'a pas observé de cas graves avant cinq ans au
moins d'exposition. On dispose d'un rapport qui décrit des cas de
lésions cornéennes apparues plusieurs années après cessation de
l'exposition à l'hydroquinone.
On ne dispose pas de données épidémiologiques suffisantes pour
évaluer la cancérogénicité de l'hydroquinone chez l'homme.
8. Effets sur les autres êtres vivants au laboratoire et dans leur
milieu naturel
Pour expliquer le comportement écotoxicologique de
l'hydroquinone il faut se rapporter à ses propriétés
physico-chimiques, et notamment à sa sensibilité à la lumière, au pH
et à l'oxygène dissous. L'écotoxicité de l'hydroquinone, qui est
généralement élevée (par exemple < 1 mg/litre pour les organismes
aquatiques), varie d'une espèce à l'autre.
Les algues, les levures, les champignons et les plantes en
général sont moins sensibles à l'hydroquinone que les autres
organismes habituellement utilisés pour les épreuves toxicologiques.
Toutefois,au sein d'un même groupe toxonomique, la sensibilité des
différentes espèces à l'hydroquinone peut varier d'un facteur 1000.
RESUMEN
1. Identidad, propiedades físicas y químicas y métodos analíticos
La hidroquinona (1,4-bencenodiol; C6H4(OH)2) es una
sustancia cristalina blanca en estado puro, cuyo punto de fusión es
de 173-174 °C. Su peso específico es de 1,332 a 15 °C, y su presión
de vapor, de 2,4 x 10-3Pa (1,8 x 10-5 mmHg) a 25 °C. Es muy
hidrosoluble (70 g/litro a 25 °C), y el logaritmo de su coeficiente
de reparto n-octanol/agua es de 0,59. Por lo que se refiere a los
disolventes orgánicos, su solubilidad varía entre el 57% para el
etanol, y menos del 0,1% para el benceno. La hidroquinona es
combustible si se calienta previamente. Es un agente reductor, que
se oxida reversiblemente transformándose en semiquinona y quinona.
Se pueden obtener muestras de la hidroquinona presente en el
aire capturándola, bien sea en un disolvente o sobre una membrana-
filtro de éster de celulosa mixto.
Para analizar los niveles de hidroquinona se utilizan técnicas
de valorimetría, espectrofotometría o, muy a menudo, cromatografía.
2. Fuentes de exposición humana y ambiental
La hidroquinona está presente en forma tanto libre como
conjugada en bacterias, plantas y algunos animales. Varios países la
producen industrialmente. En 1979, la capacidad mundial total de
producción de esta sustancia superaba las 40 000 toneladas, mientras
que en 1992 fue de aproximadamente 35 000 toneladas. Es ampliamente
utilizada como agente reductor, como medio de revelado fotográfico,
como antioxidante o estabilizador de ciertos materiales que se
polimerizan en presencia de radicales libres, y como producto
químico intermedio en la producción de antioxidantes, agentes
antiozono, productos agroquímicos y polímeros. La hidroquinona se
emplea también en la elaboración de productos cosméticos y
preparados médicos.
3. Transporte, distribución y transformación en el medio ambiente
La hidroquinona presente en el medio ambiente procede de la
actividad del hombre o forma parte de productos naturales de las
plantas y animales.
Debido a sus propiedades fisicoquímicas, la hidroquinona
liberada en el medio penetra sobre todo en los compartimientos de
agua. Se degrada como resultado de procesos tanto fotoquímicos como
biológicos, por lo que no persiste en el medio. No se produce
bioacumulación.
4. Niveles ambientales y exposición humana
No se han hallado datos sobre las concentraciones de
hidroquinona en la atmósfera, el suelo o el agua. Sin embargo, se ha
analizado la hidroquinona presente en la corriente principal de humo
emitida por cigarrillos sin filtro, en la que se han hallado entre
110 y 300 µg por cigarrillo, así como en el humo lateral. Se ha
hallado hidroquinona en alimentos derivados de las plantas (por
ejemplo, el germen de trigo), en el café listo para beber y en los
tes preparados a partir de las hojas de algunas bayas en que la
concentración supera a veces el 1%.
