
INTERNATIONAL PROGRAMME ON CHEMICAL SAFETY
ENVIRONMENTAL HEALTH CRITERIA 145
METHYL PARATHION
This report contains the collective views of an international group of
experts and does not necessarily represent the decisions or the stated
policy of the United Nations Environment Programme, the International
Labour Organisation, or the World Health Organization.
Published under the joint sponsorship of
the United Nations Environment Programme,
the International Labour Organisation,
and the World Health Organization
First draft prepared by Dr R.F. Hertel and co-workers,
Fraunhofer Institute of Toxicology and Aerosol
Research, Hanover, Germany
World Health Orgnization
Geneva, 1993
The International Programme on Chemical Safety (IPCS) is a
joint venture of the United Nations Environment Programme, the
International Labour Organisation, and the World Health
Organization. The main objective of the IPCS is to carry out and
disseminate evaluations of the effects of chemicals on human health
and the quality of the environment. Supporting activities include
the development of epidemiological, experimental laboratory, and
risk-assessment methods that could produce internationally
comparable results, and the development of manpower in the field of
toxicology. Other activities carried out by the IPCS include the
development of know-how for coping with chemical accidents,
coordination of laboratory testing and epidemiological studies, and
promotion of research on the mechanisms of the biological action of
chemicals.
WHO Library Cataloguing in Publication Data
Methyl parathion.
(Environmental health criteria ; 145)
1.Environmental exposure 2.Methyl parathion - adverse effects
3.Methyl parathion - poisoning 4.Methyl parathion - toxicity
I.Series
ISBN 92 4 157145 4 (NLM Classification: WA 240)
ISSN 0250-863X
The World Health Organization welcomes requests for permission
to reproduce or translate its publications, in part or in full.
Applications and enquiries should be addressed to the Office of
Publications, World Health Organization, Geneva, Switzerland, which
will be glad to provide the latest information on any changes made
to the text, plans for new editions, and reprints and translations
already available.
(c) World Health Organization 1993
Publications of the World Health Organization enjoy copyright
protection in accordance with the provisions of Protocol 2 of the
Universal Copyright Convention. All rights reserved.
The designations employed and the presentation of the material
in this publication do not imply the expression of any opinion
whatsoever on the part of the Secretariat of the World Health
Organization concerning the legal status of any country, territory,
city or area or of its authorities, or concerning the delimitation
of its frontiers or boundaries.
The mention of specific companies or of certain manufacturers'
products does not imply that they are endorsed or recommended by the
World Health Organization in preference to others of a similar
nature that are not mentioned. Errors and omissions excepted, the
names of proprietary products are distinguished by initial capital
letters.
CONTENTS
ENVIRONMENTAL HEALTH CRITERIA FOR METHYL
PARATHION
1. SUMMARY AND EVALUATION, CONCLUSIONS, RECOMMENDATIONS
1.1. Summary and evaluation
1.1.1. Exposure
1.1.2. Uptake, metabolism, and excretion
1.1.3. Effects on organisms in the environment
1.1.4. Effects on experimental animals and
in vitro test systems
1.1.5. Effects on human beings
1.2. Conclusions
1.3. Recommendations
2. IDENTITY, PHYSICAL AND CHEMICAL PROPERTIES, ANALYTICAL METHODS
2.1. Identity
2.1.1. Primary constituent
2.1.2. Technical product
2.1.2.1. Purity
2.2. Physical and chemical properties
2.3. Conversion factors
2.4. Analytical methods
2.4.1. Sampling, extraction, clean-up
2.4.1.1 Plant material (tobacco,
fruits, vegetables, crops
with low oil (fat) content)
2.4.1.2 Dairy products, products with a
high fat content (edible fats)
2.4.1.3 Blood, body fluids
2.4.1.4 Soil, sediments
2.4.1.5 Water
2.4.1.6 Air
2.4.1.7 Formulations
2.4.2. Instrumental analytical methods
2.4.2.1 Gas chromatography
2.4.2.2 High performance liquid chroma-
tography (HPLC)
2.4.2.3 Thin layer chroma-
tography (TLC)
2.4.2.4 Spectrophotometry
2.4.2.5 Polarography
2.4.2.6 Mass spectrometry
2.4.3. Detection limits
2.4.4. Confirmatory method
3. SOURCES OF HUMAN AND ENVIRONMENTAL EXPOSURE
3.1. Natural occurrence
3.2. Man-made sources
3.2.1. Production process
3.2.2. Loss into the environment
3.2.3. Production
3.2.4. World consumption
3.2.5. Formulations
3.3. Uses
4. ENVIRONMENTAL TRANSPORTATION, DISTRIBUTION, AND TRANSFORMATION
4.1. Transportation and distribution between media
4.1.1. Air
4.1.2. Water
4.1.3. Soil
4.1.4. Vegetation and wildlife
4.1.5. Entry into the food-chain
4.2. Biotransformation
4.2.1. Degradation involving biota
4.2.2. Abiotic degradation
4.2.2.1 Photodegradation
4.2.2.2 Hydrolytic degradation
4.2.3. Bioaccumulation
4.3. Interaction with other physical,
chemical, and biological factors
4.4. Ultimate fate following use
5. ENVIRONMENTAL LEVELS AND HUMAN EXPOSURE
5.1. Environmental levels
5.1.1. Air
5.1.2. Water
5.1.3. Soil
5.1.4. Food
5.1.5. Terrestrial and aquatic organisms
5.2. General population exposure
5.3. Occupational exposure during
manufacture, formulation, or use
6. KINETICS AND METABOLISM
6.1. Absorption
6.2. Distribution
6.3. Metabolic transformation
6.4. Elimination and excretion in expired air,
faeces, or urine
6.5. Retention and turnover
7. EFFECTS ON ORGANISMS IN THE ENVIRONMENT
7.1. Microorganisms
7.1.1. Bacteria and fungi
7.1.2. Algae
7.2. Aquatic animals
7.2.1. Short-term toxicity in
aquatic invertebrates
7.2.1.1 Laboratory studies on
single species
7.2.1.2 Mesocosmic studies
7.2.2. Fish
7.2.2.1 Laboratory studies on
single species
7.2.2.2 Mesocosmic studies
7.2.3. Amphibians
7.3. Terrestrial organisms
7.3.1. Plants
7.3.2. Invertebrates
7.3.3. Birds
7.3.4. Non-laboratory mammmals
8. EFFECTS ON EXPERIMENTAL ANIMALS AND IN VITRO TEST SYSTEMS
8.1. Single exposure
8.2. Skin and eye irritation, sensitization
8.3. Short-term exposures
8.4. Long-term exposures
8.5. Reproduction, embryotoxicity,
and teratogenicity
8.6. Mutagenicity related end-points
8.7. Carcinogenicity
8.8. Special studies
8.9. Factors toxicity
8.10. Mode of action
8.10.1. Inhibition of esterases
8.10.2. Possible alkylation of
biological macromolecules
8.10.3. General
9. EFFECTS ON MAN
9.1. General population exposure
9.1.1. Acute toxicity
9.1.2. Effects of short- and
long-term exposure,
controlled human studies
9.2. Occupational exposure
9.2.1. Epidemiological studies
10. PREVIOUS EVALUATIONS BY INTERNATIONAL BODIES
REFERENCES
ANNEX I
RESUME
RESUMEN
WHO TASK GROUP ON ENVIRONMENTAL HEALTH
CRITERIA FOR METHYL PARATHION
Members
Dr L.A. Albert, Consultores Ambientales Asociados, S.C.,
Xalapa, Veracruz, Mexico (Vice-Chairman)
Dr S. Dobson, Ecotoxicology and Pollution Section, Institute of
Terrestrial Ecology, Monks Wood Experimental Station, Abbots
Ripton, Huntingdon, Cambridgeshire, United Kingdom
Dr D.J. Ecobichon, Pharmacology and Therapeutics, McGill
University, Montreal, Canada (Chairman)
Dr R.F. Hertel, Fraunhofer Institute of Toxicology & Aerosol
Research, Hanover, Germany (Co-rapporteur)
Dr S.K. Kashyap, National Institute of Occupational Health,
Meghaninagar, Ahmedabad, India
Dr I. Nordgren, Department of Toxicology, Karolinska Institute,
Stockholm, Sweden
Dr K.C. Swentzel, Toxicology Branch II, Health Effects Division,
US Environmental Protection Agency, Washington, DC, USA
(Co-rapporteur)
Dr M. Tasheva, Department of Toxicology, Institute of Hygiene and
Occupational Health, Medical Academy, Sofia, Bulgaria
Dr L. Varnagy, Department of Agrochemical Hygiene, University of
Agricultural Sciences, Institute for Plant Protection,
Keszthely, Hungary
Observers
Dr W. Flucke, Bayer AG, Fachbereich Toxikologie, Institut für
Toxikologie Landwirtschaft, Wuppertal, Germany
Secretariat
Dr K.W. Jager, International Programme on Chemical Safety, World
Health Organization, Geneva, Switzerland (Secretary)
Dr E. Matos, Unit of Carcinogen Identification and Evaluation,
International Agency for Research on Cancer (IARC), Lyon,
France
NOTE TO READERS OF THE CRITERIA
MONOGRAPHS
Every effort has been made to present information in the
criteria monographs as accurately as possible without unduly
delaying their publication. In the interest of all users of the
environmental health criteria monographs, readers are kindly
requested to communicate any errors that may have occurred to the
Director of the International Programme on Chemical Safety, World
Health Organization, Geneva, Switzerland, in order that they may be
included in corrigenda.
* * *
A detailed data profile and a legal file can be obtained from
the International Register of Potentially Toxic Chemicals, Palais
des Nations, 1211 Geneva 10, Switzerland (Telephone No. 7988400 -
7985850).
NOTE: The proprietary information contained in this monograph
cannot replace documentation for registration purposes, because the
latter has to be closely linked to the source, the manufacturing
route, and the purity/impurities of the substance to be registered.
The data should be used in accordance with paragraphs 82-84 and
recommendations paragraph 90 of the Second FAO Government
Consultation (1982).
ENVIRONMENTAL HEALTH CRITERIA FOR METHYL
PARATHION
A WHO Task Group on Environmental Health Criteria for Methyl
Parathion met at the World Health Organization, Geneva from 19 to
23 August 1991. Dr K.W. Jager, IPCS, welcomed the participants on
behalf of Dr M. Mercier, Director of the IPCS, and the three IPCS
cooperating organizations (UNEP/ILO/WHO). The Group reviewed and
revised the draft and made an evaluation of the risks for human
health and the environment from exposure to methyl parathion.
The first draft of the EHC on methyl parathion was prepared by
Dr R.F. Hertel and his co-workers of the Fraunhofer Institute of
Toxicology and Aerosol Research in Hanover, Germany. The same group
assisted in the preparation of the second draft, incorporating
comments received following circulation of the first drafts to the
IPCS contact points for Environmental Health Criteria monographs.
Dr K.W. Jager of the IPCS Central Unit was responsible for the
scientific content of the monograph, and Mrs M.O. Head of Oxford
for the editing.
The efforts of all who helped in the preparation and
finalization of the monograph are gratefully acknowledged.
1. SUMMARY AND EVALUATION, CONCLUSIONS, RECOMMENDATIONS
1.1 Summary and evaluation
1.1.1 Exposure
Methyl parathion is an organophosphorus insecticide that was
first synthesized in the 1940s. It is relatively insoluble in water,
poorly soluble in petroleum ether and mineral oils, and readily
soluble in most organic solvents. Pure methyl parathion consists of
white crystals; technical methyl parathion is a light tan colour with
a garlic-like odour. It is thermally unstable and undergoes fast
decomposition above pH 8.
Gas chromatography, with either alkali flame ionization (AFID) or
flame photometric (FPD) detectors, is the most common method for the
determination of methyl parathion. Detection limits range from 0.01 to
0.1 µg/litre in water, and from 0.1 to 1 ng/m3 in air. HPLC and TLC
are also useful methods of detection.
The distribution of methyl parathion in air, water, soil, and
organisms in the environment is influenced by several physical,
chemical, and biological factors.
Studies using model ecosystems and mathematical modelling
indicate that methyl parathion partitions mainly into the air and soil
in the environment with lesser amounts going to plants and animals.
There is virtually no movement through soil and neither the parent
compound nor its breakdown products will normally reach ground water.
Methyl parathion in air mainly arises from the spraying of the
compound, though some volatilization occurs with the evaporation of
water from leaves and the soil surface. Background atmospheric levels
of methyl parathion in agricultural areas range from not detectable to
about 70 ng/m3. Air concentrations after spraying have been shown to
decline rapidly over 3 days reaching background levels after about 9
days. Levels in river water (in laboratory studies) declined to 80% of
the initial concentration after 1 h and 10% after 1 week. Methyl
parathion is retained longer in soil than in air or water, though
retention is greatly influenced by soil type; sandy soil can lose
residues of the compound more rapidly than loams. Residues on plant
surfaces and within leaves decline rapidly with half lives of the
order of a few hours; complete loss of methyl parathion occurs within
about 6-7 days.
Animals can degrade methyl parathion and eliminate the
degradation products within a very short time. This is slower in lower
vertebrates and invertebrates than in mammals and birds.
Bioconcentration factors are low and the accumulated methyl parathion
levels transitory.
By far the most important route for the environmental degradation
of methyl parathion is microbial degradation. Loss of the compound in
the field and in model ecosystems is more rapid than that predicted
from laboratory studies. This is because of the variety of
microorganisms capable of degrading the compound in different habitats
and circumstances. The presence of sediment or plant surfaces, which
increases the microbial populations, increases the rate of breakdown
of methyl parathion.
Methyl parathion can undergo oxidative degradation, to the less
stable methyl paraoxon, by ultraviolet radiation (UVR) or sunlight;
sprayed films degrade under UVR with a half-life of about 40 h.
However, the contribution of photolysis to total loss in an aquatic
system has been estimated to be only 4%. Hydrolysis of methyl
parathion also occurs and is more rapid under alkaline conditions.
High salinity also favours hydrolysis of the compound. Half-lives of
a few minutes were recorded in strongly reducing sediments, though
methyl parathion is more stable when sorbed on other sediments.
In towns in the centre of agricultural areas of the USA, methyl
parathion concentrations in air varied with season and peaked in
August or September; maximum levels in surveys were mainly in the
range of 100-800 ng/m3 during the growing season. Concentrations in
natural waters of agricultural areas in the USA ranged up to 0.46
µg/litre, with highest levels in summer. There are only small numbers
of published reports on residues of methyl parathion in food
throughout the world. In the USA, residues of methyl parathion in food
have generally been reported at very low levels with few individual
samples exceeding maximum residue limits (MRLs). Only trace residue
levels of methyl parathion were detected in the total dietary studies
reported. Methyl parathion residues were highest in leafy (up to 2
mg/kg) and root (up to 1 mg/kg) vegetables in market basket surveys in
the USA between 1966 and 1969. Food preparation, cooking, and storage
all cause decomposition of methyl parathion residues further reducing
exposure of humans. Raw vegetables and fruits may contain higher
residues after misuse.
The production, formulation, handling, and use of methyl
parathion as an insecticide are the principal potential sources of
exposure of humans. Skin contact and, to a lesser degree, inhalation
are the main routes of exposure of workers.
In a study on farm spray-men (with unprotected workers using
ultra-low-volume (ULV) handsprays) an intake of 0.4-13 mg of methyl
parathion per 24 h was calculated from the excreted p-nitrophenol in
the urine. Early re-entry into treated crops is a further source of
exposure.
The general population may be exposed to air-, water-, and
food-borne residues of methyl parathion as a consequence of
agricultural or forestry practices, the misuse of the agent resulting
in the contamination of fields, crops, water, and air through
off-target spraying.
1.1.2 Uptake, metabolism, and excretion
Methyl parathion is readily absorbed via all routes of exposure
(oral, dermal, inhalation) and is rapidly distributed to the tissues
of the body. Maximum concentrations in various organs were detected
1-2 h after treatment. Conversion of methyl parathion to methyl
paraoxon occurs within minutes of administration. A mean terminal
half-life of 7.2 h was determined in dogs following intravenous (i.v.)
administration of methyl parathion. The liver is the primary organ of
metabolism and detoxification. Methyl parathion or methyl paraoxon are
mainly detoxified in the liver through oxidation, hydrolysis, and
demethylation or dearylation with reduced glutathione (GSH). The
reaction products are O-methyl O-p-nitrophenyl phosphorothioate or
dimethyl phosphorothioic or dimethylphosphoric acids and
p-nitrophenol. Therefore, it is possible to estimate exposure by
measuring the urinary excretion of p-nitrophenol; urinary excretion
of p-nitrophenol by human volunteers was 60% within 4 h and
approximately 100% within 24 h. The metabolism of methyl parathion is
important for species selective toxicity, and the development of
resistance. The elimination of methyl parathion and metabolic products
occurs primarily via the urine. Studies conducted on mice with
radiolabelled (32P-methyl parathion) revealed 75% of radioactivity in
the urine and up to 10% radioactivity in the faeces after 72 h.
1.1.3 Effects on organisms in the environment
Microorganisms can use methyl parathion as a carbon source and
studies on a natural community showed that concentrations of up to 5
mg/litre increased biomass and reproductive activity. Bacteria and
actinomycetes showed a positive effect of methyl parathion while fungi
and yeasts were less able to utilize the compound. A 50% inhibition of
growth of a diatom occurred at about 5 mg/litre. Cell growth of
unicellular green algae was reduced by between 25 and 80 µg methyl
parathion/litre. Populations of algae became tolerant after exposure
for several weeks.
Methyl parathion is highly toxic for aquatic invertebrates with
most LC50s ranging from < 1 µg to about 40 µg/litre. A few
arthropod species are less susceptible. The no-effect level for the
water flea (Daphnia magna) is 1.2 µg/litre. Molluscs are much less
susceptible with LC50s ranging between 12 and 25 mg/litre.
Most fish species in both fresh and sea water have LC50s of
between 6 and 25 mg/litre with a few species substantially more or
less sensitive to methyl parathion. The acute toxicity for amphibians
is similar to that for fish.
Population effects have been seen on communities of aquatic
invertebrates in experimental ponds treated with methyl parathion. The
concentrations needed to cause these effects would occur only with
overspraying of water bodies and, even then, would last for only a
short time. Population effects are, therefore, unlikely to be seen in
the field. Kills of aquatic invertebrates would be unlikely to lead to
lasting effects.
Care should be taken to avoid overspraying of ponds, rivers, and
lakes, when using methyl parathion. The compound should never be
sprayed under windy conditions.
Methyl parathion is a non-selective insecticide that kills
beneficial species as readily as pests. Kills of bees have been
reported following spraying of methyl parathion. Incidents concerning
bees were more severe with methyl parathion than with other
insecticides. Africanized honey bees are more tolerant of methyl
parathion than European strains.
Methyl parathion was moderately toxic for birds in laboratory
studies, with acute oral LD50s ranging between 3 and 8 mg/kg body
weight. Dietary LC50s ranged from 70 to 680 mg/kg diet. There is no
indication that birds would be adversely affected from recommended
usage in the field.
Extreme care must be taken to time methyl parathion spraying to
avoid adverse effects on honey bees.
1.1.4 Effects on experimental animals and in vitro test systems
Oral LD50 values of methyl parathion in rodents range from 3 to
35 mg/kg body weight, and dermal LD50 values, from 44 to 67 mg/kg
body weight.
Methyl parathion poisoning causes the usual organophosphate
cholinergic signs attributed to accumulation of acetylcholine at nerve
endings. Methyl parathion becomes toxic when it is metabolized to
methyl paraoxon. This conversion is very rapid. No indications of
organophosphorous-induced, delayed neuropathy (OPIDN) have been
observed.
Technical methyl parathion was found not to have any primary eye
or skin irritating potential.
In short-term toxicity studies, using various routes of
administration on the rat, dog, and rabbit, inhibition of plasma, red
blood cell, and brain ChE, and related cholinergic signs were
observed. In a 12-week feeding study on dogs, the no-observed-
adverse-effect level (NOAEL) was 5 mg/kg diet (equivalent to 0.1 mg/kg
body weight per day). In a 3-week dermal toxicity study on rabbits,
the no-observed-effect-level (NOEL) was 10 mg/kg body weight daily.
Inhalation exposure for 3 weeks indicated a NOEL of 0.9 mg/m3 air.
At 2.6 mg/m3, only slight inhibition of plasma ChE was observed.
Long-term toxicity/carcinogenicity studies were carried out on
mice and rats. The NOEL for rats was 0.1 mg/kg body weight per day,
based on ChE inhibition. There is no evidence of carcino genicity in
mice and rats, following long-term exposure. In another 2-year study
on rats, however, there was evidence of a peripheral neurotoxic effect
at a dose of 50 mg/kg diet.
Methyl parathion has been reported to have DNA-alkylating
properties in vitro. The results of most of the in vitro
genotoxicity studies on both bacterial and mammalian cells were
positive, while 6 in vivo studies using 3 different test systems
produced equivocal results.
In reproduction studies, at toxic dose levels (ChE inhibition),
there were no consistent effects on litter size, number of litters,
pup survival rates, and lactation performance. No primary teratogenic
or embryotoxic effects were noted.
1.1.5 Effects on human beings
Several cases of acute methyl parathion poisoning have been
reported. Signs and symptoms are those characteristic of systemic
poisoning by cholinesterase-inhibiting organophosphorous compounds.
They include peripheral and central cholinergic nervous system
manifestations appearing as rapidly as a few minutes after exposure.
In case of dermal exposure, symptoms may increase in severity for more
than one day and may last several days.
Studies on volunteers, following repeated, long-term exposures,
suggest that there is a decrease in blood cholinesterase activities
without clinical manifestations.
No cases of organophosphorous-induced, delayed peripheral
neuropathy (OPIDN) have been reported. Neuro-psychiatric sequelae have
been reported in cases of multiple exposure to pesticides including
methyl parathion.
An increase in chromosomal aberrations has been reported in cases
of acute intoxications.
No human data were available to evaluate the teratogenic and
reproductive effects of methyl parathion.
The available epidemiological studies deal with multiple exposure
to pesticides and it is not possible to evaluate the effects of
long-term exposure to methyl parathion.
1.2 Conclusions
Methyl parathion is a highly toxic organophosphorus ester
insecticide. Overexposure from handling during manufacture, use,
and/or accidental or intentional ingestion may cause severe or fatal
poisoning. Methyl parathion formulations may, or may not, be
irritating to the eyes or to the skin, but are readily absorbed. As a
consequence, hazardous exposures may occur without warning.
Methyl parathion is not persistent in the environment. It is not
bioconcentrated and is not transferred through food-chains. It is
degraded rapidly by many microorganisms and other forms of wild life.
This insecticide is likely to cause damage to ecosystems only in
instances of heavy over-exposure resulting from misuse or accidental
spills; however, pollinators and other beneficial insects are at risk
from spraying with methyl parathion.
Exposure of the general population to methyl parathion residues
occurs predominantly via food. If good agricultural practices are
followed, the Acceptable Daily Intake (0-0.02 mg/kg body weight),
established by FAO/WHO, will not be exceeded. Dermal exposure may
also occur through accidental contact with foliar residues in sprayed
fields or in areas adjacent to spraying operations as a consequence of
off-target loss of the chemical.
With good work practices, hygienic measures, and safety
precautions, methyl parathion is unlikely to present a hazard for
those occupationally exposed.
1.3 Recommendations
* For the health and welfare of workers and the general population,
the handling and application of methyl parathion should be
entrusted only to competently supervised and well-trained
applicators, who must follow adequate safety measures and use the
chemical according to good application practices.
* The manufacture, formulation, agricultural use, and disposal of
methyl parathion should be carefully managed to minimize
contamination of the environment.
* Regularly exposed workers should receive appropriate monitoring
and health evaluation.
* To minimize risks for all individuals, a 48-h interval between
the spraying and re-entry into any sprayed area is recommended.
* Pre-harvest intervals should be established and enforced by
national authorities.
* In view of the high toxicity of methyl parathion, this agent
should not be considered for use in hand-applied, ULV spraying
practices.
* Do not overspray water bodies. Choose spraying times to avoid
killing pollinating insects.
* Information on the health status of workers exposed only to
methyl parathion (i.e., in manufacture, formulation) should be
published, in order to better evaluate the risks of this chemical
for human health.
* More definitive studies should be conducted on residues of methyl
parathion in fresh foods.
* A more definitive genotoxic assessment of methyl parathion should
be conducted.
2. IDENTITY, PHYSICAL AND CHEMICAL PROPERTIES, ANALYTICAL METHODS
2.1 Identity
2.1.1 Primary constituent
Molecular formula: C8H10NO5PS
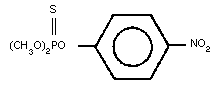 Relative molecular mass: 263.23
Common names: methyl parathion
accepted by
ESA (Entomological Society of
America)
JMAF (Japanese Ministry of
Agriculture, Fisheries and Food)
WHO (World Health Organization)
parathion-methyl
accepted by
BSI (British Standards Institution)
ISO (International Organization for
Standardization)
metaphos
accepted by the USSR
CAS chemical name: O,O-dimethyl O-(4-nitro-phenyl)
phosphorothioate
IUPAC systematic name: O,O-Dimethyl O-4-nitrophenylphos-
phorothioate
CAS registry number: 298-00-0
RTECS number: TG 0175000
EINECS number: 206-050-1
EEC number: 015-035-00-7
Common synonyms:
Demethylfenitrothion; dimethyl para-nitrophenyl
monothiophosphate; O,O-dimethyl O-( para-nitrophenyl)
phosphorothioate; dimethyl para-nitrophenyl phosphorothionate;
dimethyl 4-nitrophenyl phosphorothionate; O,O-dimethyl
O-(para-nitrophenyl) thionophosphate; dimethyl
para-nitrophenyl thiophosphate;
O,O-dimethyl- O-(para-nitrophenyl) thiophosphate; dimethyl
parathion; ENT 17292; metaphos; methyl-parathion; methylthiophos;
MPT; NCI CO2971; parathion methyl homolog; phosphorothioic acid
O,O-dimethyl O-(4-nitro-phenyl) ester; phosphorothioic acid
O,O-dimethyl O-(para-nitrophenyl) ester BAY 11405; 8056 HC;
E601
2.1.2 Technical product
Major trade names:
A-Gro; Azofos; Azophos; Bladan M; Cekumethion; Dalf; Divithion;
Drexel Methyl Parathion 4E & 601; Dygun; Dypar; Ekatox; Folidol
M, M40 & 80; Fosferno M50; Gearphos; Mepaton; Meptox; Metacid 50;
Metacide; Metafos; Metaphos; Methyl-E 605; Methyl Fosferno;
Methylthiophos; Metron; M-Parathion; Niletar; Niran M-4; Nitran;
Nitrox; Nitrox 80; Oleovofotox; Parapest M-50; Parataf; Paratox;
Paridol; Parton M; Penncap M & MLS; Sinafid M-48; Sixty-Three
Special E.C. Insecticide; Tekwaisa; Thiophenit; Thylpar M-50;
Toll; Unidol; Vertac Methyl Parathion; technical product 80%,
Wofatox; Wolfatox.
2.1.2.1. Purity
Technical methyl parathion is available as a solution containing
80% active ingredient (a.i.), 16.7% xylene, and 3.3% inert
ingredients.
The following impurities were identified in one sample of
technical-grade methyl parathion: O,O-dimethyl- S-methyl
dithiophosphate, nitroanisol, nitro-phenol, isomers of methyl
parathion, and the dithio-analogue of methyl parathion (Warner, 1975).
2.2 Physical and chemical properties
Physical state: pure: white crystalline solid or powder
(National Fire Protection Association, 1986)
technical (80%) pure: light to dark tan
liquid (Worthing & Walker, 1987)
Melting point: 37-38 °C (The Merck Index, 1983)
35-36 °C (Worthing, 1983)
Freezing point: about 29 °C (technical product)
(Worthing & Walker, 1987)
Density/specific gravity:
1.358 at 20 °C/40 °C (d204 1.358)
(The Merck Index, 1983)
Vapour pressure: 1.3 mPa at 20 °C
(Worthing & Walker, 1987)
Octanol/water partition coefficient:
log Kow = 2.68 (measured)
log Kow = 1.81-3.43 (reported range)
(Hansch & Leo, 1987)
Water solubility: 55-60 mg/litre at 25 °C (pure)
(Midwest Research Institute, 1975;
National Research Council, 1977)
37.7 mg/litre at 19 °C (pure)
(Bowman & Sans, 1979)
57 mg/litre at 22 °C (anal. grade)
(Sanders & Seiber, 1983)
Nonaqueous solubility: soluble in ethanol, chloroform,
aliphatic solvents, and slightly
soluble in light petroleum
Volatility (pure): 0.14 mg/m3 at 20 °C (Spencer, 1982)
Odour: like rotten eggs or garlic (technical grade)
(Midwest Research Institute, 1975; Anon.,
1984)
Odour threshold: 0.0125 mg/m3 (Akhmedov, 1968)
Other properties: hydrolyses and isomerizes easily
(White-Stevens, 1971)
Half-life in aqueous solution at 20 °C, pH 1-5:
175 days (Melnikov, 1971)
2.3 Conversion factors
1 ppm methyl parathion= 10.76 mg/m3 at 25 °C, 1066 mbar
1 mg methyl parathion/m3 = 0.0929 ppm
2.4 Analytical methods
2.4.1 Sampling, extraction, clean-up
Standardized methods for the determination of various residues
are reported in the Manual of pesticide residue analysis (Thier &
Zeumer, 1987).
2.4.1.1 Plant material (tobacco, fruits, vegetables, crops with low
oil (fat) content)
(a) Extraction
Three extraction methods have mainly been used, all of which are
suitable for multiresidue analysis.
(1) Soxhlet extraction with chloroform - 10% methanol has been
proposed for field-weathered crops by Bowman (1981).
(2) Acetonitrile combined with various amounts of water has been used
by Mills et al. (1963), Wessel (1967), Osadchuk et al. (1971),
Luke et al. (1975), and Stahr et al. (1979). The plant material
is homogenized in a blender with acetonitrile, in some instances
after the addition of Celite (Nelson, 1967; Funch, 1981;).
High-moisture products (fruits and vegetables) are extracted with
pure acetonitrile while samples of dry products (hays, grains,
feedstuff) are blended with acetonitrile-water (65:35).
Extraction is followed by solvent partitioning into petroleum
ether with the addition of sodium chloride (Mills et al., 1963;
Wessel, 1967; Nelson, 1967) into dichloromethane (Funch, 1981),
and dichloromethane/hexane (10:200) (Osadchuk et al., 1971).
(3) Acetone was preferred as the solvent in particular in
multiresidue analysis by Becker (1971), Pflugmacher & Ebing
(1974), Sagredos & Eckert (1976), Becker (1979), Specht & Tillkes
(1980), Miellet (1982), Sonobe et al. (1982), Luke & Doose
(1983), Luke & Doose (1984), Ebing (1985), Andersson & Ohlin
(1986), Vogelsang & Thier (1986), Gyorfi et al. (1987), Thier &
Zeumer (1987), and Becker & Schug, (1990). In some instances,
celite was added. Depending on the water content of the sample,
water was added. In a second step, the acetone extracts were
further extracted with either dichloromethane, dichloro
methane/petroleum ether, or dichloromethane/ n-hexane. The
extract was dried over anhydrous sodium sulfate, reduced in
volume in a Kuderna-Danish concentrator, and subjected to
further clean-up.
Extraction with acetone- o-xylene (19:1) (Ross & Harvey, 1981),
toluene/hexane (75:25) (Johansson, 1978), chloroform (Ault et
al., 1979), or supercritical fluid extraction using methanol
(Capriel et al., 1986), has also been reported.
(b) Column clean-up
The published clean-up procedures are usually suitable for
multiresidue analysis. For plant material with a low fat content, 3
column clean-up procedures have been developed.
(1) The oldest method involves the use of chromatography on Florisil
(often topped with anhydrous sodium sulfate) (Mills et al., 1963;
Nelson, 1967; Schnorbus & Phillips, 1967; Wessel, 1967; Beckman
& Garber, 1969; Osadchuk et al., 1971; Luke et al., 1975;
Johansson, 1978; Gretch & Rosen, 1984, 1987). Although it has
been claimed that organo phosphorous pesticides are partially
lost during Florisil clean-up (Luke et al., 1975), high
recoveries (usually > 80 %) have been reported for methyl
parathion. Various solvents and solvent mixtures are used for
chromatography on Florisil including: diethylether/petroleum
ether, ethyl ether/hexane, and acetone/toluene,
diethylether/petroleum ether being the most frequently used.
Fractionation is achieved by increasing successively the
diethylether content. Florisil clean-up is usually used for a
combined clean-up of organochlorine and organophosphorous
pesticides. Luke et al. (1975) reported that gas chromatography
(GC) with a thermionic detector was sufficiently selective to
detect organophosphorous pesticides without Florisil clean-up.
(2) Alternatively, clean-up of pesticides in multiresidue analysis
has been achieved by chromatography on charcoal (Becker, 1971,
1979; Miellet, 1982; Sonobe et al., 1982; Luke & Doose, 1984;
Ebing, 1985; Gyorfi et al., 1987). To this end, charcoal is mixed
with silica gel (1:15) (and sometimes also celite or magnesia).
In most instances, elution is achieved with mixtures of
dichloromethane/acetone/toluene (e.g., 5:1:1) (Ebing, 1985; Thier
& Zeumer, 1987). Recoveries are high (often > 90 %). Charcoal
clean-up is particularly suited for dry products (< 10 % water).
The simultaneous clean-up of organochlorine and organo-
phosphorous pesticides is also possible with chromatography on
charcoal.
(3) In recent years, a clean-up of pesticides in multiresidue
analysis by gel permeation chromatography (GPC) has become
popular (Pflugmacher & Ebing, 1974; Ault et al., 1979; Specht &
Tillkes, 1980; Andersson & Ohlin, 1986; Vogelgesang & Thier,
1986; Steinwandter, 1988). The stationary phase consists, in most
instances, of Bio Beads SX3 (a polystyrene gel). Ethyl
acetate/cyclohexane (1:1), dichloromethane/cyclohexane (1:1) and,
more recently, acetone/cyclohexane (3:1) have been used as
elution mixtures. Gel permeation chromatography is mainly used to
protect the GC column and the GC detector against contam-
ination. GPC removes material of higher relative molecular mass.
Recoveries > 85% have been reported. Frequently, GPC is combined
with the additional purification step of silica gel
chromatography (Specht & Tillkes, 1980; Andersson & Ohlin, 1986;
Vogelsaifng & Thier, 1986) where elution is achieved with
toluene/hexane (35:65), followed by toluene and acetone/toluene,
with increasing acetone content. However, while the additional
clean-up by silica gel column chromatography is important when
organo chlorine pesticides are present, it is not necessary for
organophosphorous pesticides if analysis is performed by gas
chromatography with flame photometric detection.
2.4.1.2 Dairy products, products with a high fat content (edible
fats)
Clean-up techniques for products with a high fat content have
been reviewed by Waters (1990). Florisil column chromatography and gel
permeation chromatography are also suited for a clean-up of samples
with a high fat content. In addition, clean-up using normal phase HPLC
has been reported (Gillespie & Waters, 1986). Fat is dissolved in
n-hexane and fractionated on silica gel HPLC using
dichloromethane/hexane as solvent. However, complete separation
ofmethyl parathion from the fat is not achieved. As an alternative,
fat is adsorbed on aluminum oxide (Luke & Doose, 1984) or on Calflo E
(calcium silicate) (Specht, 1978; Thier & Zeumer, 1987). Finally, a
sweep codistillation clean-up of edible oils has been reported by
Storherr et al. (1967) and Watts & Storherr (1967). This method has
been standardized also for plant material (Thier & Zeumer, 1987).
After extraction of the sample with ethyl acetate, the concentrated
extract is injected into a heated glass column packed with glass wool
or glass beads followed by the injection of ethyl acetate or petroleum
ether in a nitrogen stream. The nitrogen carrier gas sweeps the
volatile component through the tube to a condensing bath and through
an Arnakrom scrubber tube to a collection tube. Sweep codistillation
may be followed by a further Florisil clean-up.
The extraction and clean-up of vegetable oil can be speeded up by
performing extraction and clean-up in one step using a system of three
ready-to-use cartridges in series (Extralut-3, Sep-Pack silicade1 and
Sep-Pack C18) where the assembled columns are eluted with
acetonitril (saturated with n-hexane) (Di Muccio et al., 1990).
2.4.1.3 Blood, body fluids
Methyl parathion is extracted from blood with hexane or benzene
and analysed without further clean-up (Gabica et al., 1971; De Potter
et al., 1978). No extraction is necessary if methyl parathion is
determined by polarography (Zietek, 1976).
Measurement of the urinary metabolites and the cholinesterase
activity were used to supervise the exposure of workers coming into
contact with methyl parathion or parathion and to observe their
elimination in cases of poisoning (see section 5.3) (Elliot et al.,
1960; Arterberry et al., 1961; Shafic & Enos, 1969; Wolfe et al.,
1970; Ware et al., 1974b; NIOSH, 1976).
2.4.1.4 Soil, sediments
Methyl parathion is extracted from soil with acetone,
acetone/ n-hexane or hexane/isopropanol (Schutzmann et al., 1971;
Agishev et al., 1977; Garrido & Monteoliva, 1981; Wegman et al., 1984;
Kjoelholt, 1985). It is partitioned in a second step into
dichloromethane. While several authors determine the pesticides
without further clean-up, additional silica gel adsorption
chromatography has been used by Wegman et al., (1984) and Kjoelholt
(1985). The recovery of methyl parathion is 70-85%.
When sediments are analysed, elemental sulfur represents a
particular problem. Kjoelholt et al. separated the sulfur by tetra
butylammonium hydrogensulfate (Kjoelholt, 1985), while Schutzmann et
al. (1971) refluxed the sediment extract with Raney copper.
For the extraction, the sediment mixed with sand and sodium
sulfate can be placed into a column and eluted using acetone :
dichloromethane (1:1) (Belisle & Swineford, 1988).
2.4.1.5 Water
Extraction and concentration of methyl parathion from water is
achieved either by liquid/liquid extraction (Kawahara et al., 1967;
Pionke et al., 1968; Mestres et al., 1969; Konrad et al., 1969; Zweig
& Devine, 1969; Schutzmann et al., 1971; Coburn & Chau, 1974; Chmil et
al., 1978; Chernyak & Oradovskii, 1980; Miller et al., 1981; Spingarn
et al., 1982; Bruchet et al., 1984; Albanis et al., 1986; Li & Wang,
1987; Brodesser & Schoeler, 1987), or by adsorption on polymeric
material (Paschal et al., 1977; Le Bel et al., 1979; Agostiano et al.,
1983; Xue, 1984; Clark et al., 1985). Various solvents have been used
for solvent extractions including: diethyl ether/hexane (1:1),
benzene, petroleum ether, hexane/isopropanol; chloroform,
dichloromethane, and ethyl acetate. Recoveries have been high (in
most instances > 90 %). If the liquid/liquid extraction is scaled up
using a "Goulden large sample extractor" and 120 litre of water,
detection limits may be lower by a factor of about 150 compared with
1-litre samples (i.e., a detection limit of 2.5 ng/litre (ppt) has
been achieved for methyl parathion) (Foster & Rogerson, 1990). The
extraction efficiency can be further improved by continuous
liquid-liquid extraction, which allows the use of non-polar solvents
as n-pentane (Bruchet et al., 1984; Brodesser & Schoeler, 1987).
Water samples are frequently analysed for pesticides without further
clean-up, while Florisil clean-up has been used in some instances
(Mestres et al., 1969; Miller et al., 1981).
High concentration factors are achieved, if methyl parathion (and
other pesticides) are adsorbed on polymeric material, such as XAD-2
(Paschal et al., 1977; Le Bel et al., 1979), XAD-4 (Xue et al., 1984),
Tenax (Agostiano et al., 1983) or Porapack Q (Clark et al., 1985).
Elution from XAD is achieved with diethyl ether, acetone/hexane
(15:85), diethyl ether-hexane (85:15). Recoveries are >90 %. If Tenax
is used, both solvent elution (diethyl ether) or thermoelution can be
used to desorb the pesticides. Solid-phase extraction (using C-18
cartridges) will become the method of choice for the rapid extraction
of organophosphorous insecticides from water (Swineford & Belisle,
1989; Sherma & Bretschneider, 1990).
2.4.1.6 Air
Most methods for the determination of pesticides in air have been
developed as multiresidue methods. Pesticides in air are either
absorbed in liquids or adsorbed on polymeric material. Thus,
pesticides may be trapped in ethylene glycol, which is subsequently
extracted with dichloromethane (Tessari & Spencer, 1971; Sherma &
Shafik, 1975) or they may be trapped on glass beads coated with
cottonseed oil (Compton, 1973). Further clean-up is achieved by silica
gel or Florisil column chromatography.
Among the solid polymeric material used to trap pesticides,
polyurethane foam (PUF) is by far the most popular (Lewis et al.,
1977; Rice et al., 1977; Lewis & McLeod, 1982; Lewis & Jackson, 1982;
Belashova et al., 1983; Beine, 1987). Air can be collected both with
low-volume (approx. 4 litre/min) or high-volume samplers (up to 250
litre/min). PUF can be reused after careful cleaning (e.g., with 5%
diethyl ether in n-hexane). In some instances, Tenax, Chromosorb
102, or Porapack R is sandwiched between PUF plugs to enhance the
collection efficiency. Collection efficiencies in excess of 80% have
been reported for methyl parathion. A filter may be added to remove
particulate matter (Lewis et al., 1977). Methyl parathion is usually
determined without further clean-up. Finally, XAD-4 (Wehner et al.,
1984) and silica gel (Klisenko & Girenko, 1980; Liang & Zhang, 1986)
have been used as solid trapping materials.
2.4.1.7 Formulations
When analysing formulations, the determination of by-products and
impurities is an important objective. A variety of instrumental
techniques have been used for the analysis of formulations including:
gas chromatography (Jackson, 1976; Jackson, 1977a), high performance
liquid chromatography (Jackson, 1977b), infrared analysis (Goza,
1972), P-31-nuclear magnetic resonance spectroscopy (Greenhalgh et
al., 1983), and spectrophotometry after alkaline hydrolysis to
p-nitrophenol (Blanco & Sanchez, 1989). An inter laboratory study
has been carried out using both GC (Jackson, 1977a) and HPLC (Jackson,
1977b). With both methods, coefficients of variation of 1.7% have been
determined. The instrumental techniques are described below.
2.4.2 Instrumental analytical methods
2.4.2.1 Gas chromatography
Gas chromatrophic (GC) methods for the determination of
pesticides (including methyl parathion) have been reviewed by Ebing
(1987).
Organophosphorous pesticides, including methyl parathion, are
sufficiently volatile and thermally stable to be amenable to gas
chromatography and it is by far the most important method for the
determination of methyl parathion. This technique provides the good
resolution necessary for multiresidue analysis. Moreover, very
sensitive and specific detectors are available, in particular for the
analysis of organophosphorous pesticides.
(a) Detectors
The two most widely used detectors for organophosphorous
pesticides are the alkali flame ionization detector (AFID) and
variations of this detector (thermionic detector (Patterson, 1982),
nitrogen-phosphorous detector) and the flame photometric detector
(FPD) (Bowman, 1981). The AFID makes use of the phenomenon that the
flame ionization detector yields enhanced response to nitrogen- and
phosphorus-containing compounds, in the presence of alkali metal
salts. The detection limit is in the low picogram range. The detector
discriminates against other compounds 30-50 fold. The flame
photometric detector (FPD) operates with a cool, hydrogen rich flame
for the detection of phosphorus- and sulfur-containing compounds,
which form POH and S2 species. These species emit light at 526 nm
(POH) and 394 nm (S2), which is monitored by using interference
filters and a photomultiplier. The detector is easy to operate and
results are reproducible. The detector is highly specific. The
response of 100 ng of parathion is 130 000 times greater than that of
an equal amount of aldrin. Furthermore, It is of advantage that any
solvent can be used with the detector. For the determination of methyl
parathion the P mode is the method of choice, though the S mode can
also be used (sensitivity 10 times lower) as methyl parathion contains
both P and S atoms.
Finally, the electron capture detector (ECD) is sometimes used
for the analysis of methyl parathion as it responds not only to the
P=S moiety, but in particular to the NO2 group.
(b) Columns
A definite identification of a pesticide by its retention time on
one column is not possible. Analysis on at least one further column
with a stationary phase of different polarity is necessary to confirm
the identity of a compound.
Packed columns are frequently used for pesticide residue
analysis, though resolution is substantially poorer compared with
capillary columns and identification of the pesticides is less
specific. Solid supports are usually of the Chromosorb W type. In some
instances, Gaschrom Q has also been used. A large variety of
stationary phases, used either alone or in admixture, have been
employed. The most frequently used phases are DC 200, QF-1, OV 17,
OV-101, OV-210, and SE-30. Relative retention times for many
stationary phases have been reported by several authors for a large
variety of pesticides (up to 600 compounds including other industrial
chemicals) (Bowman & Beroza, 1967; Ambrus et al., 1981b; Daldrup et
al., 1981; Prinsloo & de Beer, 1987; Saxton, 1987; Suprock & Vinopal,
1987; Omura et al., 1990).
Packed column GC allows the separation of only a limited number
of pesticides. Capillary columns exhibit a considerably better
separation efficiency than packed columns. Such capillary columns have
been used by several authors for methyl parathion analysis (Krijgsman
& van den Kamp, 1976; Ripley & Braun, 1983; Stan & Goebel, 1983;
Ebing, 1985; Andersson & Ohlin, 1986; Vogelsang & Thier, 1986).
Retention time data on a SE-30 capillary column have been reported
(Ripley & Braun, 1983). Several injection techniques for capillary
columns have been compared (Stan & Goebel, 1984; Stan & Mueller,
1988). Cold splitless (PTV) injection appears to be best suited for
organophosphorous pesticide analysis. The resolution can be further
improved by applying two-dimensional capillary gas chromatography
using two columns of different polarity (Stan & Mrowetz, 1983).
2.4.2.2 High performance liquid chromatography (HPLC)
The main advantage of HPLC is its ability to analyse compounds
that are heat labile, such as phenylurea and carbamates. As stated
above, organophosphorous pesticides including methyl parathion are
sufficiently heat stable for analysis using gas chromatography and
there is no direct need to use HPLC. Thus, relatively few studies
dealing with the HPLC analysis of methyl parathion have been reported.
HPLC analysis has been achieved using reversed phase
chromatography, with acetonitrile/water (60:40) (Funch, 1981), or
methanol/acetic acid/water (32:0.6: 47.4) as solvents, and UV-
detection (Zhao & Wang, 1984). HPLC conditions for 166 pesticides
including methyl parathion were reported by Lawrence & Turton (1978).
Retention data of 560 pesticides and other industrial chemicals have
been published by Daldrup et al. (1981, 1982) using two gradient
systems.
Sharma et al. (1990) developed a method for the rapid
quantitative analysis of organophosphorus (including methyl parathion)
and carbamate pesticides using HPLC and refractive index detection.
HPLC appears to be particularly suited for the analysis of polar
metabolites of methyl parathion (Abe et al., 1979).
Fluorogenic labelling of organophosphorous pesticides leads to an
improvement in sensitivity. Such labelling can be achieved by
hydrolysis of the compounds to the corresponding phenols and
derivatization with dansyl chloride (5-dimethylamino-naphthalene-1-
sulfonyl chloride) (Lawrence et al., 1976). Besides the UV and
fluorescence detector, electrochemical detectors have been used for
the detection of methyl parathion using amperometric detection in the
reductive mode (Bratin et al., 1981; Clark et al., 1985) or polaro-
graphic detection (Koen & Huber, 1970). Acetonitrile/water with
additional acetate buffer is used as solvent. The response is similar
to the UV detector, but there is less interference from the plant
material (Clark et al., 1985).
2.4.2.3 Thin layer chromatography (TLC)
Thin layer chromatography is well suited for the analysis
organophosphorous pesticides, even if it is not as specific as GC
(Kawahara et al., 1967; Schütz & Schindler, 1974; Thielemann, 1974;
Katkar & Barve, 1976; Lawrence et al., 1976; Curini et al., 1980;
Daldrup et al., 1981; Pfeiffer & Stahr, 1982; Korsos & Lantos, 1984).
Usually, silica gel G plates are used with a variety of solvent or
solvent mixtures. These include benzene, chloroform/cyclohexane,
n-hexane/acetone, chloroform/benzene, dichloro-methane/acetone.
Silver nitrate is frequently used as spray reagent, which, in the
presence of organophosphorous pesticides, leads to white spots against
a black background (Pfeiffer & Stahr, 1982).
As an alternative, an enzymatic reaction has been frequently
applied to detect organophosphorous compounds on TLC plates (Mueller,
1973; Leshev & Talanov, 1977; Ambrus et al., 1981a; Bhaskar & Kumar,
1981; Devi et al., 1982). This method makes use of the fact that
cholinesterase (from horse serum or cow liver) hydrolises 1-naphthyl
acetate to 1-naphthol, which reacts either with Fast Blue Salt B or
p-nitrobenzenediazoniumfluoroborate to form a coloured complex. If
methyl parathion is inhibiting the enzyme reaction, white spots on a
red or orange background appear. The sensitivity may be enhanced if
methyl parathion is oxidized to methylparaoxon by reaction with
bromine or hydrogen peroxide.
2.4.2.4 Spectrophotometry
Colorimetric methods, which were of importance during the early
years of organophosphorous pesticide analysis, have largely been
replaced by chromatographic methods.
The inhibition of cholinesterase by organophosphorous pesticides,
described above, is also the basis of a photometric method (Archer &
Zweig, 1959; Kumar & Ramasundari, 1980; Bhaskar & Kumar, 1982, 1984;
Kumar, 1985). Sadar et al. (1970) made use of the fact that
cholinesterase hydrolyses the non fluorescent N-methyl-
indoxylacetate to the highly fluorescent indoxyl. This reaction is
again inhibited by methyl parathion.
In another spectrophotometric method, methyl parathion is treated
with hydroxylamine hydrochloride and sodium nitroprusside, under
alkaline conditions, to form a water-soluble, coloured complex (Sastry
& Vijaya, 1986). The method is rapid and accurate and can be used for
formulations and for residues in fruits and vegetables.
2.4.2.5 Polarography
Polarography and various modifications of this method, i.e., the
"differential pulse polarography" (DPP), have been used repeatedly to
determine methyl parathion and other organophosphorous compounds with
a nitro group (Nangniot, 1966; Gajan, 1969; Kheifets et al., 1976;
Zietek, 1976; Smyth & Osteryoung, 1978; Kheifets et al., 1980; Khan,
1988; Reddy & Reddy, 1989). The method allows the simultanous
determination of parathion, methyl parathion, paraoxon, EPN, and the
metabolite 4-nitrophenol (Zietek, 1976) in blood, without prior
extraction. Polarography has been proposed as confirmatory method for
the determination of methyl parathion (and three further pesticides).
A collaborative study of 10 laboratories showed a coeffient of
variation of 15-16% (Gajan, 1969). In addition the method was applied
to water analysis (Kheifets et al., 1976, 1980; Bourquet et al.,
1989). Bourquet et al. (1989) showed a 20-50 increase in sensitivity
when "adsorptive stripping voltametry" was used instead of DPP.
2.4.2.6 Mass spectrometry
Coupled gas chromatography/electron impact mass spectrometry
(GC/MS) is a particularly valuable method for confirming pesticide
residues in various environmental samples. Methyl parathion shows an
abundant m/z=109, 125, and 263 (M+.) under electron impact
conditions (Mestres et al., 1977; Wilkins, 1990). Under positive ion
chemical ionization mass spectrometry (methane), the protonatic
molecule is the most abundant ion (m/z 264) while the structure
specific fragment at m/z 125 is due to (CH3O)2 P=S+ (8.8%)
(Holmstead & Casida, 1974). The negative ion chemical ionization
spectrum shows the typical thiophenolate fragment at m/z=154
(-S-C6H4-NO2) (Nielsen, 1985).
In addition, field ionization (FI) and field desorption (FD) mass
spectrometry have been applied repeatedly in the determination of of
methyl parathion (Damico et al., 1969; Klisenko et al., 1981; Schulten
& Sun, 1981; Golovatyi et al., 1982). The FD spectra show little
fragmentation and, thus, are not well suited for environmental
analysis. Among the newer mass spectrometric techniques, tandem mass
spectrometry (MS/MS) shows more promise for organophosphorous
pesticide analysis, as this technique enhances the selectivity of the
method and thus may reduce the necessary clean-up. Under MS/MS
conditions (chemical ionization), the protonated molecule forms an
abundant fragment at m/z 125 ((CH3O)2 P=S+) (Hummel & Yost, 1986;
Roach & Andrzejewski, 1987).
HPLC/MS of methyl parathion has been demonstrated (De Wit et al.,
1987; Betowski & Jones, 1988; Farran et al., 1990). As this method is
more difficult to handle and less sensitive and reproducible than
GC/MS, there is no need to use it in routine analysis, except when
other thermally labile pesticides are to be determined together with
organophosphorous compounds.
2.4.3 Detection limits
Detection limits are rarely reported. When plant material was
analysed, the detection limit for the overall method (extraction,
clean-up, analysis) was 10-100 µg/kg when gas chromatography with AFID
or FPD was used. In water analysis, substantially better detection
limits were achieved (usually 0.01-0.1 µg/litre), which may be further
reduced if a large-scale extractor is used (Foster & Rogerson, 1990).
In air analysis, detection limits have been reported to be 0.1-1
ng/m3.
2.4.4 Confirmatory method
A confirmatory derivatization method was proposed by Lee et al.
(1984). Following hydrolysis with KOH, 4-nitrophenol was derivatized
with pentafluoro benzyl bromide to the corresponding ether. Analysis
is carried out by GC with ECD. Levels as low as 0.01 ppb can be
confirmed.
Table 1. Sampling, extraction, clean-up, and determination of methyl parathiona
Matrix Sampling, extraction, clean-up Analytical Recovery (%) Detection limitb References
method (µg/kg or litre)
fruits, extr.: acetonitrile, GC (ECD, TID) 86-92 n.r. Wessel (1967)
vegetables part.: petroleum ether, TLC
clean-up: Florisil
plant material, extr.: propylene carbonate, GC (ECD, TID) 82-95 n.r. Schnorbus &
dairy products clean-up: Florisil Phillips (1967)
fruits, extr.: acetonitrile, GC (ECD) 90-98 n.r. Osadchuck et al.
vegetables, part.: dichloromethane + hexane, (1971)
fat, oil clean-up: Florisil
vegetables extr.: acetone, GC (ECD, TID) 93 (celery) n.r. Luke et al. (1975)
part.: dichloromethane/petroleum
ether,
clean-up: Florisil
apples extr.: toluene + n-hexane, GC (ECD) 93 1-20 Johansson (1978)
clean-up: Florisil
vegetables autom. extraction + n.r. 91-104 n.r. Gretch & Rosen
clean-up: Florisil (pepper) (1984)
food extr.: acetone, GC n.r. n.r. Specht & Tillkes
part.: dichloromethane, (1980)
clean-up: GPC + silica gel
Table 1 (continued)
Matrix Sampling, extraction, clean-up Analytical Recovery (%) Detection limitb References
method (µg/kg or litre)
fruits, extr.: acetone, GC (ECD,FPD, > 80 10-100 Andersson &
vegetables part.: dichloromethane hexane, TID) Ohlin (1986)
clean-up: GPC and silica gel
vegetables, extr.: trichloromethane, GC (FPD) 93-105 n.r. Ault et al. (1979)
fruits, clean-up: GPC
crops
vegetables extr.: acetone, GC (TID) 85-95 n.r. Pflugmacher &
part.: dichloromethane, Ebing (1974)
clean-up: GPC
- clean-up: GPC n.r. n.r. n.r. Steinwandter
(1988)
- clean-up: cellulose column n.r. 82 n.r. Stahr et al. (1979)
fruits, extr.: acetonitrile, HPLC (UV 280) 77-87 10 Funch (1981)
vegetables part.: dichloromethane
honey bees, extr.: acetone o-xylene GC (FPD) 92-101 1 Ross & Harvey
beewax, pollen (1981)
plants, soil extr.: supercritical methanol GC (ECD, AFID) 38 n.r. Capriel et al.
(1986)
tobacco extr.: hexane/acetone, GC (FPD) 99-104 20 Sagredos & Eckert
clean-up: alumina (1976)
Table 1 (continued)
Matrix Sampling, extraction, clean-up Analytical Recovery (%) Detection limitb References
method (µg/kg or litre)
vegetables extr.: acetone, GC (ECD,TID, n.r. n.r. Gyorfi et al.
part.: dichloromethane, FPD) (1987)
clean-up: charcoal
plant material extr.: acetone, GC (AFID, ECD) 92-103 n.r. Becker (1971)
part.: dichloromethane
plant material extr.: acetone, GC (ECD, AFID) 92-103 n.r. Becker (1979)
part.: dichloromethane,
clean-up: charcoal
plant material extr.: acetone, HPLC n.r. n.r. Miellet (1982)
clean-up: charcoal/Florisil
barley, malt, extr.: acetone or acetonitrile, GC (FPD) 82 30 Sonobe et al.
hops part.: hexane, (1982)
clean-up: charcoal
low moisture extr.: acetone, GC (FPD) 93 n.r. Luke & Doose
products part.: dichloromethane/petrol, (1983)
(pepper) ether,
clean-up: charcoal
ready-to-eat extr.: acetone GC (ECD, TID) n.r. 0.7-1.8 Vogelsang & Thier
foods part.: dichloromethane, (1986)
clean-up: + GPC silica gel
honey bees extr.: acetone GC (ECD) 91 15 Ebing (1985)
clean-up: charcoal
Table 1 (continued)
Matrix Sampling, extraction, clean-up Analytical Recovery (%) Detection limitb References
method (µg/kg or litre)
milk, oilseeds fat adsorbed on alumina GC (ECD, FPD) n.r. 80 Luke & Doose
extr.: acetonitrile, (1984)
part.: petroleum ether
fat ad.: of fat on Calflo E n.r. n.r. Specht (1978)
edible oils sweep co-distillation GC (TID) 95 10 (mg/kg) Storherr et al.
(1967)
edible oils extr.: petroleum ether, GC(FPD) 83-107 n.r. Gillespie &
clean-up: HPLC Walters (1989)
milk sweep co-distillation GC (TID) > 87 n.r. Watts & Storherr
(1967)
blood extr.: n-hexane GC (FPD) n.r. 3 Gabica et al.
(1971)
serum extr.: benzene GC (AFID) 69 2 De Potter et al.
(1978)
blood no extr. polarography 7x10-8 mol Zietek (1976)
soil extr.: acetone/hexane GC (TID) n.r. n.r. Agishev et al.
(1977)
soil extr.: acetone/hexane TLC (silica n.r. n.r. Garrido &
gel) Monteoliva (1981)
Table 1 (continued)
Matrix Sampling, extraction, clean-up Analytical Recovery (%) Detection limitb References
method (µg/kg or litre)
soil, sediment extr.: acetone/hexane, GC (AFID) 71 0.17 Kjoelholt (1985)
clean-up: ad. chrom.
soil extr.: acetone, GC (TID) 78-85 5 Wegman et al.
part.: dichloromethane, (1984)
clean-up: silica gel
soil, water, extr.: hexane/isopropanol, GC (ECD) 45 n.r. Schutzmann et al.
sediment desulfurization with Raney copper (1971)
water diethylether/hexane or benzene/ GC (ECD) n.r. n.r. Kawahara et al.
n-C6, (1967)
clean-up: TLC
water extr.: benzene GC (TID) 95 n.r. Pionke et al.
(1968)
water extr.: benzene GC 92-101 0.001 (?) Konrad et al.
(1969)
water extr.: petroleum ether GC 98 0.04 Zweig & Devine
(1969)
water extr.: trichloromethane TLC 60-95 1 Chmil et al.
(1978)
water extr.: trichloromethane GC(TID) n.r. 0.01 Chernyak &
Oradovskii (1980)
Table 1 (continued)
Matrix Sampling, extraction, clean-up Analytical Recovery (%) Detection limitb References
method (µg/kg or litre)
water/ extr.: at pH 11: dichloromethane; GC/MS 60-85 5 Spingarn et al.
wastewater at pH 2: dichloromethane (1982)
water extr.: dichloromethane/hexane, GC (ECD) n.r. n.r. Albanis et al.
clean-up: Florisil (1986)
water extr.: ethylacetate GC (FPD) 85-91 0.08 ng(abs.) Li & Wang (1987)
wastewater extr.: dichloromethane, GC (FPD) 90 0.75 Miller et al.
clean-up: Florisil (1981)
water extr.: petroleum ether, GC (ECD) n.r. 0.5 Mestres et al.
clean-up: Florisil (1969)
water extr.: dichloromethane GC/MS 75 n.r. Bruchet et al.
(continuous) liquid-liquid) (1984)
water extr.: n-pentane (continous GC (TID) 90 0.01 Brodesser &
liquid-liquid) Schoeler (1987)
water hydrolysis KOH, derivat. penta GC (ECD) 95 0.1 Coburn & Chau
fluoro-benzylbromide, (1974)
clean-up: silica gel
water ad.: on Tenax, thermoelution GC (FID/ECD) 62 0.01 Agostiano et al.
(1983)
water, run-off ad.: XAD-2, HPLC (rev. 99 2 Paschal et al.
water elut.: diethylether phase, UV) (1977)
Table 1 (continued)
Matrix Sampling, extraction, clean-up Analytical Recovery (%) Detection limitb References
method (µg/kg or litre)
water, ad.: XAD-2, GC (TID, FID) 93-100 15 pg(abs.) Le Bel et al.
drinking-water elut.: acetone/hexane (1979)
water ad.: XAD-4, GC n.r. n.r. Xue (1984)
elut.: diethylether/hexane
water ad.: Porapack Q, HPLC (rev. 96-105 < 1 Clark et al. (1985)
elut.: acetonitrile phase
electro-chem.)
water ad.: C-18, TLC n.r. 0.2 ng(abs.) Sherma &
elut.: ethyl acetate Bretschneider
(1990)
water ad.: C-18, acetone GC (FPD) > 79 n.r. Swineford &
Belisle (1989)
water extr.: dichloromethane GC/MS 48 0.0025 Foster & Rogerson
(large-scale extractor) (1990)
air ab.: ethylene-glycol, GC (FPD) 87-97 n.r. Sherma & Shafik
extr.: dichloromethane, (1975)
clean-up: silica gel
air ab.: cotton seed oil coated glass GC (FPD) 91 0.04 ng/m3 Compton (1973)
beads,
clean-up: Florisil
air clothscreen with ethylene glycol, GC (ECD/FPD) 93 n.r. Tessari & Spencer
extr.: acetone/hexane, (1971)
clean-up: alumina + Florisil
Table 1 (continued)
Matrix Sampling, extraction, clean-up Analytical Recovery (%) Detection limitb References
method (µg/kg or litre)
air ad.: silica gel, activated GC (ECD/FPD) n.r. 1 ng (abs.) Klisenko &
charcoal Girenko (1980)
air ad.: silica gel GC (FPD) 101-104 30 pg (abs.) Liang & Zhang
(1986)
air ad.: XAD-4, GC (ECD, TID) 74 1-3 ng/m3 Wehner et al.
elut: ethylacetate, (1984)
clean-up: HPLC
air ad.: PUF, GC (ECD) 100 n.r. Rice et al. (1977)
elut: petroleum ether
air ad.: PUF (high volume sampler) GC (ECD, FPD) 86 0.1 ng/m3 Lewis et al.
(1977)
air ad.: PUF (low volume sampler), GC (ECD, FPD) 80 20 ng/m3 Lewis & MacLeod
elut: diethylether/hexane (1982)
air ad.: PUF/other polymers (high GC 72-91 n.r. Lewis & Jackson
volume sampler) (1982)
air ad.: PUF, n.r. n.r. n.r. Belashova et al.
elut.: trichloromethane or (1983)
acetaldehyde
air ad.: Tenax, GC (FID) n.r. 2.5 µg/m3 Beine (1987)
elut.: toluene
formulations - GC or HPLC - - Jackson (1976)
Table 1 (continued)
Matrix Sampling, extraction, clean-up Analytical Recovery (%) Detection limitb References
method (µg/kg or litre)
formulations - GC - - Jackson (1977a)
formulations - HPLC - - Jackson (1977b)
formulations - IR - - Goza (1972)
formulations - P-31 NMR - - Greenhalgh et al.
(1983)
formulations hydrolysis to p-nitrophenol Spectr. - - Blanco & Sanchez
(1989)
a Abbreviations: GC = gas chromatography, TLC = thin-layer chromatography, GPC = gel
permeation chromatography, MS = mass spectrometry, HPLC = high performance liquid
chromatography, NMR = nuclear magnetic resonance, IR = infrared spectroscopy,
ECD = electron capture detector, FID = flame ionization detector, AFID = alkali flame
ionization detector, FPD = flame photometric detector, TID = thermionic detector,
UV = ultraviolet detector, spectr. = spectrophotometry, extr. = extraction, part. = partitioning,
ad. = adsorption, ab. = absorption, elut. = elution, n.r. = not reported, (abs.) = absolute.
b µg/kg or litre unless stated otherwise.
Table 2. Methods used in the determination of methyl parathion
Method Detection limit Remarks References
HPLC (UV) n.r. analysis Abe et al. (1979)
of metabolism
HPLC (UV) n.r. in mixtures Zhao & Wang
(rev. phase, methanol/ (1984)
acetic acid)
HPLC n.r. review on HPLC Lawrence & Turton (1978)
methods
HPLC (fluorescence) 10-20 µg (abs.)- deriv. with dansyl Lawrence et al. (1976)
chloride
HPLC 1. acetonitrile n.r. retention times of Daldrup et al. (1982)
2 acetonitrile/phosphoric 560 compounds
acid KH2PO4/H2O
HPLC 1. acetonitrile n.r. retention times of Daldrup et al. (1981)
2 acetonitrile/phosphoric 570 compounds
acid KH2PO4/H2O
HPLC (rev. phase, 10 µg/kg fruits and vegetables Funch (1981)
acetonitrile/H2O)
Table 2 (continued)
Method Detection limit Remarks References
HPLC (rev. phase, 1 µg/kg reduction amperometric Clark et al. (1985)
acetonitrile/0.01 KC1 detection
0.03 M potassium (vegetables, water)
acetate/H20)
HPLC (rev. phase, n.r. electrochemical Bratin et al. (1981)
acetonitrile/sodium detection
acetate/H2O)
HPLC rev. phase (H2O 30 µg/kg polarographic Koen & Huber (1970)
ethyl alcohol/acetic detection
acid/NaOH)
GC < 2 ng TID Patterson (1982)
GC n.r. retention times of Daldrup et al. (1981)
570 compounds
GC (TID) 20 µg/kg retention times Ambrus et al. (1981a,b)
GC n.r. retention times of Saxton (1987)
600 compounds
GC n.r. retention times of Prinsloo & de Beer (1987)
42 pesticides
n.r. retention times of Suprock & Vinopal (1987)
78 pesticides
Table 2 (continued)
Method Detection limit Remarks References
GC n.r. retentions times of Bowman & Beroza (1967)
20 OP-pesticides
(milk, corn silage)
GC n.r. two dimensional Stan & Mrowetz (1983)
GC
GC (FPD) 100 pg capillary columns, Krijgsman & Van de Kamp (1976)
relative retention
times
GC (ECD, TID) n.r. capillary columns, Stan & Goebel (1983)
simultaneous
detection of ECD, TID
GC n.r. retention times Ripley & Braun (1983)
of 194 pesticides
GC < 0.1 ng relative retention Omura et al. (1990)
times of 40 pesticides
on 11 phases
Table 2 (continued)
Method Detection limit Remarks References
GC (ECD) n.r. hydrolysis of Lee et al. (1984)
methyl parathion
to 4-nitrophenol,
derivat.
penta-fluorobenzylbromide
Clean-up: silica gel
TLC (silica gel G) n.r. detection with GC Kawahara et al. (1967)
TLC (silica gel) 0.1 µg 4 solvent mixtures, Schütz & Schindler (1974)
reduct. to amines
TLC (silica gel) 0.06-0.6 µg saponification and Thielemann (1974)
reduct. to
p-amino-phenol
TLC (silica gel G) n.r. elut.: n-hexane/acetone Katkar & Barve (1976)
TLC (silica gel) n.r. 17 solvent systems, Curini et al. (1980)
spray reagent: AgNO3
TLC (silica gel) n.r. elut.: 1.methanol/NH3H2O Daldrup et al. (1981)
2. dichloromethane/
acetone
TLC (silica gel) n.r. elut.: n-heptane/acetone Pfeiffer & Stahr (1982)
Table 2 (continued)
Method Detection limit Remarks References
TLC (silica gel) elut.: petroleum ether/ Korsos & Lantos (1984)
diethylether, two
dimensional TLC
TLC n.r. elut.: benzene/acetone, Mueller (1973)
detect. enzymatic
reaction
TLC (silica gel/ elut.: 4 solvent Leshchev & Talanov (1977)
starch) mixtures, milk,
feed, animal tissue,
extr: acetone, detect.
enzymatic reaction
TLC (silica gel G) n.r. detect. enzymatic Bhaskar & Kumar (1981)
reaction
TLC (silica gel G) 5 µg (abs.) elut.: dichloromethane Ambrus et al. (1981a,b)
or ethyl acetate,
detect. enzymatic
reaction
TLC n.r. detect. enzymatic Devi et al. (1982)
reaction
Table 2 (continued)
Method Detection limit Remarks References
polarography 140 µg/kg oscillographic Nangniot (1966)
polarography,
pesticide
residues
polarography 10 µg/kg single sweep Gajan (1969)
oscillographic
polarography,
non-fatty foods
polarography n.r. differential Kheifets et al. (1976)
oscillographic
polarography
(water)
polarography 7x10-6 mol/litre methyl parathion and Zietek (1976)
metabolites in blood
polarography 10-8 mol/litre - Smyth & Osteryoung (1978)
polarography n.r. adsorptive stripping Bourquet et al. (1988)
polarography n.r. - Kahn (1988)
polarography 3.9.10-9 mol/litre polargaraphy, diff. Reddy & Reddy (1989)
pulse polargraphy
cyclic voltametry
Table 2 (continued)
Method Detection limit Remarks References
differ. n.r. water Kheifets et al. (1980)
chronoamperometry
spectrophotometry n.r. enzymatic reaction Kumar (1985)
(cholinesterase,
Fast Blue B)
spectrophotometry n.r. reduction to amine, Sastry & Vijaya (1986)
formation of a
coloured complex
spectrophotometry n.r. reaction with 3-methyl- Sastry & Vijaya (1987)
2-benzothiazolinone
spectrophotometry n.r. hydrolysis to Ramakrishna & Ramachandran
4-nitro-phenol (1978)
a Abbreviations: GC = gas chromatography, HPLC = high performance liquid chromatography, TLC= thin layer chromatography,
ECD = electron capture detector, TID = thermionic detector, FPD = flame photometric detector, UV = ultraviolet
detector, elut. = elution, n.r. = not reported, (abs.) = absolute.
3. SOURCES OF HUMAN AND ENVIRONMENTAL EXPOSURE
3.1 Natural occurrence
Natural occurrence of methyl parathion is unlikely.
3.2 Man-made sources
3.2.1 Production process
Methyl parathion is a representative of the highly active
insecticides, the thiophosphorus esters, developed in the 1940s by
Schrader, a German chemist. Methyl parathion was introduced as a
commercial chemical in 1949. It is synthesized by the reaction of
O,O-dimethyl phosphoro-chloridothioate with the sodium salt of 4-
nitrophenol (Schrader, 1963).
Relative molecular mass: 263.23
Common names: methyl parathion
accepted by
ESA (Entomological Society of
America)
JMAF (Japanese Ministry of
Agriculture, Fisheries and Food)
WHO (World Health Organization)
parathion-methyl
accepted by
BSI (British Standards Institution)
ISO (International Organization for
Standardization)
metaphos
accepted by the USSR
CAS chemical name: O,O-dimethyl O-(4-nitro-phenyl)
phosphorothioate
IUPAC systematic name: O,O-Dimethyl O-4-nitrophenylphos-
phorothioate
CAS registry number: 298-00-0
RTECS number: TG 0175000
EINECS number: 206-050-1
EEC number: 015-035-00-7
Common synonyms:
Demethylfenitrothion; dimethyl para-nitrophenyl
monothiophosphate; O,O-dimethyl O-( para-nitrophenyl)
phosphorothioate; dimethyl para-nitrophenyl phosphorothionate;
dimethyl 4-nitrophenyl phosphorothionate; O,O-dimethyl
O-(para-nitrophenyl) thionophosphate; dimethyl
para-nitrophenyl thiophosphate;
O,O-dimethyl- O-(para-nitrophenyl) thiophosphate; dimethyl
parathion; ENT 17292; metaphos; methyl-parathion; methylthiophos;
MPT; NCI CO2971; parathion methyl homolog; phosphorothioic acid
O,O-dimethyl O-(4-nitro-phenyl) ester; phosphorothioic acid
O,O-dimethyl O-(para-nitrophenyl) ester BAY 11405; 8056 HC;
E601
2.1.2 Technical product
Major trade names:
A-Gro; Azofos; Azophos; Bladan M; Cekumethion; Dalf; Divithion;
Drexel Methyl Parathion 4E & 601; Dygun; Dypar; Ekatox; Folidol
M, M40 & 80; Fosferno M50; Gearphos; Mepaton; Meptox; Metacid 50;
Metacide; Metafos; Metaphos; Methyl-E 605; Methyl Fosferno;
Methylthiophos; Metron; M-Parathion; Niletar; Niran M-4; Nitran;
Nitrox; Nitrox 80; Oleovofotox; Parapest M-50; Parataf; Paratox;
Paridol; Parton M; Penncap M & MLS; Sinafid M-48; Sixty-Three
Special E.C. Insecticide; Tekwaisa; Thiophenit; Thylpar M-50;
Toll; Unidol; Vertac Methyl Parathion; technical product 80%,
Wofatox; Wolfatox.
2.1.2.1. Purity
Technical methyl parathion is available as a solution containing
80% active ingredient (a.i.), 16.7% xylene, and 3.3% inert
ingredients.
The following impurities were identified in one sample of
technical-grade methyl parathion: O,O-dimethyl- S-methyl
dithiophosphate, nitroanisol, nitro-phenol, isomers of methyl
parathion, and the dithio-analogue of methyl parathion (Warner, 1975).
2.2 Physical and chemical properties
Physical state: pure: white crystalline solid or powder
(National Fire Protection Association, 1986)
technical (80%) pure: light to dark tan
liquid (Worthing & Walker, 1987)
Melting point: 37-38 °C (The Merck Index, 1983)
35-36 °C (Worthing, 1983)
Freezing point: about 29 °C (technical product)
(Worthing & Walker, 1987)
Density/specific gravity:
1.358 at 20 °C/40 °C (d204 1.358)
(The Merck Index, 1983)
Vapour pressure: 1.3 mPa at 20 °C
(Worthing & Walker, 1987)
Octanol/water partition coefficient:
log Kow = 2.68 (measured)
log Kow = 1.81-3.43 (reported range)
(Hansch & Leo, 1987)
Water solubility: 55-60 mg/litre at 25 °C (pure)
(Midwest Research Institute, 1975;
National Research Council, 1977)
37.7 mg/litre at 19 °C (pure)
(Bowman & Sans, 1979)
57 mg/litre at 22 °C (anal. grade)
(Sanders & Seiber, 1983)
Nonaqueous solubility: soluble in ethanol, chloroform,
aliphatic solvents, and slightly
soluble in light petroleum
Volatility (pure): 0.14 mg/m3 at 20 °C (Spencer, 1982)
Odour: like rotten eggs or garlic (technical grade)
(Midwest Research Institute, 1975; Anon.,
1984)
Odour threshold: 0.0125 mg/m3 (Akhmedov, 1968)
Other properties: hydrolyses and isomerizes easily
(White-Stevens, 1971)
Half-life in aqueous solution at 20 °C, pH 1-5:
175 days (Melnikov, 1971)
2.3 Conversion factors
1 ppm methyl parathion= 10.76 mg/m3 at 25 °C, 1066 mbar
1 mg methyl parathion/m3 = 0.0929 ppm
2.4 Analytical methods
2.4.1 Sampling, extraction, clean-up
Standardized methods for the determination of various residues
are reported in the Manual of pesticide residue analysis (Thier &
Zeumer, 1987).
2.4.1.1 Plant material (tobacco, fruits, vegetables, crops with low
oil (fat) content)
(a) Extraction
Three extraction methods have mainly been used, all of which are
suitable for multiresidue analysis.
(1) Soxhlet extraction with chloroform - 10% methanol has been
proposed for field-weathered crops by Bowman (1981).
(2) Acetonitrile combined with various amounts of water has been used
by Mills et al. (1963), Wessel (1967), Osadchuk et al. (1971),
Luke et al. (1975), and Stahr et al. (1979). The plant material
is homogenized in a blender with acetonitrile, in some instances
after the addition of Celite (Nelson, 1967; Funch, 1981;).
High-moisture products (fruits and vegetables) are extracted with
pure acetonitrile while samples of dry products (hays, grains,
feedstuff) are blended with acetonitrile-water (65:35).
Extraction is followed by solvent partitioning into petroleum
ether with the addition of sodium chloride (Mills et al., 1963;
Wessel, 1967; Nelson, 1967) into dichloromethane (Funch, 1981),
and dichloromethane/hexane (10:200) (Osadchuk et al., 1971).
(3) Acetone was preferred as the solvent in particular in
multiresidue analysis by Becker (1971), Pflugmacher & Ebing
(1974), Sagredos & Eckert (1976), Becker (1979), Specht & Tillkes
(1980), Miellet (1982), Sonobe et al. (1982), Luke & Doose
(1983), Luke & Doose (1984), Ebing (1985), Andersson & Ohlin
(1986), Vogelsang & Thier (1986), Gyorfi et al. (1987), Thier &
Zeumer (1987), and Becker & Schug, (1990). In some instances,
celite was added. Depending on the water content of the sample,
water was added. In a second step, the acetone extracts were
further extracted with either dichloromethane, dichloro
methane/petroleum ether, or dichloromethane/ n-hexane. The
extract was dried over anhydrous sodium sulfate, reduced in
volume in a Kuderna-Danish concentrator, and subjected to
further clean-up.
Extraction with acetone- o-xylene (19:1) (Ross & Harvey, 1981),
toluene/hexane (75:25) (Johansson, 1978), chloroform (Ault et
al., 1979), or supercritical fluid extraction using methanol
(Capriel et al., 1986), has also been reported.
(b) Column clean-up
The published clean-up procedures are usually suitable for
multiresidue analysis. For plant material with a low fat content, 3
column clean-up procedures have been developed.
(1) The oldest method involves the use of chromatography on Florisil
(often topped with anhydrous sodium sulfate) (Mills et al., 1963;
Nelson, 1967; Schnorbus & Phillips, 1967; Wessel, 1967; Beckman
& Garber, 1969; Osadchuk et al., 1971; Luke et al., 1975;
Johansson, 1978; Gretch & Rosen, 1984, 1987). Although it has
been claimed that organo phosphorous pesticides are partially
lost during Florisil clean-up (Luke et al., 1975), high
recoveries (usually > 80 %) have been reported for methyl
parathion. Various solvents and solvent mixtures are used for
chromatography on Florisil including: diethylether/petroleum
ether, ethyl ether/hexane, and acetone/toluene,
diethylether/petroleum ether being the most frequently used.
Fractionation is achieved by increasing successively the
diethylether content. Florisil clean-up is usually used for a
combined clean-up of organochlorine and organophosphorous
pesticides. Luke et al. (1975) reported that gas chromatography
(GC) with a thermionic detector was sufficiently selective to
detect organophosphorous pesticides without Florisil clean-up.
(2) Alternatively, clean-up of pesticides in multiresidue analysis
has been achieved by chromatography on charcoal (Becker, 1971,
1979; Miellet, 1982; Sonobe et al., 1982; Luke & Doose, 1984;
Ebing, 1985; Gyorfi et al., 1987). To this end, charcoal is mixed
with silica gel (1:15) (and sometimes also celite or magnesia).
In most instances, elution is achieved with mixtures of
dichloromethane/acetone/toluene (e.g., 5:1:1) (Ebing, 1985; Thier
& Zeumer, 1987). Recoveries are high (often > 90 %). Charcoal
clean-up is particularly suited for dry products (< 10 % water).
The simultaneous clean-up of organochlorine and organo-
phosphorous pesticides is also possible with chromatography on
charcoal.
(3) In recent years, a clean-up of pesticides in multiresidue
analysis by gel permeation chromatography (GPC) has become
popular (Pflugmacher & Ebing, 1974; Ault et al., 1979; Specht &
Tillkes, 1980; Andersson & Ohlin, 1986; Vogelgesang & Thier,
1986; Steinwandter, 1988). The stationary phase consists, in most
instances, of Bio Beads SX3 (a polystyrene gel). Ethyl
acetate/cyclohexane (1:1), dichloromethane/cyclohexane (1:1) and,
more recently, acetone/cyclohexane (3:1) have been used as
elution mixtures. Gel permeation chromatography is mainly used to
protect the GC column and the GC detector against contam-
ination. GPC removes material of higher relative molecular mass.
Recoveries > 85% have been reported. Frequently, GPC is combined
with the additional purification step of silica gel
chromatography (Specht & Tillkes, 1980; Andersson & Ohlin, 1986;
Vogelsaifng & Thier, 1986) where elution is achieved with
toluene/hexane (35:65), followed by toluene and acetone/toluene,
with increasing acetone content. However, while the additional
clean-up by silica gel column chromatography is important when
organo chlorine pesticides are present, it is not necessary for
organophosphorous pesticides if analysis is performed by gas
chromatography with flame photometric detection.
2.4.1.2 Dairy products, products with a high fat content (edible
fats)
Clean-up techniques for products with a high fat content have
been reviewed by Waters (1990). Florisil column chromatography and gel
permeation chromatography are also suited for a clean-up of samples
with a high fat content. In addition, clean-up using normal phase HPLC
has been reported (Gillespie & Waters, 1986). Fat is dissolved in
n-hexane and fractionated on silica gel HPLC using
dichloromethane/hexane as solvent. However, complete separation
ofmethyl parathion from the fat is not achieved. As an alternative,
fat is adsorbed on aluminum oxide (Luke & Doose, 1984) or on Calflo E
(calcium silicate) (Specht, 1978; Thier & Zeumer, 1987). Finally, a
sweep codistillation clean-up of edible oils has been reported by
Storherr et al. (1967) and Watts & Storherr (1967). This method has
been standardized also for plant material (Thier & Zeumer, 1987).
After extraction of the sample with ethyl acetate, the concentrated
extract is injected into a heated glass column packed with glass wool
or glass beads followed by the injection of ethyl acetate or petroleum
ether in a nitrogen stream. The nitrogen carrier gas sweeps the
volatile component through the tube to a condensing bath and through
an Arnakrom scrubber tube to a collection tube. Sweep codistillation
may be followed by a further Florisil clean-up.
The extraction and clean-up of vegetable oil can be speeded up by
performing extraction and clean-up in one step using a system of three
ready-to-use cartridges in series (Extralut-3, Sep-Pack silicade1 and
Sep-Pack C18) where the assembled columns are eluted with
acetonitril (saturated with n-hexane) (Di Muccio et al., 1990).
2.4.1.3 Blood, body fluids
Methyl parathion is extracted from blood with hexane or benzene
and analysed without further clean-up (Gabica et al., 1971; De Potter
et al., 1978). No extraction is necessary if methyl parathion is
determined by polarography (Zietek, 1976).
Measurement of the urinary metabolites and the cholinesterase
activity were used to supervise the exposure of workers coming into
contact with methyl parathion or parathion and to observe their
elimination in cases of poisoning (see section 5.3) (Elliot et al.,
1960; Arterberry et al., 1961; Shafic & Enos, 1969; Wolfe et al.,
1970; Ware et al., 1974b; NIOSH, 1976).
2.4.1.4 Soil, sediments
Methyl parathion is extracted from soil with acetone,
acetone/ n-hexane or hexane/isopropanol (Schutzmann et al., 1971;
Agishev et al., 1977; Garrido & Monteoliva, 1981; Wegman et al., 1984;
Kjoelholt, 1985). It is partitioned in a second step into
dichloromethane. While several authors determine the pesticides
without further clean-up, additional silica gel adsorption
chromatography has been used by Wegman et al., (1984) and Kjoelholt
(1985). The recovery of methyl parathion is 70-85%.
When sediments are analysed, elemental sulfur represents a
particular problem. Kjoelholt et al. separated the sulfur by tetra
butylammonium hydrogensulfate (Kjoelholt, 1985), while Schutzmann et
al. (1971) refluxed the sediment extract with Raney copper.
For the extraction, the sediment mixed with sand and sodium
sulfate can be placed into a column and eluted using acetone :
dichloromethane (1:1) (Belisle & Swineford, 1988).
2.4.1.5 Water
Extraction and concentration of methyl parathion from water is
achieved either by liquid/liquid extraction (Kawahara et al., 1967;
Pionke et al., 1968; Mestres et al., 1969; Konrad et al., 1969; Zweig
& Devine, 1969; Schutzmann et al., 1971; Coburn & Chau, 1974; Chmil et
al., 1978; Chernyak & Oradovskii, 1980; Miller et al., 1981; Spingarn
et al., 1982; Bruchet et al., 1984; Albanis et al., 1986; Li & Wang,
1987; Brodesser & Schoeler, 1987), or by adsorption on polymeric
material (Paschal et al., 1977; Le Bel et al., 1979; Agostiano et al.,
1983; Xue, 1984; Clark et al., 1985). Various solvents have been used
for solvent extractions including: diethyl ether/hexane (1:1),
benzene, petroleum ether, hexane/isopropanol; chloroform,
dichloromethane, and ethyl acetate. Recoveries have been high (in
most instances > 90 %). If the liquid/liquid extraction is scaled up
using a "Goulden large sample extractor" and 120 litre of water,
detection limits may be lower by a factor of about 150 compared with
1-litre samples (i.e., a detection limit of 2.5 ng/litre (ppt) has
been achieved for methyl parathion) (Foster & Rogerson, 1990). The
extraction efficiency can be further improved by continuous
liquid-liquid extraction, which allows the use of non-polar solvents
as n-pentane (Bruchet et al., 1984; Brodesser & Schoeler, 1987).
Water samples are frequently analysed for pesticides without further
clean-up, while Florisil clean-up has been used in some instances
(Mestres et al., 1969; Miller et al., 1981).
High concentration factors are achieved, if methyl parathion (and
other pesticides) are adsorbed on polymeric material, such as XAD-2
(Paschal et al., 1977; Le Bel et al., 1979), XAD-4 (Xue et al., 1984),
Tenax (Agostiano et al., 1983) or Porapack Q (Clark et al., 1985).
Elution from XAD is achieved with diethyl ether, acetone/hexane
(15:85), diethyl ether-hexane (85:15). Recoveries are >90 %. If Tenax
is used, both solvent elution (diethyl ether) or thermoelution can be
used to desorb the pesticides. Solid-phase extraction (using C-18
cartridges) will become the method of choice for the rapid extraction
of organophosphorous insecticides from water (Swineford & Belisle,
1989; Sherma & Bretschneider, 1990).
2.4.1.6 Air
Most methods for the determination of pesticides in air have been
developed as multiresidue methods. Pesticides in air are either
absorbed in liquids or adsorbed on polymeric material. Thus,
pesticides may be trapped in ethylene glycol, which is subsequently
extracted with dichloromethane (Tessari & Spencer, 1971; Sherma &
Shafik, 1975) or they may be trapped on glass beads coated with
cottonseed oil (Compton, 1973). Further clean-up is achieved by silica
gel or Florisil column chromatography.
Among the solid polymeric material used to trap pesticides,
polyurethane foam (PUF) is by far the most popular (Lewis et al.,
1977; Rice et al., 1977; Lewis & McLeod, 1982; Lewis & Jackson, 1982;
Belashova et al., 1983; Beine, 1987). Air can be collected both with
low-volume (approx. 4 litre/min) or high-volume samplers (up to 250
litre/min). PUF can be reused after careful cleaning (e.g., with 5%
diethyl ether in n-hexane). In some instances, Tenax, Chromosorb
102, or Porapack R is sandwiched between PUF plugs to enhance the
collection efficiency. Collection efficiencies in excess of 80% have
been reported for methyl parathion. A filter may be added to remove
particulate matter (Lewis et al., 1977). Methyl parathion is usually
determined without further clean-up. Finally, XAD-4 (Wehner et al.,
1984) and silica gel (Klisenko & Girenko, 1980; Liang & Zhang, 1986)
have been used as solid trapping materials.
2.4.1.7 Formulations
When analysing formulations, the determination of by-products and
impurities is an important objective. A variety of instrumental
techniques have been used for the analysis of formulations including:
gas chromatography (Jackson, 1976; Jackson, 1977a), high performance
liquid chromatography (Jackson, 1977b), infrared analysis (Goza,
1972), P-31-nuclear magnetic resonance spectroscopy (Greenhalgh et
al., 1983), and spectrophotometry after alkaline hydrolysis to
p-nitrophenol (Blanco & Sanchez, 1989). An inter laboratory study
has been carried out using both GC (Jackson, 1977a) and HPLC (Jackson,
1977b). With both methods, coefficients of variation of 1.7% have been
determined. The instrumental techniques are described below.
2.4.2 Instrumental analytical methods
2.4.2.1 Gas chromatography
Gas chromatrophic (GC) methods for the determination of
pesticides (including methyl parathion) have been reviewed by Ebing
(1987).
Organophosphorous pesticides, including methyl parathion, are
sufficiently volatile and thermally stable to be amenable to gas
chromatography and it is by far the most important method for the
determination of methyl parathion. This technique provides the good
resolution necessary for multiresidue analysis. Moreover, very
sensitive and specific detectors are available, in particular for the
analysis of organophosphorous pesticides.
(a) Detectors
The two most widely used detectors for organophosphorous
pesticides are the alkali flame ionization detector (AFID) and
variations of this detector (thermionic detector (Patterson, 1982),
nitrogen-phosphorous detector) and the flame photometric detector
(FPD) (Bowman, 1981). The AFID makes use of the phenomenon that the
flame ionization detector yields enhanced response to nitrogen- and
phosphorus-containing compounds, in the presence of alkali metal
salts. The detection limit is in the low picogram range. The detector
discriminates against other compounds 30-50 fold. The flame
photometric detector (FPD) operates with a cool, hydrogen rich flame
for the detection of phosphorus- and sulfur-containing compounds,
which form POH and S2 species. These species emit light at 526 nm
(POH) and 394 nm (S2), which is monitored by using interference
filters and a photomultiplier. The detector is easy to operate and
results are reproducible. The detector is highly specific. The
response of 100 ng of parathion is 130 000 times greater than that of
an equal amount of aldrin. Furthermore, It is of advantage that any
solvent can be used with the detector. For the determination of methyl
parathion the P mode is the method of choice, though the S mode can
also be used (sensitivity 10 times lower) as methyl parathion contains
both P and S atoms.
Finally, the electron capture detector (ECD) is sometimes used
for the analysis of methyl parathion as it responds not only to the
P=S moiety, but in particular to the NO2 group.
(b) Columns
A definite identification of a pesticide by its retention time on
one column is not possible. Analysis on at least one further column
with a stationary phase of different polarity is necessary to confirm
the identity of a compound.
Packed columns are frequently used for pesticide residue
analysis, though resolution is substantially poorer compared with
capillary columns and identification of the pesticides is less
specific. Solid supports are usually of the Chromosorb W type. In some
instances, Gaschrom Q has also been used. A large variety of
stationary phases, used either alone or in admixture, have been
employed. The most frequently used phases are DC 200, QF-1, OV 17,
OV-101, OV-210, and SE-30. Relative retention times for many
stationary phases have been reported by several authors for a large
variety of pesticides (up to 600 compounds including other industrial
chemicals) (Bowman & Beroza, 1967; Ambrus et al., 1981b; Daldrup et
al., 1981; Prinsloo & de Beer, 1987; Saxton, 1987; Suprock & Vinopal,
1987; Omura et al., 1990).
Packed column GC allows the separation of only a limited number
of pesticides. Capillary columns exhibit a considerably better
separation efficiency than packed columns. Such capillary columns have
been used by several authors for methyl parathion analysis (Krijgsman
& van den Kamp, 1976; Ripley & Braun, 1983; Stan & Goebel, 1983;
Ebing, 1985; Andersson & Ohlin, 1986; Vogelsang & Thier, 1986).
Retention time data on a SE-30 capillary column have been reported
(Ripley & Braun, 1983). Several injection techniques for capillary
columns have been compared (Stan & Goebel, 1984; Stan & Mueller,
1988). Cold splitless (PTV) injection appears to be best suited for
organophosphorous pesticide analysis. The resolution can be further
improved by applying two-dimensional capillary gas chromatography
using two columns of different polarity (Stan & Mrowetz, 1983).
2.4.2.2 High performance liquid chromatography (HPLC)
The main advantage of HPLC is its ability to analyse compounds
that are heat labile, such as phenylurea and carbamates. As stated
above, organophosphorous pesticides including methyl parathion are
sufficiently heat stable for analysis using gas chromatography and
there is no direct need to use HPLC. Thus, relatively few studies
dealing with the HPLC analysis of methyl parathion have been reported.
HPLC analysis has been achieved using reversed phase
chromatography, with acetonitrile/water (60:40) (Funch, 1981), or
methanol/acetic acid/water (32:0.6: 47.4) as solvents, and UV-
detection (Zhao & Wang, 1984). HPLC conditions for 166 pesticides
including methyl parathion were reported by Lawrence & Turton (1978).
Retention data of 560 pesticides and other industrial chemicals have
been published by Daldrup et al. (1981, 1982) using two gradient
systems.
Sharma et al. (1990) developed a method for the rapid
quantitative analysis of organophosphorus (including methyl parathion)
and carbamate pesticides using HPLC and refractive index detection.
HPLC appears to be particularly suited for the analysis of polar
metabolites of methyl parathion (Abe et al., 1979).
Fluorogenic labelling of organophosphorous pesticides leads to an
improvement in sensitivity. Such labelling can be achieved by
hydrolysis of the compounds to the corresponding phenols and
derivatization with dansyl chloride (5-dimethylamino-naphthalene-1-
sulfonyl chloride) (Lawrence et al., 1976). Besides the UV and
fluorescence detector, electrochemical detectors have been used for
the detection of methyl parathion using amperometric detection in the
reductive mode (Bratin et al., 1981; Clark et al., 1985) or polaro-
graphic detection (Koen & Huber, 1970). Acetonitrile/water with
additional acetate buffer is used as solvent. The response is similar
to the UV detector, but there is less interference from the plant
material (Clark et al., 1985).
2.4.2.3 Thin layer chromatography (TLC)
Thin layer chromatography is well suited for the analysis
organophosphorous pesticides, even if it is not as specific as GC
(Kawahara et al., 1967; Schütz & Schindler, 1974; Thielemann, 1974;
Katkar & Barve, 1976; Lawrence et al., 1976; Curini et al., 1980;
Daldrup et al., 1981; Pfeiffer & Stahr, 1982; Korsos & Lantos, 1984).
Usually, silica gel G plates are used with a variety of solvent or
solvent mixtures. These include benzene, chloroform/cyclohexane,
n-hexane/acetone, chloroform/benzene, dichloro-methane/acetone.
Silver nitrate is frequently used as spray reagent, which, in the
presence of organophosphorous pesticides, leads to white spots against
a black background (Pfeiffer & Stahr, 1982).
As an alternative, an enzymatic reaction has been frequently
applied to detect organophosphorous compounds on TLC plates (Mueller,
1973; Leshev & Talanov, 1977; Ambrus et al., 1981a; Bhaskar & Kumar,
1981; Devi et al., 1982). This method makes use of the fact that
cholinesterase (from horse serum or cow liver) hydrolises 1-naphthyl
acetate to 1-naphthol, which reacts either with Fast Blue Salt B or
p-nitrobenzenediazoniumfluoroborate to form a coloured complex. If
methyl parathion is inhibiting the enzyme reaction, white spots on a
red or orange background appear. The sensitivity may be enhanced if
methyl parathion is oxidized to methylparaoxon by reaction with
bromine or hydrogen peroxide.
2.4.2.4 Spectrophotometry
Colorimetric methods, which were of importance during the early
years of organophosphorous pesticide analysis, have largely been
replaced by chromatographic methods.
The inhibition of cholinesterase by organophosphorous pesticides,
described above, is also the basis of a photometric method (Archer &
Zweig, 1959; Kumar & Ramasundari, 1980; Bhaskar & Kumar, 1982, 1984;
Kumar, 1985). Sadar et al. (1970) made use of the fact that
cholinesterase hydrolyses the non fluorescent N-methyl-
indoxylacetate to the highly fluorescent indoxyl. This reaction is
again inhibited by methyl parathion.
In another spectrophotometric method, methyl parathion is treated
with hydroxylamine hydrochloride and sodium nitroprusside, under
alkaline conditions, to form a water-soluble, coloured complex (Sastry
& Vijaya, 1986). The method is rapid and accurate and can be used for
formulations and for residues in fruits and vegetables.
2.4.2.5 Polarography
Polarography and various modifications of this method, i.e., the
"differential pulse polarography" (DPP), have been used repeatedly to
determine methyl parathion and other organophosphorous compounds with
a nitro group (Nangniot, 1966; Gajan, 1969; Kheifets et al., 1976;
Zietek, 1976; Smyth & Osteryoung, 1978; Kheifets et al., 1980; Khan,
1988; Reddy & Reddy, 1989). The method allows the simultanous
determination of parathion, methyl parathion, paraoxon, EPN, and the
metabolite 4-nitrophenol (Zietek, 1976) in blood, without prior
extraction. Polarography has been proposed as confirmatory method for
the determination of methyl parathion (and three further pesticides).
A collaborative study of 10 laboratories showed a coeffient of
variation of 15-16% (Gajan, 1969). In addition the method was applied
to water analysis (Kheifets et al., 1976, 1980; Bourquet et al.,
1989). Bourquet et al. (1989) showed a 20-50 increase in sensitivity
when "adsorptive stripping voltametry" was used instead of DPP.
2.4.2.6 Mass spectrometry
Coupled gas chromatography/electron impact mass spectrometry
(GC/MS) is a particularly valuable method for confirming pesticide
residues in various environmental samples. Methyl parathion shows an
abundant m/z=109, 125, and 263 (M+.) under electron impact
conditions (Mestres et al., 1977; Wilkins, 1990). Under positive ion
chemical ionization mass spectrometry (methane), the protonatic
molecule is the most abundant ion (m/z 264) while the structure
specific fragment at m/z 125 is due to (CH3O)2 P=S+ (8.8%)
(Holmstead & Casida, 1974). The negative ion chemical ionization
spectrum shows the typical thiophenolate fragment at m/z=154
(-S-C6H4-NO2) (Nielsen, 1985).
In addition, field ionization (FI) and field desorption (FD) mass
spectrometry have been applied repeatedly in the determination of of
methyl parathion (Damico et al., 1969; Klisenko et al., 1981; Schulten
& Sun, 1981; Golovatyi et al., 1982). The FD spectra show little
fragmentation and, thus, are not well suited for environmental
analysis. Among the newer mass spectrometric techniques, tandem mass
spectrometry (MS/MS) shows more promise for organophosphorous
pesticide analysis, as this technique enhances the selectivity of the
method and thus may reduce the necessary clean-up. Under MS/MS
conditions (chemical ionization), the protonated molecule forms an
abundant fragment at m/z 125 ((CH3O)2 P=S+) (Hummel & Yost, 1986;
Roach & Andrzejewski, 1987).
HPLC/MS of methyl parathion has been demonstrated (De Wit et al.,
1987; Betowski & Jones, 1988; Farran et al., 1990). As this method is
more difficult to handle and less sensitive and reproducible than
GC/MS, there is no need to use it in routine analysis, except when
other thermally labile pesticides are to be determined together with
organophosphorous compounds.
2.4.3 Detection limits
Detection limits are rarely reported. When plant material was
analysed, the detection limit for the overall method (extraction,
clean-up, analysis) was 10-100 µg/kg when gas chromatography with AFID
or FPD was used. In water analysis, substantially better detection
limits were achieved (usually 0.01-0.1 µg/litre), which may be further
reduced if a large-scale extractor is used (Foster & Rogerson, 1990).
In air analysis, detection limits have been reported to be 0.1-1
ng/m3.
2.4.4 Confirmatory method
A confirmatory derivatization method was proposed by Lee et al.
(1984). Following hydrolysis with KOH, 4-nitrophenol was derivatized
with pentafluoro benzyl bromide to the corresponding ether. Analysis
is carried out by GC with ECD. Levels as low as 0.01 ppb can be
confirmed.
Table 1. Sampling, extraction, clean-up, and determination of methyl parathiona
Matrix Sampling, extraction, clean-up Analytical Recovery (%) Detection limitb References
method (µg/kg or litre)
fruits, extr.: acetonitrile, GC (ECD, TID) 86-92 n.r. Wessel (1967)
vegetables part.: petroleum ether, TLC
clean-up: Florisil
plant material, extr.: propylene carbonate, GC (ECD, TID) 82-95 n.r. Schnorbus &
dairy products clean-up: Florisil Phillips (1967)
fruits, extr.: acetonitrile, GC (ECD) 90-98 n.r. Osadchuck et al.
vegetables, part.: dichloromethane + hexane, (1971)
fat, oil clean-up: Florisil
vegetables extr.: acetone, GC (ECD, TID) 93 (celery) n.r. Luke et al. (1975)
part.: dichloromethane/petroleum
ether,
clean-up: Florisil
apples extr.: toluene + n-hexane, GC (ECD) 93 1-20 Johansson (1978)
clean-up: Florisil
vegetables autom. extraction + n.r. 91-104 n.r. Gretch & Rosen
clean-up: Florisil (pepper) (1984)
food extr.: acetone, GC n.r. n.r. Specht & Tillkes
part.: dichloromethane, (1980)
clean-up: GPC + silica gel
Table 1 (continued)
Matrix Sampling, extraction, clean-up Analytical Recovery (%) Detection limitb References
method (µg/kg or litre)
fruits, extr.: acetone, GC (ECD,FPD, > 80 10-100 Andersson &
vegetables part.: dichloromethane hexane, TID) Ohlin (1986)
clean-up: GPC and silica gel
vegetables, extr.: trichloromethane, GC (FPD) 93-105 n.r. Ault et al. (1979)
fruits, clean-up: GPC
crops
vegetables extr.: acetone, GC (TID) 85-95 n.r. Pflugmacher &
part.: dichloromethane, Ebing (1974)
clean-up: GPC
- clean-up: GPC n.r. n.r. n.r. Steinwandter
(1988)
- clean-up: cellulose column n.r. 82 n.r. Stahr et al. (1979)
fruits, extr.: acetonitrile, HPLC (UV 280) 77-87 10 Funch (1981)
vegetables part.: dichloromethane
honey bees, extr.: acetone o-xylene GC (FPD) 92-101 1 Ross & Harvey
beewax, pollen (1981)
plants, soil extr.: supercritical methanol GC (ECD, AFID) 38 n.r. Capriel et al.
(1986)
tobacco extr.: hexane/acetone, GC (FPD) 99-104 20 Sagredos & Eckert
clean-up: alumina (1976)
Table 1 (continued)
Matrix Sampling, extraction, clean-up Analytical Recovery (%) Detection limitb References
method (µg/kg or litre)
vegetables extr.: acetone, GC (ECD,TID, n.r. n.r. Gyorfi et al.
part.: dichloromethane, FPD) (1987)
clean-up: charcoal
plant material extr.: acetone, GC (AFID, ECD) 92-103 n.r. Becker (1971)
part.: dichloromethane
plant material extr.: acetone, GC (ECD, AFID) 92-103 n.r. Becker (1979)
part.: dichloromethane,
clean-up: charcoal
plant material extr.: acetone, HPLC n.r. n.r. Miellet (1982)
clean-up: charcoal/Florisil
barley, malt, extr.: acetone or acetonitrile, GC (FPD) 82 30 Sonobe et al.
hops part.: hexane, (1982)
clean-up: charcoal
low moisture extr.: acetone, GC (FPD) 93 n.r. Luke & Doose
products part.: dichloromethane/petrol, (1983)
(pepper) ether,
clean-up: charcoal
ready-to-eat extr.: acetone GC (ECD, TID) n.r. 0.7-1.8 Vogelsang & Thier
foods part.: dichloromethane, (1986)
clean-up: + GPC silica gel
honey bees extr.: acetone GC (ECD) 91 15 Ebing (1985)
clean-up: charcoal
Table 1 (continued)
Matrix Sampling, extraction, clean-up Analytical Recovery (%) Detection limitb References
method (µg/kg or litre)
milk, oilseeds fat adsorbed on alumina GC (ECD, FPD) n.r. 80 Luke & Doose
extr.: acetonitrile, (1984)
part.: petroleum ether
fat ad.: of fat on Calflo E n.r. n.r. Specht (1978)
edible oils sweep co-distillation GC (TID) 95 10 (mg/kg) Storherr et al.
(1967)
edible oils extr.: petroleum ether, GC(FPD) 83-107 n.r. Gillespie &
clean-up: HPLC Walters (1989)
milk sweep co-distillation GC (TID) > 87 n.r. Watts & Storherr
(1967)
blood extr.: n-hexane GC (FPD) n.r. 3 Gabica et al.
(1971)
serum extr.: benzene GC (AFID) 69 2 De Potter et al.
(1978)
blood no extr. polarography 7x10-8 mol Zietek (1976)
soil extr.: acetone/hexane GC (TID) n.r. n.r. Agishev et al.
(1977)
soil extr.: acetone/hexane TLC (silica n.r. n.r. Garrido &
gel) Monteoliva (1981)
Table 1 (continued)
Matrix Sampling, extraction, clean-up Analytical Recovery (%) Detection limitb References
method (µg/kg or litre)
soil, sediment extr.: acetone/hexane, GC (AFID) 71 0.17 Kjoelholt (1985)
clean-up: ad. chrom.
soil extr.: acetone, GC (TID) 78-85 5 Wegman et al.
part.: dichloromethane, (1984)
clean-up: silica gel
soil, water, extr.: hexane/isopropanol, GC (ECD) 45 n.r. Schutzmann et al.
sediment desulfurization with Raney copper (1971)
water diethylether/hexane or benzene/ GC (ECD) n.r. n.r. Kawahara et al.
n-C6, (1967)
clean-up: TLC
water extr.: benzene GC (TID) 95 n.r. Pionke et al.
(1968)
water extr.: benzene GC 92-101 0.001 (?) Konrad et al.
(1969)
water extr.: petroleum ether GC 98 0.04 Zweig & Devine
(1969)
water extr.: trichloromethane TLC 60-95 1 Chmil et al.
(1978)
water extr.: trichloromethane GC(TID) n.r. 0.01 Chernyak &
Oradovskii (1980)
Table 1 (continued)
Matrix Sampling, extraction, clean-up Analytical Recovery (%) Detection limitb References
method (µg/kg or litre)
water/ extr.: at pH 11: dichloromethane; GC/MS 60-85 5 Spingarn et al.
wastewater at pH 2: dichloromethane (1982)
water extr.: dichloromethane/hexane, GC (ECD) n.r. n.r. Albanis et al.
clean-up: Florisil (1986)
water extr.: ethylacetate GC (FPD) 85-91 0.08 ng(abs.) Li & Wang (1987)
wastewater extr.: dichloromethane, GC (FPD) 90 0.75 Miller et al.
clean-up: Florisil (1981)
water extr.: petroleum ether, GC (ECD) n.r. 0.5 Mestres et al.
clean-up: Florisil (1969)
water extr.: dichloromethane GC/MS 75 n.r. Bruchet et al.
(continuous) liquid-liquid) (1984)
water extr.: n-pentane (continous GC (TID) 90 0.01 Brodesser &
liquid-liquid) Schoeler (1987)
water hydrolysis KOH, derivat. penta GC (ECD) 95 0.1 Coburn & Chau
fluoro-benzylbromide, (1974)
clean-up: silica gel
water ad.: on Tenax, thermoelution GC (FID/ECD) 62 0.01 Agostiano et al.
(1983)
water, run-off ad.: XAD-2, HPLC (rev. 99 2 Paschal et al.
water elut.: diethylether phase, UV) (1977)
Table 1 (continued)
Matrix Sampling, extraction, clean-up Analytical Recovery (%) Detection limitb References
method (µg/kg or litre)
water, ad.: XAD-2, GC (TID, FID) 93-100 15 pg(abs.) Le Bel et al.
drinking-water elut.: acetone/hexane (1979)
water ad.: XAD-4, GC n.r. n.r. Xue (1984)
elut.: diethylether/hexane
water ad.: Porapack Q, HPLC (rev. 96-105 < 1 Clark et al. (1985)
elut.: acetonitrile phase
electro-chem.)
water ad.: C-18, TLC n.r. 0.2 ng(abs.) Sherma &
elut.: ethyl acetate Bretschneider
(1990)
water ad.: C-18, acetone GC (FPD) > 79 n.r. Swineford &
Belisle (1989)
water extr.: dichloromethane GC/MS 48 0.0025 Foster & Rogerson
(large-scale extractor) (1990)
air ab.: ethylene-glycol, GC (FPD) 87-97 n.r. Sherma & Shafik
extr.: dichloromethane, (1975)
clean-up: silica gel
air ab.: cotton seed oil coated glass GC (FPD) 91 0.04 ng/m3 Compton (1973)
beads,
clean-up: Florisil
air clothscreen with ethylene glycol, GC (ECD/FPD) 93 n.r. Tessari & Spencer
extr.: acetone/hexane, (1971)
clean-up: alumina + Florisil
Table 1 (continued)
Matrix Sampling, extraction, clean-up Analytical Recovery (%) Detection limitb References
method (µg/kg or litre)
air ad.: silica gel, activated GC (ECD/FPD) n.r. 1 ng (abs.) Klisenko &
charcoal Girenko (1980)
air ad.: silica gel GC (FPD) 101-104 30 pg (abs.) Liang & Zhang
(1986)
air ad.: XAD-4, GC (ECD, TID) 74 1-3 ng/m3 Wehner et al.
elut: ethylacetate, (1984)
clean-up: HPLC
air ad.: PUF, GC (ECD) 100 n.r. Rice et al. (1977)
elut: petroleum ether
air ad.: PUF (high volume sampler) GC (ECD, FPD) 86 0.1 ng/m3 Lewis et al.
(1977)
air ad.: PUF (low volume sampler), GC (ECD, FPD) 80 20 ng/m3 Lewis & MacLeod
elut: diethylether/hexane (1982)
air ad.: PUF/other polymers (high GC 72-91 n.r. Lewis & Jackson
volume sampler) (1982)
air ad.: PUF, n.r. n.r. n.r. Belashova et al.
elut.: trichloromethane or (1983)
acetaldehyde
air ad.: Tenax, GC (FID) n.r. 2.5 µg/m3 Beine (1987)
elut.: toluene
formulations - GC or HPLC - - Jackson (1976)
Table 1 (continued)
Matrix Sampling, extraction, clean-up Analytical Recovery (%) Detection limitb References
method (µg/kg or litre)
formulations - GC - - Jackson (1977a)
formulations - HPLC - - Jackson (1977b)
formulations - IR - - Goza (1972)
formulations - P-31 NMR - - Greenhalgh et al.
(1983)
formulations hydrolysis to p-nitrophenol Spectr. - - Blanco & Sanchez
(1989)
a Abbreviations: GC = gas chromatography, TLC = thin-layer chromatography, GPC = gel
permeation chromatography, MS = mass spectrometry, HPLC = high performance liquid
chromatography, NMR = nuclear magnetic resonance, IR = infrared spectroscopy,
ECD = electron capture detector, FID = flame ionization detector, AFID = alkali flame
ionization detector, FPD = flame photometric detector, TID = thermionic detector,
UV = ultraviolet detector, spectr. = spectrophotometry, extr. = extraction, part. = partitioning,
ad. = adsorption, ab. = absorption, elut. = elution, n.r. = not reported, (abs.) = absolute.
b µg/kg or litre unless stated otherwise.
Table 2. Methods used in the determination of methyl parathion
Method Detection limit Remarks References
HPLC (UV) n.r. analysis Abe et al. (1979)
of metabolism
HPLC (UV) n.r. in mixtures Zhao & Wang
(rev. phase, methanol/ (1984)
acetic acid)
HPLC n.r. review on HPLC Lawrence & Turton (1978)
methods
HPLC (fluorescence) 10-20 µg (abs.)- deriv. with dansyl Lawrence et al. (1976)
chloride
HPLC 1. acetonitrile n.r. retention times of Daldrup et al. (1982)
2 acetonitrile/phosphoric 560 compounds
acid KH2PO4/H2O
HPLC 1. acetonitrile n.r. retention times of Daldrup et al. (1981)
2 acetonitrile/phosphoric 570 compounds
acid KH2PO4/H2O
HPLC (rev. phase, 10 µg/kg fruits and vegetables Funch (1981)
acetonitrile/H2O)
Table 2 (continued)
Method Detection limit Remarks References
HPLC (rev. phase, 1 µg/kg reduction amperometric Clark et al. (1985)
acetonitrile/0.01 KC1 detection
0.03 M potassium (vegetables, water)
acetate/H20)
HPLC (rev. phase, n.r. electrochemical Bratin et al. (1981)
acetonitrile/sodium detection
acetate/H2O)
HPLC rev. phase (H2O 30 µg/kg polarographic Koen & Huber (1970)
ethyl alcohol/acetic detection
acid/NaOH)
GC < 2 ng TID Patterson (1982)
GC n.r. retention times of Daldrup et al. (1981)
570 compounds
GC (TID) 20 µg/kg retention times Ambrus et al. (1981a,b)
GC n.r. retention times of Saxton (1987)
600 compounds
GC n.r. retention times of Prinsloo & de Beer (1987)
42 pesticides
n.r. retention times of Suprock & Vinopal (1987)
78 pesticides
Table 2 (continued)
Method Detection limit Remarks References
GC n.r. retentions times of Bowman & Beroza (1967)
20 OP-pesticides
(milk, corn silage)
GC n.r. two dimensional Stan & Mrowetz (1983)
GC
GC (FPD) 100 pg capillary columns, Krijgsman & Van de Kamp (1976)
relative retention
times
GC (ECD, TID) n.r. capillary columns, Stan & Goebel (1983)
simultaneous
detection of ECD, TID
GC n.r. retention times Ripley & Braun (1983)
of 194 pesticides
GC < 0.1 ng relative retention Omura et al. (1990)
times of 40 pesticides
on 11 phases
Table 2 (continued)
Method Detection limit Remarks References
GC (ECD) n.r. hydrolysis of Lee et al. (1984)
methyl parathion
to 4-nitrophenol,
derivat.
penta-fluorobenzylbromide
Clean-up: silica gel
TLC (silica gel G) n.r. detection with GC Kawahara et al. (1967)
TLC (silica gel) 0.1 µg 4 solvent mixtures, Schütz & Schindler (1974)
reduct. to amines
TLC (silica gel) 0.06-0.6 µg saponification and Thielemann (1974)
reduct. to
p-amino-phenol
TLC (silica gel G) n.r. elut.: n-hexane/acetone Katkar & Barve (1976)
TLC (silica gel) n.r. 17 solvent systems, Curini et al. (1980)
spray reagent: AgNO3
TLC (silica gel) n.r. elut.: 1.methanol/NH3H2O Daldrup et al. (1981)
2. dichloromethane/
acetone
TLC (silica gel) n.r. elut.: n-heptane/acetone Pfeiffer & Stahr (1982)
Table 2 (continued)
Method Detection limit Remarks References
TLC (silica gel) elut.: petroleum ether/ Korsos & Lantos (1984)
diethylether, two
dimensional TLC
TLC n.r. elut.: benzene/acetone, Mueller (1973)
detect. enzymatic
reaction
TLC (silica gel/ elut.: 4 solvent Leshchev & Talanov (1977)
starch) mixtures, milk,
feed, animal tissue,
extr: acetone, detect.
enzymatic reaction
TLC (silica gel G) n.r. detect. enzymatic Bhaskar & Kumar (1981)
reaction
TLC (silica gel G) 5 µg (abs.) elut.: dichloromethane Ambrus et al. (1981a,b)
or ethyl acetate,
detect. enzymatic
reaction
TLC n.r. detect. enzymatic Devi et al. (1982)
reaction
Table 2 (continued)
Method Detection limit Remarks References
polarography 140 µg/kg oscillographic Nangniot (1966)
polarography,
pesticide
residues
polarography 10 µg/kg single sweep Gajan (1969)
oscillographic
polarography,
non-fatty foods
polarography n.r. differential Kheifets et al. (1976)
oscillographic
polarography
(water)
polarography 7x10-6 mol/litre methyl parathion and Zietek (1976)
metabolites in blood
polarography 10-8 mol/litre - Smyth & Osteryoung (1978)
polarography n.r. adsorptive stripping Bourquet et al. (1988)
polarography n.r. - Kahn (1988)
polarography 3.9.10-9 mol/litre polargaraphy, diff. Reddy & Reddy (1989)
pulse polargraphy
cyclic voltametry
Table 2 (continued)
Method Detection limit Remarks References
differ. n.r. water Kheifets et al. (1980)
chronoamperometry
spectrophotometry n.r. enzymatic reaction Kumar (1985)
(cholinesterase,
Fast Blue B)
spectrophotometry n.r. reduction to amine, Sastry & Vijaya (1986)
formation of a
coloured complex
spectrophotometry n.r. reaction with 3-methyl- Sastry & Vijaya (1987)
2-benzothiazolinone
spectrophotometry n.r. hydrolysis to Ramakrishna & Ramachandran
4-nitro-phenol (1978)
a Abbreviations: GC = gas chromatography, HPLC = high performance liquid chromatography, TLC= thin layer chromatography,
ECD = electron capture detector, TID = thermionic detector, FPD = flame photometric detector, UV = ultraviolet
detector, elut. = elution, n.r. = not reported, (abs.) = absolute.
3. SOURCES OF HUMAN AND ENVIRONMENTAL EXPOSURE
3.1 Natural occurrence
Natural occurrence of methyl parathion is unlikely.
3.2 Man-made sources
3.2.1 Production process
Methyl parathion is a representative of the highly active
insecticides, the thiophosphorus esters, developed in the 1940s by
Schrader, a German chemist. Methyl parathion was introduced as a
commercial chemical in 1949. It is synthesized by the reaction of
O,O-dimethyl phosphoro-chloridothioate with the sodium salt of 4-
nitrophenol (Schrader, 1963).
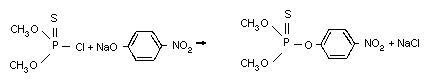 3.2.2 Loss into the environment
Emissions of methyl parathion during the production process can
be disregarded when compared with those from its use as an
insecticide. The air emission from a factory in the USA was reported
to be around 0.1% of the production level (Archer et al., 1978). The
major losses of this insecticide are directly caused by spraying, and
evaporation from water surfaces, leaves, and from the soil (Woodrow et
al., 1977).
3.2.3 Production
According to the European Directory of Agrochemical Products
(1986) and the Directory of World Chemical Producers (1990), methyl
parathion is produced throughout the world by many companies. World
production in 1966 was 31 700 tonnes, including 14 800 tonnes produced
in the USA.
In Table 3, selected countries producing methyl parathion are
listed together with their production capacities (Bayer, 1988).
Table 3. Methyl parathion production capacities in different
countriesa
Country Production capacity in
tonnes/year
Brazil 3000
Denmark 15 000
German Democratic Republic 3500
Mexico 8000
India 3000
China 40 000
USSR 5000-10 000
a From: Bayer (1988).
3.2.4 World consumption
Recent data from Bayer concerning the consumption of the active
ingredient only are reported in Table 4 (Bayer, 1988).
Table 4. Methyl parathion consumption in tonnes in some areas of the
worlda
Region 1984 1985 1986
Africa 191 308 152
North America 2 045 2 776 2 932
South America 9 135 6 555 5 587
Asia, New Zealand, 2 757 3 028 2 620
Australia
Western Europe 894 1 087 1 019
Total 15 022 13 754 12 310
aFrom: Bayer (1988).
In 1984, the USA exported 3010 tonnes of methyl parathion (HSDB,
1990).
3.2.5 Formulations
Methyl parathion is used in following formulations:
(1) emulsifiable concentrates (EC) with 19.5%, 40%, 50%, 60% active
ingredient (a.i.)
(2) wettable powders containing 40% a.i.
(3) dusts 1.5%, 2%, and 3% methyl parathion,
(4) microencapsulated methyl parathion, and
(5) ready-to-use liquid (less than 1% a.i.).
The usual carriers are: petroleum solvents and clay carriers
(such as propargite).
Combinations are available containing parathion, omethoate,
tetradifon, prothoate, and petroleum oil.
3.3 Uses
Methyl parathion is a broad-spectrum insecticide with
non-systemic contact and stomach action. The normal method of
application is foliar spraying by aircraft or ground equipment. Data
from 1971 show that most methyl parathion was used for protecting
cotton fields (Table 5).
Table 5. Methyl parathion consumption pattern (1971)a
Protection of consumption (%)
cotton 83
soybeans 8
grain including corn 5
wheat 2
tobacco, peanuts, vegetables, and citrus fruits 2
aFrom: HSDB (1990).
Only foliar application of methyl parathion is known. It is used
as a contact insecticide and acaricide. There are different routes of
application depending on the type of plant to be protected and the
organisms killed. The recommended application rate is 0.5-1 kg a.i./ha
for vegetables, 1-2 kg/ha for cereals, 1.5-6 kg/ha for fruit trees,
2-5 kg/ha for citrus fruits, and 0.12-1.0 kg/ha for cotton.
4. ENVIRONMENTAL TRANSPORTATION, DISTRIBUTION, AND TRANSFORMATION
4.1 Transportation and distribution between media
The transportation and distribution of methyl parathion in air,
water, soil, fauna, and flora are influenced by several physical,
chemical, and biological parameters. The transportation and fate of
methyl parathion were studied by Gile & Gillett (1981). They used the
simulated ecosystem developed at the Corvallis Environmental Research
Laboratory of the US EPA (Gillett & Gile, 1976). A 16-h daily light
cycle with an average of 27 000 lx at the soil surface was used. The
temperatures varied from 18 °C at night to 30 °C during the day. The
ecological compartment was ventilated with 10 litre air/min. The
simulated ecosystem included alfalfa (Medicago sativa) and
perennial ryegrass (Lolium perenne). Twenty days after planting,
different representative kinds of invertebrates (earthworms,
nematodes, garden snails) were added to the microcosms. Ten days
later, radioactive labelled 14C-methyl parathion (50 µCi) was
applied at rates of 0.3, 0.6, and 2.4 kg/ha. One week following the
methyl parathion application, a gravid gray-tailed vole (Microtus
canicaudus) was placed in the model ecosystem. The relative 14C
mass balance of the study is shown in the Table 6.
Most radioactivity was found in the upper 5 cm of soil. A
comparable experiment with p-nitrophenol showed a lower soil content
and no residues in the groundwater as well.
Crossland & Elgar (1983) used a mathematical model to predict the
dispersion and degradation of methyl parathion in freshwater ponds.
Basic assumptions of the model were that loss processes could be
adequately described in terms of simple partition phenomena and
first-order rate kinetics. Predictions of the model were compared with
experimentally-obtained data for concentrations of methyl parathion in
water and sediment. They started with a concentration of 100 µg methyl
parathion/litre pond water. At the limit of the analytical method
(0.005 µg/g), they could not find any residues of methyl parathion, 16
days after treatment. The authors described the degradation by a
pseudo first order rate constant that was temperature-dependent.
Since the degradation of methyl parathion in distilled water (pH not
given) was faster than expected and the bacteria concentration was
only 106/litre, a sediment-catalysed hydrolysis was supposed.
Crossland & Bennett (1984) compared degradation of methyl parathion in
experimental ponds and laboratory aquaria. Degradation was faster in
the natural ponds and faster than predicted from simple mathematical
models. Addition of plants, sediment, or sediment with plants, to the
laboratory aquaria increased the rate of breakdown of methyl
parathion; sediment had the greatest effect reducing half-life from
300 h in water alone to 90-140 h. These findings support the
investigation of Goedicke & Winkler (1976), who considered, from their
testing of the persistence of different formulations of methyl
parathion in soils, that the compound would not contaminate
groundwater, if applied at suggested rates and intervals.
Table 6. 14C mass balance of methyl parathion in a model ecosystema
Samples Application rate of methyl parathion
0.3 kg/ha 0.6 kg/ha 2.4 kg/ha
air 57b 46 33
soil 30 30 28
groundwater 0.0 0.1 0.0
plants 12 23 38
animals 1.0 0.6 1.1
a From: Gillett & Gile (1976).
b %.
4.1.1 Air
Most of this insecticide is directly liberated by spraying.
However, a perceptible amount is released simultaneously with
evaporation from water surfaces, leaves, or soil (Woodrow et al.,
1977).
Air samples were analysed after the application of methyl
parathion at a concentration of 1.12 kg/ha (Jackson & Lewis, 1978).
The conventional emulsifiable concentrate was compared with an
encapsulated formulation. The filter collection efficiency was
determined to be 105% and the extraction efficiency was 92%. During
the experimental period, the temperature varied from 18 to 34 °C at an
average relative humidity of 72%. The results of the analysis of the
air samples collected in tobacco-growing areas of North Carolina are
shown in Table 7.
Table 7. Concentration of methyl parathion in the air after applicationa
Time (days) Methyl parathion (mg/m3)
emulsifiable concentrate encapsulated formulation
0 7.408 3.783
1 3.338 0.330
3 0.584 0.107
6 0.036 0.025
6 0.054 0.019
9 0.013 0.016
a From: Jackson & Lewis (1978).
Since the usual atmospheric levels of methyl parathion in the
surroundings of agricultural areas range from not detectable to 71
ng/m3, Jackson & Lewis (1978) discussed the possibility that the
concentrations measured on day 9 may have been the result of the
background level in the air of the heavily treated areas
The atmospheric concentration of methyl parathion after spraying
in the Kalinin District, Tashkent Province of the Uzbek USSR, during
July and August, was determined by Akhmedov (1968). He found that the
concentrations measured were dependent on the size of the area of
methyl parathion application, the time of application, the
temperature, and the wind velocity. In addition, the odour threshold
was estimated, and effects on the brain electrical activity,
resorption action, dark adaptation, and the light sensitivity of the
eyes were studied.
After the aerial treatment of forests, Vrochinsky & Makovsky
(1977) measured the following concentrations of methyl parathion in
the air (Table 8).
The concentrations of methyl parathion increased in foggy
conditions because of the adsorption of the compound on the surface of
water aerosols (Goncharuk et al., (1988).
Table 8. Methyl parathion in air after spraying forestsa
Time (days) Methyl parathion (mg/m3)
0 0.12
1 0.05
5 0.024
10 0.0015
a From: Vrochinsky & Makovsky (1977).
14C-Methyl parathion was subjected to simulated rainfall (total
amount: 2.5, 25, and 38 mm/h) after application of 177 µg ai/cm2 to
an octadecylsilane/trimethylsilane-treated glass slide. The amounts of
14C remaining after washoff were 56%, 6%, and 2% respectively; thus,
methyl parathion shows a high rate of washoff (Cohen & Steinmetz,
1986).
4.1.2 Water
Various mechanisms exist for the transportation of methyl
parathion following its application to aquatic environments,
including: application-associated losses, volatilization, wind
erosion, rinsing by rain into groundwater, and transportation as a
soil-methyl parathion complex.
Eichelberger & Lichtenberg (1971) estimated the water pollution
factor by investigating the persistence of methyl parathion in river
water. They used a sealed glass jar containing river water and methyl
parathion and applied sunlight and artificial fluorescent light. The
initial concentration of methyl parathion was 10 µg/litre (Table 9):
Badawy & El-Dib (1984) found that methyl parathion was more
stable in water of high salinity, such as sea water, than in fresh
water.
Table 9. Persistence of methyl parathion in river watera
Time % of the initial concentration (10 µg/litre)
1 hour 80b
1 week 25
2 weeks 10
4 weeks 0
a Adapted from: Eichelberger & Lichtenberg (1971).
b Recoveries were rounded off to the nearest 5%.
Because of a collision between two ships in the Mediterranean Sea
near Port-Said, Egypt, the sea became contaminated with more than
10 000 kg methyl parathion. Maximum methyl parathion concentrations
(96 µlitre/litre) were found 50 m in the drifting direction (surface
current, wind). In general, the concentration decreased with distance
and time and reached the detection limit up to 80 days after the
accident. The residues in sediment gradually increased during the
first 20 days (concentration factor 49.5) (Badawy et al., 1984).
Crossland et al. (1986) gave mathematical tools for calculating
the fate of chemicals in aquatic systems (because of the importance of
the degradation of methyl parathion in water, see also section 4.2).
4.1.3 Soil
Lichtenstein (1975) incorporated an emulsifiable concentration of
methyl parathion into the upper 5 inches of a silt loam at a rate of
3.1 mg/kg). One month after treatment, 3.5% of the methyl parathion
could be detected in the soil. The author showed that percolating
water transported metabolites vertically as well as horizontally.
Methyl parathion moved less than 20 cm in a loamy soil following an
annual precipitation of 1500 mm (Haque & Freed, 1974).
Bound residues of [ring-14C] methyl parathion in a silt loam
were monitored during an incubation period of 49 days (Gerstl &
Helling, 1985). After this period, 54% of the initial 14C remained
in the soil; of this, 13% was soxhlet-extractable with methanol and
87% was bound residue. Several treatments indicated that bound
residues of methyl parathion are not easily released (i.e., converted
to an extractable form), but that they are slowly mineralized to
CO2.
A simulated spillage of emulsifiable or microencapsulated
formulations of methyl parathion on soil (sandy loam; pH ranging from
6.6 to 7.8, with a mean of 7.2) was studied for 45 months by Butler
and coworkers (1981). The uptake of the insecticide was studied in
five different experiments. The soil was contaminated with: a) 51%
emulsifiable concentrate formulation (E.C.), b) dilute drum rinse of
E.C., c) 22% microencapsulated formulation (M.C.), d) dilute drum
rinse of M.C., and e) a solid cake of M.C. microencapsulated
formulation of the initial values (Table 10). At 45 months, soil
residues of methyl parathion had decreased by 64% for emulsifiable
concentrate spills, and 68% for the soil beneath the microencapsulated
cake; the residue in the cake itself only decreased by 31% (Table 10).
Soil residue concentrations from the simulated drum rinses (Table 10)
were very low by 45 months (emulsifiable concentrate) and by one year
(microencapsulated formulation).
Performing laboratory experiments, Davidson et al. (1980) showed
that, at low application rates (24.5 mg/kg), methyl parathion was
non-persistent in soils (Webster & Cecil) but was persistent following
application of large quantities (10015 mg/kg). Therefore, it is
impossible to predict the behaviour of methyl parathion at high
applications rates on the basis of results following low application
rates.
4.1.4 Vegetation and wildlife
Residue levels of methyl parathion on foliage depend on the
formulation, the method of application, humidity, rain, temperature,
dust levels etc. Kido et al. (1975) investigated surface and internal
residue levels of methyl parathion on grape leaves treated with methyl
parathion sprays (at the rate of 0.84 kg a.i./ha.); 90.2% of the
initial surface residue was lost from the leaves one day after
application. The major portion, over 60%, of the total residues was
found in the internal portion of the leaves, and over 99% of the total
residues had been lost, 5 days after application. Overhead sprinkler
irrigation of the vines had only a slight, or no, effect on the
reduction of methyl parathion residues (Kido et al., 1975). The
residual life of methyl parathion on cotton can be extended by
Table 10. Persistence of methyl parathion in sandy loam soil and
in solid cake material following contamination of the soil
with different formulations of methyl parathiona
Time Mean concentrations of Methyl parathion (mg/kg)
(months)
E.C.b E.C. M.C.c M.C. M.C.
(51%) (rinse) (22%) (rinse) (cake)
0 48 900 17 600 30 800 2 140 379 000
1 33 700 10 800 14 200 940 258 000
3 25 300 7 000 17 100 550 305 000
12 20 900 3 800 20 000 0.15 87 500
20 20 800 1 400 13 300 230 149 000
45 17 500 130 9 800 n.r.d 262 000
a Modified from: Butler et al. (1981).
b E.C. = emulsifiable concentrate.
c M.C. = microencapsulated formulation.
d n.r. = not recorded.
application at dusk rather than dawn. For example, methyl parathion
decreased to less than 50% after 4 h in sunlight, but only to 84%
after the same time at night (Ware et al., 1980). The persistence of
methyl parathion following application to cotton was also increased by
combining it with molasses (Ware et al., 1980), toxaphene (Buck et
al., 1980; Ware et al., 1980; Bigley et al., 1981), camphene (Bigley
et al., 1981), or cedar oil (Bigley et al., 1981). Ware et al. (1983)
compared surface residues of methyl parathion on cotton foliage. When
applied to cotton fields (at 1.1 kg/ha) as a typical, low-volume spray
diluted with water versus ultra-low-volume (ULV) application using
vegetable oil as the carrier. Forty-eight hours after application as
an aqueous dilution, 1.8 % of the initial residue remained compared
with 7.2 % after application as ULV. Cole et al. (1986) sprayed methyl
parathion 4E (EC) in either water or water-crop oil (6:1) at 8 litres
of a 1.8% dilution/ha on a 5 ha plot of cotton using a pawnee
airplane. The residues found in the leaves sprayed with the mixture
containing crop oil were higher than those in water-sprayed leaves in
all samples collected after the treatment (Table 11).
Table 11. Comparison of methyl parathion residues in cotton leaves
treated with water sprays and with water-oil spraysa
Days after treatment Methyl parathion concentration
water water-oil formulation
1 14.80±8.74b 27.70±7.99
2 9.17±7.15 9.68±4.29
3 2.30±0.89 7.48±2.85
4 1.52±0.31 8.70±4.58
5 1.96±1.49 5.97±2.61
a From: Cole et al. (1986).
b mg/kg mean ± SE.
The drift from a commercial aerial application of methyl
parathion was quantified by Draper & Street (1981) by determining leaf
surface residues of methyl parathion in a treated alfalfa field and an
adjoining non-target pasture (with quackgrass, Agropyron repens, as
predominant species). Four hours after the pesticide spraying by plane
(0.27 kg/litre emulsifiable concentrate; 0.7 litre/ha; in the morning)
2.8 mg methyl parathion/kg were present as foliar residues in the
target field, and 0.26 mg/kg, in the untreated non-target pasture. At
both places, the foliar residues of the parent compound dissipated
rapidly with time.
The time-dependent decrease in the residues of 2 different
formulations of methyl parathion applied to tobacco plants was
evaluated. Methyl parathion in either the emulsifiable or the
encapsulated form was applied at rates of 0, 0.56, and 1.12 kg/ha.
Samples were collected before spraying and within 10 min of the
application. It was observed that the encapsulated formulation of
methyl parathion did not decompose as fast as the emulsifiable form
(Leidy et al., 1977). Varis (1972) tried to determine the influence of
plant growth on the loss of methyl parathion residues in sugar beet
seedlings. Methyl parathion was applied as a dust formulation (1.5%)
at 20 kg/ha, 14 days after sowing. The residue methyl parathion
concentration in the plants decreased to about 50% within 24 h. Within
6 days, the methyl parathion residue was reduced by 90%, 73% reduction
being due to plant growth.
Fuhremann & Lichtenstein (1978) performed experiments with
unextractable, soil-bound residues of radioactive labelled methyl
parathion and measured the potential pick up of the 14C-containing
residues. Earthworms (Lumbricus spp.) and oat (Avena sativa L.)
plants were able to release and incorporate some soil-bound,
14C-ring-labelled methyl parathion. Oat plants were found to release
more chemical from the soil than the earthworms.
Following applications of insecticides (including methyl
parathion) to nearby sugarcane or cotton fields, alterations in brain
acetylcholinesterase activity were found in birds living in brushland
within the Lower Rio Grande Valley of South Texas (Custer & Mitchell,
1987). These alterations might have resulted from exposure during the
use of agricultural fields as feeding or resting sites.
4.1.5 Entry into the food-chain
Methyl parathion hydrolyses faster than parathion. Because of the
physical and chemical properties of methyl parathion, its pollution
potential seems to be very small. Therefore, the most probable entry
into the food-chain seems to be directly via residues on vegetables or
crops.
Since animals can degrade methyl parathion and excrete the
degradation products within a very short time, a risk from eating meat
seems to be unlikely. However, there may be an additional hazard from
methyl parathion bound to glucosides (Dorough, 1978).
4.2 Biotransformation
4.2.1 Degradation involving biota
Both field and laboratory studies have been conducted on the
degradation of methyl parathion were. Data suggest that biodegrad
ation is the major degradative pathway in eutrophic systems, whereas
absorption, photolysis, and hydrolysis are more important in
oligotrophic systems.
The half-lives of methyl parathion residues reported in the
literature for plants were relatively short, but varied with ambient
conditions (see also section 4.1.4).
Singh et al. (1978) recorded half-lives of methyl parathion
applied to urd (Phaseolus mungo Roxb.) and pea ( Pisum sativum (L)
var. arvense Poir.) at the rate of 0.63 and 1.25 kg a.i. per ha,
respectively. Half-lives were 1.7 and 2.5 days for urd and 2.0 and 2.7
days for pea, respectively. Foliar residues of methyl parathion on
alfalfa treated by aircraft (0.27 kg/litre, emulsifiable concentrate)
dissipated showing a first-order half-life of 12 h. This calculation
is based on initial slopes of semi-logarithmic plots (Draper & Street,
1981). The authors, however, noted that dissipation kinetics appeared
to be greater than first-order. The times required for a 50% reduction
in methyl parathion residues in cotton foliage were determined to be
4.4-5.4 h (emulsifiable concentrate) or 28.1 h (encapsulated
formulation) following application at a rate of 0.28 kg/ha (Smith et
al., 1987). Based on data previously reported by Ware et al. (1974a)
following application of methyl parathion to cotton (1.12 kg/ha),
half-lives of 12 h (emulsifiable concentrate) and 70 h (encapsulated
formulation) were calculated (Smith et al., 1987). In another study
using emulsifiable concentrate formulations of methyl parathion at a
rate of 1.15 kg/ha, a 50% disappearance time of 2-4 h was calculated
for methyl parathion on cotton plants (Willis et al., 1985). A
half-life of 0.96 days was described for methyl parathion residues
(initial concentration = 0.4 µg/cm) on apple leaf surfaces (Goedicke,
1989).
A single report is available on the persistence of methyl
parathion in a submerged aquatic macrophyte (Hydrilla verticilla)
and a fish (carp), both initially exposed to 3.8 mg methyl
parathion/litre. The first order half-lives were 7.9 and 5.4 days,
respectively (Sabharwal & Belsare, 1986).
The half-life of methyl parathion in a soil (not characterized in
detail) has been reported to be about 45 days (Menzie, 1972). In
another study it was calculated to be as short as 2.7 days (Singh et
al., 1978), possibly due to the high pH of the soil (pH = 8.6) and
temperature (28 °C-33 °C). Half-lives of 12 and 22 days were measured
for methyl parathion in 2 soils (pH = 6.1 and 5.5, respectively) when
incubated at 22 °C (Möllhoff, 1981). Concentrations of methyl
parathion in a loamy sand soil (pH = 5.3) decreased from a level of
about 5 mg/kg to 0.3 mg/kg during a period of 57 days (Goedicke &
Winkler, 1976). Thirty days following treatment, 3.1% of initial
residues of methyl parathion were found in a soil (clay?) of a field
treated with 5.6 kg/ha (Lichtenstein & Schulz, 1964) (see also section
4.1.3).
During an incubation study under aerobic conditions, methyl
parathion was degraded mainly to CO2 and 4-nitrophenol, and, to a
minor extent, to desmethyl parathion (Möllhoff, 1981). Methyl
parathion may be degraded in the environment by: a ) hydrolysis to
p-nitrophenol and dimethylthiophosphoric acid; or b ) nitro-group
reduction to methyl aminoparathion (e.g., Sharmila et al., 1988).
Hydrolysis can be both chemical and microbial while nitro-group
reduction is essentially microbial. Generally, hydrolysis is the major
pathway in nonflooded soil while methyl parathion is degraded mainly
by nitro-group reduction in predominantly anaerobic systems, such as
flooded soil (Ou et al., 1983; Ou, 1985; Adhya et al., 1987).
In a few instances, hydrolysis is the major or only pathway of
methyl parathion degradation in soils, even under flooded conditions
(Ou, 1985). Adhya et al. (1987) evaluated the influence of different
physical and chemical characteristics on the persistence of methyl
parathion in 5 tropical soils under flooded and nonflooded conditions.
They found that nitro-group reduction was the major pathway of methyl
parathion degradation in 4 out of 5 of the soils under flooded
conditions, while, in one soil (Sukinda-soil), degradation of methyl
parathion proceeded exclusively by hydrolysis, even under flooded
conditions. The latter finding was confirmed by Sharmila et al.
(1989a).
A temperature-dependent shift from nitro-group reduction (at
25 °C) to predominantly hydrolysis (at 35 °C) occurred in a flooded
alluvial soil; both pathways were mediated microbially (Sharmila et
al., 1988). The addition of yeast extract also influenced the
degradation pathway of methyl parathion by bacterial cultures in
enriched flooded alluvial and laterite (Sukinda) soils (Sharmila et
al., 1989b). Low redox potential in a flooded soil favoured
degradation by nitro-group reduction, whereas hydrolysis was
concomitant with a more positive potential (Adhya et al., 1981a).
Adhya et al. (1981b) reported studies on sulfur-containing
anaerobic ecosystems, such as oceanic sediments, which they supposed
could serve as a potential sink for pesticides. They found that methyl
parathion was decomposed in acid, sulfur-containing soils and soils
with a low sulfate content to aminomethyl parathion; however, no
decomposition occurred under aerobic conditions. Demethylation could
be demonstrated in anaerobic sulfate soils.
Evidence for microbial participation was provided by the fact
that sterilization of the enriched soil samples increased the
stability of methyl parathion in soil (Adhya et al., 1981a). The
authors reported a very rapid reduction of the nitro group of methyl
parathion by equilibration with a soil incubated with rice straw under
flooding. Sterilization of this soil preparation prevented this rapid
reduction. The degradation of methyl parathion and its metabolite
p-nitrophenol in flooded alluvial soil is given in Table 12.
It appeared from this study, that the degradation of the
metabolite p-nitrophenol is more rapid than the decomposition of
methyl parathion.
Table 12. Degradation of methyl parathion and its metabolite p-nitrophenol
in flooded alluvial soila
Days after methyl µg of compound recovered/20 g of soil
parathion addition
methyl parathion p-nitrophenol
0 485.3 0
0.5 428.1 trace
1 333.7 120.0
2 219.8 98.6
3 185.6 72.0
6 95.5 0
12 58.2 0
a From: Adhya et al. (1981a).
Isolated mixed bacterial cultures from soil utilized methyl
parathion and parathion as a sole carbon source (Chaudhry et al.,
1988). Pseudomonas sp. was capable of hydrolysing methyl parathion
and parathion to p-nitrophenol but needed another carbon source for
growth. The optimum pH range for enzymatic hydrolysis by this
bacterium was from 7.5 to 9.5. In view of the instability of methyl
parathion in alkaline solutions, it is not clear whether the
hydrolysis noted was or was not partially due to the pH of the
solution rather than wholly due to bacterial action. The thermal
optimum was between 35 °C and 40 °C. Flavobacterium sp. culture was
able to metabolize p-nitrophenol by degrading it to nitrite and to
use it for growth. The DNAs from Pseudomonas sp. and from the mixed
culture showed homology with the organophosphate degradation gene from
a previously reported parathion-hydrolysing bacterium, Flavobacterium
sp. Ou & Sharma (1989) showed that methyl parathion is extensively
degraded by a mixed bacterial culture and a Bacillus sp. to its
final oxidation products carbon dioxide and water, whilst a
Pseudomonas sp. isolated from the mixed culture could degrade the
hydrolysis product p-nitrophenol. A Flavobacterium sp. isolated
from flooded soil was able to hydrolyse methyl parathion, but a
Pseudomonas sp. from flooded soil was not (Adhya et al., 1981c). The
transformation of methyl parathion by pure cultures of Flavobacterium
sp. followed multiphasic kinetics (Lewis et al., 1985).
A different result was described by Arndt et al. (1981) for
microorganisms in compost. They added 70 mg of methyl parathion
dissolved in 20 ml ethyl acetate to 1.2 kg of grass (40%), apples
(23%), potatoes (17%), yoghourt (13%), and bread (7%). After
composting this mixture for 7 days, no degradation product of methyl
parathion was found. The recovery rate was 95%. The authors concluded
that the insecticide could accumulate in the compost under the
conditions tested, but it could not be excluded that this result was
affected by the ethyl acetate.
The concentration of methyl parathion (applied at 0.28 kg/ha) in
a lake (Clear Lake, California, USA) dropped from 0.50 µg/litre to
0.28 µg/litre, measured 8 and 48 h, respectively, after treatment
(Apperson et al., 1976). After a third application (total 3 X 0.28
kg/ha) the residue level of methyl parathion was 5.4 µg/litre, and 7
days later, 2 µg/litre (Apperson et al., 1976). Eichelberger &
Lichtenberg (1971) found that 90% of methyl parathion in river water
was degraded during a period of 2 weeks, whereas there was no
degradation in distilled water. The latter finding may be pH related,
since Cowart et al. (1971) noted 50% hydrolysis of the pesticide after
14 days in distilled water at pH 6. Under field conditions, in the
presence of sediment and aquatic plants, degradation is accelerated
and persistence is lower. Dortland (1980) showed that persistence
decreased by a factor of 2-3 when sediment and plants were added to
the aquatic microcosm. When considering the aquatic ecosystem as a
whole (which includes adsorption on sediments and adsorption on, and
incorporation in, aquatic biota) a fair estimate of the persistence of
methyl parathion in the water column seemed to be 2-3 days (Walker,
1978). This value was recorded in microcosm studies and field
experiments in both freshwater and estuarine aquatic environments.
Predicted half-life values in rivers, ponds, eutrophic lakes, and
oligotrophic lakes were reported to be 0.6, 27.3, 28.3, and 151.6 h,
respectively (Smith et al., 1978). Methyl parathion was degraded with
a half-life of 28 h in sediment collected from a field site and with
a half-life of 7 h in microbial mats derived from laboratory mesocosms
(Newton et al., 1990). The half-lives of methyl parathion in the water
and sediment of a carp pond were 5.7 days and 5.0 days, respectively
(initial residues: 3.77 mg/litre in water and 0.52 mg/kg in soil)
(Sabharwal & Belsare, 1986). It should be emphasized that the
persistence values reported depend not only on the type of biotope but
also on the abiotic conditions, i.e., temperature, pH, and salinity,
as pointed out, for example, by Badawy & El-Dib (1984).
Holm et al. (1983) found in their model ecosystem that the
sediment type had no observable effect on the degradation of methyl
parathion and that it depended primarily on the communities of
microorganisms. These communities and their ability to degrade methyl
parathion did not change with different sediment types. The microbial
degradation rate constants in an aquatic channel microcosmos ranged
from 2.7 X 10-6/s to 6.9 X 10-6/s. This was significantly higher
than the rate constants determined for abiotic degradation. Cripe et
al. (1987) modified the river die-away test for determining the
biodegradability of organic substances and tested the degradation
products for their toxicity. Because of their sensitivity, mysids and
daphnids were used for testing the toxicity of the degradation
products. This test showed a rapid, sediment-mediated biodegradation
of methyl parathion.
The biodegration rate of methyl parathion was compared in 3 types
of test systems composed of sediment and water collected from various
estuarine sites (Van Veld & Spain, 1983). Generally, methyl parathion
degradation was fastest in intact sediment/water cores, followed by
sediment/water shake flasks, and was slowest in water shake flasks.
Lewis & Holm (1981) determined the transformation rate of methyl
parathion by "aufwuchs" microorganisms, i.e., aquatic microbial growth
attached to submerged surfaces or suspended in streamers or mats.
"Aufwuchs" fungi, protozoa, and algae did not transform methyl
parathion, but bacteria rapidly transformed it.
Lewis et al. (1984) examined the effects of microbial community
interactions on methyl parathion transformation rates. They found
either stimulation or inhibition of bacterial transformation rates in
the presence of various cultures, filtrates, or exudates of algae,
fungi, or other bacteria.
The biotic and abiotic degradation rates of methyl parathion in
water and sediment samples over a 3-year period was studied by
Pritchard et al. (1987). The aim of their study was to find the reason
for the different degradation rates reported for methyl parathion, but
the divergences in biodegration could not be assigned to any single
factor. The predominant degradation in an aerobic system appears to be
the biological hydrolysis, producing p-nitrophenol.
Phosphatases are an important group of enzymes involved in the
breakdown of methyl parathion (Portier & Meyers, 1982; Portier et al.,
1983). A proposed pathway for the breakdown of methyl parathion in
aquatic systems is given by Bourquin et al. (1979) in Fig. 1.
Methyl parathion is degraded by bacteria in soil, but more slowly
by bacteria in water. Crossland et al. (1986) estimated the rate of
biodegradation of methyl parathion using a mathematical model.
Sorption on sediment was the dominant process for loss of methyl
parathion from the water compartment. The rate of biodegradation in
sediment (4.0 µmol/litre per h) greatly exceeded that of sorption on
sediment (0.02-0.05 µmol/litre per h) and, therefore, the sediment
compartment may be considered a sink for methyl parathion.
The complete decomposition of methyl parathion into innocuous
compounds can be realized by planktonic and attached microorganisms
(Lassiter et al., 1986). The metabolite p-nitrophenol can be further
metabolized by algae, as reported by Werner & Pawlitz (1978).
4.2.2 Abiotic degradation
Data on the abiotic degradation of methyl parathion are presented
in Table 13.
4.2.2.1 Photodegradation
When exposed to UV radiation or sunlight, methyl parathion
undergoes oxidative degradation. The degradation rate constant of
methyl parathion sprayed as a film (0.67 µg/cm2) and exposed to 300
nm light was reported to be 46.6 X 10-7/s, corresponding to a
half-life of 41.2 h (Chen et al., 1984). In a stationary reactor, the
half-life of methyl parathion dissolved in an aqueous solution (pH=7)
was 72 min after radiation with a Hg low pressure lamp (at 254 nm)
(Hicke & Thiemann, 1987). Methyl parathion has been shown to be one of
the most light-sensitive insecticides. Baker & Applegate (1970, 1974)
showed photodegradation of methyl parathion using light in the
spectral range 300-400 nm (Table 13); methyl paraoxon, the active
cholinesterase inhibitor, was produced. Although photodegradation of
methyl parathion in the terrestrial compartment of the environment may
be important, it plays only a minor role in aquatic media (Env. Res.
Lab., 1981). The first-order transformation rate for photolysis upon
exposure to daylight fluorescent lamps was low compared to hydrolysis
and, in particular, compared to microbial degradation in an aquatic
channel microcosm (Holm et al., 1983). The loss of methyl parathion
through photolysis was estimated to be 4%.
Nevertheless, it seems that sunlight may reduce the half-life of
methyl parathion considerably. Schimmel et al. (1983) reported a
half-life of 6.3 days for a 1 mg methyl parathion/litre solution
exposed to sunlight. In darkness, with the same test conditions, the
half-life was 18 days. Like parathion, the photoreaction of methyl
parathion was accelerated in the presence of green and blue green
algae (Zepp & Schlotzhauer, 1983).
3.2.2 Loss into the environment
Emissions of methyl parathion during the production process can
be disregarded when compared with those from its use as an
insecticide. The air emission from a factory in the USA was reported
to be around 0.1% of the production level (Archer et al., 1978). The
major losses of this insecticide are directly caused by spraying, and
evaporation from water surfaces, leaves, and from the soil (Woodrow et
al., 1977).
3.2.3 Production
According to the European Directory of Agrochemical Products
(1986) and the Directory of World Chemical Producers (1990), methyl
parathion is produced throughout the world by many companies. World
production in 1966 was 31 700 tonnes, including 14 800 tonnes produced
in the USA.
In Table 3, selected countries producing methyl parathion are
listed together with their production capacities (Bayer, 1988).
Table 3. Methyl parathion production capacities in different
countriesa
Country Production capacity in
tonnes/year
Brazil 3000
Denmark 15 000
German Democratic Republic 3500
Mexico 8000
India 3000
China 40 000
USSR 5000-10 000
a From: Bayer (1988).
3.2.4 World consumption
Recent data from Bayer concerning the consumption of the active
ingredient only are reported in Table 4 (Bayer, 1988).
Table 4. Methyl parathion consumption in tonnes in some areas of the
worlda
Region 1984 1985 1986
Africa 191 308 152
North America 2 045 2 776 2 932
South America 9 135 6 555 5 587
Asia, New Zealand, 2 757 3 028 2 620
Australia
Western Europe 894 1 087 1 019
Total 15 022 13 754 12 310
aFrom: Bayer (1988).
In 1984, the USA exported 3010 tonnes of methyl parathion (HSDB,
1990).
3.2.5 Formulations
Methyl parathion is used in following formulations:
(1) emulsifiable concentrates (EC) with 19.5%, 40%, 50%, 60% active
ingredient (a.i.)
(2) wettable powders containing 40% a.i.
(3) dusts 1.5%, 2%, and 3% methyl parathion,
(4) microencapsulated methyl parathion, and
(5) ready-to-use liquid (less than 1% a.i.).
The usual carriers are: petroleum solvents and clay carriers
(such as propargite).
Combinations are available containing parathion, omethoate,
tetradifon, prothoate, and petroleum oil.
3.3 Uses
Methyl parathion is a broad-spectrum insecticide with
non-systemic contact and stomach action. The normal method of
application is foliar spraying by aircraft or ground equipment. Data
from 1971 show that most methyl parathion was used for protecting
cotton fields (Table 5).
Table 5. Methyl parathion consumption pattern (1971)a
Protection of consumption (%)
cotton 83
soybeans 8
grain including corn 5
wheat 2
tobacco, peanuts, vegetables, and citrus fruits 2
aFrom: HSDB (1990).
Only foliar application of methyl parathion is known. It is used
as a contact insecticide and acaricide. There are different routes of
application depending on the type of plant to be protected and the
organisms killed. The recommended application rate is 0.5-1 kg a.i./ha
for vegetables, 1-2 kg/ha for cereals, 1.5-6 kg/ha for fruit trees,
2-5 kg/ha for citrus fruits, and 0.12-1.0 kg/ha for cotton.
4. ENVIRONMENTAL TRANSPORTATION, DISTRIBUTION, AND TRANSFORMATION
4.1 Transportation and distribution between media
The transportation and distribution of methyl parathion in air,
water, soil, fauna, and flora are influenced by several physical,
chemical, and biological parameters. The transportation and fate of
methyl parathion were studied by Gile & Gillett (1981). They used the
simulated ecosystem developed at the Corvallis Environmental Research
Laboratory of the US EPA (Gillett & Gile, 1976). A 16-h daily light
cycle with an average of 27 000 lx at the soil surface was used. The
temperatures varied from 18 °C at night to 30 °C during the day. The
ecological compartment was ventilated with 10 litre air/min. The
simulated ecosystem included alfalfa (Medicago sativa) and
perennial ryegrass (Lolium perenne). Twenty days after planting,
different representative kinds of invertebrates (earthworms,
nematodes, garden snails) were added to the microcosms. Ten days
later, radioactive labelled 14C-methyl parathion (50 µCi) was
applied at rates of 0.3, 0.6, and 2.4 kg/ha. One week following the
methyl parathion application, a gravid gray-tailed vole (Microtus
canicaudus) was placed in the model ecosystem. The relative 14C
mass balance of the study is shown in the Table 6.
Most radioactivity was found in the upper 5 cm of soil. A
comparable experiment with p-nitrophenol showed a lower soil content
and no residues in the groundwater as well.
Crossland & Elgar (1983) used a mathematical model to predict the
dispersion and degradation of methyl parathion in freshwater ponds.
Basic assumptions of the model were that loss processes could be
adequately described in terms of simple partition phenomena and
first-order rate kinetics. Predictions of the model were compared with
experimentally-obtained data for concentrations of methyl parathion in
water and sediment. They started with a concentration of 100 µg methyl
parathion/litre pond water. At the limit of the analytical method
(0.005 µg/g), they could not find any residues of methyl parathion, 16
days after treatment. The authors described the degradation by a
pseudo first order rate constant that was temperature-dependent.
Since the degradation of methyl parathion in distilled water (pH not
given) was faster than expected and the bacteria concentration was
only 106/litre, a sediment-catalysed hydrolysis was supposed.
Crossland & Bennett (1984) compared degradation of methyl parathion in
experimental ponds and laboratory aquaria. Degradation was faster in
the natural ponds and faster than predicted from simple mathematical
models. Addition of plants, sediment, or sediment with plants, to the
laboratory aquaria increased the rate of breakdown of methyl
parathion; sediment had the greatest effect reducing half-life from
300 h in water alone to 90-140 h. These findings support the
investigation of Goedicke & Winkler (1976), who considered, from their
testing of the persistence of different formulations of methyl
parathion in soils, that the compound would not contaminate
groundwater, if applied at suggested rates and intervals.
Table 6. 14C mass balance of methyl parathion in a model ecosystema
Samples Application rate of methyl parathion
0.3 kg/ha 0.6 kg/ha 2.4 kg/ha
air 57b 46 33
soil 30 30 28
groundwater 0.0 0.1 0.0
plants 12 23 38
animals 1.0 0.6 1.1
a From: Gillett & Gile (1976).
b %.
4.1.1 Air
Most of this insecticide is directly liberated by spraying.
However, a perceptible amount is released simultaneously with
evaporation from water surfaces, leaves, or soil (Woodrow et al.,
1977).
Air samples were analysed after the application of methyl
parathion at a concentration of 1.12 kg/ha (Jackson & Lewis, 1978).
The conventional emulsifiable concentrate was compared with an
encapsulated formulation. The filter collection efficiency was
determined to be 105% and the extraction efficiency was 92%. During
the experimental period, the temperature varied from 18 to 34 °C at an
average relative humidity of 72%. The results of the analysis of the
air samples collected in tobacco-growing areas of North Carolina are
shown in Table 7.
Table 7. Concentration of methyl parathion in the air after applicationa
Time (days) Methyl parathion (mg/m3)
emulsifiable concentrate encapsulated formulation
0 7.408 3.783
1 3.338 0.330
3 0.584 0.107
6 0.036 0.025
6 0.054 0.019
9 0.013 0.016
a From: Jackson & Lewis (1978).
Since the usual atmospheric levels of methyl parathion in the
surroundings of agricultural areas range from not detectable to 71
ng/m3, Jackson & Lewis (1978) discussed the possibility that the
concentrations measured on day 9 may have been the result of the
background level in the air of the heavily treated areas
The atmospheric concentration of methyl parathion after spraying
in the Kalinin District, Tashkent Province of the Uzbek USSR, during
July and August, was determined by Akhmedov (1968). He found that the
concentrations measured were dependent on the size of the area of
methyl parathion application, the time of application, the
temperature, and the wind velocity. In addition, the odour threshold
was estimated, and effects on the brain electrical activity,
resorption action, dark adaptation, and the light sensitivity of the
eyes were studied.
After the aerial treatment of forests, Vrochinsky & Makovsky
(1977) measured the following concentrations of methyl parathion in
the air (Table 8).
The concentrations of methyl parathion increased in foggy
conditions because of the adsorption of the compound on the surface of
water aerosols (Goncharuk et al., (1988).
Table 8. Methyl parathion in air after spraying forestsa
Time (days) Methyl parathion (mg/m3)
0 0.12
1 0.05
5 0.024
10 0.0015
a From: Vrochinsky & Makovsky (1977).
14C-Methyl parathion was subjected to simulated rainfall (total
amount: 2.5, 25, and 38 mm/h) after application of 177 µg ai/cm2 to
an octadecylsilane/trimethylsilane-treated glass slide. The amounts of
14C remaining after washoff were 56%, 6%, and 2% respectively; thus,
methyl parathion shows a high rate of washoff (Cohen & Steinmetz,
1986).
4.1.2 Water
Various mechanisms exist for the transportation of methyl
parathion following its application to aquatic environments,
including: application-associated losses, volatilization, wind
erosion, rinsing by rain into groundwater, and transportation as a
soil-methyl parathion complex.
Eichelberger & Lichtenberg (1971) estimated the water pollution
factor by investigating the persistence of methyl parathion in river
water. They used a sealed glass jar containing river water and methyl
parathion and applied sunlight and artificial fluorescent light. The
initial concentration of methyl parathion was 10 µg/litre (Table 9):
Badawy & El-Dib (1984) found that methyl parathion was more
stable in water of high salinity, such as sea water, than in fresh
water.
Table 9. Persistence of methyl parathion in river watera
Time % of the initial concentration (10 µg/litre)
1 hour 80b
1 week 25
2 weeks 10
4 weeks 0
a Adapted from: Eichelberger & Lichtenberg (1971).
b Recoveries were rounded off to the nearest 5%.
Because of a collision between two ships in the Mediterranean Sea
near Port-Said, Egypt, the sea became contaminated with more than
10 000 kg methyl parathion. Maximum methyl parathion concentrations
(96 µlitre/litre) were found 50 m in the drifting direction (surface
current, wind). In general, the concentration decreased with distance
and time and reached the detection limit up to 80 days after the
accident. The residues in sediment gradually increased during the
first 20 days (concentration factor 49.5) (Badawy et al., 1984).
Crossland et al. (1986) gave mathematical tools for calculating
the fate of chemicals in aquatic systems (because of the importance of
the degradation of methyl parathion in water, see also section 4.2).
4.1.3 Soil
Lichtenstein (1975) incorporated an emulsifiable concentration of
methyl parathion into the upper 5 inches of a silt loam at a rate of
3.1 mg/kg). One month after treatment, 3.5% of the methyl parathion
could be detected in the soil. The author showed that percolating
water transported metabolites vertically as well as horizontally.
Methyl parathion moved less than 20 cm in a loamy soil following an
annual precipitation of 1500 mm (Haque & Freed, 1974).
Bound residues of [ring-14C] methyl parathion in a silt loam
were monitored during an incubation period of 49 days (Gerstl &
Helling, 1985). After this period, 54% of the initial 14C remained
in the soil; of this, 13% was soxhlet-extractable with methanol and
87% was bound residue. Several treatments indicated that bound
residues of methyl parathion are not easily released (i.e., converted
to an extractable form), but that they are slowly mineralized to
CO2.
A simulated spillage of emulsifiable or microencapsulated
formulations of methyl parathion on soil (sandy loam; pH ranging from
6.6 to 7.8, with a mean of 7.2) was studied for 45 months by Butler
and coworkers (1981). The uptake of the insecticide was studied in
five different experiments. The soil was contaminated with: a) 51%
emulsifiable concentrate formulation (E.C.), b) dilute drum rinse of
E.C., c) 22% microencapsulated formulation (M.C.), d) dilute drum
rinse of M.C., and e) a solid cake of M.C. microencapsulated
formulation of the initial values (Table 10). At 45 months, soil
residues of methyl parathion had decreased by 64% for emulsifiable
concentrate spills, and 68% for the soil beneath the microencapsulated
cake; the residue in the cake itself only decreased by 31% (Table 10).
Soil residue concentrations from the simulated drum rinses (Table 10)
were very low by 45 months (emulsifiable concentrate) and by one year
(microencapsulated formulation).
Performing laboratory experiments, Davidson et al. (1980) showed
that, at low application rates (24.5 mg/kg), methyl parathion was
non-persistent in soils (Webster & Cecil) but was persistent following
application of large quantities (10015 mg/kg). Therefore, it is
impossible to predict the behaviour of methyl parathion at high
applications rates on the basis of results following low application
rates.
4.1.4 Vegetation and wildlife
Residue levels of methyl parathion on foliage depend on the
formulation, the method of application, humidity, rain, temperature,
dust levels etc. Kido et al. (1975) investigated surface and internal
residue levels of methyl parathion on grape leaves treated with methyl
parathion sprays (at the rate of 0.84 kg a.i./ha.); 90.2% of the
initial surface residue was lost from the leaves one day after
application. The major portion, over 60%, of the total residues was
found in the internal portion of the leaves, and over 99% of the total
residues had been lost, 5 days after application. Overhead sprinkler
irrigation of the vines had only a slight, or no, effect on the
reduction of methyl parathion residues (Kido et al., 1975). The
residual life of methyl parathion on cotton can be extended by
Table 10. Persistence of methyl parathion in sandy loam soil and
in solid cake material following contamination of the soil
with different formulations of methyl parathiona
Time Mean concentrations of Methyl parathion (mg/kg)
(months)
E.C.b E.C. M.C.c M.C. M.C.
(51%) (rinse) (22%) (rinse) (cake)
0 48 900 17 600 30 800 2 140 379 000
1 33 700 10 800 14 200 940 258 000
3 25 300 7 000 17 100 550 305 000
12 20 900 3 800 20 000 0.15 87 500
20 20 800 1 400 13 300 230 149 000
45 17 500 130 9 800 n.r.d 262 000
a Modified from: Butler et al. (1981).
b E.C. = emulsifiable concentrate.
c M.C. = microencapsulated formulation.
d n.r. = not recorded.
application at dusk rather than dawn. For example, methyl parathion
decreased to less than 50% after 4 h in sunlight, but only to 84%
after the same time at night (Ware et al., 1980). The persistence of
methyl parathion following application to cotton was also increased by
combining it with molasses (Ware et al., 1980), toxaphene (Buck et
al., 1980; Ware et al., 1980; Bigley et al., 1981), camphene (Bigley
et al., 1981), or cedar oil (Bigley et al., 1981). Ware et al. (1983)
compared surface residues of methyl parathion on cotton foliage. When
applied to cotton fields (at 1.1 kg/ha) as a typical, low-volume spray
diluted with water versus ultra-low-volume (ULV) application using
vegetable oil as the carrier. Forty-eight hours after application as
an aqueous dilution, 1.8 % of the initial residue remained compared
with 7.2 % after application as ULV. Cole et al. (1986) sprayed methyl
parathion 4E (EC) in either water or water-crop oil (6:1) at 8 litres
of a 1.8% dilution/ha on a 5 ha plot of cotton using a pawnee
airplane. The residues found in the leaves sprayed with the mixture
containing crop oil were higher than those in water-sprayed leaves in
all samples collected after the treatment (Table 11).
Table 11. Comparison of methyl parathion residues in cotton leaves
treated with water sprays and with water-oil spraysa
Days after treatment Methyl parathion concentration
water water-oil formulation
1 14.80±8.74b 27.70±7.99
2 9.17±7.15 9.68±4.29
3 2.30±0.89 7.48±2.85
4 1.52±0.31 8.70±4.58
5 1.96±1.49 5.97±2.61
a From: Cole et al. (1986).
b mg/kg mean ± SE.
The drift from a commercial aerial application of methyl
parathion was quantified by Draper & Street (1981) by determining leaf
surface residues of methyl parathion in a treated alfalfa field and an
adjoining non-target pasture (with quackgrass, Agropyron repens, as
predominant species). Four hours after the pesticide spraying by plane
(0.27 kg/litre emulsifiable concentrate; 0.7 litre/ha; in the morning)
2.8 mg methyl parathion/kg were present as foliar residues in the
target field, and 0.26 mg/kg, in the untreated non-target pasture. At
both places, the foliar residues of the parent compound dissipated
rapidly with time.
The time-dependent decrease in the residues of 2 different
formulations of methyl parathion applied to tobacco plants was
evaluated. Methyl parathion in either the emulsifiable or the
encapsulated form was applied at rates of 0, 0.56, and 1.12 kg/ha.
Samples were collected before spraying and within 10 min of the
application. It was observed that the encapsulated formulation of
methyl parathion did not decompose as fast as the emulsifiable form
(Leidy et al., 1977). Varis (1972) tried to determine the influence of
plant growth on the loss of methyl parathion residues in sugar beet
seedlings. Methyl parathion was applied as a dust formulation (1.5%)
at 20 kg/ha, 14 days after sowing. The residue methyl parathion
concentration in the plants decreased to about 50% within 24 h. Within
6 days, the methyl parathion residue was reduced by 90%, 73% reduction
being due to plant growth.
Fuhremann & Lichtenstein (1978) performed experiments with
unextractable, soil-bound residues of radioactive labelled methyl
parathion and measured the potential pick up of the 14C-containing
residues. Earthworms (Lumbricus spp.) and oat (Avena sativa L.)
plants were able to release and incorporate some soil-bound,
14C-ring-labelled methyl parathion. Oat plants were found to release
more chemical from the soil than the earthworms.
Following applications of insecticides (including methyl
parathion) to nearby sugarcane or cotton fields, alterations in brain
acetylcholinesterase activity were found in birds living in brushland
within the Lower Rio Grande Valley of South Texas (Custer & Mitchell,
1987). These alterations might have resulted from exposure during the
use of agricultural fields as feeding or resting sites.
4.1.5 Entry into the food-chain
Methyl parathion hydrolyses faster than parathion. Because of the
physical and chemical properties of methyl parathion, its pollution
potential seems to be very small. Therefore, the most probable entry
into the food-chain seems to be directly via residues on vegetables or
crops.
Since animals can degrade methyl parathion and excrete the
degradation products within a very short time, a risk from eating meat
seems to be unlikely. However, there may be an additional hazard from
methyl parathion bound to glucosides (Dorough, 1978).
4.2 Biotransformation
4.2.1 Degradation involving biota
Both field and laboratory studies have been conducted on the
degradation of methyl parathion were. Data suggest that biodegrad
ation is the major degradative pathway in eutrophic systems, whereas
absorption, photolysis, and hydrolysis are more important in
oligotrophic systems.
The half-lives of methyl parathion residues reported in the
literature for plants were relatively short, but varied with ambient
conditions (see also section 4.1.4).
Singh et al. (1978) recorded half-lives of methyl parathion
applied to urd (Phaseolus mungo Roxb.) and pea ( Pisum sativum (L)
var. arvense Poir.) at the rate of 0.63 and 1.25 kg a.i. per ha,
respectively. Half-lives were 1.7 and 2.5 days for urd and 2.0 and 2.7
days for pea, respectively. Foliar residues of methyl parathion on
alfalfa treated by aircraft (0.27 kg/litre, emulsifiable concentrate)
dissipated showing a first-order half-life of 12 h. This calculation
is based on initial slopes of semi-logarithmic plots (Draper & Street,
1981). The authors, however, noted that dissipation kinetics appeared
to be greater than first-order. The times required for a 50% reduction
in methyl parathion residues in cotton foliage were determined to be
4.4-5.4 h (emulsifiable concentrate) or 28.1 h (encapsulated
formulation) following application at a rate of 0.28 kg/ha (Smith et
al., 1987). Based on data previously reported by Ware et al. (1974a)
following application of methyl parathion to cotton (1.12 kg/ha),
half-lives of 12 h (emulsifiable concentrate) and 70 h (encapsulated
formulation) were calculated (Smith et al., 1987). In another study
using emulsifiable concentrate formulations of methyl parathion at a
rate of 1.15 kg/ha, a 50% disappearance time of 2-4 h was calculated
for methyl parathion on cotton plants (Willis et al., 1985). A
half-life of 0.96 days was described for methyl parathion residues
(initial concentration = 0.4 µg/cm) on apple leaf surfaces (Goedicke,
1989).
A single report is available on the persistence of methyl
parathion in a submerged aquatic macrophyte (Hydrilla verticilla)
and a fish (carp), both initially exposed to 3.8 mg methyl
parathion/litre. The first order half-lives were 7.9 and 5.4 days,
respectively (Sabharwal & Belsare, 1986).
The half-life of methyl parathion in a soil (not characterized in
detail) has been reported to be about 45 days (Menzie, 1972). In
another study it was calculated to be as short as 2.7 days (Singh et
al., 1978), possibly due to the high pH of the soil (pH = 8.6) and
temperature (28 °C-33 °C). Half-lives of 12 and 22 days were measured
for methyl parathion in 2 soils (pH = 6.1 and 5.5, respectively) when
incubated at 22 °C (Möllhoff, 1981). Concentrations of methyl
parathion in a loamy sand soil (pH = 5.3) decreased from a level of
about 5 mg/kg to 0.3 mg/kg during a period of 57 days (Goedicke &
Winkler, 1976). Thirty days following treatment, 3.1% of initial
residues of methyl parathion were found in a soil (clay?) of a field
treated with 5.6 kg/ha (Lichtenstein & Schulz, 1964) (see also section
4.1.3).
During an incubation study under aerobic conditions, methyl
parathion was degraded mainly to CO2 and 4-nitrophenol, and, to a
minor extent, to desmethyl parathion (Möllhoff, 1981). Methyl
parathion may be degraded in the environment by: a ) hydrolysis to
p-nitrophenol and dimethylthiophosphoric acid; or b ) nitro-group
reduction to methyl aminoparathion (e.g., Sharmila et al., 1988).
Hydrolysis can be both chemical and microbial while nitro-group
reduction is essentially microbial. Generally, hydrolysis is the major
pathway in nonflooded soil while methyl parathion is degraded mainly
by nitro-group reduction in predominantly anaerobic systems, such as
flooded soil (Ou et al., 1983; Ou, 1985; Adhya et al., 1987).
In a few instances, hydrolysis is the major or only pathway of
methyl parathion degradation in soils, even under flooded conditions
(Ou, 1985). Adhya et al. (1987) evaluated the influence of different
physical and chemical characteristics on the persistence of methyl
parathion in 5 tropical soils under flooded and nonflooded conditions.
They found that nitro-group reduction was the major pathway of methyl
parathion degradation in 4 out of 5 of the soils under flooded
conditions, while, in one soil (Sukinda-soil), degradation of methyl
parathion proceeded exclusively by hydrolysis, even under flooded
conditions. The latter finding was confirmed by Sharmila et al.
(1989a).
A temperature-dependent shift from nitro-group reduction (at
25 °C) to predominantly hydrolysis (at 35 °C) occurred in a flooded
alluvial soil; both pathways were mediated microbially (Sharmila et
al., 1988). The addition of yeast extract also influenced the
degradation pathway of methyl parathion by bacterial cultures in
enriched flooded alluvial and laterite (Sukinda) soils (Sharmila et
al., 1989b). Low redox potential in a flooded soil favoured
degradation by nitro-group reduction, whereas hydrolysis was
concomitant with a more positive potential (Adhya et al., 1981a).
Adhya et al. (1981b) reported studies on sulfur-containing
anaerobic ecosystems, such as oceanic sediments, which they supposed
could serve as a potential sink for pesticides. They found that methyl
parathion was decomposed in acid, sulfur-containing soils and soils
with a low sulfate content to aminomethyl parathion; however, no
decomposition occurred under aerobic conditions. Demethylation could
be demonstrated in anaerobic sulfate soils.
Evidence for microbial participation was provided by the fact
that sterilization of the enriched soil samples increased the
stability of methyl parathion in soil (Adhya et al., 1981a). The
authors reported a very rapid reduction of the nitro group of methyl
parathion by equilibration with a soil incubated with rice straw under
flooding. Sterilization of this soil preparation prevented this rapid
reduction. The degradation of methyl parathion and its metabolite
p-nitrophenol in flooded alluvial soil is given in Table 12.
It appeared from this study, that the degradation of the
metabolite p-nitrophenol is more rapid than the decomposition of
methyl parathion.
Table 12. Degradation of methyl parathion and its metabolite p-nitrophenol
in flooded alluvial soila
Days after methyl µg of compound recovered/20 g of soil
parathion addition
methyl parathion p-nitrophenol
0 485.3 0
0.5 428.1 trace
1 333.7 120.0
2 219.8 98.6
3 185.6 72.0
6 95.5 0
12 58.2 0
a From: Adhya et al. (1981a).
Isolated mixed bacterial cultures from soil utilized methyl
parathion and parathion as a sole carbon source (Chaudhry et al.,
1988). Pseudomonas sp. was capable of hydrolysing methyl parathion
and parathion to p-nitrophenol but needed another carbon source for
growth. The optimum pH range for enzymatic hydrolysis by this
bacterium was from 7.5 to 9.5. In view of the instability of methyl
parathion in alkaline solutions, it is not clear whether the
hydrolysis noted was or was not partially due to the pH of the
solution rather than wholly due to bacterial action. The thermal
optimum was between 35 °C and 40 °C. Flavobacterium sp. culture was
able to metabolize p-nitrophenol by degrading it to nitrite and to
use it for growth. The DNAs from Pseudomonas sp. and from the mixed
culture showed homology with the organophosphate degradation gene from
a previously reported parathion-hydrolysing bacterium, Flavobacterium
sp. Ou & Sharma (1989) showed that methyl parathion is extensively
degraded by a mixed bacterial culture and a Bacillus sp. to its
final oxidation products carbon dioxide and water, whilst a
Pseudomonas sp. isolated from the mixed culture could degrade the
hydrolysis product p-nitrophenol. A Flavobacterium sp. isolated
from flooded soil was able to hydrolyse methyl parathion, but a
Pseudomonas sp. from flooded soil was not (Adhya et al., 1981c). The
transformation of methyl parathion by pure cultures of Flavobacterium
sp. followed multiphasic kinetics (Lewis et al., 1985).
A different result was described by Arndt et al. (1981) for
microorganisms in compost. They added 70 mg of methyl parathion
dissolved in 20 ml ethyl acetate to 1.2 kg of grass (40%), apples
(23%), potatoes (17%), yoghourt (13%), and bread (7%). After
composting this mixture for 7 days, no degradation product of methyl
parathion was found. The recovery rate was 95%. The authors concluded
that the insecticide could accumulate in the compost under the
conditions tested, but it could not be excluded that this result was
affected by the ethyl acetate.
The concentration of methyl parathion (applied at 0.28 kg/ha) in
a lake (Clear Lake, California, USA) dropped from 0.50 µg/litre to
0.28 µg/litre, measured 8 and 48 h, respectively, after treatment
(Apperson et al., 1976). After a third application (total 3 X 0.28
kg/ha) the residue level of methyl parathion was 5.4 µg/litre, and 7
days later, 2 µg/litre (Apperson et al., 1976). Eichelberger &
Lichtenberg (1971) found that 90% of methyl parathion in river water
was degraded during a period of 2 weeks, whereas there was no
degradation in distilled water. The latter finding may be pH related,
since Cowart et al. (1971) noted 50% hydrolysis of the pesticide after
14 days in distilled water at pH 6. Under field conditions, in the
presence of sediment and aquatic plants, degradation is accelerated
and persistence is lower. Dortland (1980) showed that persistence
decreased by a factor of 2-3 when sediment and plants were added to
the aquatic microcosm. When considering the aquatic ecosystem as a
whole (which includes adsorption on sediments and adsorption on, and
incorporation in, aquatic biota) a fair estimate of the persistence of
methyl parathion in the water column seemed to be 2-3 days (Walker,
1978). This value was recorded in microcosm studies and field
experiments in both freshwater and estuarine aquatic environments.
Predicted half-life values in rivers, ponds, eutrophic lakes, and
oligotrophic lakes were reported to be 0.6, 27.3, 28.3, and 151.6 h,
respectively (Smith et al., 1978). Methyl parathion was degraded with
a half-life of 28 h in sediment collected from a field site and with
a half-life of 7 h in microbial mats derived from laboratory mesocosms
(Newton et al., 1990). The half-lives of methyl parathion in the water
and sediment of a carp pond were 5.7 days and 5.0 days, respectively
(initial residues: 3.77 mg/litre in water and 0.52 mg/kg in soil)
(Sabharwal & Belsare, 1986). It should be emphasized that the
persistence values reported depend not only on the type of biotope but
also on the abiotic conditions, i.e., temperature, pH, and salinity,
as pointed out, for example, by Badawy & El-Dib (1984).
Holm et al. (1983) found in their model ecosystem that the
sediment type had no observable effect on the degradation of methyl
parathion and that it depended primarily on the communities of
microorganisms. These communities and their ability to degrade methyl
parathion did not change with different sediment types. The microbial
degradation rate constants in an aquatic channel microcosmos ranged
from 2.7 X 10-6/s to 6.9 X 10-6/s. This was significantly higher
than the rate constants determined for abiotic degradation. Cripe et
al. (1987) modified the river die-away test for determining the
biodegradability of organic substances and tested the degradation
products for their toxicity. Because of their sensitivity, mysids and
daphnids were used for testing the toxicity of the degradation
products. This test showed a rapid, sediment-mediated biodegradation
of methyl parathion.
The biodegration rate of methyl parathion was compared in 3 types
of test systems composed of sediment and water collected from various
estuarine sites (Van Veld & Spain, 1983). Generally, methyl parathion
degradation was fastest in intact sediment/water cores, followed by
sediment/water shake flasks, and was slowest in water shake flasks.
Lewis & Holm (1981) determined the transformation rate of methyl
parathion by "aufwuchs" microorganisms, i.e., aquatic microbial growth
attached to submerged surfaces or suspended in streamers or mats.
"Aufwuchs" fungi, protozoa, and algae did not transform methyl
parathion, but bacteria rapidly transformed it.
Lewis et al. (1984) examined the effects of microbial community
interactions on methyl parathion transformation rates. They found
either stimulation or inhibition of bacterial transformation rates in
the presence of various cultures, filtrates, or exudates of algae,
fungi, or other bacteria.
The biotic and abiotic degradation rates of methyl parathion in
water and sediment samples over a 3-year period was studied by
Pritchard et al. (1987). The aim of their study was to find the reason
for the different degradation rates reported for methyl parathion, but
the divergences in biodegration could not be assigned to any single
factor. The predominant degradation in an aerobic system appears to be
the biological hydrolysis, producing p-nitrophenol.
Phosphatases are an important group of enzymes involved in the
breakdown of methyl parathion (Portier & Meyers, 1982; Portier et al.,
1983). A proposed pathway for the breakdown of methyl parathion in
aquatic systems is given by Bourquin et al. (1979) in Fig. 1.
Methyl parathion is degraded by bacteria in soil, but more slowly
by bacteria in water. Crossland et al. (1986) estimated the rate of
biodegradation of methyl parathion using a mathematical model.
Sorption on sediment was the dominant process for loss of methyl
parathion from the water compartment. The rate of biodegradation in
sediment (4.0 µmol/litre per h) greatly exceeded that of sorption on
sediment (0.02-0.05 µmol/litre per h) and, therefore, the sediment
compartment may be considered a sink for methyl parathion.
The complete decomposition of methyl parathion into innocuous
compounds can be realized by planktonic and attached microorganisms
(Lassiter et al., 1986). The metabolite p-nitrophenol can be further
metabolized by algae, as reported by Werner & Pawlitz (1978).
4.2.2 Abiotic degradation
Data on the abiotic degradation of methyl parathion are presented
in Table 13.
4.2.2.1 Photodegradation
When exposed to UV radiation or sunlight, methyl parathion
undergoes oxidative degradation. The degradation rate constant of
methyl parathion sprayed as a film (0.67 µg/cm2) and exposed to 300
nm light was reported to be 46.6 X 10-7/s, corresponding to a
half-life of 41.2 h (Chen et al., 1984). In a stationary reactor, the
half-life of methyl parathion dissolved in an aqueous solution (pH=7)
was 72 min after radiation with a Hg low pressure lamp (at 254 nm)
(Hicke & Thiemann, 1987). Methyl parathion has been shown to be one of
the most light-sensitive insecticides. Baker & Applegate (1970, 1974)
showed photodegradation of methyl parathion using light in the
spectral range 300-400 nm (Table 13); methyl paraoxon, the active
cholinesterase inhibitor, was produced. Although photodegradation of
methyl parathion in the terrestrial compartment of the environment may
be important, it plays only a minor role in aquatic media (Env. Res.
Lab., 1981). The first-order transformation rate for photolysis upon
exposure to daylight fluorescent lamps was low compared to hydrolysis
and, in particular, compared to microbial degradation in an aquatic
channel microcosm (Holm et al., 1983). The loss of methyl parathion
through photolysis was estimated to be 4%.
Nevertheless, it seems that sunlight may reduce the half-life of
methyl parathion considerably. Schimmel et al. (1983) reported a
half-life of 6.3 days for a 1 mg methyl parathion/litre solution
exposed to sunlight. In darkness, with the same test conditions, the
half-life was 18 days. Like parathion, the photoreaction of methyl
parathion was accelerated in the presence of green and blue green
algae (Zepp & Schlotzhauer, 1983).
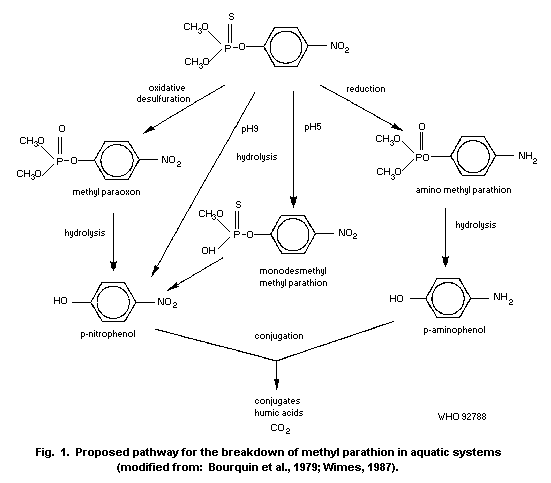 Table 13. Abiotic degradation of methyl parathion
Transformation Time Experimental conditions Light Initial concentration Conversion References
process
temp (°C) pH (mg/litre) (%)
Hydrolysis in 24 h a 6 0.26 8.8 Cowart et al. (1971)
distilled water 7 days a 6 0.26 32.0 Cowart et al. (1971)
14 days a 6 0.26 50.5 Cowart et al. (1971)
21 days a 6 0.26 73.8 Cowart et al. (1971)
28 days a 6 0.26 100 Cowart et al. (1971)
Hydrolysis in 31.7 days 10 1-5 a 50 Mühlmann & Schrader
distilled water 12.5 h 40 1-5 a 50 (1957)
Mühlmann & Schrader
(1957)
Hydrolysis in 8.4 h 70 6 6 50 Ruzicka et al. (1967)
ethanol buffer
Hydrolysis in 4 h 37.5 12 a 64-73 Jaglan & Gunther
0.01 M NaOH (1970)
Table 13 (continued)
Transformation Time Experimental conditions Light Initial concentration Conversion References
process
temp (°C) pH (mg/litre) (%)
UV-degradation 2 h 30 350 nm 0.1 39 Baker & Applegate
of pure product 4 h 30 350 nm 0.1 65 (1974)
6 h 30 350 nm 0.1 82 Baker & Applegate
8 h 30 350 nm 0.1 91 (1974)
Baker & Applegate
(1974)
Baker & Applegate
(1974)
Temperature- 2 h 35 dark 0.1 9 Baker & Applegate
degradation of 4 h 35 dark 0.1 8 (1974)
pure product 6 h 35 dark 0.1 24 Baker & Applegate
8 h 35 dark 0.1 31 (1974)
Baker & Applegate
(1974)
Baker & Applegate
(1974)
a No data given.
Exposure of methyl parathion to sunlight resulted in the
formation of trace levels of O, O, S-trimethyl phosphorothioate and
trimethylphosphate (Chukwudebe et al., 1989).
According to Sauvegrain (1980), methyl parathion seems to be
oxidized by oxidizing agents, i.e., ozone and chlorine. Methyl
parathion treatment with ozone eliminated 80-100% of the compound. The
oxidation of methyl parathion leads to methyl paraoxon, which is
further transformed into p-nitrophenol.
4.2.2.2 Hydrolytic degradation
The half-life of methyl parathion in an aqueous solution (20 °C,
pH 1-5) was reported to be 175 days (Melnikov, 1971). At a
concentration of 0.03 mol/litre (pH 10), sodium perborate greatly
accelerated the degradation of methyl parathion (Qian et al., 1985).
The half-life in the presence of perborate was 12 min, while the rate
was too slow to be measurable when the same concentration of sodium
carbonate was added. Badawy & El-Dib (1984) also found that the
degradation of methyl parathion occurred much more rapidly under
alkaline (pH 8.5) than under neutral (pH 7.0) or acidic (pH 0.5)
conditions. The rate of degradation was also positively correlated
with salinity.
Although chemical hydrolysis occurs in the aquatic environment,
this degradation reaction plays only a limited role in the
disappearance of methyl parathion. In an aquatic channel microcosm,
only 7% of degradation of the pesticide was attributed to chemical
hydrolysis (Holm et al., 1983). In a sterile, seawater-sediment
system, methyl parathion remained for 7 days whereas, in a
corresponding nonsterile system, 100% of the compound was degraded
within this period (Env. Res. Lab., 1981).
Methyl paraoxon, the more toxic oxygen analogue of methyl
parathion is also chemically hydrolysed. According to Jaglan & Gunther
(1970), the chemical hydrolysis of methyl paraoxon is much faster than
that of methyl parathion, because of the presence of oxygen in the
oxon, which makes the phosphorus more susceptible to attack by the
hydroxide ion. At pH 8.5 (37.5 °C), approximately 35% of methyl
paraoxon was hydrolysed within 16 h compared with about 5% for methyl
parathion.
The hydrolysis products of methyl parathion and methyl paraoxon
are dimethyl phosphorothioic acid or dimethyl phosphoric acid and
p-nitrophenol. These compounds are less toxic than the parent
compounds, thus hydrolysis is detoxifying (Thuma et al., 1983).
Pritchard et al. (1987) reported that there was no biotic
degradation of methyl parathion in seawater, i.e., "the rate resulting
from the substraction of the sterile rate from the nonsterile rate was
not significantly different from zero".
Several research groups investigated the binding of methyl
parathion on soils as well as the soil catalysed degradation of methyl
parathion. Saltzman et al. (1976) and Mingelgrin et al. (1977)
analysed the influences of different water contents and cations on the
kaolinite-catalysed degradation of methyl parathion; when adsorbed on
kaolinite, methyl parathion seems to be more stable than parathion.
A concentration of 10% Ca-kaolinite catalysed the degradation of
methyl parathion most efficiently.
Wolfe et al. (1986) studied the influences of pH and redox
transformations on the detoxification of methyl parathion in soils,
quantitatively. The disappearence of methyl parathion could be
described by first-order kinetics. Amino methyl parathion was
identified as a reaction product. Half-lives in the range of a few
minutes were measured in strongly reducing sediments, thus, confirming
the data of Gambrell et al. (1984). It was suggested that more
information about the effect of sediment sorption was needed for
further studies on the reaction kinetics.
4.2.3 Bioaccumulation
Temporary accumulation (up to 10 days) occurred following an
aerial spraying of pine and deciduous forest with methyl parathion (3
kg 20% solution/ha in April and 1 kg 40% solution/ha in September),
which led to higher levels of methyl parathion in the tissues of a
variety of vertebrates compared with the concentrations in soil,
water, and plants (Fedorenko et al., 1981).
Takimoto et al. (1984) reported bioaccumulation of methyl
parathion in killifish (Oryzias latipes). Bioaccumulation factors
of 88-fold (postlarva) to 540-fold (female adult) were found in the
killifish. Residues in the bluegill sunfish (Lepomis macrochirus),
exposed to methyl parathion treatments in a lake, varied from 11 to
110 µg/kg, corresponding to bioaccumulation factors of 28-39 (Apperson
et al., 1976).
Sabharwal & Belsare (1986) added 4 mg methyl parathion/litre to
the water of a carp-rearing pond and measured the methyl parathion
concentrations in the water, soil, macrophytes, and carps over a
period of 35 days. The methyl parathion limits of detection in water,
soil, macrophytes, and fish were 0.0066, 0.12, 0.0478, and 0.0746
mg/kg respectively (see Table 14).
There was an accumulation of methyl parathion in the soil,
macrophytes, and fish, whereas the compound degraded immediately in
water. The bioaccumulation in the carp peaked at 3 days.
Table 14. The persistence of methyl parathion in water, soil,
macrophytes, and fisha
Time (days) methyl parathion concentration (mg/kg)
water soil macrophytes fish
0 3.77 0.52 1.2 0.52
1 3.15 2.28 14.41 10.26
3 2.16 1.5 11.73 26.17
7 1.50 - 8.98 11.74
14 0.60 - 4.16 5.67
21 0.28 - 2.24 2.06
28 ndb ndb 1.42 0.83
35 ndb ndb 0.73 0.48
a From: Sabharwal & Belsare (1986).
b nd = not detectable.
Using a mean Kow value of 2.55, and on the basis of the log
Kow/log bio-concentration regression curve for fathead minnows, the
estimated bioconcentration factor was reported to be 22 (Env. Res.
Lab., 1981). According to Zitko & McLeese (1980), the expected
bioconcentration factor in aquatic biota for methyl parathion is
estimated to be 20.
Crossland & Bennett (1984) using a range of published log Kow
values estimated that bioaccumulation factors would be between 2.5 and
84.
Accumulation of methyl parathion does not occur in the blood of
mammals. After ingestion, it is rapidly absorbed and the blood
concentration reaches a maximum 1-3 h following ingestion and,
thereafter, decreases. Although a significant portion of methyl
parathion is found in the bile, it is present in all organs (see also
section 6.2).
4.3 Interaction with other physical, chemical, and biological
factors
Methyl parathion shows interactions with the following
substances: adrenocorticoids, anaesthetics, tricyclic antidepressive
agents, antihistamines, atropine, barbiturates, clofibrate,
colistimethate, corticosteroids, curare, decamethonium, dexpanthenol,
fluorophosphate, hexamethonium, kanamycin, morphine, muscle relaxants,
anticholinesterases, neomycin, parasympathomimetics, phenothiazines,
polymyxin, pralidoxime, procainamide, streptomycin, succinylcholin,
sympathomimetics, d-tubocurarine (Martin, 1978).
A significant increase in the toxicity of oxygen analogues of
organophosphorus insecticides to house flies was observed following
treatment with polychlorinated biphenyl (PCB) (Aroclor 1248)
(Fuhremann, 1980). Detergents increased the hydrolysis of
organophosphates, such as methyl parathion (Peterka & Cerna, 1988).
Yang & Sun (1977) found an inversely proportional correlation between
fish toxicity and the partition coefficient of different insecticides,
including methyl parathion.
DEF ( S,S,S-tributyltrithiophosphate), a defoliant, enhanced the
toxic effect of methyl parathion in the fish (Gambusia affinis)
(Fabacher, 1976).
4.4 Ultimate fate following use
The ultimate fate of methyl parathion depends on the degradation
pathways. The most important one is chemical as well as biological
hydrolysis; the others are oxidative desulfurisation, nitro reduction,
and photodegradation. Important degradation products are methyl
paraoxon, dimethylthiophosphoric acid, dimethylphosphoric acid, and
p-nitrophenol.
5. ENVIRONMENTAL LEVELS AND HUMAN EXPOSURE
5.1 Environmental levels
5.1.1 Air
In a pilot study, Stanley et al. (1971) measured methyl parathion
concentrations of up to 129 ng/m3 in air samples collected in the
USA (Stoneville). The technique used for air sampling was that of
Miles et al. (1970).
In Tennessee, USA, average hourly concentrations of methyl
parathion in air were < 0.57 ng/m3 (maximum, 2.9 ng/m3) at a site
located one mile south-east of a methyl parathion plant and one mile
west of a plant producing the nematocide ethoprophos
( O-ethyl- S,S-dipropyl phosphorodithioate), and < 0.64 ng/m3
(maximum, 5.1 ng/m3) at another site located one mile north of a
methyl parathion plant. Particulate samples collected from the 2 sites
contained < 0.086 ng methyl parathion/m3 (Foster, 1974).
In the USA, maximum atmospheric levels were detected of 29.6
ng/m3 in Alabama, 5.4 ng/m3 in Florida, and 129 ng/m3 in
Mississippi (Midwest Research Institute, 1975). Methyl parathion was
found in air samples in the Mississippi Delta, one of the highest
pesticide usage areas in the USA, because of the intensive cotton
production, at a maximal concentration of 2060 ng/m3 (Arthur et al.,
1976). The average monthly concentrations of methyl parathion peaked
in August or September with levels varying from 111.7 ng/m3
(September 1972) to 791.1 ng/m3 (September 1973).
In another study, airborne residues of methyl parathion and
methyl paraoxon were determined after the use of methyl parathion on
rice in the Sacramento valley in California, USA (Seiber et al.,
1989). Sampling was conducted on the roof tops of public buildings in
4 towns in 2 counties where methyl parathion was used in significant
quantities, and in a reference area where no use occurred. Daily
maximum average concentrations were 25.7 ng/m3 for methyl parathion
and 3.1 ng/m3 for methyl paraoxon. The range in averages for all
sites in the vicinity of usage during springtime 1986 was 0.2-6.2
ng/m3 for methyl parathion and < 0.5-0.8 ng/m3 for methyl
paraoxon. With one exception, the background samples did not show any
methyl parathion above the detection limit.
Methyl parathion and methyl paraoxon concentrations measured in
the condensate from coastal fog near Monterey (California, USA) ranged
between 0.046 and 0.43 µg/litre and between 0.039 and 0.49 µg/litre,
respectively. The oxon to thion ratios were 0.28-2.6, and thion to
oxon conversion appeared to take place during atmospheric transport
from agricultural to the nonagricultural areas (Schomburg et al.,
1991).
In the Kalinin District, Tashkent Province, the Uzbek SSR (USSR),
during July and August, the concentrations of methyl parathion in the
air after spraying with 30% emulsion, measured at 500, 750, and 1000
m from the place of the treatment, were 0.055- 0.08, 0.01-0.02, and
0-0.008 mg/m3, respectively (Akhmedov, 1968).
Tessari & Spencer (1971) analysed indoor and outdoor air samples,
collected monthly for a year, at the homes of families where the head
of the household was occupationally exposed to pesticides. A nylon
chiffon cloth screen was exposed to the atmosphere for 5 days and the
absorbed pesticides were extracted and analysed using a column
chromatography method. The authors found methyl parathion in 13 out of
52 samples, at an average concentration of 1.04 µg/m3. The range was
0.04-9.4 µg/m3. The values obtained from outdoor sampling were much
smaller, 3 out of 53 samples containing 0.35 µg methyl parathion/m3
with a range of 0.15-0.71 µg/m3.
5.1.2 Water
Methyl parathion concentrations of up to 0.23 µg/litre were found
in selected Western streams of the USA in 1968-71 (Schulze et al.,
1973).
In 1970, methyl parathion was detected in 3 out of 18 surface
drain effluent water samples in California, USA, at concentrations of
10-190 ng/kg, and, in 8 out of 60 subsurface drain effluent water
samples, at concentrations of 10-170 ng/kg (Midwest Research
Institute, 1975).
In water samples from 10 sites in the Cape Fear River Basin in
North Carolina, USA, taken monthly between July 1974 and June 1975
(except October), maximum concentrations of methyl parathion in
dissolved fractions and in particulate-associated fractions were 468
ng/litre and 123 ng/litre, respectively (Pfaender et al., 1977).
Methyl parathion was detected in waste water from a parathion
production plant in the USA at levels of 2.0 mg/litre in pre-treatment
water and < 0.004 mg/litre in post-treatment water (Marcus et al.,
1978).
Methyl parathion residues in major Mississippi stream systems
(USA), monitored during 1972-73, ranged between 0.08 and 0.46 µg/litre
(Leard et al., 1980).
In one station at the Negro River Basin (Argentina), methyl
parathion was detected at a concentration of 0.034 µg/litre in March
1986, which is the end of the summer season in South America (Natale
et al., 1988).
In a study on the Ionnina basin and Kalamas river (Greece), from
September 1984 to October 1985, a seasonal fluctuation was found in
the concentration of methyl parathion, with a maximum during the
summer and a minimum during the winter (Albanis et al., 1986). The
mean concentration in the lake Pamvotis (Greece) was 7.7 ng methyl
parathion/litre in July. The natural outlet of the lake is the Kalamas
River, where a maximum concentration of 32 ng methyl parathion/litre
was found. With the exception of the river, the other analyses showed
much lower concentrations of methyl parathion. The results of this
study show very clearly the seasonal influence of the application of
this pesticide on natural water concentrations.
Normally, the methyl parathion concentration in the River Rhine
is below the limit of detection and the Sandoz accident on 1 November
1987 did not affect the wells of the waterworks. A maximum value
measured in the Rhine during the second half of 1986 was higher (<
0.05 mg/m3) than that following this accident (Winter & Lindner,
1987).
Methyl parathion was detected in Hungarian surface waters only
once between 1977 and 1986 (concentration not given), which
corresponded to a sampling frequency of 0.14% (Csernatoni et al.,
1988).
5.1.3 Soil
In 1969, 76 samples of onions and the soils in which they had
been grown were collected in the 10 major onion-producing states of
the USA for analysis of the pesticide residues. The limit of
quantification of methyl parathion was 0.01 mg/kg. Methyl parathion
was found in a range of 0.09-1.9 mg/kg in 11.8% of the soil samples.
No residues were detected in the onion samples (Wiersma et al., 1972).
Methyl parathion was found at levels of 0.09-1.90 mg/kg in soil
samples from onion-producing States in the USA (Midwest Research
Institute, 1975). In cropland soil (South Dakota, USA), the
concentration of methyl parathion was 0.01 mg/kg soil (Carey et al.,
1979).
5.1.4 Food
Renvall et al. (1975) reported pesticide analyses of fruits and
vegetables on the Swedish market from July 1967 to April 1973. Methyl
parathion belonged to the most frequently occurring pesticides with a
rate of 6%. Levels in 4 out of 207 oranges analysed, 1 out of 37
lemons, 4 out of 69 grapefruits, and, 2 out of 29 clementines or
mandarins exceeded 0.11 mg/kg. In a more recent study, in the Swedish
monitoring programme during the period 1981-84, methyl parathion was
found in apples, celery, grapes, lemons, lettuce, limes, mandarins,
oranges, pears, and plums. One out of 74 celeries analysed (imported),
1 out of 238 lemons (imported), 1 out of 248 lettuces (domestic), 5
out of 421 mandarins (imported), and, 8 out of 917 oranges (imported)
exceeded the Swedish maximum residue limits of 0.1-0.5 mg methyl
parathion/kg (Andersson, 1986).
In a study on the presence of organophosphorus insecticide
residues in Mexican food, methyl parathion residues were found in
market samples of avocados, rice, strawberries, and tomatoes, with
respectively 6, 4, 3, and 5 positive samples out of 10. The average
concentrations were 0.3, 0.8, 0.5, and 0.5 mg/kg, respectively (Albert
et al., 1979).
A report on pesticide residues in the United Kingdom (1982-85)
gave a residue level for methyl parathion in lemons of 0.3 mg/kg
(MAFF, 1986). In a more recent report, no methyl parathion was found
in cooking apples and in imported apples with a reporting limit of
determination of 0.1 mg/kg; however, a concentration of 0.08 mg methyl
parathion/kg was found in one sample of lemons from Spain (MAFF,
1990).
Methyl parathion was detected in citrus fruits in France at
levels of 0.003-1.25 mg/kg (Mestres et al., 1977). Lamontagne (1978)
found methyl parathion in concentrations of 0.311 mg/kg in fruit and
0.87-2.12 mg/kg in greenhouse plants in France. Branca & Quaglino
(1988) found methyl parathion at a residue level of 0.036 mg/kg in one
out of 34 samples of French potatoes imported into Italy.
Pesticide residue levels were analysed during 1968-69 in samples
of ready-to-eat foods from 30 markets in 24 different cities with
populations of between 50 000 and more than 1 000 000 in the USA. The
limit of determination was 0.05 mg/kg. Methyl parathion was found
infrequently (1 X Boston, 1 X Los Angeles, 2 X Minneapolis) in
concentrations of 0.008, traces, 0.001, and 0.025 mg/kg in leafy
vegetables and 0.033 mg/kg in grain (Boston) (Corneliussen, 1970).
From June 1971 to July 1972, methyl parathion was detected in 7 out of
420 samples of ready-to-eat foods. The concentrations found in leafy
vegetables ranged from a trace to 0.010 mg/kg. In one sample of fruit
(type not given), a concentration of 0.007 mg/kg was found (Boston)
(Manske & Johnson, 1975). In the report of the Food and Drug
Administration, 5 samples of leafy vegetables containing methyl
parathion residues are mentioned. The concentrations ranged from a
trace to 0.003 mg/kg (Johnson & Manske, 1976). In "market-basket"
surveys conducted by the US Food and Drug Administration in 1966-69,
methyl parathion was detected in leafy and stem vegetables at levels
of 0-2.00 mg/kg, and, in root vegetables, at levels of 0-1.0 mg/kg
(Midwest Research Institute, 1975). Johnson et al. (1981) did not find
any methyl parathion in infant and toddler Total Diet Studies (TDS) in
the USA in 1975-76. In the adult TDS in the USA in 1973-74, trace
residue levels were found in leafy vegetables, but none in fruit
(Manske & Johnson, 1977). "Dislodgable" methyl parathion residues were
found on sweet corn in the USA at levels of 0-0.14 µg/cm2, one and
two days after application of the pesticide (Wicker et al., 1979).
Soybeans analysed in 1979 showed levels of 1-40 mg methyl parathion/kg
and soybean forage analysed at intervals of 1-14 days after treatment,
0.3-6.6 mg methyl parathion/kg. Levels of 0.1-0.3 mg methyl
parathion/kg were measured in 12 samples of cottonseed (FAO, 1985).
Samples of standing agricultural crops were analysed in 1971
during the National Pesticide Monitoring Programme in the USA (Carey
et al., 1978). Levels of methyl parathion detected in samples of
alfalfa, field orn (kernels), cotton, cotton stalks, and mixed hay
ranged from 0.02 to 4.57 mg/kg dry weight.
During a TDS in Canada in 1972, Smith et al. (1975) found methyl
parathion residues in leafy vegetables from Winnipeg at an average
level of 0.012 mg/kg.
In a TDS in New Zealand during 1971-73, methyl parathion was
found in one sample of leafy vegetables at a level of 0.15 mg/kg in
1973, in one sample of root vegetables at the level of 0.26 mg/kg in
1972, and in 4 samples of citrus fruit at an average level of 0.20
mg/kg and a maximum level of 1.4 mg/kg during each of the years
1971-73. In 1971, 3 samples of pip fruit contained, on average, 0.03
mg/kg, and, in 1972, one sample of stone fruit contained 0.25 mg/kg.
Some of these figures exceeded the New Zealand tolerances (Love et
al., 1974). In 1974, methyl parathion was detected at levels of
0.003-0.007 mg/kg in fruit and 0.002-0.008 mg/kg in tinned food from
Auckland and Wellington, New Zealand (Dick et al., 1978).
The loss of methyl parathion in food during heating and storage
was confirmed by Elkins et al. (1972). The samples were analysed
before, and after, standardized heat treatment. Spinach and apricots
were fortified separately with methyl parathion. The spinach samples
were heated for 66 min at 122 °C and the apricot samples were heated
for 50 min at 103 °C. The initial concentration of methyl parathion in
the spinach samples was 0.88 mg/kg. It disappeared completely after
heating. The methyl parathion level in the apricot samples was 0.85
mg/kg, but this decreased to 46% of this level after heating. The
detection limit was less than 0.005 mg/kg. A further decomposition can
be expected during the storage of preserved food. Generally, methyl
parathion residues in fruit decomposed very rapidly, except in the
waxy skin of apples and in the oil vessels of olives (Stoll, 1982).
Rippel et al. (1970) found remarkable differences in the
degradation of methyl parathion in packaged citrus juice, depending on
the kind of package surface. The rate of decrease of the methyl
parathion residues was insignificant in glass containers. It was
substantially higher in packages with tin-layer surfaces than in
packages with painted protective surfaces, since the tin layers
reduced the nitro group of the methyl parathion.
5.1.5 Terrestrial and aquatic organisms
Methyl parathion is rapidly metabolized in most organisms,
resulting in low bioconcentration factors after acute exposure. There
are few studies of residues of methyl parathion in organisms in the
environment, but those conducted have consistently shown low methyl
parathion residues.
Methyl parathion was detected in tissue samples from estuarine
fish at a mean level of 47 µg/kg (Butler & Schutzmann, 1978). It has
been detected at a concentration of 59 µg/kg in the ovaries of spotted
sea trout (Cynoscion nebulosus), collected in Texas, USA (Midwest
Research Institute, 1975).
Methyl parathion was detected in 34 out of 55 suspectedly
poisoned apiaries examined in Connecticut (USA) in 1983-85 (Anderson
& Wojtas, 1986). Concentrations of methyl parathion found in dead bees
and in brood comb ranged from 0.04 to 5.8 mg/kg.
5.2 General population exposure
The general population can come into contact with methyl
parathion via air, water, or food. Average methyl parathion intake
from food in the USA during 1988 was estimated to range from 0.1 to
0.2 ng/kg per day in 3 different age groups (FDA, 1989). Draper &
Street (1981) estimated that a 70-kg male living in a residence
adjacent (50 yards) to an alfalfa field sprayed with methyl parathion
at a rate of 0.19 kg a.i./ha would be exposed to a total dermal dose
of 0.38 mg. Within a pesticide monitoring programme in the USA, based
on the analysis of 6990 samples collected from the general population
via the National Center for Health Statistics 1976-80,
para-nitrophenol as an indicator for exposure to methyl and ethyl
parathion was detected in 2.4% of urine samples from 12 to 74-year-old
persons (Carey & Kutz, 1985).
5.3 Occupational exposure during manufacture, formulation, or use
There is a special risk for farm workers, since incidents of
poisonings and illnesses during the mixing, loading, and application
of methyl parathion have been reported. Exposure may also occur during
the cleaning and repair of equipment and during early re-entry into
fields. According to NIOSH (1976), 150 000 workers in the USA (field
workers, aerial application personnel, mixer and blender operators,
tractor tank loaders, ground applicator vehicle drivers, field
inspectors, and warehouse personnel) are conceivably exposed to methyl
parathion. A maximum air concentration of methyl parathion was
estimated to be 1.77 µg/m3. The exposure to methyl parathion was
estimated by Hayes (1971) for workers checking cotton for insect
damage as 0.7 mg/h via skin contact and < 0.01 mg/h through
inhalation (NIOSH, 1976).
Davis et al. (1981) estimated that workers in apple orchards
sprayed with methyl parathion would be exposed to dermal doses ranging
from 0.055 mg to 3.1 mg, with the amount varying with time after
spraying and the formulation of the pesticide. Two field studies were
carried out by Kummer & Van Sittert (1986) to evaluate the health risk
for the farm workers. In a number of cases, the men involved in
hand-held ULV-spraying wore very little clothing and did not stop
spraying, when it was too windy. Another possible contamination risk
was the filling of bottles from larger (25-litre) containers, and the
repairing and cleaning of the equipment with unprotected hands.
However, no signs of acute poisoning could be observed in any of the
persons involved in these studies. The urine was collected in spot
samples in one of the studies and in 24-h samples in the other.