Los aficionados a la fotografía pueden verse expuestos a la
hidroquinona por vía cutánea o por inhalación; sin embargo, no se
dispone de datos sobre niveles de exposición. Otra causa de
exposición cutánea es el uso de productos cosméticos o médicos con
hidroquinona, como los utilizados para aclarar el color de la piel.
Los países de la Comunidad Europea (CE) han restringido su empleo en
los productos cosméticos a un máximo del 2%. En los Estados Unidos,
la Administración de Alimentos y Medicamentos ha propuesto
concentraciones de entre 1,5% y 2% para los productos de maquillaje
decolorante. Algunos medicamentos de venta con receta contienen
concentraciones de hasta un 4%, y en ciertos países se venden
cosméticos decolorantes que contienen concentraciones incluso
mayores.
Son pocos los datos disponibles sobre la hidroquinona por lo
que se refiere a la vigilancia de la higiene industrial. Según se ha
señalado, el valor promedio de las concentraciones que este producto
alcanza en el aire durante su fabricación y transformación oscila
entre 0,13 y 0,79 mg/m3. El límite de la exposición atmosférica
ocupacional (promedio ponderado por el tiempo) oscila entre 0,5 y 2
mg/m3, según los países.
5. Cinética y metabolismo
La hidroquinona es rápida y ampliamente absorbida en el tubo
digestivo y la tráquea de los animales. La absorción cutánea es más
lenta, pero se ve acelerada por vehículos como los alcoholes. La
hidroquinona se distribuye de forma rápida y generalizada por los
tejidos. Es metabolizada en p-benzoquinona y otros productos de
oxidación, y la detoxificación se produce por conjugación,
formándose los derivados monoglucurónido, monosulfato y
mercaptúrico. La excreción de la hidroquinona y de sus metabolitos
es rápida y se produce principalmente a través de la orina.
La hidroquinona y/o sus derivados reaccionan con distintos
componentes biológicos, como macromoléculas o moléculas de bajo peso
molecular, y tienen efectos sobre el metabolismo celular.
6. Efectos en mamíferos de laboratorio y en los sistemas in vitro
Los valores de la DL50 por vía oral en varias especies
animales oscilan entre 300 y 1300 mg/kg de peso corporal. En el
gato, sin embargo, la DL50 varía entre 42 y 86 mg/kg de peso
corporal. La exposición aguda a altos niveles de hidroquinona tiene
efectos graves sobre el sistema nervioso central (SNC), que abarcan
desde la hiperexcitabilidad, pasando por temblores, convulsiones y
coma, hasta la muerte. A dosis subletales esos efectos son
reversibles. Se ha calculado que la DL50 por vía cutánea es de >
3800 mg/kg en roedores. No se dispone de los valores de la CL50
por inhalación.
Un preparado de hidroquinona al 2% aplicado a conejos mediante
una única prueba del parche tuvo unos efectos irritantes de 1,22 (en
una escala de 0 a 4). La aplicación tópica diaria durante tres
semanas de hidroquinona al 2% o 5% en una emulsión de aceite/agua
sobre la piel depilada de cobayos negros provocó despigmentación,
cambios inflamatorios y espesamiento de la epidermis. La
despigmentación fue más marcada con las concentraciones mayores, y
los cobayos hembra se revelaron más sensibles que los machos.
Las pruebas de sensibilización realizadas en cobayos han
provocado respuestas de carácter entre débil y fuerte, según los
métodos o vehículos empleados. Las reacciones más intensas fueron
las observadas en la prueba de maximización aplicada a cobayos. Se
observó también en estos animales una sensibilización cruzada de
casi el 100% entre la hidroquinona y el p-metoxifenol, pero los
indicios de reacción cruzada con la p-fenilendiamina, el ácido
sulfanílico y la p-benzoquinona fueron sólo limitados.