Methyl parathion absorption could be verified from its metabolites in
the spraymen's urine. Average levels of urinary nitrophenol (mg/g
creatinine) for 6 supervisors and 2 groups of sprayers were reported
to be 0.08 (range of 0.05-0.20), 0.38 (range of 0.04-1.38), and 0.13
(range of 0.06-0.44), respectively. An intake of 0.4-13 mg methyl
parathion was calculated from the excreted p-nitrophenol.
Since investigations showed that clothing worn by agricultural
workers became contaminated with methyl parathion following
application and that the laundering of contaminated clothing with
uncontaminated fabrics resulted in the transfer of the methyl
parathion residue, recommendations were made that contaminated fabrics
should not be washed with regular family laundry. Suggestions for the
procedure of laundering were made by Easley et al. (1981) and Laughlin
et al. (1981). The most effective procedure was using a pre-rinse
programme and a detergent together with sodium hypochlorite (NaOCl) as
a bleach. Laughlin & Gold (1989) discussed further aspects of
laundering protective clothing contaminated with methyl parathion.
Fluorocarbon soil repellent finishes on such protective clothing
decrease pesticide absorption, but may hinder pesticide removal in
laundering. Storage of laundered garments at 20 °C with air flow
and/or at high humidity levels was recommended to dissipate residues
of methyl parathion.
Ware et al. (1974b) suggested that serum insecticide levels,
serum and red blood cell cholinesterase activities, and urinary
excretion of p-nitrophenol should be investigated, because they are
more effective for evaluating the possible potential poisoning hazard
than the analysis of skin and clothing contamination. The safety of
re-entering cotton fields 24 h following application of methyl
parathion was tested. Methyl parathion was applied at 1.12 kg a.i./ha.
During the application, the temperature ranged from 30 to 38 °C. The
foliar residues decreased from 1.6 mg/m2, 24 h following methyl
parathion treatment, to 0.9 mg/m2, 6 h later. No methyl parathion
was detectable in the serum of the volunteers. The 48-h urinary
excretion of p-nitrophenol ranged from 0.15 to 1.20 mg. Serum
cholinesterase levels varied within normal intervals whereas the red
blood cell cholinesterase levels showed a temporary, but not
pronounced, depression of about 5-7%. The amounts of methyl parathion
and methyl paraoxon extracted from clothing and hand surfaces are
shown in Table 15.
During the working period, the mean air concentration was 0.2 ng
methyl parathion/litre, of which, 1.2 µg methyl parathion was inhaled
over 5 h. From all these data, it was concluded that a 24-h interval
is safe for methyl parathion in this form of application.
Munn et al. (1985) collected human exposure samples from workers
and dependants wearing nylon gloves, as well as environmental samples,
during the onion harvest season of 1982 in Colorado, USA. Children in
agricultural settings normally accompany their parents to the fields,
as part of a family unit, the young children playing in this
environment and older children helping their parents in the fields.
Munn et al. (1985) recorded the length of time the gloves were worn,
and the age and sex of the participants. No association between age
and methyl parathion levels was found. The urine samples collected
prior to their leaving the field did not contain detectable levels of
methyl parathion. This could be because the nylon gloves reduced the
absorption of organophosphate residues by about 90%.
Table 15. Extracted residues of methyl parathion and methyl paraoxon
following a 5-h working perioda
Extract from: Methyl parathion Methyl paraoxon
residue (mg) residue (mg)
Hands 0.2 0.5
Shirts 0.2 4.0
mep.5s 1.7 39.0
a From: Ware et al. (1974b).
6. KINETICS AND METABOLISM
6.1 Absorption
Methyl parathion can be absorbed through the digestive tract, the
skin, and the respiratory tract (White-Stevens, 1971).
The primary routes of exposure are via skin contact with
contaminated plants or material, and via inhalation. Severe accidental
intoxications of humans have occurred.
The absorption of methyl parathion from the digestive tract is
rapid, and it appears in the bloodstream immediately after oral
intake. Studies on guinea-pigs were performed to analyse the rate of
absorption of radioactive labelled (32P) methyl parathion. One
minute after dosage, it could be detected in various organs. The
maximum level was found 1-2 h after treatment. The liver showed a
remarkably high concentration (Gar et al., 1958).
Miyamoto et al. (1963) administered 50 mg 32P-labelled methyl
parathion/kg body weight to guinea-pigs or 1.5 mg/kg body weight to
rats, by stomach tube. Maximum concentrations in the blood and brain
were reached 1-3 h after treatment. An oral dose of 50 mg methyl
parathion/kg resulted in no detectable levels of methyl parathion in
either the brain or blood after 3 min, but, after 6-8 min, at which
point lethal effects occurred, levels of methyl parathion increased to
182 ng/ml in plasma and to 137 ng/g in brain (Yamamoto et al., 1981).
6.2 Distribution
Accumulation of methyl parathion was observed in tissues. The
highest concentrations were found in the lung and the liver (NRC,
1977). Transplacental transport of methyl parathion is discussed in
section 8.5.
Total radioactive residues recovered in the 12 tissues analysed
(excluding the gastrointestinal tract) from rats given a single oral
dose of 5 mg C-14-methyl parathion/kg body weight were about 11% of
the administered dose, 1 h after treatment, declining to 0.3% at 24 h,
about 0.1% at 48 h, and to only 0.04%, 6 days later. The kidney had
the highest relative activity up to 8 h after treatment. The
14C-activity in the plasma was initially about 5 times higher than
that in the erythrocytes. However, from day 2 to day 6 after dosing,
the 14C-activity in the erythrocytes was greater than that in plasma
and remained constant (Weber et al., 1979).
Sultatos et al. (1990) measured the partition coefficient for
methyl parathion between mouse liver and blood by either equilibrium
dialysis or a perfusion technique and obtained values of 9.5 and 16.4
respectively.
In a kinetic study on mongrel dogs of both sexes, Braeckman et
al. (1980) found a rapid decrease in serum methyl parathion
concentrations during the first few hours. The authors injected methyl
parathion intravenously in doses of 1, 3, 10, and 30 mg/kg body
weight. The dogs were pretreated with 1-5 mg atropine/kg body weight,
10 min before injecting methyl parathion. The blood samples were taken
for up to 160 h. Besides quantifying serum levels of methyl parathion,
the authors also measured serum cholinesterase activity at the 2
highest concentrations of methyl parathion. The determination of serum
methyl parathion concentrations was performed according to De Potter
et al. (1978). The cholinesterase activity decreased within 30 min to
its lowest value, i.e., 40% of the normal level in dogs receiving 10
mg/kg body weight and 25% in dogs receiving 30 mg/kg. The first rapid
fall in the methyl parathion concentration after injection was due to
distribution and elimination. A slower decrease in serum methyl
parathion concentrations at higher doses was the result of dee
compartment linear kinetics. This is in line with observations of
Tilstone et al. (1979), who found a rebound effect after a
haemoperfusion.
6.3 Metabolic transformation
Organic nitro compounds, orally administered to ruminants, will
undergo reduction of the nitro groups to amino groups. This reaction
takes place in the rumen (Karlog et al., 1978).
The metabolism of methyl parathion in rodents is illustrated in
Fig. 2.
Because of the importance of a first pass through the liver for
the metabolism of methyl parathion, there is a distinct difference
between the oral and intravenous toxicity (Morgan et al., 1977;
Braeckman et al., 1983). Conversion of methyl parathion to its toxic
metabolite, methyl paraoxon, may occur within minutes following oral
administration (Yamamoto et al., 1983).
Mouse liver, perfused with methyl parathion, released the toxic
metabolite methyl paraoxon into the effluate. Mouse whole blood
rapidly detoxified the methyl paraoxon formed (Sultatos, 1987).
Table 13. Abiotic degradation of methyl parathion
Transformation Time Experimental conditions Light Initial concentration Conversion References
process
temp (°C) pH (mg/litre) (%)
Hydrolysis in 24 h a 6 0.26 8.8 Cowart et al. (1971)
distilled water 7 days a 6 0.26 32.0 Cowart et al. (1971)
14 days a 6 0.26 50.5 Cowart et al. (1971)
21 days a 6 0.26 73.8 Cowart et al. (1971)
28 days a 6 0.26 100 Cowart et al. (1971)
Hydrolysis in 31.7 days 10 1-5 a 50 Mühlmann & Schrader
distilled water 12.5 h 40 1-5 a 50 (1957)
Mühlmann & Schrader
(1957)
Hydrolysis in 8.4 h 70 6 6 50 Ruzicka et al. (1967)
ethanol buffer
Hydrolysis in 4 h 37.5 12 a 64-73 Jaglan & Gunther
0.01 M NaOH (1970)
Table 13 (continued)
Transformation Time Experimental conditions Light Initial concentration Conversion References
process
temp (°C) pH (mg/litre) (%)
UV-degradation 2 h 30 350 nm 0.1 39 Baker & Applegate
of pure product 4 h 30 350 nm 0.1 65 (1974)
6 h 30 350 nm 0.1 82 Baker & Applegate
8 h 30 350 nm 0.1 91 (1974)
Baker & Applegate
(1974)
Baker & Applegate
(1974)
Temperature- 2 h 35 dark 0.1 9 Baker & Applegate
degradation of 4 h 35 dark 0.1 8 (1974)
pure product 6 h 35 dark 0.1 24 Baker & Applegate
8 h 35 dark 0.1 31 (1974)
Baker & Applegate
(1974)
Baker & Applegate
(1974)
a No data given.
Exposure of methyl parathion to sunlight resulted in the
formation of trace levels of O, O, S-trimethyl phosphorothioate and
trimethylphosphate (Chukwudebe et al., 1989).
According to Sauvegrain (1980), methyl parathion seems to be
oxidized by oxidizing agents, i.e., ozone and chlorine. Methyl
parathion treatment with ozone eliminated 80-100% of the compound. The
oxidation of methyl parathion leads to methyl paraoxon, which is
further transformed into p-nitrophenol.
4.2.2.2 Hydrolytic degradation
The half-life of methyl parathion in an aqueous solution (20 °C,
pH 1-5) was reported to be 175 days (Melnikov, 1971). At a
concentration of 0.03 mol/litre (pH 10), sodium perborate greatly
accelerated the degradation of methyl parathion (Qian et al., 1985).
The half-life in the presence of perborate was 12 min, while the rate
was too slow to be measurable when the same concentration of sodium
carbonate was added. Badawy & El-Dib (1984) also found that the
degradation of methyl parathion occurred much more rapidly under
alkaline (pH 8.5) than under neutral (pH 7.0) or acidic (pH 0.5)
conditions. The rate of degradation was also positively correlated
with salinity.
Although chemical hydrolysis occurs in the aquatic environment,
this degradation reaction plays only a limited role in the
disappearance of methyl parathion. In an aquatic channel microcosm,
only 7% of degradation of the pesticide was attributed to chemical
hydrolysis (Holm et al., 1983). In a sterile, seawater-sediment
system, methyl parathion remained for 7 days whereas, in a
corresponding nonsterile system, 100% of the compound was degraded
within this period (Env. Res. Lab., 1981).
Methyl paraoxon, the more toxic oxygen analogue of methyl
parathion is also chemically hydrolysed. According to Jaglan & Gunther
(1970), the chemical hydrolysis of methyl paraoxon is much faster than
that of methyl parathion, because of the presence of oxygen in the
oxon, which makes the phosphorus more susceptible to attack by the
hydroxide ion. At pH 8.5 (37.5 °C), approximately 35% of methyl
paraoxon was hydrolysed within 16 h compared with about 5% for methyl
parathion.
The hydrolysis products of methyl parathion and methyl paraoxon
are dimethyl phosphorothioic acid or dimethyl phosphoric acid and
p-nitrophenol. These compounds are less toxic than the parent
compounds, thus hydrolysis is detoxifying (Thuma et al., 1983).
Pritchard et al. (1987) reported that there was no biotic
degradation of methyl parathion in seawater, i.e., "the rate resulting
from the substraction of the sterile rate from the nonsterile rate was
not significantly different from zero".
Several research groups investigated the binding of methyl
parathion on soils as well as the soil catalysed degradation of methyl
parathion. Saltzman et al. (1976) and Mingelgrin et al. (1977)
analysed the influences of different water contents and cations on the
kaolinite-catalysed degradation of methyl parathion; when adsorbed on
kaolinite, methyl parathion seems to be more stable than parathion.
A concentration of 10% Ca-kaolinite catalysed the degradation of
methyl parathion most efficiently.
Wolfe et al. (1986) studied the influences of pH and redox
transformations on the detoxification of methyl parathion in soils,
quantitatively. The disappearence of methyl parathion could be
described by first-order kinetics. Amino methyl parathion was
identified as a reaction product. Half-lives in the range of a few
minutes were measured in strongly reducing sediments, thus, confirming
the data of Gambrell et al. (1984). It was suggested that more
information about the effect of sediment sorption was needed for
further studies on the reaction kinetics.
4.2.3 Bioaccumulation
Temporary accumulation (up to 10 days) occurred following an
aerial spraying of pine and deciduous forest with methyl parathion (3
kg 20% solution/ha in April and 1 kg 40% solution/ha in September),
which led to higher levels of methyl parathion in the tissues of a
variety of vertebrates compared with the concentrations in soil,
water, and plants (Fedorenko et al., 1981).
Takimoto et al. (1984) reported bioaccumulation of methyl
parathion in killifish (Oryzias latipes). Bioaccumulation factors
of 88-fold (postlarva) to 540-fold (female adult) were found in the
killifish. Residues in the bluegill sunfish (Lepomis macrochirus),
exposed to methyl parathion treatments in a lake, varied from 11 to
110 µg/kg, corresponding to bioaccumulation factors of 28-39 (Apperson
et al., 1976).
Sabharwal & Belsare (1986) added 4 mg methyl parathion/litre to
the water of a carp-rearing pond and measured the methyl parathion
concentrations in the water, soil, macrophytes, and carps over a
period of 35 days. The methyl parathion limits of detection in water,
soil, macrophytes, and fish were 0.0066, 0.12, 0.0478, and 0.0746
mg/kg respectively (see Table 14).
There was an accumulation of methyl parathion in the soil,
macrophytes, and fish, whereas the compound degraded immediately in
water. The bioaccumulation in the carp peaked at 3 days.
Table 14. The persistence of methyl parathion in water, soil,
macrophytes, and fisha
Time (days) methyl parathion concentration (mg/kg)
water soil macrophytes fish
0 3.77 0.52 1.2 0.52
1 3.15 2.28 14.41 10.26
3 2.16 1.5 11.73 26.17
7 1.50 - 8.98 11.74
14 0.60 - 4.16 5.67
21 0.28 - 2.24 2.06
28 ndb ndb 1.42 0.83
35 ndb ndb 0.73 0.48
a From: Sabharwal & Belsare (1986).
b nd = not detectable.
Using a mean Kow value of 2.55, and on the basis of the log
Kow/log bio-concentration regression curve for fathead minnows, the
estimated bioconcentration factor was reported to be 22 (Env. Res.
Lab., 1981). According to Zitko & McLeese (1980), the expected
bioconcentration factor in aquatic biota for methyl parathion is
estimated to be 20.
Crossland & Bennett (1984) using a range of published log Kow
values estimated that bioaccumulation factors would be between 2.5 and
84.
Accumulation of methyl parathion does not occur in the blood of
mammals. After ingestion, it is rapidly absorbed and the blood
concentration reaches a maximum 1-3 h following ingestion and,
thereafter, decreases. Although a significant portion of methyl
parathion is found in the bile, it is present in all organs (see also
section 6.2).
4.3 Interaction with other physical, chemical, and biological
factors
Methyl parathion shows interactions with the following
substances: adrenocorticoids, anaesthetics, tricyclic antidepressive
agents, antihistamines, atropine, barbiturates, clofibrate,
colistimethate, corticosteroids, curare, decamethonium, dexpanthenol,
fluorophosphate, hexamethonium, kanamycin, morphine, muscle relaxants,
anticholinesterases, neomycin, parasympathomimetics, phenothiazines,
polymyxin, pralidoxime, procainamide, streptomycin, succinylcholin,
sympathomimetics, d-tubocurarine (Martin, 1978).
A significant increase in the toxicity of oxygen analogues of
organophosphorus insecticides to house flies was observed following
treatment with polychlorinated biphenyl (PCB) (Aroclor 1248)
(Fuhremann, 1980). Detergents increased the hydrolysis of
organophosphates, such as methyl parathion (Peterka & Cerna, 1988).
Yang & Sun (1977) found an inversely proportional correlation between
fish toxicity and the partition coefficient of different insecticides,
including methyl parathion.
DEF ( S,S,S-tributyltrithiophosphate), a defoliant, enhanced the
toxic effect of methyl parathion in the fish (Gambusia affinis)
(Fabacher, 1976).
4.4 Ultimate fate following use
The ultimate fate of methyl parathion depends on the degradation
pathways. The most important one is chemical as well as biological
hydrolysis; the others are oxidative desulfurisation, nitro reduction,
and photodegradation. Important degradation products are methyl
paraoxon, dimethylthiophosphoric acid, dimethylphosphoric acid, and
p-nitrophenol.
5. ENVIRONMENTAL LEVELS AND HUMAN EXPOSURE
5.1 Environmental levels
5.1.1 Air
In a pilot study, Stanley et al. (1971) measured methyl parathion
concentrations of up to 129 ng/m3 in air samples collected in the
USA (Stoneville). The technique used for air sampling was that of
Miles et al. (1970).
In Tennessee, USA, average hourly concentrations of methyl
parathion in air were < 0.57 ng/m3 (maximum, 2.9 ng/m3) at a site
located one mile south-east of a methyl parathion plant and one mile
west of a plant producing the nematocide ethoprophos
( O-ethyl- S,S-dipropyl phosphorodithioate), and < 0.64 ng/m3
(maximum, 5.1 ng/m3) at another site located one mile north of a
methyl parathion plant. Particulate samples collected from the 2 sites
contained < 0.086 ng methyl parathion/m3 (Foster, 1974).
In the USA, maximum atmospheric levels were detected of 29.6
ng/m3 in Alabama, 5.4 ng/m3 in Florida, and 129 ng/m3 in
Mississippi (Midwest Research Institute, 1975). Methyl parathion was
found in air samples in the Mississippi Delta, one of the highest
pesticide usage areas in the USA, because of the intensive cotton
production, at a maximal concentration of 2060 ng/m3 (Arthur et al.,
1976). The average monthly concentrations of methyl parathion peaked
in August or September with levels varying from 111.7 ng/m3
(September 1972) to 791.1 ng/m3 (September 1973).
In another study, airborne residues of methyl parathion and
methyl paraoxon were determined after the use of methyl parathion on
rice in the Sacramento valley in California, USA (Seiber et al.,
1989). Sampling was conducted on the roof tops of public buildings in
4 towns in 2 counties where methyl parathion was used in significant
quantities, and in a reference area where no use occurred. Daily
maximum average concentrations were 25.7 ng/m3 for methyl parathion
and 3.1 ng/m3 for methyl paraoxon. The range in averages for all
sites in the vicinity of usage during springtime 1986 was 0.2-6.2
ng/m3 for methyl parathion and < 0.5-0.8 ng/m3 for methyl
paraoxon. With one exception, the background samples did not show any
methyl parathion above the detection limit.
Methyl parathion and methyl paraoxon concentrations measured in
the condensate from coastal fog near Monterey (California, USA) ranged
between 0.046 and 0.43 µg/litre and between 0.039 and 0.49 µg/litre,
respectively. The oxon to thion ratios were 0.28-2.6, and thion to
oxon conversion appeared to take place during atmospheric transport
from agricultural to the nonagricultural areas (Schomburg et al.,
1991).
In the Kalinin District, Tashkent Province, the Uzbek SSR (USSR),
during July and August, the concentrations of methyl parathion in the
air after spraying with 30% emulsion, measured at 500, 750, and 1000
m from the place of the treatment, were 0.055- 0.08, 0.01-0.02, and
0-0.008 mg/m3, respectively (Akhmedov, 1968).
Tessari & Spencer (1971) analysed indoor and outdoor air samples,
collected monthly for a year, at the homes of families where the head
of the household was occupationally exposed to pesticides. A nylon
chiffon cloth screen was exposed to the atmosphere for 5 days and the
absorbed pesticides were extracted and analysed using a column
chromatography method. The authors found methyl parathion in 13 out of
52 samples, at an average concentration of 1.04 µg/m3. The range was
0.04-9.4 µg/m3. The values obtained from outdoor sampling were much
smaller, 3 out of 53 samples containing 0.35 µg methyl parathion/m3
with a range of 0.15-0.71 µg/m3.
5.1.2 Water
Methyl parathion concentrations of up to 0.23 µg/litre were found
in selected Western streams of the USA in 1968-71 (Schulze et al.,
1973).
In 1970, methyl parathion was detected in 3 out of 18 surface
drain effluent water samples in California, USA, at concentrations of
10-190 ng/kg, and, in 8 out of 60 subsurface drain effluent water
samples, at concentrations of 10-170 ng/kg (Midwest Research
Institute, 1975).
In water samples from 10 sites in the Cape Fear River Basin in
North Carolina, USA, taken monthly between July 1974 and June 1975
(except October), maximum concentrations of methyl parathion in
dissolved fractions and in particulate-associated fractions were 468
ng/litre and 123 ng/litre, respectively (Pfaender et al., 1977).
Methyl parathion was detected in waste water from a parathion
production plant in the USA at levels of 2.0 mg/litre in pre-treatment
water and < 0.004 mg/litre in post-treatment water (Marcus et al.,
1978).
Methyl parathion residues in major Mississippi stream systems
(USA), monitored during 1972-73, ranged between 0.08 and 0.46 µg/litre
(Leard et al., 1980).
In one station at the Negro River Basin (Argentina), methyl
parathion was detected at a concentration of 0.034 µg/litre in March
1986, which is the end of the summer season in South America (Natale
et al., 1988).
In a study on the Ionnina basin and Kalamas river (Greece), from
September 1984 to October 1985, a seasonal fluctuation was found in
the concentration of methyl parathion, with a maximum during the
summer and a minimum during the winter (Albanis et al., 1986). The
mean concentration in the lake Pamvotis (Greece) was 7.7 ng methyl
parathion/litre in July. The natural outlet of the lake is the Kalamas
River, where a maximum concentration of 32 ng methyl parathion/litre
was found. With the exception of the river, the other analyses showed
much lower concentrations of methyl parathion. The results of this
study show very clearly the seasonal influence of the application of
this pesticide on natural water concentrations.
Normally, the methyl parathion concentration in the River Rhine
is below the limit of detection and the Sandoz accident on 1 November
1987 did not affect the wells of the waterworks. A maximum value
measured in the Rhine during the second half of 1986 was higher (<
0.05 mg/m3) than that following this accident (Winter & Lindner,
1987).
Methyl parathion was detected in Hungarian surface waters only
once between 1977 and 1986 (concentration not given), which
corresponded to a sampling frequency of 0.14% (Csernatoni et al.,
1988).
5.1.3 Soil
In 1969, 76 samples of onions and the soils in which they had
been grown were collected in the 10 major onion-producing states of
the USA for analysis of the pesticide residues. The limit of
quantification of methyl parathion was 0.01 mg/kg. Methyl parathion
was found in a range of 0.09-1.9 mg/kg in 11.8% of the soil samples.
No residues were detected in the onion samples (Wiersma et al., 1972).
Methyl parathion was found at levels of 0.09-1.90 mg/kg in soil
samples from onion-producing States in the USA (Midwest Research
Institute, 1975). In cropland soil (South Dakota, USA), the
concentration of methyl parathion was 0.01 mg/kg soil (Carey et al.,
1979).
5.1.4 Food
Renvall et al. (1975) reported pesticide analyses of fruits and
vegetables on the Swedish market from July 1967 to April 1973. Methyl
parathion belonged to the most frequently occurring pesticides with a
rate of 6%. Levels in 4 out of 207 oranges analysed, 1 out of 37
lemons, 4 out of 69 grapefruits, and, 2 out of 29 clementines or
mandarins exceeded 0.11 mg/kg. In a more recent study, in the Swedish
monitoring programme during the period 1981-84, methyl parathion was
found in apples, celery, grapes, lemons, lettuce, limes, mandarins,
oranges, pears, and plums. One out of 74 celeries analysed (imported),
1 out of 238 lemons (imported), 1 out of 248 lettuces (domestic), 5
out of 421 mandarins (imported), and, 8 out of 917 oranges (imported)
exceeded the Swedish maximum residue limits of 0.1-0.5 mg methyl
parathion/kg (Andersson, 1986).
In a study on the presence of organophosphorus insecticide
residues in Mexican food, methyl parathion residues were found in
market samples of avocados, rice, strawberries, and tomatoes, with
respectively 6, 4, 3, and 5 positive samples out of 10. The average
concentrations were 0.3, 0.8, 0.5, and 0.5 mg/kg, respectively (Albert
et al., 1979).
A report on pesticide residues in the United Kingdom (1982-85)
gave a residue level for methyl parathion in lemons of 0.3 mg/kg
(MAFF, 1986). In a more recent report, no methyl parathion was found
in cooking apples and in imported apples with a reporting limit of
determination of 0.1 mg/kg; however, a concentration of 0.08 mg methyl
parathion/kg was found in one sample of lemons from Spain (MAFF,
1990).
Methyl parathion was detected in citrus fruits in France at
levels of 0.003-1.25 mg/kg (Mestres et al., 1977). Lamontagne (1978)
found methyl parathion in concentrations of 0.311 mg/kg in fruit and
0.87-2.12 mg/kg in greenhouse plants in France. Branca & Quaglino
(1988) found methyl parathion at a residue level of 0.036 mg/kg in one
out of 34 samples of French potatoes imported into Italy.
Pesticide residue levels were analysed during 1968-69 in samples
of ready-to-eat foods from 30 markets in 24 different cities with
populations of between 50 000 and more than 1 000 000 in the USA. The
limit of determination was 0.05 mg/kg. Methyl parathion was found
infrequently (1 X Boston, 1 X Los Angeles, 2 X Minneapolis) in
concentrations of 0.008, traces, 0.001, and 0.025 mg/kg in leafy
vegetables and 0.033 mg/kg in grain (Boston) (Corneliussen, 1970).
From June 1971 to July 1972, methyl parathion was detected in 7 out of
420 samples of ready-to-eat foods. The concentrations found in leafy
vegetables ranged from a trace to 0.010 mg/kg. In one sample of fruit
(type not given), a concentration of 0.007 mg/kg was found (Boston)
(Manske & Johnson, 1975). In the report of the Food and Drug
Administration, 5 samples of leafy vegetables containing methyl
parathion residues are mentioned. The concentrations ranged from a
trace to 0.003 mg/kg (Johnson & Manske, 1976). In "market-basket"
surveys conducted by the US Food and Drug Administration in 1966-69,
methyl parathion was detected in leafy and stem vegetables at levels
of 0-2.00 mg/kg, and, in root vegetables, at levels of 0-1.0 mg/kg
(Midwest Research Institute, 1975). Johnson et al. (1981) did not find
any methyl parathion in infant and toddler Total Diet Studies (TDS) in
the USA in 1975-76. In the adult TDS in the USA in 1973-74, trace
residue levels were found in leafy vegetables, but none in fruit
(Manske & Johnson, 1977). "Dislodgable" methyl parathion residues were
found on sweet corn in the USA at levels of 0-0.14 µg/cm2, one and
two days after application of the pesticide (Wicker et al., 1979).
Soybeans analysed in 1979 showed levels of 1-40 mg methyl parathion/kg
and soybean forage analysed at intervals of 1-14 days after treatment,
0.3-6.6 mg methyl parathion/kg. Levels of 0.1-0.3 mg methyl
parathion/kg were measured in 12 samples of cottonseed (FAO, 1985).
Samples of standing agricultural crops were analysed in 1971
during the National Pesticide Monitoring Programme in the USA (Carey
et al., 1978). Levels of methyl parathion detected in samples of
alfalfa, field orn (kernels), cotton, cotton stalks, and mixed hay
ranged from 0.02 to 4.57 mg/kg dry weight.
During a TDS in Canada in 1972, Smith et al. (1975) found methyl
parathion residues in leafy vegetables from Winnipeg at an average
level of 0.012 mg/kg.
In a TDS in New Zealand during 1971-73, methyl parathion was
found in one sample of leafy vegetables at a level of 0.15 mg/kg in
1973, in one sample of root vegetables at the level of 0.26 mg/kg in
1972, and in 4 samples of citrus fruit at an average level of 0.20
mg/kg and a maximum level of 1.4 mg/kg during each of the years
1971-73. In 1971, 3 samples of pip fruit contained, on average, 0.03
mg/kg, and, in 1972, one sample of stone fruit contained 0.25 mg/kg.
Some of these figures exceeded the New Zealand tolerances (Love et
al., 1974). In 1974, methyl parathion was detected at levels of
0.003-0.007 mg/kg in fruit and 0.002-0.008 mg/kg in tinned food from
Auckland and Wellington, New Zealand (Dick et al., 1978).
The loss of methyl parathion in food during heating and storage
was confirmed by Elkins et al. (1972). The samples were analysed
before, and after, standardized heat treatment. Spinach and apricots
were fortified separately with methyl parathion. The spinach samples
were heated for 66 min at 122 °C and the apricot samples were heated
for 50 min at 103 °C. The initial concentration of methyl parathion in
the spinach samples was 0.88 mg/kg. It disappeared completely after
heating. The methyl parathion level in the apricot samples was 0.85
mg/kg, but this decreased to 46% of this level after heating. The
detection limit was less than 0.005 mg/kg. A further decomposition can
be expected during the storage of preserved food. Generally, methyl
parathion residues in fruit decomposed very rapidly, except in the
waxy skin of apples and in the oil vessels of olives (Stoll, 1982).
Rippel et al. (1970) found remarkable differences in the
degradation of methyl parathion in packaged citrus juice, depending on
the kind of package surface. The rate of decrease of the methyl
parathion residues was insignificant in glass containers. It was
substantially higher in packages with tin-layer surfaces than in
packages with painted protective surfaces, since the tin layers
reduced the nitro group of the methyl parathion.
5.1.5 Terrestrial and aquatic organisms
Methyl parathion is rapidly metabolized in most organisms,
resulting in low bioconcentration factors after acute exposure. There
are few studies of residues of methyl parathion in organisms in the
environment, but those conducted have consistently shown low methyl
parathion residues.
Methyl parathion was detected in tissue samples from estuarine
fish at a mean level of 47 µg/kg (Butler & Schutzmann, 1978). It has
been detected at a concentration of 59 µg/kg in the ovaries of spotted
sea trout (Cynoscion nebulosus), collected in Texas, USA (Midwest
Research Institute, 1975).
Methyl parathion was detected in 34 out of 55 suspectedly
poisoned apiaries examined in Connecticut (USA) in 1983-85 (Anderson
& Wojtas, 1986). Concentrations of methyl parathion found in dead bees
and in brood comb ranged from 0.04 to 5.8 mg/kg.
5.2 General population exposure
The general population can come into contact with methyl
parathion via air, water, or food. Average methyl parathion intake
from food in the USA during 1988 was estimated to range from 0.1 to
0.2 ng/kg per day in 3 different age groups (FDA, 1989). Draper &
Street (1981) estimated that a 70-kg male living in a residence
adjacent (50 yards) to an alfalfa field sprayed with methyl parathion
at a rate of 0.19 kg a.i./ha would be exposed to a total dermal dose
of 0.38 mg. Within a pesticide monitoring programme in the USA, based
on the analysis of 6990 samples collected from the general population
via the National Center for Health Statistics 1976-80,
para-nitrophenol as an indicator for exposure to methyl and ethyl
parathion was detected in 2.4% of urine samples from 12 to 74-year-old
persons (Carey & Kutz, 1985).
5.3 Occupational exposure during manufacture, formulation, or use
There is a special risk for farm workers, since incidents of
poisonings and illnesses during the mixing, loading, and application
of methyl parathion have been reported. Exposure may also occur during
the cleaning and repair of equipment and during early re-entry into
fields. According to NIOSH (1976), 150 000 workers in the USA (field
workers, aerial application personnel, mixer and blender operators,
tractor tank loaders, ground applicator vehicle drivers, field
inspectors, and warehouse personnel) are conceivably exposed to methyl
parathion. A maximum air concentration of methyl parathion was
estimated to be 1.77 µg/m3. The exposure to methyl parathion was
estimated by Hayes (1971) for workers checking cotton for insect
damage as 0.7 mg/h via skin contact and < 0.01 mg/h through
inhalation (NIOSH, 1976).
Davis et al. (1981) estimated that workers in apple orchards
sprayed with methyl parathion would be exposed to dermal doses ranging
from 0.055 mg to 3.1 mg, with the amount varying with time after
spraying and the formulation of the pesticide. Two field studies were
carried out by Kummer & Van Sittert (1986) to evaluate the health risk
for the farm workers. In a number of cases, the men involved in
hand-held ULV-spraying wore very little clothing and did not stop
spraying, when it was too windy. Another possible contamination risk
was the filling of bottles from larger (25-litre) containers, and the
repairing and cleaning of the equipment with unprotected hands.
However, no signs of acute poisoning could be observed in any of the
persons involved in these studies. The urine was collected in spot
samples in one of the studies and in 24-h samples in the other.
Methyl parathion absorption could be verified from its metabolites in
the spraymen's urine. Average levels of urinary nitrophenol (mg/g
creatinine) for 6 supervisors and 2 groups of sprayers were reported
to be 0.08 (range of 0.05-0.20), 0.38 (range of 0.04-1.38), and 0.13
(range of 0.06-0.44), respectively. An intake of 0.4-13 mg methyl
parathion was calculated from the excreted p-nitrophenol.
Since investigations showed that clothing worn by agricultural
workers became contaminated with methyl parathion following
application and that the laundering of contaminated clothing with
uncontaminated fabrics resulted in the transfer of the methyl
parathion residue, recommendations were made that contaminated fabrics
should not be washed with regular family laundry. Suggestions for the
procedure of laundering were made by Easley et al. (1981) and Laughlin
et al. (1981). The most effective procedure was using a pre-rinse
programme and a detergent together with sodium hypochlorite (NaOCl) as
a bleach. Laughlin & Gold (1989) discussed further aspects of
laundering protective clothing contaminated with methyl parathion.
Fluorocarbon soil repellent finishes on such protective clothing
decrease pesticide absorption, but may hinder pesticide removal in
laundering. Storage of laundered garments at 20 °C with air flow
and/or at high humidity levels was recommended to dissipate residues
of methyl parathion.
Ware et al. (1974b) suggested that serum insecticide levels,
serum and red blood cell cholinesterase activities, and urinary
excretion of p-nitrophenol should be investigated, because they are
more effective for evaluating the possible potential poisoning hazard
than the analysis of skin and clothing contamination. The safety of
re-entering cotton fields 24 h following application of methyl
parathion was tested. Methyl parathion was applied at 1.12 kg a.i./ha.
During the application, the temperature ranged from 30 to 38 °C. The
foliar residues decreased from 1.6 mg/m2, 24 h following methyl
parathion treatment, to 0.9 mg/m2, 6 h later. No methyl parathion
was detectable in the serum of the volunteers. The 48-h urinary
excretion of p-nitrophenol ranged from 0.15 to 1.20 mg. Serum
cholinesterase levels varied within normal intervals whereas the red
blood cell cholinesterase levels showed a temporary, but not
pronounced, depression of about 5-7%. The amounts of methyl parathion
and methyl paraoxon extracted from clothing and hand surfaces are
shown in Table 15.
During the working period, the mean air concentration was 0.2 ng
methyl parathion/litre, of which, 1.2 µg methyl parathion was inhaled
over 5 h. From all these data, it was concluded that a 24-h interval
is safe for methyl parathion in this form of application.
Munn et al. (1985) collected human exposure samples from workers
and dependants wearing nylon gloves, as well as environmental samples,
during the onion harvest season of 1982 in Colorado, USA. Children in
agricultural settings normally accompany their parents to the fields,
as part of a family unit, the young children playing in this
environment and older children helping their parents in the fields.
Munn et al. (1985) recorded the length of time the gloves were worn,
and the age and sex of the participants. No association between age
and methyl parathion levels was found. The urine samples collected
prior to their leaving the field did not contain detectable levels of
methyl parathion. This could be because the nylon gloves reduced the
absorption of organophosphate residues by about 90%.
Table 15. Extracted residues of methyl parathion and methyl paraoxon
following a 5-h working perioda
Extract from: Methyl parathion Methyl paraoxon
residue (mg) residue (mg)
Hands 0.2 0.5
Shirts 0.2 4.0
mep.5s 1.7 39.0
a From: Ware et al. (1974b).
6. KINETICS AND METABOLISM
6.1 Absorption
Methyl parathion can be absorbed through the digestive tract, the
skin, and the respiratory tract (White-Stevens, 1971).
The primary routes of exposure are via skin contact with
contaminated plants or material, and via inhalation. Severe accidental
intoxications of humans have occurred.
The absorption of methyl parathion from the digestive tract is
rapid, and it appears in the bloodstream immediately after oral
intake. Studies on guinea-pigs were performed to analyse the rate of
absorption of radioactive labelled (32P) methyl parathion. One
minute after dosage, it could be detected in various organs. The
maximum level was found 1-2 h after treatment. The liver showed a
remarkably high concentration (Gar et al., 1958).
Miyamoto et al. (1963) administered 50 mg 32P-labelled methyl
parathion/kg body weight to guinea-pigs or 1.5 mg/kg body weight to
rats, by stomach tube. Maximum concentrations in the blood and brain
were reached 1-3 h after treatment. An oral dose of 50 mg methyl
parathion/kg resulted in no detectable levels of methyl parathion in
either the brain or blood after 3 min, but, after 6-8 min, at which
point lethal effects occurred, levels of methyl parathion increased to
182 ng/ml in plasma and to 137 ng/g in brain (Yamamoto et al., 1981).
6.2 Distribution
Accumulation of methyl parathion was observed in tissues. The
highest concentrations were found in the lung and the liver (NRC,
1977). Transplacental transport of methyl parathion is discussed in
section 8.5.
Total radioactive residues recovered in the 12 tissues analysed
(excluding the gastrointestinal tract) from rats given a single oral
dose of 5 mg C-14-methyl parathion/kg body weight were about 11% of
the administered dose, 1 h after treatment, declining to 0.3% at 24 h,
about 0.1% at 48 h, and to only 0.04%, 6 days later. The kidney had
the highest relative activity up to 8 h after treatment. The
14C-activity in the plasma was initially about 5 times higher than
that in the erythrocytes. However, from day 2 to day 6 after dosing,
the 14C-activity in the erythrocytes was greater than that in plasma
and remained constant (Weber et al., 1979).
Sultatos et al. (1990) measured the partition coefficient for
methyl parathion between mouse liver and blood by either equilibrium
dialysis or a perfusion technique and obtained values of 9.5 and 16.4
respectively.
In a kinetic study on mongrel dogs of both sexes, Braeckman et
al. (1980) found a rapid decrease in serum methyl parathion
concentrations during the first few hours. The authors injected methyl
parathion intravenously in doses of 1, 3, 10, and 30 mg/kg body
weight. The dogs were pretreated with 1-5 mg atropine/kg body weight,
10 min before injecting methyl parathion. The blood samples were taken
for up to 160 h. Besides quantifying serum levels of methyl parathion,
the authors also measured serum cholinesterase activity at the 2
highest concentrations of methyl parathion. The determination of serum
methyl parathion concentrations was performed according to De Potter
et al. (1978). The cholinesterase activity decreased within 30 min to
its lowest value, i.e., 40% of the normal level in dogs receiving 10
mg/kg body weight and 25% in dogs receiving 30 mg/kg. The first rapid
fall in the methyl parathion concentration after injection was due to
distribution and elimination. A slower decrease in serum methyl
parathion concentrations at higher doses was the result of dee
compartment linear kinetics. This is in line with observations of
Tilstone et al. (1979), who found a rebound effect after a
haemoperfusion.
6.3 Metabolic transformation
Organic nitro compounds, orally administered to ruminants, will
undergo reduction of the nitro groups to amino groups. This reaction
takes place in the rumen (Karlog et al., 1978).
The metabolism of methyl parathion in rodents is illustrated in
Fig. 2.
Because of the importance of a first pass through the liver for
the metabolism of methyl parathion, there is a distinct difference
between the oral and intravenous toxicity (Morgan et al., 1977;
Braeckman et al., 1983). Conversion of methyl parathion to its toxic
metabolite, methyl paraoxon, may occur within minutes following oral
administration (Yamamoto et al., 1983).
Mouse liver, perfused with methyl parathion, released the toxic
metabolite methyl paraoxon into the effluate. Mouse whole blood
rapidly detoxified the methyl paraoxon formed (Sultatos, 1987).
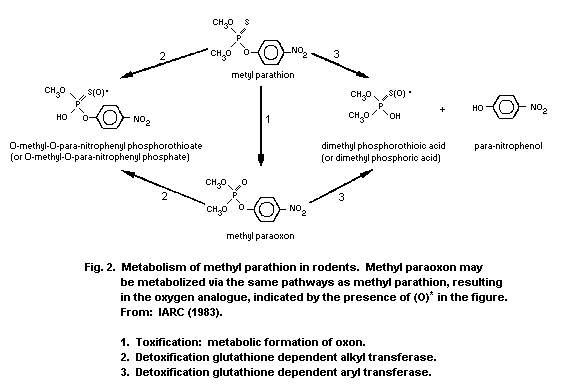 A reduction of the cellular concentration of reduced glutathione
(GSH) influences mitosis, mobility, and other GSH-dependent cell
functions. Glutathione S-transferases are mainly located in the
cytosol and display overlapping substrate specificity. They also show
peroxidase activity and prevent the peroxidation of membrane lipids.
The interaction of methyl parathion with GSH or with the glutathione
S-transferases therefore is important not only for the non-oxidative
detoxification of the insecticide, but also for species-selective
toxicity, and the development of resistance. Placental and fetal human
glutathione S-transferase catalysed the dealkylation of methyl
parathion exclusively to demethyl parathion via O-dealkylation
(Radulovic et al., 1986; 1987).
Only after the metabolic formation of methyl paraoxon by liver
microsomal oxidases does the substance become toxic. Therefore, this
is an activation reaction. Methyl parathion and methyl paraoxon are
mainly detoxified by conjugation with GSH (Hennighausen, 1984).
Detoxification is achieved by degradation reactions, that involve
either demethylation or dearylation. The resulting desmethyl compounds
and dimethyl phosphoric acids are essentially nontoxic (NRC, 1977).
These detoxification reactions are due to the glutathione-dependent
alkyl and aryl transferases; the reaction products are
O-methyl- O-p-nitrophenyl phosphorothioate (or
O-methyl- O-p-nitrophenyl phosphate) or dimethyl phosphorothioic
acid (or dimethyl phosphoric acid) and p-nitrophenol. In addition,
hydrolysis of methylparaoxon by tissue arylesterases may occur. Thus,
it is possible to follow an exposure to methyl parathion by measuring
the urinary excretion of p-nitrophenol (Benke & Murphy, 1975).
However, prior depletion of glutathione by acetaminophen (Costa
& Murphy, 1984) or diethyl maleate (Sultatos & Woods, 1988) has little
effect on the toxicity of methyl parathion in the mouse, indicating
that perhaps glutathione does not play a significant role in the
detoxification of methyl parathion.
The amount of the active toxic compound (methyl paraoxon) that
will be produced after exposure to methyl parathion, depends on the
kinetics of the oxidation of methyl parathion and on the kinetics of
the detoxification reactions. Dealkylation is important at high
dosages (Plapp & Casida, 1958). This enzyme system was found in the
supernatant of the liver homogenate. The main metabolites were
demethyl parathion (80%) and demethyl paraoxon (Fukami & Shihido,
1963; Shihido & Fukami, 1963).
The same major metabolites were generated when rat liver
microsomes metabolized methyl parathion: demethyl paraoxon, methyl
paraoxon, i.e., dimethyl phosphate, dimethyl phosphorothioate, and
p-nitrophenol. When rats were treated with methyl parathion,
dimethyl phosphoric acid was excreted in the urine together with
O-methyl and O,O-dimethyl paraoxon (Menzie, 1974).
Adult rats have an increasing capacity to metabolize the oxygen
analogue by both oxidative and hydrolytic pathways (Benke & Murphy,
1975).
Willems et al. (1980) calculated the high serum clearance of
methyl parathion from their intravenous studies on dogs to be 2.1
litre/kg per h.
Malaysian prawns (Macrobrachium rosenbergii) as well as ridgeback
prawns (Sicyonia ingentis) decomposed methyl parathion readily to
p-nitrophenol and p-nitrophenyl conjugates. The dominant way of
detoxification was the formation of ß-glycosides and sulfate esters
(Foster & Crosby, 1987).
The metabolism of methyl parathion in humans is similar to that
reported in experimental animals (Fig. 3) (Benke & Murphy, 1975;
Morgan et al., 1977). The liver is the primary organ for
detoxification and metabolism (Nakatsugawa et al., 1968, 1969). The
main metabolites recovered from urine following administration of
methyl parathion to human subjects were also p-nitrophenol and
dimethyl phosphate. Eight hours after application, p-nitrophenol
excretion was nearly complete. Methyl paraoxon was hydrolysed to
dimethyl phosphate and an amount representing 12% of the administered
dose was excreted. Its excretion was more protracted than that of
p-nitrophenol (Morgan et al., 1977).
Rao & McKinley (1969) found remarkable differences in the rates
of metabolism of methyl parathion by liver homogenates from male and
female chickens. The rate of the oxidative desulfurating system of the
male liver homogenates was substantially higher than that of the
homogenates of female chicken livers; however, the rates of the
demethylating system showed no differences. Also no sexually
determined differences of the oxidative or the demethylating system
were found in the liver homogenates of rats, guinea-pigs, or monkeys.
6.4 Elimination and excretion in expired air, faeces, urine
After an oral dose of 32P-methyl parathion to mice (17 mg/kg),
75% of the radioactivity was found after 72 h as metabolites in the
urine and up to 10% was eliminated in the faeces (Hollingworth et al.,
1967).
In male rats, treated with a single oral dose of 14C-methyl
parathion (benzene ring-labelled) at 0.1, 1 or 5 mg/kg body weight and
in female rats given a single oral dose of 1 mg/kg body weight, over
99% of the administered dose was eliminated in the urine and the
faeces within 48 h. Elimination in the faeces accounted for only 5-7%
after 1 or 5 mg/kg body weight, but amounted to about 20% after 0.1
mg/kg body weight Male rats treated with an intravenous dose of 1 mg
methyl parathion/kg body weight eliminated about 99% of the
administered radioactivity in the urine within 48 h, and
approximately 1% of the dose in the faeces (Table 16, Weber et al.,
1979).
A reduction of the cellular concentration of reduced glutathione
(GSH) influences mitosis, mobility, and other GSH-dependent cell
functions. Glutathione S-transferases are mainly located in the
cytosol and display overlapping substrate specificity. They also show
peroxidase activity and prevent the peroxidation of membrane lipids.
The interaction of methyl parathion with GSH or with the glutathione
S-transferases therefore is important not only for the non-oxidative
detoxification of the insecticide, but also for species-selective
toxicity, and the development of resistance. Placental and fetal human
glutathione S-transferase catalysed the dealkylation of methyl
parathion exclusively to demethyl parathion via O-dealkylation
(Radulovic et al., 1986; 1987).
Only after the metabolic formation of methyl paraoxon by liver
microsomal oxidases does the substance become toxic. Therefore, this
is an activation reaction. Methyl parathion and methyl paraoxon are
mainly detoxified by conjugation with GSH (Hennighausen, 1984).
Detoxification is achieved by degradation reactions, that involve
either demethylation or dearylation. The resulting desmethyl compounds
and dimethyl phosphoric acids are essentially nontoxic (NRC, 1977).
These detoxification reactions are due to the glutathione-dependent
alkyl and aryl transferases; the reaction products are
O-methyl- O-p-nitrophenyl phosphorothioate (or
O-methyl- O-p-nitrophenyl phosphate) or dimethyl phosphorothioic
acid (or dimethyl phosphoric acid) and p-nitrophenol. In addition,
hydrolysis of methylparaoxon by tissue arylesterases may occur. Thus,
it is possible to follow an exposure to methyl parathion by measuring
the urinary excretion of p-nitrophenol (Benke & Murphy, 1975).
However, prior depletion of glutathione by acetaminophen (Costa
& Murphy, 1984) or diethyl maleate (Sultatos & Woods, 1988) has little
effect on the toxicity of methyl parathion in the mouse, indicating
that perhaps glutathione does not play a significant role in the
detoxification of methyl parathion.
The amount of the active toxic compound (methyl paraoxon) that
will be produced after exposure to methyl parathion, depends on the
kinetics of the oxidation of methyl parathion and on the kinetics of
the detoxification reactions. Dealkylation is important at high
dosages (Plapp & Casida, 1958). This enzyme system was found in the
supernatant of the liver homogenate. The main metabolites were
demethyl parathion (80%) and demethyl paraoxon (Fukami & Shihido,
1963; Shihido & Fukami, 1963).
The same major metabolites were generated when rat liver
microsomes metabolized methyl parathion: demethyl paraoxon, methyl
paraoxon, i.e., dimethyl phosphate, dimethyl phosphorothioate, and
p-nitrophenol. When rats were treated with methyl parathion,
dimethyl phosphoric acid was excreted in the urine together with
O-methyl and O,O-dimethyl paraoxon (Menzie, 1974).
Adult rats have an increasing capacity to metabolize the oxygen
analogue by both oxidative and hydrolytic pathways (Benke & Murphy,
1975).
Willems et al. (1980) calculated the high serum clearance of
methyl parathion from their intravenous studies on dogs to be 2.1
litre/kg per h.
Malaysian prawns (Macrobrachium rosenbergii) as well as ridgeback
prawns (Sicyonia ingentis) decomposed methyl parathion readily to
p-nitrophenol and p-nitrophenyl conjugates. The dominant way of
detoxification was the formation of ß-glycosides and sulfate esters
(Foster & Crosby, 1987).
The metabolism of methyl parathion in humans is similar to that
reported in experimental animals (Fig. 3) (Benke & Murphy, 1975;
Morgan et al., 1977). The liver is the primary organ for
detoxification and metabolism (Nakatsugawa et al., 1968, 1969). The
main metabolites recovered from urine following administration of
methyl parathion to human subjects were also p-nitrophenol and
dimethyl phosphate. Eight hours after application, p-nitrophenol
excretion was nearly complete. Methyl paraoxon was hydrolysed to
dimethyl phosphate and an amount representing 12% of the administered
dose was excreted. Its excretion was more protracted than that of
p-nitrophenol (Morgan et al., 1977).
Rao & McKinley (1969) found remarkable differences in the rates
of metabolism of methyl parathion by liver homogenates from male and
female chickens. The rate of the oxidative desulfurating system of the
male liver homogenates was substantially higher than that of the
homogenates of female chicken livers; however, the rates of the
demethylating system showed no differences. Also no sexually
determined differences of the oxidative or the demethylating system
were found in the liver homogenates of rats, guinea-pigs, or monkeys.
6.4 Elimination and excretion in expired air, faeces, urine
After an oral dose of 32P-methyl parathion to mice (17 mg/kg),
75% of the radioactivity was found after 72 h as metabolites in the
urine and up to 10% was eliminated in the faeces (Hollingworth et al.,
1967).
In male rats, treated with a single oral dose of 14C-methyl
parathion (benzene ring-labelled) at 0.1, 1 or 5 mg/kg body weight and
in female rats given a single oral dose of 1 mg/kg body weight, over
99% of the administered dose was eliminated in the urine and the
faeces within 48 h. Elimination in the faeces accounted for only 5-7%
after 1 or 5 mg/kg body weight, but amounted to about 20% after 0.1
mg/kg body weight Male rats treated with an intravenous dose of 1 mg
methyl parathion/kg body weight eliminated about 99% of the
administered radioactivity in the urine within 48 h, and
approximately 1% of the dose in the faeces (Table 16, Weber et al.,
1979).
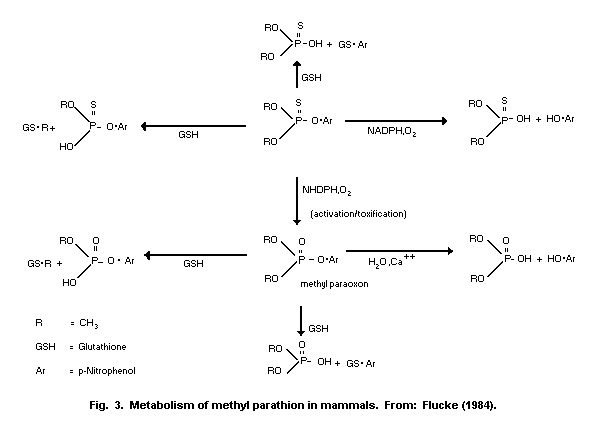 Table 16. Elimination of 14C-labelled methyl parathion in ratsa,b
Doses Route of No. of Urine Faeces Balance
(mg/kg) administration rats (%) (%) (%)
0.1 oral 5 79.8±11 19.4±5. 99.2
3
1 oral 4 93.6±2.6 6.3±1.1 99.9
1 iv 5 99.0±3.8 0.8±0.1 99.8
1 oral 4 93.3±5.1 6.6±2.7 99.9
5 oral 5 94.7±6.0 5.1±0.5 99.8
a Adapted from: Weber et al. (1979).
b iv = intravenously.
The kinetics of the toxic metabolite of methyl parathion, methyl
paraoxon, were studied in conscious dogs (De Schryver et al., 1987).
Thirty min before performing the test, the dogs received atropine as
protection against intoxication. Methyl paraoxon was administered
intravenously (2.5 mg/kg body weight) or orally (15 mg/kg body
weight). The distribution of an intravenous dose was very fast.
The elimination was fitted by using a one-compartment model. The
average half-life was determined to be 9.7 min, the average volume of
distribution 1.76 litre/kg, and the average plasma clearance 126 ml/kg
per min. Within 3-16 min, the maximal plasma concentration (927-2905
µg/litre) was reached following oral application. The bioavailability
ranged from 5 to 71%. The hepatic extraction in anaesthetized dogs
varied at a high level of 70-92%. From comparison of the urinary
excretion as p-nitrophenol after intravenous (87 and 97%) and oral
(63 and 60%) administration of methylparaoxon, the gastrointestinal
absorption seemed to be about 60%. It was assumed, that the kinetics
were linear in this dose range.
The concentration of the main metabolites paranitrophenol (PNP)
and dimethylphosphate (DMP) in the urine of 4 human volunteers,
following 2 days of ingestion of 2 or 4 mg methyl parathion, is shown
in Table 17. Unmetabolized traces of methyl parathion were also found
in the urine, which was collected after 4-, 8-, and 24-h (Morgan et
al., 1977). The urinary excretion of nitrophenol was 60% within 4 h,
86% within 8 h, and approximately 100% within 24 h following
ingestion. Table 17 shows the dependence of urinary metabolite
excretion on methyl parathion dosage.
Table 17. p-Nitrophenol (PNP) and dimethylphosphate (DMP) concentrations in
24-h urine samples collected from human volunteers following
administration of 2 or 4 mg methyl parathiona
PNP DMP
mean range mean range
a) 2 mg methyl parathion
urinary 0.13 0.08-0.20 0.06 0.02-0.11
concentration
(mg/litre)
24-h excretion (mg) 0.29 0.14-0.43 0.12 0.07-0.16
excretion per g 0.16 0.10-0.23 0.06 0.03-0.10
creatinine (mg/g)
b) 4 mg methyl parathion
urinary 0.34 0.16-0.61 0.14 0.05-0.23
concentration
(mg/litre)
24-h excretion (mg) 0.58 0.34-0.88 0.23 0.12-0.41
excretion per g 0.31 0.15-0.42 0.13 0.06-0.20
creatinine (mg/g)
a Adapted from: Morgan et al. (1977).
6.5 Retention and turnover
Braeckman et al. (1980) injected 10 mg methyl parathion per kg
body weight intravenously into dogs, recorded the uptake of methyl
parathion, and determined a harmonic mean terminal half-life of 7.2 h.
Five hours after the injection, the concentration decreased to 30% of
the initial value. Primarily, the peripheral body compartments
contained this residual methyl parathion. The excretion was completed
within 35 h.
The velocity of the excretion of the main metabolites after oral
or intravenous application was similar. However, the bioavaibility
after oral intake was reduced by first-pass extraction by the liver
compared with the intravenous application. Methyl parathion was shown
to bind to a great extent (90%) to plasma proteins in both dogs and
humans (Braeckman et al., 1983).
7. EFFECTS ON ORGANISMS IN THE ENVIRONMENT
7.1 Microorganisms
7.1.1 Bacteria and fungi
Soil concentrations of methyl parathion of 5 mg/kg or more were
found to reduce microbial reductive potential (Reddy & Gambrell,
1985).
In biotests for sanitary control of water samples, growth
inhibition of Escherichia coli by several toxicants was studied in
a liquid medium (Vogel-Bonner medium, supplemented with thymine and
glucose at 37 °C). The minimal concentrations of methyl parathion that
significantly increased growth rate and doubling time of E. coli
were reported to be 62.5 mg/litre and 125 mg/litre; the bacterium
used the compound as a carbon source (Espigares et al., 1990).
Portier et al. (1983) tested the effects of methyl parathion (1.5
or 5 mg/litre) on the reproduction of aquatic microorganisms from
drainage basins in laboratory experiments, using static or
flow-through approaches (28 °C; pH 7.5; 22%; 22 days; or 28 °C; pH
7.2; 0%; 24 days). In bacteria and Actinomycetes , methyl parathion
had a positive effect on the development. In fungi and yeasts, slight
negative effects were found that were related to the test conditions
rather than to the toxicant concentration. In general, a concentration
of up to 5 mg methyl parathion/litre resulted in increased activity
and biomass production in a microbial community, being used as carbon
source by the microorganisms (Portier & Meiers, 1982).
Bhunia et al. (1991) cultured Nostoc muscorum , a blue-green
alga (Cyanobacterium), which is a major nitrogen-fixing organism in
tropical soil, with methyl parathion at 5, 10, 20, or 35 mg/litre.
Only the highest concentration significantly reduced the growth of the
cells in culture. However, the chlorophyll- a contents of the
cultures were marginally reduced at 5 mg methyl parathion/litre and
substantially reduced at 10 mg/litre. Nitrogenase activity was reduced
to < 50% of control levels at 10 mg/litre.
7.1.2 Algae
The 96-h EC50, i.e., the calculated concentration of methyl
parathion that would inhibit growth by 50% of the diatom Skeletonema
costatum , ranged between 5.0 and 5.3 mg/litre (Walsh & Alexander,
1980; Walsh et al., 1987).
Exposure of cultures of Chlorella protothecoides to 26-80 µg
methyl parathion/litre resulted in decreases in cell growth, as
measured by cell count, and chlorophyll and protein contents (Saroja-
Subbaraj & Bose, 1982; Saroja-Subbaraj & Bose, 1983a). These effects
were correlated with a reduction in photosynthetic electron transfer
(Saroja-Subbaraj & Bose, 1983a; Saroja-Subbaraj & Bose, 1983b).
Recovery from the effect on photosynthesis occurred after removal of
the pesticide. Tolerance to the effect of methyl parathion on cell
growth occurred for several weeks after exposure (Saroja-
Subbaraj & Bose, 1984).
In a natural phytoplankton community, addition of 1 mg methyl
parathion/litre led to a 5% decrease in the productivity (Butler,
1964).
An algal bloom (species not specified) in a methyl
parathion-treated pond was suggested to have been induced by the
mortality of herbivorous mayfly larvae and Daphnia (Crossland & Elgar,
1983).
7.2 Aquatic animals
The acute effects of methyl parathion on aquatic animals in
laboratory studies are presented in Table 18. The data show that the
sensitivity of aquatic animals to methyl parathion varies considerably
between species.
LC50 values of more than 1 mg/litre have been found for some
freshwater biota (molluscs, fish, and amphibians). Insect sensitivity
to methyl parathion depends not only on the species but on the life
stage. In general, instar I larvae are more affected than instar IV
larvae. Apperson et al. (1978) showed that larvae may develop a
resistance to methyl parathion. Both freshwater and marine
crustaceans are sensitive to methyl parathion with EC50 values
ranging from 0.002 to 0.050 mg/litre. In general, copepods were less
sensitive than decapods in laboratory tests.
Many laboratory studies have been performed on the acute toxicity
of methyl parathion for fish. The following symptoms of methyl
parathion poisoning can be expected to occur in fish: darkening of the
skin, hyperactivity, body tremors, lethargy, jerky swimming, scalosis,
loss of equilibrium, opercular or gaping paralysis, and death (Rao et
al., 1967; Anees, 1975; Midwest Research Institute, 1975). One
response that may be considered to be somewhat characteristic of acute
methyl parathion poisoning in fish is the extreme forward position of
the pectoral and pelvic fins (Midwest Research Institute, 1975;
Srivastava & Singh, 1981).
Table 18. Acute effects of methyl parathion on aquatic animals in laboratory studies
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
MOLLUSCA
Freshwater mussel
Lamellidens 48 st.c m, LC50 20 000 - Moorthy et al. (1983)
marginalis
Lamellidens m, LC50 25 000 - Moorthy et al. (1983)
marginilis
Lamellidens 20 g 48 st. m, LC50 23 400 - Rao et al. (1983)
marginalis
Eastern oyster
Crassostrea larvae 48 st.; natural seawater d, EC50 12 000 P: 99% Mayer (1997)
virginice 25 °C s: TEG
Marine hard clam
Mercenaria adult 96 St.: wellwater; no effect 25 000 s: acetone Mayer (1987)
mercenaria 24°/oo d 20 °C
pH8
Nassa dosoleta adult 96 st.; wellwater; no effect 25 000 s: acetone Meyer (1987
24°/oo d 20 °C
pH8
Table 18 (continued)
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
ANNELIDA
(Estuerine)
Branchiura - 72 st.; 4.4 °C m, 100% 4000 P: techn.gr. Naqvi (1973)
sowerbyi s: acetone
Branchiura - 72 st.; 21 °C m, 0% 4000 P: techn.gr. Naqvi (1973)
sowerbyi s: acetone
- 72 st.; 32.2 °C m, 100% 4000 P: techn.gr. Naqvi (1973)
s: acetone
CRUSTACEA
(Freshwater)
Water flea
Daphnia adult 24 st.; dechlorinated i, LC50 2.4 P: 93.8% Stephenson &
longispira tap-water; s: acetone Kane (1984)
19.5 °C; H 250e
Daphnia < 24 h 24 st.; dechlorinated i, LC50 4.1 P: 93.8% Stephenson &
magna old tap-water; s: acetone Kane (1984)
19.5 °C; H 250
Daphnia adult 24 - i, LC50 5.4 P: 93.8% Stephenson &
magna s: acetone Kane (1984)
Daphnia < 24 h 48 st.; artificial i, LC50 7.8-9.1 P: 99% Dortland (1980)
magna old water; 18 °C s: acetone
Table 18 (continued)
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
Daphnia first 48 st.; reconst. i. LC50 0.14 P: 98.7 Mayer & Ellersieck
magnia instar water; 21 °C s: acetone (1986)
pH 7.2-7.5;
H40-50
Daphnia - 3 st.; 24 °C m, LC50 8.5 - Nishiuchi & Hashimoto,
pulex (1967)
Moira macrocopa - 3 st.; 24 °C m, LC50 5.5 - Nishiuchi &
Hashimoto (1967)
Simocephalus first 48 st.; reconst. i, LC50 0.37 P: 98.7% Mayer & Ellersieck
secrultus instar water; 15 °C s: acetone (1986)
larva pH 7.2-7.5
H 40-50
Scud
Gammarus adult 96 st.; reconst. m, LC50 3.8 P: 98.7% Mayer & Ellersieck
fasciatus water; 15 °C s: acetone (1986)
pH 7.2-7.5;
H 40-50
Table 18 (continued)
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
Field crab
Oziotelphusa - 48 st.; tap-water; m, LC50 1000 P: techn.gr. Reddy et al. (1986a)
senex senex pH 7.3; 30 °C s: acetone
DO 6.2g; H 38
Crayfish
Orconectes adult 96 st.: reconst. m, LC50 15 P: 98.7% Mayer & Ellersieck
nais water; 15 °C s: acetone (1986)
pH 7.2-7.5;
H 162-272
Procambarus 2.5- 96 st.; tap-water; m, LC50 3 P: techn.gr. Cheeh et al. (1980)
acutus 3.5 cm pH 8.4; H 100
from clean area 1.2- 48 st.; tap-water; m, LC50 2.4 P: techn.gr. Albaugh (1972)
1.5 cm pH 8.7; H 10 as acetone
from treated 1.2- 48 st.; tap-water; m, LC50 3.4 P: techn.gr. Albaugh (1972)
area 1.5 cm pH 8.7; H 10 s: acetone
Procambarus 8.9 cm 36 st.; distilled m, LC50 41 P: 51% Chang & Lange (1967)
clarkii water
22.2-25-5 °C
Procambarus 8.9 cm 24 at.; tap-water m, LC50 50 P: tech.gr. Muncy & Oliver (1963)
clarkii 16-32 °C
pH 7.6
Table 18 (continued)
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
Procambarus 8.9 cm 48 st.; tap-water m, LC50 40 P: tech.gr Muncy & Oliver (1963)
clarkii 16-32 °C
pH 7.6
Procambarus 8.9 cm 72 st.; tap-water m, LC50 40 P: tech.gr. Muncy & Oliver (1963)
clarkii 16-32 °C
pH 7.6
ESTUARINE
AND MARINE
Copepod
Acartia tonsa - 96 st.; natural m, LC50 28 P: 99% Mayer (1987)
seawater; 22°/oo s: TEG
22 °C; pH 8.1-
8.2; DO 7-7.6
Acartia tonsa adult 96 st.; syntheric m, LC50 890 P: 80% Khattat & Farley
seawater; 22°/oo (1976)
17 °C
Table 18 (continued)
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
Sand shrimp
Crangon 2.6 cm 24 st.; wellwater m, LC50 11 s: acetone Eisler (1969)
septemspinosa 0.25 g 24°/oo; 20 °C; pH 8
DO 7.1-7.7
Crangon 2.6 cm 48 st.; wellwater m. LC50 3 s: acetone Eisler (1969)
septemspinosa 0.25 g 24°/oo; 20 °C; pH 8
DO 7.1-7.7
Crangon 2.6 cm 96 st.; wellwater m, LC50 2 s: acetone Eisler (1969)
septemspinosa 0.25 g 24°/oo; 20 °C; pH 8
DO 7.1-7.7
Mysid shrimp
Mysidopsis 24 h 96 st.; natural m, LC50 0.98 P: 99% Mayer (1987)
bahia old seawater; 20°/oo; s: TEG
25 °C;
DO 4.3-5.5
Mysidopsis 24 h 96 st.; natural no effect 0.32 P: 99% Mayer (1987)
bahia old seawater; 20°/oo; s: TEG
25 °C;
DO 4.3-5.5
Mysidopsis 24 h 96 flow-through m, LC50 0.77 P: 99% Mayer (1987)
bahia old natural seawater; s: TEG
20°/oo; 25 °C;
DO 4.3-5.5
Table 18 (continued)
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
Mysidopsis < 24 h 96 flow-through m, LC50 0.78 P: 99% Mayer (1987)
bahia old 14°/oo; 19.5 °C; s: TEG
Mysidopsis juvenile 96 flow-through m, LC50 0.77 s: TEG Nimmo et al. (1981)
bahia 22-28 °C
Mysidopsis juvenile 96 flow-through MATCg 0.11-0.16 s: TEG Nimmo et al. (1981)
bahia 22-28 °C
Hermit crab
Pagurus 3.5 mm 24 st.; wellwater m, LC50 23 s: acetone Eisler (1969)
longicarpus 0.28 g 24°/oo; 20 °C; ph 8;
DO 7.1-7.7
Pagurus 3.5 mm 48 st.; wellwater m, LC50 7 s: acetone Eisler (1969)
longicarpus 0.28 g 24°/oo; 20 °C; ph 8;
DO 7.1-7.7
Pagurus 3.5 mm 96 st.; wellwater m, LC50 7 s: acetone Eisler (1969)
longicarpus 0.28 g 24°/oo; 20 °C; ph 8;
DO 7.1-7.7
Table 18 (continued)
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
Crab
Portunus Zo÷e IV 24 25 °C m, LC50 0.17-0.5 - Hirayama & Tamaoi
trituberculatus stage (1980)
Grass shrimp
Palaemonetes 31 mm 24 st.; wellwater; m, LC50 15 s: acetone Eisler (1969)
vulgaris 0.47 g 24°/oo; 20 °C; ph 8;
DO 7.1-7.7
Palaemonetes 31 mm 48 st.; wellwater; m, LC50 10 s: acetone Eisler (1969)
vulgaris 0.47 g 24°/oo; 20 °C; ph 8;
DO 7.1-7.7
Palaemonetes 31 mm 96 st.; wellwater; m, LC50 3 s: acetone Eisler (1969)
vulgaris 0.47 g 24°/oo; 20 °C; ph 8;
DO 7.1-7.7
Brown shrimp
Penaeus aztecus adult 24 flow-through m, LC50 5.5 s: acetone Butler (1964)
29°/oo; 25 °C
Penaeus aztecus adult 48 flow-through m, LC50 5.5 s: acetone Butler (1964)
29°/oo; 25 °C
Table 18 (continued)
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
Pink shrimp
Penaeus duorerum - - flow-through m, LC50 1.9 s: acetone Schoor & Brausch,
17-31 °/oo + TEG (1980)
7.6-28.8 °C
Penaeus duorarum post- 96 flow-through m, LC50 1.2 s: TEG Mayer (1987)
larvae natural seawater P: 99%
20 °/oo; 25 °C
Japanese shrimp
Peneaus post- 24 25 °C m, LC50 0.5-0.9 - Hirayama & Tamaoi
japonicus larve (1980)
Shrimp
Penaeus post- 96 st.; natural seawater m, LC50 1.4 s: TEG Mayer (1987)
stylirostris larvae 20 °/oo; 25 °C; P: 99%
DO 5.6-6.3
Penaeus adult 96 st.; 15 °/oo; m, LC50 148 - Reddy & Rao (1986)
monodon 23 °C; pH 7.3
Penaeus adult 96 st.; 15 °/oo; m, LC50 98 - Reddy & Rao (1986)
indicus 23 °C; pH 7.3
Table 18 (continued)
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
Penaeus (inter-molt) 48 st.; seawater; m, LC50 95-Reddy & Rao (1986)
indicus 2.5 g 15 °/oo; 23 °C
pH 7.1
Metapenaeus adult 96 st.; 15 °/oo m, LC50 102 - Reddy & Rao (1986)
monoceros 23 °C; pH 7.3
Metapenaeus (inter-molt) 48 st.; seawater; m, LC50 120- Reddy & Rao (1988)
monoceros 2.5 g 15 °/oo; 23 °C;
pH 7.1
Metapenaeus adult 96 st.; 15 °/oo m, LC50 115 - Reddy & Rao (1986)
dopsoni 23 °C; pH 7.3
INSECTA
Mosquito
Culex piplens 4th 24 st.; 28 °C; m, LC50 30 P: 98.2% Yasuno et al. (1965)
instar deionized s: ethanol
larva water
Culex piplens 4th 24 st.; 28 °C; m, LC50 2000 P: 98.2% Yasuno et al. (1965)
instar polluted s: ethanol
larva water
Culex piplens 4th 96 st.; 28 °C; m, LC50 30 P: 98.2% Yasuno et al. (1965)
instar deionized s: ethanol
larva water
Table 18 (continued)
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
Culex pipiens 4th 96 st.; 28 °C; m, LC50 80 000 P: 98.2% Yasuno et al. (1965)
instar polluted s: ethanol
larva water
Chactorus 1st 24 st.; lake m, LC50 1.6 P: techn.gr. Apperson et al.
astictopus instar water; 25 °C s: acetone (1978)
larva (1962 exper.)
Chactorus 4th 24 st.; lake m, LC50 30 P: techn.gr. Apperson et al.
astictopus instar water; 25 °C s: acetone (1978)
larva (1962 exper.)
Chactorus 1st 24 st.; lake m, LC50 18 P: techn.gr. Apperson et al.
astictopus instar water; 25 °C s: acetone (1978)
larva (1978 exper.)
Chactorus 4th 24 st.; lake m, LC50 85 P: techn.gr. Apperson et al.
astictopus instar water; 25 °C s: acetone (1978)
larva (1978 exper.)
Table 18 (continued)
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
Damselfly
Ischnura larva 96 st.; reconst. m, LC50 33 P: 98.7% Mayer & Ellersiack
venticalus water; 15 °C s: acetone (1987)
pH 7.2-7.5
H 167-272
FISH
(Freshwater)
Betta adult 120 tap-water; 25 °C m, LC50 7500-8000 s: haxana Walsh & Hanselka
splendens pH 7-7.4 (1972
Goldfish
Carassius 0.6- 96 st.; reconst. m, LC50 9000 P: 80% Mayer & Ellersieck
auratus 1.7 g water; 18 °C; s: acetone (1986)
pH 7.1
Carassius 4.6 cm 24 st.; dest. m, LC50 14 000 P: 80% Pickering et al.
auratus 1.2 g water; 25 °C; s: acetone (1962)
pH 7.4-7.5
H 20; DO 4-8
Carassius 4.6 cm 48 st.; distilled m, LC50 12 000 P: 80% Pickering et al.
auratus 1.2 g water; 25 °C; s: acetone (1962)
pH 7.4-7.5
H 20; DO 4-8
Table 18 (continued)
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
Carassius 4.6 cm 96 st.; distilled m, LC50 12 000 P: 80% Pickering et al.
auratus 1.2 g water; 25 °C; s: acetone (1962)
pH 7.4-7.5
H 20; DO 4-8
Golden carp
Cyprinus - 48 st.; 24 °C m, LC50 > 10 000 P: 80% Nishiuchi &
auratus s: acetone Hashimoto (1967)
Carp
Cyprinus < 1 year 24 st.; 20 °C m, LC50 27 600 P: 80% Rehwoldt et al.
carpio pH 7.2; DO 6; s: acetone (1977)
H 50
Cyprinus < 1 year 48 st.; 20 °C m, LC50 21 200 P: 80% Rehwoldt et al.
carpio pH 7.2; DO 6; s: acetone (1977)
H 50
Cyprinus < 1 year 96 st.; 20 °C m, LC50 14 800 P: 80% Rahwoldt et al.
carpio pH 7.2; DO 6; s: acetone (1977)
H 50
Table 18 (continued)
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
Cyprinus 35 g 48 st.; 20 °C m, LC50 12 000 P: 80% Nagaratnamma &
carpio pH 7.2; DO 6; s: acetone Ramamurthi (1982)
H 50
Cyprinus 0.6- 96 st.; reconst. m, LC50 7130 P: 80% Mayer & Ellersieck
carpio 1.7 g water; 18 °C s: acetone (1986)
pH 7.1
Cyprinus 0.6 g 96 st.; reconst. m, LC50 8900 P: techn.gr. Johncon & Finley
carpio 1.7 g water; 18 °C s: acetone (1980)
pH 7.2-7.5
H 40-50
Cyprinus 0.6 g 48 st.; 24 °C m, LC50 > 10 000 P: techn.gr. Nishiuchi &
carpio s: acetone Hashimoto (1967)
Banded killifish
Fundulus < 1 year 24 st.; 20 °C m, LC50 24 900 P: techn.gr. Rehwoldt et al.
diaplanus pH 7.2; DO 6; s: acetone (1977)
H 50
Fundulus < 1 year 48 st.; 20 °C m, LC50 18 600 P: techn.gr. Rehwoldt et al.
diaplanus pH 7.2; DO 6; s: acetone (1977)
H 50
Fundulus < 1 year 96 st.; 20 °C m, LC50 15 200 P: techn.gr. Rehwoldt et al.
diaplanus pH 7.2; DO 6; s: acetone (1977)
H50
Table 18 (continued)
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
Mosquito fish
Gambusia adult 48 st.; dechlorinated m, LC50 13 480 P: 99% Chambers &
affinis non tap-water s: Yarbrough (1974)
resistent methoxy-ethanol
Gambusia adult 48 st.; dechlorinated m, LC50 17 480 P: 99% Chambers &
affinis non tap-water s: Yarbrough (1974)
resistent methoxy-ethanol
Catfish
Heteropneustes adult 96 24 °C; pH 7,7; m, LC50 7000 s: acetone Srivastava & Singh
fossilis (fem) DO 6; H 117 (1981)
Heteropneustes 16 cm 24 st.; 23 °C; m, LC50 9400 s: acetone Singh & Srivastava
fossilis 35 g pH 7.7; DO (1982)
6.1; H 115
Heteropneustes 16 cm 48 st.; 23 °C; m, LC50 8600 s: acetone Singh & Srivastava
fossilis 35 g pH 7.7; DO (1982)
6.1; H 115
Heteropneustes 16 cm 72 st.; 23 °C; m, LC50 8000 s: acetone Singh & Srivastava
fossilis 35 g pH 7.7; DO (1982)
6.1; H 115
Table 18 (continued)
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
Heteropneustes 16 cm 96 st.; 23 °C; m, LC50 7000 s: acetone Singh & Srivastava
fossilis 35 g pH 7.7; DO (1982)
6.1; H 115
Black Bullhead
Ictalurus melas 0.6- 96 st.; reconst. m, LC50 6640 P: 80% Mayer & Ellersieck
1.7 g water; 18 °C s: acetone (1986)
pH 7.1
Catfish
Mystus cavasius 6-8 cm 96 26-30 °C m, LC50 5900 - Murty & Ramani (1982)
7g
Channel Catfish
Ictalurus 1.4 g 96 st.; reconst. m, LC50 5240 P. techn.gr. Mayer & Ellersieck
punctatus water; 18 °C s: acetone (1986)
pH 7.2-7.5; H 40-50
Guppy (Poecilia
reticulata)
Lebistes 6 mon. 24 st.; distilled m, LC50 11 000 P. 80% Pickering et al.
reticulatus water; 25 °C s: acetone (1962)
pH 7.4-7.5;
H 20, DO 4-8
Table 18 (continued)
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
Lebistes 6 mon. 48 st.; distilled m, LC50 9800 P. 80% Pickering et al.
reticulatus water; 25 °C s: acetone (1962)
pH 7.4-7.5;
H 20, DO 4-8
Lebistes 6 mon. 96 st.; distilled m, LC50 9800 P. 80% Pickering et al.
reticulatus water; 25 °C s: acetone (1962)
pH 7.4-7.5;
H 20, DO 4-8
Lebistes < 1 year 24 st.; 20 °C m, LC50 12 200 P. 80% Rehwoldt et al.
reticulatus pH 7.2; DO 6 s: acetone (1977)
H 20
Lebistes < 1 year 48 st.; 20 °C m, LC50 9400 P. 80% Rehwoldt et al.
reticulatus pH 7.2: DO 6 s: acetone (1977)
H 20
Lebistes < 1 year 96 st.; 20 °C m. LC50 6200 P. 80% Rehwoldt et al.
reticulatus pH 7.2; DO 6 s: acetone (1977)
H 20
Table 18 (continued)
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
Green sunfish
Lepomis 0.8 g 96 st.; reconst. m, LC50 6860 P: techn.gr. Mayer (1987)
cyanellus water; 17 °C s: acetone
pH 7.2-7.5;
H 40-50
Lepomis 0.8 g 48 st.; tap-water m, LC50 > 5000 P: techn.gr. Minchew & Ferguson
cyanellus 20 °C s: acetone (1969)
Pumpkinseed
Lepomis 40-50 g 24 injection, st. m, LD50 > 2500 P: 99% Benke et al. (1974)
gibbosus s: corn oil
Lepomis < 1 year 24 st.: 20 °C m, LD50 4900 P: 99% Rehwoldt et al.
gibbosus pH 7.2; DO 6; s: corn oil (1977)
H 50
Lepomis < 1 year 48 st.: 20 °C m, LD50 3600 P: 99% Rehwoldt et al.
gibbosus pH 7.2; DO 6; s: corn oil (1977)
H 50
Lepomis < 1 year 96 st.: 20 °C m, LD50 3600 P: 99% Rehwoldt et al.
gibbosus pH 7.2; DO 6; s: corn oil (1977)
H 50
Table 18 (continued)
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
Bluegill sunfish
Lepomis fingerling 24 st.; reconst. m, LC50 6470 P: 44.6% McCann & Jasper
machrochirus water; 18 °C s: water (1972)
pH 7; H 17
Lepomis 0.6- 96 st.; reconst. m, LC50 5720 P: 80% Macak & McAllister
machrochirus 1.7 g water; 18 °C s: acetone (1970)
pH 7.1
Lepomis 4-6 cm 24 st.; distilled m, LC50 9800 P: 80% Pickering et al.
machrochirus 1.2 g water; 25 °C s: acetone (1962)
pH 7.4-7.5
H 20; DO 4-8
Lepomis 4-6 cm 48 st.; distilled m, LC50 8600 P: 80% Pickering et al.
machrochirus 1.2 g water; 25 °C s: acetone (1962)
pH 7.4-7.5
H 20; DO 4-8
Lepomis 4-6 cm 96 st.; distilled m, LC50 2400 P: 80% Pickering et al.
machrochirus 1.2 g water; 25 °C s: acetone (1962)
pH 7.2-7.5;
H 20; DO 4-6
Table 18 (continued)
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
Lepomis 1 g 96 st.; reconst. m, LC50 4380 P: techn.gr. Mayer & Ellersieck
machrochirus water; 17 °C s: acetone (1986)
pH 7.2-7.5
H 40-50
Lepomis 0.6- 96 st.; reconst. m, LC50 5170 P: 80% Macek & McAllister
machrochirus 1.7 g water; 18 °C s: acetone (1970)
pH 7.1
Largemouth bass
Micropterus 0.6- 96 st.; reconst. m, LC50 5220 P: 80% Mayer & Ellersieck
salmoides 1.7 g water; 18 °C s: acetone (1986)
pH 7.1
Mystus cavasius - 96 - m, LC50 5900 - Murty & Ramani
(1982)
Golden shiner
Notamigonus - 48 st.; tap-water: m, LC50 > 5000 P: techn.gr. Minchew & Ferguson
chrysoleuces 20 °C s: acetone (1969)
Coho salmon
Oncorhynchus 0.6- 96 st.; reconst. m, LC50 5300 P: 80% Meyer & Ellersieck
kisutch 1.7 g water; 13 °C s: acetone (1986)
pH 7.1
Table 18 (continued)
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
Medaka
Oryzias latipes - 48 er.; 24 °C m, LC50 7500 P: techn.gr, Nishiuchi &
s: acetone Hashimoto (1967)
Yellow perch
Perca 1.4 g 96 st.; reconst. m, LC50 3060 P: techn.gr. Mayer & Ellersieck
flavescens water; 18 °C s: acetone (1986)
pH 7.2-7.5;
H 40-50
Punti
Puntius 6- 24 st.; 27.9 °C m, LC50 2900 P: 50% Rao et al. (1967)
puckelli 8.5 cm pH 8.3; H 130
Puntius 6- 48 st.; 27.9 °C m, LC50 2700 P: 50% Rao et al. (1967)
puckelli 8.5 cm pH 8.3; H 130
Puntius 6- 96 st.; 27.9 =C m, LC50 2100 P: 50% Rao et al. (1967)
puckelli 8.5 cm pH 8.3; H 130
Table 18 (continued)
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
Fathead minnow
Pimephales 1.2 g 96 st.; reconst. m, LC50 8300 P: techn.gr. Mayer & Ellersieck
promelas water; 18 °C s: acetone (1986)
pH 7.2-7.5; H40-50
Pimephales - 48 flow-through m, LC50 7400 P: 98.5% Solon & Nair (1970)
promelas s: acetone
Pimephales - 96 flow-through m, LC50 3750 P: 98.5% Solon & Nair (1970)
promelas s: acetone
Pimephales newly 96 st.; sterilized m, LC50 4460 P: 80% Jarvinen & Tanner
promelas hatched water; 25 °C (1982)
larvae pH 7.4-7.8;
DO 6.5-8.4; H 64
Pimephales newly 96 st.; sterilized m, LC50 1220 P: 80% Jarvinen & Tanner
promelas hatched water; 25 °C DO 6.5-8.4; H stock (1982)
larvae pH 7.4-7.8; 64 solution aged
11 weeks
Pimephales newly 96 st.; sterilized m, LC50 8170 P: 80% Jarvinen & Tanner
promelas hatched water; 25 °C controlled- (1982)
larvae pH 7.4-7.8; release
DO 6.5-8.4; H 64 formulation
Table 18 (continued)
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
Pimephales newly 96 st.; sterilized m, LC50 3470 P: 80% Jarvinen & Tanner
promelas hatched water; 25 °C controlled (1982)
larvae pH 7.4-7.8; release
DO 6.5-8.4; H 64 formulation
for 11 weeks
Pimephales newly 96 flow-through; m, LC50 5360 P: 80% Jarvinen & Tanner
promelas hatched sterilized water (1982)
larvae 25 °C; pH 7.4-
7.8; DO 6.5-8.4;
H 64
Pimephales newly 96 flow-through; m, LC50 6910 P: 80% Jarvinen & Tanner
promelas hatched sterilized water controlled (1882)
larvae 25 °C; pH release
7.4-7.8; formulation
DO 6.5-8.4;
H 64
Pimephales 4-6 cm 24 st.; distilled m, LC50 13 000 P: 80% Pickering et al.
promelas 1-2 g water; 25 °C s: acetone (1862)
pH 7.4-7.5;
H 20; DO 4-8
Table 18 (continued)
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
Pimephales 4-6 cm 48 st.; distilled m, LC50 9800 P: 80% Pickering et al.
promelas 1-2 g water; 25 °C s: acetone (1962)
pH 7.4-7.5;
H 20; DO 4-8
Pimephales 4-6 cm 96 st.; distilled m, LC50 9500 P: 80% Pickering et al.
promelas 1-2 g water; 25 °C s: acetone (1962)
pH 7.4-7.5;
H 20; DO 4-8
White perch
Roccus < 1 year 24 st.; 12 °C m, LC50 22 400 P: 80% Rehwoldt et al.
americanus pH 7.2 s: acetone (1977)
Roccus < 1 year 46 st.; 12 °C m, LC50 18 600 P: 80% Rehwoldt et al.
americanus pH 7.2 s: acetone (1977)
Roccus < 1 year 96 st.; 12 °C m, LC50 14 000 P: 60% Rehwoldt et al.
americanus pH 7.2 s: acetone (1977)
Cutthroat trout
Salmo clarki 0.2 g 96 st.; reconst. m, LC50 1850 P: techn.gr. Mayer & Ellersieck
water; 12 °C s: acetone (1996)
pH 7.2-7.5;
H 162-272
Table 18 (continued)
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
Rainbow trout
(Oncorhynchus
mykiss)
Salmo 1.1 g 96 st.; reconst. m, LC50 3700 P: techn.gr. Mayer & Ellersieck
gairdneri water; 12 °C; s: acetone (1986)
pH 7.2-7.5;
H 162-272
Salmo 0.6- 96 st.; reconst. m, LC50 2750 P: 80% Macek & McAllister
gairdneri 1.7 g water; 13 °C; s: acetone (1970)
pH 7.1
Salmo 24 mm 96 st.; 12 °C m, LC50 2800 P: 76.8% Palawski et al.
gairdneri (1983)
Brown trout
Salmo trutta 0.6- 96 st,; reconst, m, LC50 4750 P: 80% Mayer & Ellersieck
1.7 g pH 7.1 s: acetone (1986)
water; 13 °C
Table 18 (continued)
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
Brook trout
Salvelinus 0.5 g 96 st.; reconst. m, LC50 3780 P: techn.gr. Mayer & Ellersieck
fontinalis water; 12 °C s: acetone (1986)
pH 7.2-7.5;
H 40-50
Northern pike
Esox lucius 0.4 g 24 st.; 18 °C m, LC50 760 P: techn.gr. Mayer & Ellersieck
pH 7.1; H 44 s: acetone (1986)
Tilapia
tilapia - 48 st.; 26-28 °C m, LC50 266 P: techn.gr. Rao & Rao (1983)
mossambica pH 7; H 140 s:
2-methoxyethanol
ESTUARINE
AND MARINE
American eel
Anguilla 59 24 st.; underground m, LC50 27 600 P: act, Eisler (1970a)
rostrata mm-0.14 g wellwater; ingredient
24%0; 20 °C; s: acetone
pH 8; DO 7.1-7.7
Table 18 (continued)
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
Anguilla 59 48 st.; underground m, LC50 22 400 P: act. Eisler (1970a)
rostrata am-0. 14 g wellwater; ingredient
24°/oo; 20 °C; s: acetone
pH 8; DO 7.1-7.7
Anguilla 59 96 st.; underground m, LC50 16 900 P: act. Eisler (1970e)
rostrate am-0.14 g wellwater; ingredient
24°/oo; 20 °C; s: acetone
pH 8; DO 7.1-7.7
Anguilla < 1 year 24 st.; 20 °C. m, LC50 42 600 P: act. Rehwoldt et al.
rostrata pH 7.2; DO 6; ingredient (1977)
H 50 s: acetone
Anguilla < 1 year 48 st.; 20 °C. m, LC50 37 200 P: act. Rehwoldt et al.
rostrata pH 7.2; DO 6; ingredient (1977)
H 50 s: acetone
Anguilla < 1 year 96 st.; 20 °C. m, LC50 6300 P: act. Rehwoldt et al.
rostrata pH 7.2; DO 6; ingredient (1977)
H 50 s: acetone
Table 18 (continued)
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
Sheephead
minnow
Cyprinodon 28 days 96 st.; natural m, LC50 12 000 P: 99% Mayer (1987)
variegatus old sea-water; s: TEG
20°/oo; 25 °C;
DO 4.6-5.7
Cyprinodon 28 days 96 st.; natural no effect 10 000 P: 99% Mayer (1987)
variegatus old sea-water; s: TEG
20°/oo; 25 °C;
DO 4.6-5.7
Mummichog
Fundulus 55 mm 24 st.; underground m, LC50 > 85 100 P: act. Eisler (1970a)
heteroclitus 1.7 g wellwater; 24°/oo; ingredient
20 °C; pH 8; s: acetone
DO 7.1-7.7
Fundulus 55 mm 48 st.; underground m, LC50 85 200 P: act. Eisler (1970a)
heteroclitus 1.7 g wallwater; 24°/oo; ingredient
20 °C; pH 8; s: acetone
DO 7.1-7.7
Fundulus 55 mm 96 st.; underground m, LC50 58 000 P: act. Eisler (1970a)
heteroclitus 1.7 g wellwater; 24°/oo; ingredient
20 °C; pH 8; s: acetone
DO 7.1-7.7
Table 18 (continued)
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
Fundulus 42 mm 96 st.: underground m, LC50 8000 P: act. Eisler (1970b)
heteroclitus wellwater; 24°/oo; ingredient
20 °C; pH 8; s: acetone
DO 7.1-7.7
Fundulus 42 mm 96 st.; underground m, LC50 4000 P: act. Eisler (1970b)
heteroclitus ( + 240h wellwater; 24°/oo; ingredient
observation) 20 °C; pH 8; s: acetone
DO 7. 1-7.7
Fundulus 42 mm 96 st.; underground m, LC50 1210 solution Eisler (1970b)
heteroclitus wellwater; 24°/oo; aged for
20 °C; pH 8; 96 h
DO 7.1-7.7
Fundulus 42 mm 96 10 °C 20% M 8000 P: act. Eisler (1970b)
heteroclitus ingredient
s: acetone
Fundulus 42 mm 96 15 °C 10% M 8000 P: act. Eisler (1970b)
heteroclitus ingredient
s: acetone
Table 18 (continued)
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
Fundulus 42 mm 96 36 °/oo 100% M 8000 P: act. Eisler (1970b)
heteroclitus ingredient
s: acetone
Striped killifish
Fundulus 84 mm 24 st.; underground m, LC50 29 000 P: act. Eisler (1970a)
majalis 6.5 g wellwater; 24°/oo; ingredient
20 °C; pH 8; s: acetone
DO 7.1-7.7
Fundulus 84 mm 48 st.; underground m, LC50 19 400 P: act. Eisler (1970a)
majalis 6.5 g wellwater; 24°/oo; ingredient
20 °C; pH 8; s: acetone
DO 7.1-7.7
Fundulus 84 mm 24 st.; underground m, LC50 13 800 P: act. Eisler (1970a)
majalis 6.5 g wellwater; 24°/oo; ingredient
20 °C; pH 8; s: acetone
DO 7.1-7.7
Table 18 (continued)
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
Fundulus 42 mm 96 20 °C 50% M 8000 P: act. Eisler (1970b)
heteroclitus ingredient
s: acetone
Fundulus 42 mm 96 25 °C 100% M 8000 P: act. Eisler (1970b)
heteroclitus ingredient
s: acetone
Fundulus 42 mm 96 30 °C 100% M 8000 P: act. Eisler (1970b)
heteroclitus ingredient
s: acetone
Fundulus 42 mm 96 12 °/oo 0% M 8000 P: act. Eisler (1970b)
heteroclitus ingredient
s: acetone
Fundulus 42 mm 96 18 °/oo 0% M 8000 P: act. Eisler (1970b)
heteroclitus ingredient
s: acetone
Fundulus 42 mm 96 24 °/oo 10% M 8000 P: act. Eisler (1970b)
heteroclitus ingredient
s: acetone
Fundulus 42 mm 96 30 °/oo 70% M 8000 P: act. Eisler (1970b)
heteroclitus ingredient
s: acetone
Table 18 (continued)
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
Spot
Leiostomus 84 mm 96 st.; natural m, LC50 93 P: 99% Mayer (1987)
xanthurus 6.5 g seawater; 2°/oo; s: TEG
25 °C; DO 3.2-4.5
Leiostomus 84 mm 96 st.; natural no effect 56 P: 99% Mayer (1987)
xanthurus 6.5 g seawater; 2°/oo; s: TEG
25 °C; DO 3.2-4.5
Leiostomus 84 mm 96 flow-through m, LC50 59 P: 99% Mayer (1987)
xanthurus 6.5 g 20°/oo; 25 °C s: TEG
Atlantic silverside
Menidia 50 mm 24 st.; underground m, LC50 24 800 P: act. Eisler (1970a)
menidia 0.8 g. wellwater; 24°/oo; ingredient
20 °Cr pH 8r s: acetone
DO 7.1-7.7
Menidia 50 mm 48 at.; underground m, LC50 21 900 P: act. Eisler (1970a)
menidia 0.8 g wellwater; 24°/oo; ingredient
20 °Cr pH 8; s: acetone
DO 7.1-7.7
Menidia 50 mm 96 st.; underground m, LC50 5700 P: act. Eisler (1970a)
menidia 0.8 g wellwater; 24°/oo; ingredient
20 °C; pH 8; s: acetone
DO 7.1-7.7
Table 18 (continued)
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
Striped bass
Morone 1 year 24 st.; 20 °C; m, LC50 16 800 P: act. Rehwoldt et al.
saxatilis pH 7.2; DO 6; ingredient (1977)
H 50 s: acetone
Morone 1 year 48 st.; 20 °C; m, LC50 14 200 P: act. Rehwoldt et al.
saxatilis pH 7.2; DO 6; ingredient (1977)
H 50 s: acetone
Morone 1 year 96 st.; 20 °C; m, LC50 14 000 P: act. Rehwoldt et al.
saxatilis pH 7.2; DO 6; ingredient (1977)
H 50 s: acetone
Morone adult 96 interm. flow m, LC50 790 P: 99% Earnest (1970)
saxatilis 12.8 °
Morone juvenile 96 flow-through m, LC50 790 P: 86% Korn & Earnest
saxatilis 13 °C; 30 °/oo s: ethanol (1974)
Black mullet
Mugil cephalus 48 mm 24 st.: underground m, LC50 39 000 P: act. Eisler (1970a)
0.78 g wellwater; 24 °/oo ingredient
20 °C; pH 8; acetone
DO 7.1-7.7
Table 18 (continued)
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
Mugil cephalus 48 mm 48 st.: underground m, LC50 26 300 P: act. Eisler (1970a)
0.78 g wellwater; 24 °/oo ingredient
20 °C; pH 8; acetone
DO 7.1-7.7
Mugil cephalus 48 mm 96 st.: underground m, LC50 5200 P: act. Eisler (1970a)
0.78 g wellwater; 24 °/oo ingredient
20 °C; pH 8; acetone
DO 7.1-7.7
Northern puffer
Sphaeroides 196 mm 24 st.; underground m, LC50 100 000 P: act.. Eisler (1970a)
maculatus 153 g wellwater; 24 °/oo ingredient
20 °C; pH 8; s: acetone
DO 7.1-7.7
Sphaeroides 196 mm 48 st.; underground m, LC50 91 000 P: act. Eisler (1970a)
maculatus 153 g wellwater; 24 °/oo ingredient
20 °C; pH 8; s: acetone
DO 7.1-7.7
Sphaeroides 196 mm 96 st.; underground m, LC50 75 800 P: act. Eisler (1970a)
maculatus 153 g wellwater; 24 °/oo ingredient
20 °C; pH 8; s: acetone
DO 7.1-7.7
Table 18 (continued)
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
Bluehead
Thalassoma 90 mm 24 st.; underground m, LC~ 98 000 P. act. Eisler (1970a)
bifasciatum 7 g wellwater; 24 °/oo ingredient
20 °C; pH 8;
DO 7.1-7.7
Thalassoma 90 mm 48 st.; underground m, LC50 88 000 P. act. Eisler (1970a)
bifasciatum 7 g wellwater; 24 °/oo ingredient
20 °C; pH 8;
DO 7.1-7.7
Thalassoma 90 mm 96 st.; underground m, LC50 12 300 P. enct. Eislar (197Oa)
bifasciatum 7 g wellwater; 24 °/oo ingredient
20 °C; pH 8; s: acetone
DO 7.1-7.7
AMPHIBIA
Rana adult 96 st.; tap-water: m, LC50 8000 Mudgall & Patil
cyanophlyctis 23 °C; pH 7.3-7.8; (1987)
(male) H 60-70; DO 6.7-7.9
Rana adult 96 st.; tap-water: m, LC50 11 500 Mudgall & Patil
cyanophlyctis 23 °C; pH 7.3-7.8; (1987)
(female) H 60-70; DO 6.7-7.9
Table 18 (continued)
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
Western chorus
frog
Bendacris tadpole 96 st.; 15 °C; m, LC50 3700 Mayer & Ellersieck
triseriata pH 7.1; H 44 (1986)
a Criterion: m = mortality; i = immobilization; d = development; % M = % mortality.
b P = purity; s = solvent; TEG = triethyiene glycol; techn.gr. = technical grade.
c st. = static.
d °/oo = salinity.
e H = hardness in mg/litre CaCO3.
f DO = dissolved oxygen in mg O2/litre.
g MATC = maximum acceptable toxic concentration.
Murty et al. (1984) state that the lowest concentration causing
irreversible effects in the fish Mystus carasius after a 1-h exposure
was 15 mg/litre.
7.2.1 Short-term toxicity in aquatic invertebrates
7.2.1.1 Laboratory studies on single species
Exposure of the freshwater mussel (Lamellidens marginalis) to
sublethal (8 mg/litre) concentrations resulted in a transient increase
(at 12 h) followed by a decrease (at 24-72 h) in the rate of
respiration (Moorthy et al., 1984). Exposure of this species to
concentrations ranging from 10 to 50 mg/litre resulted in a
concentration-dependent decrease in heart rate (Rao et al., 1983a).
For crustaceans, long-term toxicity levels appear to be of the
same magnitude as acute: a no-effect level on the reproduction of
Daphnia magna was 0.0012 mg methyl parathion/litre after 21 days
(artificial water, 18 °C; Dortland, 1980).
Exposure of the freshwater crab (Oziotelphusa senex senex) to
sublethal levels of methyl parathion (0.1-1 mg/litre) resulted in
complete inhibition of molt, a delay in the onset of molt, or a
decrease in the percentage of molting animals (Reddy et al., 1985). A
decrease in the carbohydrate content and increase in acid phosphatase
activity in both the hepatopancreas and muscle also occurred (Reddy et
al., 1986a; 1986b).
Eisler (1970a,b) found a 20% increase in mortality in Nassa
docoleta after 10 days' exposure to 25 mg/litre (well water with a
salinity of 24o/oo, 20°C, pH 8) in.
Exposure of prawns (Penaeus indicus or Metapenaeus monoceros)
to sublethal concentrations of methyl parathion resulted in a
concentration-dependent inhibition of acetylcholinesterase activity,
which recovered in 7 days (Reddy & Rao, 1988). An increase in tissue
levels of ammonia, urea, and glutamine, apparently resulted from the
increased production of ammonia from purines and glutamate (Reddy et
al., 1988; Reddy & Rao, 1990a). There was also an increase in tissue
levels of fatty acids and cholesterol (Reddy & Rao, 1989), while the
activity of alkaline phosphatase in the hepatopancreas was inhibited,
and the acid phosphatase activity, enhanced (Reddy & Rao, 1990b).
Changes in hepatic glycogen content and haemolymph glucose levels were
observed after 5 days of sublethal methyl parathion exposure (Reddy &
Rao, 1990b).
Cripe et al. (1981) tested the stamina of mysid shrimp
(Mysidopsis bahia) in swimming against a water current in the
presence of methyl parathion. Concentrations of 0.10 and 0.31 µg/litre
did not affect maximum sustained speeds of the shrimp, but they
were significantly reduced on exposure to 0.58 µg/litre.
7.2.1.2 Mesocosmic studies
After treatment of ponds with methyl parathion, the effects on
daphnids were similar to those observed in the laboratory. However,
indirect biological effects occurred that could not be predicted on
the basis of laboratory tests. For example, the observed increase in
populations of the crustacean Diaptomus sp. in treated ponds was
attributed to the mortality of competitors (Daphnia spp.) and
predators ( Cyclops and aquatic insects) (Crossland & Elgar, 1983).
Generally, recovery of zooplankton occurred soon after the end of
treatment of ponds (Apperson et al., 1976; Crossland & Elgar, 1983).
The numbers of free-swimming Diptera and Ephemeroptera were
significantly reduced compared with controls, as were the benthic
chironomid larvae in ponds treated at 100 µg/litre. Seventy days after
treatment, there was evidence of recovery of populations of
chironimids and Ephemeroptera , with full recovery 90 days after
treatment (Crossland & Elgar, 1983).
7.2.2 Fish
7.2.2.1 Laboratory studies on single species
Jarvinen & Tanner (1982) conducted a long-term mortality study on
the fish Pimephales promelas (flow through conditions, sterile
water, 25 °C, pH 7.4-7.8, 46 mg CaCO3/litre, 6.5-8.4 mg dissolved
O2/litre). Methyl parathion concentrations of 0.59-0.77 mg/litre
induced increased mortality after 32 days. No effects on mortality
were found at 0.38 mg/litre for the technical grade product and 0.59
mg/litre for the controlled release formulation. Mortality in rainbow
trout (Salmo gairdneri) increased to 98% after exposure to 2.8 mg
technical grade methyl parathion/litre (wellwater, 12 °C, pH 7.5, 272
mg CaCO3/litre) for 96 h, followed by 7 days of observation
(Palawski et al., 1983).
Exposure of the tilapia fish (Tilapia mossambica) to methyl
parathion at a concentration of 0.09 mg/litre for 48 h resulted in a
decrease in various anions and cations in tissues (Rao et al., 1983b),
and in inhibition of acetylcholinesterase (20-60%) and ATPase (10-14%)
activities. The activities of aspartate and alanine amino-transferase
in muscle, gill, liver, and brain increased by 12-31% and 9-31%,
respectively (Rao & Rao, 1984a; 1984b). Concentrations of
carbohydrate and glycogen decreased in the tissues examined (Rao &
Rao, 1983). Levels of soluble protein and the activity of
glucose-6-phosphate dehydrogenase, a key enzyme of the hexose
monophosphate shunt, in muscle, gill, and liver, were increased (Rao
& Rao, 1987). Changes in carbohydrate metabolism were also observed in
the freshwater fish Clarias batrachus , when exposed to sublethal
concentrations of methyl parathion (7 mg/litre) for 48 and 96 h (Rani
et al., 1989). There were significant decreases in glycogen (liver)
and in pyruvate (liver, brain, gill) contents and increases in glucose
(gill) and lactate (liver, brain, gill) levels, and the specific
activities of several enzymes were inhibited.
Exposure of the catfish (Channa punctatus) to 52 µg methyl
parathion/litre resulted in the elevation of serum triiodothyronine
(T3) as well as depression of brain acetylcholinesterase activity
(Ghosh et al., 1989). This low dose of methyl parathion also impaired
the regulation of gonadal function by gonadotropic hormone and gonado
tropin-releasing hormone in Channa punctatus (Ghosh et al., 1990).
The inhibiting effect was also seen under field conditions where water
concentrations of methyl parathion amounted to 0.239 µg/litre (Ghosh
et al., 1990).
Exposure to sublethal doses of 0.1 mg methyl parathion/litre
(corresponding to 1/5th of the LC50 values) for 75 days produced
severe ovarian damage in the carp minnow (Rasbora daniconius)
(Rastogi & Kulshrestha, 1990). Effects included diminished growth of
ovaries and histopathological changes in immature, maturing, and
mature oocytes.
Sublethal concentrations of methyl parathion (1.2 mg/litre)
induced behavioural abnormalities in the juveniles of the fish
Cyprinus carpio , such as imbalance, increased opercular movement
and irritation (Babu et al., 1986). Exposed juveniles, when
transferred to pesticide-free medium, showed rapid recovery.
Little et al. (1990) exposed rainbow trout (Oncorhynchus mykiss)
to methyl parathion at 0.01 or 0.1 mg/litre and measured various
behavioural parameters. Swimming capacity (as cm/s) was unaffected at
any concentration tested, though spontaneous swimming activity was
significantly reduced at both exposures. Number of prey (Daphnia)
consumed was reduced, even at the lower exposure (0.01 mg/litre), but
the percentage of daphnia consumed and the strike frequency of the
fish on daphnia were only affected at 0.1 mg/litre. The capacity of
the trout to escape from a predator was only reduced at 0.1 mg/litre.
In a static system (well water, salinity: 24o/oo, 20 °C, pH 8),
with the fish Fundulus heteroclitus, the LC50 was 0.96 mg/litre
after exposure for 10 days or 4 mg/litre after exposure for 4 days
followed by 10 days in clean water (Eisler, 1970b).
7.2.2.2 Mesocosmic studies
In a methyl parathion-treated experimental pond, a high mortality
rate was observed in rainbow trout, 37 days after treatment, which was
associated with depression of the concentration of dissolved oxygen to
less than 3 mg/litre, and decay of large amounts of algal biomass
(Crossland & Elgar, 1983; see also section 7.3).
7.2.3 Amphibians
After application of methyl parathion to Rana cyanophlyctis,
Mudgall & Patil (1987) found increased levels of glycogen in muscles,
liver, and kidney, compared with control animals. On the basis of the
marked elevated glycogen concentration in the kidney, it was concluded
that the kidneys were the main target organ.
The effects of metacid (DDT + 50% w/w methyl parathion) on the
development of the Indian bullfrog (Rana tigrina) were determined
by Mohanty-Hejmadi & Dutta (1981). Threshold concentrations for
adverse effects on eggs, feeding stage, and limb bud stage tadpoles
ranged from 0.00005% to 0.004% metacid. These levels were much lower
than the recommended dosage for the field application of metacid
(0.15%).
7.3 Terrestrial organisms
7.3.1 Plants
Methyl parathion has been found to have phytotoxic effects in
diverse crops, such as cotton (Gossypium hirsutum) (Brown et al.,
1962; Roark et al., 1963; Youngman et al., 1989, 1990) and lettuce
(Lactuca sativa) (Toscana et al., 1982; Johnson et al., 1983;
Youngman et al., 1989).
Swamy & Veeresh (1987) found a reduction in lipid synthesis in
methyl parathion-treated seeds of Sorghum sp., 24 h after
germination. An increase in lipid production with a substantial
elevation in unsaturated fatty acids was observed in methyl
parathion-treated sorghum, 120 h after germination. The same effect
occurred in 48-h seedlings, which were treated with the degradation
products of methyl parathion. From this, it was concluded that the
time-related reversal effect of methyl parathion is triggered by the
pesticide degradation products themselves.
Exposure of sorghum seeds to methyl parathion for 1 h before
germination resulted in an accumulation of proline in the seedlings
and a reduction in growth, without affecting the water content.
Residues of methyl parathion in the soil also influenced seed
germination and seedling growth (Deshpande & Swamy, 1987).
7.3.2 Invertebrates
Poisoning of bees has been reported after incorrect application
of methyl parathion on windy days (Bubien, 1971).
Analysis of dead honey bees (Apis mellifera; Hymenoptera) for
pesticide residues, during 1983-85 in the USA, showed that the health
of colonies, poisoned with methyl parathion (Penncap-M) or with a
combination of methyl parathion and other insecticides, was often
severely affected, whereas colonies contaminated by insecticides other
than methyl parathion often recovered (Anderson & Wojtas, 1986).
Acute toxicity values were established for acetone formulations
of methyl parathion applied topically to workers of Africanized and
European honey bees (Apis mellifera) (Danka et al., 1986). The
LC50 values of 0.32 µg and 0.17 µg/bee, respectively, showed the
greater tolerance to methyl parathion of Africanized bees compared
with European bees.
Jepson (1989) calculated a hazard ratio (ratio of contact LD50
at 0.11 µg/bee to the application rate of the pesticide at 500 g
a.i./ha) for methyl parathion in honey bees of 8937 (using the method
of Smart & Stevenson, 1982). Values of the hazard ratio greater than
50 are usually considered to indicate danger for bees. Along with
azinphos methyl, methyl parathion has a very high indication of
danger for bees from field spraying. Although the intrinsic toxicity
for bees is as high for other pesticides, such as the pyrethroids, the
hazard ratio is lower, since application rates of these pesticides are
also lower.
Methyl parathion applied to small barriered plots of spring wheat
at 1000 g a.i./ha did not have any apparent adverse effects on leaf
litter decomposition and on earthworm populations (species not
differentiated). Effects on individual earthworm species could not be
demonstrated, because of statistically insufficient numbers of mature
specimens collected (Shires, 1985).
Methyl parathion has adverse effects on many different beneficial
insects. It was placed in the highest class of toxicity (score 4 in a
classification of 1-4) for Chrysopa (Plannipennia), Coccinellidae
(Coleoptera), and Hymenoptera (Entomophaga) (Höbaus, 1987). Side
effects on the predator mite Phytoseiulus persimilis were placed in
class 3 (Kniehase & Zoebelein, 1990).
Thompson & Gore (1972) assessed the toxicity of methyl parathion
(95-99% purity) for Folsomia candida (Collembola) by direct contact
in a spray tower and when applied to soil. In the direct-contact
study, a 0.01% methyl parathion solution caused a 100% mortality of
the collembola, 24 h after being treated. A 100% mortality rate also
occurred in soil (Plainfield sand) treated with 0.5 mg methyl
parathion/kg dry weight soil after a 24-h exposure.
Methyl parathion (0.05%) sprayed on coconut leaflets was found to
be highly toxic to the parasitoid fauna (Hymenoptera; Ento-mophaga )
of a coconut coccid (Opisina arenosella; Homoptera). The mortality
of the caged insects was assessed 24 h after introduction of leaflets
and after longer periods (Jalaluddin & Mohanasundaram, 1989).
Flanders et al. (1984) conducted a field study of methyl
parathion (sprayed at recommended rates of 0.84 kg a.i./ha, in an
encapsulated formulation, on soybeans) effects on Pediobius
foveolatus , a parasitoid of the Mexican bean beetle Epilachnia
varivestis . The pupae within parasitized beetles were unaffected by
the insecticide and emerged normally. However, residues of methyl
parathion on the plants killed 100% of the adult parasites emerging
within 1 day, and 50% of those emerging within 3 days of the spraying.
By 9 days after spraying, the mortality of emerging parasite adults
was no longer affected by residues.
Walker et al. (1985) examined the effects of methyl parathion,
used at 0.6 kg a.i./ha on rice fields in Louisiana, on the survival
and reproduction of parasitic nematodes (Romanomermis culcivorax),
introduced into the fields to control mosquito larvae. There were not
any adverse effects of the insecticide on the nematodes.
Only a few cases of resistance to methyl parathion have been
reported among arthropod parasites or predators. The reports refer to
the braconid Bracon mellitor (Hymenoptera), a parasite of the boll
weevil (Anthonomous grandii), which developed low levels of
resistance after 5 or more generations of selection in the laboratory,
and to field populations of the coccinellid Coleomegilla maculata
(Coleoptera) taken from cotton fields, treated extensively with methyl
parathion for 2 decades (Croft, 1977).
One week after application of methyl parathion (1000 g a.i./ha)
on small barriered plots of spring wheat, the number of predatory
beetles (mainly 4 species of Carabidae and 3 genera of Staphylinidae)
fell to about 10% of that in the untreated control plot. Recovery
occurred between 4 and 6 weeks after application, but a further fall
in numbers of predatory beetles was observed 8-12 weeks after
application (Shires, 1985). This second reduction was attributed to an
indirect effect of the treatments, causing removal of the predators'
food supply (mainly cereal aphids).
7.3.3 Birds
The acute lethal toxicities of methyl parathion for birds are
compiled in Table 19.
Percutaneous administration of methyl parathion was more toxic
for young mallard ducks (Anas platyrhynchos) than oral (dietary)
administration (Hudson et al., 1979).
Studies on mallard ducks (Anas platyrhynchos) have shown that
methyl parathion can affect the brood-rearing phase by increasing
mortality and causing behavioural changes (Fairbrother et al., 1988).
At least 40% of young ducklings exposed to sub-lethal oral doses of
methyl parathion (4 mg/kg body weight) died within 40 min in outdoor
enclosures. Several activities (swimming, preening, feeding) of
mothers and ducklings were changed in treated broods. Ducks (Anas
platyrhynchos; A. discors; Aix sponsa) nesting in agricultural
fields aerially treated with methyl parathion (1.4 kg a.i./ha) had a
higher average daily rate of duckling losses than those nesting in
untreated fields (Brewer et al., 1988).
Spraying of methyl parathion at 1.4 kg a.i./ha did not
significantly reduce the hatchability of starling (Sturnus vulgaris)
eggs and the number of young fledglings per nest. However,
collectively, the number of fledglings from the treated field was
significantly lower than that from the control field (Robinson et al.,
1988).
Buerger et al. (1991) dosed wild bobwhite quail (Colinus
virginianus) with methyl parathion at 0, 2, 4, or 6 mg/kg body
weight by oral intubation and then released them into the wild. The
birds were monitored for 14 days by radio telemetry. Only the birds
receiving 6 mg methyl parathion/kg body weight showed significantly
reduced survival and this was the result of predation rather than
overt toxicity. Activity was not affected by any treatment. Survivors
did not show any inhibition of brain cholinesterase activity after 14
days, compared with controls.
Bennett et al. (1991) examined parameters of reproductive success
in mallard ducks exposed to a dietary concentration of methyl
parathion of 400 mg/kg. The female mallards were fed the methyl
parathion diet at different stages of egg laying and incubation.
Numbers of hatchlings per nest were 61%, 43%, and 58% of controls for
birds exposed during egg laying, early incubation, and late
incubation, respectively. Daily egg production was reduced during the
treatment period, though 4 out of 10 hens resumed egg laying after
treatment was terminated.
A dose-dependent inhibition of brain and plasma cholinesterase,
hyperglycaemia, and elevated corticosterone concentrations were
observed in the American kestrel (Falco sparverius) exposed to oral
doses of up to 3 mg methyl parathion/kg body weight (Rattner &
Franson, 1984).
Table 19. Acute lethal toxicities of methyl parathion for birds
Species Age Oral LD50 Dietary LC50 References
(mg/kg body weighta) mg/kgb
Mallard duck 5 days 8 Fairbrother et al.
(Anas platyrhynchos) (1988)
3 months 10 Hudson et al. (1984)
adult 6.6
Mallard duck 10 days 682 Hill et al. (1975)
(Anas platyrhynchos)
Mallard duck 5 days 336 Hill et al. (1975)
(Anas platyrhynchos)
Kestrel > 8 3.08 Rattner & Franson
(Falco sparverius) months (1984)
Bobwhite quail 14 days 90 Hill et al. (1975)
(Colinus virginianus)
Table 19 (continued)
Species Age Oral LD50 Dietary LC50 References
(mg/kg body weighta) mg/kgb
Bobwhite quail 14 days 91 Bennet (1989)
(Colinus virginianus)
Japanese quail 14 days 79 Hill et al. (1975)
(Coturnix coturnix japonica)
Japanese quail
(Coturnix coturnix japonica) 14 days 69 Hill & Camardese
(1986)
Ring-necked pheasant 10 days 91 Hill et al. (1975)
(Phasianus colchicus)
Red-winged blackbird - 10 Schafer (1972)
(Agelaius phoeniceus)
a intubation of a single dose.
b 8 days - standard test. 5 days feeding followed by 3 days observation.
Egg production in Japanese quail was inhibited and hatchability
reduced at 60 mg/kg (NRC, 1977).
Methyl parathion-induced mortality following long-term ingestion
was generally due to anorexia. Grackles (Quiscalus quiscula) had
lost 28-36% of their initial body weight, when they died. No fat was
visible and the muscles were reduced on the sternum. There was an
increase in mortality at relatively constant intake rate of methyl
parathion observed between May and August, which was related to an
increase in natural activity within this time. It was concluded, that
median lethal dietary concentrations are relative and depend on the
anorexic and physiological condition of wild birds (Grue, 1982). The
mean brain AChE activity of grackles (Quiscalus mexicanus) was
significantly inhibited more than that of white-winged doves (Zenaida
asiatica) and that of mourning doves (Zenaida macroura) after
applications of EPN (phenylphosphonothioic acid O-ethyl O-p-nitro-
phenyl ester) and methyl parathion (Custer & Mitchell, 1987).
Free-living, female red-winged blackbirds (Agelaius phoeniceus)
were captured on their nests and given oral doses of 2.4 or 4.2
methyl parathion mg/kg body weight and released immediately after
dosing. Although methyl parathion caused ataxia, lacrimation, and
lethargy and significantly depressed cholinesterase activity (> 35%)
at 4.2 mg/kg, there were no apparent adverse effects on incubation
behaviour and nesting success (Meyers et al., 1990).
Depressed brain acetylcholinesterase activity was also observed
in 2 bird species (red-winged blackbird, (Agelaius phoeniceus), and
dickcissel (Spiza americana)) inhabiting wheat fields treated with
methyl parathion (0.67 kg a.i./ha). Maximal inhibition occurred 5 days
after pesticide application. Enzyme activity levels returned to near
normal levels by the tenth day following application. Cholinesterase
inhibition for dickcissels and red-winged blackbirds differed
significantly (74% versus 40%), and these differences could not be
explained by the diets of the 2 species, as they were similar
(Niethammer & Baskett, 1983).
A subacute oral dose of 3.5 mg methyl parathion/kg per day
resulted in inhibition of brain cholinesterase (average decrease of
36%) in nuthatches (Sitta carolinensis) after 3-7 days exposure
(Herbert et al., 1989).
7.3.4 Non-laboratory mammmals
Two wild rodent species (Sigmodon hispudus and Mus musculus)
were found to have a higher mortality rate and to recover more slowly
from exposure to methyl parathion at oral doses of 14-80 mg/kg,
compared with laboratory rodents (Roberts et al., 1988). Clark (1986)
reported a greater tolerance to methyl parathion in little brown bats
(Myotis lucifugus) compared with wild mice (Mus musculus): the
24-h oral LD50 value (372 mg/kg body weight) of methyl parathion for
little brown bats was 8.5 times the LD50 value for mice (44 mg/kg
body weight). A loss of coordination was observed in 50% of the
animals that were still alive 24 h after the treatment. The poisoned
bats could be more easily captured by predators. The threshold of the
coordination loss was about 1/3 of the LD50 value. In toxicity
tests, mink (Mustela vison) rejected methyl parathion-treated diets
and appeared to die from starvation rather than from methyl parathion
poisoning (Aulerich et al., 1987).
8. EFFECTS ON EXPERIMENTAL ANIMALS AND IN VITRO TEST SYSTEMS
The inhibition by methyl parathion of acetylcholinesterase at
nerve endings results in an accumulation of endogenous acetylcholine,
as evidenced by peripheral and central cholinergic nervous system
signs (Taylor, 1980).
Toxic effects include profuse salivation, lacrimation, nasal
discharge, colic, diarrhoea, pupil constriction, excessive sweating,
coughing, vomiting, frequent urination, anxiety, restlessness,
hyperactivity, and hyperkinesis.
A more complete treatise on the effects of organophosphorus
insecticides in general, especially their short- and long-term effects
on the nervous system, can be found in Environmental Health Criteria
63: Organophosphorus insecticides - A general introduction (WHO,
1986).
8.1 Single exposure
Toxicological data on methyl parathion are summarized by Taylor
(1980) and Flucke (1984).
The acute toxicity values in a number of species following the
oral (Table 20), dermal (Table 21), inhalational (Table 22), and
intraperitoneal (Table 23) administration of methyl parathion show
lethal doses of about 3-400 mg/kg for the oral route, 40-300 mg/kg for
the dermal route, 3.5-72 mg/kg for the intraperitoneal route, and
30-300 mg/m3 for inhalation exposure. The acute subcutaneous LD50s
for methyl parathion in rats and mice were 6 and 18 mg/kg body weight,
respectively (Krueger & Casida, 1957; RTECS, 1991); the acute
intravenous LD50 was reported to be 4.1-14.5 mg/kg body weight in
rats, 2.3-13 mg/kg body weight in mice, and 50 mg/kg body weight in
guinea-pigs (NIOSH, 1976).
Izmirova et al. (1984) found an abrupt reduction in the blood and
brain cholinesterase and acetylcholinesterase activities in albino
rats in the 30th and 90th min after a single oral administration of 32
mg methyl parathion/kg. The blood cholinesterase activity was reduced
by 71% and the brain acetylcholinesterase activity by 54%. Twenty-four
hours after administration, the cholinesterase activity was higher
than that in the controls.
Table 20. Acute oral toxicity
Animal (sex)a LD50 (mg/kg body References
weight)
rat (m) fasted 2.9 Heimann (1982)
rat (f) fasted 3.2 Heimann (1982)
rat 6 Bayer AG (1988); RTECS
(1991)
rat (m) 7.4 Flucke & Kimmerle (1977)
rat (f) nonfasted 9.3 Heimann (1982)
rat (m) nonfasted 10.8 Heimann (1982)
rat (m) 11.7 Kimmerle (1975)
rat (m) 14 Gaines (1960, 1969)
rat (f) 24 Gaines (1960, 1969)
rat 35 Kagan (1971)
mouse 23 RTECS (1991)
mouse 14.5 Haley et al. (1975)
mouse 33.1 Mundy et al. (1978)
mouse 21.8 Mundy et al. (1978)
mouse 19.5 Haley et al. (1975)
rabbit (m) fasted 19 Heimann (1982)
rabbit (f) fasted 19.4 Heimann (1982)
rabbit 420 RTECS (1991)
Table 20. (cont'd) Acute oral toxicity
Animal (sex)a LD50 (mg/kg body References
weight)
guinea-pig 1270 RTECS (1991)
guinea-pig 417 NIOSH (1976)
dog 90 Hirschelmann & Bekemeier
(1975)
a m= male, f= female.
Table 21. Acute dermal toxicity
Animal, Duration of Ld50 (mg/kg Ld100 References
sexa exposure (h) body weight)
rat 1b 63 RTECS
(1991)
rat (m,f) - 67 Gaines
(1960,
1969)
rat (m) 24 46 Heimann
(1982)
rat (f) 24 44 Heimann
(1982)
rabbit (m) 6 1270 (pure) Deichmann
et al. (1952)
rabbit (m) 6 350-780
(technical Deichmann
grade) et al. (1952)
rabbit (m) 6 420 (pure, in Deichmann
corn oil) et al. (1952)
rabbit (m) 6 2500 (pure, Deichmann
suspended in et al. (1952)
water)
rabbit - 300 RTECS
(1991)
a m = male, f = female.
b no data given.
Table 22. Acute inhalation toxicity
Animal Duration of LC50 References
(sex)a exposure (h) (mg/m3 air)
rat 1 120 RTECS (1991)
rat 1 34 Molnar & Paksy
(1978)
rat (m) 1 200 Kimmerle & Lorke
(1968)
rat (m) 1 260 Thyssen (1979)
rat (f) 1 320 Thyssen (1979)
rat (m) 4 120 Kimmerle & Lorke
(1968)
rat (m) 4 185 Thyssen (1979)
rat (f) 4 170 Thyssen (1979)
mouse 4 120 RTECS (1991)
a m = male f= female.
Table 23. Acute intraperitoneal toxicity
Animal LD50(mg/kg body weight) References
rat 3.5 Du Bois & Coon (1952)
rat adult 5.8 Brodeur & Du Bois (1963)
rat juvenile 3.5 Brodeur & Du Bois (1963)
rat 7 Kimmerle (1975)
mouse 9.3 Kimmerle (1975)
mouse 11.0 Benke et al. (1974)
mouse 6.4 Kamienski & Murphy (1971)
mouse 8.2 Mirer et al. (1977)
mouse 72 Goyer & Cheymol (1967)
Dogs that received 10 or 30 mg methyl parathion/kg body weight
intravenously showed minimal activity of the plasma cholinesterases,
30 min after treatment. Sixteen hours after the injection of 10 mg
methyl parathion/kg body weight, the enzyme activities had returned to
their pre-injection values. However, following treatment with 30 mg
methyl parathion/kg body weight, it took 7 days for complete recovery
(Braeckman et al., 1980).
After i.p. injection of 2.4 mg Wofatox (methyl parathion), Karcsu
et al. (1981) observed complete inhibition of the histochemically
detectable acetylcholinesterase activity in the central nervous system
of the rat. Partial enzyme inhibition was found in the motor neurons
and in the striated muscles. Ultrastructural changes in the myocardium
of the rats were also confirmed.
8.2 Skin and eye irritation, sensitization
The skin of rabbits exposed to methyl parathion for 4 or 6 h did
not show perceptible signs of irritation (concentrations up to
LD100, Table 21, Deichmann et al., 1952). Similar results were
obtained by Hecht & Wirth (1950) and by Heimann (1982) in their
studies on rats.
The irritation potential of methyl parathion on the rabbit skin
and eye was studied according the OECD guidelines for the testing of
chemicals (Nos. 404 and 405). It was concluded that methyl parathion
had no primary irritating potential (Pauluhn, 1983).
8.3 Short-term exposures
Groups of Wistar albino rats were exposed to methyl parathion
aerosol concentrations of 0.9, 2.6, and 9.7 mg/m3 air for 6 h/day,
5 days/week for 3 consecutive weeks. No mortality occurred. Plasma and
brain cholinesterase levels were significantly depressed in the
highest dose group. At 2.6 mg/m3, slight inhibition of plasma ChE
occurred (Thyssen & Mohr, 1982).
Groups of New Zealand white rabbits were administered methyl
parathion (purity 96.3%) dermally at dose levels of 10, 50, and 250
mg/kg body weight, applied for 5 days/week over 3 weeks. The site was
left uncovered for 6 h and then it was cleaned with soap and water.
There was a dose-related inhibition of erythrocyte and brain
cholinesterases in the 50 and 250 mg/kg dose groups. Plasma ChE was
also significantly depressed in the highest dose group; these animals
presented signs of cholinergic poisoning and 5 out of 6 animals died
(Mihail & Vogel, 1984).
A 12-week dietary study at 5, 20, and 50 mg methyl parathion/kg
was performed on male and female dogs. The doses corresponded to 0.1,
0.4, and 1.0 mg/kg body weight per day. A significant decrease in
plasma cholinesterase activity was observed only at 50 mg/kg diet
(Williams et al., 1959).
8.4 Long-term exposures
Kazakova et al. (1974) fed chicken and cattle daily with 2.5 mg
methyl parathion/kg body weight for one year. No changes in health
status and food intake were observed. In pigs and cows, 10 mg/kg body
weight led to irritation, depression, miosis, salivation, intensified
peristaltics, and diarrhoea.
Rats fed diets containing 40 mg methyl parathion/kg for 2 years,
and mice (females: fed up to 125 mg/kg, males: fed up to 77 mg/kg) did
not display any cholinergic toxicity (NCI, 1979).
In a 2- year study (combined long-term/carcinogenicity), 500 rats
(50 male, 50 female per dose, 100 controls) were fed diets containing
0, 2, 10, or 50 mg methyl parathion/kg. The intake of active
ingredient was 0, 0.144, 0.713, 4.917 mg/kg body weight per day
(females). The highest dose led to retardation of growth, increase in
mortality, inhibition of cholinesterase-activity in plasma,
erythrocytes, and brain, reduction of haemoglobin, and haematocrit,
and an increase in reticulocytes, after 2 years. Female rats showed a
reduction in plasma proteins and a reversible increase in urea in
plasma and protein in urine. At 10 mg/kg diet, the cholinesterase
activity in plasma and red blood cells was inhibited. Male rats also
showed reduced cholinesterase activity in the brain. Extensive
histopathological examinations (cardiovascular, respiratory, and
urogenital systems, digestive tract, organs, and glands) did not
exhibit any substance-related changes. No toxic effects were found at
the lowest dose (Bomhard et al., 1981; Schilde & Bomhard, 1984).
Sixty rats per sex and group were fed diets containing methyl
parathion at concentrations of 0.5, 5, or 50 mg/kg for 2 years.
Sciatic nerve preparations from 1 out of 5 males in the low-dose group
and 1 out of 5 in the mid-dose group reportedly showed moderate
degenerative changes. In the high-dose group (50 mg/kg diet), sciatic
nerve preparations from treated males showed a loss of myelinated
fibres. These animals also showed more myelin degeneration and Schwann
cell proliferation. Similar, less severe changes were seen with a
lower incidence, in males fed 5.0 or 0.5 mg/kg per day males and in
the controls. Only 1 rat in the low-dose group and 1 in the mid-dose
group had more severe changes than the controls; however, 4 high-dose
males showed more severe changes. No obvious differences were seen in
the females. Haemoglobin, haematocrit, and RBCs were slightly reduced
in mid-and high-dose males, and moderately reduced in high-dose
females (Daly, 1983).
8.5 Reproduction, embryotoxicity, and teratogenicity
Dosages of 4 or 6 mg methyl parathion/kg body weight were
injected intraperitoneally into pregnant, female albino Holzmann rats.
The injection was made on day 9 or day 15 using an ethanol-propylene
glycol vehicle. It was found, that the fetal, cerebral, cortical
cholinesterase activity was reduced, indicating the transplacental
passage of the organophosphate. Large subcutaneous haematomas also
occurred; however, no significant developmental defects were noticed
(Fish, 1966). Ackermann (1974) also reported that there was no
placental barrier for methyl parathion.
A 3-generation study was performed by the Woodard Research
Corporation in 1966. This unpublished report was reviewed by Anon.,
FAO/WHO (1969). Rats received diets of 0, 10, or 30 mg methyl
parathion per kg diet. A sporadic reduction in the litter size of
groups was observed (30 mg/kg: F2alpha, F2ß, F3alpha; 10 mg/kg:
F1ß), also a delayed growth of litters until weaning (30 mg/kg:
F2alpha, F3alpha, F3ß; 10 mg/kg: F1ß), a reduced rate of
survival of the litters (30 mg/kg: F1alpha, F1ß, F2alpha); 10
mg/kg: F3alpha), and an increased rate of stillbirths (30 mg/kg:
F1ß, F3alpha).
Another 3-generation study was performed by the Midwest Research
Institute (1975). Rats received 0, 10, or 30 mg methyl parathion/kg
diet (corresponding to 0, 0.5, or 1.5 mg methyl parathion/kg body
weight); 2 litters of each generation were evaluated. No adverse
effects on growth, survival, or reproduction were observed at the 30
mg/kg level, however, the 10 mg/kg level caused a reduction in the
postnatal survival in weaning rats in the F1ß and F3alpha
generations. Similar results were found in the 3-generation study of
Löser & Eiben (1982). Rats (male and female, SPF-Wistar W 74 strain,
5-6 weeks old) were fed a diet containing technical methyl parathion
(95% pure) at 2, 10, or 50 mg/kg for 77 days and then mated. The
no-effect level in this study was 2 mg/kg diet. A dose of 50 mg methyl
parathion/kg caused reductions in neonatal weights and litter size,
and delayed body weight gains, while 10 mg/kg caused sporadic
reductions in litter size (F2alpha, F3alpha), delayed growth of
litters until weaning (F1alpha, F2alpha, F2ß), and a reduced
rate of survival of the litters.
Single doses of 3, 30, or 100 µg methyl parathion (in 10% DMSO)
were administered subgerminally into chicken eggs on day 2 and
intra-amniotically on days 3 and 4. These doses did not induce any
specific malformations. Embryotoxicity was noted at the 2 higher doses
(30 and 100 µg) (Benes & Jelinek, 1979). These findings were confirmed
by estimating the embryotoxicity range and parameters (Jelinek et al.,
1985). Doses of up to 55 µlitre Wofatox 50EC/kg egg reduced
haematocrit, glucose, cholesterol, and AChE activity and increased
aspartate aminotransferase and lactate dehydrogenase values in blood
samples of chicken embryos (Somlyay et al., 1989). The injection of 2
different concentrations of methyl parathion (13 and 135 mg/kg egg)
into pheasant eggs resulted in increased mortality and in an increased
incidence of skeletal deformities in the survivors (Varnagy et al.,
1984; Déli & Varnagy, 1985; Varnagy & Déli, 1985). Biochemical studies
on muscle samples from chicken embryos (eggs treated with 0.4% or
4.0% solution of Wofatox 50EC) showed decreased creatine kinase
activity, decreased creatine, creatine-phosphate, and Mg2+ (in
cervical muscle only) contents, and increased creatinine, Ca2+, and
Mg2+ (in femoral muscle only) values (Déli et al., 1985). Scanning
electron microscopic examination of the cartilage in chicken embryos
showed degeneration of collagen structure and chondrocytes at a high
insecticide concentration (eggs treated with 0.4% or 4.0% solution of
Wofatox 50EC) (Varnagy et al., 1988). Analysis of the protein pattern
of the cervical muscles of 18-day-old embryos, treated with 0.4%
methyl parathion solution showed decreases in alpha-actinin,
alpha-tubulin, ß-tubulin, and gamma-proteins (Déli & Kiss, 1988).
Studies on chickens showed that methyl parathion at 1-10
µmol/litre had no or only little effect on the adenylate cyclase in
the embryo muscle. Comparable results were obtained with rats using
the plasma membrane adenylate cyclase in rat livers, even at 100
µmol/litre. In the presence of adenylate cyclase-stimulating agents,
additional activation of methyl parathion was observed; it enhanced
the stimulating activity of GTP and isoproterenol together, but not
alone. Methyl parathion is soluble, but not metabolized, in plasma
membranes, so it may alter cellular levels of cAMP, and, thus, cell
growth (Déli & Kiss, 1986).
At very high doses (20 or 60 mg/kg body weight), methyl parathion
injected intraperitoneally in ICR-CL mice on day 10 of pregnancy,
caused convulsions, hypersalivation, ataxia, and tremor. At the higher
dose, 5 out of the 14 litters died. This dose caused reduced neonatal
weight, an increase in the occurrence of cleft palate, and an
increased incidence of cervical ribs in the fetuses. At the lower
dose, cleft palate, and a statistically non-significant increase in
the number of cervical ribs and underdeveloped sternebrae were
observed (Tanimura et al., 1967).
A single intraperitoneal injection of 5, 10, or 15 mg/kg body
weight was administered to Wistar rats on day 12 of pregnancy; signs
of toxicity and reduced body weight were observed with 15 mg/kg, but
there was no evidence of teratogenicity at any of the doses (Tanimura
et al., 1967).
On 6 alternate days between days 5 and 15 of pregnancy, 3 groups
of rats received orally 0.1, 1, or 3 mg methyl parathion/kg body
weight. Another group of rats received 3 mg methyl parathion/kg body
weight on 8 alternate days between days 5 and 19 of pregnancy. No
teratogenic effects were observed; however, the high doses caused
increased resorptions and decreased fetal body weight (Fuchs et al.,
1976).
Methyl parathion was administered orally by gavage, to groups of
female rats from day 6 to day 15 of gestation at dose levels of 0.1,
0.3, or 1 mg/kg body weight. Weight gain in the mothers and a slight
retardation in growth in the fetuses were noted at the highest dose
level. Methyl parathion was not toxic for the embryo or fetus and no
teratogenic effects were apparent (Machemer, 1977a).
Groups of 24-26 rats received intravenous injections of 0, 0.03,
0.1, or 0.3 mg methyl parathion/kg body weight per day from day 6 to
day 15 of pregnancy. On day 20, the fetuses were evaluated. No
treatment-related effects were found (Machemer, 1977b).
No signs of embryotoxicity or teratogenicity were found in
rabbits that received 0.3, 1.0, or 3.0 mg methyl parathion/kg body
weight on days 6-18 of pregnancy (Renhof, 1984).
Daily intraperitoneal administration of methyl parathion (1 or
1.5 mg/kg body weight) to rats during days 6-19 of gestation resulted
in decreases in both maternal and fetal protein synthesis (Gupta et
al., 1984). The effect was dose dependent, and was greater on day 19
than on day 15 of gestation; it was also greater in fetal than in
maternal tissues. The same dosage regimen resulted in a postnatal
decrease in acetylcholinesterase activity and muscarinic receptor
binding. Recovery of acetylcholinesterase activity to near normal
levels occurred by day 28 in the low-dose offspring, but not in the
high-dose weanlings (Gupta et al., 1985).
8.6 Mutagenicity and related end-points
Methyl parathion has been reported to have DNA-alkylating
properties. Mutagenicity test results have been both positive and
negative. The results of most of the in vitro mutagenicity studies
with both bacterial and mammalian cells were positive; the in vivo
studies produced equivocal results. A survey is given in Table 24.
8.7 Carcinogenicity
The carcinogenicity of methyl parathion was studied in mice by
the National Cancer Institute (NCI) in 1979. Groups of 50,
six-week-old female B6C3F1 mice received diets containing either
62.5 or 125 mg methyl parathion/kg for 102 weeks. For 37 weeks, 2
groups of 50 male mice received diets containing either 62.5 or 125 mg
methyl parathion/kg, which was reduced then to 20 or 50 mg/kg for
another 65 weeks. Untreated matched groups of 20 males and 20 females
were used as a control. From all groups, 80-86% were still alive at
the end of the study. There was no statistically significant increase
in tumour incidence.
The NCI (1979) also studied the carcinogenicity of methyl
parathion in rats. Groups of 50 female and male Fischer 344 rats (6
weeks old) received separate diets containing 20 or 40 mg methyl
parathion/kg for 105 weeks. As matched controls, 20 male and 20 female
rats remained untreated. Only 46% of the high-dose females survived,
but 78% high-dose males, 74% low-dose males, 82% low-dose females, 85%
control males, and 95% control females were still alive at the end of
the study. No statistically significant increase in tumour rates was
found.
Male and female rats in groups of 50 were fed for 2 years with
diets containing 2, 10, or 50 mg methyl parathion/kg. No toxic effects
were found at the low dose (see section 8.4). No morphological changes
due to the insecticide were detected. No carcinogenic effects of
methyl parathion were observed (Bomhard et al., 1981; Schilde &
Bomhard, 1984).
Table 24. Mutagenicity tests of methyl parathion
Test Species Dose levels Metabolic Results Reference
activation
Microorganism
Gene mutation tests S. typhimurium _a +/- - Simmon et al. (1977)
Ames TA100, TA1535 +/- Carrere et al. (1978)
TA1536, TA1537
TA 1538
Ames S.typhimurium 250-1250 µg/plate + - Rashid & Mumma (1984)
TA100
Ames S. typhimurium 250-1250 pg/plate + - Rashid & Mumma (1984)
TA98, TA1535,
TA1537, TA1538
Ames S. typhimurium Herbold (1986)
TA1535 > 1000/µg/plate +/- +
TA 100 > 500/µg/plate +/- +
TA1537, TA1538 20-2500/µg/plate +/-
Reverse mutations E. coli WP2 and 1 crystal or microdrop - - Dean (1972)
WP2uvrA 250-2500/µg/plate +/- - Simmon et al. (1977)
Rashid & Mumma (1984)
Forward mutations E. coli 1 x 10-2mol/litre - + Mohn (1973)
streptomycin/ Wild (1975)
5-methyl-tryptophane
resistance
Table 24 (continued)
Test Species Dose levels Metabolic Results Reference
activation
ade-6 forward mutation Sacharomyces 11-228 mmol/litre - - Gilot-Delhalle et al.
pombe (1983)
Recombinogenic Aspergillus 2 mg - - Morpugo et al. (1977)
activity/point nidulans
mutation
(8-Aza-guanine resistance
Streptomyces _a - - Carere et al. (1978)
coelicolor
Insects
Sex link recessive Drosophila 1.25 x 10-5% w/w + Tripathy et al. (1987)
lethal test melanogaster 6.3 x 10-6% w/w
Larvae 24,48,72 h 24-48 and 72 h
old exposure
Mammals
Thymidine kinase Mouse Iymphoma _a -/+ + Jones et al. (1982)
locus ceils L51784
DNA effects Chinese hamster 20 and 40 µg/ml _a + Chen et al. (1981 )
chromatid ovary cells V79 28-72 h
exchange (in vitro)
Table 24 (continued)
Test Species Dose levels Metabolic Results Reference
activation
Human lymphold 20 µg/ml - + Sobti et al. (1982)
cells (LAZ-007)
Human lymphold 20 and 40/µg/ml _a + (for Chen et al. (1981)
cells 835 M and 28-72 h 20/µg/ml)
Jeff cells
Sister chromatid Human lymphocytes 36-181.8 µmol/litre - + (dose Singh et al. (1987)
exchange (SCE) dependent)
Unscheduled DNA Human fetal lung _a +/- - Simmon et al. (1977)
synthesis WI 38 fibroblast
Human lymphoid - - Huang (1973)
cells up to 50/µg
B411-4 6-50 h
RMPI - 1788
RMPI - 7191
Mouse (in vivo) 10 mg/kg ip - Degraeve & Moutschen
bone marrow (1984)
germ cells
Mouse, Swiss 9.4, 18.8, 37.5, + (dose Mathew et al. (1990)
bone marrow 75 mg/kg body weight, oral dependent)
Table 24 (continued)
Test Species Dose levels Metabolic Results Reference
activation
Chromosomal aberrations Rat bone marrow 0.5 mg/kg body weight + (dose Malhi & Grover (1987)
cells (in vivo) 1 mg/kg 1.95%
2 mg/kg 9.26%
5 days/week for 16.86%
7 weeks dependent)
Micronucleus Wistar rat 1, 2 and 4 mg/kg ip + (dose Grover & Mahli (1985)
(in vivo) dependent)
Micronucleus Mouse 5-10 mg/kg body weight, - Herbold (1986)
orally, daily,
for 2 days
Micronucleus Mouse, Swiss 9.4, 18.8, 37.5, 75 + (dose Mathew et al. (1990)
mg/kg body weight, orally dependent)
Dominant lethal Mouse, male 20 mg/kg diet - Simmon et al. (1977)
ICR/SIM 40 mg/kg for
80 mg/kg 7 weeks
Dominant lethal Mice, male Q 0.15 mg/litre (daily) in - Degraeve et al. (1984)
strain drinking-water, 5-7
weeks
a = no information given.
8.8 Special studies
Seven white New Zealand rabbits per group were fed 0, 0.036,
0.162, 0.519, or 1.479 mg methyl parathion/kg body weight per day, for
8 weeks. A dose-dependent increasing atrophy of the thymus cortex and
a reduced, delayed-type hypersensitivity response (DTH) were found
(Street & Sharma, 1975). In a preliminary study, Fan et al. (1981)
examined the effects of methyl parathion on immunological responses to
S. typhimurium infection in mice. Mortality rates among infected
animals fed 0.08, 0.3, or 0.7 mg methyl parathion/kg body weight
(duration "extending beyond 2 weeks") were determined and protection
by vaccination was examined. Dose-related increases in mortality were
seen in unvaccinated mice and protection by immunization was
decreased. These limited findings were reviewed by Sharma & Reddy
(1987) and Thomas & House (1989).
Methyl parathion was found by Barnes & Denz (1953) not to cause
delayed neuropathy in their hen test. However, Nagymajitenyi et al.
(1988) found neurotoxic effects on the central and peripheral nervous
systems in both acute and short-term studies on CFY rats, in which the
conduction velocity of the peripheral nerves, muscle function
(ischidiacus nerve/gastrocnemius muscle), and EEG activity were
measured. In the short-term studies, the rats were given 0.44 mg
methyl parathion/kg body weight for 5 days/week for 6 weeks; in the
acute study, the rats received 0.4 mg/kg, orally.
Lipid metabolism in rats was investigated by Hasan & Ahmad Khan,
(1985). The rats received daily intraperitoneal doses of 1.0, 1.5, or
2.0 mg methyl parathion/kg body weight for 7 days. The concentrations
of total lipids, phospholipids, and cholesterol increased in a
dose-related manner in the cerebral hemisphere, cerebellum, brain
stem, and spinal cord. Lipid peroxidation increased in the CNS with
the exception of the cerebellum.
Khan & Hasan (1988) studied changes in the levels of ganglio
sides and glycogen of the cerebral hemisphere, cerebellum, brain stem,
and spinal cord following intraperitoneal injection of methyl
parathion (1, 1.5, or 2 mg/kg body weight) in 24 rats for 7 days. A
dose-related depletion in the concentration of gangliosides and
glycogen content were discernible in all regions of the CNS.
Preweanling, male, rat pups were exposed daily through
subcutaneous injection to parathion (1.3 or 1.9 mg/kg body weight) or
the vehicle (corn oil) on postnatal days 5-20, a period critical for
the development of behavioural and biochemical parameters of the
cholinergic nervous system. This exposure resulted in dose-dependent
reductions in acetylcholinesterase activity and muscarinic receptor
binding in the cortex. During the preweanling period, there were no
differences among the groups in most reflex measures, eye opening, or
incisor eruption. Postweanling behavioural assessment revealed small
deficits in tests of spatial memory in both the T-maze and radial arm
maze. There were no differences in neuromuscular abilities or
spontaneous activity measures (Stamper et al., 1988).
The behavioural effects of short-term exposure of male Wistar
rats to methyl parathion (1/50 or 1/100 of LD50, orally, for 6
weeks) were studied. Open-field (OF) and elevated plus-maze (EPM)
tasks were used to decide whether or not the compound could affect
behaviour. Significant effects were measured in OF activity during the
first minute, on the activity of crossing outer squares, increasing
latencies to leave centre, start of rearing, grooming, and defecation.
EPM parameters showed an increased amount of time spent in the open
arms and a clear tendency to enter open arms more frequently. The
defecation rate in the EPM was significantly decreased (Schultz et
al., 1990).
8.9 Factors modifying toxicity
Methyl parathion becomes toxic only after metabolic
transformation to the oxon analogue, methyl paraoxon, by liver
microsomal oxidation. The microsomal enzymes metabolize methyl
parathion in 2 ways in vitro : a) oxidation to methyl paraoxon,
and b) degradation to dimethyl phosphorothiotic acid and
p-nitrophenol. NADPH and O2 are necessary for both reactions,
indicating that these are oxidative processes (Nakatsugawa et al.,
1968).
Piperonyl butoxide inhibits the mixed function oxidase activity
of the microsomal fraction of liver cells. Therefore, it inhibits both
oxidative activation of methyl parathion and detoxification, but not
the dealkylation reactions due to glutathione- S-alkyltransferase. At
a dosage of 400 mg/kg body weight, piperonyl butoxide antagonized the
toxic effects of methyl parathion in mice when given 1 h before the
mice received the insecticide. The intraperitoneal toxicity of methyl
parathion was reduced 40-fold (Kamienski & Murphy, 1971; Levine &
Murphy, 1977a,b; Mirer et al., 1977). Diethyl maleate reduced the
glutathione content of the liver by 80%. This agent increased the
acute toxicity of methyl parathion by the inhibition of
glutathione-dependent detoxification (Mirer et al., 1977).
Pap et al. (1976) showed that methyl parathion was less toxic in
rats with a thioacetamide-induced liver cirrhosis than in normal rats.
After activating the microsomal enzymes in the liver with sodium
phenobarbital or norandrostenolone phenylpropionate, the cirrhotic
rats showed a normal susceptibility to methyl parathion, indicating
the involvement of liver microsomes in the activation of methyl
parathion. Treatment of normal rats with chloramphenicol could
increase their survival time after poisoning with methyl parathion.
Lead nitrate (Pb(NO3)2) reduced the toxicity of methyl
parathion due to an increase in the carboxylesterase-dependent
metabolism of the insecticide (Hapke et al., 1978).
A single oral dose of 5 or 10 mg methyl parathion/kg body weight
resulted in decreases in the cholinesterase activity in rats of 43.6%
or 72.3%, respectively. However, rats pretreated on 5 successive days
with a combination of 7 mg gentamycin/kg body weight and 20 mg
rifamycin/kg body weight showed a remarkable protection against the
toxic effects of methyl parathion. The toxic signs were minimal; the
rats showed no, or only transient, signs of poisoning, and no
convulsions were be observed in the rats that had been pretreated. The
combination of these 2 drugs significantly prevented the methyl
parathion-induced inhibition of cholinesterase in plasma and of the
liver carboxylesterase. Gentamycin or rifamycin alone did not have any
effect. Youssef et al. (1987) demonstrated, that gentamicin and
rifamycin inhibited the formation of the oxidation product of methyl
parathion, methyl paraoxon, in the liver and skeletal muscle. Both
substances potentiated the rate of urinary p-nitrophenol excretion
within 48 h of the methyl parathion application. Pretreatment with
rifamycin influenced the rate of liver glutathion reduction, whereas
gentamicin did not show this effect.
Male rats were treated with a single i.p. dose of 5 mg methyl
parathion/kg. Pretreatment with memantine hydrochloride (18 mg/kg,
i.p.), 30 min before methyl parathion administration, and atropine
sulfate (16 mg/kg, i.p.), 15 min before, significantly reduced
(P <0.01) the inhibition of acetylcholinesterase (Gupta & Kadel,
1990).
Pretreatment with cimetidine, which suppresses the hepatic
microsomal oxidative metabolism, decreased the toxicity of methyl
parathion in rats and mice (Joshi & Thornburg, 1986).
Fuchs et al. (1986) showed that the LD50 in rats increased by
19-24% after simultaneous oral administration of 0.5 g humic acids/kg
body weight and methyl parathion. It was supposed that the absorption
of methyl parathion from the digestive tract decreased as a result of
the intake of the humic acids.
Sultatos (1987) perfused mouse livers in situ with methyl
parathion. The acute toxicity of methyl parathion in mice was
antagonized by pretreatment with phenobarbital, daily, for 4 days (80
mg/kg, i.p.). This effect was due to hepatic microsomal activation and
resulted in an increased clearance of methyl parathion. Similar
results were obtained by Du Bois & Kinoshita (1968), Du Bois (1969,
1971), and Murphy (1980).
The influence of temperature on the toxicity of methyl parathion
in mice was studied by Nomiyama et al. (1980). They found median
lethal doses (i.p.) of 14 mg/kg body weight at 8 °C, 44 mg/kg body
weight at 22 °C, and 35 mg/kg body weight at 38 °C.
An influence of age on the toxicity and metabolism of methyl
parathion was observed by Benke & Murphy (1975) in rats. Rats became
much less sensitive to poisoning with methyl parathion with increasing
age. The effect was explained by a presumable increase in the
detoxification processes as the GSH-dependent (glutathion-dependent)
dealkylation. Methyl parathion dealkylation rates increased directly
with age for both sexes.
Carbon disulfide pretreatment, 1 h before administration of 10 mg
methyl parathion/kg body weight to mice did not significantly affect
the methyl parathion toxicity (Yasoshima & Masuda, 1986).
Prior depletion of glutathione by acetaminophen (600 mg/kg, i.p.,
Costa & Murphy, 1984) or by diethyl maleate (1 ml/kg, i.p., Sultatos
& Woods, 1988) had little effect on the toxicity of methyl parathion
(2.5 mg/kg body weight and up to 55 mg/kg body weight i.p.,
respectively) in the mouse.
Interactions of organophosphorus pesticides and several
pyrethroid insecticides were reported by Gaughan et al. (1980).
Following an intraperitoneal injection of organophosphorus pesticides
in mice, they found pronounced inhibition of the liver microsomal
esterase, which hydrolyses trans-permethrin. Methyl parathion did not
potentiate the toxicity of deltamethrin (Audegond et al., 1989).
Equitoxic oral or i.p. combinations of methyl parathion with other
organophosphorus insecticides (amiton, coumaphos, crufomat,
dimethoate, dioxathion, disulfoton, fensulfothion, ethyl parathion,
phosphamidon, trichlofon) caused only subadditive or additive effects
on the LD50 values (Du Bois, 1961; West et al., 1961; Du Bois &
Kinoshita, 1963; Frawley et al., 1963; Sanderson & Edson, 1964;
McCollister et al., 1968; Flucke & Kimmerle (1977). Williams et al.
(1957) found additive effects in their testing of oral combinations of
methyl parathion with demeton, EPN, malathion, or ethyl parathion in
dogs.
Mice pretreated with 50-300 mg diethyl dithiocarbamate per kg
body weight displayed a remarkable reduction in the acute toxicity of
methyl parathion. The toxicity was up to 10 times less. Lange &
Wiezorek (1975) explained this observation by an effect of the
dithiocarbamate on the microsomal oxidases and, thus, on the
metabolism of methyl parathion. Another explanation is that compounds
that temporarily occupy the active site of acetylcholinesterase
prevent phosphorylation of the enzyme until there has been time for
destruction of the organic phosphorus compound by A-esterases (Hayes
& Laws, 1991).
Dithiocarb reduced the toxicity of methyl parathion in mice
markedly, when applied 30 min before the methyl parathion. No effect
was observed when dithiocarb and methyl parathion were applied
simultaneously (Lange et al., 1977).
Orlando et al. (1972) found that pretreatment with quinidine
sulfate had an inhibitory effect on the toxic action of i.v. injection
of methyl parathion in rabbits. This could be demonstrated by
electrocardiography. Quinidine sulfate reduced the influence of methyl
parathion on the nicotinic-and muscarinic-type receptors.
The effect of methyl parathion on monoamine oxidase activity
(MAO) in rat brain mitochondria was investigated by Nag & Nandi
(1987). In vitro methyl parathion reduced the MAO significantly;
however, in vivo , the effect was negligible.
Methyl parathion is an inhibitor of malate dehydrogenase in the
mitochondria of liver and skeletal muscle. There was also an
inhibitory effect on plasmatic malate dehydrogenase and lactate
dehydrogenase in the liver (Tripathi & Shukla, 1988).
8.10 Mode of action
The mode of action of organophosphorus insecticides, such as
methyl parathion, is described in Environmental Health Criteria 63
(WHO, 1986).
8.10.1 Inhibition of esterases
The primary biochemical effect associated with toxicity caused by
organophosphorus pesticides is inhibition of acetylcholinesterase
(AChE). The normal function of AChE is to terminate neurotransmission
due to acetylcholine, liberated at cholinergic nerve endings in
response to nervous stimuli. Loss of AChE activity may lead to a range
of effects resulting from excessive nervous stimulation and
culminating in respiratory failure and death. The chemistry of the
inhibition of AChE and of many other esterases (e.g., NTE and liver
carboxyesterases, which are discussed elsewhere) by these chemicals is
similar and is given in schematic form in Fig. 4. Following the
formation of a Michaelis complex (reaction 1), a specific serine
residue in the protein is phosphorylated with loss of the leaving
group X (reaction 2). Two further reactions are possible: reaction 3
(reactivation) may occur spontaneously at a rate that is dependent on
the nature of the attached group and on the protein and is also
dependent on the influence of pH and of added nucleophilic reagents,
such as oximes, which may catalyse reactivation. Reaction 4 ("aging")
involves cleavage of an R-O-P-bond with the loss of R and the
formation of a charged monosubstituted phosphoric acid residue still
attached to protein. The reaction is called "aging" because it is time
dependent, and the product is no longer responsive to nucleophilic
reactivating agents, such as some oximes. Since therapy of
organophosphorus compound poisoning is, in part, dependent on the
reactivating power of oximes, understanding of the "aging" reaction is
important. Pseudocholinesterase (ChE), which is present in blood
plasma and nervous tissue, but has no known physiological function, is
inhibited by organophosphorus compounds in a similar way to AChE, but
the specificity of the 2 enzymes is different. Though no toxic effect
arises as a result of inhibition of pseudoChE, measures of its
inhibition can be made for monitoring purposes.
Table 16. Elimination of 14C-labelled methyl parathion in ratsa,b
Doses Route of No. of Urine Faeces Balance
(mg/kg) administration rats (%) (%) (%)
0.1 oral 5 79.8±11 19.4±5. 99.2
3
1 oral 4 93.6±2.6 6.3±1.1 99.9
1 iv 5 99.0±3.8 0.8±0.1 99.8
1 oral 4 93.3±5.1 6.6±2.7 99.9
5 oral 5 94.7±6.0 5.1±0.5 99.8
a Adapted from: Weber et al. (1979).
b iv = intravenously.
The kinetics of the toxic metabolite of methyl parathion, methyl
paraoxon, were studied in conscious dogs (De Schryver et al., 1987).
Thirty min before performing the test, the dogs received atropine as
protection against intoxication. Methyl paraoxon was administered
intravenously (2.5 mg/kg body weight) or orally (15 mg/kg body
weight). The distribution of an intravenous dose was very fast.
The elimination was fitted by using a one-compartment model. The
average half-life was determined to be 9.7 min, the average volume of
distribution 1.76 litre/kg, and the average plasma clearance 126 ml/kg
per min. Within 3-16 min, the maximal plasma concentration (927-2905
µg/litre) was reached following oral application. The bioavailability
ranged from 5 to 71%. The hepatic extraction in anaesthetized dogs
varied at a high level of 70-92%. From comparison of the urinary
excretion as p-nitrophenol after intravenous (87 and 97%) and oral
(63 and 60%) administration of methylparaoxon, the gastrointestinal
absorption seemed to be about 60%. It was assumed, that the kinetics
were linear in this dose range.
The concentration of the main metabolites paranitrophenol (PNP)
and dimethylphosphate (DMP) in the urine of 4 human volunteers,
following 2 days of ingestion of 2 or 4 mg methyl parathion, is shown
in Table 17. Unmetabolized traces of methyl parathion were also found
in the urine, which was collected after 4-, 8-, and 24-h (Morgan et
al., 1977). The urinary excretion of nitrophenol was 60% within 4 h,
86% within 8 h, and approximately 100% within 24 h following
ingestion. Table 17 shows the dependence of urinary metabolite
excretion on methyl parathion dosage.
Table 17. p-Nitrophenol (PNP) and dimethylphosphate (DMP) concentrations in
24-h urine samples collected from human volunteers following
administration of 2 or 4 mg methyl parathiona
PNP DMP
mean range mean range
a) 2 mg methyl parathion
urinary 0.13 0.08-0.20 0.06 0.02-0.11
concentration
(mg/litre)
24-h excretion (mg) 0.29 0.14-0.43 0.12 0.07-0.16
excretion per g 0.16 0.10-0.23 0.06 0.03-0.10
creatinine (mg/g)
b) 4 mg methyl parathion
urinary 0.34 0.16-0.61 0.14 0.05-0.23
concentration
(mg/litre)
24-h excretion (mg) 0.58 0.34-0.88 0.23 0.12-0.41
excretion per g 0.31 0.15-0.42 0.13 0.06-0.20
creatinine (mg/g)
a Adapted from: Morgan et al. (1977).
6.5 Retention and turnover
Braeckman et al. (1980) injected 10 mg methyl parathion per kg
body weight intravenously into dogs, recorded the uptake of methyl
parathion, and determined a harmonic mean terminal half-life of 7.2 h.
Five hours after the injection, the concentration decreased to 30% of
the initial value. Primarily, the peripheral body compartments
contained this residual methyl parathion. The excretion was completed
within 35 h.
The velocity of the excretion of the main metabolites after oral
or intravenous application was similar. However, the bioavaibility
after oral intake was reduced by first-pass extraction by the liver
compared with the intravenous application. Methyl parathion was shown
to bind to a great extent (90%) to plasma proteins in both dogs and
humans (Braeckman et al., 1983).
7. EFFECTS ON ORGANISMS IN THE ENVIRONMENT
7.1 Microorganisms
7.1.1 Bacteria and fungi
Soil concentrations of methyl parathion of 5 mg/kg or more were
found to reduce microbial reductive potential (Reddy & Gambrell,
1985).
In biotests for sanitary control of water samples, growth
inhibition of Escherichia coli by several toxicants was studied in
a liquid medium (Vogel-Bonner medium, supplemented with thymine and
glucose at 37 °C). The minimal concentrations of methyl parathion that
significantly increased growth rate and doubling time of E. coli
were reported to be 62.5 mg/litre and 125 mg/litre; the bacterium
used the compound as a carbon source (Espigares et al., 1990).
Portier et al. (1983) tested the effects of methyl parathion (1.5
or 5 mg/litre) on the reproduction of aquatic microorganisms from
drainage basins in laboratory experiments, using static or
flow-through approaches (28 °C; pH 7.5; 22%; 22 days; or 28 °C; pH
7.2; 0%; 24 days). In bacteria and Actinomycetes , methyl parathion
had a positive effect on the development. In fungi and yeasts, slight
negative effects were found that were related to the test conditions
rather than to the toxicant concentration. In general, a concentration
of up to 5 mg methyl parathion/litre resulted in increased activity
and biomass production in a microbial community, being used as carbon
source by the microorganisms (Portier & Meiers, 1982).
Bhunia et al. (1991) cultured Nostoc muscorum , a blue-green
alga (Cyanobacterium), which is a major nitrogen-fixing organism in
tropical soil, with methyl parathion at 5, 10, 20, or 35 mg/litre.
Only the highest concentration significantly reduced the growth of the
cells in culture. However, the chlorophyll- a contents of the
cultures were marginally reduced at 5 mg methyl parathion/litre and
substantially reduced at 10 mg/litre. Nitrogenase activity was reduced
to < 50% of control levels at 10 mg/litre.
7.1.2 Algae
The 96-h EC50, i.e., the calculated concentration of methyl
parathion that would inhibit growth by 50% of the diatom Skeletonema
costatum , ranged between 5.0 and 5.3 mg/litre (Walsh & Alexander,
1980; Walsh et al., 1987).
Exposure of cultures of Chlorella protothecoides to 26-80 µg
methyl parathion/litre resulted in decreases in cell growth, as
measured by cell count, and chlorophyll and protein contents (Saroja-
Subbaraj & Bose, 1982; Saroja-Subbaraj & Bose, 1983a). These effects
were correlated with a reduction in photosynthetic electron transfer
(Saroja-Subbaraj & Bose, 1983a; Saroja-Subbaraj & Bose, 1983b).
Recovery from the effect on photosynthesis occurred after removal of
the pesticide. Tolerance to the effect of methyl parathion on cell
growth occurred for several weeks after exposure (Saroja-
Subbaraj & Bose, 1984).
In a natural phytoplankton community, addition of 1 mg methyl
parathion/litre led to a 5% decrease in the productivity (Butler,
1964).
An algal bloom (species not specified) in a methyl
parathion-treated pond was suggested to have been induced by the
mortality of herbivorous mayfly larvae and Daphnia (Crossland & Elgar,
1983).
7.2 Aquatic animals
The acute effects of methyl parathion on aquatic animals in
laboratory studies are presented in Table 18. The data show that the
sensitivity of aquatic animals to methyl parathion varies considerably
between species.
LC50 values of more than 1 mg/litre have been found for some
freshwater biota (molluscs, fish, and amphibians). Insect sensitivity
to methyl parathion depends not only on the species but on the life
stage. In general, instar I larvae are more affected than instar IV
larvae. Apperson et al. (1978) showed that larvae may develop a
resistance to methyl parathion. Both freshwater and marine
crustaceans are sensitive to methyl parathion with EC50 values
ranging from 0.002 to 0.050 mg/litre. In general, copepods were less
sensitive than decapods in laboratory tests.
Many laboratory studies have been performed on the acute toxicity
of methyl parathion for fish. The following symptoms of methyl
parathion poisoning can be expected to occur in fish: darkening of the
skin, hyperactivity, body tremors, lethargy, jerky swimming, scalosis,
loss of equilibrium, opercular or gaping paralysis, and death (Rao et
al., 1967; Anees, 1975; Midwest Research Institute, 1975). One
response that may be considered to be somewhat characteristic of acute
methyl parathion poisoning in fish is the extreme forward position of
the pectoral and pelvic fins (Midwest Research Institute, 1975;
Srivastava & Singh, 1981).
Table 18. Acute effects of methyl parathion on aquatic animals in laboratory studies
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
MOLLUSCA
Freshwater mussel
Lamellidens 48 st.c m, LC50 20 000 - Moorthy et al. (1983)
marginalis
Lamellidens m, LC50 25 000 - Moorthy et al. (1983)
marginilis
Lamellidens 20 g 48 st. m, LC50 23 400 - Rao et al. (1983)
marginalis
Eastern oyster
Crassostrea larvae 48 st.; natural seawater d, EC50 12 000 P: 99% Mayer (1997)
virginice 25 °C s: TEG
Marine hard clam
Mercenaria adult 96 St.: wellwater; no effect 25 000 s: acetone Mayer (1987)
mercenaria 24°/oo d 20 °C
pH8
Nassa dosoleta adult 96 st.; wellwater; no effect 25 000 s: acetone Meyer (1987
24°/oo d 20 °C
pH8
Table 18 (continued)
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
ANNELIDA
(Estuerine)
Branchiura - 72 st.; 4.4 °C m, 100% 4000 P: techn.gr. Naqvi (1973)
sowerbyi s: acetone
Branchiura - 72 st.; 21 °C m, 0% 4000 P: techn.gr. Naqvi (1973)
sowerbyi s: acetone
- 72 st.; 32.2 °C m, 100% 4000 P: techn.gr. Naqvi (1973)
s: acetone
CRUSTACEA
(Freshwater)
Water flea
Daphnia adult 24 st.; dechlorinated i, LC50 2.4 P: 93.8% Stephenson &
longispira tap-water; s: acetone Kane (1984)
19.5 °C; H 250e
Daphnia < 24 h 24 st.; dechlorinated i, LC50 4.1 P: 93.8% Stephenson &
magna old tap-water; s: acetone Kane (1984)
19.5 °C; H 250
Daphnia adult 24 - i, LC50 5.4 P: 93.8% Stephenson &
magna s: acetone Kane (1984)
Daphnia < 24 h 48 st.; artificial i, LC50 7.8-9.1 P: 99% Dortland (1980)
magna old water; 18 °C s: acetone
Table 18 (continued)
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
Daphnia first 48 st.; reconst. i. LC50 0.14 P: 98.7 Mayer & Ellersieck
magnia instar water; 21 °C s: acetone (1986)
pH 7.2-7.5;
H40-50
Daphnia - 3 st.; 24 °C m, LC50 8.5 - Nishiuchi & Hashimoto,
pulex (1967)
Moira macrocopa - 3 st.; 24 °C m, LC50 5.5 - Nishiuchi &
Hashimoto (1967)
Simocephalus first 48 st.; reconst. i, LC50 0.37 P: 98.7% Mayer & Ellersieck
secrultus instar water; 15 °C s: acetone (1986)
larva pH 7.2-7.5
H 40-50
Scud
Gammarus adult 96 st.; reconst. m, LC50 3.8 P: 98.7% Mayer & Ellersieck
fasciatus water; 15 °C s: acetone (1986)
pH 7.2-7.5;
H 40-50
Table 18 (continued)
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
Field crab
Oziotelphusa - 48 st.; tap-water; m, LC50 1000 P: techn.gr. Reddy et al. (1986a)
senex senex pH 7.3; 30 °C s: acetone
DO 6.2g; H 38
Crayfish
Orconectes adult 96 st.: reconst. m, LC50 15 P: 98.7% Mayer & Ellersieck
nais water; 15 °C s: acetone (1986)
pH 7.2-7.5;
H 162-272
Procambarus 2.5- 96 st.; tap-water; m, LC50 3 P: techn.gr. Cheeh et al. (1980)
acutus 3.5 cm pH 8.4; H 100
from clean area 1.2- 48 st.; tap-water; m, LC50 2.4 P: techn.gr. Albaugh (1972)
1.5 cm pH 8.7; H 10 as acetone
from treated 1.2- 48 st.; tap-water; m, LC50 3.4 P: techn.gr. Albaugh (1972)
area 1.5 cm pH 8.7; H 10 s: acetone
Procambarus 8.9 cm 36 st.; distilled m, LC50 41 P: 51% Chang & Lange (1967)
clarkii water
22.2-25-5 °C
Procambarus 8.9 cm 24 at.; tap-water m, LC50 50 P: tech.gr. Muncy & Oliver (1963)
clarkii 16-32 °C
pH 7.6
Table 18 (continued)
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
Procambarus 8.9 cm 48 st.; tap-water m, LC50 40 P: tech.gr Muncy & Oliver (1963)
clarkii 16-32 °C
pH 7.6
Procambarus 8.9 cm 72 st.; tap-water m, LC50 40 P: tech.gr. Muncy & Oliver (1963)
clarkii 16-32 °C
pH 7.6
ESTUARINE
AND MARINE
Copepod
Acartia tonsa - 96 st.; natural m, LC50 28 P: 99% Mayer (1987)
seawater; 22°/oo s: TEG
22 °C; pH 8.1-
8.2; DO 7-7.6
Acartia tonsa adult 96 st.; syntheric m, LC50 890 P: 80% Khattat & Farley
seawater; 22°/oo (1976)
17 °C
Table 18 (continued)
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
Sand shrimp
Crangon 2.6 cm 24 st.; wellwater m, LC50 11 s: acetone Eisler (1969)
septemspinosa 0.25 g 24°/oo; 20 °C; pH 8
DO 7.1-7.7
Crangon 2.6 cm 48 st.; wellwater m. LC50 3 s: acetone Eisler (1969)
septemspinosa 0.25 g 24°/oo; 20 °C; pH 8
DO 7.1-7.7
Crangon 2.6 cm 96 st.; wellwater m, LC50 2 s: acetone Eisler (1969)
septemspinosa 0.25 g 24°/oo; 20 °C; pH 8
DO 7.1-7.7
Mysid shrimp
Mysidopsis 24 h 96 st.; natural m, LC50 0.98 P: 99% Mayer (1987)
bahia old seawater; 20°/oo; s: TEG
25 °C;
DO 4.3-5.5
Mysidopsis 24 h 96 st.; natural no effect 0.32 P: 99% Mayer (1987)
bahia old seawater; 20°/oo; s: TEG
25 °C;
DO 4.3-5.5
Mysidopsis 24 h 96 flow-through m, LC50 0.77 P: 99% Mayer (1987)
bahia old natural seawater; s: TEG
20°/oo; 25 °C;
DO 4.3-5.5
Table 18 (continued)
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
Mysidopsis < 24 h 96 flow-through m, LC50 0.78 P: 99% Mayer (1987)
bahia old 14°/oo; 19.5 °C; s: TEG
Mysidopsis juvenile 96 flow-through m, LC50 0.77 s: TEG Nimmo et al. (1981)
bahia 22-28 °C
Mysidopsis juvenile 96 flow-through MATCg 0.11-0.16 s: TEG Nimmo et al. (1981)
bahia 22-28 °C
Hermit crab
Pagurus 3.5 mm 24 st.; wellwater m, LC50 23 s: acetone Eisler (1969)
longicarpus 0.28 g 24°/oo; 20 °C; ph 8;
DO 7.1-7.7
Pagurus 3.5 mm 48 st.; wellwater m, LC50 7 s: acetone Eisler (1969)
longicarpus 0.28 g 24°/oo; 20 °C; ph 8;
DO 7.1-7.7
Pagurus 3.5 mm 96 st.; wellwater m, LC50 7 s: acetone Eisler (1969)
longicarpus 0.28 g 24°/oo; 20 °C; ph 8;
DO 7.1-7.7
Table 18 (continued)
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
Crab
Portunus Zo÷e IV 24 25 °C m, LC50 0.17-0.5 - Hirayama & Tamaoi
trituberculatus stage (1980)
Grass shrimp
Palaemonetes 31 mm 24 st.; wellwater; m, LC50 15 s: acetone Eisler (1969)
vulgaris 0.47 g 24°/oo; 20 °C; ph 8;
DO 7.1-7.7
Palaemonetes 31 mm 48 st.; wellwater; m, LC50 10 s: acetone Eisler (1969)
vulgaris 0.47 g 24°/oo; 20 °C; ph 8;
DO 7.1-7.7
Palaemonetes 31 mm 96 st.; wellwater; m, LC50 3 s: acetone Eisler (1969)
vulgaris 0.47 g 24°/oo; 20 °C; ph 8;
DO 7.1-7.7
Brown shrimp
Penaeus aztecus adult 24 flow-through m, LC50 5.5 s: acetone Butler (1964)
29°/oo; 25 °C
Penaeus aztecus adult 48 flow-through m, LC50 5.5 s: acetone Butler (1964)
29°/oo; 25 °C
Table 18 (continued)
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
Pink shrimp
Penaeus duorerum - - flow-through m, LC50 1.9 s: acetone Schoor & Brausch,
17-31 °/oo + TEG (1980)
7.6-28.8 °C
Penaeus duorarum post- 96 flow-through m, LC50 1.2 s: TEG Mayer (1987)
larvae natural seawater P: 99%
20 °/oo; 25 °C
Japanese shrimp
Peneaus post- 24 25 °C m, LC50 0.5-0.9 - Hirayama & Tamaoi
japonicus larve (1980)
Shrimp
Penaeus post- 96 st.; natural seawater m, LC50 1.4 s: TEG Mayer (1987)
stylirostris larvae 20 °/oo; 25 °C; P: 99%
DO 5.6-6.3
Penaeus adult 96 st.; 15 °/oo; m, LC50 148 - Reddy & Rao (1986)
monodon 23 °C; pH 7.3
Penaeus adult 96 st.; 15 °/oo; m, LC50 98 - Reddy & Rao (1986)
indicus 23 °C; pH 7.3
Table 18 (continued)
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
Penaeus (inter-molt) 48 st.; seawater; m, LC50 95-Reddy & Rao (1986)
indicus 2.5 g 15 °/oo; 23 °C
pH 7.1
Metapenaeus adult 96 st.; 15 °/oo m, LC50 102 - Reddy & Rao (1986)
monoceros 23 °C; pH 7.3
Metapenaeus (inter-molt) 48 st.; seawater; m, LC50 120- Reddy & Rao (1988)
monoceros 2.5 g 15 °/oo; 23 °C;
pH 7.1
Metapenaeus adult 96 st.; 15 °/oo m, LC50 115 - Reddy & Rao (1986)
dopsoni 23 °C; pH 7.3
INSECTA
Mosquito
Culex piplens 4th 24 st.; 28 °C; m, LC50 30 P: 98.2% Yasuno et al. (1965)
instar deionized s: ethanol
larva water
Culex piplens 4th 24 st.; 28 °C; m, LC50 2000 P: 98.2% Yasuno et al. (1965)
instar polluted s: ethanol
larva water
Culex piplens 4th 96 st.; 28 °C; m, LC50 30 P: 98.2% Yasuno et al. (1965)
instar deionized s: ethanol
larva water
Table 18 (continued)
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
Culex pipiens 4th 96 st.; 28 °C; m, LC50 80 000 P: 98.2% Yasuno et al. (1965)
instar polluted s: ethanol
larva water
Chactorus 1st 24 st.; lake m, LC50 1.6 P: techn.gr. Apperson et al.
astictopus instar water; 25 °C s: acetone (1978)
larva (1962 exper.)
Chactorus 4th 24 st.; lake m, LC50 30 P: techn.gr. Apperson et al.
astictopus instar water; 25 °C s: acetone (1978)
larva (1962 exper.)
Chactorus 1st 24 st.; lake m, LC50 18 P: techn.gr. Apperson et al.
astictopus instar water; 25 °C s: acetone (1978)
larva (1978 exper.)
Chactorus 4th 24 st.; lake m, LC50 85 P: techn.gr. Apperson et al.
astictopus instar water; 25 °C s: acetone (1978)
larva (1978 exper.)
Table 18 (continued)
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
Damselfly
Ischnura larva 96 st.; reconst. m, LC50 33 P: 98.7% Mayer & Ellersiack
venticalus water; 15 °C s: acetone (1987)
pH 7.2-7.5
H 167-272
FISH
(Freshwater)
Betta adult 120 tap-water; 25 °C m, LC50 7500-8000 s: haxana Walsh & Hanselka
splendens pH 7-7.4 (1972
Goldfish
Carassius 0.6- 96 st.; reconst. m, LC50 9000 P: 80% Mayer & Ellersieck
auratus 1.7 g water; 18 °C; s: acetone (1986)
pH 7.1
Carassius 4.6 cm 24 st.; dest. m, LC50 14 000 P: 80% Pickering et al.
auratus 1.2 g water; 25 °C; s: acetone (1962)
pH 7.4-7.5
H 20; DO 4-8
Carassius 4.6 cm 48 st.; distilled m, LC50 12 000 P: 80% Pickering et al.
auratus 1.2 g water; 25 °C; s: acetone (1962)
pH 7.4-7.5
H 20; DO 4-8
Table 18 (continued)
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
Carassius 4.6 cm 96 st.; distilled m, LC50 12 000 P: 80% Pickering et al.
auratus 1.2 g water; 25 °C; s: acetone (1962)
pH 7.4-7.5
H 20; DO 4-8
Golden carp
Cyprinus - 48 st.; 24 °C m, LC50 > 10 000 P: 80% Nishiuchi &
auratus s: acetone Hashimoto (1967)
Carp
Cyprinus < 1 year 24 st.; 20 °C m, LC50 27 600 P: 80% Rehwoldt et al.
carpio pH 7.2; DO 6; s: acetone (1977)
H 50
Cyprinus < 1 year 48 st.; 20 °C m, LC50 21 200 P: 80% Rehwoldt et al.
carpio pH 7.2; DO 6; s: acetone (1977)
H 50
Cyprinus < 1 year 96 st.; 20 °C m, LC50 14 800 P: 80% Rahwoldt et al.
carpio pH 7.2; DO 6; s: acetone (1977)
H 50
Table 18 (continued)
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
Cyprinus 35 g 48 st.; 20 °C m, LC50 12 000 P: 80% Nagaratnamma &
carpio pH 7.2; DO 6; s: acetone Ramamurthi (1982)
H 50
Cyprinus 0.6- 96 st.; reconst. m, LC50 7130 P: 80% Mayer & Ellersieck
carpio 1.7 g water; 18 °C s: acetone (1986)
pH 7.1
Cyprinus 0.6 g 96 st.; reconst. m, LC50 8900 P: techn.gr. Johncon & Finley
carpio 1.7 g water; 18 °C s: acetone (1980)
pH 7.2-7.5
H 40-50
Cyprinus 0.6 g 48 st.; 24 °C m, LC50 > 10 000 P: techn.gr. Nishiuchi &
carpio s: acetone Hashimoto (1967)
Banded killifish
Fundulus < 1 year 24 st.; 20 °C m, LC50 24 900 P: techn.gr. Rehwoldt et al.
diaplanus pH 7.2; DO 6; s: acetone (1977)
H 50
Fundulus < 1 year 48 st.; 20 °C m, LC50 18 600 P: techn.gr. Rehwoldt et al.
diaplanus pH 7.2; DO 6; s: acetone (1977)
H 50
Fundulus < 1 year 96 st.; 20 °C m, LC50 15 200 P: techn.gr. Rehwoldt et al.
diaplanus pH 7.2; DO 6; s: acetone (1977)
H50
Table 18 (continued)
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
Mosquito fish
Gambusia adult 48 st.; dechlorinated m, LC50 13 480 P: 99% Chambers &
affinis non tap-water s: Yarbrough (1974)
resistent methoxy-ethanol
Gambusia adult 48 st.; dechlorinated m, LC50 17 480 P: 99% Chambers &
affinis non tap-water s: Yarbrough (1974)
resistent methoxy-ethanol
Catfish
Heteropneustes adult 96 24 °C; pH 7,7; m, LC50 7000 s: acetone Srivastava & Singh
fossilis (fem) DO 6; H 117 (1981)
Heteropneustes 16 cm 24 st.; 23 °C; m, LC50 9400 s: acetone Singh & Srivastava
fossilis 35 g pH 7.7; DO (1982)
6.1; H 115
Heteropneustes 16 cm 48 st.; 23 °C; m, LC50 8600 s: acetone Singh & Srivastava
fossilis 35 g pH 7.7; DO (1982)
6.1; H 115
Heteropneustes 16 cm 72 st.; 23 °C; m, LC50 8000 s: acetone Singh & Srivastava
fossilis 35 g pH 7.7; DO (1982)
6.1; H 115
Table 18 (continued)
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
Heteropneustes 16 cm 96 st.; 23 °C; m, LC50 7000 s: acetone Singh & Srivastava
fossilis 35 g pH 7.7; DO (1982)
6.1; H 115
Black Bullhead
Ictalurus melas 0.6- 96 st.; reconst. m, LC50 6640 P: 80% Mayer & Ellersieck
1.7 g water; 18 °C s: acetone (1986)
pH 7.1
Catfish
Mystus cavasius 6-8 cm 96 26-30 °C m, LC50 5900 - Murty & Ramani (1982)
7g
Channel Catfish
Ictalurus 1.4 g 96 st.; reconst. m, LC50 5240 P. techn.gr. Mayer & Ellersieck
punctatus water; 18 °C s: acetone (1986)
pH 7.2-7.5; H 40-50
Guppy (Poecilia
reticulata)
Lebistes 6 mon. 24 st.; distilled m, LC50 11 000 P. 80% Pickering et al.
reticulatus water; 25 °C s: acetone (1962)
pH 7.4-7.5;
H 20, DO 4-8
Table 18 (continued)
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
Lebistes 6 mon. 48 st.; distilled m, LC50 9800 P. 80% Pickering et al.
reticulatus water; 25 °C s: acetone (1962)
pH 7.4-7.5;
H 20, DO 4-8
Lebistes 6 mon. 96 st.; distilled m, LC50 9800 P. 80% Pickering et al.
reticulatus water; 25 °C s: acetone (1962)
pH 7.4-7.5;
H 20, DO 4-8
Lebistes < 1 year 24 st.; 20 °C m, LC50 12 200 P. 80% Rehwoldt et al.
reticulatus pH 7.2; DO 6 s: acetone (1977)
H 20
Lebistes < 1 year 48 st.; 20 °C m, LC50 9400 P. 80% Rehwoldt et al.
reticulatus pH 7.2: DO 6 s: acetone (1977)
H 20
Lebistes < 1 year 96 st.; 20 °C m. LC50 6200 P. 80% Rehwoldt et al.
reticulatus pH 7.2; DO 6 s: acetone (1977)
H 20
Table 18 (continued)
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
Green sunfish
Lepomis 0.8 g 96 st.; reconst. m, LC50 6860 P: techn.gr. Mayer (1987)
cyanellus water; 17 °C s: acetone
pH 7.2-7.5;
H 40-50
Lepomis 0.8 g 48 st.; tap-water m, LC50 > 5000 P: techn.gr. Minchew & Ferguson
cyanellus 20 °C s: acetone (1969)
Pumpkinseed
Lepomis 40-50 g 24 injection, st. m, LD50 > 2500 P: 99% Benke et al. (1974)
gibbosus s: corn oil
Lepomis < 1 year 24 st.: 20 °C m, LD50 4900 P: 99% Rehwoldt et al.
gibbosus pH 7.2; DO 6; s: corn oil (1977)
H 50
Lepomis < 1 year 48 st.: 20 °C m, LD50 3600 P: 99% Rehwoldt et al.
gibbosus pH 7.2; DO 6; s: corn oil (1977)
H 50
Lepomis < 1 year 96 st.: 20 °C m, LD50 3600 P: 99% Rehwoldt et al.
gibbosus pH 7.2; DO 6; s: corn oil (1977)
H 50
Table 18 (continued)
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
Bluegill sunfish
Lepomis fingerling 24 st.; reconst. m, LC50 6470 P: 44.6% McCann & Jasper
machrochirus water; 18 °C s: water (1972)
pH 7; H 17
Lepomis 0.6- 96 st.; reconst. m, LC50 5720 P: 80% Macak & McAllister
machrochirus 1.7 g water; 18 °C s: acetone (1970)
pH 7.1
Lepomis 4-6 cm 24 st.; distilled m, LC50 9800 P: 80% Pickering et al.
machrochirus 1.2 g water; 25 °C s: acetone (1962)
pH 7.4-7.5
H 20; DO 4-8
Lepomis 4-6 cm 48 st.; distilled m, LC50 8600 P: 80% Pickering et al.
machrochirus 1.2 g water; 25 °C s: acetone (1962)
pH 7.4-7.5
H 20; DO 4-8
Lepomis 4-6 cm 96 st.; distilled m, LC50 2400 P: 80% Pickering et al.
machrochirus 1.2 g water; 25 °C s: acetone (1962)
pH 7.2-7.5;
H 20; DO 4-6
Table 18 (continued)
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
Lepomis 1 g 96 st.; reconst. m, LC50 4380 P: techn.gr. Mayer & Ellersieck
machrochirus water; 17 °C s: acetone (1986)
pH 7.2-7.5
H 40-50
Lepomis 0.6- 96 st.; reconst. m, LC50 5170 P: 80% Macek & McAllister
machrochirus 1.7 g water; 18 °C s: acetone (1970)
pH 7.1
Largemouth bass
Micropterus 0.6- 96 st.; reconst. m, LC50 5220 P: 80% Mayer & Ellersieck
salmoides 1.7 g water; 18 °C s: acetone (1986)
pH 7.1
Mystus cavasius - 96 - m, LC50 5900 - Murty & Ramani
(1982)
Golden shiner
Notamigonus - 48 st.; tap-water: m, LC50 > 5000 P: techn.gr. Minchew & Ferguson
chrysoleuces 20 °C s: acetone (1969)
Coho salmon
Oncorhynchus 0.6- 96 st.; reconst. m, LC50 5300 P: 80% Meyer & Ellersieck
kisutch 1.7 g water; 13 °C s: acetone (1986)
pH 7.1
Table 18 (continued)
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
Medaka
Oryzias latipes - 48 er.; 24 °C m, LC50 7500 P: techn.gr, Nishiuchi &
s: acetone Hashimoto (1967)
Yellow perch
Perca 1.4 g 96 st.; reconst. m, LC50 3060 P: techn.gr. Mayer & Ellersieck
flavescens water; 18 °C s: acetone (1986)
pH 7.2-7.5;
H 40-50
Punti
Puntius 6- 24 st.; 27.9 °C m, LC50 2900 P: 50% Rao et al. (1967)
puckelli 8.5 cm pH 8.3; H 130
Puntius 6- 48 st.; 27.9 °C m, LC50 2700 P: 50% Rao et al. (1967)
puckelli 8.5 cm pH 8.3; H 130
Puntius 6- 96 st.; 27.9 =C m, LC50 2100 P: 50% Rao et al. (1967)
puckelli 8.5 cm pH 8.3; H 130
Table 18 (continued)
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
Fathead minnow
Pimephales 1.2 g 96 st.; reconst. m, LC50 8300 P: techn.gr. Mayer & Ellersieck
promelas water; 18 °C s: acetone (1986)
pH 7.2-7.5; H40-50
Pimephales - 48 flow-through m, LC50 7400 P: 98.5% Solon & Nair (1970)
promelas s: acetone
Pimephales - 96 flow-through m, LC50 3750 P: 98.5% Solon & Nair (1970)
promelas s: acetone
Pimephales newly 96 st.; sterilized m, LC50 4460 P: 80% Jarvinen & Tanner
promelas hatched water; 25 °C (1982)
larvae pH 7.4-7.8;
DO 6.5-8.4; H 64
Pimephales newly 96 st.; sterilized m, LC50 1220 P: 80% Jarvinen & Tanner
promelas hatched water; 25 °C DO 6.5-8.4; H stock (1982)
larvae pH 7.4-7.8; 64 solution aged
11 weeks
Pimephales newly 96 st.; sterilized m, LC50 8170 P: 80% Jarvinen & Tanner
promelas hatched water; 25 °C controlled- (1982)
larvae pH 7.4-7.8; release
DO 6.5-8.4; H 64 formulation
Table 18 (continued)
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
Pimephales newly 96 st.; sterilized m, LC50 3470 P: 80% Jarvinen & Tanner
promelas hatched water; 25 °C controlled (1982)
larvae pH 7.4-7.8; release
DO 6.5-8.4; H 64 formulation
for 11 weeks
Pimephales newly 96 flow-through; m, LC50 5360 P: 80% Jarvinen & Tanner
promelas hatched sterilized water (1982)
larvae 25 °C; pH 7.4-
7.8; DO 6.5-8.4;
H 64
Pimephales newly 96 flow-through; m, LC50 6910 P: 80% Jarvinen & Tanner
promelas hatched sterilized water controlled (1882)
larvae 25 °C; pH release
7.4-7.8; formulation
DO 6.5-8.4;
H 64
Pimephales 4-6 cm 24 st.; distilled m, LC50 13 000 P: 80% Pickering et al.
promelas 1-2 g water; 25 °C s: acetone (1862)
pH 7.4-7.5;
H 20; DO 4-8
Table 18 (continued)
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
Pimephales 4-6 cm 48 st.; distilled m, LC50 9800 P: 80% Pickering et al.
promelas 1-2 g water; 25 °C s: acetone (1962)
pH 7.4-7.5;
H 20; DO 4-8
Pimephales 4-6 cm 96 st.; distilled m, LC50 9500 P: 80% Pickering et al.
promelas 1-2 g water; 25 °C s: acetone (1962)
pH 7.4-7.5;
H 20; DO 4-8
White perch
Roccus < 1 year 24 st.; 12 °C m, LC50 22 400 P: 80% Rehwoldt et al.
americanus pH 7.2 s: acetone (1977)
Roccus < 1 year 46 st.; 12 °C m, LC50 18 600 P: 80% Rehwoldt et al.
americanus pH 7.2 s: acetone (1977)
Roccus < 1 year 96 st.; 12 °C m, LC50 14 000 P: 60% Rehwoldt et al.
americanus pH 7.2 s: acetone (1977)
Cutthroat trout
Salmo clarki 0.2 g 96 st.; reconst. m, LC50 1850 P: techn.gr. Mayer & Ellersieck
water; 12 °C s: acetone (1996)
pH 7.2-7.5;
H 162-272
Table 18 (continued)
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
Rainbow trout
(Oncorhynchus
mykiss)
Salmo 1.1 g 96 st.; reconst. m, LC50 3700 P: techn.gr. Mayer & Ellersieck
gairdneri water; 12 °C; s: acetone (1986)
pH 7.2-7.5;
H 162-272
Salmo 0.6- 96 st.; reconst. m, LC50 2750 P: 80% Macek & McAllister
gairdneri 1.7 g water; 13 °C; s: acetone (1970)
pH 7.1
Salmo 24 mm 96 st.; 12 °C m, LC50 2800 P: 76.8% Palawski et al.
gairdneri (1983)
Brown trout
Salmo trutta 0.6- 96 st,; reconst, m, LC50 4750 P: 80% Mayer & Ellersieck
1.7 g pH 7.1 s: acetone (1986)
water; 13 °C
Table 18 (continued)
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
Brook trout
Salvelinus 0.5 g 96 st.; reconst. m, LC50 3780 P: techn.gr. Mayer & Ellersieck
fontinalis water; 12 °C s: acetone (1986)
pH 7.2-7.5;
H 40-50
Northern pike
Esox lucius 0.4 g 24 st.; 18 °C m, LC50 760 P: techn.gr. Mayer & Ellersieck
pH 7.1; H 44 s: acetone (1986)
Tilapia
tilapia - 48 st.; 26-28 °C m, LC50 266 P: techn.gr. Rao & Rao (1983)
mossambica pH 7; H 140 s:
2-methoxyethanol
ESTUARINE
AND MARINE
American eel
Anguilla 59 24 st.; underground m, LC50 27 600 P: act, Eisler (1970a)
rostrata mm-0.14 g wellwater; ingredient
24%0; 20 °C; s: acetone
pH 8; DO 7.1-7.7
Table 18 (continued)
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
Anguilla 59 48 st.; underground m, LC50 22 400 P: act. Eisler (1970a)
rostrata am-0. 14 g wellwater; ingredient
24°/oo; 20 °C; s: acetone
pH 8; DO 7.1-7.7
Anguilla 59 96 st.; underground m, LC50 16 900 P: act. Eisler (1970e)
rostrate am-0.14 g wellwater; ingredient
24°/oo; 20 °C; s: acetone
pH 8; DO 7.1-7.7
Anguilla < 1 year 24 st.; 20 °C. m, LC50 42 600 P: act. Rehwoldt et al.
rostrata pH 7.2; DO 6; ingredient (1977)
H 50 s: acetone
Anguilla < 1 year 48 st.; 20 °C. m, LC50 37 200 P: act. Rehwoldt et al.
rostrata pH 7.2; DO 6; ingredient (1977)
H 50 s: acetone
Anguilla < 1 year 96 st.; 20 °C. m, LC50 6300 P: act. Rehwoldt et al.
rostrata pH 7.2; DO 6; ingredient (1977)
H 50 s: acetone
Table 18 (continued)
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
Sheephead
minnow
Cyprinodon 28 days 96 st.; natural m, LC50 12 000 P: 99% Mayer (1987)
variegatus old sea-water; s: TEG
20°/oo; 25 °C;
DO 4.6-5.7
Cyprinodon 28 days 96 st.; natural no effect 10 000 P: 99% Mayer (1987)
variegatus old sea-water; s: TEG
20°/oo; 25 °C;
DO 4.6-5.7
Mummichog
Fundulus 55 mm 24 st.; underground m, LC50 > 85 100 P: act. Eisler (1970a)
heteroclitus 1.7 g wellwater; 24°/oo; ingredient
20 °C; pH 8; s: acetone
DO 7.1-7.7
Fundulus 55 mm 48 st.; underground m, LC50 85 200 P: act. Eisler (1970a)
heteroclitus 1.7 g wallwater; 24°/oo; ingredient
20 °C; pH 8; s: acetone
DO 7.1-7.7
Fundulus 55 mm 96 st.; underground m, LC50 58 000 P: act. Eisler (1970a)
heteroclitus 1.7 g wellwater; 24°/oo; ingredient
20 °C; pH 8; s: acetone
DO 7.1-7.7
Table 18 (continued)
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
Fundulus 42 mm 96 st.: underground m, LC50 8000 P: act. Eisler (1970b)
heteroclitus wellwater; 24°/oo; ingredient
20 °C; pH 8; s: acetone
DO 7.1-7.7
Fundulus 42 mm 96 st.; underground m, LC50 4000 P: act. Eisler (1970b)
heteroclitus ( + 240h wellwater; 24°/oo; ingredient
observation) 20 °C; pH 8; s: acetone
DO 7. 1-7.7
Fundulus 42 mm 96 st.; underground m, LC50 1210 solution Eisler (1970b)
heteroclitus wellwater; 24°/oo; aged for
20 °C; pH 8; 96 h
DO 7.1-7.7
Fundulus 42 mm 96 10 °C 20% M 8000 P: act. Eisler (1970b)
heteroclitus ingredient
s: acetone
Fundulus 42 mm 96 15 °C 10% M 8000 P: act. Eisler (1970b)
heteroclitus ingredient
s: acetone
Table 18 (continued)
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
Fundulus 42 mm 96 36 °/oo 100% M 8000 P: act. Eisler (1970b)
heteroclitus ingredient
s: acetone
Striped killifish
Fundulus 84 mm 24 st.; underground m, LC50 29 000 P: act. Eisler (1970a)
majalis 6.5 g wellwater; 24°/oo; ingredient
20 °C; pH 8; s: acetone
DO 7.1-7.7
Fundulus 84 mm 48 st.; underground m, LC50 19 400 P: act. Eisler (1970a)
majalis 6.5 g wellwater; 24°/oo; ingredient
20 °C; pH 8; s: acetone
DO 7.1-7.7
Fundulus 84 mm 24 st.; underground m, LC50 13 800 P: act. Eisler (1970a)
majalis 6.5 g wellwater; 24°/oo; ingredient
20 °C; pH 8; s: acetone
DO 7.1-7.7
Table 18 (continued)
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
Fundulus 42 mm 96 20 °C 50% M 8000 P: act. Eisler (1970b)
heteroclitus ingredient
s: acetone
Fundulus 42 mm 96 25 °C 100% M 8000 P: act. Eisler (1970b)
heteroclitus ingredient
s: acetone
Fundulus 42 mm 96 30 °C 100% M 8000 P: act. Eisler (1970b)
heteroclitus ingredient
s: acetone
Fundulus 42 mm 96 12 °/oo 0% M 8000 P: act. Eisler (1970b)
heteroclitus ingredient
s: acetone
Fundulus 42 mm 96 18 °/oo 0% M 8000 P: act. Eisler (1970b)
heteroclitus ingredient
s: acetone
Fundulus 42 mm 96 24 °/oo 10% M 8000 P: act. Eisler (1970b)
heteroclitus ingredient
s: acetone
Fundulus 42 mm 96 30 °/oo 70% M 8000 P: act. Eisler (1970b)
heteroclitus ingredient
s: acetone
Table 18 (continued)
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
Spot
Leiostomus 84 mm 96 st.; natural m, LC50 93 P: 99% Mayer (1987)
xanthurus 6.5 g seawater; 2°/oo; s: TEG
25 °C; DO 3.2-4.5
Leiostomus 84 mm 96 st.; natural no effect 56 P: 99% Mayer (1987)
xanthurus 6.5 g seawater; 2°/oo; s: TEG
25 °C; DO 3.2-4.5
Leiostomus 84 mm 96 flow-through m, LC50 59 P: 99% Mayer (1987)
xanthurus 6.5 g 20°/oo; 25 °C s: TEG
Atlantic silverside
Menidia 50 mm 24 st.; underground m, LC50 24 800 P: act. Eisler (1970a)
menidia 0.8 g. wellwater; 24°/oo; ingredient
20 °Cr pH 8r s: acetone
DO 7.1-7.7
Menidia 50 mm 48 at.; underground m, LC50 21 900 P: act. Eisler (1970a)
menidia 0.8 g wellwater; 24°/oo; ingredient
20 °Cr pH 8; s: acetone
DO 7.1-7.7
Menidia 50 mm 96 st.; underground m, LC50 5700 P: act. Eisler (1970a)
menidia 0.8 g wellwater; 24°/oo; ingredient
20 °C; pH 8; s: acetone
DO 7.1-7.7
Table 18 (continued)
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
Striped bass
Morone 1 year 24 st.; 20 °C; m, LC50 16 800 P: act. Rehwoldt et al.
saxatilis pH 7.2; DO 6; ingredient (1977)
H 50 s: acetone
Morone 1 year 48 st.; 20 °C; m, LC50 14 200 P: act. Rehwoldt et al.
saxatilis pH 7.2; DO 6; ingredient (1977)
H 50 s: acetone
Morone 1 year 96 st.; 20 °C; m, LC50 14 000 P: act. Rehwoldt et al.
saxatilis pH 7.2; DO 6; ingredient (1977)
H 50 s: acetone
Morone adult 96 interm. flow m, LC50 790 P: 99% Earnest (1970)
saxatilis 12.8 °
Morone juvenile 96 flow-through m, LC50 790 P: 86% Korn & Earnest
saxatilis 13 °C; 30 °/oo s: ethanol (1974)
Black mullet
Mugil cephalus 48 mm 24 st.: underground m, LC50 39 000 P: act. Eisler (1970a)
0.78 g wellwater; 24 °/oo ingredient
20 °C; pH 8; acetone
DO 7.1-7.7
Table 18 (continued)
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
Mugil cephalus 48 mm 48 st.: underground m, LC50 26 300 P: act. Eisler (1970a)
0.78 g wellwater; 24 °/oo ingredient
20 °C; pH 8; acetone
DO 7.1-7.7
Mugil cephalus 48 mm 96 st.: underground m, LC50 5200 P: act. Eisler (1970a)
0.78 g wellwater; 24 °/oo ingredient
20 °C; pH 8; acetone
DO 7.1-7.7
Northern puffer
Sphaeroides 196 mm 24 st.; underground m, LC50 100 000 P: act.. Eisler (1970a)
maculatus 153 g wellwater; 24 °/oo ingredient
20 °C; pH 8; s: acetone
DO 7.1-7.7
Sphaeroides 196 mm 48 st.; underground m, LC50 91 000 P: act. Eisler (1970a)
maculatus 153 g wellwater; 24 °/oo ingredient
20 °C; pH 8; s: acetone
DO 7.1-7.7
Sphaeroides 196 mm 96 st.; underground m, LC50 75 800 P: act. Eisler (1970a)
maculatus 153 g wellwater; 24 °/oo ingredient
20 °C; pH 8; s: acetone
DO 7.1-7.7
Table 18 (continued)
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
Bluehead
Thalassoma 90 mm 24 st.; underground m, LC~ 98 000 P. act. Eisler (1970a)
bifasciatum 7 g wellwater; 24 °/oo ingredient
20 °C; pH 8;
DO 7.1-7.7
Thalassoma 90 mm 48 st.; underground m, LC50 88 000 P. act. Eisler (1970a)
bifasciatum 7 g wellwater; 24 °/oo ingredient
20 °C; pH 8;
DO 7.1-7.7
Thalassoma 90 mm 96 st.; underground m, LC50 12 300 P. enct. Eislar (197Oa)
bifasciatum 7 g wellwater; 24 °/oo ingredient
20 °C; pH 8; s: acetone
DO 7.1-7.7
AMPHIBIA
Rana adult 96 st.; tap-water: m, LC50 8000 Mudgall & Patil
cyanophlyctis 23 °C; pH 7.3-7.8; (1987)
(male) H 60-70; DO 6.7-7.9
Rana adult 96 st.; tap-water: m, LC50 11 500 Mudgall & Patil
cyanophlyctis 23 °C; pH 7.3-7.8; (1987)
(female) H 60-70; DO 6.7-7.9
Table 18 (continued)
Species Life Test Experimental Criterion Concentration Remarksb References
stage period conditions effect (µg/litre)
(h) measureda
Western chorus
frog
Bendacris tadpole 96 st.; 15 °C; m, LC50 3700 Mayer & Ellersieck
triseriata pH 7.1; H 44 (1986)
a Criterion: m = mortality; i = immobilization; d = development; % M = % mortality.
b P = purity; s = solvent; TEG = triethyiene glycol; techn.gr. = technical grade.
c st. = static.
d °/oo = salinity.
e H = hardness in mg/litre CaCO3.
f DO = dissolved oxygen in mg O2/litre.
g MATC = maximum acceptable toxic concentration.
Murty et al. (1984) state that the lowest concentration causing
irreversible effects in the fish Mystus carasius after a 1-h exposure
was 15 mg/litre.
7.2.1 Short-term toxicity in aquatic invertebrates
7.2.1.1 Laboratory studies on single species
Exposure of the freshwater mussel (Lamellidens marginalis) to
sublethal (8 mg/litre) concentrations resulted in a transient increase
(at 12 h) followed by a decrease (at 24-72 h) in the rate of
respiration (Moorthy et al., 1984). Exposure of this species to
concentrations ranging from 10 to 50 mg/litre resulted in a
concentration-dependent decrease in heart rate (Rao et al., 1983a).
For crustaceans, long-term toxicity levels appear to be of the
same magnitude as acute: a no-effect level on the reproduction of
Daphnia magna was 0.0012 mg methyl parathion/litre after 21 days
(artificial water, 18 °C; Dortland, 1980).
Exposure of the freshwater crab (Oziotelphusa senex senex) to
sublethal levels of methyl parathion (0.1-1 mg/litre) resulted in
complete inhibition of molt, a delay in the onset of molt, or a
decrease in the percentage of molting animals (Reddy et al., 1985). A
decrease in the carbohydrate content and increase in acid phosphatase
activity in both the hepatopancreas and muscle also occurred (Reddy et
al., 1986a; 1986b).
Eisler (1970a,b) found a 20% increase in mortality in Nassa
docoleta after 10 days' exposure to 25 mg/litre (well water with a
salinity of 24o/oo, 20°C, pH 8) in.
Exposure of prawns (Penaeus indicus or Metapenaeus monoceros)
to sublethal concentrations of methyl parathion resulted in a
concentration-dependent inhibition of acetylcholinesterase activity,
which recovered in 7 days (Reddy & Rao, 1988). An increase in tissue
levels of ammonia, urea, and glutamine, apparently resulted from the
increased production of ammonia from purines and glutamate (Reddy et
al., 1988; Reddy & Rao, 1990a). There was also an increase in tissue
levels of fatty acids and cholesterol (Reddy & Rao, 1989), while the
activity of alkaline phosphatase in the hepatopancreas was inhibited,
and the acid phosphatase activity, enhanced (Reddy & Rao, 1990b).
Changes in hepatic glycogen content and haemolymph glucose levels were
observed after 5 days of sublethal methyl parathion exposure (Reddy &
Rao, 1990b).
Cripe et al. (1981) tested the stamina of mysid shrimp
(Mysidopsis bahia) in swimming against a water current in the
presence of methyl parathion. Concentrations of 0.10 and 0.31 µg/litre
did not affect maximum sustained speeds of the shrimp, but they
were significantly reduced on exposure to 0.58 µg/litre.
7.2.1.2 Mesocosmic studies
After treatment of ponds with methyl parathion, the effects on
daphnids were similar to those observed in the laboratory. However,
indirect biological effects occurred that could not be predicted on
the basis of laboratory tests. For example, the observed increase in
populations of the crustacean Diaptomus sp. in treated ponds was
attributed to the mortality of competitors (Daphnia spp.) and
predators ( Cyclops and aquatic insects) (Crossland & Elgar, 1983).
Generally, recovery of zooplankton occurred soon after the end of
treatment of ponds (Apperson et al., 1976; Crossland & Elgar, 1983).
The numbers of free-swimming Diptera and Ephemeroptera were
significantly reduced compared with controls, as were the benthic
chironomid larvae in ponds treated at 100 µg/litre. Seventy days after
treatment, there was evidence of recovery of populations of
chironimids and Ephemeroptera , with full recovery 90 days after
treatment (Crossland & Elgar, 1983).
7.2.2 Fish
7.2.2.1 Laboratory studies on single species
Jarvinen & Tanner (1982) conducted a long-term mortality study on
the fish Pimephales promelas (flow through conditions, sterile
water, 25 °C, pH 7.4-7.8, 46 mg CaCO3/litre, 6.5-8.4 mg dissolved
O2/litre). Methyl parathion concentrations of 0.59-0.77 mg/litre
induced increased mortality after 32 days. No effects on mortality
were found at 0.38 mg/litre for the technical grade product and 0.59
mg/litre for the controlled release formulation. Mortality in rainbow
trout (Salmo gairdneri) increased to 98% after exposure to 2.8 mg
technical grade methyl parathion/litre (wellwater, 12 °C, pH 7.5, 272
mg CaCO3/litre) for 96 h, followed by 7 days of observation
(Palawski et al., 1983).
Exposure of the tilapia fish (Tilapia mossambica) to methyl
parathion at a concentration of 0.09 mg/litre for 48 h resulted in a
decrease in various anions and cations in tissues (Rao et al., 1983b),
and in inhibition of acetylcholinesterase (20-60%) and ATPase (10-14%)
activities. The activities of aspartate and alanine amino-transferase
in muscle, gill, liver, and brain increased by 12-31% and 9-31%,
respectively (Rao & Rao, 1984a; 1984b). Concentrations of
carbohydrate and glycogen decreased in the tissues examined (Rao &
Rao, 1983). Levels of soluble protein and the activity of
glucose-6-phosphate dehydrogenase, a key enzyme of the hexose
monophosphate shunt, in muscle, gill, and liver, were increased (Rao
& Rao, 1987). Changes in carbohydrate metabolism were also observed in
the freshwater fish Clarias batrachus , when exposed to sublethal
concentrations of methyl parathion (7 mg/litre) for 48 and 96 h (Rani
et al., 1989). There were significant decreases in glycogen (liver)
and in pyruvate (liver, brain, gill) contents and increases in glucose
(gill) and lactate (liver, brain, gill) levels, and the specific
activities of several enzymes were inhibited.
Exposure of the catfish (Channa punctatus) to 52 µg methyl
parathion/litre resulted in the elevation of serum triiodothyronine
(T3) as well as depression of brain acetylcholinesterase activity
(Ghosh et al., 1989). This low dose of methyl parathion also impaired
the regulation of gonadal function by gonadotropic hormone and gonado
tropin-releasing hormone in Channa punctatus (Ghosh et al., 1990).
The inhibiting effect was also seen under field conditions where water
concentrations of methyl parathion amounted to 0.239 µg/litre (Ghosh
et al., 1990).
Exposure to sublethal doses of 0.1 mg methyl parathion/litre
(corresponding to 1/5th of the LC50 values) for 75 days produced
severe ovarian damage in the carp minnow (Rasbora daniconius)
(Rastogi & Kulshrestha, 1990). Effects included diminished growth of
ovaries and histopathological changes in immature, maturing, and
mature oocytes.
Sublethal concentrations of methyl parathion (1.2 mg/litre)
induced behavioural abnormalities in the juveniles of the fish
Cyprinus carpio , such as imbalance, increased opercular movement
and irritation (Babu et al., 1986). Exposed juveniles, when
transferred to pesticide-free medium, showed rapid recovery.
Little et al. (1990) exposed rainbow trout (Oncorhynchus mykiss)
to methyl parathion at 0.01 or 0.1 mg/litre and measured various
behavioural parameters. Swimming capacity (as cm/s) was unaffected at
any concentration tested, though spontaneous swimming activity was
significantly reduced at both exposures. Number of prey (Daphnia)
consumed was reduced, even at the lower exposure (0.01 mg/litre), but
the percentage of daphnia consumed and the strike frequency of the
fish on daphnia were only affected at 0.1 mg/litre. The capacity of
the trout to escape from a predator was only reduced at 0.1 mg/litre.
In a static system (well water, salinity: 24o/oo, 20 °C, pH 8),
with the fish Fundulus heteroclitus, the LC50 was 0.96 mg/litre
after exposure for 10 days or 4 mg/litre after exposure for 4 days
followed by 10 days in clean water (Eisler, 1970b).
7.2.2.2 Mesocosmic studies
In a methyl parathion-treated experimental pond, a high mortality
rate was observed in rainbow trout, 37 days after treatment, which was
associated with depression of the concentration of dissolved oxygen to
less than 3 mg/litre, and decay of large amounts of algal biomass
(Crossland & Elgar, 1983; see also section 7.3).