Un estudio de toxicidad oral realizado durante seis semanas con
ratas macho F-344 reveló la aparición de nefropatía y proliferación
de las células renales. En estudios de sobrealimentación forzada
mediante sonda esofágica llevados a cabo durante 13 semanas con
ratas F-344 y ratones B6C3F1, se observaron en las ratas signos de
nefrotoxicidad a dosis de 100 y 200 mg/kg, temblores y convulsiones
a dosis de 200 mg/kg, así como una disminución del aumento del peso
corporal tanto en las ratas como en los ratones. Las dosis de 400
mg/kg resultaron letales para las ratas. En los ratones sometidos
durante 13 semanas a dosis de 400 mg/kg se observaron temblores,
convulsiones y lesiones del epitelio gástrico. La exposición de
ratas Sprague Dawley a hidroquinona durante 13 semanas dio lugar a
una atenuación del aumento del peso corporal y la aparición de
signos de afección del SNC a la dosis de 200 mg/kg. Estos signos
neurológicos se observaron también a la dosis de 64 mg/kg de peso
corporal, pero no así a la de 20 mg/kg.
La hidroquinona inyectada por vía subcutánea redujo la
fecundidad de las ratas macho y prolongó el ciclo menstrual de las
ratas hembra. Esa observación no se reprodujo en cambio en los
estudios de administración oral (un estudio de dominancia letal y
otro efectuado con dos generaciones). En un estudio sobre el
desarrollo de la rata, dosis orales de 300 mg/kg de peso corporal
tuvieron un ligero efecto tóxico en las madres y provocaron una
disminución del peso corporal del feto. En el conejo, el nivel sin
efectos observados (NOEL) por lo que se refiere a la toxicidad
materna fue de 25 mg/kg al día, y de 75 mg/kg al día en lo que
respecta a la toxicidad ontogénica. En un estudio de los efectos
sobre la reproducción realizado con dos generaciones de ratas, la
hidroquinona no tuvo efecto alguno a dosis orales de hasta 150 mg/kg
de peso corporal al día. El nivel sin efectos adversos observados
(NOAEL) fue de 15 mg/kg al día por lo que hace a la toxicidad
materna, y de 150 mg/kg al día en lo referente a los efectos sobre
la reproducción observados a lo largo de dos generaciones.
La hidroquinona tiene un efecto inductor sobre los micronúcleos
in vivo e in vitro. Se han observado aberraciones cromosómicas
estructurales y numéricas in vitro y tras la administración
intraperitoneal in vivo. Se ha demostrado además la inducción de
mutaciones genéticas, intercambio de cromátides hermanas y lesiones
del ADN in vitro. La hidroquinona causó aberraciones cromosómicas
en las células germinales de ratones macho, del mismo orden de
magnitud que en las células de médula ósea de esa misma especie tras
inyección intraperitoneal. En una prueba de dominancia letal llevada
a cabo con ratas macho tratadas por vía oral no se observó ningún
efecto de inducción de mutaciones de las células germinales.
En un estudio de dos años de duración, la administración oral
de hidroquinona provocó una incidencia dosisdependiente de adenomas
de las células tubulares renales en ratas macho F-344/N. La
incidencia fue estadísticamente significativa en el grupo tratado
con la dosis más alta; en los machos sometidos a esa dosis se halló
también hiperplasia de las células tubulares renales. En las ratas
hembra se observó un aumento dosisdependiente de la incidencia de
leucemia de células mononucleares. En los ratones hembra B6C3F1 se
observó una incidencia significativamente mayor de adenomas
hepatocelulares. En otro estudio, la hidroquinona (presente a un
nivel del 0,8% en la ingesta alimentaria) provocó un aumento
significativo de la incidencia de hiperplasia epitelial de la papila
renal y un aumento significativo de la incidencia de adenomas e
hiperplasia de los túbulos renales en las ratas macho. En las ratas
hembra no se observó ningún incremento de la incidencia de leucemia
de células mononucleares. En los ratones, la incidencia de
hiperplasia de las células escamosas del epitelio del antro cardiaco
aumentó significativamente en los dos sexos. En los ratones macho se
observó un aumento significativo de la incidencia de adenomas
hepatocelulares, así como de hiperplasia tubular renal. Se observó
también un reducido número de adenomas de células renales.