7.2.3 Amphibians
After application of methyl parathion to Rana cyanophlyctis,
Mudgall & Patil (1987) found increased levels of glycogen in muscles,
liver, and kidney, compared with control animals. On the basis of the
marked elevated glycogen concentration in the kidney, it was concluded
that the kidneys were the main target organ.
The effects of metacid (DDT + 50% w/w methyl parathion) on the
development of the Indian bullfrog (Rana tigrina) were determined
by Mohanty-Hejmadi & Dutta (1981). Threshold concentrations for
adverse effects on eggs, feeding stage, and limb bud stage tadpoles
ranged from 0.00005% to 0.004% metacid. These levels were much lower
than the recommended dosage for the field application of metacid
(0.15%).
7.3 Terrestrial organisms
7.3.1 Plants
Methyl parathion has been found to have phytotoxic effects in
diverse crops, such as cotton (Gossypium hirsutum) (Brown et al.,
1962; Roark et al., 1963; Youngman et al., 1989, 1990) and lettuce
(Lactuca sativa) (Toscana et al., 1982; Johnson et al., 1983;
Youngman et al., 1989).
Swamy & Veeresh (1987) found a reduction in lipid synthesis in
methyl parathion-treated seeds of Sorghum sp., 24 h after
germination. An increase in lipid production with a substantial
elevation in unsaturated fatty acids was observed in methyl
parathion-treated sorghum, 120 h after germination. The same effect
occurred in 48-h seedlings, which were treated with the degradation
products of methyl parathion. From this, it was concluded that the
time-related reversal effect of methyl parathion is triggered by the
pesticide degradation products themselves.
Exposure of sorghum seeds to methyl parathion for 1 h before
germination resulted in an accumulation of proline in the seedlings
and a reduction in growth, without affecting the water content.
Residues of methyl parathion in the soil also influenced seed
germination and seedling growth (Deshpande & Swamy, 1987).
7.3.2 Invertebrates
Poisoning of bees has been reported after incorrect application
of methyl parathion on windy days (Bubien, 1971).
Analysis of dead honey bees (Apis mellifera; Hymenoptera) for
pesticide residues, during 1983-85 in the USA, showed that the health
of colonies, poisoned with methyl parathion (Penncap-M) or with a
combination of methyl parathion and other insecticides, was often
severely affected, whereas colonies contaminated by insecticides other
than methyl parathion often recovered (Anderson & Wojtas, 1986).
Acute toxicity values were established for acetone formulations
of methyl parathion applied topically to workers of Africanized and
European honey bees (Apis mellifera) (Danka et al., 1986). The
LC50 values of 0.32 µg and 0.17 µg/bee, respectively, showed the
greater tolerance to methyl parathion of Africanized bees compared
with European bees.
Jepson (1989) calculated a hazard ratio (ratio of contact LD50
at 0.11 µg/bee to the application rate of the pesticide at 500 g
a.i./ha) for methyl parathion in honey bees of 8937 (using the method
of Smart & Stevenson, 1982). Values of the hazard ratio greater than
50 are usually considered to indicate danger for bees. Along with
azinphos methyl, methyl parathion has a very high indication of
danger for bees from field spraying. Although the intrinsic toxicity
for bees is as high for other pesticides, such as the pyrethroids, the
hazard ratio is lower, since application rates of these pesticides are
also lower.
Methyl parathion applied to small barriered plots of spring wheat
at 1000 g a.i./ha did not have any apparent adverse effects on leaf
litter decomposition and on earthworm populations (species not
differentiated). Effects on individual earthworm species could not be
demonstrated, because of statistically insufficient numbers of mature
specimens collected (Shires, 1985).
Methyl parathion has adverse effects on many different beneficial
insects. It was placed in the highest class of toxicity (score 4 in a
classification of 1-4) for Chrysopa (Plannipennia), Coccinellidae
(Coleoptera), and Hymenoptera (Entomophaga) (Höbaus, 1987). Side
effects on the predator mite Phytoseiulus persimilis were placed in
class 3 (Kniehase & Zoebelein, 1990).
Thompson & Gore (1972) assessed the toxicity of methyl parathion
(95-99% purity) for Folsomia candida (Collembola) by direct contact
in a spray tower and when applied to soil. In the direct-contact
study, a 0.01% methyl parathion solution caused a 100% mortality of
the collembola, 24 h after being treated. A 100% mortality rate also
occurred in soil (Plainfield sand) treated with 0.5 mg methyl
parathion/kg dry weight soil after a 24-h exposure.
Methyl parathion (0.05%) sprayed on coconut leaflets was found to
be highly toxic to the parasitoid fauna (Hymenoptera; Ento-mophaga )
of a coconut coccid (Opisina arenosella; Homoptera). The mortality
of the caged insects was assessed 24 h after introduction of leaflets
and after longer periods (Jalaluddin & Mohanasundaram, 1989).
Flanders et al. (1984) conducted a field study of methyl
parathion (sprayed at recommended rates of 0.84 kg a.i./ha, in an
encapsulated formulation, on soybeans) effects on Pediobius
foveolatus , a parasitoid of the Mexican bean beetle Epilachnia
varivestis . The pupae within parasitized beetles were unaffected by
the insecticide and emerged normally. However, residues of methyl
parathion on the plants killed 100% of the adult parasites emerging
within 1 day, and 50% of those emerging within 3 days of the spraying.
By 9 days after spraying, the mortality of emerging parasite adults
was no longer affected by residues.
Walker et al. (1985) examined the effects of methyl parathion,
used at 0.6 kg a.i./ha on rice fields in Louisiana, on the survival
and reproduction of parasitic nematodes (Romanomermis culcivorax),
introduced into the fields to control mosquito larvae. There were not
any adverse effects of the insecticide on the nematodes.
Only a few cases of resistance to methyl parathion have been
reported among arthropod parasites or predators. The reports refer to
the braconid Bracon mellitor (Hymenoptera), a parasite of the boll
weevil (Anthonomous grandii), which developed low levels of
resistance after 5 or more generations of selection in the laboratory,
and to field populations of the coccinellid Coleomegilla maculata
(Coleoptera) taken from cotton fields, treated extensively with methyl
parathion for 2 decades (Croft, 1977).
One week after application of methyl parathion (1000 g a.i./ha)
on small barriered plots of spring wheat, the number of predatory
beetles (mainly 4 species of Carabidae and 3 genera of Staphylinidae)
fell to about 10% of that in the untreated control plot. Recovery
occurred between 4 and 6 weeks after application, but a further fall
in numbers of predatory beetles was observed 8-12 weeks after
application (Shires, 1985). This second reduction was attributed to an
indirect effect of the treatments, causing removal of the predators'
food supply (mainly cereal aphids).
7.3.3 Birds
The acute lethal toxicities of methyl parathion for birds are
compiled in Table 19.
Percutaneous administration of methyl parathion was more toxic
for young mallard ducks (Anas platyrhynchos) than oral (dietary)
administration (Hudson et al., 1979).
Studies on mallard ducks (Anas platyrhynchos) have shown that
methyl parathion can affect the brood-rearing phase by increasing
mortality and causing behavioural changes (Fairbrother et al., 1988).
At least 40% of young ducklings exposed to sub-lethal oral doses of
methyl parathion (4 mg/kg body weight) died within 40 min in outdoor
enclosures. Several activities (swimming, preening, feeding) of
mothers and ducklings were changed in treated broods. Ducks (Anas
platyrhynchos; A. discors; Aix sponsa) nesting in agricultural
fields aerially treated with methyl parathion (1.4 kg a.i./ha) had a
higher average daily rate of duckling losses than those nesting in
untreated fields (Brewer et al., 1988).
Spraying of methyl parathion at 1.4 kg a.i./ha did not
significantly reduce the hatchability of starling (Sturnus vulgaris)
eggs and the number of young fledglings per nest. However,
collectively, the number of fledglings from the treated field was
significantly lower than that from the control field (Robinson et al.,
1988).
Buerger et al. (1991) dosed wild bobwhite quail (Colinus
virginianus) with methyl parathion at 0, 2, 4, or 6 mg/kg body
weight by oral intubation and then released them into the wild. The
birds were monitored for 14 days by radio telemetry. Only the birds
receiving 6 mg methyl parathion/kg body weight showed significantly
reduced survival and this was the result of predation rather than
overt toxicity. Activity was not affected by any treatment. Survivors
did not show any inhibition of brain cholinesterase activity after 14
days, compared with controls.
Bennett et al. (1991) examined parameters of reproductive success
in mallard ducks exposed to a dietary concentration of methyl
parathion of 400 mg/kg. The female mallards were fed the methyl
parathion diet at different stages of egg laying and incubation.
Numbers of hatchlings per nest were 61%, 43%, and 58% of controls for
birds exposed during egg laying, early incubation, and late
incubation, respectively. Daily egg production was reduced during the
treatment period, though 4 out of 10 hens resumed egg laying after
treatment was terminated.
A dose-dependent inhibition of brain and plasma cholinesterase,
hyperglycaemia, and elevated corticosterone concentrations were
observed in the American kestrel (Falco sparverius) exposed to oral
doses of up to 3 mg methyl parathion/kg body weight (Rattner &
Franson, 1984).
Table 19. Acute lethal toxicities of methyl parathion for birds
Species Age Oral LD50 Dietary LC50 References
(mg/kg body weighta) mg/kgb
Mallard duck 5 days 8 Fairbrother et al.
(Anas platyrhynchos) (1988)
3 months 10 Hudson et al. (1984)
adult 6.6
Mallard duck 10 days 682 Hill et al. (1975)
(Anas platyrhynchos)
Mallard duck 5 days 336 Hill et al. (1975)
(Anas platyrhynchos)
Kestrel > 8 3.08 Rattner & Franson
(Falco sparverius) months (1984)
Bobwhite quail 14 days 90 Hill et al. (1975)
(Colinus virginianus)
Table 19 (continued)
Species Age Oral LD50 Dietary LC50 References
(mg/kg body weighta) mg/kgb
Bobwhite quail 14 days 91 Bennet (1989)
(Colinus virginianus)
Japanese quail 14 days 79 Hill et al. (1975)
(Coturnix coturnix japonica)
Japanese quail
(Coturnix coturnix japonica) 14 days 69 Hill & Camardese
(1986)
Ring-necked pheasant 10 days 91 Hill et al. (1975)
(Phasianus colchicus)
Red-winged blackbird - 10 Schafer (1972)
(Agelaius phoeniceus)
a intubation of a single dose.
b 8 days - standard test. 5 days feeding followed by 3 days observation.
Egg production in Japanese quail was inhibited and hatchability
reduced at 60 mg/kg (NRC, 1977).
Methyl parathion-induced mortality following long-term ingestion
was generally due to anorexia. Grackles (Quiscalus quiscula) had
lost 28-36% of their initial body weight, when they died. No fat was
visible and the muscles were reduced on the sternum. There was an
increase in mortality at relatively constant intake rate of methyl
parathion observed between May and August, which was related to an
increase in natural activity within this time. It was concluded, that
median lethal dietary concentrations are relative and depend on the
anorexic and physiological condition of wild birds (Grue, 1982). The
mean brain AChE activity of grackles (Quiscalus mexicanus) was
significantly inhibited more than that of white-winged doves (Zenaida
asiatica) and that of mourning doves (Zenaida macroura) after
applications of EPN (phenylphosphonothioic acid O-ethyl O-p-nitro-
phenyl ester) and methyl parathion (Custer & Mitchell, 1987).
Free-living, female red-winged blackbirds (Agelaius phoeniceus)
were captured on their nests and given oral doses of 2.4 or 4.2
methyl parathion mg/kg body weight and released immediately after
dosing. Although methyl parathion caused ataxia, lacrimation, and
lethargy and significantly depressed cholinesterase activity (> 35%)
at 4.2 mg/kg, there were no apparent adverse effects on incubation
behaviour and nesting success (Meyers et al., 1990).
Depressed brain acetylcholinesterase activity was also observed
in 2 bird species (red-winged blackbird, (Agelaius phoeniceus), and
dickcissel (Spiza americana)) inhabiting wheat fields treated with
methyl parathion (0.67 kg a.i./ha). Maximal inhibition occurred 5 days
after pesticide application. Enzyme activity levels returned to near
normal levels by the tenth day following application. Cholinesterase
inhibition for dickcissels and red-winged blackbirds differed
significantly (74% versus 40%), and these differences could not be
explained by the diets of the 2 species, as they were similar
(Niethammer & Baskett, 1983).
A subacute oral dose of 3.5 mg methyl parathion/kg per day
resulted in inhibition of brain cholinesterase (average decrease of
36%) in nuthatches (Sitta carolinensis) after 3-7 days exposure
(Herbert et al., 1989).
7.3.4 Non-laboratory mammmals
Two wild rodent species (Sigmodon hispudus and Mus musculus)
were found to have a higher mortality rate and to recover more slowly
from exposure to methyl parathion at oral doses of 14-80 mg/kg,
compared with laboratory rodents (Roberts et al., 1988). Clark (1986)
reported a greater tolerance to methyl parathion in little brown bats
(Myotis lucifugus) compared with wild mice (Mus musculus): the
24-h oral LD50 value (372 mg/kg body weight) of methyl parathion for
little brown bats was 8.5 times the LD50 value for mice (44 mg/kg
body weight). A loss of coordination was observed in 50% of the
animals that were still alive 24 h after the treatment. The poisoned
bats could be more easily captured by predators. The threshold of the
coordination loss was about 1/3 of the LD50 value. In toxicity
tests, mink (Mustela vison) rejected methyl parathion-treated diets
and appeared to die from starvation rather than from methyl parathion
poisoning (Aulerich et al., 1987).
8. EFFECTS ON EXPERIMENTAL ANIMALS AND IN VITRO TEST SYSTEMS
The inhibition by methyl parathion of acetylcholinesterase at
nerve endings results in an accumulation of endogenous acetylcholine,
as evidenced by peripheral and central cholinergic nervous system
signs (Taylor, 1980).
Toxic effects include profuse salivation, lacrimation, nasal
discharge, colic, diarrhoea, pupil constriction, excessive sweating,
coughing, vomiting, frequent urination, anxiety, restlessness,
hyperactivity, and hyperkinesis.
A more complete treatise on the effects of organophosphorus
insecticides in general, especially their short- and long-term effects
on the nervous system, can be found in Environmental Health Criteria
63: Organophosphorus insecticides - A general introduction (WHO,
1986).
8.1 Single exposure
Toxicological data on methyl parathion are summarized by Taylor
(1980) and Flucke (1984).
The acute toxicity values in a number of species following the
oral (Table 20), dermal (Table 21), inhalational (Table 22), and
intraperitoneal (Table 23) administration of methyl parathion show
lethal doses of about 3-400 mg/kg for the oral route, 40-300 mg/kg for
the dermal route, 3.5-72 mg/kg for the intraperitoneal route, and
30-300 mg/m3 for inhalation exposure. The acute subcutaneous LD50s
for methyl parathion in rats and mice were 6 and 18 mg/kg body weight,
respectively (Krueger & Casida, 1957; RTECS, 1991); the acute
intravenous LD50 was reported to be 4.1-14.5 mg/kg body weight in
rats, 2.3-13 mg/kg body weight in mice, and 50 mg/kg body weight in
guinea-pigs (NIOSH, 1976).
Izmirova et al. (1984) found an abrupt reduction in the blood and
brain cholinesterase and acetylcholinesterase activities in albino
rats in the 30th and 90th min after a single oral administration of 32
mg methyl parathion/kg. The blood cholinesterase activity was reduced
by 71% and the brain acetylcholinesterase activity by 54%. Twenty-four
hours after administration, the cholinesterase activity was higher
than that in the controls.
Table 20. Acute oral toxicity
Animal (sex)a LD50 (mg/kg body References
weight)
rat (m) fasted 2.9 Heimann (1982)
rat (f) fasted 3.2 Heimann (1982)
rat 6 Bayer AG (1988); RTECS
(1991)
rat (m) 7.4 Flucke & Kimmerle (1977)
rat (f) nonfasted 9.3 Heimann (1982)
rat (m) nonfasted 10.8 Heimann (1982)
rat (m) 11.7 Kimmerle (1975)
rat (m) 14 Gaines (1960, 1969)
rat (f) 24 Gaines (1960, 1969)
rat 35 Kagan (1971)
mouse 23 RTECS (1991)
mouse 14.5 Haley et al. (1975)
mouse 33.1 Mundy et al. (1978)
mouse 21.8 Mundy et al. (1978)
mouse 19.5 Haley et al. (1975)
rabbit (m) fasted 19 Heimann (1982)
rabbit (f) fasted 19.4 Heimann (1982)
rabbit 420 RTECS (1991)
Table 20. (cont'd) Acute oral toxicity
Animal (sex)a LD50 (mg/kg body References
weight)
guinea-pig 1270 RTECS (1991)
guinea-pig 417 NIOSH (1976)
dog 90 Hirschelmann & Bekemeier
(1975)
a m= male, f= female.
Table 21. Acute dermal toxicity
Animal, Duration of Ld50 (mg/kg Ld100 References
sexa exposure (h) body weight)
rat 1b 63 RTECS
(1991)
rat (m,f) - 67 Gaines
(1960,
1969)
rat (m) 24 46 Heimann
(1982)
rat (f) 24 44 Heimann
(1982)
rabbit (m) 6 1270 (pure) Deichmann
et al. (1952)
rabbit (m) 6 350-780
(technical Deichmann
grade) et al. (1952)
rabbit (m) 6 420 (pure, in Deichmann
corn oil) et al. (1952)
rabbit (m) 6 2500 (pure, Deichmann
suspended in et al. (1952)
water)
rabbit - 300 RTECS
(1991)
a m = male, f = female.
b no data given.
Table 22. Acute inhalation toxicity
Animal Duration of LC50 References
(sex)a exposure (h) (mg/m3 air)
rat 1 120 RTECS (1991)
rat 1 34 Molnar & Paksy
(1978)
rat (m) 1 200 Kimmerle & Lorke
(1968)
rat (m) 1 260 Thyssen (1979)
rat (f) 1 320 Thyssen (1979)
rat (m) 4 120 Kimmerle & Lorke
(1968)
rat (m) 4 185 Thyssen (1979)
rat (f) 4 170 Thyssen (1979)
mouse 4 120 RTECS (1991)
a m = male f= female.
Table 23. Acute intraperitoneal toxicity
Animal LD50(mg/kg body weight) References
rat 3.5 Du Bois & Coon (1952)
rat adult 5.8 Brodeur & Du Bois (1963)
rat juvenile 3.5 Brodeur & Du Bois (1963)
rat 7 Kimmerle (1975)
mouse 9.3 Kimmerle (1975)
mouse 11.0 Benke et al. (1974)
mouse 6.4 Kamienski & Murphy (1971)
mouse 8.2 Mirer et al. (1977)
mouse 72 Goyer & Cheymol (1967)
Dogs that received 10 or 30 mg methyl parathion/kg body weight
intravenously showed minimal activity of the plasma cholinesterases,
30 min after treatment. Sixteen hours after the injection of 10 mg
methyl parathion/kg body weight, the enzyme activities had returned to
their pre-injection values. However, following treatment with 30 mg
methyl parathion/kg body weight, it took 7 days for complete recovery
(Braeckman et al., 1980).
After i.p. injection of 2.4 mg Wofatox (methyl parathion), Karcsu
et al. (1981) observed complete inhibition of the histochemically
detectable acetylcholinesterase activity in the central nervous system
of the rat. Partial enzyme inhibition was found in the motor neurons
and in the striated muscles. Ultrastructural changes in the myocardium
of the rats were also confirmed.
8.2 Skin and eye irritation, sensitization
The skin of rabbits exposed to methyl parathion for 4 or 6 h did
not show perceptible signs of irritation (concentrations up to
LD100, Table 21, Deichmann et al., 1952). Similar results were
obtained by Hecht & Wirth (1950) and by Heimann (1982) in their
studies on rats.
The irritation potential of methyl parathion on the rabbit skin
and eye was studied according the OECD guidelines for the testing of
chemicals (Nos. 404 and 405). It was concluded that methyl parathion
had no primary irritating potential (Pauluhn, 1983).
8.3 Short-term exposures
Groups of Wistar albino rats were exposed to methyl parathion
aerosol concentrations of 0.9, 2.6, and 9.7 mg/m3 air for 6 h/day,
5 days/week for 3 consecutive weeks. No mortality occurred. Plasma and
brain cholinesterase levels were significantly depressed in the
highest dose group. At 2.6 mg/m3, slight inhibition of plasma ChE
occurred (Thyssen & Mohr, 1982).
Groups of New Zealand white rabbits were administered methyl
parathion (purity 96.3%) dermally at dose levels of 10, 50, and 250
mg/kg body weight, applied for 5 days/week over 3 weeks. The site was
left uncovered for 6 h and then it was cleaned with soap and water.
There was a dose-related inhibition of erythrocyte and brain
cholinesterases in the 50 and 250 mg/kg dose groups. Plasma ChE was
also significantly depressed in the highest dose group; these animals
presented signs of cholinergic poisoning and 5 out of 6 animals died
(Mihail & Vogel, 1984).
A 12-week dietary study at 5, 20, and 50 mg methyl parathion/kg
was performed on male and female dogs. The doses corresponded to 0.1,
0.4, and 1.0 mg/kg body weight per day. A significant decrease in
plasma cholinesterase activity was observed only at 50 mg/kg diet
(Williams et al., 1959).
8.4 Long-term exposures
Kazakova et al. (1974) fed chicken and cattle daily with 2.5 mg
methyl parathion/kg body weight for one year. No changes in health
status and food intake were observed. In pigs and cows, 10 mg/kg body
weight led to irritation, depression, miosis, salivation, intensified
peristaltics, and diarrhoea.
Rats fed diets containing 40 mg methyl parathion/kg for 2 years,
and mice (females: fed up to 125 mg/kg, males: fed up to 77 mg/kg) did
not display any cholinergic toxicity (NCI, 1979).
In a 2- year study (combined long-term/carcinogenicity), 500 rats
(50 male, 50 female per dose, 100 controls) were fed diets containing
0, 2, 10, or 50 mg methyl parathion/kg. The intake of active
ingredient was 0, 0.144, 0.713, 4.917 mg/kg body weight per day
(females). The highest dose led to retardation of growth, increase in
mortality, inhibition of cholinesterase-activity in plasma,
erythrocytes, and brain, reduction of haemoglobin, and haematocrit,
and an increase in reticulocytes, after 2 years. Female rats showed a
reduction in plasma proteins and a reversible increase in urea in
plasma and protein in urine. At 10 mg/kg diet, the cholinesterase
activity in plasma and red blood cells was inhibited. Male rats also
showed reduced cholinesterase activity in the brain. Extensive
histopathological examinations (cardiovascular, respiratory, and
urogenital systems, digestive tract, organs, and glands) did not
exhibit any substance-related changes. No toxic effects were found at
the lowest dose (Bomhard et al., 1981; Schilde & Bomhard, 1984).
Sixty rats per sex and group were fed diets containing methyl
parathion at concentrations of 0.5, 5, or 50 mg/kg for 2 years.
Sciatic nerve preparations from 1 out of 5 males in the low-dose group
and 1 out of 5 in the mid-dose group reportedly showed moderate
degenerative changes. In the high-dose group (50 mg/kg diet), sciatic
nerve preparations from treated males showed a loss of myelinated
fibres. These animals also showed more myelin degeneration and Schwann
cell proliferation. Similar, less severe changes were seen with a
lower incidence, in males fed 5.0 or 0.5 mg/kg per day males and in
the controls. Only 1 rat in the low-dose group and 1 in the mid-dose
group had more severe changes than the controls; however, 4 high-dose
males showed more severe changes. No obvious differences were seen in
the females. Haemoglobin, haematocrit, and RBCs were slightly reduced
in mid-and high-dose males, and moderately reduced in high-dose
females (Daly, 1983).
8.5 Reproduction, embryotoxicity, and teratogenicity
Dosages of 4 or 6 mg methyl parathion/kg body weight were
injected intraperitoneally into pregnant, female albino Holzmann rats.
The injection was made on day 9 or day 15 using an ethanol-propylene
glycol vehicle. It was found, that the fetal, cerebral, cortical
cholinesterase activity was reduced, indicating the transplacental
passage of the organophosphate. Large subcutaneous haematomas also
occurred; however, no significant developmental defects were noticed
(Fish, 1966). Ackermann (1974) also reported that there was no
placental barrier for methyl parathion.
A 3-generation study was performed by the Woodard Research
Corporation in 1966. This unpublished report was reviewed by Anon.,
FAO/WHO (1969). Rats received diets of 0, 10, or 30 mg methyl
parathion per kg diet. A sporadic reduction in the litter size of
groups was observed (30 mg/kg: F2alpha, F2ß, F3alpha; 10 mg/kg:
F1ß), also a delayed growth of litters until weaning (30 mg/kg:
F2alpha, F3alpha, F3ß; 10 mg/kg: F1ß), a reduced rate of
survival of the litters (30 mg/kg: F1alpha, F1ß, F2alpha); 10
mg/kg: F3alpha), and an increased rate of stillbirths (30 mg/kg:
F1ß, F3alpha).
Another 3-generation study was performed by the Midwest Research
Institute (1975). Rats received 0, 10, or 30 mg methyl parathion/kg
diet (corresponding to 0, 0.5, or 1.5 mg methyl parathion/kg body
weight); 2 litters of each generation were evaluated. No adverse
effects on growth, survival, or reproduction were observed at the 30
mg/kg level, however, the 10 mg/kg level caused a reduction in the
postnatal survival in weaning rats in the F1ß and F3alpha
generations. Similar results were found in the 3-generation study of
Löser & Eiben (1982). Rats (male and female, SPF-Wistar W 74 strain,
5-6 weeks old) were fed a diet containing technical methyl parathion
(95% pure) at 2, 10, or 50 mg/kg for 77 days and then mated. The
no-effect level in this study was 2 mg/kg diet. A dose of 50 mg methyl
parathion/kg caused reductions in neonatal weights and litter size,
and delayed body weight gains, while 10 mg/kg caused sporadic
reductions in litter size (F2alpha, F3alpha), delayed growth of
litters until weaning (F1alpha, F2alpha, F2ß), and a reduced
rate of survival of the litters.
Single doses of 3, 30, or 100 µg methyl parathion (in 10% DMSO)
were administered subgerminally into chicken eggs on day 2 and
intra-amniotically on days 3 and 4. These doses did not induce any
specific malformations. Embryotoxicity was noted at the 2 higher doses
(30 and 100 µg) (Benes & Jelinek, 1979). These findings were confirmed
by estimating the embryotoxicity range and parameters (Jelinek et al.,
1985). Doses of up to 55 µlitre Wofatox 50EC/kg egg reduced
haematocrit, glucose, cholesterol, and AChE activity and increased
aspartate aminotransferase and lactate dehydrogenase values in blood
samples of chicken embryos (Somlyay et al., 1989). The injection of 2
different concentrations of methyl parathion (13 and 135 mg/kg egg)
into pheasant eggs resulted in increased mortality and in an increased
incidence of skeletal deformities in the survivors (Varnagy et al.,
1984; Déli & Varnagy, 1985; Varnagy & Déli, 1985). Biochemical studies
on muscle samples from chicken embryos (eggs treated with 0.4% or
4.0% solution of Wofatox 50EC) showed decreased creatine kinase
activity, decreased creatine, creatine-phosphate, and Mg2+ (in
cervical muscle only) contents, and increased creatinine, Ca2+, and
Mg2+ (in femoral muscle only) values (Déli et al., 1985). Scanning
electron microscopic examination of the cartilage in chicken embryos
showed degeneration of collagen structure and chondrocytes at a high
insecticide concentration (eggs treated with 0.4% or 4.0% solution of
Wofatox 50EC) (Varnagy et al., 1988). Analysis of the protein pattern
of the cervical muscles of 18-day-old embryos, treated with 0.4%
methyl parathion solution showed decreases in alpha-actinin,
alpha-tubulin, ß-tubulin, and gamma-proteins (Déli & Kiss, 1988).
Studies on chickens showed that methyl parathion at 1-10
µmol/litre had no or only little effect on the adenylate cyclase in
the embryo muscle. Comparable results were obtained with rats using
the plasma membrane adenylate cyclase in rat livers, even at 100
µmol/litre. In the presence of adenylate cyclase-stimulating agents,
additional activation of methyl parathion was observed; it enhanced
the stimulating activity of GTP and isoproterenol together, but not
alone. Methyl parathion is soluble, but not metabolized, in plasma
membranes, so it may alter cellular levels of cAMP, and, thus, cell
growth (Déli & Kiss, 1986).
At very high doses (20 or 60 mg/kg body weight), methyl parathion
injected intraperitoneally in ICR-CL mice on day 10 of pregnancy,
caused convulsions, hypersalivation, ataxia, and tremor. At the higher
dose, 5 out of the 14 litters died. This dose caused reduced neonatal
weight, an increase in the occurrence of cleft palate, and an
increased incidence of cervical ribs in the fetuses. At the lower
dose, cleft palate, and a statistically non-significant increase in
the number of cervical ribs and underdeveloped sternebrae were
observed (Tanimura et al., 1967).
A single intraperitoneal injection of 5, 10, or 15 mg/kg body
weight was administered to Wistar rats on day 12 of pregnancy; signs
of toxicity and reduced body weight were observed with 15 mg/kg, but
there was no evidence of teratogenicity at any of the doses (Tanimura
et al., 1967).
On 6 alternate days between days 5 and 15 of pregnancy, 3 groups
of rats received orally 0.1, 1, or 3 mg methyl parathion/kg body
weight. Another group of rats received 3 mg methyl parathion/kg body
weight on 8 alternate days between days 5 and 19 of pregnancy. No
teratogenic effects were observed; however, the high doses caused
increased resorptions and decreased fetal body weight (Fuchs et al.,
1976).
Methyl parathion was administered orally by gavage, to groups of
female rats from day 6 to day 15 of gestation at dose levels of 0.1,
0.3, or 1 mg/kg body weight. Weight gain in the mothers and a slight
retardation in growth in the fetuses were noted at the highest dose
level. Methyl parathion was not toxic for the embryo or fetus and no
teratogenic effects were apparent (Machemer, 1977a).
Groups of 24-26 rats received intravenous injections of 0, 0.03,
0.1, or 0.3 mg methyl parathion/kg body weight per day from day 6 to
day 15 of pregnancy. On day 20, the fetuses were evaluated. No
treatment-related effects were found (Machemer, 1977b).
No signs of embryotoxicity or teratogenicity were found in
rabbits that received 0.3, 1.0, or 3.0 mg methyl parathion/kg body
weight on days 6-18 of pregnancy (Renhof, 1984).
Daily intraperitoneal administration of methyl parathion (1 or
1.5 mg/kg body weight) to rats during days 6-19 of gestation resulted
in decreases in both maternal and fetal protein synthesis (Gupta et
al., 1984). The effect was dose dependent, and was greater on day 19
than on day 15 of gestation; it was also greater in fetal than in
maternal tissues. The same dosage regimen resulted in a postnatal
decrease in acetylcholinesterase activity and muscarinic receptor
binding. Recovery of acetylcholinesterase activity to near normal
levels occurred by day 28 in the low-dose offspring, but not in the
high-dose weanlings (Gupta et al., 1985).
8.6 Mutagenicity and related end-points
Methyl parathion has been reported to have DNA-alkylating
properties. Mutagenicity test results have been both positive and
negative. The results of most of the in vitro mutagenicity studies
with both bacterial and mammalian cells were positive; the in vivo
studies produced equivocal results. A survey is given in Table 24.
8.7 Carcinogenicity
The carcinogenicity of methyl parathion was studied in mice by
the National Cancer Institute (NCI) in 1979. Groups of 50,
six-week-old female B6C3F1 mice received diets containing either
62.5 or 125 mg methyl parathion/kg for 102 weeks. For 37 weeks, 2
groups of 50 male mice received diets containing either 62.5 or 125 mg
methyl parathion/kg, which was reduced then to 20 or 50 mg/kg for
another 65 weeks. Untreated matched groups of 20 males and 20 females
were used as a control. From all groups, 80-86% were still alive at
the end of the study. There was no statistically significant increase
in tumour incidence.
The NCI (1979) also studied the carcinogenicity of methyl
parathion in rats. Groups of 50 female and male Fischer 344 rats (6
weeks old) received separate diets containing 20 or 40 mg methyl
parathion/kg for 105 weeks. As matched controls, 20 male and 20 female
rats remained untreated. Only 46% of the high-dose females survived,
but 78% high-dose males, 74% low-dose males, 82% low-dose females, 85%
control males, and 95% control females were still alive at the end of
the study. No statistically significant increase in tumour rates was
found.
Male and female rats in groups of 50 were fed for 2 years with
diets containing 2, 10, or 50 mg methyl parathion/kg. No toxic effects
were found at the low dose (see section 8.4). No morphological changes
due to the insecticide were detected. No carcinogenic effects of
methyl parathion were observed (Bomhard et al., 1981; Schilde &
Bomhard, 1984).
Table 24. Mutagenicity tests of methyl parathion
Test Species Dose levels Metabolic Results Reference
activation
Microorganism
Gene mutation tests S. typhimurium _a +/- - Simmon et al. (1977)
Ames TA100, TA1535 +/- Carrere et al. (1978)
TA1536, TA1537
TA 1538
Ames S.typhimurium 250-1250 µg/plate + - Rashid & Mumma (1984)
TA100
Ames S. typhimurium 250-1250 pg/plate + - Rashid & Mumma (1984)
TA98, TA1535,
TA1537, TA1538
Ames S. typhimurium Herbold (1986)
TA1535 > 1000/µg/plate +/- +
TA 100 > 500/µg/plate +/- +
TA1537, TA1538 20-2500/µg/plate +/-
Reverse mutations E. coli WP2 and 1 crystal or microdrop - - Dean (1972)
WP2uvrA 250-2500/µg/plate +/- - Simmon et al. (1977)
Rashid & Mumma (1984)
Forward mutations E. coli 1 x 10-2mol/litre - + Mohn (1973)
streptomycin/ Wild (1975)
5-methyl-tryptophane
resistance
Table 24 (continued)
Test Species Dose levels Metabolic Results Reference
activation
ade-6 forward mutation Sacharomyces 11-228 mmol/litre - - Gilot-Delhalle et al.
pombe (1983)
Recombinogenic Aspergillus 2 mg - - Morpugo et al. (1977)
activity/point nidulans
mutation
(8-Aza-guanine resistance
Streptomyces _a - - Carere et al. (1978)
coelicolor
Insects
Sex link recessive Drosophila 1.25 x 10-5% w/w + Tripathy et al. (1987)
lethal test melanogaster 6.3 x 10-6% w/w
Larvae 24,48,72 h 24-48 and 72 h
old exposure
Mammals
Thymidine kinase Mouse Iymphoma _a -/+ + Jones et al. (1982)
locus ceils L51784
DNA effects Chinese hamster 20 and 40 µg/ml _a + Chen et al. (1981 )
chromatid ovary cells V79 28-72 h
exchange (in vitro)
Table 24 (continued)
Test Species Dose levels Metabolic Results Reference
activation
Human lymphold 20 µg/ml - + Sobti et al. (1982)
cells (LAZ-007)
Human lymphold 20 and 40/µg/ml _a + (for Chen et al. (1981)
cells 835 M and 28-72 h 20/µg/ml)
Jeff cells
Sister chromatid Human lymphocytes 36-181.8 µmol/litre - + (dose Singh et al. (1987)
exchange (SCE) dependent)
Unscheduled DNA Human fetal lung _a +/- - Simmon et al. (1977)
synthesis WI 38 fibroblast
Human lymphoid - - Huang (1973)
cells up to 50/µg
B411-4 6-50 h
RMPI - 1788
RMPI - 7191
Mouse (in vivo) 10 mg/kg ip - Degraeve & Moutschen
bone marrow (1984)
germ cells
Mouse, Swiss 9.4, 18.8, 37.5, + (dose Mathew et al. (1990)
bone marrow 75 mg/kg body weight, oral dependent)
Table 24 (continued)
Test Species Dose levels Metabolic Results Reference
activation
Chromosomal aberrations Rat bone marrow 0.5 mg/kg body weight + (dose Malhi & Grover (1987)
cells (in vivo) 1 mg/kg 1.95%
2 mg/kg 9.26%
5 days/week for 16.86%
7 weeks dependent)
Micronucleus Wistar rat 1, 2 and 4 mg/kg ip + (dose Grover & Mahli (1985)
(in vivo) dependent)
Micronucleus Mouse 5-10 mg/kg body weight, - Herbold (1986)
orally, daily,
for 2 days
Micronucleus Mouse, Swiss 9.4, 18.8, 37.5, 75 + (dose Mathew et al. (1990)
mg/kg body weight, orally dependent)
Dominant lethal Mouse, male 20 mg/kg diet - Simmon et al. (1977)
ICR/SIM 40 mg/kg for
80 mg/kg 7 weeks
Dominant lethal Mice, male Q 0.15 mg/litre (daily) in - Degraeve et al. (1984)
strain drinking-water, 5-7
weeks
a = no information given.
8.8 Special studies
Seven white New Zealand rabbits per group were fed 0, 0.036,
0.162, 0.519, or 1.479 mg methyl parathion/kg body weight per day, for
8 weeks. A dose-dependent increasing atrophy of the thymus cortex and
a reduced, delayed-type hypersensitivity response (DTH) were found
(Street & Sharma, 1975). In a preliminary study, Fan et al. (1981)
examined the effects of methyl parathion on immunological responses to
S. typhimurium infection in mice. Mortality rates among infected
animals fed 0.08, 0.3, or 0.7 mg methyl parathion/kg body weight
(duration "extending beyond 2 weeks") were determined and protection
by vaccination was examined. Dose-related increases in mortality were
seen in unvaccinated mice and protection by immunization was
decreased. These limited findings were reviewed by Sharma & Reddy
(1987) and Thomas & House (1989).
Methyl parathion was found by Barnes & Denz (1953) not to cause
delayed neuropathy in their hen test. However, Nagymajitenyi et al.
(1988) found neurotoxic effects on the central and peripheral nervous
systems in both acute and short-term studies on CFY rats, in which the
conduction velocity of the peripheral nerves, muscle function
(ischidiacus nerve/gastrocnemius muscle), and EEG activity were
measured. In the short-term studies, the rats were given 0.44 mg
methyl parathion/kg body weight for 5 days/week for 6 weeks; in the
acute study, the rats received 0.4 mg/kg, orally.
Lipid metabolism in rats was investigated by Hasan & Ahmad Khan,
(1985). The rats received daily intraperitoneal doses of 1.0, 1.5, or
2.0 mg methyl parathion/kg body weight for 7 days. The concentrations
of total lipids, phospholipids, and cholesterol increased in a
dose-related manner in the cerebral hemisphere, cerebellum, brain
stem, and spinal cord. Lipid peroxidation increased in the CNS with
the exception of the cerebellum.
Khan & Hasan (1988) studied changes in the levels of ganglio
sides and glycogen of the cerebral hemisphere, cerebellum, brain stem,
and spinal cord following intraperitoneal injection of methyl
parathion (1, 1.5, or 2 mg/kg body weight) in 24 rats for 7 days. A
dose-related depletion in the concentration of gangliosides and
glycogen content were discernible in all regions of the CNS.
Preweanling, male, rat pups were exposed daily through
subcutaneous injection to parathion (1.3 or 1.9 mg/kg body weight) or
the vehicle (corn oil) on postnatal days 5-20, a period critical for
the development of behavioural and biochemical parameters of the
cholinergic nervous system. This exposure resulted in dose-dependent
reductions in acetylcholinesterase activity and muscarinic receptor
binding in the cortex. During the preweanling period, there were no
differences among the groups in most reflex measures, eye opening, or
incisor eruption. Postweanling behavioural assessment revealed small
deficits in tests of spatial memory in both the T-maze and radial arm
maze. There were no differences in neuromuscular abilities or
spontaneous activity measures (Stamper et al., 1988).
The behavioural effects of short-term exposure of male Wistar
rats to methyl parathion (1/50 or 1/100 of LD50, orally, for 6
weeks) were studied. Open-field (OF) and elevated plus-maze (EPM)
tasks were used to decide whether or not the compound could affect
behaviour. Significant effects were measured in OF activity during the
first minute, on the activity of crossing outer squares, increasing
latencies to leave centre, start of rearing, grooming, and defecation.
EPM parameters showed an increased amount of time spent in the open
arms and a clear tendency to enter open arms more frequently. The
defecation rate in the EPM was significantly decreased (Schultz et
al., 1990).
8.9 Factors modifying toxicity
Methyl parathion becomes toxic only after metabolic
transformation to the oxon analogue, methyl paraoxon, by liver
microsomal oxidation. The microsomal enzymes metabolize methyl
parathion in 2 ways in vitro : a) oxidation to methyl paraoxon,
and b) degradation to dimethyl phosphorothiotic acid and
p-nitrophenol. NADPH and O2 are necessary for both reactions,
indicating that these are oxidative processes (Nakatsugawa et al.,
1968).
Piperonyl butoxide inhibits the mixed function oxidase activity
of the microsomal fraction of liver cells. Therefore, it inhibits both
oxidative activation of methyl parathion and detoxification, but not
the dealkylation reactions due to glutathione- S-alkyltransferase. At
a dosage of 400 mg/kg body weight, piperonyl butoxide antagonized the
toxic effects of methyl parathion in mice when given 1 h before the
mice received the insecticide. The intraperitoneal toxicity of methyl
parathion was reduced 40-fold (Kamienski & Murphy, 1971; Levine &
Murphy, 1977a,b; Mirer et al., 1977). Diethyl maleate reduced the
glutathione content of the liver by 80%. This agent increased the
acute toxicity of methyl parathion by the inhibition of
glutathione-dependent detoxification (Mirer et al., 1977).
Pap et al. (1976) showed that methyl parathion was less toxic in
rats with a thioacetamide-induced liver cirrhosis than in normal rats.
After activating the microsomal enzymes in the liver with sodium
phenobarbital or norandrostenolone phenylpropionate, the cirrhotic
rats showed a normal susceptibility to methyl parathion, indicating
the involvement of liver microsomes in the activation of methyl
parathion. Treatment of normal rats with chloramphenicol could
increase their survival time after poisoning with methyl parathion.
Lead nitrate (Pb(NO3)2) reduced the toxicity of methyl
parathion due to an increase in the carboxylesterase-dependent
metabolism of the insecticide (Hapke et al., 1978).
A single oral dose of 5 or 10 mg methyl parathion/kg body weight
resulted in decreases in the cholinesterase activity in rats of 43.6%
or 72.3%, respectively. However, rats pretreated on 5 successive days
with a combination of 7 mg gentamycin/kg body weight and 20 mg
rifamycin/kg body weight showed a remarkable protection against the
toxic effects of methyl parathion. The toxic signs were minimal; the
rats showed no, or only transient, signs of poisoning, and no
convulsions were be observed in the rats that had been pretreated. The
combination of these 2 drugs significantly prevented the methyl
parathion-induced inhibition of cholinesterase in plasma and of the
liver carboxylesterase. Gentamycin or rifamycin alone did not have any
effect. Youssef et al. (1987) demonstrated, that gentamicin and
rifamycin inhibited the formation of the oxidation product of methyl
parathion, methyl paraoxon, in the liver and skeletal muscle. Both
substances potentiated the rate of urinary p-nitrophenol excretion
within 48 h of the methyl parathion application. Pretreatment with
rifamycin influenced the rate of liver glutathion reduction, whereas
gentamicin did not show this effect.
Male rats were treated with a single i.p. dose of 5 mg methyl
parathion/kg. Pretreatment with memantine hydrochloride (18 mg/kg,
i.p.), 30 min before methyl parathion administration, and atropine
sulfate (16 mg/kg, i.p.), 15 min before, significantly reduced
(P <0.01) the inhibition of acetylcholinesterase (Gupta & Kadel,
1990).
Pretreatment with cimetidine, which suppresses the hepatic
microsomal oxidative metabolism, decreased the toxicity of methyl
parathion in rats and mice (Joshi & Thornburg, 1986).
Fuchs et al. (1986) showed that the LD50 in rats increased by
19-24% after simultaneous oral administration of 0.5 g humic acids/kg
body weight and methyl parathion. It was supposed that the absorption
of methyl parathion from the digestive tract decreased as a result of
the intake of the humic acids.
Sultatos (1987) perfused mouse livers in situ with methyl
parathion. The acute toxicity of methyl parathion in mice was
antagonized by pretreatment with phenobarbital, daily, for 4 days (80
mg/kg, i.p.). This effect was due to hepatic microsomal activation and
resulted in an increased clearance of methyl parathion. Similar
results were obtained by Du Bois & Kinoshita (1968), Du Bois (1969,
1971), and Murphy (1980).
The influence of temperature on the toxicity of methyl parathion
in mice was studied by Nomiyama et al. (1980). They found median
lethal doses (i.p.) of 14 mg/kg body weight at 8 °C, 44 mg/kg body
weight at 22 °C, and 35 mg/kg body weight at 38 °C.
An influence of age on the toxicity and metabolism of methyl
parathion was observed by Benke & Murphy (1975) in rats. Rats became
much less sensitive to poisoning with methyl parathion with increasing
age. The effect was explained by a presumable increase in the
detoxification processes as the GSH-dependent (glutathion-dependent)
dealkylation. Methyl parathion dealkylation rates increased directly
with age for both sexes.
Carbon disulfide pretreatment, 1 h before administration of 10 mg
methyl parathion/kg body weight to mice did not significantly affect
the methyl parathion toxicity (Yasoshima & Masuda, 1986).
Prior depletion of glutathione by acetaminophen (600 mg/kg, i.p.,
Costa & Murphy, 1984) or by diethyl maleate (1 ml/kg, i.p., Sultatos
& Woods, 1988) had little effect on the toxicity of methyl parathion
(2.5 mg/kg body weight and up to 55 mg/kg body weight i.p.,
respectively) in the mouse.
Interactions of organophosphorus pesticides and several
pyrethroid insecticides were reported by Gaughan et al. (1980).
Following an intraperitoneal injection of organophosphorus pesticides
in mice, they found pronounced inhibition of the liver microsomal
esterase, which hydrolyses trans-permethrin. Methyl parathion did not
potentiate the toxicity of deltamethrin (Audegond et al., 1989).
Equitoxic oral or i.p. combinations of methyl parathion with other
organophosphorus insecticides (amiton, coumaphos, crufomat,
dimethoate, dioxathion, disulfoton, fensulfothion, ethyl parathion,
phosphamidon, trichlofon) caused only subadditive or additive effects
on the LD50 values (Du Bois, 1961; West et al., 1961; Du Bois &
Kinoshita, 1963; Frawley et al., 1963; Sanderson & Edson, 1964;
McCollister et al., 1968; Flucke & Kimmerle (1977). Williams et al.
(1957) found additive effects in their testing of oral combinations of
methyl parathion with demeton, EPN, malathion, or ethyl parathion in
dogs.
Mice pretreated with 50-300 mg diethyl dithiocarbamate per kg
body weight displayed a remarkable reduction in the acute toxicity of
methyl parathion. The toxicity was up to 10 times less. Lange &
Wiezorek (1975) explained this observation by an effect of the
dithiocarbamate on the microsomal oxidases and, thus, on the
metabolism of methyl parathion. Another explanation is that compounds
that temporarily occupy the active site of acetylcholinesterase
prevent phosphorylation of the enzyme until there has been time for
destruction of the organic phosphorus compound by A-esterases (Hayes
& Laws, 1991).
Dithiocarb reduced the toxicity of methyl parathion in mice
markedly, when applied 30 min before the methyl parathion. No effect
was observed when dithiocarb and methyl parathion were applied
simultaneously (Lange et al., 1977).
Orlando et al. (1972) found that pretreatment with quinidine
sulfate had an inhibitory effect on the toxic action of i.v. injection
of methyl parathion in rabbits. This could be demonstrated by
electrocardiography. Quinidine sulfate reduced the influence of methyl
parathion on the nicotinic-and muscarinic-type receptors.
The effect of methyl parathion on monoamine oxidase activity
(MAO) in rat brain mitochondria was investigated by Nag & Nandi
(1987). In vitro methyl parathion reduced the MAO significantly;
however, in vivo , the effect was negligible.
Methyl parathion is an inhibitor of malate dehydrogenase in the
mitochondria of liver and skeletal muscle. There was also an
inhibitory effect on plasmatic malate dehydrogenase and lactate
dehydrogenase in the liver (Tripathi & Shukla, 1988).
8.10 Mode of action
The mode of action of organophosphorus insecticides, such as
methyl parathion, is described in Environmental Health Criteria 63
(WHO, 1986).
8.10.1 Inhibition of esterases
The primary biochemical effect associated with toxicity caused by
organophosphorus pesticides is inhibition of acetylcholinesterase
(AChE). The normal function of AChE is to terminate neurotransmission
due to acetylcholine, liberated at cholinergic nerve endings in
response to nervous stimuli. Loss of AChE activity may lead to a range
of effects resulting from excessive nervous stimulation and
culminating in respiratory failure and death. The chemistry of the
inhibition of AChE and of many other esterases (e.g., NTE and liver
carboxyesterases, which are discussed elsewhere) by these chemicals is
similar and is given in schematic form in Fig. 4. Following the
formation of a Michaelis complex (reaction 1), a specific serine
residue in the protein is phosphorylated with loss of the leaving
group X (reaction 2). Two further reactions are possible: reaction 3
(reactivation) may occur spontaneously at a rate that is dependent on
the nature of the attached group and on the protein and is also
dependent on the influence of pH and of added nucleophilic reagents,
such as oximes, which may catalyse reactivation. Reaction 4 ("aging")
involves cleavage of an R-O-P-bond with the loss of R and the
formation of a charged monosubstituted phosphoric acid residue still
attached to protein. The reaction is called "aging" because it is time
dependent, and the product is no longer responsive to nucleophilic
reactivating agents, such as some oximes. Since therapy of
organophosphorus compound poisoning is, in part, dependent on the
reactivating power of oximes, understanding of the "aging" reaction is
important. Pseudocholinesterase (ChE), which is present in blood
plasma and nervous tissue, but has no known physiological function, is
inhibited by organophosphorus compounds in a similar way to AChE, but
the specificity of the 2 enzymes is different. Though no toxic effect
arises as a result of inhibition of pseudoChE, measures of its
inhibition can be made for monitoring purposes.
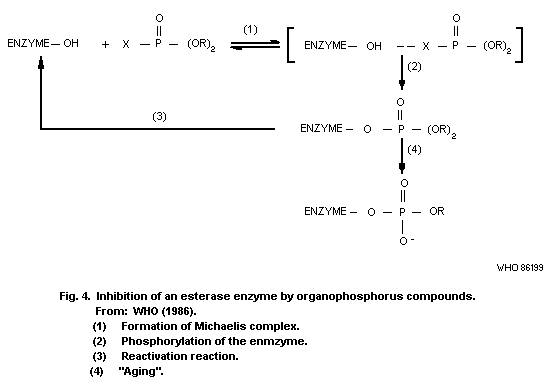 8.10.2 Possible alkylation of biological macromolecules
It has been shown, under laboratory conditions, that some
organophosphates can react with, and alkylate, the reagent
4-nitro-benzylpyridine (Preussmann et al., 1969). The study was
interpreted to imply that the in vivo alkylating potential of some
pesticides was similar to that of the known mutagens, dimethyl sulfate
and methyl methanesulfonate. Furthermore, Löfroth et al. (1969)
derived a substrate constant (a logarithmic measure of alkylating
ability) of 0.75 for dichlorvos, which is intermediate between those
known for methyl and ethyl methanesulfonates. Concern over the
possible mutagenic and carcinogenic potential of organophosphorus
compounds on the basis of the above data was misplaced, since
alternative reactions were not considered. Compared with the carbon
atom of the alkyl group, the phosphorus atom is markedly more
electron-deficient and susceptible to attack by nucleophiles. Analysis
by Bedford & Robinson (1972) of the data of Löfroth et al. (1969)
revealed that the proposed rates of alkylation by hard nucleophiles
were probably combined rates of phosphorylation and alkylation, and
that phosphorylation was the totally dominant reaction in the case of
the hydroxide ion. The comparison with known mutagens was therefore
inappropriate. Two factors detract further from the toxicological
significance of the alkylation studies. The first is that mammalian
tissues (plasma, liver, etc.) contain active enzymes that catalyse the
phosphorylation of water by the organophosphorus esters. Viewed
inversely, these enzymes (often called A-esterases) catalyse the
hydrolysis of the organophosphorus esters, thereby rapidly reducing
circulating levels of hazardous material. Secondly, the comparative
rate of reaction of most of these pesticides with AChE is many orders
greater than their rate of alkylation of the typical nucleophile
4-nitrobenzylpyridine: for dichlorvos, the ratio of rates was
1 x 107 in favour of the inhibitory phosphorylation of AChE
(Aldridge & Johnson, 1977). It follows that, at low exposure levels,
in vivo phosphorylation of AChE and other esterases will be the
dominant reaction with negligible uncatalysed alkylation of genetic
material. Indeed, no such alkylation has been detected in sensitive
in vivo studies designed to check this point (Wooder et al., 1977).
Some catalysed alkylations of glutathione by organosphorus compounds
are known to occur in vivo , but these are essentially
detoxification reactions.
8.10.3 General
Following lethal amounts of methyl parathion, hypotension,
bradycardia, bronchoconstriction, and bronchial fluid accumulation
occur with the inability of respiratory muscles to work. Cyanosis and
central respiratory depression can be observed. In less severe cases
of intoxication, bradycardia, muscle rigidity, muscle hypotonia,
bronchial spasm, and constriction dominate (Meyer-Jones et al., 1977).
9. EFFECTS ON MAN
The only confirmed effects on humans of exposure to methyl
parathion are the signs and symptoms characteristic of systemic
poisoning by cholinesterase-inhibiting organophosphorus compounds,
observed in case studies. The results of oral ingestion studies
performed by Rider et al. (1969, 1970, 1971) suggest that
manifestations of acute methyl parathion toxicity are absent in humans
whose erythrocyte cholinesterase activity has been reduced to as
little as 45% of their pre-exposure baselines (see section 9.1.2).
The effects of methyl parathion exposure on human beings were
compiled in 1976 by NIOSH. Details are given by Hayes & Laws (1991).
WHO (1986) summarized the signs and symptoms of organo-phosphate
insecticide poisoning as follows:
(a) Muscarinic manifestations
- increased bronchial secretion, excessive sweating,
salivation, and lacrimation;
- pinpoint pupils, bronchoconstriction, abdominal cramps
(vomiting and diarrhoea); and
- bradycardia.
(b) Nicotinic manifestations
- fasciculation of fine muscles and, in more severe
cases, of the diaphragm and respiratory muscles; and
- tachycardia.
(c) Central nervous system manifestations
- headache, dizziness, restlessness, and anxiety;
- mental confusion, convulsions, and coma; and
- depression of the respiratory centre.
All these signs and symptoms can occur in different combinations
and can vary in time of onset, sequence, and duration, depending on
the chemical, dose, and route of exposure. Mild poisoning might
include muscarinic and nicotinic signs only. Severe cases always show
central nervous system involvement; the clinical picture is dominated
by respiratory failure, sometimes leading to pulmonary oedema, due to
the combination of the above-mentioned signs and symptoms.
9.1 General population exposure
The general population may be exposed to air-, water-, and
food-borne residues of methyl parathion as a consequence of
agricultural/forestry practices, the misuse of the agent, and
contamination of field crops, water, and air by off-target loss.
Lisi et al. (1986, 1987) studied the allergic potential of methyl
parathion in 200 persons. No significant sensitization to methyl
parathion was found.
9.1.1 Acute toxicity
Several cases of methyl parathion poisoning have been reported
throughout the world; these have been reviewed by Hayes & Laws (1991).
Human manifestations of acute poisoning by methyl parathion are
comparable with those described in experimental animals (Durham &
Hayes, 1962; Fazekas & Rengei, 1965; Hayes & Laws, 1991).
In cases of fatal methyl parathion poisoning, gross and
microscopic alterations occur in all the organs (brain, lung, heart,
liver, kidneys, spleen, vascular walls, perivascular areas). Fazekas
(1971) already saw alterations due to methyl parathion-poisoning, 2 h
after the poisoning. Ember et al. (1970) found a high content of
vitamin A in the liver in 5 cases of suicide with methyl parathion.
Van Bao et al. (1974) reported an increase in chromosome
aberrations in the lymphocytes of 4 patients who had suffered acute
methyl parathion poisoning as a result of attempted suicide. The
increase in chromosome aberrations was detected only in cell cultures
carried out 1 month after their admission to hospital. No significant
changes were found, compared with controls, 6 months later.
9.1.2 Effects of short- and long-term exposure, controlled human
studies
Five male volunteers received 3.0 mg methyl parathion per day for
28 days, then 3.5 mg methyl parathion for 28 days, and 4.0 mg methyl
parathion for 43 days. No symptoms of poisoning or effects on the
plasma or red blood cell cholinesterases could be noticed (Moeller &
Rider, 1961).
In another study, 3 groups of 5 volunteers each received 4.5 mg
methyl parathion daily, for 30 days, then 5.0 g for 29 days or 5.5 mg
for 28 days, followed by 6.0 mg for 29 days or 6.5 mg for 35 days, and
finally 7.0 mg for 24 days. In no case was significant inhibition of
the plasma or red blood cell cholinesterase activity found (Moeller &
Rider, 1962).
Morgan et al. (1977) studied the cholinesterase activities in 4
human volunteers, who received 2 or 4 mg methyl parathion on 5
successive days. These doses did not cause any depression of plasma
and red blood cell cholinesterase activity.
Rider et al. (1969) reported studies on human volunteers to
determine the level of minimal toxicity of methyl parathion. For 30
days, 5 volunteers received capsules containing methyl parathion, with
the dose increasing daily, and 2 received capsules containing corn
oil. Depression in plasma cholinesterase activity (15%) was observed
at an oral dosage of 11.0 mg per day, while, at higher dosages up to
and including 19 mg per day, no significant cholinesterase depression
was observed. No significant changes in the blood cell count, urine
analysis, or the prothrombin times occurred, nor was there any
evidence of toxic effects.
After 4 weeks with daily doses of 24 mg methyl parathion, 2 out
of 5 volunteers showed inhibition of plasma and red blood cell
cholinesterase activities with decreases of 24 or 23% for plasma and
27 or 55% for red blood cells (Rider et al., 1970).
Five volunteers received doses increasing from 14 to 20 mg methyl
parathion per day, orally, for 6 days. No inhibition of the
cholinesterases was found. However, doses of 28 or 30 mg methyl
parathion caused a decrease in the cholinesterase activities of about
37% (Rider et al., 1971).
Two male volunteers received, orally, 2 or 4 mg methyl parathion
per day. No influence of methyl parathion on neurophysiological
parameters was found, and there was no inhibition of the plasma or red
blood cell cholinesterase activity (Rodnitzky et al., 1978).
9.2 Occupational exposure
The production, formulation, handling, and use as an insecticide
of methyl parathion are potential sources of exposure. Skin contact or
inhalation are the main hazards for workers. The main hazard for the
general population is the ingestion of contaminated food. Wind-drift
during spraying may be a health risk, since Kummer & Van Sittert
(1986) observed that, in a number of cases, the spraymen did not stop
spraying, when it was too windy.
The analysis of 375 pesticide poisonings in Bulgaria during
1965-68 showed that 82.5% of all cases were due to organophosphates.
Six of the intoxications were attributed to methyl parathion. A large
number of poisonings, usually mild, occurred not in applicators
directly engaged in plant protection but in other agricultural workers
when they entered a previously sprayed crop area for further
cultivation and hand-harvesting (Kaloyanova-Simeonova, 1970).
Hatcher & Wiseman (1969) reported 16 cases of methyl parathion
intoxication among 118 organophosphorus insecticide poisonings of farm
workers that occurred in the lower Rio Grande Valley (Texas) in 1968.
Toxicity following dermal exposure was predominant.
Neuropsychiatric sequelae from occupational exposure to
organophosphorus pesticides have been reported (Dille & Smith, 1964).
However, the patients had been exposed to other pesticides besides
methyl parathion.
Data on chromosomal aberrations due to methyl parathion are
scarce. Data from persons who had worked with various pesticides were
presented by Yoder et al., 1973 (positive finding); Rupa et al., 1989
(positive finding); and Nehéz et al., 1988 (positive finding in farm
workers in the open field, but not in those in enclosed spaces like
greenhouses). Van Bao et al. (1974) found chromosome aberrations in
one case of an agricultural worker, accidentally exposed to methyl
parathion (without exposure data).
De Cassia Stocco et al. (1982) reported data from subjects
exposed to methyl parathion and DDT at a formulation plant near of Sao
Paulo, Brazil. No increased frequency of chromosome aberrations was
found in the lymphocyte cultures of 15 healthy male workers (with
blood cholinesterase level < 75% of presumably the normal mean
levels), who were exposed repeatedly or long-term to methyl parathion
for durations ranging from 1 week to 7 years, but who had intermittent
periods of non-exposure.
Richter et al. (1986) investigated the risk of exposure to methyl
parathion spray drift in the workers in 3 kibbutzim. The
cholinesterase levels were measured in 36 agricultural workers and 25
residents from the same kibbutzim. No effects due to the methyl
parathion spray drift exposure were observed in the field workers or
in the residents.
9.2.1 Epidemiological studies
There are no epidemiological studies on effects related only to
methyl parathion exposure.
10. PREVIOUS EVALUATIONS BY INTERNATIONAL BODIES
The FAO/WHO Joint Meeting on Pesticide Residues (JMPR) evaluated
methyl parathion in 1968, 1972, 1975, 1979, 1980, and 1984 (FAO/WHO,
1969, 1973, 1976, 1980, 1981 and 1985). The acceptable daily intake
for man (ADI) was estimated at 0-0.02 mg/kg body weight in 1984. This
was based on levels causing no toxicological effects of:
- 2 mg/kg diet, equivalent to 0.1 mg/kg body weight in the
rat; and
- 0.3 mg/kg body weight per day in man.
The FAO/WHO Codex Alimentarius Commission (FAO/WHO, 1986)
recommended Maximum Residue Limits (MRLs) in several food commodities,
ranging from 0.05 to 0.2 mg/kg as follows:
Commodity MRL (mg/kg)
Cantaloupe 0.2
Cole crops 0.2
Cottonseed oil 0.05
Cucumbers 0.2
Fruit, other 0.2
Hops (dry cones) 0.05a
Melons 0.2
Sugar beets 0.05a
Tea (fermented and dried) 0.2
Tomatoes 0.2
a Levels at, or about, the limit of determination.
The International Agency for Research on Cancer (IARC) evaluated
methyl parathion in 1982 and in 1987 (IARC, 1983, 1987), and concluded
that the available data do not provide evidence that methyl parathion
is carcinogenic to experimental animals. No data on humans were
available. The available data provide no evidence that methyl
parathion is likely to present a carcinogenic risk for humans.
WHO (1990) classified technical methyl parathion as "extremely
hazardous" in normal use, based on an oral LD50 in the rat of 14
mg/kg. WHO/FAO (1975) issued a data sheet on methyl parathion (No. 7).
REFERENCES
ABE, T., FUJIMOTO, Y., TATSUNO, T., & FUKAMI, J. (1979) Separation of
methyl parathion and fenitrothion metabolites by liquid
chromatography. Bull. environ. Contam. Toxicol., 22: 791-795.
ACKERMANN, H. (1974) [Transfer of organophosphorous insecticides to
the embryo formation of PO derivatives and development of toxic
symptoms.] Tagungsber. Akad. Landwirtsch., 126: 23-29 (in German).
ADHYA, T.K., SUDHAKAR-BARIK, & SETHUNATHAN, N. (1981a) Stability of
commercial formulation of fenitrothion, methyl parathion, and
parathion in anaerobic soils. J. Agric. Food Chem., 29: 90-93.
ADHYA, T.K., SUDHAKAR-BARIK, & SETHUNATHAN, N. (1981b) Fate of
fenitrothion, methyl parathion, and parathion in anoxic
sulfur-containing soil systems. Pest. Biochem. Physiol., 16: 14-20.
ADHYA, T.K., SUDHAKAR-BARIK, & SETHUNATHAN, N. (1981c) Hydrolysis of
selected organophosphorus insecticides by two bacteria isolated from
flooded soil. J. appl. Bacteriol., 50: 167-172.
ADHYA, T.K., WAHID, P.A., & SETHUNATHAN, N. (1987) Persistence and
biodegradation of selected organophosphorus insecticides in flooded
versus non-flooded soils. Biol. Fertil. Soils, 4(1): 36-40.
AGISHEV, M.K., TSUKERMAN, V.G., & KUTSENKO, A.A. (1977) [Methods for
determining pesticide residues in soil.] Khim. sel'sk. Khoz., 15:
52-54 (in Russian).
AGOSTIANO, A., CASELLI, M., & PROVENZANO, M.R. (1983) Analysis of
pesticides and other organic pollutants by preconcentration and
chromatographic techniques. Water Air Soil Pollut., 19: 309-320.
AKHMEDOV, B.K. (1968) The hygienic significance of metaphos as an
atmospheric pollutant. Gig. i Sanit., 33: 10-15.
ALBANIS, T.A., POMONIS, P.J., & SDOUKOS, A.T. (1986) Organophosphorus
and carbamate pesticide residues in the aquatic system of Ioannina
basin and Kalamas River (Greece). Chemosphere, 15: 1023-1034.
ALBAUGH, D.W. (1972) Insecticide tolerances of two crayfish
populations (Procambarus acutus) in south-central Texas. Bull.
environ. Contam. Toxicol., 8(6): 334-338.
ALBERT, L., GONZALEZ, M., & MARTINEZ-DEWANE, M.G. (1979)
[Organophosphorus insecticides. I. Residues of organophosphorus
insecticides in some Mexican foods.] Rev. Soc. Quim. Méx., 23(4):
189-197 (in Spanish).
ALDRIDGE, W.N. & JOHNSON, M.K. (1977) Mechanisms and structure
activity relationships providing a high safety factor for
anti-cholinesterase insecticides. In: Proceedings of the British Crop
Protection Conference, Brighton, November 1977, Croydon, British Crop
Protection Council, pp. 721-729.
AMBRUS, A., HARGITAL, E., KAROLY, G., FULOP, A., & LANTOS, J. (1981a)
General method for determination of pesticide residues in samples of
plant origin, soil, and water. II. Thin layer chromatographic
determination. J. Assoc. Off. Anal. Chem., 64: 743-748.
AMBRUS, A., VISI, E., ZAKAR, F., HARGITAI, E., SZABO, L., & PAPA, A.
(1981b) General method for determination of pesticide residues in
samples of plant origin, soil, and water. III. Gas chromatographic
analysis and confirmation. J. Assoc. Off. Anal. Chem., 64: 749-768.
ANDERSON, J.F. & WOJTAS, M.A. (1986) Honey bees (Hymenoptera: Apidae)
contaminated with pesticides and polychlorinated byphenyls. J. Econ.
Entomol., 79: 1200-1205.
ANDERSSON, A. (1986) Monitoring and biased sampling of pesticide
residues in fruits and vegetables. Methods and results, 1981-1984. Var
Föda, 38(Suppl. 1): 8-55.
ANDERSSON, A. & OHLIN, B. (1986) A capillary gas chromatographic
multiresidue method for determination of pesticides in fruits and
vegetables. Var Föda, 38(Suppl. 2): 79-109.
ANEES, M.A. (1975) Acute toxicity of four organophosphorus
insecticides to a freshwater teleost Channa punctatus (Bloch). Pak. J.
Zool., 7: 135-141.
ANON (1984) Hazardous chemical data: Vol. 2. Chris. Washington, DC, US
Department of Transportation.
APPERSON, C.S., ELSTON, R., & CASTLE, W. (1976) Biological effects
and persistence of methyl parathion in Clear Lake, California.
Environ. Entomol., 5: 1116-1120.
APPERSON, C.S., YOWS, D., & MADISON, C. (1978) Resistance to methyl
parathion in Chaoborus astcoptus. J. Econ. Entomol., 71(5): 772-773.
ARCHER, T.E. & ZWEIG, G. (1959) Direct colorimetric analysis of
cholinesterase-inhibitions with indophenyl acetate. J. Agric. Food
Chem., 7: 1-78.
ARCHER, S.R., McCURLEY, W.R., & RAWLINGS, G.D. (1978) Source
assessment: pesticide manufacturing air emissions - overview and
prioritization. Washington, DC, US Environmental Protection Agency, p.
135 (EPA-600/2-78-004d).
ARNDT, W., SCHINDLBECK, E., PARLAR, H., & KORTE, F. (1981) [Reaction
of the organophosphate insecticides during rubbish composting.]
Chemosphere, 10: 1035-1040 (in German).
ARTERBERRY, J.D., DURHAM, W.F., ELLIOT, J.W., & WOLFE, H.R. (1961)
Exposure to parathion. Arch. environ. Health, 3: 112-121.
ARTHUR, R.D., CAIN, J.D., & BARRANTINE, B.F. (1976) Atmospheric levels
of pesticides in Mississippi delta. Bull. environ. Contam. Toxicol.,
15: 129- 134.
AUDEGOND, L., CATEZ, D., FOULHOUX, P., FOURNEX, R., LE RUMEUR, C.,
L'HOTELLIER, M., & STEPNIEWSKI, J.P. (1989) Potentialisation de la
toxicité de la deltaméthrine par les insecticides organophosphorés. J.
Toxicol. clin. Exp., 9: 163-176.
AULERICH, R.J., RINGER, R.K., & SAFRONOFF, J. (1987) Primary and
secondary toxicity of warfarin, sodium monofluoroacetate, and methyl
parathion in mink. Arch. environ. Contam. Toxicol., 16: 357-366.
AULT, J.A., SCHOFIELD, C.M., JOHNSON, L.D., & WALTZ, R.H. (1979)
Automated gel permeation chromatographic preparation of vegetables,
fruits, and crops for organophosphate residue determination utilizing
flame photometric detection. J. Agric. Food Chem., 27: 825-828.
BABU, S.B.R., JAYASUNDARAMMA, B., & RAMAMURTHI, R. (1986) Behavioral
abnormalities of juveniles of the fish Cyprinus carpio exposed to
methyl parathion. Environ. Ecol., 4: 95-97.
BADAWY, M.I. & EL-DIB, M.A. (1984) Persistence and fate of methyl
parathion in sea water. Bull. environ. Contam. Toxicol., 33: 40-49.
BADAWY, M.I., EL-DIB, M.A. & OSAMA, A.A. (1984) Spill of methyl
parathion in the Mediterranean Sea: A case study at Port-Said, Egypt.
Bull. environ. Contam. Toxicol., 32: 469-477.
BAKER, R.D. & APPLEGATE, H.G. (1970) Effect of temperature and
ultraviolet radiation on the persistence of methyl parathion and DDT
in soils. Agron. J., 62: 509-512.
BAKER, R.D. & APPLEGATE, H.G. (1974) Effect of ultraviolet radiation
on the persistence of pesticides. Texas J. Sci., 5: 53-59.
BARNES, J.M. & DENZ, F.A. (1953) Experimental demyelination with
organo-phosphorous compounds. J. Pathol. Bacteriol., 65: 597-605.
BECKER, G. (1971) [Simultaneous gas chromatographic determination of
chlorinated hydrocarbons and phosphates in plant material.] Dtsch.
Lebensm.- Rundsch., 67: 125-126 (in German).
BECKER, G. (1979) [Multimethod for simultaneous detection of 75 plant
treatment preparations on plant material.] Dtsch. Lebensm.-Rundsch.,
75: 148-152 (in German).
BECKER, G. & SCHUG, P. (1990) [A micromethod for the rapid
determination of chlorinated hydrocarbons and phosphate esters in
plant-derived foods.] Dtsch. Lebensm. Rundsch., 86(8): 239-242 (in
German).
BECKMAN, H. & GARBER, D. (1969) Pesticide residues. Recovery of 65
organophosphorus pesticides from Florisil with a new solvent-elution
system. J. Assoc. Off. Anal. Chem., 52: 286-293.
BEDFORD, C.T. & ROBINSON, J. (1972) The alkylating properties of
organophosphates. Xenobiotica, 2: 307-337.
BEINE, H. (1987) [Phosphoric acid esters and related compounds -
environmental significance and determination in air.] Essen, Immission
Protection Institute for the Land of North Rhine-Westphalia, 31 pp.
(LIS Report No., 69) (in German with English summary).
BELASHOVA, I.G., KHOKHOL'KOVA, G.A., & KLISENKO, M.A. (1983)
[Selection of environmental air sampler containing pesticide vapors.]
Gig. Tr. Prof. Zabol., 54-56 (in Russian).
BELISLE, A.A. & SWINEFORD, D.M. (1988) Simple specific analysis of
organophosphorus and carbamate pesticides in sediments using column
extraction and gas chromatography. Environ. Toxicol. Chem., 7:
749-752.
BENES, V. & JELINEK, R. (1979) Embryotoxicity of parathion methyl with
the Chick Embryotoxicity Screening Test (CHEST). In: Pesticide
residues in food: 1979 evaluations - The monographs. Rome, Food and
Agriculture Organization of the United Nations, pp. 367-368 (FAO Plant
Production and Protection Paper 20, suppl.).
BENKE, G.M. & MURPHY, S.D. (1975) The influence of age on the
toxicity and metabolism of methyl parathion and parathion in male and
female rats. Toxicol. appl. Pharmacol., 31: 254-269.
BENKE, G.M., CHEEVER, K.L., MIRER, F.E., & MURPHY, S.D. (1974)
Comparative toxicity, anticholinesterase action and metabolism of
methyl-parathion and parathion in sunfish and mice. Toxicol. appl.
Pharmacol., 28(1): 97-109.
BENNETT, R.S. (1989) Role of dietary choices in the ability of
bobwhite to discriminate between insecticide-treated and untreated
food. Environ. Toxicol. Chem., 8: 731-738.
BENNETT, R.S., WILLIAMS, B.A., SCHMEDDING, D.W., & BENNETT, J.K.
(1991) Effects of dietary exposure to methyl parathion on egg laying
and incubation in mallards. Environ. Toxicol. Chem., 10: 501-507.
BETOWSKI, L.D. & JONES, T.L. (1988) Analysis of organophosphorus
pesticide samples by high-performance liquid chromatography/mass
spectrometry and high-performance liquid chromatography/mass
spectrometry/mass spectrometry. Environ. Sci. Technol., 22: 1430-1434.
BHASKAR, S.U. & KUMAR, N.V.N. (1981) Thin layer chromatographic
determination of methyl parathion as paraoxon by cholinesterase
inhibition. J. Assoc. Off. Anal. Chem., 64: 1312-1314.
BHUNIA, A.K, BASU, N.K., ROY, D., CHAKRABARTI, A., & BANERJEE, S.K.
(1991) Growth, chlorophyll content, nitrogen-fixing ability, and
certain metabolic activities of Nostoc muscorum : effect of methyl
parathion and benthiocarb. Bull. environ. Contam. Toxicol., 47: 43-50.
BHASKAR, S.U. & KUMAR, N.V.N. (1982) Selection of enzyme sources for
improved sensitivity in enzymic determination of organophosphorus
pesticides. J. Assoc. Off. Anal. Chem., 65: 1297-1298.
BHASKAR, S.U. & KUMAR, N.V.N. (1984) A rapid colorimetric method for
determination of methyl parathion residues. Pesticides, 18: 17-18.
BIGLEY, W.S., PLAPP, F.W. Jr, HANNA, R.L., & HARDING, J.A. (1981)
Effect of toxaphene, camphene, and cedar oil on methyl parathion
residues on cotton. Bull. environ. Contam. Toxicol., 27: 90-94.
BLANCO, C.C. & SANCHEZ, F.G. (1989) Derivative spectrophotometric
determination of the organophosphate insecticide methyl parathion in
commercial formulations. Analusis, 17(1-2): 80-86.
BOMHARD, E., LOSER, E., & SCHILDE, B. (1981) [E 605-methyl (methyl
parathion). Chronic toxicological studies in rats (2-year feeding
trial).] Wuppertal-Elberfeld, Bayer AG, Institute of Toxicology
(Unpublished Report No. 9889, submitted to WHO by Bayer AG,
Leverkusen, Germany) (in German).
BOURQUE, C.L., DUGUAY, M.M., & GAUTREAU, Z.M. (1989) The determination
of reducible pesticides by adsorptive stripping voltammetry. Int. J.
environ. Stud., 37: 187-197.
BOURQUIN, A.W., GARNAS, R.L., PRITCHARD, P.H., WILKES, F.G., CRIPE,
C.R., & RUBINSTEIN, N.I. (1979) Interdependent microsomes for the
assessment of pollutants in the marine environment. Int. J. environ.
Stud., 13: 131-140.
BOWMAN, M.C. (1981) Analysis of organophosphorus pesticides. In: Moye
H.A., ed. Analysis of pesticide residues, New York, Basel, Marcel
Dekker, pp. 263-332.
BOWMAN, M.C. & BEROZA, M. (1967) Temperature-programmed gas
chromatography of 20 phosphorus-containing insecticides on 4 different
columns and its application to the analysis of milk and corn silage.
J. Assoc. Off. Anal. Chem., 50: 1228-1236.
BOWMAN, B.T. & SANS, W.W. (1979) The aqueous solubility of
twenty-seven insecticides and related compounds. J. Environ. Sci.
Health, B14: 625-634.
BRAECKMAN, R.A., GODEFROOT, M.G., BLONDEEL, G.M., & BELPAIRE, F.M.
(1980) Kinetic analysis of the fate of methyl parathion in the dog.
Arch. Toxicol., 43: 263-271.
BRAECKMAN, R.A., AUDENAERT, F., WILLEMS, J.L., BELPAIRE, F.M., &
BOGAERT, M.G. (1983) Toxicokinetics of methyl parathion and parathion
in the dog after intravenous and oral administration. Arch. Toxicol.,
54: 71-82.
BRANCA, P. & QUAGLINO, P. (1988) [Presence of antiparasite pesticides
in imported potatoes.] Ind. Aliment., 27: 427-431 (in Italian).
BRATIN, K., KISSINGER, P.T., & BRUNTLETT, C.S. (1981) Reductive mode
thin-layer amperometric detector for liquid chromatography. J. Liq.
Chromatogr., 4: 1777-1795.
BREWER, L.W., DRIVER, C.J., KENDALL, R.J., ZENIER, C., & LACHER, T.E.
Jr. (1988) Effects of methyl parathion in ducks and duck broods.
Environ. Toxicol. Chem., 7: 375-379.
BRODESSER, J. & SCHOELER, H.F. (1987) An improved extraction method
for the quantitative analysis of pesticides in water. Zbl. Bakteriol.
Mikrobiol. Hyg. Ser. B, 185: 183-185.
BRODEUR, J. & DU BOIS, K.P. (1963) Comparison of acute toxicity of
anticholinesterase insecticides to weanling and adult male rats. Proc.
Soc. Exp. Biol. Med., 114: 509-511.
BROWN, L.C., CATHEY, G.W., & LINCOLN, C. (1962) Growth and development
of cotton as affected by toxaphene-DDT, methyl parathion, and calcium
arsenate. J. Econ. Entomol., 55: 298-301.
BRUCHET, A., COGNET, L., & MALLEVIALLE, J. (1984) Continuous
composite sampling and analysis of pesticides in water. Water Res.,
18: 1401-1409.
BUBIEN, Z. (1971) [Intoxication of animals with phosphoorganic
insecticides.] Med. Wet., 27(10): 611-612 (in Polish with English
summary).
BUCK, N.A., ESTESEN, B.J., & WARE, G.W. (1980) Dislodgeable
insecticide residues on cotton foliage: fenvalarate, permethrin,
sulprofos, chlorpyrifos, methyl parathion, EPN, oxamyl, and
profenofos. Bull. environ. Contam. Toxicol., 24: 283-288.
BUERGER, T.T, KENDALL, R.J., MUELLER, B.S., DE VOS, T., & WILLIAMS,
B.A. (1991) Effects of methyl parathion on northern bobwhite
survivability. Environ. Toxicol. Chem., 10: 527-532.
BUTLER, P.A. (1964) Commercial fishery investigation. Washington, DC,
US Department of the Interior, Fish and Wildlife Service, pp. 5-28
(Circular No. 199).
BUTLER, P.A. & SCHUTZMANN, R.L. (1978) Residues of pesticides and PCBs
in estuarine fish, 1972-1976. National Pesticide Monitoring Program.
Pestic. Monit. J., 12: 51-59.
BUTLER, L.C., STAIFF, D.C., & DAVIES, J.E. (1981) Methyl parathion
persistence in soil following simulated spillage. Arch. environ.
Contam. Toxicol., 10: 451-458.
CAPRIEL, P, HAISCH, A., & SHAHAMAT, U. (1986) Supercritical methanol:
an efficacious technique for the extraction of bound pesticide
residues from soil and plant samples. J. Agric. Food Chem., 34: 70-73.
CARERE, A., ORTALI, V.A., CARDAMONE, G., & MORPURGO, G. (1978)
Mutagenicity of dichlorvos and other structurally related pesticides
in Salmonella and Streptomyces. Chem.-Biol. Interact., 22:
297-308.
CAREY, A.E. & KUTZ, F.W. (1985) Trends in ambient concentrations of
agrochemicals in humans and the environment of the United States.
Environ. Monit. Assess., 5: 155-163.
CAREY, A.E., GOWEN, J.A., TAI, H., MITCHELL, W.G., & WIERSMA, B.
(1978) Soils, pesticide residue levels in soils and crops, 1971 -
National Soils Monitoring Program (III). Pestic. Monit. J., 12:
117-136.
CAREY, A.E., GOWEN, J.A., TAI, H., MITCHELL, W.G., & WIERSMA, G.B.
(1979) Pesticide residue levels in soils and crops from 37 states,
1972. National Soils Monitoring Program (IV). Pestic. Monit. J., 12:
209-229.
CHAMBERS, J.E. & YARBROUGH, J.D. (1974) Parathion and methyl-parathion
toxicity to insecticide-resistant and susceptible mosquitofish
Gambusia affinis. Bull. environ. Contam. Toxicol., 11(4): 315-320.
CHANG, C.S. & LANGE, W.H. (1967) Laboratory and field evaluation of
selected pesticides for control of the red crayfish in California rice
fields. J. Econ. Entomol., 60(21): 473-477.
CHAUDHRY, G.R., ALI A.N., & WHEELER, W.B. (1988) Isolation of a methyl
parathion-degrading Pseudomonas sp. that possesses DNA homologous to
the opd gene from a Flavobacterium sp. Appl. environ. Microbiol.,
54(2): 288-293.
CHEAH, M.L., AVAULT, J.W. Jr, & GRAVES, J.B. (1980) Some effects of
rice pesticides on crawfish. Louisiana. Agric., 23(2): 8-9, 11.
CHEMICAL INFORMATION SERVICES, LTD (1988) Directory of world chemical
producers: 1989/90 edition. Oceanside, New York, Chemical Information
Services, Ltd, Publisher.
CHEN, H.H., HSUEH, J.L., SIRIANNI, S.R., & HUANG, C.C. (1981)
Induction of sister-chromatid exchanges and cell cycle delay in
cultured mammalian cells treated with eight organophosphorus
pesticides. Mutat. Res., 88: 307-316.
CHEN, Z., ZABIK, M.J., & LEAVITT, R.A. (1984) Comparative study of
thin film photodegradative rates for 36 pesticides. Ind. Eng. Chem.
Prod. Res. Dev., 23: 5-11.
CHERNYAK, S.M. & ORADOVSKII, S.G. (1980) [Determination of
organochlorine and organophosphorus pesticides in sea water by
gas-liquid chromatography.] Metody Issled. Org. Veshchestva Okeane,
1980: 291-303 (in Russian).
CHMIL, V.D., GRAHL, K., & STOTTMEISTER, E. (1978) Chromatographic
methods of determining residual amounts of pesticides in water. Zh.
Anal. Khim., 33: 2420-2425.
CHUKWUDEBE, A., MARCH, R.B., OTHMAN, M., & FUKUTO, T.R. (1989)
Formation of trialkyl phosphorothioate esters from organophosphorus
insecticides after exposure to either ultraviolet light or sunlight.
J. Agric. Food Chem., 37: 539-545.
CLARK, D.R. Jr (1986) Toxicity of methyl parathion to bats: mortality
and coordination loss. Environ. Toxicol. Chem., 5(2): 191-195.
CLARK, G.J., GOODIN, R.R., & SMILEY, J.W. (1985) Comparison of
ultraviolet and reductive amperometric detection for the determination
of ethyl and methyl parathion in green vegetables and surface water
using high-performance liquid chromatography. Anal. Chem., 57:
2223-2228.
COBURN, J.A. & CHAU, A.S.Y. (1974) Confirmation of pesticide residue
identity. VIII. Organophosphorus pesticides. J. Assoc. Off. Anal.
Chem., 57: 1272- 1278.
COHEN, M.L. & STEINMETZ, W.D. (1986) Foliar washoff of pesticides by
rainfall. Environ. Sci. Technol., 20: 521-523.
COLE, C.L., MCCASLAND, W.E., & DACUS, S.C. (1986) The persistence of
selected insecticides used in water and in water-oil sprays as related
to worker re-entry. Southwest. Entomol., 11(Suppl.): 83-87.
COMPTON, B. (1973) Analysis of pesticides in air. Prog. Anal. Chem.,
5: 133-152.
CORNELIUSSEN, P.E. (1970) Residues in food and feed. Pesticide
residues in total diet samples (V). Pestic. Monit. J., 4: 89-105.
COSTA, L.G. & MURPHY, S.D. (1984) Interaction between acetaminophen
and organophosphates in mice. Res. Commun. Chem. Pathol. Pharmacol.,
44: 389-400.
COWART, R.P, BONNER, F.L., & EPPS, E.A. Jr (1971) Rate of hydrolysis
of seven organophosphate pesticides. Bull. environ. Contam. Toxicol.,
6: 231-234.
CRIPE, G.M., NIMMO, D.R., & HAMAKER, T.L. (1981) Effects of two
organophosphate pesticides on swimming stamina of the mysid
Mysidopsis bahia. In: Vernberg, J.,Calabrese, A., Thurberg, F.P.,
& Vernberg, W.B., ed. Biological monitoring of marine pollutants. New
York, London, San Francisco, Academic Press, pp. 21-36.
CRIPE, C.R., WALTER, W.W., PRITCHARD, P.H., & BOURQUIN, A.W. (1987) A
shake-flask test for estimation of biodegradability of toxic organic
substances in the aquatic environment. Ecotoxicol. environ. Saf.,
14(3): 239-251.
CROFT, B.A. (1977) Resistance in arthropod predators and parasites.
In: Watson, D.L. & Brown, A.W.A., ed. Pesticide management and
insecticide resistance. New York, London, San Francisco, Academic
Press, pp. 377-393.
CROSSLAND, N.O. & BENNETT, D. (1984) Fate and biological effects of
methyl parathion in outdoor ponds and laboratory aquaria, I: Fate.
Ecotoxicol. Environ. Saf., 8: 471-481.
CROSSLAND, N.O. & ELGAR, K.E. (1983) Fate and biological effects of
insecticides in ponds. In: Matsunaka, S., Hutson, D.H., & Murphy,
S.D., ed. IUPAC. Pesticide chemistry: human welfare and the
environment. Proceedings of the 5th International Congress of
Pesticide Chemistry, Kyoto, Japan, 29 August-4 September 1982. Volume
3: Mode of action, metabolism and toxicology, Oxford, New York,
Pergamon Press, pp. 551-556.
CROSSLAND, N.O., BENNETT, D., & WOLFF, C.J.M. (1986) Evaluation of
models used to assess the fate of chemicals in aquatic systems.
Pestic. Sci., 17: 297-304.
CSERNATONI, M., GYORFI, L., & HARGITAL, E. (1988) Results of pesticide
analyses monitoring programme in surface waters in Hungary between
1977 and 1986. In: Abbou, R., ed. Hazardous waste: detection, control,
treatment. Amsterdam, Oxford, New York, Elsevier Science Publishers,
pp. 861-868.
CURINI, M., LAGANA, A., PETRONIO, B.M., & RUSSO, M.V. (1980)
Determination of organophosphorus pesticides by thin-layer
chromatography. Talanta, 27: 45-48.
CUSTER, T.W. & MITCHELL, C.A. (1987) Exposure to insecticides of
brushland wildlife within the Lower Rio Grande Valley, Texas, USA.
Environ. Pollut., 45(3): 207-220.
DALDRUP, T., SUSANTO, F., & MICHALKE, P. (1981) [Combination of TLC,
GLC (OV 1 and OV 17) and HPLC (RP 18) for a rapid detection of drugs.]
Fresenius Z. Anal. Chem., 308: 413-427 (in German).
DALDRUP, T., MICHALKE, P., & BOEHME, W. (1982) A screening test for
pharmaceuticals, drugs and insecticides with reversed-phase liquid
chromatography - retention data of 560 compounds. Chromatogr. Newsl.,
10: 1-7.
DALY, I.W.A. (1983) Evaluation report for US-EPA of a two-year chronic
feeding study of methyl parathion in rats. East Millstone, New Jersey,
Biodynamics Inc. (Unpublished report, submitted to WHO by Bayer AG,
Leverkusen, Germany).
DAMICO, J.N., BARRON, R.P, & SPHON, J.A. (1969) Field ionization
spectrum of some pesticidal and other biologically significant
compounds. Int. J. mass Spectrom. ion Phys., 2: 161-182.
DANKA, R.G., RINDERER, T.E., HELLMICH, II, R.L., & COLLINS, A.M.
(1986) Comparative toxicities of four topically applied insecticides
to Africanized and European Honey Bees (Hymenoptera: Apidae). J. Econ.
Entomol., 79: 18-21.
DAVIDSON, J.H., RAO, P.S.C, OU, L.T., WHEELER, W.B., & ROTHWELL, F.F.
(1980) Adsorption, movement, and biological degradation of large
concentrations of selected pesticides in soils. Gainesville, Florida,
Florida University.
DAVIS, J.E., STAIFF, D.C., BUTLER, L.C., & STEVENS, E.R. (1981)
Potential exposure to dislodgeable residues after application of two
formulations of methyl parathion to apple trees. Bull. environ.
Contam. Toxicol., 27: 95-100.
DEAN, B.J. (1972) The mutagenic effects of organophosphorus pesticides
on micro-organisms. Arch. Toxicol., 30: 67-74.
DE CASSIA STOCCO, R., BECAK, W., GAETA, R., & NAZARETH RABELLOGAY, M.
(1982) Cytogenetic study of workers exposed to methylparathion. Mutat.
Res., 103: 71-76.
DEGRAEVE, N. & MOUTSCHEN, J. (1984) Absence of genetic and cytogenetic
effects in mice treated by the organophosphorus insecticide parathion,
its methyl analogue, and paraoxon. Toxicology, 32: 177-183.
DEGRAEVE, N., CHOLLET, M.C., & MOUTSCHEN, J. (1984) Cytogenic and
genetic effects of subchronic treatment with organophosphorus
insecticides. Arch. Toxicol., 56: 66-73.
DEICHMANN, W.B., PUGLIESE, W., & CASSIDY, B.S. (1952) Effects of
dimethyl and diethyl paranitrophenyl thiophosphate on experimental
animals. Ind. Hyg. occup. Med., 5: 44-51.
DELI, E. & KISS, Z. (1986) The effect of organophosphorous insecticide
Wofatox 50 EC on the adenylate cyclase activity of chicken embryo
muscle. Biochem. Pharmacol., 35: 1603-1605.
DELI, E. & KISS, Z. (1988) Effect of parathion and methylparathion on
protein content of chicken embryo muscle in vivo. Biochem.
Pharmacol., 37: 3251-3256.
DELI, E. & VARNAGY, L. (1985) Teratological examination of Wofatox 50
EC (50% methyl parathion) on pheasant embryos. Anat. Anz. Jena, 158:
237-240.
DELI, E., SOMLYAY, I., & VARNAGY, L. (1985) Biochemical study of
muscle samples from chicken embryos affected by Wofatox 50 EC. Arch.
Toxicol, 8: 277-279.
DE POTTER, M., MULLER, R., & WILLEMS, J. (1978) A method for the
determination of some organophosphorus insecticides in human serum.
Chromatographia, 11: 220-222.
DE SCHRYVER, E., DE REU, L., BELPAIRE, F., & WILLEMS, J. (1987)
Toxicokinetics of methyl paraoxon in the dog. Arch. Toxicol., 59:
319-322.
DESHPANDE, A.A. & SWAMY, G.S. (1987) Induction of proline accumulation
by methyl parathion in sorghum ( Sorghum bicolor L.). Curr. Sci., 56:
1068-1070.
DEVI, Y.P., KUMAR, N.V., & HARAN, N.V.H. (1982) Survey of pesticide
residue analysis in water resources and paddy straw samples from some
farms of the Nellore district of Andhra Pradesh. Pollut. Res., 1:
21-23.
DE WIT, J.S.M., PARKER, C.E., TOMER, KB., & JORGENSON, J.W. (1987)
Direct coupling of open-tubular liquid chromatography with mass
spectrometry. Anal. Chem., 59: 2400-2404.
DICK, G.L., HEENAN, M.P., LOVE, J.L., UDY, P.B., & DAVIDSON, F. (1978)
Survey of trace elements and pesticide residues in the New Zealand
diet. 2. Organochlorine and organophosphorus pesticide residue
content. N. Z. J. Sci., 21: 71-78.
DILLE, J.R. & SMITH, P.W. (1964) Central nervous system effects of
chronic exposure to organophosphate insecticides. Aerosp. Med., 35:
475-478.
DI MUCCIO, A., AUSILI, A., VERGORI, L., CAMONI, I., DOMMARCO, R. &
GAMBETTI, L. (1990) Single step multi-cartridge clean-up for
organophosphate pesticide residue determination in vegetable oil
extracts by gas chromatography. Analyst. 115: 1167-1169.
DOROUGH, H.W. (1978) Conjugation reactions of pesticides and their
metabolites with sugars. In: Advances in pesticide science. Symposia
papers presented at the Fourth International Congress of Pesticide
Chemistry, Zurich, Switzerland, 24-28 July 1978. Part 3: Biochemistry
of pests and mode of action of pesticides; pesticide degradation;
pesticide residues; formulation chemistry. Oxford, New York, Pergamon
Press, pp. 526-536.
DORTLAND, R.J. (1980) Toxicological evaluation of parathion and
azinphosmethyl in freshwater model ecosystems. Wageningen, Centre for
Agricultural Publishing and Documentation (PUDOC), 111 pp.
(Agricultural Research Report No. 898).
DRAPER, W.M. & STREET, J.C. (1981) Drift from a commercial, aerial
application of methyl and ethyl parathion: an estimation of human
exposure. Bull. environ. Contam. Toxicol., 26: 530-536.
DU BOIS, K.P. (1961) Potentiation of the toxicity of organophosphorous
compounds. Adv. Pest. Control Res., 4: 117-151.
DU BOIS, K.P. (1969) Combined effects of pesticides. Can. Med. Assoc.
J., 100: 173-179.
DU BOIS, K.P. & COON, J.M. (1952) Toxicology of organic
phosphorus-containing insecticides to mammals. Ind. Hyg. occup. Med.,
6: 9-13.
DU BOIS, K.P. & KINOSHITA, F.K. (1968) Influence of induction of
hepatic microsomal enzymes by phenobarbital on toxicity of organic
phosphate insecticides (33402). Proc. Soc. Exp. Biol. Med., 129:
699-702.
DURHAM, W.F. & HAYES, W.J. Jr (1962) Organic phosphorus poisoning and
its therapy. Arch. environ. Health, 6: 21-47.
EARNEST, R. (1970) Effects of pesticides on aquatic animals in the
estuarine and marine environment. In: Progress in sport fishery
research, 1970, Washington, DC, US Department of the Interior, Bureau
of Sport Fisheries and Wildlife, pp. 10-13.
EASLEY, C.B., LAUGHLIN, J.M., GOLD, R.E., & TUPY, D.R. (1981) Methyl
parathion removal from denim fabrics by selected laundry procedures.
Bull. environ. Contam. Toxicol., 27: 101-108.
EBING, W. (1985) [Multimethod for the determination of pesticide
residues in dead honeybees. I. Chlorine and phosphorus insecticides.]
Fresenius Z. Anal. Chem., 321: 45-48 (in German).
EBING, W. (1987) [Gas chromatography of pesticides. Tabular literature
abstracts, Series XV.] Berlin-Dahlem, Federal Biological Institute for
Agriculture and Forestry (Report No. 236) (in German).
EICHELBERGER, J.W. & LICHTENBERG, J.J. (1971) Persistence of
pesticides in river water. Environ. Sci. Technol., 5: 541-544.
EISLER, R. (1969) Acute toxicities of insecticides to marine decapod
crustaceans. Crustaceana, 16(3): 300-310.
EISLER, R. (1970a) 45. Factors affecting pesticide-induced toxicity in
an estuarine fish. Washington, DC, US Department of the Interior, Fish
and Wildlife Service, Bureau of Sport Fisheries and Wildlife, 20 pp.
(Technical Papers of the Bureau of Sport Fisheries and Wildlife No.
45/46).
EISLER, R. (1970b) 46. Acute toxicities of organochlorine and
organophosphorus insecticides to estuarine fishes. Washington, DC, US
Department of the Interior, Fish and Wildlife Service, Bureau of Sport
Fisheries and Wildlife, 12 pp. (Technical Papers of the Bureau of
Sport Fisheries and Wildlife No. 45/46).
ELKINS, E.R., FARROW, R.P., & KIM, E.S. (1972) The effect of heat
processing and storage on pesticide residues in spinach and apricots.
J. Agric. Food Chem., 20: 286-291.
ELLIOTT, J.W., WALKER, K.C., PENICK, A.E., & DURHAM, W.F. (1960) A
sensitive procedure for urinary p-nitrophenol determination as a
measure of exposure to parathion. J. Agric. Food Chem., 8: 111-113.
EMBER, M., MINDSZENTY, L., RENGEI, B., & GAL, G. (1970) Changes of
vitamin A in blood and liver of organophosphorus poisoned suicides.
Res. Commun. Chem. Pathol. Pharmacol., 1: 561-571.
ESPIGARES, M., ROMAN, I., GONZALEZ ALONSO, J.M., DE LUIS, B., YESTE,
F., & GALVEZ, R. (1990) Proposal and application of an ecotoxicity
biotest based on Escherichia coli. J. appl. Toxicol., 10: 443-446.
FABACHER, D.L. (1976) Toxicity of endrin and endrin-methyl parathion
formulation to largemouth bass fingerlings. Bull Environ. Contam.
Toxicol., 16: 376-378.
FAIRBROTHER, A., MEYERS, S.M., & BENNETT, R.S. (1988) Changes in
mallard hen and brood behaviors in response to methyl
parathion-induced illness of ducklings. Environ. Toxicol. Chem., 7:
499-503.
FAN, A.M-M. (1981) Effects of pesticides on immune competency:
influence of methyl parathion and carbofuran on immunologic responses
to Salmonella typhimurium infection. Diss. Abstr. Int., 41(08):
2962-B.
FAO (1985) Second Government Consultation on International
Harmonization of Pesticide Registration Requirements, Rome, 11-15
October, 1982. Rome, Food and Agriculture Organization of the United
Nations.
FAO/WHO (1969) 1968 Evaluation of some pesticide residues in food,
Geneva, World Health Organization (FAO PL: 1968/M/9/1; WHO Food
Add./69.35).
FAO/WHO (1973) 1972 Evaluation of some pesticide residues in food,
Geneva, World Health Organization (AGP: 1972/M/9/1; WHO Pesticide
Residues Series No. 2).
FAO/WHO (1976) 1975 Evaluation of some pesticide residues in food,
Geneva, World Health Organization (AGP: 1975/M/13; WHO Pesticide
Residues Series No. 5).
FAO/WHO (1980) 1979 Evaluation of some pesticide residues in food,
Rome, Food and Agriculture Organization of the United Nations (FAO
Plant Production and Protection Paper 20 Sup).
FAO/WHO (1981) 1980 Evaluation of some pesticide residues in food,
Rome, Food and Agriculture Organization of the United Nations (FAO
Plant Production and Protection Paper 26 Sup).
FAO/WHO (1985) 1984 Evaluation of some pesticide residues in food,
Rome, Food and Agriculture Organization of the United Nations (FAO
Plant Production and Protection Paper 67).
FAO/WHO (1986) Codex maximum limits for pesticide residues, 3rd ed.
Rome, Codex Alimentarius Commission, Food and Agriculture Organization
of the United Nations (CAC/Vol XIII).
FARRAN, A., CORTINA, J.L., DE PABLO, J., & BARCELO, D. (1990) On-line
continuous flow extraction system in liquid chromatography with
ultraviolet and mass spectrometric detection for the determination of
selected organic pollutants. Anal. chim. Acta, 234: 119-126.
FAZEKAS, I.G. (1971) [Concerning the macroscopic and microscopic
changes after Wofatox poisoning (methyl parathion).] Z. Rechstmed.,
68: 189-194 (in German).
FAZEKAS, I.G. & RENGEI, B. (1967) [Content of methyl parathion in
human organs after fatal Wofatox-poisoning.] Arch. Toxikol., 22:
381-386 (in German).
FEDORENKO, A.P., ALEEVA, L.V., & TITOK, V.A. (1981) Organophosphorus
pesticide accumulation in warm-blooded animals after chemical
treatment of the forest. Vestn. Zool., 4: 89-92.
FDA (1988) Food and Drug Administration pesticide program. Residues
in food. Washington, DC, Food and Drug Administration, 24 pp.
FISH, S.A. (1966) Organophosphorus cholinesterase inhibitors and fetal
development. J. Obstet. Gynecol., 96: 1148-1154.
FLANDERS, R.V., BLEDSOE, L.W., & EDWARDS, C.R. (1984) Effects of
insecticides on Pediobus faveolatus (Hymenoptera: Eulophidae), a
parasitoid of the Mexican bean beetle (Coleoptera: Coccinellidae).
Environ. Entomol., 13: 902-906.
FLUCKE, W. (1984) [Methyl parathion - Summary toxicological
information.] Wuppertal-Elberfeld, Bayer AG, Institute of Toxicology
(Unpublished report submitted to WHO by Bayer AG, Leverkusen, Germany)
(in German).
FLUCKE, W. & KIMMERLE, G. (1977) [Methyl parathion and ethyl
parathion, studies on the acute toxicity of the two active
substances.] Wuppertal-Elberfeld, Bayer AG, Institute of Toxicology
(Unpublished report No. 6823, submitted to WHO by Bayer AG,
Leverkusen, Germany) (in German).
FOSTER, G.D. & CROSBY, G.D. (1987) Comparative metabolism of
nitroaromatic compounds in freshwater, brackish water and marine
decapod crustaceans. Xenobiotica, 17(12): 1393-1404.
FOSTER, G.D. & ROGERSON, P.F. (1990) Enhanced preconcentration of
pesticides from water using the Goulden large-sample extractor. Int.
J. environ. anal. Chem., 41: 105-117.
FOSTER, R.L. (1974) Detection and measurement of ambient
organophosphate pesticides. Proc. Annu. Ind. Air Pollut. Control
Conf., 4: 66-98.
FRAWLEY, J.P., WEIR, R., TUSSIG, T., DU BOIS, K.P., & CALANDRA, J.C.
(1963) Toxicologic investigations on Delnav. Toxicol. appl.
Pharmacol., 5: 605-624.
FUCHS, V., GOLBE, S., KUHNERT, M., & OSSWALD, F. (1976) Studies into
prenatal toxic action of parathion methyl on Wistar rats and
comparison with prenatal toxicity of cyclophosphamide and trypan blue.
Arch. exp. vet. Med., 30: 343-350.
FUCHS, V., KUEHNERT, M., & GOLBS, S. (1986) Detoxifying action of
humic acids on selected contaminants. Veterinärmedizin., 41: 712-713.
FUHREMANN, T.W. (1980) Environmental behavior of insecticides. Diss.
Abstr. Int. B., 40(7): 3075-3076.
FUHREMANN, T.W. & LICHTENSTEIN, E.P. (1978) Release of soil-bound
methyl (14C)parathion residues and their uptake by earthworms and
oat plants. J. Agric. Food Chem., 26(3): 605-610.
FUKAMI, J. & SHIHIDO, T. (1963) Studies on the selective toxicities
of organic phosphorous insecticides (III). Botyu Kagaku, 28: 77-81.
FUNCH, F.H. (1981) Analysis of residues of seven pesticides in some
fruits and vegetables by high-pressure liquid chromatography. Z.
Lebensm.-Unters. Forsch, 173: 95-98.
GABICA, J., WYLLIE, J., WATSON, M., & BENSON, W.W. (1971) Example of
flame photometric analysis for methyl parathion in rat whole blood and
brain tissue. Anal. Chem., 43: 1102-1105.
GAINES, T.B. (1960) The acute toxicity of pesticides to rats.
Toxicol. appl. Pharmacol., 2: 88-99.
GAINES, T.B. (1969) Acute toxicity of pesticides. Toxicol. appl.
Pharmacol., 14: 515-534.
GAJAN, R.J. (1969) Collaborative study of confirmative procedures by
single-sweep oscillographic polarography for the determination of
organophosphorus pesticide residues in nonfatty foods. J. Assoc. Off.
Anal. Chem., 52: 811-817.
GAMBRELL, R.P., TAYLOR, B.A., REDDY, K.S., & PATRICK, W.H. Jr (1984)
Fate of selected toxic compounds under controlled redox potential and
pH conditions in soil and sediment-water systems. Athens, Georgia, US
Environmental Protection Agency, Office of Research and Development,
Environmental Research Laboratory.
GAR, K.A., SAZONOVA, N.A., FADEEV, Y.N., VLADIMIROVA, I.L., &
GOLUBEVA, Z.Z. (1958) [Incorporation and excretion of dimethyl
4-nitrophenyl thiophosphate in guinea pigs.] Org. Insektofungits.
Gerbits., 1958: 93-105 (in Russian).
GARRIDO, S.J.J. & MONTEOLIVA, H.M. (1981) [Qualitative and
quantitative determination of residues of diazinon, dimethoate,
ethion, malathion, methyl parathion and parathion in soil extracts by
thin-layer chromatography, and confirmation with gas chromatography.]
An. Edafol. Agrobiol., 40: 1787-1798 (in Spanish).
GAUGHAN, L.C., ENGEL, J.L., & CASIDA, J.E. (1980) Pesticide
interactions: effects of organophosphorus pesticides on the
metabolism, toxicity, and persistence of selected pyrethroid
insecticides. Pestic. Biochem. Physiol., 14: 81-85.
GERSTL, Z. & HELLING, C.S. (1985) Fate of bound methyl parathion
residues in soils as affected by agronomic practices. Soil Biol.
Biochem., 17(5): 667-673.
GHOSH, P., BHATTACHARYA, S., & BHATTACHARYA, S. (1989) Impact of
nonlethal levels of metacid-50 and carbaryl on thyroid function and
cholinergic system of Channa punctatus. Biomed. environ. Sci., 2:
92-97.
GHOSH, P., BHATTACHARYA, S., & BHATTACHARYA, S. (1990) Impairment of
the regulation of gonadal function in Channa punctatus by metacid-50
and carbaryl under laboratory and field conditions. Biomed. environ.
Sci., 3: 106-112.
GILE, J.D. & GILLETT, J.W. (1981) Transport and fate of
organophosphate insecticides in a laboratory model. J. Agric. Food
Chem., 29: 616-621.
GILLESPIE, A.M. & WALTERS, S.M. (1986) HPLC silica column
fractionation of pesticides and PCB from butterfat. J. liq.
Chromatogr., 9: 2111-2141.
GILLESPIE, A.M. & WALTERS, S.M. (1989) Semi-preparative reverse phase
HLPC fractionation of pesticides from edible fats and oils. J. liq.
Chromatogr., 12(9): 1687-1703.
GILLETT, J.W. & GILE, J.D. (1976) Pesticide fate in terrestrial
laboratory ecosystems. Int. J. environ. Stud., 10: 15-22.
GILOT-DELHALLE, J., COLIZZI, A., MOUTSCHEN, J., & MOUTSCHEN-DAHMEN, M.
(1983) Mutagenicity of some organophosphorus compounds at the ade 6
locus of Schizosaccharomyces pombe. Mutat. Res., 117: 139-148.
GOEDICKE, H.-J. (1989) [Exposure to residues on leaf surfaces
following the use of organophosphorus insecticides in high-intensity
apple growing.] Z. Gesamte Hyg., 35: 533-535 (in German).
GOEDICKE, H.-J. & WINKLER, R. (1976) [On the residue of methyl
parathion in soil.] Nachrichtenbl. Pflanzenschutz., 30(5): 100-101
(in German). GOLOVATYI, V.G., KOROL, E.N., KLISENKO, M.A., & GIRENKO,
D.B. (1982) FDMS study of pesticides. Eur. J. Mass Spectrom.
biochem., Med. environ. Res., 2: 97-100.
GONCHARUK, E.I., LIPATOVA, T.E., & FILATOVA, I.N. (1988) Conditions
for elevation of pesticide content in the atmospheric boundary layer
of agricultural fields. Gig. i Sanit., 1: 25-27.
GOYER, R. & CHEYMOL, J. (1967) Contribution à l'étude pharmacologique
de quelques dérivés soufrés de l'acide orthophosphorique trisubstitué.
II. Essais de toxicité. Rev. Can. Biol., 26: 35-41.
GOZA, S.W. (1972) Infrared analysis of pesticide formulations. J.
Assoc. Off. Anal. Chem., 55: 913-917.
GREENHALGH, R., BLACKWELL, B.A., PRESON, C.M., & MURRAY, W.J. (1983)
Phosphorus-31 nuclear magnetic resonance analysis of technical
organophosphorus insecticides for toxic contaminants. J. Agric. Food
Chem., 31: 710-713.
GRETCH, F.M. & ROSEN, J.D. (1984) Automated sample cleanup for
pesticide multiresidue analysis. Part II. Design and evaluation of
column chromatographs module. J. Assoc. Off. Anal. Chem., 67: 783-789.
GRETCH, F.M. & ROSEN, J.D. (1987) Automated sample cleanup for
pesticide multiresidue analysis. Part III. Evaluation of complete
system for screening subtolerance residues in vegetables. J. Assoc.
Off. Anal. Chem., 70: 109-111.
GROVER, I.S. & MALHI, P.K. (1985) Genotoxic effects of some
organophosphorus insecticides. I. Induction of micronuclei in bone
marrow cells in rats. Mutat. Res., 155: 131-134.
GROVER, I.S. & MALHI, P.K. (1987) Genotoxic effects of some
organophosphorus pesticides. II. In vivo chromosomal aberration
bioassay in bone marrow cells in rat. Mutat. Res., 188: 45-51.
GRUE, C.E. (1982) Response of common grackles to dietary
concentrations of four organophosphate pesticides. Arch. environ.
Contam. Toxicol., 11: 617-626.
GUPTA, R.C. & KADEL, W.L. (1990) Methyl parathion acute toxicity:
prophylaxis and therapy with memantine and atropine. Arch. int.
Pharmacodyn., 305: 208-221.
GUPTA, R.C., THORNBURG, J.F., STEDMAN, D.B., & WELSCH, F. (1984)
Effect of subchronic administration of methyl parathion on in vivo
protein synthesis in pregnant rats and their conceptuses1,2.
Toxicol. appl. Pharmacol., 92: 457-468.
GUPTA, R.C., RECH, R.H., LOVELL, K.L., WELSCH, F., & THORNBURG, J.E.
(1985) Brain cholinergic, behavioral, and morphological development in
rats exposed in utero to methylparathion. Toxicol. appl.
Pharmacol., 77: 405-413.
GYORFI, L., AMBRUS, A., & BOLYGO, E. (1987) Optimization of
determination and clean-up parameters for sensitive multiresidue
analysis of pesticides. In: Greenhalgh, P. & Roberts, T.R., ed.
IUPAC. Pesticide science and biotechnology. Proceedings of the Sixth
International Congress on Pesticide Chemistry, Ottawa, Canada, 10-15
August 1986. Oxford, London, Blackwell Scientific Publications, pp.
353-356.
HALEY, T.J., FARMER, J.H., HARMON, J.R., & DOOLEY, K.L. (1975)
Estimation of the LD1 and extrapolation of the LD0,1 for five
organothiophosphate pesticides. Eur. J. Toxicol., 8: 229-235.
HANSCH, C. & LEO, A. (1987) The log p database. Claremont, California,
Pomona College, 268 pp.
HAPKE, H.-J., YOUSSEF, S., & OMER, O.H. (1978) [Modification of the
alkylphosphate toxicity in lead contaminated rats.] Dtsch. Tierärztl.
Wochenschr., 85: 321-456 (in German).
HAQUE, R. & FREED, V.H. (1974) Behavior of pesticides in the
environment: Environmental chemodynamics. Residue Rev., 52: 89-116.
HASAN, M. & AHMAD KHAN, N. (1985) Methyl parathion-induced
dose-related alteration in lipid levels and lipid peroxidation in
various regions of rat brain and spinal cord. Ind. J. exp. Biol., 23:
141-144.
HATCHER, J.S. & WISEMAN, R.L. (1969) Epidemiology of pesticide
poisoning in the lower Rio Grande Valley in 1968. Texas Med., 65:
40-43.
HAYES, W.J. Jr (1971) Studies on exposure during the use of
anticholinesterase pesticides. Bull. World Health Organ., 44:
272-288.
HAYES, W.J. Jr & LAWS, E.R. Jr (1991) Handbook of pesticide
toxicology. New York, London, San Francisco, Academic Press, vol. 1-3.
HECHT, G. & WIRTH, W. (1950) [The pharmacology of the phosphoric acid
ester derivatives of thiophoric acid.] Arch. exp. Pharmakol., 211:
264-277 (in German).
HEIMANN, K.G. (1982) [E 120 (methyl parathion): studies of acute oral
and acute dermal toxicity.] Wuppertal-Elberfeld, Bayer AG, Institute
of Toxicology (Unpublished report No. 10897, submitted to WHO by
Bayer AG, Leverkusen, Germany) (in German).
HENNIGHAUSEN, G. (1984) [On the toxicological significance of the
interaction of foreign compounds with glutathione and glutathione
S-transferases.] Z. Gesamle Hyg., 30(11): 603-606 (in German with
English summary).
HERBERT, G.B., PETERLE, T.J., & GRUBB, T.C. (1989) Chronic dose
effects of methyl parathion on nuthatches: cholinesterase and
ptilochronology. Bull. environ. Contam. Toxicol., 42: 471-475.
HERBOLD, B. (1986) [E 120, C.N. methyl parathion-salmonella/microsome
test to evaluate for point mutagenic effect.] Wuppertal-Elberfeld,
Bayer AG, Institute of Toxicology (Unpublished report No. 15306,
submitted to WHO by Bayer AG, Leverkusen, Germany) (in German).
HICKE, K. & THIEMANN, W. (1987) [The decomposition of selected
phosphoric acid esters by UV-irradiation.] Vom Wasser, 69: 85-94 (in
German).
HILL, E.F. & CAMARDESE, M.B. (1986) Lethal dietary toxicities of
environmental contaminants and pesticides to Coturnix. Washington,
DC, US Department of the Interior Fish and Wildlife Service, pp.
100-101 (Fish and Wildlife Technical Report No. 2).
HILL, E.F., HEATH, R.G., SPANN, J.W., & WILLIAMS, J.D. (1975) Lethal
dietary toxicities of environmental pollutants to birds. Washington,
DC, US Department of the Interior Fish and Wildlife Service, pp. 16,
27 (Fish and Wildlife Technical Report No. 191).
HIRAYAMA, K. & TAMAOI, S. (1980) Acute toxicity of methylparathion and
diazinon (pesticide) to larvae of kuruma praure Penaeus japonicus and
of swimming crab Portunus trityberculatus. Bull. Jpn Soc. Sci. Fish.,
46: 117-123.
HIRSCHELMANN, R. & BEKEMEIER, H. (1975) Acute toxicity of demephion
and parathion methyl in dogs. In: Proceedings of the European Society
of Toxicology. Amsterdam Excerpta Medica Foundation, pp. 273-275.
HOLLINGWORTH, R.M., METCALF, R.L., & FUKUTO, T.R. (1967) The
selectivity of sumithion compared with methyl parathion. Metabolism in
the white mouse. J. Agric. Food Chem., 15: 242-249.
HOLM, H.W., KOLLIG, H.P., & PAYNE, W.R. Jr (1983) Fate of methyl
parathion in aquatic channel mirocosms. Environ. Toxicol. Chem., 2:
169-176.
HOLMSTEAD, R.L. & CASIDA, J.E. (1974) Chemical ionization mass
spectrometry of organophosphorus insecticides. J. Assoc. Off. Anal.
Chem., 57: 1050-1055.
HUANG, C.C. (1973) Effect on growth but not on chromosomes of the
mammalian cells after treatment with three organophosphorus
insecticides. Proc. Soc. Exp. Biol. Med., 142: 36-40.
HUDSON, R.H., HAEGELE, M.A., & TUCKER, R.K. (1979) Acute oral and
percutaneous toxicity of pesticides to mallards: correlations with
mammalian toxicity data. Toxicol. appl. Pharmacol., 47: 451-460.
HUDSON, R.H., TUCKER, R.K., & HAEGELE, M.A. (1984) Handbook of
toxicity of pesticides to wildlife, 2nd ed. Washington, DC, US
Department of the Interior, Fish and Wildlife Service, pp. 52-53
(Fish and Wildlife Publication, No. 153).
HUMMEL, S.V. & YOST, R.A. (1986) Tandem mass spectrometry of
organophosphate and carbamate pesticides. Org. mass Spectrom., 21:
785-791.
IARC (1983) Methyl parathion. In: Miscellaneous pesticides. Lyon,
International Agency for Research on Cancer, pp. 131-152 (IARC
Monographs on the evaluation of the carcinogenic risk of chemicals to
humans, Volume 30).
IARC (1987) Methyl parathion. In: Overall evaluations of
carcinogenicity: an updating of IARC monographs, Volumes 2 to 42.
Lyon, International Agency for Research on Cancer, pp. 392 (IARC
Monographs on the evaluation of the carcinogenic risk of chemicals to
humans, Supplement 7).
IZMIROVA, N., SHALASH, S., & KALOYANOVA, F. (1984) [Dynamics of
cholinesterase inhibition in methylparathion poisoning.] Probl. Khig.,
9: 42-49 (in Bulgarian).
JACKSON, E.R. (1976) High-performance and gas-liquid chromatography of
methyl parathion formulations. J. Assoc. Off. Anal. Chem., 59:
740-744.
JACKSON, E.R. (1977a) Collaborative study of gas-liquid
chromatographic method for determining methyl parathion. J. Assoc. Off
Anal. Chem., 60: 720-723.
JACKSON, E.R. (1977b) Collaborative study of high pressure liquid
chromatographic method for determining methyl parathion. J. Assoc.
Off. Anal. Chem., 60: 724-727.
JACKSON, M.D. & LEWIS, R.G. (1978) Volatilization of two methyl
parathion formulations from treated fields. Bull. environ. Contam.
Toxicol., 20: 793-796.
JAGLAN, P.S. & GUNTHER, F.A. (1970) Comparison of hydrolysis rates of
methyl parathion and methyl paraoxon by gas liquid chromatography and
spectrometry. J. chromatogr. Sci., 8: 483-485.
JALALUDDIN, M. & MOHANASUNDARAM, M. (1989) Residual toxicity of four
insecticides recommended for control of coconut coccids on the
parasitoid fauna of Opisina arenosella WLK. Entomol., 14: 199-202.
JARVINEN, A.W. & TANNER, D.K. (1982) Effects of selected controlled
release and corresponding unformulated technical grade pesticides to
the fathead minnow Pimephales promelas. Environ. Pollut., Ser. A,
27(3): 179-195.
JELINEK, R., PETERKA, M., & RYCHTER, Z. (1985) Chick embryotoxicity
screening test - 130 substances tested. Indian J. exp. Biol., 23:
588-595.
JOHANSSON, C.E. (1978) A multiresidue analytical method for
determining organochlorine, organophosphorus, dinitrophenyl and
carbamate pesticides in apples. Pestic. Sci., 9: 313-322.
JOHNSON, R.D. & FINLEY, M.T. (1980) Handbook of acute toxicity of
chemicals to fish and aquatic invertebrates. Summaries of toxicity
tests conducted at Columbia National Fisheries Research Laboratory,
1965-78. Washington, DC, US Department of the Interior Fish and
Wildlife Service, 98 pp. (Resource Publication 137).
JOHNSON, R.D. & MANSKE, D.D. (1976) Residues in food and feed.
Pesticide residues in total diet samples (IX). Pestic. Monit. J., 9:
157-169.
JOHNSON, R.D., MANSKE, D.D., NEW, D.H., & PODREBARAC, D.S. (1981) Food
and feed - pesticide, heavy metal, and other chemical residues in
infant and toddler. Pestic. Monit. J., 15: 39-50.
JOHNSON, M.W., WELTER, S.C., TOSCANO, N.C., & IWATA, Y. (1983) Lettuce
yield reductions correlated with methyl parathion use. J. Econ.
Entomol., 76: 1390-1394.
JONES, D.C.L., SIMMON, V.F., MORTELMANS, K.E., MITCHELL, A.D., EVANS,
E.L., JOTZ, M.M., RICCIO, E.S., ROBINSON, D.E., & KIRKHART, B.A.
(1982) In vitro mutagenicity studies of environmental chemicals.
Research Triangle Park, North Carolina, US Environmental Protection
Agency, Office of Research and Development, Health Effects Research
Laboratory (EPA-600/1-84-003; PB84-138973).
JOSHI, U.M. & THORNBURG, J.E. (1986) Interactions between cimetidine,
methylparathion, and parathion. J. Toxicol. environ. Health, 19:
337-344.
KAGAN, J.S. (1971) [Topical questions about the toxicology of
phosphororganic insecticides.] Ernährungsforschung, 14: 503-514 (in
German).
KALOYANOVA-SIMEONOVA, F.P. (1970) Prevention of intoxication by
pesticides in Bulgaria. In: Proceedings of the Fourth International
Congress on Rural Medicine, Usuda (Tokyo), 1969. Tokyo, Japanese
Association of Rural Medicine, pp. 35-37.
KAMIENSKI, F.X. & MURPHY, S.D. (1971) Biphasic effects of
methylenedioxyphenyl synergists on the action of hexobarbital and
organophosphate insecticides in mice. Toxicol. appl. Pharmacol., 18:
883-894.
KARCSU, S., TOTH, L., MAROSI, G., & SIMON, Z. (1981) Analysis of
histochemical changes due to acute wofatox poisoning in a model
experiment. Morphol. Igazsagugyi Orv. Sz., 21: 289-296.
KARLOG, O., NIELSEN, P., & RASMUSSEN, F. (1978) Toxicokinetics. Arch.
Toxicol., 1(Suppl.): 55-67.
KATKAR, H.N. & BARVE, V.P. (1976) A new spray reagent for the
identification and determination of organophosphorus insecticides by
thin layer chromatography. Curr. Sci., 45: 662-664.
KAWAHARA, F.K., LICHTENBERG, J.J., & EICHELBERGER, J.W. (1967)
Thin-layer and gas chromatographic analysis of parathion and methyl
parathion in the presence of chlorinated hydrocarbons. J. Water
Pollut. Control Fed., 39: 446-457.
KAZAKOVA, M.V., YAKUB, G.G., & MANDRIK, F.I. (1974) Quantitative
detection of metaphos (methyl parathion) in feeds and its effect on
animals. Veterinariya (Moscow), 2: 107.
KHAN, M.S. (1988) An electrolytic reduction method for the
determination of fenitrothion and methyl paration. Pak. J. Sci. Ind.
Res., 31: 20-22.
KHAN, M.S. & HASAN, M. (1988) Dose-related neurochemical changes in
the levels of gangliosides and glycogen in various regions of the rat
brain and spinal cord following methyl parathion administration. Exp.
Pathol., 35: 61-65.
KHATTAT, F.H. & FARLEY, S. (1976) Acute toxicity of certain pesticides
to Acartia tonsa Dana. Narangansett, Rhode Island, US Environmental
Protection Agency, Office of Research and Development, Environmental
Research Laboratory, 23 pp. (EPA-600/3-76-033).
KHEIFETS, L.Y., SOBINA, N.A., & ROMANOV, N.A. (1976) [Use of
differential oscillopolarography for determining substances with
limitable concentrations in water.] Probl. Okhr. Vod, 7: 21-24 (in
Russian).
KHEIFETS, L.Y., ROMANOV, N.A., & SOBINA, N.A. (1980) [Analytical
possibility of differential chronoamperometry with dropping
electrodes.] Zh. Anal. Khim., 35: 874-879 (in Russian).
KIDO, H., BAILEY, J.B., MCCALLEY, N.F., YATES, W.E., & COWDEN, R.E.
(1975) The effect of overhead sprinkler irrigation on methyl parathion
residue on grape leaves. Bull. environ. Contam. Toxicol., 14: 209-213.
KIMMERLE, G. (1975) [Testing of DDT for combined effect with methyl
parathion and fenitrothion in acute tests on rats.]
Wuppertal-Elberfeld, Bayer AG, Institute of Toxicology (Unpublished
report No. 5591, submitted to WHO by Bayer AG, Leverkusen, Germany)
(in German).
KIMMERLE, G. & LORKE, D. (1968) [Toxicology of insecticidal phosphoric
acid esters.] Pflanzenschutz.-Nachr. BAYER, 21: 111-142 (in German).
KJOELHOLT, J. (1985) Determination of trace amounts of
organophosphorus pesticides and related compounds in soils and
sediments using capillary gas chromatography and a
nitrogen-phosphorus detector. J. Chromatogr., 325: 231- 238.
KLISENKO, M.A. & GIRENKO, D.B. (1980) [Gas-chromatographic
determination of organophosphorus pesticides in air.] Gig. i Sanit.
1980(9): 57-60 (in Russian).
KLISENKO, M.A., GIRENKO, D.B., GOLOVATYI, V.G., SHPAKOVSKII, I.V., &
KOROL, E.N. (1981) [Field-desorption mass spectrometry in pesticide
analysis.] Khim. Sel'sk. Khoz. 1981: 59-61 (in Russian).
KNIEHASE, U. & ZOEBELEIN, G. (1990) [Testing the effects of pesticides
on the predator mite Phytoseiulus persimilis Ath.-Hen. by means of
a new laboratory method approaching to the practice.] Anz.
Schädlingskd. Pflanzenschutz. Umweltschutz., 63: 105-113 (in German).
KOEN, J.G. & HUBER, J.F.K. (1970) A rapid method for residue analysis
by column liquid chromatography with polarographic detection.
Application to the determination of parathion and methylparathion on
crops. Anal. chim. Acta, 51: 303-307.
KONRAD, J.G., PIONKE, H.B., & CHESTERS, G. (1969) Extraction of
organochlorine and organophosphate insecticides from lake waters.
Analyst, 94: 490-492.
KORN, S. & EARNEST, R. (1974) Acute toxicity of twenty insecticides to
striped bass, Morone saxatilis. Calif. Fish Game, 60(3): 128-131.
KORSOS, I. & LANTOS, J. (1984) [Thin-layer chromatographic
identification of pesticides.] Novenyvedelem (Budapest), 20: 30-34 (in
Hungarian).
KRIJGSMAN, W. & VAN DE KAMP, C.G. (1976) Analysis of organophosphorus
pesticides by capillary gas chromatography with flame photometric
detection. J. Chromatogr., 117: 201-205.
KRUEGER, H.R. & CASIDA, J.E. (1957) Toxicity of fifteen
organophosphorus insecticides to several insect species and to rats.
J. Econ. Entomol., 50: 356-358.
KUMAR, N.V.N. (1985) Chromatographic and enzymic methods as applied to
environmental monitoring of pesticide residues in some districts of
Andhra Pradesh. In: Trivedy, R.K. & Goel, P.K. ed. Current pollution
research in India. Kared, India, Environmental Publications, pp.
163-184.
KUMAR, N.V.N. & RAMASUNDARI, M. (1980) Colorimetric determination of
methyl parathion and oxygen analog. J. Assoc. Off. Anal. Chem., 63:
536-538.
KUMMER R. & VAN SITTERT, N.J. (1986) Field studies on health effects
from the application of two organophosphorus insecticide formulations
by hand-held ULV to cotton. Toxicol. Lett., 33: 7-24.
LAMONTAGNE, J.C. (1978) Résidues de produits phytosanitaires dans les
fruits et légumes. C. R. Séances Acad, Agric. France, 64: 923-931.
LANGE, P. & WIEZOREK, W.D. (1975) Effects of diethyldithiocarbamate on
acute toxicity and acetylcholinesterase inhibition by methylparathion
in mice. Acta biol. med. Germ., 34: 427-433.
LANGE, P., TIEFENBACH, B., & WIEZOREK, W.D. (1977) Effect of
dithiocarb on acute toxicity and acetylcholinesterase inhibition by
different organophosphates in mice. Proc. Eur. Soc. Toxicol., 18:
284-285.
LASSITER, R.R., PARRISH, R.S., & BURNS, L.A. (1986) Decomposition by
planktonic and attached microorganisms improves chemical fate models.
Environ. Toxicol. Chem., 5: 29-39.
LAUGHLIN, J. & GOLD, R.E. (1989) Evaporative dissipation of methyl
parathion from laundered protective apparel fabrics. Bull. environ.
Contam. Toxicol., 42: 566-573.
LAUGHLIN, J.M., EASLEY, C.B., GOLD, R.E., & TUPY, D.R. (1981) Methyl
parathion transfer from contaminated fabrics to subsequent laundry and
to laundry equipment. Bull. environ. Contam. Toxicol., 27: 518-523.
LAWRENCE, J.F. & TURTON, D. (1978) High performance liquid
chromatographic data to 166 pesticides. J. Chromatogr., 159: 207-226.
LAWRENCE, J.F., RENAULT, C., & FREI, R.W. (1976) Fluorogenic labelling
of organophosphate pesticides with dansyl chloride. Application to
residue analysis by high-pressure liquid chromatography and thin-layer
chromatography. J. Chromatogr., 121: 343-351.
LEARD, R.L., GRANTHAM, B.J., & PESSONEY, G.F. (1980) Use of selected
freshwater bivalves for monitoring organochlorine pesticide residues
in major Mississippi stream systems, 1972-1973. Pestic. Monit. J., 14:
47-52.
LE BEL, G.L., WILLIAMS, D.T., GRIFFITH, G., & BENOIT, F.M. (1979)
Isolation and concentration of organophosphorus pesticides from
drinking water at the ng/L level, using macroreticular resin. J.
Assoc. Off. Anal. Chem., 62: 241-249.
LEE, H.B., WENG, L.D., & CHAU, A.S.Y. (1984) Confirmation of pesticide
residue identity. XI. Organophosphorus pesticides. J. Assoc. Off.
Anal. Chem., 67: 553-556.
LEIDY, R.B., SHEETS, T.J., & NELSON, L.A. (1977) Residues from two
formulations of methyl parathion on flue-cured tobacco. Tob. Sci., 21:
28-31.
LESHCHEV, V.V. & TALANOV, G.A. (1977) [Determination of residues of
organophosphorus pesticides using thin-layer chromatography with
enzymic development.] Khim. Sel'sk. Khoz., 15: 46-48 (in Russian).
LEVINE, B.S. & MURPHY S.D. (1977a) Esterase inhibition and
reactivation in relation to piperonyl butoxide-phosphorothionate
interactions. Toxicol. appl. Pharmacol., 40: 379-391.
LEVINE, B.S. & MURPHY, S.D. (1977b) Effect of piperonyl butoxide on
the metabolism of dimethyl and diethyl phosphorothionate insecticides.
Toxicol. appl. Pharmacol., 40: 393-406.
LEWIS, D.L. & HOLM, H.W. (1981) Rates of transformation of methyl
parathion and diethyl phthalate by aufwuchs microorganisms. Appl.
environ. Microbiol., 42: 698-703.
LEWIS, D.L., HODSON, R.E., & FREEMAN, L.F. III (1984) Effects of
microbial community interactions on transformation rates of xenobiotic
chemicals. Appl. environ. Microbiol., 48: 561-565.
LEWIS, D.L., HODSON, R.E., & FREEMAN, L.F. III (1985) Multiphasic
kinetics for transformation of methyl parathion by Flavobacterium
species. Appl. environ. Microbiol., 50: 553-557.
LEWIS, R.G. & JACKSON, M.D. (1982) Modification and evaluation of a
high-volume air sampler for pesticides and semiolatile industrial
organic chemicals. Anal. Chem., 54: 592-594.
LEWIS, R.G. & MACLEOD, K.E. (1982) Portable sampler for pesticides and
semivolatile industrial organic chemicals in air. Anal. Chem., 54:
310-315.
LEWIS, R.G., BROWN, A.R., & JACKSON, M.D. (1977) Evaluation of
polyurethane foam for sampling of pesticides, polychorinated biphenyls
and polychlorinated naphthalenes, in ambient air. Anal. Chem., 49:
1668-1672.
LI, Y. & WANG, G. (1987) [Determination of organophosphorus pesticides
in wastewater.] Shanghai Huanjing Kexue, 6: 19-31, 42 (in Chinese).
LIANG, L. & ZHANG, G. (1986) [Gas chromatographic determination of
organophosphorus agrochemicals in the air.] Zhonghua Laodong Weisheng
Zhiyebing Zazhi, 4: 163-165 (in Chinese).
LICHTENSTEIN, E.P. (1975) Transport mechanism in soil. Metabolism and
movement of insecticides from soils into water and crop plants. Pure
appl. Chem., 42(1/2): 113-118.
LICHTENSTEIN, E.P. & SCHULZ, K.R. (1964) The effects of moisture and
microorganisms on the persistence and metabolism of some
organophosphorous insecticides in soils, with special emphasis on
parathion. J. Econ. Entomol., 57: 618-627.
LISI, P., CARAFFINI, S., & ASSALVE, D. (1986) A test series for
pesticide dermatitis. Contact Dermatitis, 15: 266-269.
LISI, P., CARAFFINI, S., & ASSALVE, D. (1987) Irritation and
sensitization potential of pesticides. Contact Dermatitis, 17:
212-218.
LITTLE, E.E., ARCHESKI, R.D., FLEROV, B.A., & KOZLOVSKAYA, V.I. (1990)
Behavioural indicators of sublethal toxicity in rainbow trout. Arch.
environ. Contam. Toxicol., 19: 380-385.
LOFROTH, G., KIM, C.H., & HUSSAIN, S. (1969) Alkylating properties of
2,2-dichlorovinyl dimethyl phosphate: a disregarded hazard. Environ.
Mutat. Soc. Newsl., 2: 21-27.
LOSER, E. & EIBEN, R. (1982) [E 605-methyl: Multigeneration tests on
rats.] Wuppertal-Elberfeld, Bayer AG, Institute of Toxicology
(Unpublished report No. 10630, submitted to WHO by Bayer AG,
Leverkusen, Germany) (in German).
LOVE, J.L., DONNELLY, R.G.C., HUGHES, J.T., & WILSON, P.D. (1974)
Pesticide residues in fruit and vegetables in New Zealand 1971-73. N.
Z. J. Sci., 17: 529-534.
LUKE, M.A. & DOOSE, G.M. (1983) A modification of the Luke
multiresidue procedure for low moisture, nonfatty products. Bull.
environ. Contam. Toxicol., 30: 110-116.
LUKE, M.A. & DOOSE, G.M. (1984) A rapid analysis for pesticides in
milk and oilseeds. Bull. environ. Contam. Toxicol., 32: 651-656.
LUKE, M.A., FROBERG, J.E., & MASUMOTO, H.T. (1975) Extraction and
cleanup of organochlorine, organophosphate, organonitrogen, and
hydrocarbon pesticides in produce for determination by gas-liquid
chromatography. J. Assoc. Off. Anal. Chem., 58: 1020-1026.
McCANN, J.A. & JASPER, R.L. (1972) Vertebral damage to bluegills
exposed to acutely toxic levels of pesticides. Trans. Am. Fish. Soc.,
101(2): 317-322.
McCOLLISTER, D.D., OLSON, K.J., ROWE, V.K., PAYNTER, O.E., WEIR, R.J.,
& DIETERICH, W.H. (1968) Toxicology of 4-tert-butyl-2-chlorophenyl
methyl methylphosphoramidate (ruelene) in laboratory animals. Food
Cosmet. Toxicol., 6: 185-198.
MACEK, K.J. & McALLISTER, W.A. (1970) Insecticide susceptibility of
some common fish family representatives. Trans. Am. Fish Soc., 99(1):
20-27.
MACHEMER, L. (1977a) [Methyl parathion. Embryotoxic or teratogenic
effects in rats after oral application.] Leverkusen, Germany, Bayer
AG, Institute for Toxicology, (Unpublished report submitted to WHO by
Bayer AG) (in German).
MACHEMER, L. (1977b) [Methyl parathion. Tests for embryotoxic and
teratogenic effects in rats after intravenous injection.]
Wuppertal-Elberfeld, Bayer AG, Institute of Toxicology (Unpublished
report No. 6812, submitted to WHO by Bayer AG, Leverkusen, Germany)
(in German).
McLEOD, K.E & LEWIS, R.G. (1982) Portable sampler for pesticides and
semivolatile industrial organic chemicals in air. Anal. Chem., 54:
310-315.
MAFF (1986) Report of the Working Party on Pesticide Residues
(1982-85). The sixteenth report of the Steering Group on Food
Surveillance. London, Ministry of Agriculture, Fisheries and Food,
Health and Safety Executive, 50 pp. (Food Surveillance Paper No. 16).
MAFF (1990) Report of the Working Party on Pesticide Residues
(1988-89). (Supplement to issue No. 8 (1990) of the Pesticide
Register). London, Ministry of Agriculture, Fisheries and Food, Health
and Safety Executive, 86 pp.
MANSKE, D.D. & JOHNSON, R.D. (1975) Residues in food and feed.
Pesticide residues in total diet samples (VIII). Pestic. Monit. J., 9:
94-105.
MANSKE, D.D. & JOHNSON, R.D. (1977) Pesticide and other chemical
residues in total diet samples (X). Pestic. Monit. J., 10: 134-148.
MARCUS, M., SPIGARELLI, J., & MILLER, H. (1978) Organic compounds in
organophosporus pesticide manufactoring wastewaters. Springfield,
Virginia, National Technical Information Service, p. 22 (PB-289821).
MARTIN, E.W. (1978) Hazards of medication. A manual on drug
interactions, contraindications, and adverse reactions with other
presribing and drug information, 2nd ed. Philadelphia, Toronto, J.B.
Lippincott Company.
MATHEW, G., ABDUL RAHIMAN, M., & VIJAYALAXMI, K.K. (1990) In vivo
genotoxic effects in mice of Metacid 50 an organophosphorus
insecticide. Mutagenesis, 5(2): 147-149.
MAYER, F.L. Jr (1987) Acute toxicity handbook of chemicals to
estuarine organisms. Gulf Breeze, Florida, US Environmental Protection
Agency, 283 pp.
MAYER, F.L. Jr & ELLERSIECK, M.R. (1986) Manual of acute toxicity:
interpretation and data base for 410 chemicals and 66 species of
freshwater animals. Washington, DC, US Department of the Interior Fish
and Wildlife Service, pp. 311 (Resource Publication 160).
MELNIKOW, N.N. (1971) Chemistry of pesticides. Berlin, Heidelberg, New
York, Springer Verlag.
MENZIE, C.M. (1972) Fate of pesticides in the environment. Ann. Rev.
Entomol., 17: 199-222.
MENZIE, C.M. (1974) Metabolism of pesticides - an update. Washington,
DC, US Department of the Interior, Fish and Wildlife Service (Report
No. 184).
MESTRES, R., LEONARDI, G., CHEVALLIER, C., & TOURTE, J. (1969)
Résidus de pesticides. XIX. Méthode de recherche des résidus de
pesticides dans les eaux naturelles. 1ère partie. Méthode d'analyse
générale. Ann. Fals. Expert. Chim., 62(685): 75-85.
MESTRES, R., CHEVALLIER, C., ESPINOZA, C., & CORNET, R. (1977) XXXIII.
Application du couplage chromatographie gazeuse spectrométrie de masse
aux méthodes de recherche et de dosage des résidus de pesticides et de
micropollutants organiques dans les matéiaux de l'environnement et
les matières alimentaires. Ann. Fals. Expert. Chim., 70: 177-188.
MEYER JONES, L., BOOTH, N.H. & McDONALD, L.E. (1977) Veterinary
pharmacology and therapeutics, 4th ed. Ames, Iowa State University
Press, pp. 1201.
MEYERS, S.M., CUMMINGS, J.L., & BENNETT, R.S. (1990) Effects of methyl
parathion on red-winged blackbird ( Agelaius phoeniceus) incubation
behavior and nesting success. Environ. Toxicol. Chem., 9: 807-813.
MIDWEST RESEARCH INSTITUTE (1975) Substitute chemical program -
initial scientific and minieconomic review of methyl parathion.
Kansas City, Missouri, Midwest Research Institute, 176 pp. (Prepared
for US Environmental Protection Agency, Criteria and Evaluation
Division, Washington, DC.) (EPA-540/1-75-004).
MIELLET, A. (1982) Dosage des résidus de pesticides par C.L.H.P. sur
plantes médicinales et aromatiques. Compte-rendu du résultat
d'applications de pesticides effectuées avec le concours du syndicat
national des plantes médicinales de Nilly-la-Forét. Ann. Fals. Expert.
Chim., 75: 369-375.
MIHAIL, F. & VOGEL, O. (1984) [E 120/methyl parathion: Subacute
dermal toxicity tests on rabbits.] Wuppertal-Elberfeld, Bayer AG,
Institute of toxicology (Unpublished report No. 12484, submitted to
WHO by Bayer AG, Leverkusen, Germany) (in German).
MILES, J.W., FETZER, L.E., & PEARCE, G.W. (1970) Collection and
determination of trace quantities of pesticides in air. Environ. Sci.
Technol., 4(5):420-425.
MILLER, H., CRAMER, P., DRINKWINE, A., SHAN, A., RISCHAN, G., & GOING,
J. (1981) Development of methods for pesticides in wastewater:
applicability of a general approach. In: Keith, L.H., ed. Advances in
the identification and analysis of organic pollutants in water. Ann
Arbor, Michigan, Ann Arbor Science Publishers, Vol. 1, pp. 115-138.
MILLS, P.A., ONLEY, J.H., & GAITHER, R.A. (1963) Rapid method for
chlorinated pesticide residues in vegetables and food. J. Am. Off.
Anal. Chem., 46: 186-191.
MINCHEW, C.D. & FERGUSON, D.E. (1969) Toxicities of six insecticides
to resistant and susceptible green sunfish and golden shiners in
static bioassays. J. Mississippi Acad. Sci., 15: 29-32.
MINGELGRIN, U., SALTZMAN, S., & YARON, B. (1977) A possible model for
the surface-induced hydrolysis of organophosphorus pesticides on
kaolinite clays. Soil Sci. Soc. Am. J., 41: 519-523.
MIRER, F.E., LEVINE, B.S., & MURPHY, S.D. (1977) Parathion and methyl
parathion toxicity and metabolism in piperonyl butoxide and diethyl
maleate pretreated mice. Chem.-Biol. Interact., 17: 99-112.
MIYAMOTO, J., SATO, Y., KADOTA, T., FUJINAMI, A., & ENDO, M. (1963)
Studies on the mode of action of organophosphorus compounds. Part I.
Metabolic fate of P32 labelled sumithion and methylparathion in
guinea pig and white rat. Agric. Biol. Chem., 27: 381-389.
MOELLER, H.C. & RIDER, J.A. (1961) Studies on the anticholinesterase
effect of parathion and methylparathion in humans. Fed. Proc., 20:
434.
MOELLER, H.C. & RIDER, J.A. (1962) Threshold of incipient toxicity to
systox and methyl parathion. Fed. Proc., 21: 451.
MOHANTY-HEJMADI, P. & DUTTA, S.K. (1981) Effects of some pesticides on
the development of the Indian bull frog Rana tigerina. Environ.
Pollut., Ser. A, 24: 145-161.
MOHN, G. (1973) 5-methyltryptophan resistance mutations in
Escherichia coli K-12. Mutagenic activity of monofunctional
alkylating agents including organophosphorus insecticides. Mutat.
Res., 20: 7-15.
MOLLHOFF, E. (1981) [Laboratory studies to metabolism of methyl
parathion in soils.] Leverkusen, Gemany, Bayer AG (Unpublished report
submitted to WHO by Bayer AG) (in German).
MOLNAR, J. & PAKSY, K.A. (1978) [Evaluation of the acute toxicity of
inhaled pesticides in experimental animals.] In: [Proceedings of the
Conference on Safety Technology in the Agricultural Use of Chemicals.]
pp. 1790-1793 (Conference paper) (in German).
MOORTHY, K.S., REDDY, B.K., CHETTY, C.S., & SWAMI, K.S. (1983)
Catalytic potential of catalase in hepatopancreas of freshwater
mussel Lamellidens marginalis during induced methyl-parathion toxic
stress. Geobios, 10(1): 42-44.
MOORTHY, K.S., REDDY, B.K., SWAMI, K.S., & CHETTY, C.S. (1984) Changes
in respiration and ionic content in tissues of freshwater mussel
exposed to methyl parathion toxicity. Toxicol. Lett., 21: 287-291.
MORGAN, D.P., HETZLER, H.L., SLACH, E.F., & LIN, L.I. (1977) Urinary
excretion of paranitrophenol and alkyl phosphates following ingestion
of methyl or ethyl parathion by human subjects. Arch. environ. Contam.
Toxicol., 6: 159-173.
MORPURGO, G., AULICINO, F., BIGNAMI, M., CONTI, L., VELCICH, A., &
MONTALENTI, S.G. (1977) Genetica - Relationship between structure and
mutagenicity of dichlorvos and other pesticides. Rend. Cl. Sci. Fis.
Mat. Nat., 62: 692-701.
MUDGALL, C.F. & PATIL, H.S. (1987) Toxic effects of dimethoate and
methyl parathion on glycogen reserves of male and female Rana. J.
Environ. Biol., 8(3): 237-244.
MUHLMANN, R. & SCHRADER, G. (1957) [Hydrolysis of the insecticdal
phosphoric acid esters.]. Z. Naturforsch., B12: 196-208 (in German).
MULLER, B. (1973) [Determination of pesticide residues in bee honey.
I. Semiquantitative thin-layer chromatographic determination of
insecticide residues in bee honey]. Nahrung, 17: 381-386 (in German).
MUNCY, R.J. & OLIVER, A.D. Jr (1963) Toxicity of ten insecticides to
the red crawfish Procambarus clarki. Trans. Am. Fish. Soc., 96(4):
428-431.
MUNDY, R.L., BOWMAN, M.C., FARMER, J.H., & HALEY, T.J. (1978)
Quantitative structure activity study of a series of substituted
O,O-dimethyl O-( p-nitrophenyl) phosphorothioates and
O-analogs. Arch. Toxicol., 41: 111-123.
MUNN, S., KEEFE, T.J., & SAVAGE, E.P. (1985) A comparative study of
pesticide exposures in adult and youth migrant field workers. Arch.
environ. Health, 40: 215-220.
MURPHY, S.D. (1980) Toxic interactions with dermal exposure to
organophosphate insecticides. Toxicol. Lett., 5: 34.
MURTHY, A. & RAMANI, A. (1982) What is an environmentally safe
pesticide? In: Christiansen, K., Koch, B., & Bro-Rasmussen, F., ed.
Chemicals in the environment. Chemicals testing and hazard ranking -
the interaction between science and administration. Proceedings of the
International Symposium, Lyngby-Copenhagen, 18-20 October 1982.
Lyngby, The Technical University of Denmark, Laboratory of
Environmental Science and Ecology, pp.323-333.
MURTY, A.S., RAMANI, A.V., CHRISTOPHER, K., & RAJABHUSHANAM, B.R.
(1984) Toxicity of methyl parathion and fensulfothion to the fish
Mystus cavasius. Environ. Pollut., A34: 37-46.
NAG, M. & NANDI, N. (1987) In vitro and in vivo effect of
organophosphate pesticides on monoamine oxidase activity in rat brain.
Biosci. Rep., 7(10): 801-804.
NAGARATNAMMA, R. & RAMAMURTHI, R. (1982) Metabolic depression in the
fresh water teleost Cyprinus carpio exposed to an organophosphate
pesticide. Curr. Sci., 51(13): 668-669.
NAGYMAJTENYI, L., DESI, I., & LORENCZ, R. (1988) Neurophysiological
markers as early signs of organophosphate neurotoxicity. Neurotoxicol.
Teratol., 10: 429-434.
NAKATSUGAWA, T., TOLMAN, N.M., & DAHM, P.A. (1968) Degradation and
activation of parathion analogs by microsomal enzymes. Biochem.
Pharmacol., 17: 1517-1528.
NAKATSUGAWA, T., TOLMAN, N.M., & DAHM, P.A. (1969) Degradation of
parathion in the rat. Biochem. Pharmacol., 18: 1103-1114.
NANGNIOT, P. (1966) Electrochemical methods for quantitative analysis
of pesticide residues. Meded. Fac. Landbouwwet. Rijksuniv., Gent, 31:
447-473.
NAQVI, S.M.Z. (1973) Toxicity of twenty-three insecticides to a
tubificed worm Branchiura sowerbyi from the Mississippi delta. J.
Econ. Entomol., 66(1): 70-74.
NATALE, O.E., GOMEZ, C.E., PECHEN de D'ANGELO, A.M., & SORIA, C.A.
(1988) Waterborne pesticides in the Negro River Basin (Argentina). In:
Abbou, R., ed. Hazardous waste: detection, control, treatment.
Amsterdam, Oxford, New York, Elsevier Science Publishers, pp.
879-907.
NATIONAL FIRE PROTECTION ASSOCIATION (1986) Fire protection guide on
hazardous materials, 9th ed. Quincy, Massachusetts, National Fire
Protection Association, pp. 49-64.
NATIONAL RESEARCH COUNCIL (1977) Drinking water and health.
Washington, DC, National Academy of Sciences, pp. 626-635 (Publication
NRC/2619).
NCI (1979) Bioassay of methyl parathion for possible carcinogenicity.
Washington, DC, US Department of Health, Education & Welfare, National
Cancer Institute (Technical Report Series No. 157; DHEW Publ. No.
(NHI) 79-1713).
NEHEZ, M., BOROS, P., FERKE, A., MOHOS, J., PALOTAS, M., VETRO, G.,
ZIMANYI, M., & DESI, I. (1988) Cytogenetic examination of people
working with agrochemicals in the southern region of Hungary. Regul.
Toxicol. Pharmacol., 8: 37-44.
NELSON, R.C. (1967) Procedure for nine organothiophosphate pesticide
residues on fruits and vegetables, using microcoulometric gas
chromatography. J. Assoc. Off. Anal. Chem., 50: 922-926.
NEWTON, T.D., GATTIE, D.K., & LEWIS, D.L. (1990) Initial test of the
benchmark chemical approach for predicting microbial transformation
rates in aquatic environments. Appl. environ. Microbiol., 56(1):
288-291.
NIELSEN, P.G. (1985) Quantitative analysis of parathion and
parathion-methyl by combined capillary column gas chromatography
negative ion chemical ionization mass spectrometry. Biomed. Mass
Spectrom., 12: 695-698.
NIETHAMMER, K.R. & BASKETT, T.S. (1983) Cholinesterase inhibition of
birds inhabiting wheat fields treated with methyl parathion and
toxaphene. Arch. environ. Contam. Toxicol., 12: 471-475.
NIMMO, W.R., HAMAKER, T.L., MATTHEWS, E., & MOORE, J.C. (1981) An
overview of the acute and chronic effects of first and second
generation pesticides on an estuarine mysid. In: Vernberg, F.J.,
Calabrese, A., Thurberg, F.P., & Vernberg, W.B., ed. Biological
monitoring of marine pollutants. Proceedings of a Symposium on
Pollution and Physiology of Marine Organisms, Milford, Connecticut,
7-9 November 1980. New York, London, Academic Press, pp. 3-19.
NIOSH (1976) Criteria for a recommended standard - occupational
exposure to methyl parathion. Cincinnati, Ohio, National Institute for
Occupational Safety and Health (DHEW 0 (NIOSH) Publication No.
77-106).
NIOSH (1980) Summary of NIOSH recommendations for occupational health
standards. Cincinnati, Ohio, National Institute for Occupational
Safety and Health.
NIOSH (1991) Registry of toxic effects of chemical substances.
Cincinnatti, Ohio, National Institute for Occupational Safety and
Health.
NISHIUCHI, Y. & HASHIMOTO, Y. (1967) Toxicity of pesticide ingredients
to some freshwater organisms. Botyu-Kagaku, 32(5): 5-11.
NOMIYAMA, K., MATSUI, K., & NOMIYAMA, H. (1980) Environmental
temperature, a factor modifying the acute toxicity of organic
solvents, heavy metals, and agricultural chemicals. Toxicol. Lett., 6:
67-70.
OMURA, M., HASHIMOTO, K., OHTA, K., IIO, T., UEDA, S., ANDO, K., &
HIRAIDE, H. (1990) Relative retention time diagram as a useful tool
for gas chromatographic analysis and electron-capture detection of
pesticides. J. Assoc. Off. Anal. Chem., 73(2): 300-306.
OOMEN, P.A. (1986) A sequential scheme for evaluating the hazard of
pesticides to bees, Apis melifera. Mede. Fac. Landbouwwet.
Rijksuniv. Gent, 51(36): 1205-1213.
ORLANDO, E., RAFFI, G.B., CASCELLA, D., DE ROSA, V., & CIPOLLA, C.
(1972) Effect of quinidine on electrocardiographic alterations during
acute poisoning by phosphoric acid esters. Lav. Um., 24: 1-13.
OSADCHUK, M., ROMACH, M., & MCCULLY, K.A. (1971) Cleanup and
separation procedures for multipesticide residue analysis in
monitoring and regulatory laboratories. In: Tahori, A.S., ed.
Pesticide chemistry. Proceedings of the 2nd International Congress on
Pesticide Chemistry, New York, Gordon & Breach, vol. 4., pp. 357-383.
OU, L.T. (1985) Methyl parathion degradation and metabolism in soil:
influence of high soil-water contents. Soil Biol. Biochem., 17:
241-243.
OU, L.-T. & SHARMA, A. (1989) Degradation of methyl parathion by a
mixed bacterial culture and a Bacillus sp. isolated from different
soils. J. Agric. Food Chem., 37: 1514-1518.
OU, L.-T., RAO, P.S.C., & DAVIDSON, J.M. (1983) Methyl parathion
degradation in soil: influence of soil-water tension. Soil Biol.
Biochem., 15: 211-215.
PALAWSKI, D., BUCKLER, D.R., & MAYER, F.L. (1983) Survival and
condition of rainbow trout (Salmo gairdneri) after acute exposures
to methyl parathion, triphenyl phosphate, and DEF. Bull. environ.
Contam. Toxicol., 30: 614-620.
PAP, A., SZABO, I., & SZARVAS, F. (1976) Effect of liver damage and of
phenobarbital, norandrostenolone, phenyl propionate or chloramphenicol
treatment of the poisoning by organic phosphoric acid esters. Kiserl.
Orvostud., 28: 172-179.
PASCHAL, D.C., BICKNELL, R., & DRESBACH, D. (1977) Determination of
ethyl and methyl parathion in runoff water with high performance
liquid chromatography. Anal. Chem., 49: 1551-1554.
PATTERSON, P.L. (1982) New uses of thermionic ionization detectors in
gas chromatography. Chromatographia, 16: 107-111.
PAULUHN, J. (1983) [Methyl parathion (E 120). Studies on the
irritant/corrosive effect on skin and eyes (rabbit).]
Wuppertal-Elberfed, Bayer AG, Institute of Toxicology (Unpublished
report No. 11712, submitted to WHO by Bayer AG, Leverkusen, Germany)
(in German).
PETERKA, V. & CERNA, V. (1988) Micellar catalysis in decomposition of
some organophosphates. Chem. Prum., 38(1): 36-39.
PFAENDER, F.K., SHUMAN, M.S., DEMPSEY, H., & HARDEN, C.W. (1977)
Monitoring heavy metals and pesticides in the Cape Fear River Basin of
North Carolina, Chapel Hill, NC. Raleigh, North Carolina, North
Carolina Water Resources Research Institute, pp. 29, 119.
PFEIFFER, R. & STAHR, H.M. (1982) Screening for organochlorine and
organophosphorus pesticides. In: Touchstone, J.C., ed. Advances in
thin layer chromatography: clinical and environmental applications.
Proceedings of the Second Biennial Symposium, Philadelphia, December
1980. New York, John Wiley and Sons, pp. 451-460.
PFLUGMACHER, J. & EBING, W. (1974) Purification of phosphoric acid
insecticide residues in vegetable extracts by gel chromatography on
Sephadex LH-20. J. Chromatogr., 93: 457-463.
PICKERING, Q.H., HENDERSON, C., & LEMKE, A.E. (1962) The toxicity of
organic phosphorus insecticides to different species of warm-water
fishes. Trans. Am. Fish. Soc., 91: 175-184.
PIONKE, H.B., KONRAD, J.G., CHESTERS, G., & ARMSTRONG, D.E. (1968)
Extraction of organochlorine and organophosphate insecticides from
lake waters. Analyst, 93: 363-367.
PLAPP, F.W. & CASIDA, J.E. (1958) Hydrolysis of the alkyl-phosphate
bond in certain dialkyl aryl phosphorothioate insecticides by rats,
cockroaches, and alkali. J. Econ. Entomol., 51(6): 800-803.
PORTIER, R.J. & MEYERS, S.P. (1982) Monitoring biotransformation and
biodegradation of xenobiotics in simulated aquatic microenvironmental
systems. Dev. Ind. Microbiol., 23: 459-475.
PORTIER, R.J., CHEN, H.M., & MEYERS, S.P. (1983) Environmental effect
and fate of selected phenols in aquatic ecosystems using microcosm
approaches. Dev. Ind. Microbiol., 24: 409-424.
PREUSSMANN, R., SCHNEIDER, H., & EPPLE, F. (1969) [Investigation of
effects of alkylating agents. II. The investigation of different
classes of alkylating agents by means of a modified colour reaction
with 4-(4-nitro-benzyl)pyridine (NBP).] Arzneimittelforschung, 7:
1059-1073 (in German).
PRINSLOO, S.M. & DE BEER, P.R. (1987) Gas chromatographic relative
retention data for pesticides on nine packed columns: II.
Organophosphorus and organochlorine pesticides, using elecron-capture
detection. J. Assoc. Off. Anal. Chem., 70: 878-888.
PRITCHARD, P.H., CRIPE, C.R., WALKER, W.W., SPAIN, J.C., & BOURQUIN,
A.W. (1987) Biotic and abiotic degradation rates of methyl parathion
in freshwater and estuarine water and sediment samples. Chemosphere,
16(7): 1509-1520.
QIAN, C., SANDERS, P.F., & SEIBER, J.N. (1985) Accelerated degradation
of organophosphorus pesticides with sodium perborate. Bull. environ.
Contam. Toxicol., 35: 682-688.
RADULOVIC, L.L., LAFERLA, J.J., & KULKARNI, A.P. (1986) Human
placental glutathione s-transferase-mediated metabolism of methyl
parathion. Biochem. Pharmacol., 35: 3473-3480.
RADULOVIC, L.L., KULKARNI, A.P., & DAUTERMAN, W.C. (1987)
Biotransformation of methyl parathion by human foetal liver
glutathione s-transferases: an in vitro study. Xenobiotica, 17:
105-114.
RAMAKRISHNA, N. & RAMACHANDRAN, B.V. (1978) Colorimetric determination
of fenitrothion and methyl parathion using a hydroxylaminolytic
procedure. J. Indian Chem. Soc., LV: 185-187.
RANI, V.J.S., VENKATESHWARLU, P., & JANAIAH, C. (1989) Changes in
carbohydrate metabolism of Clarias bactrachus (Linn) when exposed to
two organophosphorus insecticides. J. environ. Biol., 10: 197-204.
RAO, S.N. & McKINLEY, W.P. (1969) Metabolism of organophosphorus
insecticides by 0liver homogenates from different species. Can. J.
Biochem., 47: 1155-1159.
RAO, K.S.P. & RAO, K.V.R. (1983) Regulation of phosphorylases and
aldolases in tissues of the teleost Tilapia mossambica under
methyl-parathion impact. Bull. environ. Contam. Toxicol., 31(4):
474-478.
RAO, K.S.P. & RAO, K.V.R. (1984a) Tissue specific alteration of
aminotransferases and total ATPases in the fish (Tilapia mossambica)
under methyl parathion impact. Toxicol. Lett., 20: 53-57.
RAO, K.S.P. & RAO, K.V.R. (1984b) Impact of methyl parathion toxicity
and eserine inhibition of acetylcholinesterase activity in tissues of
the teleost (Tilapia mossambica) - a correlative study. Toxicol.
Lett., 22: 351-356.
RAO, K.S.P. & RAO, K.V.R. (1987) The possible role of
glucose-6-phosphate dehydrogenase in the detoxification of methyl
parathion. Toxicol. Lett., 39: 211-214.
RAO, K.V.R., RAO, K.R.S.S., & RAO, K.S.P. (1983a) Cardiac responses to
malathion and methyl parathion in the mussel, Lamellidens marginalis
(Lamark). J. environ. Biol., 4: 65-68.
RAO, K.S.P., MADHU, C., RAO, K.R.S.S., & RAO, K.V.R. (1983b) Effect of
methyl parathion on body weight, water content and ionic changes in
the teleost, Tilapia mossambica (Peters). J. Food Sci. Technol., 20:
27-29.
RAO, T.S., DUTT, S., & MANGAIAH, K. (1967) TLm values of some modern
pesticides to the fresh-water fish Puntius puckelli. Environ.
Health, 9(2): 103-109.
RASHID, K.A. & MUMMA, R.O. (1984) Genotoxicity of methyl parathion in
short-term bacterial test systems. J. environ. Sci. Health, B19:
565-577.
RASTOGI, A. & KULSHRESTHA, S.K. (1990) Effect of sublethal
doses of three pesticides on the ovary of a carp minnow Rasbora
daniconius. Bull. environ. Contam. Toxicol., 45: 742-747.
RATTNER, B.A. & FRANSON, J.C. (1984) Methyl parathion and fenvalerate
toxicity in American kestrels: acute physiological responses and
effects of cold. Can. J. Physiol. Pharmacol., 62: 787-792.
REDDY, K.S. & GAMBRELL, R.P. (1985) The rate of soil reduction as
affected by levels of methyl parathion and 2,4-D. J. environ. Sci.
Health, B20: 275-298.
REDDY, M.S. & RAO, K.V.R. (1986) Acute toxicity of insecticides to
penaeid prawns. Environ. Ecol., 4(1): 221-223.
REDDY, M.S. & RAO, K.V.R. (1988) In vivo recovery of
acetylcholinesterase activity from phosphamidon and methylparathion
induced inhibition in the nervous tissue of penaeid prawn
(Metapenaeus monoceros). Bull. environ. Contam. Toxicol., 40:
752-758.
REDDY, M.S. & RAO, K.V.R. (1989) In vivo modification of lipid
metabolism in response to phosphamidon, methylparathion and lindane
exposure in the penaeid prawn, Metapenaeus monoceros. Bull.
environ. Contam. Toxicol., 43: 603-610.
REDDY, M.S. & RAO, K.V.R. (1990a) Effects of sublethal concentrations
of phosphamidon, methyl parathion, DDT, and lindane on tissue nitrogen
metabolism in the penaeid prawn, Metapenaeus monoceros (Fabricius).
Ecotoxicol. environ. Saf., 19: 47-54.
REDDY, M.S. & RAO, K.V.R. (1990b) Methylparathion - induced
alterations in the acetylcholinesterase and phosphatases in a penaeid
prawn, Metapenaeus monoceros (Fabricius). Bull. Environ. Contam.
Toxicol., 45: 350-357.
REDDY, M.S., RAO, K.V.R., & MURTHY, B.N. (1988) Changes in nitrogen
metabolism of penaeid prawn, Penaeus indicus, during sublethal
phosphamidon and methylparathion-induced stress. Bull. environ.
Contam. Toxicol., 41: 344-351.
REDDY, P.S., BHAGYLAKSHMI, A., & RAMAMURTHI, R. (1985) Moit-inhibition
in the crab Oziotelphusa senex senex following exposure to
malathion and methyl parathion. Bull. environ. Contam. Toxicol., 35:
92-97.
REDDY, P.S., BHAGYLAKSHMI, A., & RAMAMURTHI, R. (1986a) Carbohydrate
metabolism in tissue of fresh water crab (Oziotelphusa senex senex)
exposed to methyl parathion. Bull. environ. Contam. Toxicol., 36:
204-210.
REDDY, P.S., BHAGYLAKSHMI, A., & RAMAMURTHI, R. (1986b) Changes in
acid phosphatase activity in tissues of crab, Oziotelphusa senex
senex, following exposure to methyl parathion. Bull. environ. Contam.
Toxicol., 37: 106-112.
REDDY, T.N. & REDDY, S.J. (1989) Voltammetric behaviour of some nitro
group containing pesticides. Indian J. environ. Protect., 9(8):
592-594.
REHWOLDT, R.E., KELLEY, E., & MAHONEY, M. (1977) Investigations into
the acute toxicity and some chronic effects of selected herbicides and
pesticides on several fresh water fish species. Bull. environ. Contam.
Toxicol., 18(3): 361-365.
RENHOF, M. (1984) [Methyl parathion (active principle of Folidol M):
Tests for embryotoxic effects in rabbits after oral administration.]
Wuppertal-Elberfeld, Bayer AG, Institute of toxicology (Unpublished
reports No. 12907 and 16331, submitted to WHO by Bayer AG, Leverkusen,
Germany) (in German).
RENVALL, S., LINSKOG, E., & CLEMENTZ, E. (1975) Residues of
organophosphorus pesticides in fruits and vegetables on the Swedish
market from July 1967 to April 1973. Presented at the IUPAC Third
International Congress of Pesticide Chemistry, Helsinki, 3-9 July
1974. Environ. Qual. Saf., Suppl., 3: 166-174.
RICE, C.P., OLNEY, C.E., & BIDLEMAN, T.F. (1977) Use of polyurethane
foam to collect trace amounts of chlorinated hydrocarbons and other
organics from air. WMO In: Air pollution measurement techniques.
Geneva, World Meteorological Organization, Part II, pp. 216-224
(Special Environmental Report No. 10; Publication WMO-No. 460).
RICHTER, E.D., ROSENVALD, Z., KASPI, L., LEVY, S., & GRUENER, N.
(1986) Sequential cholinesterase tests and symptoms for monitoring
organophosphate absorption in field workers and in persons exposed to
pesticide spray drift. Toxicol. Lett., 33: 25-35.
RIDER, J.A., MOELLER, H.C., PULETTI, E.J., & SWADER, J.I. (1969)
Toxicity of parathion, systox, octamethyl pyrophosphoramide, and
methyl parathion in man. Toxicol. appl. Pharmacol., 14: 603-611.
RIDER, J.A., SWADER, J.I., & PULETTI, E.J. (1970) Methyl parathion and
guthion anticholinesterase effects in human subjects. Fed. Proc., 29:
349 (Abstr. No.588).
RIDER, J.A., SWADER, J.I., & PULETTI, E.J. (1971) Anticholinesterase
toxicity studies with methyl parathion, guthion and phosdrin in human
subjects. Fed. Proc., 30: 1382.
RIPLEY, B.D. & BRAUN, H.E. (1983) Retention time data for
organochlorine, organophosphorus, and organonitrogen pesticides on
SE-30 capillary column and application of capillary gas
chromatography to pesticide residue analysis. J. Assoc. Off. Anal.
Chem., 66: 1084-1095.
RIPPEL, A., KOVAC, J., & SZOKOLAY, A. (1970) [Investigation of the
persistence of organophosphorus insecticides in citrus juices.]
Nahrung, 14(3): 223-228 (in German).
ROACH, J.A.G. & ANDRZEJEWSKI, D. (1987) Analysis for pesticide
residues by collision-induced fragmentation. In: Rosen, J.D., ed.
Application of new mass spectrometry techniques in pesticide
chemistry. New York, Chichester, Brisbane, Toronto, John Wiley and
Sons, pp. 187-210 (Chemical Analysis series, Volume 91).
ROARK, B., PFRIMMER, T.R., & MERKL, M.E. (1963) Effects of some
formulations of methyl parathion, toxaphene and DDT on the cotton
plant. Crop Sci., 3: 338-341.
ROBERTS, D.K., SILVEY, N.J., & BAILEY, E.M. Jr (1988) Brain
acetylcholinesterase activity recovery following acute methyl
parathion intoxication in two feral rodent species: comparison to
laboratory rodents. Bull. environ. Contam. Toxicol., 41: 26-35.
ROBINSON, S.C., ENDALL, R.J., & ROBINSON, R. (1988) Effects of
agricultural spraying of methyl parathion on cholinesterase activity
and reproductive success in wild starlings (Sturnus vulgaris).
Environ. Toxicol. Chem., 7(5): 343-349.
RODNITZKY, R.L., LEVIN, H.S., & MORGAN, D.P. (1978) Effects of
ingested parathion on neurobehavioral functions. Clin. Toxicol., 13:
347-359.
ROSS, B. & HARVEY, J. (1981) A rapid, inexpensive, quantitative
procedure for the extraction and analyses of Penncap-M (methyl
parathion) from honeybees ( Apis mellifera L.), beeswax, and pollen.
J. Agric. Food Chem., 29: 1095-1096.
ROYAL SOCIETY OF CHEMISTRY (1986) European directory of agrochemical
products. Volume 3: Insecticides and acaricides. Nottingham, The Royal
Society of Chemistry.
RUPA, D.S., REDDY, P.P., & REDDI, O.S. (1989) Chromosomal aberrations
in peripheral lymphocytes of cotton field workers exposed to
pesticides. Environ. Res., 49: 1-6.
RUZICKA, J.H., THOMSON, J., & WHEALS, B.B. (1967) The gas
chromatographic determination of organophosphorus pesticides. J.
Chromatogr., 31: 37-47.
SABHARWAL, A.K. & BELSARE, D.K. (1986) Persistence of methyl parathion
in a carp rearing pond. Bull. environ. Contam. Toxicol., 37: 705-709.
SADAR, M.H., KUAN, S.S., & GUIBAULT, G.G. (1970) Trace analysis of
pesticides using cholinesterase from human serum, rat liver, electric
eel, bean leaf beetle, and white fringe beetle. Anal. Chem., 42:
1770-1774.
SAGREDOS, A.N. & ECKERT, W.R. (1976) [Methods for the determination of
phytopharmaceuticals in tobacco and tobacco products. Part II.
Simultaneous determination of hexane-soluble organohosphorus
pesticides.] Beitr. Tabakforsch., 8: 447-454 (in German).
SALTZMAN, S., MINGELGRIN, U., & YARON, B. (1976) Role of water in the
hydrolysis of parathion and methylparathion on kaolinite. J. Agric.
Food Chem., 24(4): 739-743.
SANDERS, P.F. & SEIBER, J.N. (1983) A chamber for measuring
volatilization of pesticides from model soil and water disposal
systems. Chemosphere, 12: 999-1012.
SANDERSON, D.M. & EDSON, E.F. (1964) Toxicological properties of the
organophosphorus insecticide dimethoate. Br. J. ind. Med., 21: 52-64.
SAROJA-SUBBARAJ, G. & BOSE, S. (1982) Effects of methyl parathion on
the growth, cell size, pigment and protein content of Chlorella
protothecoides. Environ. Pollut., 27: 297.
SAROJA-SUBBARAJ, G. & BOSE, S. (1983a) Correlation between inhibition
of photosynthesis and growth of Chlorella treated with methyl
parathion. J. Biosci., 5: 71-78.
SAROJA-SUBBARAJ, G. & BOSE, S. (1983b) Binding characteristics of
methyl parathion to photosynthetic membranes of Chlorella. Pestic.
Biochem. Physiol., 20: 188-193.
SAROJA-SUBBARAJ, G. & BOSE, S. (1984) Recovery from methyl
parathion-induced damage of the photosynthetic apparatus in Chlorella
protothecoides. Bull. Environ. Contam. Toxicol., 32: 102-108.
SASTRY, C.S.P. & VIJAYA, D. (1986) Spectrophotometric determination of
fenitrothion and methyl parathion in insecticidal formulations. J.
Food Sci. Technol., 23: 336-338.
SASTRY, C.S.P. & VIJAYA, D. (1987) Spectrophotometric determination of
some insecticides with 3-methyl-2-benzothiazolinone hydrazone
hydrochloride. Talanta, 34: 372-374.
SAUVEGRAIN, P. (1980) Les micropolluants organiques dans les eaux
superficielles continentales. Rapport No. 1: Les pesticides
organophosphorés. Paris, Bureau National de l'Information Scientifique
et Technique.
SAXTON, W.L. (1987) Emergence temperature indexes and relative
retention times of pesticides and industrial chemicals determined by
linear programmed temperature gas chromatography. J. Chromatogr., 393:
175-194.
SAYRE, I.M. (1988) International standards for drinking water. J. Am.
Water Works Assoc., 80: 53-60.
SCHAFER, E.W. Jr (1972) The acute oral toxicity of 369 pesticidal,
pharmaceutical and other chemicals to wild birds. Toxicol. appl.
Pharmacol., 21: 315-330.
SCHILDE, B. & BOMHARD, E. (1984) [E 605-methyl (methyl parathion).
Supplementary histopathological test, further to the 2-year feeding
trial on rats. Addendum to report No. 9889.] Wuppertal-Elberfeld,
Bayer AG, Institute of Toxicology (Unpublished report No. 12559,
submitted to WHO by Bayer AG, Leverkusen, Germany) (in German).
SCHIMMEL, S.C., GARNAS, R.L., PATRICK, J.M. Jr, & MOORE, J.C. (1983)
Acute toxicity, bioconcentration and persistence of AC 222.705,
benthiocarb, chlorpyrifos, fenvalerate, methylparathion and permethrin
in the estuarine environment. J. Agric. Food Chem., 31(1): 104-113.
SCHNORBUS, R.R. & PHILLIPS, W.F. (1967) New extraction system for
residue analyses. J. Agric. Food Chem., 15: 661-666.
SCHOMBURG, C.J., GLOTFETTY, D.E., & SEIBER, J.N. (1991) Pesticide
occurrence and distribution in fog collected near Monterey,
California. Environ. Sci. Technol., 25: 155-160.
SCHOOR, W.P. & BRAUSCH, J. (1980) The inhibition of
acetylcholinesterase activity in pink shrimp Penaeus duorarum by
methyl-parathion and its oxon. Arch. environ. Contam. Toxicol., 9:
599-605.
SCHRADER, G. (1963) [The development of new insecticidal phosphoric
acid esters.] Weinheim, Verlag Chemie GmbH, pp. 223-258 (in German).
SCHULTEN, H.R. & SUN, S. E. (1981) High-resolution field desorption
mass spectrometry. Part IX. Field desorption mass spectrometry of
standard organophosphorus pesticides and their identification in waste
water. Int. J. Environ. Anal. Chem., 10: 247-263.
SCHULZ, H., DESI, I., & NAGYMAJTENYI, L. (1990) Behavioural effects of
subchronic intoxication with parathion-methyl in male Wistar rats.
Neurotoxicol. Teratol., 12: 125-127.
SCHULZE, J.A., MANIGOLD, D.B., & ANDREWS, F.L. (1973) Pesticides in
selected Western streams - 1968-71. Pestic. Monit. J., 7: 73-84.
SCHUTZ, H. & SCHINDLER, A. (1974) [Application of reaction
chromatography to chemical-toxicological analysis. Microchemical
detection and separation of sixteen nitropesticides by thin-layer
chromatography.] Fresenius Z. Anal. Chem., 270: 356-359 (in German).
SCHUTZMANN, R.L., WOODHAM, D.W., & COLLIER, C.W. (1971) Removal of
sulfur in environmental samples prior to gas chromatographic analysis
for pesticide residues. J. Assoc. Off. Anal. Chem., 54: 1117-1119.
SEIBER, J.N., McCHESNEY, M.M., & WOODROW, J.E. (1989) Airborne
residues resulting from use of methyl parathion, molinate and
thiobencarb on rice in the Sacramento Valley, California. Environ.
Toxicol. Chem., 8: 577-588.
SHAFIK, M.T. & ENOS, H.F. (1969) Determination of metabolic and
hydrolytic products of organophosphorus pesticide chemicals in human
blood and urine. J. Agric. Food Chem., 17: 1186-1189.
SHARMA, R.P. & REDDY, R.V. (1987) Toxic effects of chemicals on the
immune system. In: Haley, T.J. & Berndt, W.O., ed. Handbook of
toxicology. Washington, DC, Hemisphere Publishing Corporation, pp.
555-591.
SHARMA, V.K. JADHAV, R.K., RAO, G.J., SARAF, A.K., & CHANDRA, H.
(1990) High performance liquid chromatographic method for the analysis
of organophosphorus and carbamate pesticides. Forens. Sci. Int., 48:
21-25.
SHARMILA, M., RAMANAND, K., ADHYA, T.K., & SETHUNATHAN, N. (1988)
Temperature and the persistence of methyl parathion in a flooded soil.
Soil Biol. Biochem., 20: 399-401.
SHARMILA, M., RAMANAND, K., & SETHUNATHAN, N. (1989a) Hydrolysis of
methyl parathion in a flooded soil. Bull. environ. Contam. Toxicol.,
43: 45-51.
SHARMILA, M., RAMANAND, K., & SETHUNATHAN, N. (1989b) Effect of yeast
extract on the degradation of organophosphorus insecticides by soil
enrichment and bacterial cultures. Can. J. Microbiol., 35: 1105-1110.
SHERMA, J. & BRETSCHNEIDER, W. (1990) Determination of
organophosphorus insecticides in water by C-18 solid phase extraction
and quantitative TLC. J. liq. Chromatogr., 13(10): 1983-1989.
SHERMA, J. & SHAFIK, T.M. (1975) Multiclass, multiresidue analytical
method for determining pesticide residues in air. Arch. environ.
Contam. Toxicol., 3: 55-71.
SHIHIDO, T. & FUKAMI, J. (1963) Studies on the selective toxicities of
organic phosphorous insecticides (II), the degradation of ethyl
parathion, methyl parathion, methyl paraoxon, and sumithion in mammal,
insect and plant. Botyu-Kagaku, 28: 69-76.
SHIRES, S.W. (1985) A comparison of the effects of cypermethrin,
parathio-methyl and DDT on cereal aphids, predatory beetles,
earthworms and litter decomposition in spring wheat. Crop Prot., 4:
177-193.
SIMMON, V.F., MITCHELL, A.D., & JORGENSON, T.A. (1977) Evaluation of
selected pesticides as chemical mutagens in vitro and in vivo
studies. Menlo Park, California, Stanford Research Institute, 251 pp.
(PB-268647).
SINGH, K.S. & SINGH, A.P. (1978) Persistence of methyl and ethyl
parathion in soil and plant. Pesticides, 12: 39-41.
SINGH, N.N. & SRIVASTAVA, A.K. (1982) Toxicity of a mixture of aldrin
and formothion and other organophosphorus, organochlorine and
carbamate pesticides to the Indian catfish Heteropneustes fossilis.
Comp. Physiol. Ecol., 7(2): 115-118.
SINGH, S., LEHMANN-GRUBE, B., & GOEDDE, H.W. (1984) Cytogenetic
effects of paraoxon and methyl-parathion on cultured human
lymphocytes: SCE, clastogenic activity and cell cycle delay. Int.
Arch. occup. environ. Health, 54 (3): 195-200.
SMART, E. & STEVENSON, J.H. (1982) Laboratory estimation of toxicity
of pyrethroid insecticides to honey bees: relevance to hazard in the
field. Bee World, 63: 150-152.
SMITH, D.C., LEDUC, R., & TREMBLAY, L. (1975) Pesticide residues in
the total diet in Canada. IV. 1972 and 1973. Pestic. Sci., 6: 75-82.
SMITH, J.H., MABEY, W.R., BOHONOS, N., HOLT, B.R., LEE, S.S., CHOU,
T.-W., BOMBERGER, D.C., & MILL, T. (1978) Environmental pathways of
selected chemicals in freshwater systems, Part II: Laboratory studies.
Athens, Georgia, US Environmental Protection Agency (PB288511).
SMITH, S., WILLIS, G.H., MCDOWELL, L.L., & SOUTHWICK, L.M. (1987)
Dissipation of methyl parathion and ethyl parathion from cotton
foliage as affected by formulation. Bull. environ. Contam. Toxicol.,
39: 280-285.
SMYTH, M.R. & OSTERYOUNG, J.G. (1978) A pulse polarographic
investigation of parathion and some other nitro-containing pesticides.
Anal. chim. Acta, 96: 335-344.
SOBTI, R.C., KRISHAN, A., & PFAFFENBERGER, C.D. (1982) Cytokinetic and
cytogenetic effects of some agricultural chemicals on human lymphoid
cells in vitro: organophosphates. Mutat. Res., 102: 89-102.
SOLON, J.M. & NAIR, J.H. (1970) Effect of a sublethal concentration of
LAS (linear alkyl benzene sulfonate detergent) on the acute toxicity
of various phosphate pesticides to the fathhead minnow (Pimephales
promelas). Bull. environ. Contam. Toxicol., 5(5): 408-413.
SOMLYAY, I.M., VARNAGY, L.E., & PAVLISCSAK, Cs. (1989) Effect of
various pesticides on plasma biochemistry of chicken embryos. Mede.
Fac. Landbouwwet. Rijksuniv. Gent, 54(2a): 181-184.
SONOBE, H., SAKUMA, S., YAMADA, K., KAWASAKI, M., KATAYAMA, H., &
KUROIWA, Y. (1982) Analysis systems for trace compounds found in
barley, malt and hops. In: Proceedings of the 17th Convention, Perth,
7-12 March 1982. Sydney, Institute of Brewing, Australia and New
Zealand Section, pp. 56-69.
SPECHT, W. (1978) [Methods for the rapid work-up of large fat
quantities for analysis of pesticide residues.] Lebensmittelchem
Gerichtl. Chem., 32: 51-53 (in German).
SPECHT, W. & TILLKES, M. (1980) [Gas chromatographic determination of
pesticide residues after cleanup by gel chromatography and mini silice
gel column chromatographs.] Fresenius Z. Anal. Chem., 301: 300-307 (in
German).
SPENCER, E.Y. (1982) Guide to the chemicals used in crop protection,
7th ed. Ottawa, Agriculture Canada Research Institute, 394 pp.
(Publication No. 1093).
SPINGARN, N.E., NORTHINGTON, D.J., & PRESSELY, T. (1982) Analysis of
nonvolatile organic hazardous substances by GC/MS. J. Chromatogr.
Sci., 20: 571-574.
SRIVASTAVA, A.K. & SINGH, N.N. (1981) Effects of exposure to
methyl-parathion on carbohydrate metabolism in Indian catfish
Heteropneustes fossilis. Acta pharmacol. Toxicol., 48(1): 26-31.
STAHR, H.M., GAUL, M., HYDE, W., & MOORE, R. (1979) A cellulose column
cleanup for organophosphorus pesticides. Microchem J., 24: 97-101.
STAMPER, C.R., BALDUINI, W., MURPHY, S.D., & COSTA, L.G. (1988)
Behavioural and biochemical effects of postnatal parathion exposure in
the rat. Neurotoxicol. Teratol., 10: 261-266.
STAN, H.J. & GOEBEL, H. (1983) Automated capillary gas chromatographic
analysis of pesticide residues in food. J. Chromatogr., 268: 55-69.
STAN, H. & GOEBEL, H. (1984) Evaluation of automated splitless and
manual on-column injection techniques using capillary gas
chromatography for pesticide residue analysis. J. Chromatogr., 314:
413-420.
STAN, H.J. & MROWETZ, D. (1983) Residue analysis of organophosphorus
pesticides in food with two-dimensional gas chromatography using
capillary columns and flame photometric detection. J. high Resolut.
Chromatogr. Chromatogr. Commun., 6: 255-263.
STAN, H.J. & MUELLER, H.M. (1988) Evaluation of automated and manual
hot- splitless, cold-splitless (PTV), and on-column injection
technique using capillary gas chromatography for the analysis for
organophosphorus pesticides. J. high Resolut. Chromatogr. Chromatogr.
Commun., 11: 140-143.
STANLEY, C.W., BARNEY, J.E., II, HELTON, M.R., & YOBS, A.R. (1971)
Measurement of atmospheric levels of pesticides. Environ. Sci.
Technol., 5: 430-435.
STEINWANDTER, H. (1988) Contributions to the application of gel
chromatography in residue analysis. II. A new gel chromatographic
system using acetone for the separation of pesticide residues and
industrial chemicals. Fresenius Z. Anal. Chem., 331: 499-502.
STEPHENSON, R.R. & KANE, D.F. (1984) Persistence and effects of
chemicals in small enclosures in ponds. Arch. environ. Contam.
Toxicol., 13: 313-326.
STOLL, K. (1982) Residue breakdown in stored fruit. In: Proceedings of
the 21th International Horticultural Congress, Hamburg, 29 August-4
September 1982. The Hague, International Society for Horticultural
Science, pp. 231-235.
STORHERR, R.W., MURRAY, E.J., KLEIN, I., & ROSENBERG, L.A. (1967)
Sweep destillation cleanup of determining of organophosphate and
chlorinated hydrocarbon pesticides. J. Assoc. Off. Anal. Chem., 50:
605-615.
STREET, J.C. & SHARMA, R.P. (1975) Alteration of induced cellular and
humoral immune response by pesticides and chemicals of environmental
concern: quantitative studies of immunosuppression by DDT, aroclor
1254, carbaryl, carbofuran, and methylparathion. Toxicol. appl.
Pharmacol., 32: 587-602.
SULTATOS, L.G. (1987) The role of the liver in mediating the acute
toxicity of the pesticide methyl parathion in the mouse. Drug Metab.
Dispos., 15(5): 613-617.
SULTATOS, L.G. & WOODS, L. (1988) The role of glutathione in the
detoxification of the insecticides methyl parathion and
azinphos-methyl in the mouse. Toxicol. appl. Pharmacol., 96: 168-174.
SULTATOS, L.G., KIM, B., & WOODS, L. (1990) Evaluation of estimations
in vitro of tissue/blood distribution coefficients of
organothiophosphate insecticides. Toxicol. appl. Pharmacol., 103:
52-55.
SUPROCK, J.F. & VINOPAL, J.H. (1987) Behavior of 78 pesticides and
pesticide metabolites on four different ultra-bond gas chromatographic
columns. J. Assoc. Off. Anal. Chem., 70: 1014-1017.
SWAMY, G.S. & VEERESH, A.V. (1987) Methylparathion modulates lipid
biosynthesis in sorghum ( Sorghum bicolor L.). Moench. Pestic.
Biochem. Physiol., 28: 341-348.
SWINEFORD, D.M. & BELISLE, A.A. (1989) Analysis of trifluralin, methyl
paraoxon, methyl parathion, fenvalerate and 2,4-D dimethylamine in
pond water using solid-phase extraction. Environ. Toxicol. Chem., 8:
465-468.
TAKIMOTO, Y., OHSHIMA, M., YAMADA, H., & MIYAMOTO, J. (1984) Fate of
fenithrothion in several developmental stages of the killifish
(Oryzias latipes). Arch. environ. Contam. Toxicol., 13: 579-587.
TANIMURA, T., KATSUYA, T., & NISHIMURA, H. (1967) Embryotoxicity of
acute exposure to methyl parathion in rats and mice. Arch. environ.
Health, 15: 609-613.
TAYLOR, P. (1980) Anticholinesterase agents. In: Gilman, A.G.,
Goodman, L.S., & Gilman, A., ed. The pharmacological basis of
therapeutics, 6th ed. New York, Macmillan Publishing Co., pp.
100-119.
TESSARI, J.D. & SPENCER, D.L. (1971) Air sampling for pesticides in
the human environment. J. Assoc. Off. Anal. Chem., 54: 1376-1382.
THIELEMANN, H. (1974) [Semiquantitative thin layer chromatographic
determination of parathion-methyl.] Z. Chem., 14: 407-408 (in German).
THIER, H-P. & ZEUMER, H. (1987) [Pesticides Commission of the German
Association for the Encouragement of Research. Manual of pesticide
residue analysis.] Weinheim, VCH Verlag, vol. 1.
THOMAS, P.T. & HOUSE, R.V. (1989) Pesticide-induced modulation of the
immune system. In: Ragsdale, N.N. & Menzer, R.E., ed. Carcinogenicity
and pesticides: principles, issues and relationships. 196th National
Meeting of the American Chemical Society, Los Angeles, California,
25-30 September 1988 Washington, DC, American Chemical Society, pp.
94-106 (ACS Symposium series, 414).
THOMPSON, A.R. & GORE, F.L. (1972) Toxicity of twenty-nine
insecticides to Folsomia candida: laboratory studies. J. Econ.
Entomol., 65: 1255-1260.
THUMA, N.K., O'NEILL, P.E., BROWNLEE, S.G., & VALENTINE, R.S. (1983)
Microbial degradation of selected harzardous materials:
pentachlorophenol, hexachlorocyclopentadiene and methyl parathion,
Washington, DC, US Environmental Protection Agency, 76 pp.
(EPA-Report No. 68-03-2491).
THYSSEN, J. (1979) [E 120 (methyl parathion): Studies on acute
inhalation toxicity.] Wuppertal-Elberfeld, Bayer AG, Institute of
Toxicology (Unpublished report No. 8148, submitted to WHO by Bayer AG,
Leverkusen, Germany) (in German).
THYSSEN, J. & MOHR, U. (1982) [E 120 (methyl parathion): Subacute
inhalation test on rats - histopathological findings.]
Wuppertal-Elberfeld, Bayer AG, Institute of Toxicology (Unpublished
report No. 11302, submitted to WHO by Bayer AG, Leverkusen, Germany)
(in German).
TILSTONE, W.J., WINCHESTER, J.F., & REAVEY, P.C. (1979) The use of
pharmacokinetics principles in determining the effectiveness of
removal of toxins from blood. Clin. Pharmacokinet., 4: 23-37.
TOSCANO, N.C., SANCES, F.V., JOHNSON, M.W., & LaPRE, L.F. (1982)
Effect of various pesticides on lettuce physiology and yield. J. Econ.
Entomol., 75: 738-741.
TRIPATHI, G. & SHUKLA, S.P. (1988) Inhibition of liver and skeletal
muscle enzymes by methyl parathion. Biochem. Arch., 4: 55-61.
TRIPATHI, G. & SHUKLA, S.P. (1990) Enzymatic and ultrastructural
studies in a freshwater catfish: impact of methyl parathion. Biomed.
Environ. Sci., 3: 166-182.
TRIPATHY, N.K., DEY, L., & MAJHI, B. (1987) Genotoxicity of metacid
established through the somatic and germ line mosai assays and the
sex-linked recessive lethal test in Drosophila. Arch. Toxicol.,
61(1): 53-57.
US EPA (1981) Acephate, aldicarb, carbophenothion, DEF, EPN, ethoprop,
methyl parathion and phorate; their acute and chronic toxicity,
bioconcentration potential and persistence as related to marine
environments. Gulf Breeze, Florida, US Environmental Protection
Agency, Office of Research and Development, Environmental Research
Laboratory, 275 pp. (EPA-600/4-81-041; PB 81-244477).
VAN BAO, T, SZABO, I., RUZICSKA, P., & CZEIZEL, R. (1974) Chromosome
aberrations in patients suffering acute organic phosphate insecticide
intoxication. Humangenetik., 24: 33-57.
VAN VELD, P.A. & SPAIN, J.C. (1983) Degradation of selected xenobiotic
compounds in three types of aquatic test systems. Chemosphere, 12:
1291-1305.
VARIS, A.-L. (1972) Loss of lindane, dimethoate, and methyl parathion
residues from seedlings of sugar beet as influenced by plant growth.
Ann. Agric. Fenn., 11: 381-385.
VARNAGY, L. & DELI, E. (1985) Comparative teratological study of
insecticide Wofatox 50 EC (50% methyl parathion) on chicken and
pheasant fetuses. Anat. Anz. Jena, 158: 1-3.
VARNAGY, L., KORZENSZKY, M., & FANCSI, T. (1984) Teratological
examination of the insecticide methylparathion/Wofatox 50 EC/ on
pheasant embryos. 1. Morphological study. Vet. Res. Commun., 8:
131-139.
VARNAGY, L., DELI, E., & BAUMANN, M. (1985) Scanning electron
microscopic examination of cartilage in chicken embryos treated with
the insecticide Wofatox 50 EC. Acta vet. Hung., 36:(1-2): 117-121.
VOGELGESANG, J. & THIER, H.P. (1986) [Contributions to the analysis of
pesticide residues in ready-to-eat foods.] Z. Lebensm.-Unters.
Forsch., 182(5): 400-406 (in German).
VROCHINSKY, K.K. & MAKOVSKY, V.N. (1977) The sources of pollution and
the content of pesticides in air. In: Uses of pesticides and
environmental protection. Kiev, Vysshaya Shkola, pp. 143-154.
WALKER, T.W., MEEK, C.L., WRIGHT, V.L., & BILLODEAUX, J.S. (1985)
Susceptibility of Romanomermis culicivorax (Nematoda: Mermithidae)
post parasites to agrochemicals used in Louisiana rice production. J.
Am. Control Assoc., 1: 477-481.
WALKER, W.W. (1978) Insecticide persistence in natural sea water as
affected by salinity, temperature and sterility. Washington, DC, US
Environmental Protection Agency, Office of Research Development, 25
pp. (EPA-600/3-78-044).
WALSH, G.E. & ALEXANDER, S.V. (1980) A marine algal bioassay method:
results with pesticides and industrial wastes. Water, Air, Soil
Pollut., 13: 45-55.
WALSH, G.E., DEANS, C.H., & McLAUGHLIN, L.L. (1987) Comparison of the
EC50s of algal toxicity tests calculated by four methods. Environ.
Toxicol. Chem., 6: 767-770.
WALTERS, S.M. (1990) Clean-up techniques for pesticides in fatty
foods. Anal. chim. acta, 236: 77-82.
WARE, G.W., ESTESEN, B., & CAHILL, W.P. (1974a) Dislodgable leaf
residues of insecticides on cotton. Bull. environ. Contam. Toxicol.,
11: 434-437.
WARE, G.W., MORGAN, D.P., ESTESEN, B.J., & CAHILL, W.P. (1974b)
Establishment of reentry intervals for organophosphate-treated cotton
fields based on human data. II. Azodrin, ethyl and methyl parathion.
Arch. environ. Contam. Toxicol., 2(2): 117-129.
WARE, G.W., WATSON, T.F., ESTESEN, B., & BUCK, N.A. (1980) Effects of
molasses or toxaphene on residual life and efficacy of methyl
parathion on cotton. J. Econ. Entomol., 73: 15-17.
WARE, G.W., BUCK, N.A., & ESTESEN, B.J. (1983) Dislodgeable
insecticide residues on cotton foliage: comparison of ULV/cottonseed
oil vs. aqueous dilutions of 12 insecticides. Bull. environ. Contam.
Toxicol., 31: 551-558.
WARNER, J.S. (1975) Identification on impurities in technical-grade
pesticides. In: Substitute Chemical Programme - the first year of
progress. Proceedings of a symposium. Vol. IV: Chemical methods
workshop. Washington DC, Bureau of Commerce, National Technical
Information Service, pp. 31-65 (PB 261-007).
WATTS, R.R. & STORHERR, R.W. (1967) Sweep codistillation cleanup of
milk for determination of organophosphate and chlorinated hydrocarbon
pesticides. J. Assoc. Off. Anal. Chem., 50: 581-585.
WEBER, H., PATZSCHKE, K., & WEGNER, L.A. (1979) [Methyl
parathion-14C (benzene ring-labelled compound): Biokinetic studies
on rats.] Wuppertal-Elberfeld, Bayer AG, Pharmokinetics Institute,
Isotope Laboratory (Unpublished report No. Pharma-8722, submitted to
WHO by Bayer AG, Leverkusen, Germany) (in German).
WEGMAN, R.C.C., VAN DEN BROEK, H.H., HOFSTEE, A.W.M., & MARSMAN, J.A.
(1984) Determination of triazines, organophosphorus containing
pesticides and aromatic amines in soil samples. Meded. Fac.
Landbouwwet. Rijksuniv. Gent., 49: 1231-1239.
WEHNER, T.A., WOODROW, J.E., KIM, Y.H., & SEIBER, J.N. (1984)
Multiresidue analysis of trace organic pesticides in air. In: Keith,
L.H., ed. Identification and analysis of organic pollutants. Boston,
Butterworth, pp. 273-290.
WERNER, D. & PAWLITZ, H. (1978) Differential elimination of phenol by
diatoms and other unicellular algae from low concentrations. Bull.
environ. Contam. Toxicol., 20: 303-312.
WESSEL, J.R. (1967) Collaborative study of a method for multiple
organophosporus pesticide residues in nonfatty foods. J. Assoc. Off.
Anal. Chem., 50: 430-439.
WEST, B., VIDONE, L.B., & SHAFFER, C.B. (1961) Acute and subacute
toxicity of dimethoate. Toxicol. appl. Pharmacol., 3: 210-223.
WHITE-STEVENS, R. (1971) Pesticides in the environment, New York,
Basel, Marcel Dekker, Inc., vol. 1., parts 1 and 2.
WHO (1990) The WHO recommended classification of pesticides by hazard
and guidelines to classification, 1990-91. Geneva, World Health
Organization (Unpublished document WHO/PCS/90.1).
WHO (1986) Environmental Health Criteria, 63: Organophosphorus
insecticides: a general introduction. Geneva, World Health
Organization, 181 pp.
WHO/FAO (1975) Data sheets on pesticides No. 7, Rev. 1:
Parathion-methyl. Geneva, World Health Organization (Unpublished WHO
document VBC/05/75.7 (Rev. 1)).
WICKER, G.W., WILLIAMS, W.A., & GUTHRIE, F.E. (1979) Exposure of field
workers to organophosphorus insecticides: sweet corn and peaches.
Arch. environ. Contam. Toxicol., 8: 175-182.
WIERSMA, G.B., MITCHELL, W.G., & STANFORD, C.L. (1972) Pesticide
residues in onions and soil - 1969. Pestic. Monit. J., 5(4): 345-347.
WILD, D. (1975) Mutagenicity studies on organophosphorus insecticides.
Mutat. Res., 32: 133-150.
WILKINS, J.P.G. (1990) Rationalization of the mass spectrometric and
gas chromatographic behaviour of organophosphorus pesticides: Part 1
- Substituted phenyl phosphorothioates. Pest. Sci., 29: 163-181.
WILLEMS, J., BRAECKMAN, R., & BELPAIRE, F. (1980) Fate of methyl
parathion in the dog. Toxicol. Lett., 5(Suppl. 1): 34 (Abstract No.
038).
WILLIAMS, M.W., FUYAD, J.P., FRAWLEY, J.P., & FITZHUGH, O.G. (1957)
In vivo effects of paired combinations of five organic phosphate
insecticides. J. Agric. Food Chem., 6: 514-516.
WILLIAMS, M.W., FUYAT, H.N., & FITZHUGH, O.G. (1959) The subacute
toxicity of four organic phosphates to dogs. Toxicology, 1: 1-7.
WILLIS, G.H., McDOWELL, L.L., SOUTHWICK, L.M., & SMITH, S. (1985)
Toxaphene, methyl parathion, and fenvalerate disapperance from cotton
foliage in the mid-south. J. environ. Qual., 14(3): 446-450.
WILMES, R. (1987) Parathion-methyl: Hydrolysis studies. Leverkusen,
Germany, Bayer AG, Institute of Metabolism Research, 34 pp.
(Unpublished report No. PF 2883, submitted to WHO by Bayer AG).
WINDHOLZ, M., BUDVARI, S., BLUMETTI, R.F., & OTTERBEIN, E.S. ed.
(1983) The Merck Index, 10th ed. Rahway, New Jersey, Merck and Co.,
874 pp.
WINTER, H. & LINDNER, K. (1987) [Drinking water of river Rhine after
the "Sandoz accident".] Wasser Abwasser, 128: 525-532 (in German).
WOLFE, H.R., DURHAM, W.F., & ARMSTRONG, J.F. (1967) Exposure of
workers to pesticides. Arch. environ. Health, 14: 622-633.
WOLFE, H.R., WASH, W., DURHAM, W.F., & ARMSTRONG, J.F. (1970) Urinary
excretion of insecticide metabolites. Excretion of para-nitrophenol
and DDA as indicators of exposure to parathion. Arch. environ. Health,
21: 711-716.
WOLFE, N.L., KITCHENS, B.E., MACALDY, D.L., & GRUNDL, T.J. (1986)
Physical and chemical factors that influence the anaerobic degradation
of methyl parathion in sediment systems. Environ. Toxicol. Chem., 5:
1019-1026.
WOODER, M.F., WRIGHT, A.S., & KING, L.J. (1977) In vivo alkylation
studies with dichlorvos at practical use concentrations. Chem.-Biol.
Interact., 19: 25-46.
WOODROW, J.E., SEIBER, J.N., CROSBY, D.G., MOILANEN, K.W., SODERQUIST,
C.J., & MOURER, C. (1977) Airborne and surface residues of parathion
and its conversion products in a treated plum orchard environment.
Arch. Environ. Contam. Toxicol., 6: 175-191.
WORTHING, C.R. & HANCE, R.J. (1990) The pesticide manual, 9th ed.
Croydon, British Crop Protection Council.
XUE, J. (1984) Resin concentration method for determination of
pesticides in drinking water. Huanjing Huaxue, 3: 43-49.
YAMAMOTO, T., EGASHIRA, T., YOSHIDA, T., & KUROIWA, Y. (1983)
Comparative metabolism of fenitrothion and methylparathion in male
rats. Acta pharmacol. toxicol., 53: 96-102.
YANG, C.-F. & SUN, Y.-P. (1977) Partition distribution of insecticides
as a critical factor affecting their rates of absorption from water
and relative toxicities to fish. Arch. environ. Contam. Toxicol., 6:
325-335.
YASOSHIMA, M. & MASUDA, Y. (1986) Effect of carbon disulfide on the
anticholinesterase action of several organophosphorus insecticides in
mice. Toxicol. Lett., 32: 179-184.
YASUNO, M., HIRAKOSO, S., SASA, M., & UCHIDA, M. (1965) Inactivation
of some organophosphorus insecticides by bacteria in polluted water.
Jpn. J. exp. Med., 35(6): 545-563.
YODER, J., WATSON, M., & BESON, W.W. (1973) Lymphocyte chromosome
analysis of agricultural workers during extensive occupational
exposure to pesticides. Mutat. Res., 21: 335-340.
YOUNGMAN, R.R., TOSCANO, N.C., & GASTON, L.K. (1989) Degradation of
methyl parathion to p-nitrophenol on cotton and lettuce leaves and
its effects on plant growth. J. Econ. Entomol., 82: 1317-1322.
YOUNGMAN, R.R., LEIGH, T.F., KERBY, T.A., TOSCANO, N.C., & JACKSON,
C.E. (1990) Pesticides and cotton: effect on photosynthesis, growth,
and fruiting. J. Econ. Entomol., 83: 1549-1557.
YOUSSEF, S.H.A., EL-SAYED, M.G.A., & ATEF, M. (1987) Influence of
gentamicin and rifamycin on toxicity and biotransformation of methyl
parathione in rats. Dtsch. Tierärztl. Wochenschr., 94: 193-236.
ZEPP, R.G. & SCHLOTZHAUER, P.F. (1983) Influence of algae on
photolysis rates of chemicals in water. Environ. Sci. Technol., 17:
462-468.
ZHAO, S. & WANG, Y. (1984) [High-performance liquid chromatographic
analysis of trichlorophon and methylparathionmixed powder.] Sepu, 1:
134-135 (in Chinese).
ZIETEK, M. (1976) [Polarographic determination of parathion and
similar insecticides alongside their metabolites in blood without an
extraction procedure.] Mikrochim. Acta, 2: 249-257 (in German).
ZITKO, V. & MCLEESE, D.W. (1980) Evaluation of hazards of pesticides
used in forest spraying to the aquatic environment. St. Andrews, New
Brunswick, Biological Station (Canadian Technical Report, Fish and
Aquatic Sciences, No. 985).
ZWEIG, G. & DEVINE, J.M. (1969) Determination of organophosphorus
pesticides in water. Residue Rev., 26: 17-36.
ANNEX I. TREATMENT OF ORGANOPHOSPHATE INSECTICIDE POISONING IN MAN
(From EHC 63: Organophosphorus insecticides - a general introduction)
All cases of organophosphorus poisoning should be dealt with as
an emergency and the patient sent to hospital as quickly as possible.
Although symptoms may develop rapidly, delay in onset or a steady
increase in severity may be seen up to 48 h after ingestion of some
formulated organophosphorus insecticides.
Extensive descriptions of treatment of poisoning by
organophosphorus insecticides are given in several major references
(Kagan, 1977; Taylor, 1980; UK DHSS, 1983; Plestina, 1984) and will
also be included in the IPCS Health and Safety Guides to be prepared
for selected organophosphorus insecticides.
The treatment is based on:
(a) minimizing the absorption;
(b) general supportive treatment; and
(c) specific pharmacological treatment.
I.1 Minimizing the absorption
When dermal exposure occurs, decontamination procedures include
removal of contaminated clothes and washing of the skin with alkaline
soap or with a sodium bicarbonate solution. Particular care should be
taken in cleaning the skin area where venepuncture is performed. Blood
might be contaminated with direct-acting organophosphorus esters and,
therefore, inaccurate measures of ChE inhibition might result.
Extensive eye irrigation with water or saline should also be
performed. In the case of ingestion, vomiting might be induced, if the
patient is conscious, by the administration of ipecacuanha syrup
(10-30 ml) followed by 200 ml water. This treatment is, however,
contraindicated in the case of pesticides dissolved in hydrocarbon
solvents. Gastric lavage (with addition of bicarbonate solution or
activated charcoal) can also be performed, particularly in unconscious
patients, taking care to prevent aspiration of fluids into the lungs
(i.e., only after a tracheal tube has been put into place).
The volume of fluid introduced into the stomach should be
recorded and samples of gastric lavage frozen and stored for
subsequent chemical analysis. If the formulation of the pesticide
involved is available, it should also be stored for further analysis
(i.e., detection of toxicologically relevant impurities). A purgative
can be administered to remove the ingested compound.
I.2 General supportive treatment
Artificial respiration (via a tracheal tube) should be started at
the first sign of respiratory failure and maintained for as long as
necessary.
Cautious administration of fluids is advised, as well as general
supportive and symptomatic pharmacological treatment and absolute
rest.
I.3 Specific pharmacological treatment
I.3.1 Atropine
Atropine should be given, beginning with 2 mg iv and given at
15-30-min intervals. The dose and the frequency of atropine treatment
varies from case to case, but should maintain the patient fully
atropinized (dilated pupils, dry mouth, skin flushing, etc.).
Continuous infusion of atropine may be necessary in extreme cases and
total daily doses up to several hundred mg may be necessary during the
first few days of treatment.
I.3.2 Oxime reactivators
Cholinesterase reactivators (e.g., pralidoxime, obidoxime)
specifically restore AChE activity inhibited by organophosphates. This
is not the case with enzymes inhibited by carbamates. The treatment
should begin as soon as possible, because oximes are not effective on
"aged" phosphorylated ChEs. However, if absorption, distribution, and
metabolism are thought to be delayed for any reasons, oximes can be
administered for several days after intoxication. Effective treatment
with oximes reduces the required dose of atropine. Pralidoxime is the
most widely available oxime. A dose of 1 g pralidoxime can be given
either im or iv and repeated 2-3 times per day or, in extreme cases,
more often. If possible, blood samples should be taken for AChE
determinations before and during treatment. Skin should be carefully
cleansed before sampling. Results of the assays should influence the
decision whether to continue oxime therapy after the first 2 days.
There are indications that oxime therapy may possibly have
beneficial effects on CNS-derived symptoms.
I.3.3 Diazepam
Diazepam should be included in the therapy of all but the mildest
cases. Besides relieving anxiety, it appears to counteract some
aspects of CNS-derived symptoms that are not affected by atropine.
Doses of 10 mg sc or iv are appropriate and may be repeated as
required (Vale & Scott, 1974). Other centrally acting drugs and drugs
that may depress respiration are not recommended in the absence of
artificial respiration procedures.
I.3.4 Notes on the recommended treatment
I.3.4.1 Effects of atropine and oxime
The combined effect far exceeds the benefit of either drug
singly.
I.3.4.2 Response to atropine
The response of the eye pupil may be unreliable in cases of
organophosphorus poisoning. A flushed skin and drying of secretions
are the best guide to the effectiveness of atropinization. Although
repeated dosing may well be necessary, excessive doses at any one time
may cause toxic side-effects. Pulse-rate should not exceed 120/min.
I.3.4.3 Persistence of treatment
Some organophosphorus pesticides are very lipophilic and may be
taken into, and then released from, fat depots over a period of many
days. It is therefore quite incorrect to abandon oxime treatment after
1-2 days on the supposition that all inhibited enzyme will be aged.
Ecobichon et al. (1977) noted prompt improvement in both condition and
blood-ChEs in response to pralidoxime given on the 11th-15th days
after major symptoms of poisoning appeared due to extended exposure to
fenitrothion (a dimethyl phosphate with a short half-life for aging of
inhibited AChE).
I.3.4.4 Dosage of atropine and oxime
The recommended doses above pertain to exposures, usually for an
occupational setting, but, in the case of very severe exposure or
massive ingestion (accidental or deliberate), the therapeutic doses
may be extended considerably. Warriner et al. (1977) reported the case
of a patient who drank a large quantity of dicrotophos, in error,
while drunk. Therapeutic dosages were progressively increased up to 6
mg atropine iv every 15 min together with continuous iv infusion of
pralidoxime chloride at 0.5 g/h for 72 h, from days 3 to 6 after
intoxication. After considerable improvement, the patient relapsed and
further aggressive therapy was given at a declining rate from days 10
to 16 (atropine) and to day 23 (oxime), respectively. In total, 92 g
of pralidoxime chloride and 3912 mg of atropine were given and the
patient was discharged on the thirty-third day with no apparent
sequelae.
References to Annex I.
ECOBICHON, D.J., OZERE, R.L., REID, E., & CROCKER, J.F.S (1977) Acute
fenitrothion poisoning. Can. Med. Assoc. J., 116: 377-379.
KAGAN, JU.S. (1977) [ Toxicology of organophosphorus pesticides,]
Moscow, Meditsina, pp. 111-121, 219-233, 260-269 (in Russian).