Los estudios realizados in vivo (inyección intraperitoneal) e
in vitro con ratones han demostrado que el efecto citotóxico de la
hidroquinona se debe a que reduce la celularidad de la médula ósea y
del bazo, y a su posible efecto inmunosupresor, mediado por la
inhibición de la maduración de los linfocitos B y de la actividad
natural de las células asesinas. Los resultados indican además que
las células más afectadas por la mielotoxicidad de la hidroquinona
son quizá los macrófagos de la médula ósea. En una biovaloración
prolongada realizada con roedores no se observaron esos efectos
mielotóxicos.
En un estudio realizado durante 90 días con ratas mediante una
batería de pruebas funcionales y de observación, se advirtieron
temblores a dosis de 64 y 200 mg de hidroquinona/kg, dosis esta
última que además provocó una disminución de la actividad general.
El resultado de los análisis neuropatológicos fue negativo.
7. Efectos en el hombre
Se han notificado casos de intoxicación tras la ingestión oral
de hidroquinona sola o de productos de revelado fotográfico que
contenían dicho producto. Los principales signos de intoxicación
fueron el oscurecimiento de la orina, vómitos, dolor abdominal,
taquicardia, temblores, convulsiones y coma. Se han notificado
muertes provocadas por la ingestión de productos de revelado
fotográfico que contenían hidroquinona. En un estudio controlado
realizado con voluntarios, la ingestión de 300-500 mg de
hidroquinona diarios durante 3-5 meses no produjo cambios
patológicos observables en la sangre y la orina.
La aplicación cutánea de distintas bases que contenían
concentraciones de hidroquinona inferiores al 3% tuvo efectos
insignificantes en hombres voluntarios de distintas razas. Sin
embargo, se han notificado algunos casos que llevan a pensar que hay
cremas cutáneas decolorantes con hidroquinona al 2% que han
provocado la aparición de leucoderma, así como de ocronosis. Se han
producido casos de irritación (eritema o coloración) por
hidroquinona (en solución acuosa al 1% o en forma de crema al 5%).
También se han diagnosticado casos de dermatitis alérgica de
contacto por hidroquinona.
La exposición simultánea a hidroquinona y quinona presentes en
el aire causa irritación ocular, sensibilidad a la luz, lesiones del
epitelio corneal, úlceras corneales y trastornos visuales. En
algunos casos se han producido pérdidas considerables de visión. Se
han observado efectos irritantes a niveles de exposición de 2,25
mg/m3 o más. La exposición prolongada provoca coloración de la
conjuntiva y la córnea, así como opacidad. La exposición diaria
durante al menos dos años a concentraciones de hidroquinona de 0,05-
14,4 mg/m3 ha dado lugar al lento desarrollo de inflamación y
decoloración de la córnea y la conjuntiva; pero no se han observado
casos graves hasta transcurridos al menos 5 años. En un estudio se
describieron casos de aparición de lesiones de la córnea al cabo de
varios años de interrumpida la exposición a la hidroquinona.
No se dispone de datos epidemiológicos adecuados para evaluar
la carcinogenicidad de la hidroquinona en el hombre.
8. Efectos en otros organismos en el laboratorio y sobre el terreno
Los efectos ecotoxicológicos de la hidroquinona guardan
relación con sus propiedades fisicoquímicas, entre ellas su
sensibilidad a la luz, al pH y al oxígeno disuelto. Su ecotoxicidad,
que por lo general es alta (por ejemplo, < 1 mg/litro para los
organismos acuáticos), varía de una especie a otra.
Las algas, levaduras, hongos y plantas son menos sensibles
a la hidroquinona que los otros organismos empleados habitualmente
para evaluar la toxicidad. No obstante, dentro de un mismo grupo
taxonómico, la sensibilidad de las distintas especies a la
hidroquinona puede variar de 1 a 1000.