PLESTINA, R. (1984) Prevention, diagnosis, and treatment of
insecticide poisoning, Geneva, World Health Organization
(Unpublished document VBC/84.889).
TAYLOR, P. (1980) Anticholinesterase agents. In: Goodman, L.S. &
Gilman, A., ed. The pharmacological basis of therapeutics, 6th ed.,
New York, Macmillan Publishing Company, pp. 100-119.
UK DHSS (1983) Pesticide poisoning: notes for the guidance of medical
practitioners, London, United Kingdom Department of Health and Social
Security, pp. 41-47.
VALE, J.A. & SCOTT, G.W. (1974) Organophosphorus poisoning. Guy's
Hosp. Rep., 123: 13-25.
WARRINER, R.A., III, NIES, A.S., & HAYES, W.J., Jr (1977) Severe
organophosphate poisoning complicated by alcohol and terpentine
ingestion. Arch. environ. Health, 32: 203-205.
RESUME ET EVALUATION, CONCLUSIONS, RECOMMANDATIONS
1 Résumé et évaluation
1.1 Exposition
Le parathion-méthyl est un insecticide organophosphoré dont la
première synthèse remonte aux années 1940. Il est relativement
insoluble dans l'eau, peu soluble dans l'éther de pétrole et les
huiles minérales et facilement soluble dans la plupart des solvants
organiques. A l'état pur, il se présente sous la forme de cristaux
blancs; le parathion-méthyl technique est légèrement jaunâtre et
dégage une odeur alliacée. Il est instable à la chaleur et se
décompose rapidement au-dessus de pH 8.
La chromatographie en phase gazeuse avec détection par ionisation
de flamme alcaline (AFID) ou photométrie de flamme (FPD) est la
méthode la plus couramment utilisée pour le dosage du
parathion-méthyl. Les limites de détection dans l'eau vont de 0,01 à
0,1 µg/litre; dans l'air, elles vont de 0,1 à 1 ng/m3. La
chromatographie en phase liquide à haute performance et la
chromatographie en couche mince sont également utiles pour la
recherche du parathion-méthyl.
La distribution du parathion-méthyl dans l'air, l'eau, le sol et
les êtres vivants dépend de plusieurs facteurs physiques, chimiques et
biologiques.
Les études utilisant des modèles d'écosystèmes ainsi que des
modèles mathématiques montrent que le parathion-méthyl se partage
principalement entre l'air et le sol dans l'environnement, une plus
faible proportion se répartissant entre les végétaux et les animaux.
Il ne se déplace pratiquement pas dans le sol et ni le composé
initial, ni ses produits de décomposition n'atteignent normalement les
eaux souterraines. Le parathion-méthyl présent dans l'air provient
principalement de l'épandage de ce composé, encore qu'il puisse se
volatiliser en partie lorsque l'eau qui le contient s'évapore de la
surface des feuilles et du sol. Les niveaux atmosphériques de fond
dans les zones agricoles vont de zéro (non décelable) à environ 70
ng/m3. Les concentrations dans l'air après épandage diminuent
rapidement en trois jours pour atteindre le niveau de fond au bout
d'environ neuf jours. Dans les cours d'eau, les concentrations (études
de laboratoire) tombent à 80% de la concentration initiale au bout
d'une heure et à 10 % au bout d'une semaine. Le parathion-méthyl
demeure plus longtemps dans le sol que dans l'air ou l'eau encore que
sa rétention dépende en grande partie du type de sol; dans les sols
sableux, les résidus de parathion-méthyl disparaissent plus rapidement
que dans le terreau. Les résidus présents à la surface des plantes ou
dans les feuilles diminuent rapidement avec une demi-vie de l'ordre de
quelques heures; la disparition totale du parathion-méthyl s'effectue
en six à sept jours environ.
L'organisme animal est capable de décomposer le parathion-méthyl
et d'en éliminer les produits de dégradation en très peu de temps. Ce
processus est plus lent chez les vertébrés inférieurs et les
invertébrés que chez les mammifères et les oiseaux. Les facteurs de
bioconcentration sont faibles et le parathion-méthyl ne s'accumule que
temporairement.
C'est la dégradation microbienne qui est de loin la voie la plus
importante de dégradation du parathion-méthyl dans le milieu. Le
composé disparaît plus rapidement sur le terrain ou dans des modèles
d'écosystèmes que ne l'avaient laissé entrevoir les études de
laboratoire. Cela tient au fait qu'il existe plusieurs microorganismes
capables de décomposer cette substance dans diverses circonstances et
dans différents biotopes. La présence de sédiments ou de surfaces
végétales qui accroît les populations microbiennes, augmente la
vitesse de décomposition du parathion-méthyl.
Sous l'action du rayonnement ultra-violet ou de la lumière
solaire, le parathion-méthyl peut subir une décomposition oxydante en
paraoxon-méthyl, moins stable; après pulvérisation, le temps de
demi-décomposition par le rayonnement ultra-violet est d'environ 40
heures. Toutefois, la contribution de la photolyse à l'élimination
totale dans un système aquatique, n'est, selon les estimations, que de
4 %. L'hydrolyse du parathion-méthyl se produit également plus
rapidement en milieu alcalin. Une forte salinité favorise aussi
l'hydrolyse. En présence de sédiments fortement réducteurs, on a noté
des demi-vies de quelques minutes, encore que la sorption à d'autres
sédiments accroisse la stabilité du composé.
Dans des villes situées au centre de zones agricoles des
Etats-Unis d'Amérique, on a observé que les concentrations de
parathion-méthyl dans l'air variaient avec la saison et culminaient en
août ou septembre; les enquêtes ont révélé que les teneurs maximales
se situaient principalement dans les limites de 100 à 800 ng/m3 au
cours de la période de végétation. Dans les eaux naturelles de ces
mêmes régions des Etats-Unis, on a observé des concentrations allant
jusqu'à 0,46 µg/litre, les maxima étant atteints en été. Il n'existe
qu'un petit nombre de publications sur les résidus alimentaires de
parathion-méthyl dans le monde. Aux Etats-Unis, ces résidus se situent
en général à un très faible niveau, même si quelques échantillons
dépassent les limites maximales de résidus (LMR). Les études de ration
totale dont il est fait état dans la littérature ne font état que de
traces de résidus. C'est dans les légumes-feuilles (jusqu'à 2 mg/kg)
et les légumes racines (jusqu'à 1 mg/kg) que l'on a constaté les
résidus les plus élevés lors d'enquêtes sur le panier de la ménagère
effectuées aux Etats-Unis entre 1966 et 1969. La préparation, la
cuisson et la conservation des aliments entraînent la décomposition
des résidus de parathion-méthyl, ce qui réduit encore l'exposition des
consommateurs. En cas d'erreurs de manipulation du parathion-méthyl,
on peut trouver des résidus plus élevés dans les légumes et les fruits
crus. la production, la formulation, la manipulation et l'utilisation
du parathion-méthyl comme insecticide sont les principales sources
potentielles d'exposition humaine. C'est principalement par contact
cutané et, dans une moindre proportion, par inhalation que les
travailleurs sont exposés à cette substance.
Lors d'une étude sur des ouvriers agricoles qui pulvérisaient du
parathion-méthyl (les ouvriers non protégés procédant à un épandage
manuel de cette substance à très bas volume), on a calculé que ces
personnes absorbaient 0,4 à 13 mg de parathion-méthyl par 24 heures en
se fondant sur le dosage du p-nitrophénol dans les urines. Si les
ouvriers reviennent trop tôt sur les lieux après le traitement, ils se
trouvent encore davantage exposés.
La population générale peut être exposée à des résidus présents
dans l'air, l'eau et les aliments par suite de traitements sur les
cultures ou les forêts ou d'erreurs de manipulation (épandage en
dehors de la zone à traiter) qui entraînent la contamination des
champs, des cultures, de l'eau et de l'air.
1.2 Fixation, métabolisme et excrétion
Le parathion-méthyl est facilement absorbé par toutes les voies
d'exposition (orale, percutanée, respiratoire) et il se répand
rapidement dans les tissus de l'organisme. Les concentrations
maximales dans les divers organes ont été observées une à deux heures
après le traitement. La conversion du parathion-méthyl en
paraoxon-méthyl se produit dans les minutes qui suivent
l'administration. Après administration de parathion-méthyl par voie
intraveineuse à des chiens, on a observé une demi-vie terminale
moyenne de 7,2 heures. C'est le foie qui joue le principal rôle dans
le métabolisme et la détoxication du parathion-méthyl. Le mode
principal de détoxication du parathion-méthyl et du paraoxon-méthyl au
niveau du foie consiste en oxydation, hydrolyse et déméthylation ou
désarylation en présence de glutathion réduit (GSH). Les produits de
réaction sont le thiophosphate de o-méthyle et de o-nitrophényle
ainsi que les acides diméthylphosphorothioïque ou
diméthyl-phosphorique et le p-nitrophénol. Il est donc possible
d'évaluer l'exposition en mesurant l'excrétion urinaire du
p-nitrophénol. Chez des volontaires, l'excrétion urinaire de
p-nitrophénol était de 60 % quatre heures après l'administration et
d'environ 100 % au bout de 24 heures. Le métabolisme du
parathion-méthyl joue un rôle important dans la toxicité sélective de
ce composé pour les différentes espèces et l'apparition éventuelle
d'une résistance. L'élimination du parathion-méthyl et de ses
métabolites s'effectue principalement par la voie urinaire. Des études
menées sur des souris avec du parathion-méthyl radiomarqué au 32P
ont montré qu'au bout de 72 heures, 75 % de la radio-activité se
retrouvaient dans les urines et jusqu'à 10 % dans les matières
fécales.
1.3 Effets sur les êtres vivants dans leur milieu
naturel
Certains microorganismes peuvent utiliser le parathion-méthyl
comme source de carbone et l'étude d'une communauté naturelle a montré
que des concentrations allant jusqu'à 5 mg/litre augmentaient la
biomasse et l'activité reproductrice. L'effet est positif dans le cas
des bactéries et des actinomycètes; par contre, les champignons et les
levures sont moins capables d'utiliser ce composé. Chez une diatomée,
on a constaté une inhibition de 50 % de la croissance à une
concentration d'environ 5 mg/litre. Chez des algues vertes
unicellulaires, la croissance a été réduite par des concentrations
comprises entre 25 et 80 µg de parathion-méthyl par litre. Les
populations d'algues devenaient tolérantes au parathion-méthyl après
quelques semaines d'exposition.
Le parathion-méthyl est extrêmement toxique pour les invertébrés
aquatiques, la CL50 étant plupart du temps comprise entre <1 µg et
environ 40 µg/litre. Quelques espèces d'arthropodes sont moins
sensibles. Pour la daphnie (Daphnia magna) la concentration sans
effet observable est de 1,2 µg/litre. Les mollusques sont beaucoup
moins sensibles, puisque leur CL50 varie de 12 à 25 mg/litre.
La plupart des espèces de poissons d'eau douce ou de mer ont une
CL50 comprise entre 6 et 25 mg/litre, quelques espèces étant
nettement plus ou nettement moins sensibles au composé. La toxicité
aiguë est comparable chez les amphibiens et les poissons.
Le traitement au parathion-méthyl de mares expérimentales a
permis d'en observer les effets sur l'effectif des communautés
d'invertébrés aquatiques. Seul un épandage sur les étendues d'eau
serait susceptible d'engendrer les concentrations nécessaires à
l'apparition de ces effets et encore, seraient-ils de courte durée.
Une décimation des populations d'invertébrés est donc improbable en
situation réelle. En cas de mortalité chez les invertébrés, les effets
ne seraient probablement pas de longue durée.
Il convient dont de veiller à ne pas procéder à des épandages sur
les mares, cours d'eau et lacs. Le parathion-méthyl ne doit jamais
être épandu lorsque le vent souffle.
Le parathion-méthyl est un insecticide non-sélectif qui détruit
les espèces utiles tout autant que les ravageurs. On a fait état de
mortalité parmi des abeilles à la suite d'épandages de
parathion-méthyl. Ce genre d'accidents est plus grave avec le
parathion-méthyl qu'avec d'autres insecticides. Les abeilles adaptées
à l'Afrique supportent mieux le parathion-méthyl que les souches
européennes.
Le parathion-méthyl s'est révélé modérément toxique pour les
oiseaux au laboratoire, la DL50 aiguë par voie orale allant de 3 à
8 mg/kg de poids corporel. Par la voie alimentaire, la CL50 allait
de 70 à 680 mg/kg de nourriture. Rien n'indique que les oiseaux aient
à souffrir du parathion-méthyl lorsqu'il est épandu conformément aux
recommandations.
On veillera tout particulièrement à l'horaire des épandages pour
éviter tout effet nocif sur les abeilles.
1.4 Effets sur les animaux d'expérience et les systèmes d'épreuve
in vitro
La DL50 par voie orale varie chez les rongeurs de 3 à 35 mg/kg
de poids corporel et la DL50 par voie percutanée, de 44 à 67 mg/kg
de poids corporel.
L'intoxication par le parathion-méthyl engendre les effets
cholinergiques habituels des organophosphorés que l'on peut attribuer
à l'accumulation d'acétylcholine au niveau des terminaisons nerveuses.
La toxicité du parathion-méthyl est due à sa métabolisation en
paraoxon-méthyl. Cette conversion est très rapide. Aucun signe de
neuropathie retardée induite par les organophosphorés n'a été relevé.
Le parathion-méthyl technique n'a aucun effet irritant sur l'oeil
ni la peau.
Lors d'études de toxicité à court terme utilisant diverses voies
d'administration et portant sur des rats, des chiens et des lapins, on
a observé une inhibition de la cholinestérase du plasma, des
érythrocytes et du cerveau ainsi qu'un certain nombre de signes liés
aux effets cholinergiques. Lors d'une étude d'alimentation de 12
semaines sur des chiens, on a obtenu, pour la dose sans effet nocif
observable, une valeur de 5 mg/kg de nourriture (soit l'équivalent de
0,1 mg/kg de poids corporel par jour). Lors d'une étude de toxicité
par voie percutanée, effectuée pendant trois semaines sur des lapins,
on a obtenu une dose sans effet observable de 10 mg/kg de poids
corporel par jour. Lorsque les animaux étaient exposés par la voie
respiratoire pendant trois semaines, la dose sans effet observable
était de 0,9 mg/m3 d'air. A la dose de 2,6 mg/m3, on n'a observé
qu'une légère inhibition de la cholinestérase plasmatique.
Des études de cancérogénicité et de toxicité à long terme ont été
effectuées sur des souris et des rats. Pour les rats, la dose sans
effet observable basée sur l'inhibition de la cholinestérase était de
0,1 mg/kg de poids corporel par jour. Les résultats de ces études
n'ont fait ressortir aucun signe de cancérogénicité, ni chez les
souris ni chez les rats. Dans une autre étude de deux ans effectuée
sur des rats, on a toutefois relevé les signes d'un effet neurotoxique
périphérique à la dose de 50 mg/kg de nourriture.
Le parathion-méthyl serait capable de provoquer l'alkylation de
l'ADN in vitro. La plupart des études de génotoxicité in vitro
portant sur des cellules bactériennes et mammaliennes ont donné des
résultats positifs, alors que six études in vivo portant sur trois
systèmes d'épreuve différents ont donné des résultats ambigus.
Les études portant sur la reproduction avec administration de
doses toxiques (inhibition de la cholinestérase) n'ont pas produit
d'effets systématiques sur la taille des portées et leur nombre, le
taux de survie des petits ni la lactation. Aucun effet tératogène ou
embryotoxique direct n'a été observé.
1.5 Effets sur l'homme
Plusieurs cas d'intoxication aiguë par le parathion-méthyl ont
été signalés. Les symptômes sont caractéristiques d'une intoxication
générale par les anticholinestérasiques organophosphorés. Il s'agit
d'effets nerveux cholinergiques au niveau périphérique et au niveau
central qui apparaissent dans les minutes qui suivent l'exposition. En
cas d'exposition par voie percutanée, les symptômes peuvent s'aggraver
pendant plus d'une journée et durer plusieurs jours.
Des études sur des volontaires soumis à des expositions répétées
de longue durée ont montré que l'activité cholinestérasique du sang
diminuait sans provoquer de manifestations cliniques.
Aucun cas de neuropathie périphérique retardée induite par les
organophosphorés n'a été signalé. Dans un certain nombre de cas
d'exposition multiple à des pesticides et notamment à du
parathion-méthyl, on a observé des séquelles neurospychiatriques.
Une augmentation du nombre des aberrations chromosomiques a été
signalée dans des cas d'intoxication aiguë.
On ne possède aucune donnée obtenue sur l'homme qui puisse
permettre d'évaluer les effets tératogènes du parathion-méthyl ou ses
effets sur la reproduction.
Les études épidémiologiques disponibles sont consacrées à des
expositions multiples aux pesticides et il n'est pas possible d'en
déduire les effets qu'une exposition de longue durée au
parathion-méthyl pourrait entraîner.
2 Conclusions
Le parathion-méthyl est un insecticide organophosphoré très
toxique. Une exposition excessive due à la manipulation de ce produit
au cours de la production, de l'utilisation ou par suite d'ingestion
accidentelle ou intentionnelle peut entraîner une intoxication grave
voire mortelle. Certaines formulations de parathion-méthyl peuvent,
selon le cas, entraîner une irritation des yeux ou de la peau mais de
toute façon, elles sont toutes facilement absorbées. On peut donc être
dangereusement exposé à cet insecticide sans s'en rendre compte.
Le parathion-méthyl ne subsiste pas dans l'environnement. Il ne
subit pas de bioconcentration et ne se transmet pas le long de la
chaîne alimentaire. Il est rapidement décomposé par un grand nombre de
microorganismes et autres éléments de la faune sauvage. Cet
insecticide peut provoquer des dégâts dans les écosystèmes, mais
seulement en cas d'exposition excessive dues à une utilisation
défectueuse ou à des déversements accidentelles. Toutefois les
insectes utiles et notamment les insectes pollinisateurs peuvent
souffrir des épandages de parathion-méthyl.
C'est principalement par l'intermédiaire des denrées alimentaires
que la population générale peut être exposée à des résidus de
parathion-méthyl. Si l'on respecte les règles de bonne pratique
agricole, il n'y a pas de raison que la dose journalière admissible
fixée par le Comité d'experts FAO/OMS soit dépassée (0-0,02 mg/kg de
poids corporel)). Il peut également y avoir exposition par voie
percutanée lors de contacts accidentels avec des résidus foliaires
dans des champs traités ou des zones voisines contaminées par des
embruns de pesticides.
Moyennant de bonnes méthodes de travail et des précautions
suffisantes en matières d'hygiène et de sécurité, le parathion-méthyl
de devrait pas présenter de danger pour ceux qui lui sont exposés de
par leur profession.
3 Recommandations
* Afin de protéger la santé et le bien-être des travailleurs et de
la population générale il ne faut confier la manipulation et
l'épandage du parathion-méthyl qu'à des personnes bien encadrées
et bien formées qui utiliseront l'insecticide en prenant les
mesures de sécurité nécessaires et se conformeront aux règles de
bonne pratique en la matière.
* La fabrication, la formulation, l'utilisation agricole et
l'élimination du parathion-méthyl doivent être conduites avec
soin afin de réduire au minimum la contamination de
l'environnement.
* Les travailleurs qui sont régulièrement exposés au parathion-
méthyl doivent bénéficier d'un suivi médical approprié.
* Afin de réduire les risques pour l'ensemble de la population, il
est recommandé de ne pas revenir sur une zone traitée avant 48
heures.
* Les autorités nationales devront fixer les délais pour les
épandages avant récolte et les faire respecter.
* En raison de la forte toxicité du parathion-méthyl, cet
insecticide ne doit pas être épandu à très bas volume à l'aide de
dispositifs à main.
* Ne pas pulvériser sur les étendues d'eau. Choisir les horaires
de manière à éviter de détruire les insectes pollinisateurs.
* Les données sur l'état de santé des travailleurs exposés
uniquement au parathion-méthyl (c'est-à-dire employés à la
fabrication et à la formulation de cet insecticide) devront être
publiées afin que l'on puisse mieux en évaluer les risques pour
la santé humaine.
* Des études à caractère plus définitif devront être menées sur les
résidus de parathion-méthyl dans les denrées alimentaires
fraîches.
* Il faudrait procéder à une évaluation plus concluante de la
génotoxicité du parathion-méthyl.
RESUMEN Y EVALUACION, CONCLUSIONES Y RECOMENDACIONES
1 Resumen y evaluación
1.1 Exposición
El metilparatión es un insecticida organofosforado que
sesintetizó por primera vez en la década de 1940. Es
relativamenteinsoluble en agua, poco soluble en éter de petróleo y
aceitesminerales y fácilmente soluble en la mayoría de los
disolventesorgánicos. El metilparatión puro se encuentra en forma de
cristalesblancos; el de calidad técnica tiene un color tostado claro
y olorparecido al del ajo. Es térmicamente inestable y se descompone
conrapidez a un pH superior a 8.
El método más común para la determinación del metilparatión esla
cromatografía de gases con un detector de ionización de llama enálcali
o bien con uno fotométrico de llama. Los límites de detecciónen el
agua oscilan entre 0,01 y 0,1 µg/litro, y en el aire entre 0,1 y 1
ng/m3. También son útiles como métodos de detección lacromatografía
líquida de alta resolución y la cromatografía en capafina.
En la distribución del metilparatión en el aire, el agua, el
sueloy los organismos del medio ambiente influyen varios factores
físicos,químicos y biológicos.
Los estudios realizados utilizando modelos de ecosistemas y
laelaboración de modelos matemáticos indican que en el medioambiente
el metilparatión se reparte principalmente entre el aire y elsuelo,
con cantidades menores en las plantas y los animales.Prácticamente no
hay desplazamiento a través del suelo, y ni elcompuesto original ni
los productos derivados de su degradaciónllegan normalmente al agua
subterránea. El metilparatión presente enel aire procede sobre todo
del rociado del compuesto, aunque seproduce cierta volatilización con
la evaporación del agua de las hojasy de la superficie del suelo. Los
niveles habituales de metilparatiónen la atmósfera en las zonas
agrícolas oscilan entre una cantidad nodetectable y unos 70 ng/m3.
Se ha observado que las concentracionesen el aire después del rociado
disminuyen con rapidez en tres días,alcanzando los niveles habituales
en unos nueve días. Laconcentración en el agua fluvial (en estudios de
laboratorio)descendió al 80% de la inicial después de una hora, y
transcurridauna semana era del 10%. El metilparatión se mantiene en el
suelomás tiempo que en el aire o el agua, aunque en la retención
influyemucho el tipo de suelo; el arenoso pierde los residuos del
compuestocon mayor rapidez que las margas. Los residuos de la
superficie delas plantas y del interior de las hojas disminuyen
rápidamente, conuna semivida del orden de unas horas; el metilparatión
desaparecetotalmente en unos 6-7 días.
Los animales pueden degradar el metilparatión y eliminar
losproductos de degradación en un período muy breve de tiempo.
Elproceso es más lento en los vertebrados inferiores y en
losinvertebrados que en los mamíferos y las aves. Los factores
debioconcentración son bajos y los niveles acumulados de
metilparatióntransitorios.
La descomposición microbiana es con diferencia el mecanismomás
importante de degradación del metilparatión en el medioambiente. La
desaparición del compuesto en el campo y enecosistemas utilizados como
modelo es más rápida de lo que habíanpermitido suponer los estudios de
laboratodeloorio. Esto se debe a lavariedad de microorganismos que son
capaces de degradarlo endistintos hábitats y circunstancias. La
presencia de sedimentos o desuperficies de plantas, que aumenta la
población microbiana, acelerael ritmo de degradación del
metilparatión.
El metilparatión puede sufrir degradación oxidativa por acciónde
la radiación ultravioleta o la luz solar, convirtiéndose enmetil
paraoxón, que es menos estable; las películas de rociado sedegradan
por acción de la radiación ultravioleta con una semivida aproximada de
40 horas. Sin embargo, se ha estimado que lacontribución de la
fotolisis a la desaparición total en un sistema acuático es sólo de un
4%. También se produce hidrólisis delmetilparatión en condiciones
alcalinas, en las que es más rápida. Lasalinidad elevada favorece
asimismo la hidrólisis del compuesto. Ensedimentos muy reductores se
registraron semividas de unos minutos, aunque el metilparatión es más
estable cuando está adsorbido sobreotros sedimentos.
En las ciudades situadas en el centro de las zonas agrícolas
delos Estados Unidos, las concentraciones de metilparatión en el
airevariaban con las estaciones y alcanzaban el punto más alto en
agostoo septiembre; los niveles máximos registrados durante los
estudios fueron fundamentalmente del orden de 100-800 ng/m3 durante
elperíodo vegetativo. Las concentraciones en el agua natural de
laszonas agrícolas de los Estados Unidos llegaron a 0,46 µg/litro,
conlos niveles más altos en el verano. Son muy pocos los
informespublicados en todo el mundo sobre los residuos de
metilparatión enlos alimentos. En los Estados Unidos, se han
notificado en generalniveles muy bajos de residuos de metilparatión
en los productos alimenticios, con un pequeño número de muestras
aisladas porencima de los límites máximos de residuos (LMR). En todos
losestudios publicados sobre la alimentación sólo se detectaron
niveles ínfimos de metilparatión. En las encuestas sobre la cesta de
lacompra realizadas en los Estados Unidos entre 1966 y 1969,
lascantidades mayores de residuos de metilparatión se encontraron en
lashortalizas de hoja (hasta 2 mg/kg) y en las de raíz (hasta 1
mg/kg). En la preparación, cocción y almacenamiento de los alimentos
se descomponen los residuos de metilparatión, reduciéndose
ulteriormente la exposición humana. Las frutas y hortalizas
sinelaborar pueden contener más residuos después de un uso indebidodel
producto.
La producción, formulación, manipulación y uso delmetilparatión
como insecticida pueden ser, en principio, fuente deexposición para
las personas. Las principales vías de exposición delos trabajadores
son el contacto cutáneo y, en menor medida, lainhalación.
En un estudio sobre personas encargadas del rociado en fincas
(trabajadores no protegidos que utilizaban rociadores manuales
devolumen ultrabajo), a partir del p-nitrofenol excretado en la
orina secalculó una ingestión de 0,4-13 mg de metilparatión cada 24
horas. También se puede sufrir exposición si se entra en los
cultivosdemasiado pronto después de tratarlos.
La población general puede estar expuesta a residuos
demetilparatión presentes en el aire, el agua y los alimentos como
consecuencia de prácticas agrícolas y forestales con un uso
indebidodel producto, que provoca la contaminación de los campos,
loscultivos, el agua y el aire debido al rociado parcial fuera del
objetivo.
1.2 Ingestión, metabolismo y excreción
El metilparatión se absorbe fácilmente por todas las vías de
exposición (oral, cutánea, respiratoria) y se distribuye con rapidez
por los tejidos del cuerpo. Se detectaron concentraciones máximas en
diversos órganos 1-2 horas después del tratamiento. Después de la
administración, la transformación del metilparatión en metilparaoxón
se produce en unos minutos. En perros se determinó una semivida
terminal media de 7,2 horas tras la administración intravenosa de
metilparatión. El hígado es el principal órgano de metabolización y
desintoxicación. El metilparatión o el metilparaoxón se destoxifican
en el hígado sobre todo mediante oxidación, hidrólisis y desmetilación
o desarilación con glutatión reducido. Los productos de la reacción
son el O-metil O-p-nitrofenilfosfotioato, o bien los ácidos
dimetilfosfotioico o dimetilfosfórico, y el p-nitrofenol. Por
consiguiente, se puede estimar la exposición midiendo la excreción
urinaria de p-nitrofenol; en voluntarios humanos fue del 60% en
cuatro horas y prácticamente del 100% en 24 horas. El metabolismo del
metilparatión es importante para la toxicidad específica selectiva y
la aparición de resistencia. Le eliminación de esta sustancia y sus
productos derivados tiene lugar primordialmente por la orina. En
estudios realizados en ratones con 32P-metilparatión (marcado
radiactivamente) se observó un 75% de radiactividad en la orina y
hasta un 10% en las heces después de 72 horas.
1.3 Efectos en los seres vivos del medio ambiente
Los microorganismos pueden utilizar el metilparatión como fuente
de carbono, y en el estudio de una comunidad natural se vio que
concentraciones de hasta 5 mg/litro aumentaban la biomasa y la
actividad reproductora. En las bacterias y los actinomicetos se
observó un efecto positivo del metilparatión, mientras que los hongos
y las levaduras tenían menor capacidad para utilizar la sustancia. Con
una concentración aproximada de 5 mg/litro se produjo una inhibición
del 50% del crecimiento de una diatomea. Concentraciones de
metilparatión comprendidas entre 25 y 80 µg/litro redujeron el
crecimiento celular de las algas clorofíceas unicelulares. Las
poblaciones de algas adquirieron tolerancia tras varias semanas de
exposición.
El metilparatión es muy tóxico para los invertebrados acuáticos,
oscilando casi siempre la CL50 entre < 1 µg y alrededor de 40
µg/litro. Hay un pequeño número de especies de artrópodos que son
menos susceptibles. El nivel sin efecto para Daphnia magna es de 1,2
µg/litro. Los moluscos son mucho menos susceptibles, con CL50 entre
12 y 25 mg/litro.
La mayoría de las especies de peces, tanto de agua dulce como de
mar, tienen una CL50 de 6 a 25 mg/litro, pero hay un pequeño número
de especies cuya sensibilidad al metilparatión es considerablemente
mayor o menor. La toxicidad aguda para los anfibios es análoga a la de
los peces.
Se han observado los efectos sobre poblaciones en las comunidades
de invertebrados acuáticos de estanques experimentales tratados con
metilparatión. Las concentraciones necesarias para producir esos
efectos se alcanzarían sólo con un rociado excesivo de las masas de
agua, e incluso en este caso durarían muy poco tiempo. Por
consiguiente, en condiciones normales no es probable que se observen
efectos sobre las poblaciones. Tampoco los es que la acción letal
sobre los invertebrados acuáticos provoque efectos duraderos.
Hay que tener cuidado para evitar un rociado excesivo de
estanques, ríos y lagos al utilizar el metilparatión. Nunca se debe
efectuar la operación con viento.
El metilparatión es un insecticida no selectivo que mata especies
beneficiosas tan fácilmente como las plagas. Se ha notificado la
muerte de abejas después de su aplicación. Sus efectos sobre esta
especie fueron más graves que los de otros insecticidas. Las abejas
africanizadas son más tolerantes al metilparatión que las razas
europeas.
El metilparatión fue moderadamente tóxico para las aves en
estudios de laboratorio, con una DL50 oral aguda comprendida entre
3 y 8 mg/kg de peso corporal. La CL50 en la dieta osciló entre 70 y
680 mg/kg de alimentos. No hay indicios de que las aves puedan verse
afectadas negativamente con la utilización recomendada en el campo.
Hay que tener el máximo cuidado al programar el rociado con
metilparatión, a fin de evitar los efectos adversos sobre las abejas.
1.4 Efectos en los animales de experimentación y en sistemas de
prueba in vitro
Los valores de la DL50 del metilparatión por vía oral en
roedores oscilan entre 3 y 35 mg/kg de peso corporal, y los valores
por vía cutánea entre 44 y 67 mg/kg de peso corporal.
El envenenamiento por metilparatión provoca los signos
colinérgicos habituales de los organofosfatos, atribuidos a la
acumulación de acetilcolina en la terminaciones nerviosas. El
metilparatión adquiere la toxicidad al metabolizarse a metilparaoxón,
en un proceso que es muy rápido. No se han observado indicios de
neuropatía retardada inducida por compuestos organofosforados.
Se ha comprobado que el metilparatión de calidad técnica no tiene
potencial de irritación primaria de los ojos o la piel.
En estudios de toxicidad de corta duración, utilizando diversas
vías de administración en ratas, perros y conejos, se observó
inhibición de la colinesterasa del plasma, los eritrocitos y el
cerebro, así como signos colinérgicos conexos. En un estudio de
alimentación durante 12 semanas con perros, el nivel sin efectos
adversos observados (NOAEL) fue de 5 mg/kg de la dieta (equivalente a
0,1 mg/kg de peso corporal al día). En un estudio de toxicidad cutánea
de tres semanas en conejos, el nivel sin efectos observados (NOEL) fue
de 10 mg/kg de peso corporal al día. La exposición por inhalación
durante tres semanas dio como resultado un NOEL de 0,9 mg/m3 de
aire. Con 2,6 mg/m3 solamente se observó una ligera inhibición de la
colinesterasa del plasma.
Se realizaron estudios de toxicidad/teratogenicidad de larga
duración con ratones y ratas. El NOEL para las ratas fue de 0,1 mg/kg
de peso corporal al día, basado en la inhibición de la colinesterasa.
No hay pruebas de carcinogenicidad en ratones y ratas tras una
exposición de larga duración. Sin embargo, en otro estudio de dos años
con ratas se detectó un efecto neurotóxico periférico con una dosis de
50 mg/kg de la dieta.
Se ha informado que el metilparatión tiene propiedades
alquilizantes del ADN in vitro. Los resultados de la mayoría de los
estudios de genotoxicidad in vitro con células tanto bacterianas
como de mamífero fueron positivos, mientras que en seis estudios in
vivo, utilizando tres sistemas de prueba distintos, los resultados
fueron equívocos.
En estudios de reproducción con niveles de dosificación tóxicos
(inhibición de la colinesterasa), no se observaron efectos constantes
sobre el tamaño de la camada, el número de partos, la tasa de
supervivencia de las crías y el rendimiento de la lactación. No se
detectó ningún efecto teratogénico o embriotóxico primario.
1.5 Efectos en la especie humana
Se han registrado varios casos de intoxicación aguda por
metilparatión. Los signos y síntomas son los característicos de la
intoxicación sistémica por compuestos organofosforados inhibidores de
la colinesterasa. Cabe mencionar entre ellos las manifestaciones del
sistema nervioso colinérgico periférico y central, que aparecen apenas
unos minutos después de la exposición. En el caso de la exposición
cutánea, la gravedad de los síntomas puede ir en aumento durante más
de un día y pueden durar varios días.
Los estudios con voluntarios sometidos a exposiciones repetidas
de larga duración parecen indicar que hay una disminución de la
actividad de la colinesterasa de la sangre, sin manifestaciones
clínicas.
No se ha informado de ningún caso de neuropatía periférica
retardada inducida por compuestos organofosforados. Se han descrito
secuelas neuropsiquiátricas en casos de exposición múltiple a
plaguicidas, entre ellos el metilparatión.
En casos de intoxicaciones agudas, se ha detectado un aumento de
las aberraciones cromosómicas.
No se dispone de datos relativos al metilparatión en la especie
humana que permitan evaluar los efectos teratogénicos y sobre la
reproducción.
Los estudios epidemiológicos disponibles se refieren a una
exposición múltiple a plaguicidas, y no es posible evaluar los efectos
de una exposición de larga duración al metilparatión.
2 Conclusiones
El metilparatión es un éster organofosfórico muy tóxico,
utilizado como insecticida. Una exposición excesiva al manejarlo
durante su fabricación y uso o por ingestión accidental o intencionada
puede ocasionar una intoxicación grave o letal. Las formulaciones de
metilparatión unas veces son irritantes y otras no para los ojos o la
piel, pero se absorben fácilmente. Por consiguiente, pueden producirse
exposiciones peligrosas sin advertirlo.
El metilparatión no se mantiene mucho tiempo en el medio
ambiente, no se produce bioconcentración y no se desplaza a través de
la cadena alimentaria. Lo degradan con rapidez numerosos
microorganismos y otros tipos de seres vivos presentes en el medio
ambiente. Este insecticida puede ocasionar daños a ecosistemas
solamente en casos de una exposición muy intensa causada por el uso
indebido o escapes accidentales; sin embargo, el rociado con
metilparatión representa un riesgo para los insectos polinizadores y
otros que son beneficiosos.
La exposición de la población general a los residuos del
metilparatión tiene lugar fundamentalmente por medio de los alimentos.
Si se siguen buenas prácticas agrícolas, no se supera la ingesta
diaria admisible (0-0,02 mg/kg de peso corporal) establecida por la
FAO/OMS. Puede haber exposición cutánea accidental por contacto con
residuos foliares en campos rociados o en zonas adyacentes a los
lugares que se están rociando, como consecuencia de pérdidas del
producto que no llegan a su objetivo.
Con buenas prácticas de trabajo, medidas higiénicas y
precauciones de seguridad, no es probable que el metilparatión
represente un riesgo para las personas con exposición profesional.
3 Recomendaciones
* Para salvaguardar la salud y el bienestar de los trabajadores y
de la población general, el manejo y la aplicación del
metilparatión sólo se debería encomendar, bajo una atenta
supervisión, a personas bien capacitadas que se ajusten a las
medidas de seguridad adecuadas y utilicen el producto de acuerdo
con las buenas prácticas de aplicación.
* Se debe prestar particular atención a la fabricación, la
formulación, el uso agrícola y la eliminación del metilparatión,
a fin de reducir al mínimo la contaminación del medio ambiente.
* Los trabajadores regularmente expuestos deberían ser objeto de
vigilancia y exámenes médicos adecuados.
* A fin de reducir al mínimo el riesgo para todas las personas, se
recomienda esperar 48 horas después del rociado antes de entrar
de nuevo en cualquier zona tratada.
* Las autoridades nacionales deberían establecer intervalos sin
tratamiento antes de la recolección y obligar a respetarlos.
* A la vista de la elevada toxicidad del metilparatión, se debe
excluir este producto de la aplicación mediante rociado de
volumen ultrabajo aplicado manualmente.
* No se han de rociar masas de agua. Hay que elegir los momentos de
la aplicación de manera que se evite la muerte de insectos
polinizadores.
* Se debe hacer pública la información relativa al estado de salud
de los trabajadores expuestos exclusivamente al metilparatión (es
decir, en la fabricación, la formulación), con objeto de evaluar
mejor los riesgos de este producto químico para la salud humana.
* Deberían llevarse a cabo estudios más definitivos sobre los
residuos de metilparatión en los alimentos frescos.
* Debería realizarse una evaluación genotóxica más definitiva del
metilparatión.
8.10.2 Possible alkylation of biological macromolecules
It has been shown, under laboratory conditions, that some
organophosphates can react with, and alkylate, the reagent
4-nitro-benzylpyridine (Preussmann et al., 1969). The study was
interpreted to imply that the in vivo alkylating potential of some
pesticides was similar to that of the known mutagens, dimethyl sulfate
and methyl methanesulfonate. Furthermore, Löfroth et al. (1969)
derived a substrate constant (a logarithmic measure of alkylating
ability) of 0.75 for dichlorvos, which is intermediate between those
known for methyl and ethyl methanesulfonates. Concern over the
possible mutagenic and carcinogenic potential of organophosphorus
compounds on the basis of the above data was misplaced, since
alternative reactions were not considered. Compared with the carbon
atom of the alkyl group, the phosphorus atom is markedly more
electron-deficient and susceptible to attack by nucleophiles. Analysis
by Bedford & Robinson (1972) of the data of Löfroth et al. (1969)
revealed that the proposed rates of alkylation by hard nucleophiles
were probably combined rates of phosphorylation and alkylation, and
that phosphorylation was the totally dominant reaction in the case of
the hydroxide ion. The comparison with known mutagens was therefore
inappropriate. Two factors detract further from the toxicological
significance of the alkylation studies. The first is that mammalian
tissues (plasma, liver, etc.) contain active enzymes that catalyse the
phosphorylation of water by the organophosphorus esters. Viewed
inversely, these enzymes (often called A-esterases) catalyse the
hydrolysis of the organophosphorus esters, thereby rapidly reducing
circulating levels of hazardous material. Secondly, the comparative
rate of reaction of most of these pesticides with AChE is many orders
greater than their rate of alkylation of the typical nucleophile
4-nitrobenzylpyridine: for dichlorvos, the ratio of rates was
1 x 107 in favour of the inhibitory phosphorylation of AChE
(Aldridge & Johnson, 1977). It follows that, at low exposure levels,
in vivo phosphorylation of AChE and other esterases will be the
dominant reaction with negligible uncatalysed alkylation of genetic
material. Indeed, no such alkylation has been detected in sensitive
in vivo studies designed to check this point (Wooder et al., 1977).
Some catalysed alkylations of glutathione by organosphorus compounds
are known to occur in vivo , but these are essentially
detoxification reactions.
8.10.3 General
Following lethal amounts of methyl parathion, hypotension,
bradycardia, bronchoconstriction, and bronchial fluid accumulation
occur with the inability of respiratory muscles to work. Cyanosis and
central respiratory depression can be observed. In less severe cases
of intoxication, bradycardia, muscle rigidity, muscle hypotonia,
bronchial spasm, and constriction dominate (Meyer-Jones et al., 1977).
9. EFFECTS ON MAN
The only confirmed effects on humans of exposure to methyl
parathion are the signs and symptoms characteristic of systemic
poisoning by cholinesterase-inhibiting organophosphorus compounds,
observed in case studies. The results of oral ingestion studies
performed by Rider et al. (1969, 1970, 1971) suggest that
manifestations of acute methyl parathion toxicity are absent in humans
whose erythrocyte cholinesterase activity has been reduced to as
little as 45% of their pre-exposure baselines (see section 9.1.2).
The effects of methyl parathion exposure on human beings were
compiled in 1976 by NIOSH. Details are given by Hayes & Laws (1991).
WHO (1986) summarized the signs and symptoms of organo-phosphate
insecticide poisoning as follows:
(a) Muscarinic manifestations
- increased bronchial secretion, excessive sweating,
salivation, and lacrimation;
- pinpoint pupils, bronchoconstriction, abdominal cramps
(vomiting and diarrhoea); and
- bradycardia.
(b) Nicotinic manifestations
- fasciculation of fine muscles and, in more severe
cases, of the diaphragm and respiratory muscles; and
- tachycardia.
(c) Central nervous system manifestations
- headache, dizziness, restlessness, and anxiety;
- mental confusion, convulsions, and coma; and
- depression of the respiratory centre.
All these signs and symptoms can occur in different combinations
and can vary in time of onset, sequence, and duration, depending on
the chemical, dose, and route of exposure. Mild poisoning might
include muscarinic and nicotinic signs only. Severe cases always show
central nervous system involvement; the clinical picture is dominated
by respiratory failure, sometimes leading to pulmonary oedema, due to
the combination of the above-mentioned signs and symptoms.
9.1 General population exposure
The general population may be exposed to air-, water-, and
food-borne residues of methyl parathion as a consequence of
agricultural/forestry practices, the misuse of the agent, and
contamination of field crops, water, and air by off-target loss.
Lisi et al. (1986, 1987) studied the allergic potential of methyl
parathion in 200 persons. No significant sensitization to methyl
parathion was found.
9.1.1 Acute toxicity
Several cases of methyl parathion poisoning have been reported
throughout the world; these have been reviewed by Hayes & Laws (1991).
Human manifestations of acute poisoning by methyl parathion are
comparable with those described in experimental animals (Durham &
Hayes, 1962; Fazekas & Rengei, 1965; Hayes & Laws, 1991).
In cases of fatal methyl parathion poisoning, gross and
microscopic alterations occur in all the organs (brain, lung, heart,
liver, kidneys, spleen, vascular walls, perivascular areas). Fazekas
(1971) already saw alterations due to methyl parathion-poisoning, 2 h
after the poisoning. Ember et al. (1970) found a high content of
vitamin A in the liver in 5 cases of suicide with methyl parathion.
Van Bao et al. (1974) reported an increase in chromosome
aberrations in the lymphocytes of 4 patients who had suffered acute
methyl parathion poisoning as a result of attempted suicide. The
increase in chromosome aberrations was detected only in cell cultures
carried out 1 month after their admission to hospital. No significant
changes were found, compared with controls, 6 months later.
9.1.2 Effects of short- and long-term exposure, controlled human
studies
Five male volunteers received 3.0 mg methyl parathion per day for
28 days, then 3.5 mg methyl parathion for 28 days, and 4.0 mg methyl
parathion for 43 days. No symptoms of poisoning or effects on the
plasma or red blood cell cholinesterases could be noticed (Moeller &
Rider, 1961).
In another study, 3 groups of 5 volunteers each received 4.5 mg
methyl parathion daily, for 30 days, then 5.0 g for 29 days or 5.5 mg
for 28 days, followed by 6.0 mg for 29 days or 6.5 mg for 35 days, and
finally 7.0 mg for 24 days. In no case was significant inhibition of
the plasma or red blood cell cholinesterase activity found (Moeller &
Rider, 1962).
Morgan et al. (1977) studied the cholinesterase activities in 4
human volunteers, who received 2 or 4 mg methyl parathion on 5
successive days. These doses did not cause any depression of plasma
and red blood cell cholinesterase activity.
Rider et al. (1969) reported studies on human volunteers to
determine the level of minimal toxicity of methyl parathion. For 30
days, 5 volunteers received capsules containing methyl parathion, with
the dose increasing daily, and 2 received capsules containing corn
oil. Depression in plasma cholinesterase activity (15%) was observed
at an oral dosage of 11.0 mg per day, while, at higher dosages up to
and including 19 mg per day, no significant cholinesterase depression
was observed. No significant changes in the blood cell count, urine
analysis, or the prothrombin times occurred, nor was there any
evidence of toxic effects.
After 4 weeks with daily doses of 24 mg methyl parathion, 2 out
of 5 volunteers showed inhibition of plasma and red blood cell
cholinesterase activities with decreases of 24 or 23% for plasma and
27 or 55% for red blood cells (Rider et al., 1970).
Five volunteers received doses increasing from 14 to 20 mg methyl
parathion per day, orally, for 6 days. No inhibition of the
cholinesterases was found. However, doses of 28 or 30 mg methyl
parathion caused a decrease in the cholinesterase activities of about
37% (Rider et al., 1971).
Two male volunteers received, orally, 2 or 4 mg methyl parathion
per day. No influence of methyl parathion on neurophysiological
parameters was found, and there was no inhibition of the plasma or red
blood cell cholinesterase activity (Rodnitzky et al., 1978).
9.2 Occupational exposure
The production, formulation, handling, and use as an insecticide
of methyl parathion are potential sources of exposure. Skin contact or
inhalation are the main hazards for workers. The main hazard for the
general population is the ingestion of contaminated food. Wind-drift
during spraying may be a health risk, since Kummer & Van Sittert
(1986) observed that, in a number of cases, the spraymen did not stop
spraying, when it was too windy.
The analysis of 375 pesticide poisonings in Bulgaria during
1965-68 showed that 82.5% of all cases were due to organophosphates.
Six of the intoxications were attributed to methyl parathion. A large
number of poisonings, usually mild, occurred not in applicators
directly engaged in plant protection but in other agricultural workers
when they entered a previously sprayed crop area for further
cultivation and hand-harvesting (Kaloyanova-Simeonova, 1970).
Hatcher & Wiseman (1969) reported 16 cases of methyl parathion
intoxication among 118 organophosphorus insecticide poisonings of farm
workers that occurred in the lower Rio Grande Valley (Texas) in 1968.
Toxicity following dermal exposure was predominant.
Neuropsychiatric sequelae from occupational exposure to
organophosphorus pesticides have been reported (Dille & Smith, 1964).
However, the patients had been exposed to other pesticides besides
methyl parathion.
Data on chromosomal aberrations due to methyl parathion are
scarce. Data from persons who had worked with various pesticides were
presented by Yoder et al., 1973 (positive finding); Rupa et al., 1989
(positive finding); and Nehéz et al., 1988 (positive finding in farm
workers in the open field, but not in those in enclosed spaces like
greenhouses). Van Bao et al. (1974) found chromosome aberrations in
one case of an agricultural worker, accidentally exposed to methyl
parathion (without exposure data).
De Cassia Stocco et al. (1982) reported data from subjects
exposed to methyl parathion and DDT at a formulation plant near of Sao
Paulo, Brazil. No increased frequency of chromosome aberrations was
found in the lymphocyte cultures of 15 healthy male workers (with
blood cholinesterase level < 75% of presumably the normal mean
levels), who were exposed repeatedly or long-term to methyl parathion
for durations ranging from 1 week to 7 years, but who had intermittent
periods of non-exposure.
Richter et al. (1986) investigated the risk of exposure to methyl
parathion spray drift in the workers in 3 kibbutzim. The
cholinesterase levels were measured in 36 agricultural workers and 25
residents from the same kibbutzim. No effects due to the methyl
parathion spray drift exposure were observed in the field workers or
in the residents.
9.2.1 Epidemiological studies
There are no epidemiological studies on effects related only to
methyl parathion exposure.
10. PREVIOUS EVALUATIONS BY INTERNATIONAL BODIES
The FAO/WHO Joint Meeting on Pesticide Residues (JMPR) evaluated
methyl parathion in 1968, 1972, 1975, 1979, 1980, and 1984 (FAO/WHO,
1969, 1973, 1976, 1980, 1981 and 1985). The acceptable daily intake
for man (ADI) was estimated at 0-0.02 mg/kg body weight in 1984. This
was based on levels causing no toxicological effects of:
- 2 mg/kg diet, equivalent to 0.1 mg/kg body weight in the
rat; and
- 0.3 mg/kg body weight per day in man.
The FAO/WHO Codex Alimentarius Commission (FAO/WHO, 1986)
recommended Maximum Residue Limits (MRLs) in several food commodities,
ranging from 0.05 to 0.2 mg/kg as follows:
Commodity MRL (mg/kg)
Cantaloupe 0.2
Cole crops 0.2
Cottonseed oil 0.05
Cucumbers 0.2
Fruit, other 0.2
Hops (dry cones) 0.05a
Melons 0.2
Sugar beets 0.05a
Tea (fermented and dried) 0.2
Tomatoes 0.2
a Levels at, or about, the limit of determination.
The International Agency for Research on Cancer (IARC) evaluated
methyl parathion in 1982 and in 1987 (IARC, 1983, 1987), and concluded
that the available data do not provide evidence that methyl parathion
is carcinogenic to experimental animals. No data on humans were
available. The available data provide no evidence that methyl
parathion is likely to present a carcinogenic risk for humans.
WHO (1990) classified technical methyl parathion as "extremely
hazardous" in normal use, based on an oral LD50 in the rat of 14
mg/kg. WHO/FAO (1975) issued a data sheet on methyl parathion (No. 7).
REFERENCES
ABE, T., FUJIMOTO, Y., TATSUNO, T., & FUKAMI, J. (1979) Separation of
methyl parathion and fenitrothion metabolites by liquid
chromatography. Bull. environ. Contam. Toxicol., 22: 791-795.
ACKERMANN, H. (1974) [Transfer of organophosphorous insecticides to
the embryo formation of PO derivatives and development of toxic
symptoms.] Tagungsber. Akad. Landwirtsch., 126: 23-29 (in German).
ADHYA, T.K., SUDHAKAR-BARIK, & SETHUNATHAN, N. (1981a) Stability of
commercial formulation of fenitrothion, methyl parathion, and
parathion in anaerobic soils. J. Agric. Food Chem., 29: 90-93.
ADHYA, T.K., SUDHAKAR-BARIK, & SETHUNATHAN, N. (1981b) Fate of
fenitrothion, methyl parathion, and parathion in anoxic
sulfur-containing soil systems. Pest. Biochem. Physiol., 16: 14-20.
ADHYA, T.K., SUDHAKAR-BARIK, & SETHUNATHAN, N. (1981c) Hydrolysis of
selected organophosphorus insecticides by two bacteria isolated from
flooded soil. J. appl. Bacteriol., 50: 167-172.
ADHYA, T.K., WAHID, P.A., & SETHUNATHAN, N. (1987) Persistence and
biodegradation of selected organophosphorus insecticides in flooded
versus non-flooded soils. Biol. Fertil. Soils, 4(1): 36-40.
AGISHEV, M.K., TSUKERMAN, V.G., & KUTSENKO, A.A. (1977) [Methods for
determining pesticide residues in soil.] Khim. sel'sk. Khoz., 15:
52-54 (in Russian).
AGOSTIANO, A., CASELLI, M., & PROVENZANO, M.R. (1983) Analysis of
pesticides and other organic pollutants by preconcentration and
chromatographic techniques. Water Air Soil Pollut., 19: 309-320.
AKHMEDOV, B.K. (1968) The hygienic significance of metaphos as an
atmospheric pollutant. Gig. i Sanit., 33: 10-15.
ALBANIS, T.A., POMONIS, P.J., & SDOUKOS, A.T. (1986) Organophosphorus
and carbamate pesticide residues in the aquatic system of Ioannina
basin and Kalamas River (Greece). Chemosphere, 15: 1023-1034.
ALBAUGH, D.W. (1972) Insecticide tolerances of two crayfish
populations (Procambarus acutus) in south-central Texas. Bull.
environ. Contam. Toxicol., 8(6): 334-338.
ALBERT, L., GONZALEZ, M., & MARTINEZ-DEWANE, M.G. (1979)
[Organophosphorus insecticides. I. Residues of organophosphorus
insecticides in some Mexican foods.] Rev. Soc. Quim. Méx., 23(4):
189-197 (in Spanish).
ALDRIDGE, W.N. & JOHNSON, M.K. (1977) Mechanisms and structure
activity relationships providing a high safety factor for
anti-cholinesterase insecticides. In: Proceedings of the British Crop
Protection Conference, Brighton, November 1977, Croydon, British Crop
Protection Council, pp. 721-729.
AMBRUS, A., HARGITAL, E., KAROLY, G., FULOP, A., & LANTOS, J. (1981a)
General method for determination of pesticide residues in samples of
plant origin, soil, and water. II. Thin layer chromatographic
determination. J. Assoc. Off. Anal. Chem., 64: 743-748.
AMBRUS, A., VISI, E., ZAKAR, F., HARGITAI, E., SZABO, L., & PAPA, A.
(1981b) General method for determination of pesticide residues in
samples of plant origin, soil, and water. III. Gas chromatographic
analysis and confirmation. J. Assoc. Off. Anal. Chem., 64: 749-768.
ANDERSON, J.F. & WOJTAS, M.A. (1986) Honey bees (Hymenoptera: Apidae)
contaminated with pesticides and polychlorinated byphenyls. J. Econ.
Entomol., 79: 1200-1205.
ANDERSSON, A. (1986) Monitoring and biased sampling of pesticide
residues in fruits and vegetables. Methods and results, 1981-1984. Var
Föda, 38(Suppl. 1): 8-55.
ANDERSSON, A. & OHLIN, B. (1986) A capillary gas chromatographic
multiresidue method for determination of pesticides in fruits and
vegetables. Var Föda, 38(Suppl. 2): 79-109.
ANEES, M.A. (1975) Acute toxicity of four organophosphorus
insecticides to a freshwater teleost Channa punctatus (Bloch). Pak. J.
Zool., 7: 135-141.
ANON (1984) Hazardous chemical data: Vol. 2. Chris. Washington, DC, US
Department of Transportation.
APPERSON, C.S., ELSTON, R., & CASTLE, W. (1976) Biological effects
and persistence of methyl parathion in Clear Lake, California.
Environ. Entomol., 5: 1116-1120.
APPERSON, C.S., YOWS, D., & MADISON, C. (1978) Resistance to methyl
parathion in Chaoborus astcoptus. J. Econ. Entomol., 71(5): 772-773.
ARCHER, T.E. & ZWEIG, G. (1959) Direct colorimetric analysis of
cholinesterase-inhibitions with indophenyl acetate. J. Agric. Food
Chem., 7: 1-78.
ARCHER, S.R., McCURLEY, W.R., & RAWLINGS, G.D. (1978) Source
assessment: pesticide manufacturing air emissions - overview and
prioritization. Washington, DC, US Environmental Protection Agency, p.
135 (EPA-600/2-78-004d).
ARNDT, W., SCHINDLBECK, E., PARLAR, H., & KORTE, F. (1981) [Reaction
of the organophosphate insecticides during rubbish composting.]
Chemosphere, 10: 1035-1040 (in German).
ARTERBERRY, J.D., DURHAM, W.F., ELLIOT, J.W., & WOLFE, H.R. (1961)
Exposure to parathion. Arch. environ. Health, 3: 112-121.
ARTHUR, R.D., CAIN, J.D., & BARRANTINE, B.F. (1976) Atmospheric levels
of pesticides in Mississippi delta. Bull. environ. Contam. Toxicol.,
15: 129- 134.
AUDEGOND, L., CATEZ, D., FOULHOUX, P., FOURNEX, R., LE RUMEUR, C.,
L'HOTELLIER, M., & STEPNIEWSKI, J.P. (1989) Potentialisation de la
toxicité de la deltaméthrine par les insecticides organophosphorés. J.
Toxicol. clin. Exp., 9: 163-176.
AULERICH, R.J., RINGER, R.K., & SAFRONOFF, J. (1987) Primary and
secondary toxicity of warfarin, sodium monofluoroacetate, and methyl
parathion in mink. Arch. environ. Contam. Toxicol., 16: 357-366.
AULT, J.A., SCHOFIELD, C.M., JOHNSON, L.D., & WALTZ, R.H. (1979)
Automated gel permeation chromatographic preparation of vegetables,
fruits, and crops for organophosphate residue determination utilizing
flame photometric detection. J. Agric. Food Chem., 27: 825-828.
BABU, S.B.R., JAYASUNDARAMMA, B., & RAMAMURTHI, R. (1986) Behavioral
abnormalities of juveniles of the fish Cyprinus carpio exposed to
methyl parathion. Environ. Ecol., 4: 95-97.
BADAWY, M.I. & EL-DIB, M.A. (1984) Persistence and fate of methyl
parathion in sea water. Bull. environ. Contam. Toxicol., 33: 40-49.
BADAWY, M.I., EL-DIB, M.A. & OSAMA, A.A. (1984) Spill of methyl
parathion in the Mediterranean Sea: A case study at Port-Said, Egypt.
Bull. environ. Contam. Toxicol., 32: 469-477.
BAKER, R.D. & APPLEGATE, H.G. (1970) Effect of temperature and
ultraviolet radiation on the persistence of methyl parathion and DDT
in soils. Agron. J., 62: 509-512.
BAKER, R.D. & APPLEGATE, H.G. (1974) Effect of ultraviolet radiation
on the persistence of pesticides. Texas J. Sci., 5: 53-59.
BARNES, J.M. & DENZ, F.A. (1953) Experimental demyelination with
organo-phosphorous compounds. J. Pathol. Bacteriol., 65: 597-605.
BECKER, G. (1971) [Simultaneous gas chromatographic determination of
chlorinated hydrocarbons and phosphates in plant material.] Dtsch.
Lebensm.- Rundsch., 67: 125-126 (in German).
BECKER, G. (1979) [Multimethod for simultaneous detection of 75 plant
treatment preparations on plant material.] Dtsch. Lebensm.-Rundsch.,
75: 148-152 (in German).
BECKER, G. & SCHUG, P. (1990) [A micromethod for the rapid
determination of chlorinated hydrocarbons and phosphate esters in
plant-derived foods.] Dtsch. Lebensm. Rundsch., 86(8): 239-242 (in
German).
BECKMAN, H. & GARBER, D. (1969) Pesticide residues. Recovery of 65
organophosphorus pesticides from Florisil with a new solvent-elution
system. J. Assoc. Off. Anal. Chem., 52: 286-293.
BEDFORD, C.T. & ROBINSON, J. (1972) The alkylating properties of
organophosphates. Xenobiotica, 2: 307-337.
BEINE, H. (1987) [Phosphoric acid esters and related compounds -
environmental significance and determination in air.] Essen, Immission
Protection Institute for the Land of North Rhine-Westphalia, 31 pp.
(LIS Report No., 69) (in German with English summary).
BELASHOVA, I.G., KHOKHOL'KOVA, G.A., & KLISENKO, M.A. (1983)
[Selection of environmental air sampler containing pesticide vapors.]
Gig. Tr. Prof. Zabol., 54-56 (in Russian).
BELISLE, A.A. & SWINEFORD, D.M. (1988) Simple specific analysis of
organophosphorus and carbamate pesticides in sediments using column
extraction and gas chromatography. Environ. Toxicol. Chem., 7:
749-752.
BENES, V. & JELINEK, R. (1979) Embryotoxicity of parathion methyl with
the Chick Embryotoxicity Screening Test (CHEST). In: Pesticide
residues in food: 1979 evaluations - The monographs. Rome, Food and
Agriculture Organization of the United Nations, pp. 367-368 (FAO Plant
Production and Protection Paper 20, suppl.).
BENKE, G.M. & MURPHY, S.D. (1975) The influence of age on the
toxicity and metabolism of methyl parathion and parathion in male and
female rats. Toxicol. appl. Pharmacol., 31: 254-269.
BENKE, G.M., CHEEVER, K.L., MIRER, F.E., & MURPHY, S.D. (1974)
Comparative toxicity, anticholinesterase action and metabolism of
methyl-parathion and parathion in sunfish and mice. Toxicol. appl.
Pharmacol., 28(1): 97-109.
BENNETT, R.S. (1989) Role of dietary choices in the ability of
bobwhite to discriminate between insecticide-treated and untreated
food. Environ. Toxicol. Chem., 8: 731-738.
BENNETT, R.S., WILLIAMS, B.A., SCHMEDDING, D.W., & BENNETT, J.K.
(1991) Effects of dietary exposure to methyl parathion on egg laying
and incubation in mallards. Environ. Toxicol. Chem., 10: 501-507.
BETOWSKI, L.D. & JONES, T.L. (1988) Analysis of organophosphorus
pesticide samples by high-performance liquid chromatography/mass
spectrometry and high-performance liquid chromatography/mass
spectrometry/mass spectrometry. Environ. Sci. Technol., 22: 1430-1434.
BHASKAR, S.U. & KUMAR, N.V.N. (1981) Thin layer chromatographic
determination of methyl parathion as paraoxon by cholinesterase
inhibition. J. Assoc. Off. Anal. Chem., 64: 1312-1314.
BHUNIA, A.K, BASU, N.K., ROY, D., CHAKRABARTI, A., & BANERJEE, S.K.
(1991) Growth, chlorophyll content, nitrogen-fixing ability, and
certain metabolic activities of Nostoc muscorum : effect of methyl
parathion and benthiocarb. Bull. environ. Contam. Toxicol., 47: 43-50.
BHASKAR, S.U. & KUMAR, N.V.N. (1982) Selection of enzyme sources for
improved sensitivity in enzymic determination of organophosphorus
pesticides. J. Assoc. Off. Anal. Chem., 65: 1297-1298.
BHASKAR, S.U. & KUMAR, N.V.N. (1984) A rapid colorimetric method for
determination of methyl parathion residues. Pesticides, 18: 17-18.
BIGLEY, W.S., PLAPP, F.W. Jr, HANNA, R.L., & HARDING, J.A. (1981)
Effect of toxaphene, camphene, and cedar oil on methyl parathion
residues on cotton. Bull. environ. Contam. Toxicol., 27: 90-94.
BLANCO, C.C. & SANCHEZ, F.G. (1989) Derivative spectrophotometric
determination of the organophosphate insecticide methyl parathion in
commercial formulations. Analusis, 17(1-2): 80-86.
BOMHARD, E., LOSER, E., & SCHILDE, B. (1981) [E 605-methyl (methyl
parathion). Chronic toxicological studies in rats (2-year feeding
trial).] Wuppertal-Elberfeld, Bayer AG, Institute of Toxicology
(Unpublished Report No. 9889, submitted to WHO by Bayer AG,
Leverkusen, Germany) (in German).
BOURQUE, C.L., DUGUAY, M.M., & GAUTREAU, Z.M. (1989) The determination
of reducible pesticides by adsorptive stripping voltammetry. Int. J.
environ. Stud., 37: 187-197.
BOURQUIN, A.W., GARNAS, R.L., PRITCHARD, P.H., WILKES, F.G., CRIPE,
C.R., & RUBINSTEIN, N.I. (1979) Interdependent microsomes for the
assessment of pollutants in the marine environment. Int. J. environ.
Stud., 13: 131-140.
BOWMAN, M.C. (1981) Analysis of organophosphorus pesticides. In: Moye
H.A., ed. Analysis of pesticide residues, New York, Basel, Marcel
Dekker, pp. 263-332.
BOWMAN, M.C. & BEROZA, M. (1967) Temperature-programmed gas
chromatography of 20 phosphorus-containing insecticides on 4 different
columns and its application to the analysis of milk and corn silage.
J. Assoc. Off. Anal. Chem., 50: 1228-1236.
BOWMAN, B.T. & SANS, W.W. (1979) The aqueous solubility of
twenty-seven insecticides and related compounds. J. Environ. Sci.
Health, B14: 625-634.
BRAECKMAN, R.A., GODEFROOT, M.G., BLONDEEL, G.M., & BELPAIRE, F.M.
(1980) Kinetic analysis of the fate of methyl parathion in the dog.
Arch. Toxicol., 43: 263-271.
BRAECKMAN, R.A., AUDENAERT, F., WILLEMS, J.L., BELPAIRE, F.M., &
BOGAERT, M.G. (1983) Toxicokinetics of methyl parathion and parathion
in the dog after intravenous and oral administration. Arch. Toxicol.,
54: 71-82.
BRANCA, P. & QUAGLINO, P. (1988) [Presence of antiparasite pesticides
in imported potatoes.] Ind. Aliment., 27: 427-431 (in Italian).
BRATIN, K., KISSINGER, P.T., & BRUNTLETT, C.S. (1981) Reductive mode
thin-layer amperometric detector for liquid chromatography. J. Liq.
Chromatogr., 4: 1777-1795.
BREWER, L.W., DRIVER, C.J., KENDALL, R.J., ZENIER, C., & LACHER, T.E.
Jr. (1988) Effects of methyl parathion in ducks and duck broods.
Environ. Toxicol. Chem., 7: 375-379.
BRODESSER, J. & SCHOELER, H.F. (1987) An improved extraction method
for the quantitative analysis of pesticides in water. Zbl. Bakteriol.
Mikrobiol. Hyg. Ser. B, 185: 183-185.
BRODEUR, J. & DU BOIS, K.P. (1963) Comparison of acute toxicity of
anticholinesterase insecticides to weanling and adult male rats. Proc.
Soc. Exp. Biol. Med., 114: 509-511.
BROWN, L.C., CATHEY, G.W., & LINCOLN, C. (1962) Growth and development
of cotton as affected by toxaphene-DDT, methyl parathion, and calcium
arsenate. J. Econ. Entomol., 55: 298-301.
BRUCHET, A., COGNET, L., & MALLEVIALLE, J. (1984) Continuous
composite sampling and analysis of pesticides in water. Water Res.,
18: 1401-1409.
BUBIEN, Z. (1971) [Intoxication of animals with phosphoorganic
insecticides.] Med. Wet., 27(10): 611-612 (in Polish with English
summary).
BUCK, N.A., ESTESEN, B.J., & WARE, G.W. (1980) Dislodgeable
insecticide residues on cotton foliage: fenvalarate, permethrin,
sulprofos, chlorpyrifos, methyl parathion, EPN, oxamyl, and
profenofos. Bull. environ. Contam. Toxicol., 24: 283-288.
BUERGER, T.T, KENDALL, R.J., MUELLER, B.S., DE VOS, T., & WILLIAMS,
B.A. (1991) Effects of methyl parathion on northern bobwhite
survivability. Environ. Toxicol. Chem., 10: 527-532.
BUTLER, P.A. (1964) Commercial fishery investigation. Washington, DC,
US Department of the Interior, Fish and Wildlife Service, pp. 5-28
(Circular No. 199).
BUTLER, P.A. & SCHUTZMANN, R.L. (1978) Residues of pesticides and PCBs
in estuarine fish, 1972-1976. National Pesticide Monitoring Program.
Pestic. Monit. J., 12: 51-59.
BUTLER, L.C., STAIFF, D.C., & DAVIES, J.E. (1981) Methyl parathion
persistence in soil following simulated spillage. Arch. environ.
Contam. Toxicol., 10: 451-458.
CAPRIEL, P, HAISCH, A., & SHAHAMAT, U. (1986) Supercritical methanol:
an efficacious technique for the extraction of bound pesticide
residues from soil and plant samples. J. Agric. Food Chem., 34: 70-73.
CARERE, A., ORTALI, V.A., CARDAMONE, G., & MORPURGO, G. (1978)
Mutagenicity of dichlorvos and other structurally related pesticides
in Salmonella and Streptomyces. Chem.-Biol. Interact., 22:
297-308.
CAREY, A.E. & KUTZ, F.W. (1985) Trends in ambient concentrations of
agrochemicals in humans and the environment of the United States.
Environ. Monit. Assess., 5: 155-163.
CAREY, A.E., GOWEN, J.A., TAI, H., MITCHELL, W.G., & WIERSMA, B.
(1978) Soils, pesticide residue levels in soils and crops, 1971 -
National Soils Monitoring Program (III). Pestic. Monit. J., 12:
117-136.
CAREY, A.E., GOWEN, J.A., TAI, H., MITCHELL, W.G., & WIERSMA, G.B.
(1979) Pesticide residue levels in soils and crops from 37 states,
1972. National Soils Monitoring Program (IV). Pestic. Monit. J., 12:
209-229.
CHAMBERS, J.E. & YARBROUGH, J.D. (1974) Parathion and methyl-parathion
toxicity to insecticide-resistant and susceptible mosquitofish
Gambusia affinis. Bull. environ. Contam. Toxicol., 11(4): 315-320.
CHANG, C.S. & LANGE, W.H. (1967) Laboratory and field evaluation of
selected pesticides for control of the red crayfish in California rice
fields. J. Econ. Entomol., 60(21): 473-477.
CHAUDHRY, G.R., ALI A.N., & WHEELER, W.B. (1988) Isolation of a methyl
parathion-degrading Pseudomonas sp. that possesses DNA homologous to
the opd gene from a Flavobacterium sp. Appl. environ. Microbiol.,
54(2): 288-293.
CHEAH, M.L., AVAULT, J.W. Jr, & GRAVES, J.B. (1980) Some effects of
rice pesticides on crawfish. Louisiana. Agric., 23(2): 8-9, 11.
CHEMICAL INFORMATION SERVICES, LTD (1988) Directory of world chemical
producers: 1989/90 edition. Oceanside, New York, Chemical Information
Services, Ltd, Publisher.
CHEN, H.H., HSUEH, J.L., SIRIANNI, S.R., & HUANG, C.C. (1981)
Induction of sister-chromatid exchanges and cell cycle delay in
cultured mammalian cells treated with eight organophosphorus
pesticides. Mutat. Res., 88: 307-316.
CHEN, Z., ZABIK, M.J., & LEAVITT, R.A. (1984) Comparative study of
thin film photodegradative rates for 36 pesticides. Ind. Eng. Chem.
Prod. Res. Dev., 23: 5-11.
CHERNYAK, S.M. & ORADOVSKII, S.G. (1980) [Determination of
organochlorine and organophosphorus pesticides in sea water by
gas-liquid chromatography.] Metody Issled. Org. Veshchestva Okeane,
1980: 291-303 (in Russian).
CHMIL, V.D., GRAHL, K., & STOTTMEISTER, E. (1978) Chromatographic
methods of determining residual amounts of pesticides in water. Zh.
Anal. Khim., 33: 2420-2425.
CHUKWUDEBE, A., MARCH, R.B., OTHMAN, M., & FUKUTO, T.R. (1989)
Formation of trialkyl phosphorothioate esters from organophosphorus
insecticides after exposure to either ultraviolet light or sunlight.
J. Agric. Food Chem., 37: 539-545.
CLARK, D.R. Jr (1986) Toxicity of methyl parathion to bats: mortality
and coordination loss. Environ. Toxicol. Chem., 5(2): 191-195.
CLARK, G.J., GOODIN, R.R., & SMILEY, J.W. (1985) Comparison of
ultraviolet and reductive amperometric detection for the determination
of ethyl and methyl parathion in green vegetables and surface water
using high-performance liquid chromatography. Anal. Chem., 57:
2223-2228.
COBURN, J.A. & CHAU, A.S.Y. (1974) Confirmation of pesticide residue
identity. VIII. Organophosphorus pesticides. J. Assoc. Off. Anal.
Chem., 57: 1272- 1278.
COHEN, M.L. & STEINMETZ, W.D. (1986) Foliar washoff of pesticides by
rainfall. Environ. Sci. Technol., 20: 521-523.
COLE, C.L., MCCASLAND, W.E., & DACUS, S.C. (1986) The persistence of
selected insecticides used in water and in water-oil sprays as related
to worker re-entry. Southwest. Entomol., 11(Suppl.): 83-87.
COMPTON, B. (1973) Analysis of pesticides in air. Prog. Anal. Chem.,
5: 133-152.
CORNELIUSSEN, P.E. (1970) Residues in food and feed. Pesticide
residues in total diet samples (V). Pestic. Monit. J., 4: 89-105.
COSTA, L.G. & MURPHY, S.D. (1984) Interaction between acetaminophen
and organophosphates in mice. Res. Commun. Chem. Pathol. Pharmacol.,
44: 389-400.
COWART, R.P, BONNER, F.L., & EPPS, E.A. Jr (1971) Rate of hydrolysis
of seven organophosphate pesticides. Bull. environ. Contam. Toxicol.,
6: 231-234.
CRIPE, G.M., NIMMO, D.R., & HAMAKER, T.L. (1981) Effects of two
organophosphate pesticides on swimming stamina of the mysid
Mysidopsis bahia. In: Vernberg, J.,Calabrese, A., Thurberg, F.P.,
& Vernberg, W.B., ed. Biological monitoring of marine pollutants. New
York, London, San Francisco, Academic Press, pp. 21-36.
CRIPE, C.R., WALTER, W.W., PRITCHARD, P.H., & BOURQUIN, A.W. (1987) A
shake-flask test for estimation of biodegradability of toxic organic
substances in the aquatic environment. Ecotoxicol. environ. Saf.,
14(3): 239-251.
CROFT, B.A. (1977) Resistance in arthropod predators and parasites.
In: Watson, D.L. & Brown, A.W.A., ed. Pesticide management and
insecticide resistance. New York, London, San Francisco, Academic
Press, pp. 377-393.
CROSSLAND, N.O. & BENNETT, D. (1984) Fate and biological effects of
methyl parathion in outdoor ponds and laboratory aquaria, I: Fate.
Ecotoxicol. Environ. Saf., 8: 471-481.
CROSSLAND, N.O. & ELGAR, K.E. (1983) Fate and biological effects of
insecticides in ponds. In: Matsunaka, S., Hutson, D.H., & Murphy,
S.D., ed. IUPAC. Pesticide chemistry: human welfare and the
environment. Proceedings of the 5th International Congress of
Pesticide Chemistry, Kyoto, Japan, 29 August-4 September 1982. Volume
3: Mode of action, metabolism and toxicology, Oxford, New York,
Pergamon Press, pp. 551-556.
CROSSLAND, N.O., BENNETT, D., & WOLFF, C.J.M. (1986) Evaluation of
models used to assess the fate of chemicals in aquatic systems.
Pestic. Sci., 17: 297-304.
CSERNATONI, M., GYORFI, L., & HARGITAL, E. (1988) Results of pesticide
analyses monitoring programme in surface waters in Hungary between
1977 and 1986. In: Abbou, R., ed. Hazardous waste: detection, control,
treatment. Amsterdam, Oxford, New York, Elsevier Science Publishers,
pp. 861-868.
CURINI, M., LAGANA, A., PETRONIO, B.M., & RUSSO, M.V. (1980)
Determination of organophosphorus pesticides by thin-layer
chromatography. Talanta, 27: 45-48.
CUSTER, T.W. & MITCHELL, C.A. (1987) Exposure to insecticides of
brushland wildlife within the Lower Rio Grande Valley, Texas, USA.
Environ. Pollut., 45(3): 207-220.
DALDRUP, T., SUSANTO, F., & MICHALKE, P. (1981) [Combination of TLC,
GLC (OV 1 and OV 17) and HPLC (RP 18) for a rapid detection of drugs.]
Fresenius Z. Anal. Chem., 308: 413-427 (in German).
DALDRUP, T., MICHALKE, P., & BOEHME, W. (1982) A screening test for
pharmaceuticals, drugs and insecticides with reversed-phase liquid
chromatography - retention data of 560 compounds. Chromatogr. Newsl.,
10: 1-7.
DALY, I.W.A. (1983) Evaluation report for US-EPA of a two-year chronic
feeding study of methyl parathion in rats. East Millstone, New Jersey,
Biodynamics Inc. (Unpublished report, submitted to WHO by Bayer AG,
Leverkusen, Germany).
DAMICO, J.N., BARRON, R.P, & SPHON, J.A. (1969) Field ionization
spectrum of some pesticidal and other biologically significant
compounds. Int. J. mass Spectrom. ion Phys., 2: 161-182.
DANKA, R.G., RINDERER, T.E., HELLMICH, II, R.L., & COLLINS, A.M.
(1986) Comparative toxicities of four topically applied insecticides
to Africanized and European Honey Bees (Hymenoptera: Apidae). J. Econ.
Entomol., 79: 18-21.
DAVIDSON, J.H., RAO, P.S.C, OU, L.T., WHEELER, W.B., & ROTHWELL, F.F.
(1980) Adsorption, movement, and biological degradation of large
concentrations of selected pesticides in soils. Gainesville, Florida,
Florida University.
DAVIS, J.E., STAIFF, D.C., BUTLER, L.C., & STEVENS, E.R. (1981)
Potential exposure to dislodgeable residues after application of two
formulations of methyl parathion to apple trees. Bull. environ.
Contam. Toxicol., 27: 95-100.
DEAN, B.J. (1972) The mutagenic effects of organophosphorus pesticides
on micro-organisms. Arch. Toxicol., 30: 67-74.
DE CASSIA STOCCO, R., BECAK, W., GAETA, R., & NAZARETH RABELLOGAY, M.
(1982) Cytogenetic study of workers exposed to methylparathion. Mutat.
Res., 103: 71-76.
DEGRAEVE, N. & MOUTSCHEN, J. (1984) Absence of genetic and cytogenetic
effects in mice treated by the organophosphorus insecticide parathion,
its methyl analogue, and paraoxon. Toxicology, 32: 177-183.
DEGRAEVE, N., CHOLLET, M.C., & MOUTSCHEN, J. (1984) Cytogenic and
genetic effects of subchronic treatment with organophosphorus
insecticides. Arch. Toxicol., 56: 66-73.
DEICHMANN, W.B., PUGLIESE, W., & CASSIDY, B.S. (1952) Effects of
dimethyl and diethyl paranitrophenyl thiophosphate on experimental
animals. Ind. Hyg. occup. Med., 5: 44-51.
DELI, E. & KISS, Z. (1986) The effect of organophosphorous insecticide
Wofatox 50 EC on the adenylate cyclase activity of chicken embryo
muscle. Biochem. Pharmacol., 35: 1603-1605.
DELI, E. & KISS, Z. (1988) Effect of parathion and methylparathion on
protein content of chicken embryo muscle in vivo. Biochem.
Pharmacol., 37: 3251-3256.
DELI, E. & VARNAGY, L. (1985) Teratological examination of Wofatox 50
EC (50% methyl parathion) on pheasant embryos. Anat. Anz. Jena, 158:
237-240.
DELI, E., SOMLYAY, I., & VARNAGY, L. (1985) Biochemical study of
muscle samples from chicken embryos affected by Wofatox 50 EC. Arch.
Toxicol, 8: 277-279.
DE POTTER, M., MULLER, R., & WILLEMS, J. (1978) A method for the
determination of some organophosphorus insecticides in human serum.
Chromatographia, 11: 220-222.
DE SCHRYVER, E., DE REU, L., BELPAIRE, F., & WILLEMS, J. (1987)
Toxicokinetics of methyl paraoxon in the dog. Arch. Toxicol., 59:
319-322.
DESHPANDE, A.A. & SWAMY, G.S. (1987) Induction of proline accumulation
by methyl parathion in sorghum ( Sorghum bicolor L.). Curr. Sci., 56:
1068-1070.
DEVI, Y.P., KUMAR, N.V., & HARAN, N.V.H. (1982) Survey of pesticide
residue analysis in water resources and paddy straw samples from some
farms of the Nellore district of Andhra Pradesh. Pollut. Res., 1:
21-23.
DE WIT, J.S.M., PARKER, C.E., TOMER, KB., & JORGENSON, J.W. (1987)
Direct coupling of open-tubular liquid chromatography with mass
spectrometry. Anal. Chem., 59: 2400-2404.
DICK, G.L., HEENAN, M.P., LOVE, J.L., UDY, P.B., & DAVIDSON, F. (1978)
Survey of trace elements and pesticide residues in the New Zealand
diet. 2. Organochlorine and organophosphorus pesticide residue
content. N. Z. J. Sci., 21: 71-78.
DILLE, J.R. & SMITH, P.W. (1964) Central nervous system effects of
chronic exposure to organophosphate insecticides. Aerosp. Med., 35:
475-478.
DI MUCCIO, A., AUSILI, A., VERGORI, L., CAMONI, I., DOMMARCO, R. &
GAMBETTI, L. (1990) Single step multi-cartridge clean-up for
organophosphate pesticide residue determination in vegetable oil
extracts by gas chromatography. Analyst. 115: 1167-1169.
DOROUGH, H.W. (1978) Conjugation reactions of pesticides and their
metabolites with sugars. In: Advances in pesticide science. Symposia
papers presented at the Fourth International Congress of Pesticide
Chemistry, Zurich, Switzerland, 24-28 July 1978. Part 3: Biochemistry
of pests and mode of action of pesticides; pesticide degradation;
pesticide residues; formulation chemistry. Oxford, New York, Pergamon
Press, pp. 526-536.
DORTLAND, R.J. (1980) Toxicological evaluation of parathion and
azinphosmethyl in freshwater model ecosystems. Wageningen, Centre for
Agricultural Publishing and Documentation (PUDOC), 111 pp.
(Agricultural Research Report No. 898).
DRAPER, W.M. & STREET, J.C. (1981) Drift from a commercial, aerial
application of methyl and ethyl parathion: an estimation of human
exposure. Bull. environ. Contam. Toxicol., 26: 530-536.
DU BOIS, K.P. (1961) Potentiation of the toxicity of organophosphorous
compounds. Adv. Pest. Control Res., 4: 117-151.
DU BOIS, K.P. (1969) Combined effects of pesticides. Can. Med. Assoc.
J., 100: 173-179.
DU BOIS, K.P. & COON, J.M. (1952) Toxicology of organic
phosphorus-containing insecticides to mammals. Ind. Hyg. occup. Med.,
6: 9-13.
DU BOIS, K.P. & KINOSHITA, F.K. (1968) Influence of induction of
hepatic microsomal enzymes by phenobarbital on toxicity of organic
phosphate insecticides (33402). Proc. Soc. Exp. Biol. Med., 129:
699-702.
DURHAM, W.F. & HAYES, W.J. Jr (1962) Organic phosphorus poisoning and
its therapy. Arch. environ. Health, 6: 21-47.
EARNEST, R. (1970) Effects of pesticides on aquatic animals in the
estuarine and marine environment. In: Progress in sport fishery
research, 1970, Washington, DC, US Department of the Interior, Bureau
of Sport Fisheries and Wildlife, pp. 10-13.
EASLEY, C.B., LAUGHLIN, J.M., GOLD, R.E., & TUPY, D.R. (1981) Methyl
parathion removal from denim fabrics by selected laundry procedures.
Bull. environ. Contam. Toxicol., 27: 101-108.
EBING, W. (1985) [Multimethod for the determination of pesticide
residues in dead honeybees. I. Chlorine and phosphorus insecticides.]
Fresenius Z. Anal. Chem., 321: 45-48 (in German).
EBING, W. (1987) [Gas chromatography of pesticides. Tabular literature
abstracts, Series XV.] Berlin-Dahlem, Federal Biological Institute for
Agriculture and Forestry (Report No. 236) (in German).
EICHELBERGER, J.W. & LICHTENBERG, J.J. (1971) Persistence of
pesticides in river water. Environ. Sci. Technol., 5: 541-544.
EISLER, R. (1969) Acute toxicities of insecticides to marine decapod
crustaceans. Crustaceana, 16(3): 300-310.
EISLER, R. (1970a) 45. Factors affecting pesticide-induced toxicity in
an estuarine fish. Washington, DC, US Department of the Interior, Fish
and Wildlife Service, Bureau of Sport Fisheries and Wildlife, 20 pp.
(Technical Papers of the Bureau of Sport Fisheries and Wildlife No.
45/46).
EISLER, R. (1970b) 46. Acute toxicities of organochlorine and
organophosphorus insecticides to estuarine fishes. Washington, DC, US
Department of the Interior, Fish and Wildlife Service, Bureau of Sport
Fisheries and Wildlife, 12 pp. (Technical Papers of the Bureau of
Sport Fisheries and Wildlife No. 45/46).
ELKINS, E.R., FARROW, R.P., & KIM, E.S. (1972) The effect of heat
processing and storage on pesticide residues in spinach and apricots.
J. Agric. Food Chem., 20: 286-291.
ELLIOTT, J.W., WALKER, K.C., PENICK, A.E., & DURHAM, W.F. (1960) A
sensitive procedure for urinary p-nitrophenol determination as a
measure of exposure to parathion. J. Agric. Food Chem., 8: 111-113.
EMBER, M., MINDSZENTY, L., RENGEI, B., & GAL, G. (1970) Changes of
vitamin A in blood and liver of organophosphorus poisoned suicides.
Res. Commun. Chem. Pathol. Pharmacol., 1: 561-571.
ESPIGARES, M., ROMAN, I., GONZALEZ ALONSO, J.M., DE LUIS, B., YESTE,
F., & GALVEZ, R. (1990) Proposal and application of an ecotoxicity
biotest based on Escherichia coli. J. appl. Toxicol., 10: 443-446.
FABACHER, D.L. (1976) Toxicity of endrin and endrin-methyl parathion
formulation to largemouth bass fingerlings. Bull Environ. Contam.
Toxicol., 16: 376-378.
FAIRBROTHER, A., MEYERS, S.M., & BENNETT, R.S. (1988) Changes in
mallard hen and brood behaviors in response to methyl
parathion-induced illness of ducklings. Environ. Toxicol. Chem., 7:
499-503.
FAN, A.M-M. (1981) Effects of pesticides on immune competency:
influence of methyl parathion and carbofuran on immunologic responses
to Salmonella typhimurium infection. Diss. Abstr. Int., 41(08):
2962-B.
FAO (1985) Second Government Consultation on International
Harmonization of Pesticide Registration Requirements, Rome, 11-15
October, 1982. Rome, Food and Agriculture Organization of the United
Nations.
FAO/WHO (1969) 1968 Evaluation of some pesticide residues in food,
Geneva, World Health Organization (FAO PL: 1968/M/9/1; WHO Food
Add./69.35).
FAO/WHO (1973) 1972 Evaluation of some pesticide residues in food,
Geneva, World Health Organization (AGP: 1972/M/9/1; WHO Pesticide
Residues Series No. 2).
FAO/WHO (1976) 1975 Evaluation of some pesticide residues in food,
Geneva, World Health Organization (AGP: 1975/M/13; WHO Pesticide
Residues Series No. 5).
FAO/WHO (1980) 1979 Evaluation of some pesticide residues in food,
Rome, Food and Agriculture Organization of the United Nations (FAO
Plant Production and Protection Paper 20 Sup).
FAO/WHO (1981) 1980 Evaluation of some pesticide residues in food,
Rome, Food and Agriculture Organization of the United Nations (FAO
Plant Production and Protection Paper 26 Sup).
FAO/WHO (1985) 1984 Evaluation of some pesticide residues in food,
Rome, Food and Agriculture Organization of the United Nations (FAO
Plant Production and Protection Paper 67).
FAO/WHO (1986) Codex maximum limits for pesticide residues, 3rd ed.
Rome, Codex Alimentarius Commission, Food and Agriculture Organization
of the United Nations (CAC/Vol XIII).
FARRAN, A., CORTINA, J.L., DE PABLO, J., & BARCELO, D. (1990) On-line
continuous flow extraction system in liquid chromatography with
ultraviolet and mass spectrometric detection for the determination of
selected organic pollutants. Anal. chim. Acta, 234: 119-126.
FAZEKAS, I.G. (1971) [Concerning the macroscopic and microscopic
changes after Wofatox poisoning (methyl parathion).] Z. Rechstmed.,
68: 189-194 (in German).
FAZEKAS, I.G. & RENGEI, B. (1967) [Content of methyl parathion in
human organs after fatal Wofatox-poisoning.] Arch. Toxikol., 22:
381-386 (in German).
FEDORENKO, A.P., ALEEVA, L.V., & TITOK, V.A. (1981) Organophosphorus
pesticide accumulation in warm-blooded animals after chemical
treatment of the forest. Vestn. Zool., 4: 89-92.
FDA (1988) Food and Drug Administration pesticide program. Residues
in food. Washington, DC, Food and Drug Administration, 24 pp.
FISH, S.A. (1966) Organophosphorus cholinesterase inhibitors and fetal
development. J. Obstet. Gynecol., 96: 1148-1154.
FLANDERS, R.V., BLEDSOE, L.W., & EDWARDS, C.R. (1984) Effects of
insecticides on Pediobus faveolatus (Hymenoptera: Eulophidae), a
parasitoid of the Mexican bean beetle (Coleoptera: Coccinellidae).
Environ. Entomol., 13: 902-906.
FLUCKE, W. (1984) [Methyl parathion - Summary toxicological
information.] Wuppertal-Elberfeld, Bayer AG, Institute of Toxicology
(Unpublished report submitted to WHO by Bayer AG, Leverkusen, Germany)
(in German).
FLUCKE, W. & KIMMERLE, G. (1977) [Methyl parathion and ethyl
parathion, studies on the acute toxicity of the two active
substances.] Wuppertal-Elberfeld, Bayer AG, Institute of Toxicology
(Unpublished report No. 6823, submitted to WHO by Bayer AG,
Leverkusen, Germany) (in German).
FOSTER, G.D. & CROSBY, G.D. (1987) Comparative metabolism of
nitroaromatic compounds in freshwater, brackish water and marine
decapod crustaceans. Xenobiotica, 17(12): 1393-1404.
FOSTER, G.D. & ROGERSON, P.F. (1990) Enhanced preconcentration of
pesticides from water using the Goulden large-sample extractor. Int.
J. environ. anal. Chem., 41: 105-117.
FOSTER, R.L. (1974) Detection and measurement of ambient
organophosphate pesticides. Proc. Annu. Ind. Air Pollut. Control
Conf., 4: 66-98.
FRAWLEY, J.P., WEIR, R., TUSSIG, T., DU BOIS, K.P., & CALANDRA, J.C.
(1963) Toxicologic investigations on Delnav. Toxicol. appl.
Pharmacol., 5: 605-624.
FUCHS, V., GOLBE, S., KUHNERT, M., & OSSWALD, F. (1976) Studies into
prenatal toxic action of parathion methyl on Wistar rats and
comparison with prenatal toxicity of cyclophosphamide and trypan blue.
Arch. exp. vet. Med., 30: 343-350.
FUCHS, V., KUEHNERT, M., & GOLBS, S. (1986) Detoxifying action of
humic acids on selected contaminants. Veterinärmedizin., 41: 712-713.
FUHREMANN, T.W. (1980) Environmental behavior of insecticides. Diss.
Abstr. Int. B., 40(7): 3075-3076.
FUHREMANN, T.W. & LICHTENSTEIN, E.P. (1978) Release of soil-bound
methyl (14C)parathion residues and their uptake by earthworms and
oat plants. J. Agric. Food Chem., 26(3): 605-610.
FUKAMI, J. & SHIHIDO, T. (1963) Studies on the selective toxicities
of organic phosphorous insecticides (III). Botyu Kagaku, 28: 77-81.
FUNCH, F.H. (1981) Analysis of residues of seven pesticides in some
fruits and vegetables by high-pressure liquid chromatography. Z.
Lebensm.-Unters. Forsch, 173: 95-98.
GABICA, J., WYLLIE, J., WATSON, M., & BENSON, W.W. (1971) Example of
flame photometric analysis for methyl parathion in rat whole blood and
brain tissue. Anal. Chem., 43: 1102-1105.
GAINES, T.B. (1960) The acute toxicity of pesticides to rats.
Toxicol. appl. Pharmacol., 2: 88-99.
GAINES, T.B. (1969) Acute toxicity of pesticides. Toxicol. appl.
Pharmacol., 14: 515-534.
GAJAN, R.J. (1969) Collaborative study of confirmative procedures by
single-sweep oscillographic polarography for the determination of
organophosphorus pesticide residues in nonfatty foods. J. Assoc. Off.
Anal. Chem., 52: 811-817.
GAMBRELL, R.P., TAYLOR, B.A., REDDY, K.S., & PATRICK, W.H. Jr (1984)
Fate of selected toxic compounds under controlled redox potential and
pH conditions in soil and sediment-water systems. Athens, Georgia, US
Environmental Protection Agency, Office of Research and Development,
Environmental Research Laboratory.
GAR, K.A., SAZONOVA, N.A., FADEEV, Y.N., VLADIMIROVA, I.L., &
GOLUBEVA, Z.Z. (1958) [Incorporation and excretion of dimethyl
4-nitrophenyl thiophosphate in guinea pigs.] Org. Insektofungits.
Gerbits., 1958: 93-105 (in Russian).
GARRIDO, S.J.J. & MONTEOLIVA, H.M. (1981) [Qualitative and
quantitative determination of residues of diazinon, dimethoate,
ethion, malathion, methyl parathion and parathion in soil extracts by
thin-layer chromatography, and confirmation with gas chromatography.]
An. Edafol. Agrobiol., 40: 1787-1798 (in Spanish).
GAUGHAN, L.C., ENGEL, J.L., & CASIDA, J.E. (1980) Pesticide
interactions: effects of organophosphorus pesticides on the
metabolism, toxicity, and persistence of selected pyrethroid
insecticides. Pestic. Biochem. Physiol., 14: 81-85.
GERSTL, Z. & HELLING, C.S. (1985) Fate of bound methyl parathion
residues in soils as affected by agronomic practices. Soil Biol.
Biochem., 17(5): 667-673.
GHOSH, P., BHATTACHARYA, S., & BHATTACHARYA, S. (1989) Impact of
nonlethal levels of metacid-50 and carbaryl on thyroid function and
cholinergic system of Channa punctatus. Biomed. environ. Sci., 2:
92-97.
GHOSH, P., BHATTACHARYA, S., & BHATTACHARYA, S. (1990) Impairment of
the regulation of gonadal function in Channa punctatus by metacid-50
and carbaryl under laboratory and field conditions. Biomed. environ.
Sci., 3: 106-112.
GILE, J.D. & GILLETT, J.W. (1981) Transport and fate of
organophosphate insecticides in a laboratory model. J. Agric. Food
Chem., 29: 616-621.
GILLESPIE, A.M. & WALTERS, S.M. (1986) HPLC silica column
fractionation of pesticides and PCB from butterfat. J. liq.
Chromatogr., 9: 2111-2141.
GILLESPIE, A.M. & WALTERS, S.M. (1989) Semi-preparative reverse phase
HLPC fractionation of pesticides from edible fats and oils. J. liq.
Chromatogr., 12(9): 1687-1703.
GILLETT, J.W. & GILE, J.D. (1976) Pesticide fate in terrestrial
laboratory ecosystems. Int. J. environ. Stud., 10: 15-22.
GILOT-DELHALLE, J., COLIZZI, A., MOUTSCHEN, J., & MOUTSCHEN-DAHMEN, M.
(1983) Mutagenicity of some organophosphorus compounds at the ade 6
locus of Schizosaccharomyces pombe. Mutat. Res., 117: 139-148.
GOEDICKE, H.-J. (1989) [Exposure to residues on leaf surfaces
following the use of organophosphorus insecticides in high-intensity
apple growing.] Z. Gesamte Hyg., 35: 533-535 (in German).
GOEDICKE, H.-J. & WINKLER, R. (1976) [On the residue of methyl
parathion in soil.] Nachrichtenbl. Pflanzenschutz., 30(5): 100-101
(in German). GOLOVATYI, V.G., KOROL, E.N., KLISENKO, M.A., & GIRENKO,
D.B. (1982) FDMS study of pesticides. Eur. J. Mass Spectrom.
biochem., Med. environ. Res., 2: 97-100.
GONCHARUK, E.I., LIPATOVA, T.E., & FILATOVA, I.N. (1988) Conditions
for elevation of pesticide content in the atmospheric boundary layer
of agricultural fields. Gig. i Sanit., 1: 25-27.
GOYER, R. & CHEYMOL, J. (1967) Contribution à l'étude pharmacologique
de quelques dérivés soufrés de l'acide orthophosphorique trisubstitué.
II. Essais de toxicité. Rev. Can. Biol., 26: 35-41.
GOZA, S.W. (1972) Infrared analysis of pesticide formulations. J.
Assoc. Off. Anal. Chem., 55: 913-917.
GREENHALGH, R., BLACKWELL, B.A., PRESON, C.M., & MURRAY, W.J. (1983)
Phosphorus-31 nuclear magnetic resonance analysis of technical
organophosphorus insecticides for toxic contaminants. J. Agric. Food
Chem., 31: 710-713.
GRETCH, F.M. & ROSEN, J.D. (1984) Automated sample cleanup for
pesticide multiresidue analysis. Part II. Design and evaluation of
column chromatographs module. J. Assoc. Off. Anal. Chem., 67: 783-789.
GRETCH, F.M. & ROSEN, J.D. (1987) Automated sample cleanup for
pesticide multiresidue analysis. Part III. Evaluation of complete
system for screening subtolerance residues in vegetables. J. Assoc.
Off. Anal. Chem., 70: 109-111.
GROVER, I.S. & MALHI, P.K. (1985) Genotoxic effects of some
organophosphorus insecticides. I. Induction of micronuclei in bone
marrow cells in rats. Mutat. Res., 155: 131-134.
GROVER, I.S. & MALHI, P.K. (1987) Genotoxic effects of some
organophosphorus pesticides. II. In vivo chromosomal aberration
bioassay in bone marrow cells in rat. Mutat. Res., 188: 45-51.
GRUE, C.E. (1982) Response of common grackles to dietary
concentrations of four organophosphate pesticides. Arch. environ.
Contam. Toxicol., 11: 617-626.
GUPTA, R.C. & KADEL, W.L. (1990) Methyl parathion acute toxicity:
prophylaxis and therapy with memantine and atropine. Arch. int.
Pharmacodyn., 305: 208-221.
GUPTA, R.C., THORNBURG, J.F., STEDMAN, D.B., & WELSCH, F. (1984)
Effect of subchronic administration of methyl parathion on in vivo
protein synthesis in pregnant rats and their conceptuses1,2.
Toxicol. appl. Pharmacol., 92: 457-468.
GUPTA, R.C., RECH, R.H., LOVELL, K.L., WELSCH, F., & THORNBURG, J.E.
(1985) Brain cholinergic, behavioral, and morphological development in
rats exposed in utero to methylparathion. Toxicol. appl.
Pharmacol., 77: 405-413.
GYORFI, L., AMBRUS, A., & BOLYGO, E. (1987) Optimization of
determination and clean-up parameters for sensitive multiresidue
analysis of pesticides. In: Greenhalgh, P. & Roberts, T.R., ed.
IUPAC. Pesticide science and biotechnology. Proceedings of the Sixth
International Congress on Pesticide Chemistry, Ottawa, Canada, 10-15
August 1986. Oxford, London, Blackwell Scientific Publications, pp.
353-356.
HALEY, T.J., FARMER, J.H., HARMON, J.R., & DOOLEY, K.L. (1975)
Estimation of the LD1 and extrapolation of the LD0,1 for five
organothiophosphate pesticides. Eur. J. Toxicol., 8: 229-235.
HANSCH, C. & LEO, A. (1987) The log p database. Claremont, California,
Pomona College, 268 pp.
HAPKE, H.-J., YOUSSEF, S., & OMER, O.H. (1978) [Modification of the
alkylphosphate toxicity in lead contaminated rats.] Dtsch. Tierärztl.
Wochenschr., 85: 321-456 (in German).
HAQUE, R. & FREED, V.H. (1974) Behavior of pesticides in the
environment: Environmental chemodynamics. Residue Rev., 52: 89-116.
HASAN, M. & AHMAD KHAN, N. (1985) Methyl parathion-induced
dose-related alteration in lipid levels and lipid peroxidation in
various regions of rat brain and spinal cord. Ind. J. exp. Biol., 23:
141-144.
HATCHER, J.S. & WISEMAN, R.L. (1969) Epidemiology of pesticide
poisoning in the lower Rio Grande Valley in 1968. Texas Med., 65:
40-43.
HAYES, W.J. Jr (1971) Studies on exposure during the use of
anticholinesterase pesticides. Bull. World Health Organ., 44:
272-288.
HAYES, W.J. Jr & LAWS, E.R. Jr (1991) Handbook of pesticide
toxicology. New York, London, San Francisco, Academic Press, vol. 1-3.
HECHT, G. & WIRTH, W. (1950) [The pharmacology of the phosphoric acid
ester derivatives of thiophoric acid.] Arch. exp. Pharmakol., 211:
264-277 (in German).
HEIMANN, K.G. (1982) [E 120 (methyl parathion): studies of acute oral
and acute dermal toxicity.] Wuppertal-Elberfeld, Bayer AG, Institute
of Toxicology (Unpublished report No. 10897, submitted to WHO by
Bayer AG, Leverkusen, Germany) (in German).
HENNIGHAUSEN, G. (1984) [On the toxicological significance of the
interaction of foreign compounds with glutathione and glutathione
S-transferases.] Z. Gesamle Hyg., 30(11): 603-606 (in German with
English summary).
HERBERT, G.B., PETERLE, T.J., & GRUBB, T.C. (1989) Chronic dose
effects of methyl parathion on nuthatches: cholinesterase and
ptilochronology. Bull. environ. Contam. Toxicol., 42: 471-475.
HERBOLD, B. (1986) [E 120, C.N. methyl parathion-salmonella/microsome
test to evaluate for point mutagenic effect.] Wuppertal-Elberfeld,
Bayer AG, Institute of Toxicology (Unpublished report No. 15306,
submitted to WHO by Bayer AG, Leverkusen, Germany) (in German).
HICKE, K. & THIEMANN, W. (1987) [The decomposition of selected
phosphoric acid esters by UV-irradiation.] Vom Wasser, 69: 85-94 (in
German).
HILL, E.F. & CAMARDESE, M.B. (1986) Lethal dietary toxicities of
environmental contaminants and pesticides to Coturnix. Washington,
DC, US Department of the Interior Fish and Wildlife Service, pp.
100-101 (Fish and Wildlife Technical Report No. 2).
HILL, E.F., HEATH, R.G., SPANN, J.W., & WILLIAMS, J.D. (1975) Lethal
dietary toxicities of environmental pollutants to birds. Washington,
DC, US Department of the Interior Fish and Wildlife Service, pp. 16,
27 (Fish and Wildlife Technical Report No. 191).
HIRAYAMA, K. & TAMAOI, S. (1980) Acute toxicity of methylparathion and
diazinon (pesticide) to larvae of kuruma praure Penaeus japonicus and
of swimming crab Portunus trityberculatus. Bull. Jpn Soc. Sci. Fish.,
46: 117-123.
HIRSCHELMANN, R. & BEKEMEIER, H. (1975) Acute toxicity of demephion
and parathion methyl in dogs. In: Proceedings of the European Society
of Toxicology. Amsterdam Excerpta Medica Foundation, pp. 273-275.
HOLLINGWORTH, R.M., METCALF, R.L., & FUKUTO, T.R. (1967) The
selectivity of sumithion compared with methyl parathion. Metabolism in
the white mouse. J. Agric. Food Chem., 15: 242-249.
HOLM, H.W., KOLLIG, H.P., & PAYNE, W.R. Jr (1983) Fate of methyl
parathion in aquatic channel mirocosms. Environ. Toxicol. Chem., 2:
169-176.
HOLMSTEAD, R.L. & CASIDA, J.E. (1974) Chemical ionization mass
spectrometry of organophosphorus insecticides. J. Assoc. Off. Anal.
Chem., 57: 1050-1055.
HUANG, C.C. (1973) Effect on growth but not on chromosomes of the
mammalian cells after treatment with three organophosphorus
insecticides. Proc. Soc. Exp. Biol. Med., 142: 36-40.
HUDSON, R.H., HAEGELE, M.A., & TUCKER, R.K. (1979) Acute oral and
percutaneous toxicity of pesticides to mallards: correlations with
mammalian toxicity data. Toxicol. appl. Pharmacol., 47: 451-460.
HUDSON, R.H., TUCKER, R.K., & HAEGELE, M.A. (1984) Handbook of
toxicity of pesticides to wildlife, 2nd ed. Washington, DC, US
Department of the Interior, Fish and Wildlife Service, pp. 52-53
(Fish and Wildlife Publication, No. 153).
HUMMEL, S.V. & YOST, R.A. (1986) Tandem mass spectrometry of
organophosphate and carbamate pesticides. Org. mass Spectrom., 21:
785-791.
IARC (1983) Methyl parathion. In: Miscellaneous pesticides. Lyon,
International Agency for Research on Cancer, pp. 131-152 (IARC
Monographs on the evaluation of the carcinogenic risk of chemicals to
humans, Volume 30).
IARC (1987) Methyl parathion. In: Overall evaluations of
carcinogenicity: an updating of IARC monographs, Volumes 2 to 42.
Lyon, International Agency for Research on Cancer, pp. 392 (IARC
Monographs on the evaluation of the carcinogenic risk of chemicals to
humans, Supplement 7).
IZMIROVA, N., SHALASH, S., & KALOYANOVA, F. (1984) [Dynamics of
cholinesterase inhibition in methylparathion poisoning.] Probl. Khig.,
9: 42-49 (in Bulgarian).
JACKSON, E.R. (1976) High-performance and gas-liquid chromatography of
methyl parathion formulations. J. Assoc. Off. Anal. Chem., 59:
740-744.
JACKSON, E.R. (1977a) Collaborative study of gas-liquid
chromatographic method for determining methyl parathion. J. Assoc. Off
Anal. Chem., 60: 720-723.
JACKSON, E.R. (1977b) Collaborative study of high pressure liquid
chromatographic method for determining methyl parathion. J. Assoc.
Off. Anal. Chem., 60: 724-727.
JACKSON, M.D. & LEWIS, R.G. (1978) Volatilization of two methyl
parathion formulations from treated fields. Bull. environ. Contam.
Toxicol., 20: 793-796.
JAGLAN, P.S. & GUNTHER, F.A. (1970) Comparison of hydrolysis rates of
methyl parathion and methyl paraoxon by gas liquid chromatography and
spectrometry. J. chromatogr. Sci., 8: 483-485.
JALALUDDIN, M. & MOHANASUNDARAM, M. (1989) Residual toxicity of four
insecticides recommended for control of coconut coccids on the
parasitoid fauna of Opisina arenosella WLK. Entomol., 14: 199-202.
JARVINEN, A.W. & TANNER, D.K. (1982) Effects of selected controlled
release and corresponding unformulated technical grade pesticides to
the fathead minnow Pimephales promelas. Environ. Pollut., Ser. A,
27(3): 179-195.
JELINEK, R., PETERKA, M., & RYCHTER, Z. (1985) Chick embryotoxicity
screening test - 130 substances tested. Indian J. exp. Biol., 23:
588-595.
JOHANSSON, C.E. (1978) A multiresidue analytical method for
determining organochlorine, organophosphorus, dinitrophenyl and
carbamate pesticides in apples. Pestic. Sci., 9: 313-322.
JOHNSON, R.D. & FINLEY, M.T. (1980) Handbook of acute toxicity of
chemicals to fish and aquatic invertebrates. Summaries of toxicity
tests conducted at Columbia National Fisheries Research Laboratory,
1965-78. Washington, DC, US Department of the Interior Fish and
Wildlife Service, 98 pp. (Resource Publication 137).
JOHNSON, R.D. & MANSKE, D.D. (1976) Residues in food and feed.
Pesticide residues in total diet samples (IX). Pestic. Monit. J., 9:
157-169.
JOHNSON, R.D., MANSKE, D.D., NEW, D.H., & PODREBARAC, D.S. (1981) Food
and feed - pesticide, heavy metal, and other chemical residues in
infant and toddler. Pestic. Monit. J., 15: 39-50.
JOHNSON, M.W., WELTER, S.C., TOSCANO, N.C., & IWATA, Y. (1983) Lettuce
yield reductions correlated with methyl parathion use. J. Econ.
Entomol., 76: 1390-1394.
JONES, D.C.L., SIMMON, V.F., MORTELMANS, K.E., MITCHELL, A.D., EVANS,
E.L., JOTZ, M.M., RICCIO, E.S., ROBINSON, D.E., & KIRKHART, B.A.
(1982) In vitro mutagenicity studies of environmental chemicals.
Research Triangle Park, North Carolina, US Environmental Protection
Agency, Office of Research and Development, Health Effects Research
Laboratory (EPA-600/1-84-003; PB84-138973).
JOSHI, U.M. & THORNBURG, J.E. (1986) Interactions between cimetidine,
methylparathion, and parathion. J. Toxicol. environ. Health, 19:
337-344.
KAGAN, J.S. (1971) [Topical questions about the toxicology of
phosphororganic insecticides.] Ernährungsforschung, 14: 503-514 (in
German).
KALOYANOVA-SIMEONOVA, F.P. (1970) Prevention of intoxication by
pesticides in Bulgaria. In: Proceedings of the Fourth International
Congress on Rural Medicine, Usuda (Tokyo), 1969. Tokyo, Japanese
Association of Rural Medicine, pp. 35-37.
KAMIENSKI, F.X. & MURPHY, S.D. (1971) Biphasic effects of
methylenedioxyphenyl synergists on the action of hexobarbital and
organophosphate insecticides in mice. Toxicol. appl. Pharmacol., 18:
883-894.
KARCSU, S., TOTH, L., MAROSI, G., & SIMON, Z. (1981) Analysis of
histochemical changes due to acute wofatox poisoning in a model
experiment. Morphol. Igazsagugyi Orv. Sz., 21: 289-296.
KARLOG, O., NIELSEN, P., & RASMUSSEN, F. (1978) Toxicokinetics. Arch.
Toxicol., 1(Suppl.): 55-67.
KATKAR, H.N. & BARVE, V.P. (1976) A new spray reagent for the
identification and determination of organophosphorus insecticides by
thin layer chromatography. Curr. Sci., 45: 662-664.
KAWAHARA, F.K., LICHTENBERG, J.J., & EICHELBERGER, J.W. (1967)
Thin-layer and gas chromatographic analysis of parathion and methyl
parathion in the presence of chlorinated hydrocarbons. J. Water
Pollut. Control Fed., 39: 446-457.
KAZAKOVA, M.V., YAKUB, G.G., & MANDRIK, F.I. (1974) Quantitative
detection of metaphos (methyl parathion) in feeds and its effect on
animals. Veterinariya (Moscow), 2: 107.
KHAN, M.S. (1988) An electrolytic reduction method for the
determination of fenitrothion and methyl paration. Pak. J. Sci. Ind.
Res., 31: 20-22.
KHAN, M.S. & HASAN, M. (1988) Dose-related neurochemical changes in
the levels of gangliosides and glycogen in various regions of the rat
brain and spinal cord following methyl parathion administration. Exp.
Pathol., 35: 61-65.
KHATTAT, F.H. & FARLEY, S. (1976) Acute toxicity of certain pesticides
to Acartia tonsa Dana. Narangansett, Rhode Island, US Environmental
Protection Agency, Office of Research and Development, Environmental
Research Laboratory, 23 pp. (EPA-600/3-76-033).
KHEIFETS, L.Y., SOBINA, N.A., & ROMANOV, N.A. (1976) [Use of
differential oscillopolarography for determining substances with
limitable concentrations in water.] Probl. Okhr. Vod, 7: 21-24 (in
Russian).
KHEIFETS, L.Y., ROMANOV, N.A., & SOBINA, N.A. (1980) [Analytical
possibility of differential chronoamperometry with dropping
electrodes.] Zh. Anal. Khim., 35: 874-879 (in Russian).
KIDO, H., BAILEY, J.B., MCCALLEY, N.F., YATES, W.E., & COWDEN, R.E.
(1975) The effect of overhead sprinkler irrigation on methyl parathion
residue on grape leaves. Bull. environ. Contam. Toxicol., 14: 209-213.
KIMMERLE, G. (1975) [Testing of DDT for combined effect with methyl
parathion and fenitrothion in acute tests on rats.]
Wuppertal-Elberfeld, Bayer AG, Institute of Toxicology (Unpublished
report No. 5591, submitted to WHO by Bayer AG, Leverkusen, Germany)
(in German).
KIMMERLE, G. & LORKE, D. (1968) [Toxicology of insecticidal phosphoric
acid esters.] Pflanzenschutz.-Nachr. BAYER, 21: 111-142 (in German).
KJOELHOLT, J. (1985) Determination of trace amounts of
organophosphorus pesticides and related compounds in soils and
sediments using capillary gas chromatography and a
nitrogen-phosphorus detector. J. Chromatogr., 325: 231- 238.
KLISENKO, M.A. & GIRENKO, D.B. (1980) [Gas-chromatographic
determination of organophosphorus pesticides in air.] Gig. i Sanit.
1980(9): 57-60 (in Russian).
KLISENKO, M.A., GIRENKO, D.B., GOLOVATYI, V.G., SHPAKOVSKII, I.V., &
KOROL, E.N. (1981) [Field-desorption mass spectrometry in pesticide
analysis.] Khim. Sel'sk. Khoz. 1981: 59-61 (in Russian).
KNIEHASE, U. & ZOEBELEIN, G. (1990) [Testing the effects of pesticides
on the predator mite Phytoseiulus persimilis Ath.-Hen. by means of
a new laboratory method approaching to the practice.] Anz.
Schädlingskd. Pflanzenschutz. Umweltschutz., 63: 105-113 (in German).
KOEN, J.G. & HUBER, J.F.K. (1970) A rapid method for residue analysis
by column liquid chromatography with polarographic detection.
Application to the determination of parathion and methylparathion on
crops. Anal. chim. Acta, 51: 303-307.
KONRAD, J.G., PIONKE, H.B., & CHESTERS, G. (1969) Extraction of
organochlorine and organophosphate insecticides from lake waters.
Analyst, 94: 490-492.
KORN, S. & EARNEST, R. (1974) Acute toxicity of twenty insecticides to
striped bass, Morone saxatilis. Calif. Fish Game, 60(3): 128-131.
KORSOS, I. & LANTOS, J. (1984) [Thin-layer chromatographic
identification of pesticides.] Novenyvedelem (Budapest), 20: 30-34 (in
Hungarian).
KRIJGSMAN, W. & VAN DE KAMP, C.G. (1976) Analysis of organophosphorus
pesticides by capillary gas chromatography with flame photometric
detection. J. Chromatogr., 117: 201-205.
KRUEGER, H.R. & CASIDA, J.E. (1957) Toxicity of fifteen
organophosphorus insecticides to several insect species and to rats.
J. Econ. Entomol., 50: 356-358.
KUMAR, N.V.N. (1985) Chromatographic and enzymic methods as applied to
environmental monitoring of pesticide residues in some districts of
Andhra Pradesh. In: Trivedy, R.K. & Goel, P.K. ed. Current pollution
research in India. Kared, India, Environmental Publications, pp.
163-184.
KUMAR, N.V.N. & RAMASUNDARI, M. (1980) Colorimetric determination of
methyl parathion and oxygen analog. J. Assoc. Off. Anal. Chem., 63:
536-538.
KUMMER R. & VAN SITTERT, N.J. (1986) Field studies on health effects
from the application of two organophosphorus insecticide formulations
by hand-held ULV to cotton. Toxicol. Lett., 33: 7-24.
LAMONTAGNE, J.C. (1978) Résidues de produits phytosanitaires dans les
fruits et légumes. C. R. Séances Acad, Agric. France, 64: 923-931.
LANGE, P. & WIEZOREK, W.D. (1975) Effects of diethyldithiocarbamate on
acute toxicity and acetylcholinesterase inhibition by methylparathion
in mice. Acta biol. med. Germ., 34: 427-433.
LANGE, P., TIEFENBACH, B., & WIEZOREK, W.D. (1977) Effect of
dithiocarb on acute toxicity and acetylcholinesterase inhibition by
different organophosphates in mice. Proc. Eur. Soc. Toxicol., 18:
284-285.
LASSITER, R.R., PARRISH, R.S., & BURNS, L.A. (1986) Decomposition by
planktonic and attached microorganisms improves chemical fate models.
Environ. Toxicol. Chem., 5: 29-39.
LAUGHLIN, J. & GOLD, R.E. (1989) Evaporative dissipation of methyl
parathion from laundered protective apparel fabrics. Bull. environ.
Contam. Toxicol., 42: 566-573.
LAUGHLIN, J.M., EASLEY, C.B., GOLD, R.E., & TUPY, D.R. (1981) Methyl
parathion transfer from contaminated fabrics to subsequent laundry and
to laundry equipment. Bull. environ. Contam. Toxicol., 27: 518-523.
LAWRENCE, J.F. & TURTON, D. (1978) High performance liquid
chromatographic data to 166 pesticides. J. Chromatogr., 159: 207-226.
LAWRENCE, J.F., RENAULT, C., & FREI, R.W. (1976) Fluorogenic labelling
of organophosphate pesticides with dansyl chloride. Application to
residue analysis by high-pressure liquid chromatography and thin-layer
chromatography. J. Chromatogr., 121: 343-351.
LEARD, R.L., GRANTHAM, B.J., & PESSONEY, G.F. (1980) Use of selected
freshwater bivalves for monitoring organochlorine pesticide residues
in major Mississippi stream systems, 1972-1973. Pestic. Monit. J., 14:
47-52.
LE BEL, G.L., WILLIAMS, D.T., GRIFFITH, G., & BENOIT, F.M. (1979)
Isolation and concentration of organophosphorus pesticides from
drinking water at the ng/L level, using macroreticular resin. J.
Assoc. Off. Anal. Chem., 62: 241-249.
LEE, H.B., WENG, L.D., & CHAU, A.S.Y. (1984) Confirmation of pesticide
residue identity. XI. Organophosphorus pesticides. J. Assoc. Off.
Anal. Chem., 67: 553-556.
LEIDY, R.B., SHEETS, T.J., & NELSON, L.A. (1977) Residues from two
formulations of methyl parathion on flue-cured tobacco. Tob. Sci., 21:
28-31.
LESHCHEV, V.V. & TALANOV, G.A. (1977) [Determination of residues of
organophosphorus pesticides using thin-layer chromatography with
enzymic development.] Khim. Sel'sk. Khoz., 15: 46-48 (in Russian).
LEVINE, B.S. & MURPHY S.D. (1977a) Esterase inhibition and
reactivation in relation to piperonyl butoxide-phosphorothionate
interactions. Toxicol. appl. Pharmacol., 40: 379-391.
LEVINE, B.S. & MURPHY, S.D. (1977b) Effect of piperonyl butoxide on
the metabolism of dimethyl and diethyl phosphorothionate insecticides.
Toxicol. appl. Pharmacol., 40: 393-406.
LEWIS, D.L. & HOLM, H.W. (1981) Rates of transformation of methyl
parathion and diethyl phthalate by aufwuchs microorganisms. Appl.
environ. Microbiol., 42: 698-703.
LEWIS, D.L., HODSON, R.E., & FREEMAN, L.F. III (1984) Effects of
microbial community interactions on transformation rates of xenobiotic
chemicals. Appl. environ. Microbiol., 48: 561-565.
LEWIS, D.L., HODSON, R.E., & FREEMAN, L.F. III (1985) Multiphasic
kinetics for transformation of methyl parathion by Flavobacterium
species. Appl. environ. Microbiol., 50: 553-557.
LEWIS, R.G. & JACKSON, M.D. (1982) Modification and evaluation of a
high-volume air sampler for pesticides and semiolatile industrial
organic chemicals. Anal. Chem., 54: 592-594.
LEWIS, R.G. & MACLEOD, K.E. (1982) Portable sampler for pesticides and
semivolatile industrial organic chemicals in air. Anal. Chem., 54:
310-315.
LEWIS, R.G., BROWN, A.R., & JACKSON, M.D. (1977) Evaluation of
polyurethane foam for sampling of pesticides, polychorinated biphenyls
and polychlorinated naphthalenes, in ambient air. Anal. Chem., 49:
1668-1672.
LI, Y. & WANG, G. (1987) [Determination of organophosphorus pesticides
in wastewater.] Shanghai Huanjing Kexue, 6: 19-31, 42 (in Chinese).
LIANG, L. & ZHANG, G. (1986) [Gas chromatographic determination of
organophosphorus agrochemicals in the air.] Zhonghua Laodong Weisheng
Zhiyebing Zazhi, 4: 163-165 (in Chinese).
LICHTENSTEIN, E.P. (1975) Transport mechanism in soil. Metabolism and
movement of insecticides from soils into water and crop plants. Pure
appl. Chem., 42(1/2): 113-118.
LICHTENSTEIN, E.P. & SCHULZ, K.R. (1964) The effects of moisture and
microorganisms on the persistence and metabolism of some
organophosphorous insecticides in soils, with special emphasis on
parathion. J. Econ. Entomol., 57: 618-627.
LISI, P., CARAFFINI, S., & ASSALVE, D. (1986) A test series for
pesticide dermatitis. Contact Dermatitis, 15: 266-269.
LISI, P., CARAFFINI, S., & ASSALVE, D. (1987) Irritation and
sensitization potential of pesticides. Contact Dermatitis, 17:
212-218.
LITTLE, E.E., ARCHESKI, R.D., FLEROV, B.A., & KOZLOVSKAYA, V.I. (1990)
Behavioural indicators of sublethal toxicity in rainbow trout. Arch.
environ. Contam. Toxicol., 19: 380-385.
LOFROTH, G., KIM, C.H., & HUSSAIN, S. (1969) Alkylating properties of
2,2-dichlorovinyl dimethyl phosphate: a disregarded hazard. Environ.
Mutat. Soc. Newsl., 2: 21-27.
LOSER, E. & EIBEN, R. (1982) [E 605-methyl: Multigeneration tests on
rats.] Wuppertal-Elberfeld, Bayer AG, Institute of Toxicology
(Unpublished report No. 10630, submitted to WHO by Bayer AG,
Leverkusen, Germany) (in German).
LOVE, J.L., DONNELLY, R.G.C., HUGHES, J.T., & WILSON, P.D. (1974)
Pesticide residues in fruit and vegetables in New Zealand 1971-73. N.
Z. J. Sci., 17: 529-534.
LUKE, M.A. & DOOSE, G.M. (1983) A modification of the Luke
multiresidue procedure for low moisture, nonfatty products. Bull.
environ. Contam. Toxicol., 30: 110-116.
LUKE, M.A. & DOOSE, G.M. (1984) A rapid analysis for pesticides in
milk and oilseeds. Bull. environ. Contam. Toxicol., 32: 651-656.
LUKE, M.A., FROBERG, J.E., & MASUMOTO, H.T. (1975) Extraction and
cleanup of organochlorine, organophosphate, organonitrogen, and
hydrocarbon pesticides in produce for determination by gas-liquid
chromatography. J. Assoc. Off. Anal. Chem., 58: 1020-1026.
McCANN, J.A. & JASPER, R.L. (1972) Vertebral damage to bluegills
exposed to acutely toxic levels of pesticides. Trans. Am. Fish. Soc.,
101(2): 317-322.
McCOLLISTER, D.D., OLSON, K.J., ROWE, V.K., PAYNTER, O.E., WEIR, R.J.,
& DIETERICH, W.H. (1968) Toxicology of 4-tert-butyl-2-chlorophenyl
methyl methylphosphoramidate (ruelene) in laboratory animals. Food
Cosmet. Toxicol., 6: 185-198.
MACEK, K.J. & McALLISTER, W.A. (1970) Insecticide susceptibility of
some common fish family representatives. Trans. Am. Fish Soc., 99(1):
20-27.
MACHEMER, L. (1977a) [Methyl parathion. Embryotoxic or teratogenic
effects in rats after oral application.] Leverkusen, Germany, Bayer
AG, Institute for Toxicology, (Unpublished report submitted to WHO by
Bayer AG) (in German).
MACHEMER, L. (1977b) [Methyl parathion. Tests for embryotoxic and
teratogenic effects in rats after intravenous injection.]
Wuppertal-Elberfeld, Bayer AG, Institute of Toxicology (Unpublished
report No. 6812, submitted to WHO by Bayer AG, Leverkusen, Germany)
(in German).
McLEOD, K.E & LEWIS, R.G. (1982) Portable sampler for pesticides and
semivolatile industrial organic chemicals in air. Anal. Chem., 54:
310-315.
MAFF (1986) Report of the Working Party on Pesticide Residues
(1982-85). The sixteenth report of the Steering Group on Food
Surveillance. London, Ministry of Agriculture, Fisheries and Food,
Health and Safety Executive, 50 pp. (Food Surveillance Paper No. 16).
MAFF (1990) Report of the Working Party on Pesticide Residues
(1988-89). (Supplement to issue No. 8 (1990) of the Pesticide
Register). London, Ministry of Agriculture, Fisheries and Food, Health
and Safety Executive, 86 pp.
MANSKE, D.D. & JOHNSON, R.D. (1975) Residues in food and feed.
Pesticide residues in total diet samples (VIII). Pestic. Monit. J., 9:
94-105.
MANSKE, D.D. & JOHNSON, R.D. (1977) Pesticide and other chemical
residues in total diet samples (X). Pestic. Monit. J., 10: 134-148.
MARCUS, M., SPIGARELLI, J., & MILLER, H. (1978) Organic compounds in
organophosporus pesticide manufactoring wastewaters. Springfield,
Virginia, National Technical Information Service, p. 22 (PB-289821).
MARTIN, E.W. (1978) Hazards of medication. A manual on drug
interactions, contraindications, and adverse reactions with other
presribing and drug information, 2nd ed. Philadelphia, Toronto, J.B.
Lippincott Company.
MATHEW, G., ABDUL RAHIMAN, M., & VIJAYALAXMI, K.K. (1990) In vivo
genotoxic effects in mice of Metacid 50 an organophosphorus
insecticide. Mutagenesis, 5(2): 147-149.
MAYER, F.L. Jr (1987) Acute toxicity handbook of chemicals to
estuarine organisms. Gulf Breeze, Florida, US Environmental Protection
Agency, 283 pp.
MAYER, F.L. Jr & ELLERSIECK, M.R. (1986) Manual of acute toxicity:
interpretation and data base for 410 chemicals and 66 species of
freshwater animals. Washington, DC, US Department of the Interior Fish
and Wildlife Service, pp. 311 (Resource Publication 160).
MELNIKOW, N.N. (1971) Chemistry of pesticides. Berlin, Heidelberg, New
York, Springer Verlag.
MENZIE, C.M. (1972) Fate of pesticides in the environment. Ann. Rev.
Entomol., 17: 199-222.
MENZIE, C.M. (1974) Metabolism of pesticides - an update. Washington,
DC, US Department of the Interior, Fish and Wildlife Service (Report
No. 184).
MESTRES, R., LEONARDI, G., CHEVALLIER, C., & TOURTE, J. (1969)
Résidus de pesticides. XIX. Méthode de recherche des résidus de
pesticides dans les eaux naturelles. 1ère partie. Méthode d'analyse
générale. Ann. Fals. Expert. Chim., 62(685): 75-85.
MESTRES, R., CHEVALLIER, C., ESPINOZA, C., & CORNET, R. (1977) XXXIII.
Application du couplage chromatographie gazeuse spectrométrie de masse
aux méthodes de recherche et de dosage des résidus de pesticides et de
micropollutants organiques dans les matéiaux de l'environnement et
les matières alimentaires. Ann. Fals. Expert. Chim., 70: 177-188.
MEYER JONES, L., BOOTH, N.H. & McDONALD, L.E. (1977) Veterinary
pharmacology and therapeutics, 4th ed. Ames, Iowa State University
Press, pp. 1201.
MEYERS, S.M., CUMMINGS, J.L., & BENNETT, R.S. (1990) Effects of methyl
parathion on red-winged blackbird ( Agelaius phoeniceus) incubation
behavior and nesting success. Environ. Toxicol. Chem., 9: 807-813.
MIDWEST RESEARCH INSTITUTE (1975) Substitute chemical program -
initial scientific and minieconomic review of methyl parathion.
Kansas City, Missouri, Midwest Research Institute, 176 pp. (Prepared
for US Environmental Protection Agency, Criteria and Evaluation
Division, Washington, DC.) (EPA-540/1-75-004).
MIELLET, A. (1982) Dosage des résidus de pesticides par C.L.H.P. sur
plantes médicinales et aromatiques. Compte-rendu du résultat
d'applications de pesticides effectuées avec le concours du syndicat
national des plantes médicinales de Nilly-la-Forét. Ann. Fals. Expert.
Chim., 75: 369-375.
MIHAIL, F. & VOGEL, O. (1984) [E 120/methyl parathion: Subacute
dermal toxicity tests on rabbits.] Wuppertal-Elberfeld, Bayer AG,
Institute of toxicology (Unpublished report No. 12484, submitted to
WHO by Bayer AG, Leverkusen, Germany) (in German).
MILES, J.W., FETZER, L.E., & PEARCE, G.W. (1970) Collection and
determination of trace quantities of pesticides in air. Environ. Sci.
Technol., 4(5):420-425.
MILLER, H., CRAMER, P., DRINKWINE, A., SHAN, A., RISCHAN, G., & GOING,
J. (1981) Development of methods for pesticides in wastewater:
applicability of a general approach. In: Keith, L.H., ed. Advances in
the identification and analysis of organic pollutants in water. Ann
Arbor, Michigan, Ann Arbor Science Publishers, Vol. 1, pp. 115-138.
MILLS, P.A., ONLEY, J.H., & GAITHER, R.A. (1963) Rapid method for
chlorinated pesticide residues in vegetables and food. J. Am. Off.
Anal. Chem., 46: 186-191.
MINCHEW, C.D. & FERGUSON, D.E. (1969) Toxicities of six insecticides
to resistant and susceptible green sunfish and golden shiners in
static bioassays. J. Mississippi Acad. Sci., 15: 29-32.
MINGELGRIN, U., SALTZMAN, S., & YARON, B. (1977) A possible model for
the surface-induced hydrolysis of organophosphorus pesticides on
kaolinite clays. Soil Sci. Soc. Am. J., 41: 519-523.
MIRER, F.E., LEVINE, B.S., & MURPHY, S.D. (1977) Parathion and methyl
parathion toxicity and metabolism in piperonyl butoxide and diethyl
maleate pretreated mice. Chem.-Biol. Interact., 17: 99-112.
MIYAMOTO, J., SATO, Y., KADOTA, T., FUJINAMI, A., & ENDO, M. (1963)
Studies on the mode of action of organophosphorus compounds. Part I.
Metabolic fate of P32 labelled sumithion and methylparathion in
guinea pig and white rat. Agric. Biol. Chem., 27: 381-389.
MOELLER, H.C. & RIDER, J.A. (1961) Studies on the anticholinesterase
effect of parathion and methylparathion in humans. Fed. Proc., 20:
434.
MOELLER, H.C. & RIDER, J.A. (1962) Threshold of incipient toxicity to
systox and methyl parathion. Fed. Proc., 21: 451.
MOHANTY-HEJMADI, P. & DUTTA, S.K. (1981) Effects of some pesticides on
the development of the Indian bull frog Rana tigerina. Environ.
Pollut., Ser. A, 24: 145-161.
MOHN, G. (1973) 5-methyltryptophan resistance mutations in
Escherichia coli K-12. Mutagenic activity of monofunctional
alkylating agents including organophosphorus insecticides. Mutat.
Res., 20: 7-15.
MOLLHOFF, E. (1981) [Laboratory studies to metabolism of methyl
parathion in soils.] Leverkusen, Gemany, Bayer AG (Unpublished report
submitted to WHO by Bayer AG) (in German).
MOLNAR, J. & PAKSY, K.A. (1978) [Evaluation of the acute toxicity of
inhaled pesticides in experimental animals.] In: [Proceedings of the
Conference on Safety Technology in the Agricultural Use of Chemicals.]
pp. 1790-1793 (Conference paper) (in German).
MOORTHY, K.S., REDDY, B.K., CHETTY, C.S., & SWAMI, K.S. (1983)
Catalytic potential of catalase in hepatopancreas of freshwater
mussel Lamellidens marginalis during induced methyl-parathion toxic
stress. Geobios, 10(1): 42-44.
MOORTHY, K.S., REDDY, B.K., SWAMI, K.S., & CHETTY, C.S. (1984) Changes
in respiration and ionic content in tissues of freshwater mussel
exposed to methyl parathion toxicity. Toxicol. Lett., 21: 287-291.
MORGAN, D.P., HETZLER, H.L., SLACH, E.F., & LIN, L.I. (1977) Urinary
excretion of paranitrophenol and alkyl phosphates following ingestion
of methyl or ethyl parathion by human subjects. Arch. environ. Contam.
Toxicol., 6: 159-173.
MORPURGO, G., AULICINO, F., BIGNAMI, M., CONTI, L., VELCICH, A., &
MONTALENTI, S.G. (1977) Genetica - Relationship between structure and
mutagenicity of dichlorvos and other pesticides. Rend. Cl. Sci. Fis.
Mat. Nat., 62: 692-701.
MUDGALL, C.F. & PATIL, H.S. (1987) Toxic effects of dimethoate and
methyl parathion on glycogen reserves of male and female Rana. J.
Environ. Biol., 8(3): 237-244.
MUHLMANN, R. & SCHRADER, G. (1957) [Hydrolysis of the insecticdal
phosphoric acid esters.]. Z. Naturforsch., B12: 196-208 (in German).
MULLER, B. (1973) [Determination of pesticide residues in bee honey.
I. Semiquantitative thin-layer chromatographic determination of
insecticide residues in bee honey]. Nahrung, 17: 381-386 (in German).
MUNCY, R.J. & OLIVER, A.D. Jr (1963) Toxicity of ten insecticides to
the red crawfish Procambarus clarki. Trans. Am. Fish. Soc., 96(4):
428-431.
MUNDY, R.L., BOWMAN, M.C., FARMER, J.H., & HALEY, T.J. (1978)
Quantitative structure activity study of a series of substituted
O,O-dimethyl O-( p-nitrophenyl) phosphorothioates and
O-analogs. Arch. Toxicol., 41: 111-123.
MUNN, S., KEEFE, T.J., & SAVAGE, E.P. (1985) A comparative study of
pesticide exposures in adult and youth migrant field workers. Arch.
environ. Health, 40: 215-220.
MURPHY, S.D. (1980) Toxic interactions with dermal exposure to
organophosphate insecticides. Toxicol. Lett., 5: 34.
MURTHY, A. & RAMANI, A. (1982) What is an environmentally safe
pesticide? In: Christiansen, K., Koch, B., & Bro-Rasmussen, F., ed.
Chemicals in the environment. Chemicals testing and hazard ranking -
the interaction between science and administration. Proceedings of the
International Symposium, Lyngby-Copenhagen, 18-20 October 1982.
Lyngby, The Technical University of Denmark, Laboratory of
Environmental Science and Ecology, pp.323-333.
MURTY, A.S., RAMANI, A.V., CHRISTOPHER, K., & RAJABHUSHANAM, B.R.
(1984) Toxicity of methyl parathion and fensulfothion to the fish
Mystus cavasius. Environ. Pollut., A34: 37-46.
NAG, M. & NANDI, N. (1987) In vitro and in vivo effect of
organophosphate pesticides on monoamine oxidase activity in rat brain.
Biosci. Rep., 7(10): 801-804.
NAGARATNAMMA, R. & RAMAMURTHI, R. (1982) Metabolic depression in the
fresh water teleost Cyprinus carpio exposed to an organophosphate
pesticide. Curr. Sci., 51(13): 668-669.
NAGYMAJTENYI, L., DESI, I., & LORENCZ, R. (1988) Neurophysiological
markers as early signs of organophosphate neurotoxicity. Neurotoxicol.
Teratol., 10: 429-434.
NAKATSUGAWA, T., TOLMAN, N.M., & DAHM, P.A. (1968) Degradation and
activation of parathion analogs by microsomal enzymes. Biochem.
Pharmacol., 17: 1517-1528.
NAKATSUGAWA, T., TOLMAN, N.M., & DAHM, P.A. (1969) Degradation of
parathion in the rat. Biochem. Pharmacol., 18: 1103-1114.
NANGNIOT, P. (1966) Electrochemical methods for quantitative analysis
of pesticide residues. Meded. Fac. Landbouwwet. Rijksuniv., Gent, 31:
447-473.
NAQVI, S.M.Z. (1973) Toxicity of twenty-three insecticides to a
tubificed worm Branchiura sowerbyi from the Mississippi delta. J.
Econ. Entomol., 66(1): 70-74.
NATALE, O.E., GOMEZ, C.E., PECHEN de D'ANGELO, A.M., & SORIA, C.A.
(1988) Waterborne pesticides in the Negro River Basin (Argentina). In:
Abbou, R., ed. Hazardous waste: detection, control, treatment.
Amsterdam, Oxford, New York, Elsevier Science Publishers, pp.
879-907.
NATIONAL FIRE PROTECTION ASSOCIATION (1986) Fire protection guide on
hazardous materials, 9th ed. Quincy, Massachusetts, National Fire
Protection Association, pp. 49-64.
NATIONAL RESEARCH COUNCIL (1977) Drinking water and health.
Washington, DC, National Academy of Sciences, pp. 626-635 (Publication
NRC/2619).
NCI (1979) Bioassay of methyl parathion for possible carcinogenicity.
Washington, DC, US Department of Health, Education & Welfare, National
Cancer Institute (Technical Report Series No. 157; DHEW Publ. No.
(NHI) 79-1713).
NEHEZ, M., BOROS, P., FERKE, A., MOHOS, J., PALOTAS, M., VETRO, G.,
ZIMANYI, M., & DESI, I. (1988) Cytogenetic examination of people
working with agrochemicals in the southern region of Hungary. Regul.
Toxicol. Pharmacol., 8: 37-44.
NELSON, R.C. (1967) Procedure for nine organothiophosphate pesticide
residues on fruits and vegetables, using microcoulometric gas
chromatography. J. Assoc. Off. Anal. Chem., 50: 922-926.
NEWTON, T.D., GATTIE, D.K., & LEWIS, D.L. (1990) Initial test of the
benchmark chemical approach for predicting microbial transformation
rates in aquatic environments. Appl. environ. Microbiol., 56(1):
288-291.
NIELSEN, P.G. (1985) Quantitative analysis of parathion and
parathion-methyl by combined capillary column gas chromatography
negative ion chemical ionization mass spectrometry. Biomed. Mass
Spectrom., 12: 695-698.
NIETHAMMER, K.R. & BASKETT, T.S. (1983) Cholinesterase inhibition of
birds inhabiting wheat fields treated with methyl parathion and
toxaphene. Arch. environ. Contam. Toxicol., 12: 471-475.
NIMMO, W.R., HAMAKER, T.L., MATTHEWS, E., & MOORE, J.C. (1981) An
overview of the acute and chronic effects of first and second
generation pesticides on an estuarine mysid. In: Vernberg, F.J.,
Calabrese, A., Thurberg, F.P., & Vernberg, W.B., ed. Biological
monitoring of marine pollutants. Proceedings of a Symposium on
Pollution and Physiology of Marine Organisms, Milford, Connecticut,
7-9 November 1980. New York, London, Academic Press, pp. 3-19.
NIOSH (1976) Criteria for a recommended standard - occupational
exposure to methyl parathion. Cincinnati, Ohio, National Institute for
Occupational Safety and Health (DHEW 0 (NIOSH) Publication No.
77-106).
NIOSH (1980) Summary of NIOSH recommendations for occupational health
standards. Cincinnati, Ohio, National Institute for Occupational
Safety and Health.
NIOSH (1991) Registry of toxic effects of chemical substances.
Cincinnatti, Ohio, National Institute for Occupational Safety and
Health.
NISHIUCHI, Y. & HASHIMOTO, Y. (1967) Toxicity of pesticide ingredients
to some freshwater organisms. Botyu-Kagaku, 32(5): 5-11.
NOMIYAMA, K., MATSUI, K., & NOMIYAMA, H. (1980) Environmental
temperature, a factor modifying the acute toxicity of organic
solvents, heavy metals, and agricultural chemicals. Toxicol. Lett., 6:
67-70.
OMURA, M., HASHIMOTO, K., OHTA, K., IIO, T., UEDA, S., ANDO, K., &
HIRAIDE, H. (1990) Relative retention time diagram as a useful tool
for gas chromatographic analysis and electron-capture detection of
pesticides. J. Assoc. Off. Anal. Chem., 73(2): 300-306.
OOMEN, P.A. (1986) A sequential scheme for evaluating the hazard of
pesticides to bees, Apis melifera. Mede. Fac. Landbouwwet.
Rijksuniv. Gent, 51(36): 1205-1213.
ORLANDO, E., RAFFI, G.B., CASCELLA, D., DE ROSA, V., & CIPOLLA, C.
(1972) Effect of quinidine on electrocardiographic alterations during
acute poisoning by phosphoric acid esters. Lav. Um., 24: 1-13.
OSADCHUK, M., ROMACH, M., & MCCULLY, K.A. (1971) Cleanup and
separation procedures for multipesticide residue analysis in
monitoring and regulatory laboratories. In: Tahori, A.S., ed.
Pesticide chemistry. Proceedings of the 2nd International Congress on
Pesticide Chemistry, New York, Gordon & Breach, vol. 4., pp. 357-383.
OU, L.T. (1985) Methyl parathion degradation and metabolism in soil:
influence of high soil-water contents. Soil Biol. Biochem., 17:
241-243.
OU, L.-T. & SHARMA, A. (1989) Degradation of methyl parathion by a
mixed bacterial culture and a Bacillus sp. isolated from different
soils. J. Agric. Food Chem., 37: 1514-1518.
OU, L.-T., RAO, P.S.C., & DAVIDSON, J.M. (1983) Methyl parathion
degradation in soil: influence of soil-water tension. Soil Biol.
Biochem., 15: 211-215.
PALAWSKI, D., BUCKLER, D.R., & MAYER, F.L. (1983) Survival and
condition of rainbow trout (Salmo gairdneri) after acute exposures
to methyl parathion, triphenyl phosphate, and DEF. Bull. environ.
Contam. Toxicol., 30: 614-620.
PAP, A., SZABO, I., & SZARVAS, F. (1976) Effect of liver damage and of
phenobarbital, norandrostenolone, phenyl propionate or chloramphenicol
treatment of the poisoning by organic phosphoric acid esters. Kiserl.
Orvostud., 28: 172-179.
PASCHAL, D.C., BICKNELL, R., & DRESBACH, D. (1977) Determination of
ethyl and methyl parathion in runoff water with high performance
liquid chromatography. Anal. Chem., 49: 1551-1554.
PATTERSON, P.L. (1982) New uses of thermionic ionization detectors in
gas chromatography. Chromatographia, 16: 107-111.
PAULUHN, J. (1983) [Methyl parathion (E 120). Studies on the
irritant/corrosive effect on skin and eyes (rabbit).]
Wuppertal-Elberfed, Bayer AG, Institute of Toxicology (Unpublished
report No. 11712, submitted to WHO by Bayer AG, Leverkusen, Germany)
(in German).
PETERKA, V. & CERNA, V. (1988) Micellar catalysis in decomposition of
some organophosphates. Chem. Prum., 38(1): 36-39.
PFAENDER, F.K., SHUMAN, M.S., DEMPSEY, H., & HARDEN, C.W. (1977)
Monitoring heavy metals and pesticides in the Cape Fear River Basin of
North Carolina, Chapel Hill, NC. Raleigh, North Carolina, North
Carolina Water Resources Research Institute, pp. 29, 119.
PFEIFFER, R. & STAHR, H.M. (1982) Screening for organochlorine and
organophosphorus pesticides. In: Touchstone, J.C., ed. Advances in
thin layer chromatography: clinical and environmental applications.
Proceedings of the Second Biennial Symposium, Philadelphia, December
1980. New York, John Wiley and Sons, pp. 451-460.
PFLUGMACHER, J. & EBING, W. (1974) Purification of phosphoric acid
insecticide residues in vegetable extracts by gel chromatography on
Sephadex LH-20. J. Chromatogr., 93: 457-463.
PICKERING, Q.H., HENDERSON, C., & LEMKE, A.E. (1962) The toxicity of
organic phosphorus insecticides to different species of warm-water
fishes. Trans. Am. Fish. Soc., 91: 175-184.
PIONKE, H.B., KONRAD, J.G., CHESTERS, G., & ARMSTRONG, D.E. (1968)
Extraction of organochlorine and organophosphate insecticides from
lake waters. Analyst, 93: 363-367.
PLAPP, F.W. & CASIDA, J.E. (1958) Hydrolysis of the alkyl-phosphate
bond in certain dialkyl aryl phosphorothioate insecticides by rats,
cockroaches, and alkali. J. Econ. Entomol., 51(6): 800-803.
PORTIER, R.J. & MEYERS, S.P. (1982) Monitoring biotransformation and
biodegradation of xenobiotics in simulated aquatic microenvironmental
systems. Dev. Ind. Microbiol., 23: 459-475.
PORTIER, R.J., CHEN, H.M., & MEYERS, S.P. (1983) Environmental effect
and fate of selected phenols in aquatic ecosystems using microcosm
approaches. Dev. Ind. Microbiol., 24: 409-424.
PREUSSMANN, R., SCHNEIDER, H., & EPPLE, F. (1969) [Investigation of
effects of alkylating agents. II. The investigation of different
classes of alkylating agents by means of a modified colour reaction
with 4-(4-nitro-benzyl)pyridine (NBP).] Arzneimittelforschung, 7:
1059-1073 (in German).
PRINSLOO, S.M. & DE BEER, P.R. (1987) Gas chromatographic relative
retention data for pesticides on nine packed columns: II.
Organophosphorus and organochlorine pesticides, using elecron-capture
detection. J. Assoc. Off. Anal. Chem., 70: 878-888.
PRITCHARD, P.H., CRIPE, C.R., WALKER, W.W., SPAIN, J.C., & BOURQUIN,
A.W. (1987) Biotic and abiotic degradation rates of methyl parathion
in freshwater and estuarine water and sediment samples. Chemosphere,
16(7): 1509-1520.
QIAN, C., SANDERS, P.F., & SEIBER, J.N. (1985) Accelerated degradation
of organophosphorus pesticides with sodium perborate. Bull. environ.
Contam. Toxicol., 35: 682-688.
RADULOVIC, L.L., LAFERLA, J.J., & KULKARNI, A.P. (1986) Human
placental glutathione s-transferase-mediated metabolism of methyl
parathion. Biochem. Pharmacol., 35: 3473-3480.
RADULOVIC, L.L., KULKARNI, A.P., & DAUTERMAN, W.C. (1987)
Biotransformation of methyl parathion by human foetal liver
glutathione s-transferases: an in vitro study. Xenobiotica, 17:
105-114.
RAMAKRISHNA, N. & RAMACHANDRAN, B.V. (1978) Colorimetric determination
of fenitrothion and methyl parathion using a hydroxylaminolytic
procedure. J. Indian Chem. Soc., LV: 185-187.
RANI, V.J.S., VENKATESHWARLU, P., & JANAIAH, C. (1989) Changes in
carbohydrate metabolism of Clarias bactrachus (Linn) when exposed to
two organophosphorus insecticides. J. environ. Biol., 10: 197-204.
RAO, S.N. & McKINLEY, W.P. (1969) Metabolism of organophosphorus
insecticides by 0liver homogenates from different species. Can. J.
Biochem., 47: 1155-1159.
RAO, K.S.P. & RAO, K.V.R. (1983) Regulation of phosphorylases and
aldolases in tissues of the teleost Tilapia mossambica under
methyl-parathion impact. Bull. environ. Contam. Toxicol., 31(4):
474-478.
RAO, K.S.P. & RAO, K.V.R. (1984a) Tissue specific alteration of
aminotransferases and total ATPases in the fish (Tilapia mossambica)
under methyl parathion impact. Toxicol. Lett., 20: 53-57.
RAO, K.S.P. & RAO, K.V.R. (1984b) Impact of methyl parathion toxicity
and eserine inhibition of acetylcholinesterase activity in tissues of
the teleost (Tilapia mossambica) - a correlative study. Toxicol.
Lett., 22: 351-356.
RAO, K.S.P. & RAO, K.V.R. (1987) The possible role of
glucose-6-phosphate dehydrogenase in the detoxification of methyl
parathion. Toxicol. Lett., 39: 211-214.
RAO, K.V.R., RAO, K.R.S.S., & RAO, K.S.P. (1983a) Cardiac responses to
malathion and methyl parathion in the mussel, Lamellidens marginalis
(Lamark). J. environ. Biol., 4: 65-68.
RAO, K.S.P., MADHU, C., RAO, K.R.S.S., & RAO, K.V.R. (1983b) Effect of
methyl parathion on body weight, water content and ionic changes in
the teleost, Tilapia mossambica (Peters). J. Food Sci. Technol., 20:
27-29.
RAO, T.S., DUTT, S., & MANGAIAH, K. (1967) TLm values of some modern
pesticides to the fresh-water fish Puntius puckelli. Environ.
Health, 9(2): 103-109.
RASHID, K.A. & MUMMA, R.O. (1984) Genotoxicity of methyl parathion in
short-term bacterial test systems. J. environ. Sci. Health, B19:
565-577.
RASTOGI, A. & KULSHRESTHA, S.K. (1990) Effect of sublethal
doses of three pesticides on the ovary of a carp minnow Rasbora
daniconius. Bull. environ. Contam. Toxicol., 45: 742-747.
RATTNER, B.A. & FRANSON, J.C. (1984) Methyl parathion and fenvalerate
toxicity in American kestrels: acute physiological responses and
effects of cold. Can. J. Physiol. Pharmacol., 62: 787-792.
REDDY, K.S. & GAMBRELL, R.P. (1985) The rate of soil reduction as
affected by levels of methyl parathion and 2,4-D. J. environ. Sci.
Health, B20: 275-298.
REDDY, M.S. & RAO, K.V.R. (1986) Acute toxicity of insecticides to
penaeid prawns. Environ. Ecol., 4(1): 221-223.
REDDY, M.S. & RAO, K.V.R. (1988) In vivo recovery of
acetylcholinesterase activity from phosphamidon and methylparathion
induced inhibition in the nervous tissue of penaeid prawn
(Metapenaeus monoceros). Bull. environ. Contam. Toxicol., 40:
752-758.
REDDY, M.S. & RAO, K.V.R. (1989) In vivo modification of lipid
metabolism in response to phosphamidon, methylparathion and lindane
exposure in the penaeid prawn, Metapenaeus monoceros. Bull.
environ. Contam. Toxicol., 43: 603-610.
REDDY, M.S. & RAO, K.V.R. (1990a) Effects of sublethal concentrations
of phosphamidon, methyl parathion, DDT, and lindane on tissue nitrogen
metabolism in the penaeid prawn, Metapenaeus monoceros (Fabricius).
Ecotoxicol. environ. Saf., 19: 47-54.
REDDY, M.S. & RAO, K.V.R. (1990b) Methylparathion - induced
alterations in the acetylcholinesterase and phosphatases in a penaeid
prawn, Metapenaeus monoceros (Fabricius). Bull. Environ. Contam.
Toxicol., 45: 350-357.
REDDY, M.S., RAO, K.V.R., & MURTHY, B.N. (1988) Changes in nitrogen
metabolism of penaeid prawn, Penaeus indicus, during sublethal
phosphamidon and methylparathion-induced stress. Bull. environ.
Contam. Toxicol., 41: 344-351.
REDDY, P.S., BHAGYLAKSHMI, A., & RAMAMURTHI, R. (1985) Moit-inhibition
in the crab Oziotelphusa senex senex following exposure to
malathion and methyl parathion. Bull. environ. Contam. Toxicol., 35:
92-97.
REDDY, P.S., BHAGYLAKSHMI, A., & RAMAMURTHI, R. (1986a) Carbohydrate
metabolism in tissue of fresh water crab (Oziotelphusa senex senex)
exposed to methyl parathion. Bull. environ. Contam. Toxicol., 36:
204-210.
REDDY, P.S., BHAGYLAKSHMI, A., & RAMAMURTHI, R. (1986b) Changes in
acid phosphatase activity in tissues of crab, Oziotelphusa senex
senex, following exposure to methyl parathion. Bull. environ. Contam.
Toxicol., 37: 106-112.
REDDY, T.N. & REDDY, S.J. (1989) Voltammetric behaviour of some nitro
group containing pesticides. Indian J. environ. Protect., 9(8):
592-594.
REHWOLDT, R.E., KELLEY, E., & MAHONEY, M. (1977) Investigations into
the acute toxicity and some chronic effects of selected herbicides and
pesticides on several fresh water fish species. Bull. environ. Contam.
Toxicol., 18(3): 361-365.
RENHOF, M. (1984) [Methyl parathion (active principle of Folidol M):
Tests for embryotoxic effects in rabbits after oral administration.]
Wuppertal-Elberfeld, Bayer AG, Institute of toxicology (Unpublished
reports No. 12907 and 16331, submitted to WHO by Bayer AG, Leverkusen,
Germany) (in German).
RENVALL, S., LINSKOG, E., & CLEMENTZ, E. (1975) Residues of
organophosphorus pesticides in fruits and vegetables on the Swedish
market from July 1967 to April 1973. Presented at the IUPAC Third
International Congress of Pesticide Chemistry, Helsinki, 3-9 July
1974. Environ. Qual. Saf., Suppl., 3: 166-174.
RICE, C.P., OLNEY, C.E., & BIDLEMAN, T.F. (1977) Use of polyurethane
foam to collect trace amounts of chlorinated hydrocarbons and other
organics from air. WMO In: Air pollution measurement techniques.
Geneva, World Meteorological Organization, Part II, pp. 216-224
(Special Environmental Report No. 10; Publication WMO-No. 460).
RICHTER, E.D., ROSENVALD, Z., KASPI, L., LEVY, S., & GRUENER, N.
(1986) Sequential cholinesterase tests and symptoms for monitoring
organophosphate absorption in field workers and in persons exposed to
pesticide spray drift. Toxicol. Lett., 33: 25-35.
RIDER, J.A., MOELLER, H.C., PULETTI, E.J., & SWADER, J.I. (1969)
Toxicity of parathion, systox, octamethyl pyrophosphoramide, and
methyl parathion in man. Toxicol. appl. Pharmacol., 14: 603-611.
RIDER, J.A., SWADER, J.I., & PULETTI, E.J. (1970) Methyl parathion and
guthion anticholinesterase effects in human subjects. Fed. Proc., 29:
349 (Abstr. No.588).
RIDER, J.A., SWADER, J.I., & PULETTI, E.J. (1971) Anticholinesterase
toxicity studies with methyl parathion, guthion and phosdrin in human
subjects. Fed. Proc., 30: 1382.
RIPLEY, B.D. & BRAUN, H.E. (1983) Retention time data for
organochlorine, organophosphorus, and organonitrogen pesticides on
SE-30 capillary column and application of capillary gas
chromatography to pesticide residue analysis. J. Assoc. Off. Anal.
Chem., 66: 1084-1095.
RIPPEL, A., KOVAC, J., & SZOKOLAY, A. (1970) [Investigation of the
persistence of organophosphorus insecticides in citrus juices.]
Nahrung, 14(3): 223-228 (in German).
ROACH, J.A.G. & ANDRZEJEWSKI, D. (1987) Analysis for pesticide
residues by collision-induced fragmentation. In: Rosen, J.D., ed.
Application of new mass spectrometry techniques in pesticide
chemistry. New York, Chichester, Brisbane, Toronto, John Wiley and
Sons, pp. 187-210 (Chemical Analysis series, Volume 91).
ROARK, B., PFRIMMER, T.R., & MERKL, M.E. (1963) Effects of some
formulations of methyl parathion, toxaphene and DDT on the cotton
plant. Crop Sci., 3: 338-341.
ROBERTS, D.K., SILVEY, N.J., & BAILEY, E.M. Jr (1988) Brain
acetylcholinesterase activity recovery following acute methyl
parathion intoxication in two feral rodent species: comparison to
laboratory rodents. Bull. environ. Contam. Toxicol., 41: 26-35.
ROBINSON, S.C., ENDALL, R.J., & ROBINSON, R. (1988) Effects of
agricultural spraying of methyl parathion on cholinesterase activity
and reproductive success in wild starlings (Sturnus vulgaris).
Environ. Toxicol. Chem., 7(5): 343-349.
RODNITZKY, R.L., LEVIN, H.S., & MORGAN, D.P. (1978) Effects of
ingested parathion on neurobehavioral functions. Clin. Toxicol., 13:
347-359.
ROSS, B. & HARVEY, J. (1981) A rapid, inexpensive, quantitative
procedure for the extraction and analyses of Penncap-M (methyl
parathion) from honeybees ( Apis mellifera L.), beeswax, and pollen.
J. Agric. Food Chem., 29: 1095-1096.
ROYAL SOCIETY OF CHEMISTRY (1986) European directory of agrochemical
products. Volume 3: Insecticides and acaricides. Nottingham, The Royal
Society of Chemistry.
RUPA, D.S., REDDY, P.P., & REDDI, O.S. (1989) Chromosomal aberrations
in peripheral lymphocytes of cotton field workers exposed to
pesticides. Environ. Res., 49: 1-6.
RUZICKA, J.H., THOMSON, J., & WHEALS, B.B. (1967) The gas
chromatographic determination of organophosphorus pesticides. J.
Chromatogr., 31: 37-47.
SABHARWAL, A.K. & BELSARE, D.K. (1986) Persistence of methyl parathion
in a carp rearing pond. Bull. environ. Contam. Toxicol., 37: 705-709.
SADAR, M.H., KUAN, S.S., & GUIBAULT, G.G. (1970) Trace analysis of
pesticides using cholinesterase from human serum, rat liver, electric
eel, bean leaf beetle, and white fringe beetle. Anal. Chem., 42:
1770-1774.
SAGREDOS, A.N. & ECKERT, W.R. (1976) [Methods for the determination of
phytopharmaceuticals in tobacco and tobacco products. Part II.
Simultaneous determination of hexane-soluble organohosphorus
pesticides.] Beitr. Tabakforsch., 8: 447-454 (in German).
SALTZMAN, S., MINGELGRIN, U., & YARON, B. (1976) Role of water in the
hydrolysis of parathion and methylparathion on kaolinite. J. Agric.
Food Chem., 24(4): 739-743.
SANDERS, P.F. & SEIBER, J.N. (1983) A chamber for measuring
volatilization of pesticides from model soil and water disposal
systems. Chemosphere, 12: 999-1012.
SANDERSON, D.M. & EDSON, E.F. (1964) Toxicological properties of the
organophosphorus insecticide dimethoate. Br. J. ind. Med., 21: 52-64.
SAROJA-SUBBARAJ, G. & BOSE, S. (1982) Effects of methyl parathion on
the growth, cell size, pigment and protein content of Chlorella
protothecoides. Environ. Pollut., 27: 297.
SAROJA-SUBBARAJ, G. & BOSE, S. (1983a) Correlation between inhibition
of photosynthesis and growth of Chlorella treated with methyl
parathion. J. Biosci., 5: 71-78.
SAROJA-SUBBARAJ, G. & BOSE, S. (1983b) Binding characteristics of
methyl parathion to photosynthetic membranes of Chlorella. Pestic.
Biochem. Physiol., 20: 188-193.
SAROJA-SUBBARAJ, G. & BOSE, S. (1984) Recovery from methyl
parathion-induced damage of the photosynthetic apparatus in Chlorella
protothecoides. Bull. Environ. Contam. Toxicol., 32: 102-108.
SASTRY, C.S.P. & VIJAYA, D. (1986) Spectrophotometric determination of
fenitrothion and methyl parathion in insecticidal formulations. J.
Food Sci. Technol., 23: 336-338.
SASTRY, C.S.P. & VIJAYA, D. (1987) Spectrophotometric determination of
some insecticides with 3-methyl-2-benzothiazolinone hydrazone
hydrochloride. Talanta, 34: 372-374.
SAUVEGRAIN, P. (1980) Les micropolluants organiques dans les eaux
superficielles continentales. Rapport No. 1: Les pesticides
organophosphorés. Paris, Bureau National de l'Information Scientifique
et Technique.
SAXTON, W.L. (1987) Emergence temperature indexes and relative
retention times of pesticides and industrial chemicals determined by
linear programmed temperature gas chromatography. J. Chromatogr., 393:
175-194.
SAYRE, I.M. (1988) International standards for drinking water. J. Am.
Water Works Assoc., 80: 53-60.
SCHAFER, E.W. Jr (1972) The acute oral toxicity of 369 pesticidal,
pharmaceutical and other chemicals to wild birds. Toxicol. appl.
Pharmacol., 21: 315-330.
SCHILDE, B. & BOMHARD, E. (1984) [E 605-methyl (methyl parathion).
Supplementary histopathological test, further to the 2-year feeding
trial on rats. Addendum to report No. 9889.] Wuppertal-Elberfeld,
Bayer AG, Institute of Toxicology (Unpublished report No. 12559,
submitted to WHO by Bayer AG, Leverkusen, Germany) (in German).
SCHIMMEL, S.C., GARNAS, R.L., PATRICK, J.M. Jr, & MOORE, J.C. (1983)
Acute toxicity, bioconcentration and persistence of AC 222.705,
benthiocarb, chlorpyrifos, fenvalerate, methylparathion and permethrin
in the estuarine environment. J. Agric. Food Chem., 31(1): 104-113.
SCHNORBUS, R.R. & PHILLIPS, W.F. (1967) New extraction system for
residue analyses. J. Agric. Food Chem., 15: 661-666.
SCHOMBURG, C.J., GLOTFETTY, D.E., & SEIBER, J.N. (1991) Pesticide
occurrence and distribution in fog collected near Monterey,
California. Environ. Sci. Technol., 25: 155-160.
SCHOOR, W.P. & BRAUSCH, J. (1980) The inhibition of
acetylcholinesterase activity in pink shrimp Penaeus duorarum by
methyl-parathion and its oxon. Arch. environ. Contam. Toxicol., 9:
599-605.
SCHRADER, G. (1963) [The development of new insecticidal phosphoric
acid esters.] Weinheim, Verlag Chemie GmbH, pp. 223-258 (in German).
SCHULTEN, H.R. & SUN, S. E. (1981) High-resolution field desorption
mass spectrometry. Part IX. Field desorption mass spectrometry of
standard organophosphorus pesticides and their identification in waste
water. Int. J. Environ. Anal. Chem., 10: 247-263.
SCHULZ, H., DESI, I., & NAGYMAJTENYI, L. (1990) Behavioural effects of
subchronic intoxication with parathion-methyl in male Wistar rats.
Neurotoxicol. Teratol., 12: 125-127.
SCHULZE, J.A., MANIGOLD, D.B., & ANDREWS, F.L. (1973) Pesticides in
selected Western streams - 1968-71. Pestic. Monit. J., 7: 73-84.
SCHUTZ, H. & SCHINDLER, A. (1974) [Application of reaction
chromatography to chemical-toxicological analysis. Microchemical
detection and separation of sixteen nitropesticides by thin-layer
chromatography.] Fresenius Z. Anal. Chem., 270: 356-359 (in German).
SCHUTZMANN, R.L., WOODHAM, D.W., & COLLIER, C.W. (1971) Removal of
sulfur in environmental samples prior to gas chromatographic analysis
for pesticide residues. J. Assoc. Off. Anal. Chem., 54: 1117-1119.
SEIBER, J.N., McCHESNEY, M.M., & WOODROW, J.E. (1989) Airborne
residues resulting from use of methyl parathion, molinate and
thiobencarb on rice in the Sacramento Valley, California. Environ.
Toxicol. Chem., 8: 577-588.
SHAFIK, M.T. & ENOS, H.F. (1969) Determination of metabolic and
hydrolytic products of organophosphorus pesticide chemicals in human
blood and urine. J. Agric. Food Chem., 17: 1186-1189.
SHARMA, R.P. & REDDY, R.V. (1987) Toxic effects of chemicals on the
immune system. In: Haley, T.J. & Berndt, W.O., ed. Handbook of
toxicology. Washington, DC, Hemisphere Publishing Corporation, pp.
555-591.
SHARMA, V.K. JADHAV, R.K., RAO, G.J., SARAF, A.K., & CHANDRA, H.
(1990) High performance liquid chromatographic method for the analysis
of organophosphorus and carbamate pesticides. Forens. Sci. Int., 48:
21-25.
SHARMILA, M., RAMANAND, K., ADHYA, T.K., & SETHUNATHAN, N. (1988)
Temperature and the persistence of methyl parathion in a flooded soil.
Soil Biol. Biochem., 20: 399-401.
SHARMILA, M., RAMANAND, K., & SETHUNATHAN, N. (1989a) Hydrolysis of
methyl parathion in a flooded soil. Bull. environ. Contam. Toxicol.,
43: 45-51.
SHARMILA, M., RAMANAND, K., & SETHUNATHAN, N. (1989b) Effect of yeast
extract on the degradation of organophosphorus insecticides by soil
enrichment and bacterial cultures. Can. J. Microbiol., 35: 1105-1110.
SHERMA, J. & BRETSCHNEIDER, W. (1990) Determination of
organophosphorus insecticides in water by C-18 solid phase extraction
and quantitative TLC. J. liq. Chromatogr., 13(10): 1983-1989.
SHERMA, J. & SHAFIK, T.M. (1975) Multiclass, multiresidue analytical
method for determining pesticide residues in air. Arch. environ.
Contam. Toxicol., 3: 55-71.
SHIHIDO, T. & FUKAMI, J. (1963) Studies on the selective toxicities of
organic phosphorous insecticides (II), the degradation of ethyl
parathion, methyl parathion, methyl paraoxon, and sumithion in mammal,
insect and plant. Botyu-Kagaku, 28: 69-76.
SHIRES, S.W. (1985) A comparison of the effects of cypermethrin,
parathio-methyl and DDT on cereal aphids, predatory beetles,
earthworms and litter decomposition in spring wheat. Crop Prot., 4:
177-193.
SIMMON, V.F., MITCHELL, A.D., & JORGENSON, T.A. (1977) Evaluation of
selected pesticides as chemical mutagens in vitro and in vivo
studies. Menlo Park, California, Stanford Research Institute, 251 pp.
(PB-268647).
SINGH, K.S. & SINGH, A.P. (1978) Persistence of methyl and ethyl
parathion in soil and plant. Pesticides, 12: 39-41.
SINGH, N.N. & SRIVASTAVA, A.K. (1982) Toxicity of a mixture of aldrin
and formothion and other organophosphorus, organochlorine and
carbamate pesticides to the Indian catfish Heteropneustes fossilis.
Comp. Physiol. Ecol., 7(2): 115-118.
SINGH, S., LEHMANN-GRUBE, B., & GOEDDE, H.W. (1984) Cytogenetic
effects of paraoxon and methyl-parathion on cultured human
lymphocytes: SCE, clastogenic activity and cell cycle delay. Int.
Arch. occup. environ. Health, 54 (3): 195-200.
SMART, E. & STEVENSON, J.H. (1982) Laboratory estimation of toxicity
of pyrethroid insecticides to honey bees: relevance to hazard in the
field. Bee World, 63: 150-152.
SMITH, D.C., LEDUC, R., & TREMBLAY, L. (1975) Pesticide residues in
the total diet in Canada. IV. 1972 and 1973. Pestic. Sci., 6: 75-82.
SMITH, J.H., MABEY, W.R., BOHONOS, N., HOLT, B.R., LEE, S.S., CHOU,
T.-W., BOMBERGER, D.C., & MILL, T. (1978) Environmental pathways of
selected chemicals in freshwater systems, Part II: Laboratory studies.
Athens, Georgia, US Environmental Protection Agency (PB288511).
SMITH, S., WILLIS, G.H., MCDOWELL, L.L., & SOUTHWICK, L.M. (1987)
Dissipation of methyl parathion and ethyl parathion from cotton
foliage as affected by formulation. Bull. environ. Contam. Toxicol.,
39: 280-285.
SMYTH, M.R. & OSTERYOUNG, J.G. (1978) A pulse polarographic
investigation of parathion and some other nitro-containing pesticides.
Anal. chim. Acta, 96: 335-344.
SOBTI, R.C., KRISHAN, A., & PFAFFENBERGER, C.D. (1982) Cytokinetic and
cytogenetic effects of some agricultural chemicals on human lymphoid
cells in vitro: organophosphates. Mutat. Res., 102: 89-102.
SOLON, J.M. & NAIR, J.H. (1970) Effect of a sublethal concentration of
LAS (linear alkyl benzene sulfonate detergent) on the acute toxicity
of various phosphate pesticides to the fathhead minnow (Pimephales
promelas). Bull. environ. Contam. Toxicol., 5(5): 408-413.
SOMLYAY, I.M., VARNAGY, L.E., & PAVLISCSAK, Cs. (1989) Effect of
various pesticides on plasma biochemistry of chicken embryos. Mede.
Fac. Landbouwwet. Rijksuniv. Gent, 54(2a): 181-184.
SONOBE, H., SAKUMA, S., YAMADA, K., KAWASAKI, M., KATAYAMA, H., &
KUROIWA, Y. (1982) Analysis systems for trace compounds found in
barley, malt and hops. In: Proceedings of the 17th Convention, Perth,
7-12 March 1982. Sydney, Institute of Brewing, Australia and New
Zealand Section, pp. 56-69.
SPECHT, W. (1978) [Methods for the rapid work-up of large fat
quantities for analysis of pesticide residues.] Lebensmittelchem
Gerichtl. Chem., 32: 51-53 (in German).
SPECHT, W. & TILLKES, M. (1980) [Gas chromatographic determination of
pesticide residues after cleanup by gel chromatography and mini silice
gel column chromatographs.] Fresenius Z. Anal. Chem., 301: 300-307 (in
German).
SPENCER, E.Y. (1982) Guide to the chemicals used in crop protection,
7th ed. Ottawa, Agriculture Canada Research Institute, 394 pp.
(Publication No. 1093).
SPINGARN, N.E., NORTHINGTON, D.J., & PRESSELY, T. (1982) Analysis of
nonvolatile organic hazardous substances by GC/MS. J. Chromatogr.
Sci., 20: 571-574.
SRIVASTAVA, A.K. & SINGH, N.N. (1981) Effects of exposure to
methyl-parathion on carbohydrate metabolism in Indian catfish
Heteropneustes fossilis. Acta pharmacol. Toxicol., 48(1): 26-31.
STAHR, H.M., GAUL, M., HYDE, W., & MOORE, R. (1979) A cellulose column
cleanup for organophosphorus pesticides. Microchem J., 24: 97-101.
STAMPER, C.R., BALDUINI, W., MURPHY, S.D., & COSTA, L.G. (1988)
Behavioural and biochemical effects of postnatal parathion exposure in
the rat. Neurotoxicol. Teratol., 10: 261-266.
STAN, H.J. & GOEBEL, H. (1983) Automated capillary gas chromatographic
analysis of pesticide residues in food. J. Chromatogr., 268: 55-69.
STAN, H. & GOEBEL, H. (1984) Evaluation of automated splitless and
manual on-column injection techniques using capillary gas
chromatography for pesticide residue analysis. J. Chromatogr., 314:
413-420.
STAN, H.J. & MROWETZ, D. (1983) Residue analysis of organophosphorus
pesticides in food with two-dimensional gas chromatography using
capillary columns and flame photometric detection. J. high Resolut.
Chromatogr. Chromatogr. Commun., 6: 255-263.
STAN, H.J. & MUELLER, H.M. (1988) Evaluation of automated and manual
hot- splitless, cold-splitless (PTV), and on-column injection
technique using capillary gas chromatography for the analysis for
organophosphorus pesticides. J. high Resolut. Chromatogr. Chromatogr.
Commun., 11: 140-143.
STANLEY, C.W., BARNEY, J.E., II, HELTON, M.R., & YOBS, A.R. (1971)
Measurement of atmospheric levels of pesticides. Environ. Sci.
Technol., 5: 430-435.
STEINWANDTER, H. (1988) Contributions to the application of gel
chromatography in residue analysis. II. A new gel chromatographic
system using acetone for the separation of pesticide residues and
industrial chemicals. Fresenius Z. Anal. Chem., 331: 499-502.
STEPHENSON, R.R. & KANE, D.F. (1984) Persistence and effects of
chemicals in small enclosures in ponds. Arch. environ. Contam.
Toxicol., 13: 313-326.
STOLL, K. (1982) Residue breakdown in stored fruit. In: Proceedings of
the 21th International Horticultural Congress, Hamburg, 29 August-4
September 1982. The Hague, International Society for Horticultural
Science, pp. 231-235.
STORHERR, R.W., MURRAY, E.J., KLEIN, I., & ROSENBERG, L.A. (1967)
Sweep destillation cleanup of determining of organophosphate and
chlorinated hydrocarbon pesticides. J. Assoc. Off. Anal. Chem., 50:
605-615.
STREET, J.C. & SHARMA, R.P. (1975) Alteration of induced cellular and
humoral immune response by pesticides and chemicals of environmental
concern: quantitative studies of immunosuppression by DDT, aroclor
1254, carbaryl, carbofuran, and methylparathion. Toxicol. appl.
Pharmacol., 32: 587-602.
SULTATOS, L.G. (1987) The role of the liver in mediating the acute
toxicity of the pesticide methyl parathion in the mouse. Drug Metab.
Dispos., 15(5): 613-617.
SULTATOS, L.G. & WOODS, L. (1988) The role of glutathione in the
detoxification of the insecticides methyl parathion and
azinphos-methyl in the mouse. Toxicol. appl. Pharmacol., 96: 168-174.
SULTATOS, L.G., KIM, B., & WOODS, L. (1990) Evaluation of estimations
in vitro of tissue/blood distribution coefficients of
organothiophosphate insecticides. Toxicol. appl. Pharmacol., 103:
52-55.
SUPROCK, J.F. & VINOPAL, J.H. (1987) Behavior of 78 pesticides and
pesticide metabolites on four different ultra-bond gas chromatographic
columns. J. Assoc. Off. Anal. Chem., 70: 1014-1017.
SWAMY, G.S. & VEERESH, A.V. (1987) Methylparathion modulates lipid
biosynthesis in sorghum ( Sorghum bicolor L.). Moench. Pestic.
Biochem. Physiol., 28: 341-348.
SWINEFORD, D.M. & BELISLE, A.A. (1989) Analysis of trifluralin, methyl
paraoxon, methyl parathion, fenvalerate and 2,4-D dimethylamine in
pond water using solid-phase extraction. Environ. Toxicol. Chem., 8:
465-468.
TAKIMOTO, Y., OHSHIMA, M., YAMADA, H., & MIYAMOTO, J. (1984) Fate of
fenithrothion in several developmental stages of the killifish
(Oryzias latipes). Arch. environ. Contam. Toxicol., 13: 579-587.
TANIMURA, T., KATSUYA, T., & NISHIMURA, H. (1967) Embryotoxicity of
acute exposure to methyl parathion in rats and mice. Arch. environ.
Health, 15: 609-613.
TAYLOR, P. (1980) Anticholinesterase agents. In: Gilman, A.G.,
Goodman, L.S., & Gilman, A., ed. The pharmacological basis of
therapeutics, 6th ed. New York, Macmillan Publishing Co., pp.
100-119.
TESSARI, J.D. & SPENCER, D.L. (1971) Air sampling for pesticides in
the human environment. J. Assoc. Off. Anal. Chem., 54: 1376-1382.
THIELEMANN, H. (1974) [Semiquantitative thin layer chromatographic
determination of parathion-methyl.] Z. Chem., 14: 407-408 (in German).
THIER, H-P. & ZEUMER, H. (1987) [Pesticides Commission of the German
Association for the Encouragement of Research. Manual of pesticide
residue analysis.] Weinheim, VCH Verlag, vol. 1.
THOMAS, P.T. & HOUSE, R.V. (1989) Pesticide-induced modulation of the
immune system. In: Ragsdale, N.N. & Menzer, R.E., ed. Carcinogenicity
and pesticides: principles, issues and relationships. 196th National
Meeting of the American Chemical Society, Los Angeles, California,
25-30 September 1988 Washington, DC, American Chemical Society, pp.
94-106 (ACS Symposium series, 414).
THOMPSON, A.R. & GORE, F.L. (1972) Toxicity of twenty-nine
insecticides to Folsomia candida: laboratory studies. J. Econ.
Entomol., 65: 1255-1260.
THUMA, N.K., O'NEILL, P.E., BROWNLEE, S.G., & VALENTINE, R.S. (1983)
Microbial degradation of selected harzardous materials:
pentachlorophenol, hexachlorocyclopentadiene and methyl parathion,
Washington, DC, US Environmental Protection Agency, 76 pp.
(EPA-Report No. 68-03-2491).
THYSSEN, J. (1979) [E 120 (methyl parathion): Studies on acute
inhalation toxicity.] Wuppertal-Elberfeld, Bayer AG, Institute of
Toxicology (Unpublished report No. 8148, submitted to WHO by Bayer AG,
Leverkusen, Germany) (in German).
THYSSEN, J. & MOHR, U. (1982) [E 120 (methyl parathion): Subacute
inhalation test on rats - histopathological findings.]
Wuppertal-Elberfeld, Bayer AG, Institute of Toxicology (Unpublished
report No. 11302, submitted to WHO by Bayer AG, Leverkusen, Germany)
(in German).
TILSTONE, W.J., WINCHESTER, J.F., & REAVEY, P.C. (1979) The use of
pharmacokinetics principles in determining the effectiveness of
removal of toxins from blood. Clin. Pharmacokinet., 4: 23-37.
TOSCANO, N.C., SANCES, F.V., JOHNSON, M.W., & LaPRE, L.F. (1982)
Effect of various pesticides on lettuce physiology and yield. J. Econ.
Entomol., 75: 738-741.
TRIPATHI, G. & SHUKLA, S.P. (1988) Inhibition of liver and skeletal
muscle enzymes by methyl parathion. Biochem. Arch., 4: 55-61.
TRIPATHI, G. & SHUKLA, S.P. (1990) Enzymatic and ultrastructural
studies in a freshwater catfish: impact of methyl parathion. Biomed.
Environ. Sci., 3: 166-182.
TRIPATHY, N.K., DEY, L., & MAJHI, B. (1987) Genotoxicity of metacid
established through the somatic and germ line mosai assays and the
sex-linked recessive lethal test in Drosophila. Arch. Toxicol.,
61(1): 53-57.
US EPA (1981) Acephate, aldicarb, carbophenothion, DEF, EPN, ethoprop,
methyl parathion and phorate; their acute and chronic toxicity,
bioconcentration potential and persistence as related to marine
environments. Gulf Breeze, Florida, US Environmental Protection
Agency, Office of Research and Development, Environmental Research
Laboratory, 275 pp. (EPA-600/4-81-041; PB 81-244477).
VAN BAO, T, SZABO, I., RUZICSKA, P., & CZEIZEL, R. (1974) Chromosome
aberrations in patients suffering acute organic phosphate insecticide
intoxication. Humangenetik., 24: 33-57.
VAN VELD, P.A. & SPAIN, J.C. (1983) Degradation of selected xenobiotic
compounds in three types of aquatic test systems. Chemosphere, 12:
1291-1305.
VARIS, A.-L. (1972) Loss of lindane, dimethoate, and methyl parathion
residues from seedlings of sugar beet as influenced by plant growth.
Ann. Agric. Fenn., 11: 381-385.
VARNAGY, L. & DELI, E. (1985) Comparative teratological study of
insecticide Wofatox 50 EC (50% methyl parathion) on chicken and
pheasant fetuses. Anat. Anz. Jena, 158: 1-3.
VARNAGY, L., KORZENSZKY, M., & FANCSI, T. (1984) Teratological
examination of the insecticide methylparathion/Wofatox 50 EC/ on
pheasant embryos. 1. Morphological study. Vet. Res. Commun., 8:
131-139.
VARNAGY, L., DELI, E., & BAUMANN, M. (1985) Scanning electron
microscopic examination of cartilage in chicken embryos treated with
the insecticide Wofatox 50 EC. Acta vet. Hung., 36:(1-2): 117-121.
VOGELGESANG, J. & THIER, H.P. (1986) [Contributions to the analysis of
pesticide residues in ready-to-eat foods.] Z. Lebensm.-Unters.
Forsch., 182(5): 400-406 (in German).
VROCHINSKY, K.K. & MAKOVSKY, V.N. (1977) The sources of pollution and
the content of pesticides in air. In: Uses of pesticides and
environmental protection. Kiev, Vysshaya Shkola, pp. 143-154.
WALKER, T.W., MEEK, C.L., WRIGHT, V.L., & BILLODEAUX, J.S. (1985)
Susceptibility of Romanomermis culicivorax (Nematoda: Mermithidae)
post parasites to agrochemicals used in Louisiana rice production. J.
Am. Control Assoc., 1: 477-481.
WALKER, W.W. (1978) Insecticide persistence in natural sea water as
affected by salinity, temperature and sterility. Washington, DC, US
Environmental Protection Agency, Office of Research Development, 25
pp. (EPA-600/3-78-044).
WALSH, G.E. & ALEXANDER, S.V. (1980) A marine algal bioassay method:
results with pesticides and industrial wastes. Water, Air, Soil
Pollut., 13: 45-55.
WALSH, G.E., DEANS, C.H., & McLAUGHLIN, L.L. (1987) Comparison of the
EC50s of algal toxicity tests calculated by four methods. Environ.
Toxicol. Chem., 6: 767-770.
WALTERS, S.M. (1990) Clean-up techniques for pesticides in fatty
foods. Anal. chim. acta, 236: 77-82.
WARE, G.W., ESTESEN, B., & CAHILL, W.P. (1974a) Dislodgable leaf
residues of insecticides on cotton. Bull. environ. Contam. Toxicol.,
11: 434-437.
WARE, G.W., MORGAN, D.P., ESTESEN, B.J., & CAHILL, W.P. (1974b)
Establishment of reentry intervals for organophosphate-treated cotton
fields based on human data. II. Azodrin, ethyl and methyl parathion.
Arch. environ. Contam. Toxicol., 2(2): 117-129.
WARE, G.W., WATSON, T.F., ESTESEN, B., & BUCK, N.A. (1980) Effects of
molasses or toxaphene on residual life and efficacy of methyl
parathion on cotton. J. Econ. Entomol., 73: 15-17.
WARE, G.W., BUCK, N.A., & ESTESEN, B.J. (1983) Dislodgeable
insecticide residues on cotton foliage: comparison of ULV/cottonseed
oil vs. aqueous dilutions of 12 insecticides. Bull. environ. Contam.
Toxicol., 31: 551-558.
WARNER, J.S. (1975) Identification on impurities in technical-grade
pesticides. In: Substitute Chemical Programme - the first year of
progress. Proceedings of a symposium. Vol. IV: Chemical methods
workshop. Washington DC, Bureau of Commerce, National Technical
Information Service, pp. 31-65 (PB 261-007).
WATTS, R.R. & STORHERR, R.W. (1967) Sweep codistillation cleanup of
milk for determination of organophosphate and chlorinated hydrocarbon
pesticides. J. Assoc. Off. Anal. Chem., 50: 581-585.
WEBER, H., PATZSCHKE, K., & WEGNER, L.A. (1979) [Methyl
parathion-14C (benzene ring-labelled compound): Biokinetic studies
on rats.] Wuppertal-Elberfeld, Bayer AG, Pharmokinetics Institute,
Isotope Laboratory (Unpublished report No. Pharma-8722, submitted to
WHO by Bayer AG, Leverkusen, Germany) (in German).
WEGMAN, R.C.C., VAN DEN BROEK, H.H., HOFSTEE, A.W.M., & MARSMAN, J.A.
(1984) Determination of triazines, organophosphorus containing
pesticides and aromatic amines in soil samples. Meded. Fac.
Landbouwwet. Rijksuniv. Gent., 49: 1231-1239.
WEHNER, T.A., WOODROW, J.E., KIM, Y.H., & SEIBER, J.N. (1984)
Multiresidue analysis of trace organic pesticides in air. In: Keith,
L.H., ed. Identification and analysis of organic pollutants. Boston,
Butterworth, pp. 273-290.
WERNER, D. & PAWLITZ, H. (1978) Differential elimination of phenol by
diatoms and other unicellular algae from low concentrations. Bull.
environ. Contam. Toxicol., 20: 303-312.
WESSEL, J.R. (1967) Collaborative study of a method for multiple
organophosporus pesticide residues in nonfatty foods. J. Assoc. Off.
Anal. Chem., 50: 430-439.
WEST, B., VIDONE, L.B., & SHAFFER, C.B. (1961) Acute and subacute
toxicity of dimethoate. Toxicol. appl. Pharmacol., 3: 210-223.
WHITE-STEVENS, R. (1971) Pesticides in the environment, New York,
Basel, Marcel Dekker, Inc., vol. 1., parts 1 and 2.
WHO (1990) The WHO recommended classification of pesticides by hazard
and guidelines to classification, 1990-91. Geneva, World Health
Organization (Unpublished document WHO/PCS/90.1).
WHO (1986) Environmental Health Criteria, 63: Organophosphorus
insecticides: a general introduction. Geneva, World Health
Organization, 181 pp.
WHO/FAO (1975) Data sheets on pesticides No. 7, Rev. 1:
Parathion-methyl. Geneva, World Health Organization (Unpublished WHO
document VBC/05/75.7 (Rev. 1)).
WICKER, G.W., WILLIAMS, W.A., & GUTHRIE, F.E. (1979) Exposure of field
workers to organophosphorus insecticides: sweet corn and peaches.
Arch. environ. Contam. Toxicol., 8: 175-182.
WIERSMA, G.B., MITCHELL, W.G., & STANFORD, C.L. (1972) Pesticide
residues in onions and soil - 1969. Pestic. Monit. J., 5(4): 345-347.
WILD, D. (1975) Mutagenicity studies on organophosphorus insecticides.
Mutat. Res., 32: 133-150.
WILKINS, J.P.G. (1990) Rationalization of the mass spectrometric and
gas chromatographic behaviour of organophosphorus pesticides: Part 1
- Substituted phenyl phosphorothioates. Pest. Sci., 29: 163-181.
WILLEMS, J., BRAECKMAN, R., & BELPAIRE, F. (1980) Fate of methyl
parathion in the dog. Toxicol. Lett., 5(Suppl. 1): 34 (Abstract No.
038).
WILLIAMS, M.W., FUYAD, J.P., FRAWLEY, J.P., & FITZHUGH, O.G. (1957)
In vivo effects of paired combinations of five organic phosphate
insecticides. J. Agric. Food Chem., 6: 514-516.
WILLIAMS, M.W., FUYAT, H.N., & FITZHUGH, O.G. (1959) The subacute
toxicity of four organic phosphates to dogs. Toxicology, 1: 1-7.
WILLIS, G.H., McDOWELL, L.L., SOUTHWICK, L.M., & SMITH, S. (1985)
Toxaphene, methyl parathion, and fenvalerate disapperance from cotton
foliage in the mid-south. J. environ. Qual., 14(3): 446-450.
WILMES, R. (1987) Parathion-methyl: Hydrolysis studies. Leverkusen,
Germany, Bayer AG, Institute of Metabolism Research, 34 pp.
(Unpublished report No. PF 2883, submitted to WHO by Bayer AG).
WINDHOLZ, M., BUDVARI, S., BLUMETTI, R.F., & OTTERBEIN, E.S. ed.
(1983) The Merck Index, 10th ed. Rahway, New Jersey, Merck and Co.,
874 pp.
WINTER, H. & LINDNER, K. (1987) [Drinking water of river Rhine after
the "Sandoz accident".] Wasser Abwasser, 128: 525-532 (in German).
WOLFE, H.R., DURHAM, W.F., & ARMSTRONG, J.F. (1967) Exposure of
workers to pesticides. Arch. environ. Health, 14: 622-633.
WOLFE, H.R., WASH, W., DURHAM, W.F., & ARMSTRONG, J.F. (1970) Urinary
excretion of insecticide metabolites. Excretion of para-nitrophenol
and DDA as indicators of exposure to parathion. Arch. environ. Health,
21: 711-716.
WOLFE, N.L., KITCHENS, B.E., MACALDY, D.L., & GRUNDL, T.J. (1986)
Physical and chemical factors that influence the anaerobic degradation
of methyl parathion in sediment systems. Environ. Toxicol. Chem., 5:
1019-1026.
WOODER, M.F., WRIGHT, A.S., & KING, L.J. (1977) In vivo alkylation
studies with dichlorvos at practical use concentrations. Chem.-Biol.
Interact., 19: 25-46.
WOODROW, J.E., SEIBER, J.N., CROSBY, D.G., MOILANEN, K.W., SODERQUIST,
C.J., & MOURER, C. (1977) Airborne and surface residues of parathion
and its conversion products in a treated plum orchard environment.
Arch. Environ. Contam. Toxicol., 6: 175-191.
WORTHING, C.R. & HANCE, R.J. (1990) The pesticide manual, 9th ed.
Croydon, British Crop Protection Council.
XUE, J. (1984) Resin concentration method for determination of
pesticides in drinking water. Huanjing Huaxue, 3: 43-49.
YAMAMOTO, T., EGASHIRA, T., YOSHIDA, T., & KUROIWA, Y. (1983)
Comparative metabolism of fenitrothion and methylparathion in male
rats. Acta pharmacol. toxicol., 53: 96-102.
YANG, C.-F. & SUN, Y.-P. (1977) Partition distribution of insecticides
as a critical factor affecting their rates of absorption from water
and relative toxicities to fish. Arch. environ. Contam. Toxicol., 6:
325-335.
YASOSHIMA, M. & MASUDA, Y. (1986) Effect of carbon disulfide on the
anticholinesterase action of several organophosphorus insecticides in
mice. Toxicol. Lett., 32: 179-184.
YASUNO, M., HIRAKOSO, S., SASA, M., & UCHIDA, M. (1965) Inactivation
of some organophosphorus insecticides by bacteria in polluted water.
Jpn. J. exp. Med., 35(6): 545-563.
YODER, J., WATSON, M., & BESON, W.W. (1973) Lymphocyte chromosome
analysis of agricultural workers during extensive occupational
exposure to pesticides. Mutat. Res., 21: 335-340.
YOUNGMAN, R.R., TOSCANO, N.C., & GASTON, L.K. (1989) Degradation of
methyl parathion to p-nitrophenol on cotton and lettuce leaves and
its effects on plant growth. J. Econ. Entomol., 82: 1317-1322.
YOUNGMAN, R.R., LEIGH, T.F., KERBY, T.A., TOSCANO, N.C., & JACKSON,
C.E. (1990) Pesticides and cotton: effect on photosynthesis, growth,
and fruiting. J. Econ. Entomol., 83: 1549-1557.
YOUSSEF, S.H.A., EL-SAYED, M.G.A., & ATEF, M. (1987) Influence of
gentamicin and rifamycin on toxicity and biotransformation of methyl
parathione in rats. Dtsch. Tierärztl. Wochenschr., 94: 193-236.
ZEPP, R.G. & SCHLOTZHAUER, P.F. (1983) Influence of algae on
photolysis rates of chemicals in water. Environ. Sci. Technol., 17:
462-468.
ZHAO, S. & WANG, Y. (1984) [High-performance liquid chromatographic
analysis of trichlorophon and methylparathionmixed powder.] Sepu, 1:
134-135 (in Chinese).
ZIETEK, M. (1976) [Polarographic determination of parathion and
similar insecticides alongside their metabolites in blood without an
extraction procedure.] Mikrochim. Acta, 2: 249-257 (in German).
ZITKO, V. & MCLEESE, D.W. (1980) Evaluation of hazards of pesticides
used in forest spraying to the aquatic environment. St. Andrews, New
Brunswick, Biological Station (Canadian Technical Report, Fish and
Aquatic Sciences, No. 985).
ZWEIG, G. & DEVINE, J.M. (1969) Determination of organophosphorus
pesticides in water. Residue Rev., 26: 17-36.
ANNEX I. TREATMENT OF ORGANOPHOSPHATE INSECTICIDE POISONING IN MAN
(From EHC 63: Organophosphorus insecticides - a general introduction)
All cases of organophosphorus poisoning should be dealt with as
an emergency and the patient sent to hospital as quickly as possible.
Although symptoms may develop rapidly, delay in onset or a steady
increase in severity may be seen up to 48 h after ingestion of some
formulated organophosphorus insecticides.
Extensive descriptions of treatment of poisoning by
organophosphorus insecticides are given in several major references
(Kagan, 1977; Taylor, 1980; UK DHSS, 1983; Plestina, 1984) and will
also be included in the IPCS Health and Safety Guides to be prepared
for selected organophosphorus insecticides.
The treatment is based on:
(a) minimizing the absorption;
(b) general supportive treatment; and
(c) specific pharmacological treatment.
I.1 Minimizing the absorption
When dermal exposure occurs, decontamination procedures include
removal of contaminated clothes and washing of the skin with alkaline
soap or with a sodium bicarbonate solution. Particular care should be
taken in cleaning the skin area where venepuncture is performed. Blood
might be contaminated with direct-acting organophosphorus esters and,
therefore, inaccurate measures of ChE inhibition might result.
Extensive eye irrigation with water or saline should also be
performed. In the case of ingestion, vomiting might be induced, if the
patient is conscious, by the administration of ipecacuanha syrup
(10-30 ml) followed by 200 ml water. This treatment is, however,
contraindicated in the case of pesticides dissolved in hydrocarbon
solvents. Gastric lavage (with addition of bicarbonate solution or
activated charcoal) can also be performed, particularly in unconscious
patients, taking care to prevent aspiration of fluids into the lungs
(i.e., only after a tracheal tube has been put into place).
The volume of fluid introduced into the stomach should be
recorded and samples of gastric lavage frozen and stored for
subsequent chemical analysis. If the formulation of the pesticide
involved is available, it should also be stored for further analysis
(i.e., detection of toxicologically relevant impurities). A purgative
can be administered to remove the ingested compound.
I.2 General supportive treatment
Artificial respiration (via a tracheal tube) should be started at
the first sign of respiratory failure and maintained for as long as
necessary.
Cautious administration of fluids is advised, as well as general
supportive and symptomatic pharmacological treatment and absolute
rest.
I.3 Specific pharmacological treatment
I.3.1 Atropine
Atropine should be given, beginning with 2 mg iv and given at
15-30-min intervals. The dose and the frequency of atropine treatment
varies from case to case, but should maintain the patient fully
atropinized (dilated pupils, dry mouth, skin flushing, etc.).
Continuous infusion of atropine may be necessary in extreme cases and
total daily doses up to several hundred mg may be necessary during the
first few days of treatment.
I.3.2 Oxime reactivators
Cholinesterase reactivators (e.g., pralidoxime, obidoxime)
specifically restore AChE activity inhibited by organophosphates. This
is not the case with enzymes inhibited by carbamates. The treatment
should begin as soon as possible, because oximes are not effective on
"aged" phosphorylated ChEs. However, if absorption, distribution, and
metabolism are thought to be delayed for any reasons, oximes can be
administered for several days after intoxication. Effective treatment
with oximes reduces the required dose of atropine. Pralidoxime is the
most widely available oxime. A dose of 1 g pralidoxime can be given
either im or iv and repeated 2-3 times per day or, in extreme cases,
more often. If possible, blood samples should be taken for AChE
determinations before and during treatment. Skin should be carefully
cleansed before sampling. Results of the assays should influence the
decision whether to continue oxime therapy after the first 2 days.
There are indications that oxime therapy may possibly have
beneficial effects on CNS-derived symptoms.
I.3.3 Diazepam
Diazepam should be included in the therapy of all but the mildest
cases. Besides relieving anxiety, it appears to counteract some
aspects of CNS-derived symptoms that are not affected by atropine.
Doses of 10 mg sc or iv are appropriate and may be repeated as
required (Vale & Scott, 1974). Other centrally acting drugs and drugs
that may depress respiration are not recommended in the absence of
artificial respiration procedures.
I.3.4 Notes on the recommended treatment
I.3.4.1 Effects of atropine and oxime
The combined effect far exceeds the benefit of either drug
singly.
I.3.4.2 Response to atropine
The response of the eye pupil may be unreliable in cases of
organophosphorus poisoning. A flushed skin and drying of secretions
are the best guide to the effectiveness of atropinization. Although
repeated dosing may well be necessary, excessive doses at any one time
may cause toxic side-effects. Pulse-rate should not exceed 120/min.
I.3.4.3 Persistence of treatment
Some organophosphorus pesticides are very lipophilic and may be
taken into, and then released from, fat depots over a period of many
days. It is therefore quite incorrect to abandon oxime treatment after
1-2 days on the supposition that all inhibited enzyme will be aged.
Ecobichon et al. (1977) noted prompt improvement in both condition and
blood-ChEs in response to pralidoxime given on the 11th-15th days
after major symptoms of poisoning appeared due to extended exposure to
fenitrothion (a dimethyl phosphate with a short half-life for aging of
inhibited AChE).
I.3.4.4 Dosage of atropine and oxime
The recommended doses above pertain to exposures, usually for an
occupational setting, but, in the case of very severe exposure or
massive ingestion (accidental or deliberate), the therapeutic doses
may be extended considerably. Warriner et al. (1977) reported the case
of a patient who drank a large quantity of dicrotophos, in error,
while drunk. Therapeutic dosages were progressively increased up to 6
mg atropine iv every 15 min together with continuous iv infusion of
pralidoxime chloride at 0.5 g/h for 72 h, from days 3 to 6 after
intoxication. After considerable improvement, the patient relapsed and
further aggressive therapy was given at a declining rate from days 10
to 16 (atropine) and to day 23 (oxime), respectively. In total, 92 g
of pralidoxime chloride and 3912 mg of atropine were given and the
patient was discharged on the thirty-third day with no apparent
sequelae.
References to Annex I.
ECOBICHON, D.J., OZERE, R.L., REID, E., & CROCKER, J.F.S (1977) Acute
fenitrothion poisoning. Can. Med. Assoc. J., 116: 377-379.
KAGAN, JU.S. (1977) [ Toxicology of organophosphorus pesticides,]
Moscow, Meditsina, pp. 111-121, 219-233, 260-269 (in Russian).
PLESTINA, R. (1984) Prevention, diagnosis, and treatment of
insecticide poisoning, Geneva, World Health Organization
(Unpublished document VBC/84.889).
TAYLOR, P. (1980) Anticholinesterase agents. In: Goodman, L.S. &
Gilman, A., ed. The pharmacological basis of therapeutics, 6th ed.,
New York, Macmillan Publishing Company, pp. 100-119.
UK DHSS (1983) Pesticide poisoning: notes for the guidance of medical
practitioners, London, United Kingdom Department of Health and Social
Security, pp. 41-47.
VALE, J.A. & SCOTT, G.W. (1974) Organophosphorus poisoning. Guy's
Hosp. Rep., 123: 13-25.
WARRINER, R.A., III, NIES, A.S., & HAYES, W.J., Jr (1977) Severe
organophosphate poisoning complicated by alcohol and terpentine
ingestion. Arch. environ. Health, 32: 203-205.
RESUME ET EVALUATION, CONCLUSIONS, RECOMMANDATIONS
1 Résumé et évaluation
1.1 Exposition
Le parathion-méthyl est un insecticide organophosphoré dont la
première synthèse remonte aux années 1940. Il est relativement
insoluble dans l'eau, peu soluble dans l'éther de pétrole et les
huiles minérales et facilement soluble dans la plupart des solvants
organiques. A l'état pur, il se présente sous la forme de cristaux
blancs; le parathion-méthyl technique est légèrement jaunâtre et
dégage une odeur alliacée. Il est instable à la chaleur et se
décompose rapidement au-dessus de pH 8.
La chromatographie en phase gazeuse avec détection par ionisation
de flamme alcaline (AFID) ou photométrie de flamme (FPD) est la
méthode la plus couramment utilisée pour le dosage du
parathion-méthyl. Les limites de détection dans l'eau vont de 0,01 à
0,1 µg/litre; dans l'air, elles vont de 0,1 à 1 ng/m3. La
chromatographie en phase liquide à haute performance et la
chromatographie en couche mince sont également utiles pour la
recherche du parathion-méthyl.
La distribution du parathion-méthyl dans l'air, l'eau, le sol et
les êtres vivants dépend de plusieurs facteurs physiques, chimiques et
biologiques.
Les études utilisant des modèles d'écosystèmes ainsi que des
modèles mathématiques montrent que le parathion-méthyl se partage
principalement entre l'air et le sol dans l'environnement, une plus
faible proportion se répartissant entre les végétaux et les animaux.
Il ne se déplace pratiquement pas dans le sol et ni le composé
initial, ni ses produits de décomposition n'atteignent normalement les
eaux souterraines. Le parathion-méthyl présent dans l'air provient
principalement de l'épandage de ce composé, encore qu'il puisse se
volatiliser en partie lorsque l'eau qui le contient s'évapore de la
surface des feuilles et du sol. Les niveaux atmosphériques de fond
dans les zones agricoles vont de zéro (non décelable) à environ 70
ng/m3. Les concentrations dans l'air après épandage diminuent
rapidement en trois jours pour atteindre le niveau de fond au bout
d'environ neuf jours. Dans les cours d'eau, les concentrations (études
de laboratoire) tombent à 80% de la concentration initiale au bout
d'une heure et à 10 % au bout d'une semaine. Le parathion-méthyl
demeure plus longtemps dans le sol que dans l'air ou l'eau encore que
sa rétention dépende en grande partie du type de sol; dans les sols
sableux, les résidus de parathion-méthyl disparaissent plus rapidement
que dans le terreau. Les résidus présents à la surface des plantes ou
dans les feuilles diminuent rapidement avec une demi-vie de l'ordre de
quelques heures; la disparition totale du parathion-méthyl s'effectue
en six à sept jours environ.
L'organisme animal est capable de décomposer le parathion-méthyl
et d'en éliminer les produits de dégradation en très peu de temps. Ce
processus est plus lent chez les vertébrés inférieurs et les
invertébrés que chez les mammifères et les oiseaux. Les facteurs de
bioconcentration sont faibles et le parathion-méthyl ne s'accumule que
temporairement.
C'est la dégradation microbienne qui est de loin la voie la plus
importante de dégradation du parathion-méthyl dans le milieu. Le
composé disparaît plus rapidement sur le terrain ou dans des modèles
d'écosystèmes que ne l'avaient laissé entrevoir les études de
laboratoire. Cela tient au fait qu'il existe plusieurs microorganismes
capables de décomposer cette substance dans diverses circonstances et
dans différents biotopes. La présence de sédiments ou de surfaces
végétales qui accroît les populations microbiennes, augmente la
vitesse de décomposition du parathion-méthyl.
Sous l'action du rayonnement ultra-violet ou de la lumière
solaire, le parathion-méthyl peut subir une décomposition oxydante en
paraoxon-méthyl, moins stable; après pulvérisation, le temps de
demi-décomposition par le rayonnement ultra-violet est d'environ 40
heures. Toutefois, la contribution de la photolyse à l'élimination
totale dans un système aquatique, n'est, selon les estimations, que de
4 %. L'hydrolyse du parathion-méthyl se produit également plus
rapidement en milieu alcalin. Une forte salinité favorise aussi
l'hydrolyse. En présence de sédiments fortement réducteurs, on a noté
des demi-vies de quelques minutes, encore que la sorption à d'autres
sédiments accroisse la stabilité du composé.
Dans des villes situées au centre de zones agricoles des
Etats-Unis d'Amérique, on a observé que les concentrations de
parathion-méthyl dans l'air variaient avec la saison et culminaient en
août ou septembre; les enquêtes ont révélé que les teneurs maximales
se situaient principalement dans les limites de 100 à 800 ng/m3 au
cours de la période de végétation. Dans les eaux naturelles de ces
mêmes régions des Etats-Unis, on a observé des concentrations allant
jusqu'à 0,46 µg/litre, les maxima étant atteints en été. Il n'existe
qu'un petit nombre de publications sur les résidus alimentaires de
parathion-méthyl dans le monde. Aux Etats-Unis, ces résidus se situent
en général à un très faible niveau, même si quelques échantillons
dépassent les limites maximales de résidus (LMR). Les études de ration
totale dont il est fait état dans la littérature ne font état que de
traces de résidus. C'est dans les légumes-feuilles (jusqu'à 2 mg/kg)
et les légumes racines (jusqu'à 1 mg/kg) que l'on a constaté les
résidus les plus élevés lors d'enquêtes sur le panier de la ménagère
effectuées aux Etats-Unis entre 1966 et 1969. La préparation, la
cuisson et la conservation des aliments entraînent la décomposition
des résidus de parathion-méthyl, ce qui réduit encore l'exposition des
consommateurs. En cas d'erreurs de manipulation du parathion-méthyl,
on peut trouver des résidus plus élevés dans les légumes et les fruits
crus. la production, la formulation, la manipulation et l'utilisation
du parathion-méthyl comme insecticide sont les principales sources
potentielles d'exposition humaine. C'est principalement par contact
cutané et, dans une moindre proportion, par inhalation que les
travailleurs sont exposés à cette substance.
Lors d'une étude sur des ouvriers agricoles qui pulvérisaient du
parathion-méthyl (les ouvriers non protégés procédant à un épandage
manuel de cette substance à très bas volume), on a calculé que ces
personnes absorbaient 0,4 à 13 mg de parathion-méthyl par 24 heures en
se fondant sur le dosage du p-nitrophénol dans les urines. Si les
ouvriers reviennent trop tôt sur les lieux après le traitement, ils se
trouvent encore davantage exposés.
La population générale peut être exposée à des résidus présents
dans l'air, l'eau et les aliments par suite de traitements sur les
cultures ou les forêts ou d'erreurs de manipulation (épandage en
dehors de la zone à traiter) qui entraînent la contamination des
champs, des cultures, de l'eau et de l'air.
1.2 Fixation, métabolisme et excrétion
Le parathion-méthyl est facilement absorbé par toutes les voies
d'exposition (orale, percutanée, respiratoire) et il se répand
rapidement dans les tissus de l'organisme. Les concentrations
maximales dans les divers organes ont été observées une à deux heures
après le traitement. La conversion du parathion-méthyl en
paraoxon-méthyl se produit dans les minutes qui suivent
l'administration. Après administration de parathion-méthyl par voie
intraveineuse à des chiens, on a observé une demi-vie terminale
moyenne de 7,2 heures. C'est le foie qui joue le principal rôle dans
le métabolisme et la détoxication du parathion-méthyl. Le mode
principal de détoxication du parathion-méthyl et du paraoxon-méthyl au
niveau du foie consiste en oxydation, hydrolyse et déméthylation ou
désarylation en présence de glutathion réduit (GSH). Les produits de
réaction sont le thiophosphate de o-méthyle et de o-nitrophényle
ainsi que les acides diméthylphosphorothioïque ou
diméthyl-phosphorique et le p-nitrophénol. Il est donc possible
d'évaluer l'exposition en mesurant l'excrétion urinaire du
p-nitrophénol. Chez des volontaires, l'excrétion urinaire de
p-nitrophénol était de 60 % quatre heures après l'administration et
d'environ 100 % au bout de 24 heures. Le métabolisme du
parathion-méthyl joue un rôle important dans la toxicité sélective de
ce composé pour les différentes espèces et l'apparition éventuelle
d'une résistance. L'élimination du parathion-méthyl et de ses
métabolites s'effectue principalement par la voie urinaire. Des études
menées sur des souris avec du parathion-méthyl radiomarqué au 32P
ont montré qu'au bout de 72 heures, 75 % de la radio-activité se
retrouvaient dans les urines et jusqu'à 10 % dans les matières
fécales.
1.3 Effets sur les êtres vivants dans leur milieu
naturel
Certains microorganismes peuvent utiliser le parathion-méthyl
comme source de carbone et l'étude d'une communauté naturelle a montré
que des concentrations allant jusqu'à 5 mg/litre augmentaient la
biomasse et l'activité reproductrice. L'effet est positif dans le cas
des bactéries et des actinomycètes; par contre, les champignons et les
levures sont moins capables d'utiliser ce composé. Chez une diatomée,
on a constaté une inhibition de 50 % de la croissance à une
concentration d'environ 5 mg/litre. Chez des algues vertes
unicellulaires, la croissance a été réduite par des concentrations
comprises entre 25 et 80 µg de parathion-méthyl par litre. Les
populations d'algues devenaient tolérantes au parathion-méthyl après
quelques semaines d'exposition.
Le parathion-méthyl est extrêmement toxique pour les invertébrés
aquatiques, la CL50 étant plupart du temps comprise entre <1 µg et
environ 40 µg/litre. Quelques espèces d'arthropodes sont moins
sensibles. Pour la daphnie (Daphnia magna) la concentration sans
effet observable est de 1,2 µg/litre. Les mollusques sont beaucoup
moins sensibles, puisque leur CL50 varie de 12 à 25 mg/litre.
La plupart des espèces de poissons d'eau douce ou de mer ont une
CL50 comprise entre 6 et 25 mg/litre, quelques espèces étant
nettement plus ou nettement moins sensibles au composé. La toxicité
aiguë est comparable chez les amphibiens et les poissons.
Le traitement au parathion-méthyl de mares expérimentales a
permis d'en observer les effets sur l'effectif des communautés
d'invertébrés aquatiques. Seul un épandage sur les étendues d'eau
serait susceptible d'engendrer les concentrations nécessaires à
l'apparition de ces effets et encore, seraient-ils de courte durée.
Une décimation des populations d'invertébrés est donc improbable en
situation réelle. En cas de mortalité chez les invertébrés, les effets
ne seraient probablement pas de longue durée.
Il convient dont de veiller à ne pas procéder à des épandages sur
les mares, cours d'eau et lacs. Le parathion-méthyl ne doit jamais
être épandu lorsque le vent souffle.
Le parathion-méthyl est un insecticide non-sélectif qui détruit
les espèces utiles tout autant que les ravageurs. On a fait état de
mortalité parmi des abeilles à la suite d'épandages de
parathion-méthyl. Ce genre d'accidents est plus grave avec le
parathion-méthyl qu'avec d'autres insecticides. Les abeilles adaptées
à l'Afrique supportent mieux le parathion-méthyl que les souches
européennes.
Le parathion-méthyl s'est révélé modérément toxique pour les
oiseaux au laboratoire, la DL50 aiguë par voie orale allant de 3 à
8 mg/kg de poids corporel. Par la voie alimentaire, la CL50 allait
de 70 à 680 mg/kg de nourriture. Rien n'indique que les oiseaux aient
à souffrir du parathion-méthyl lorsqu'il est épandu conformément aux
recommandations.
On veillera tout particulièrement à l'horaire des épandages pour
éviter tout effet nocif sur les abeilles.
1.4 Effets sur les animaux d'expérience et les systèmes d'épreuve
in vitro
La DL50 par voie orale varie chez les rongeurs de 3 à 35 mg/kg
de poids corporel et la DL50 par voie percutanée, de 44 à 67 mg/kg
de poids corporel.
L'intoxication par le parathion-méthyl engendre les effets
cholinergiques habituels des organophosphorés que l'on peut attribuer
à l'accumulation d'acétylcholine au niveau des terminaisons nerveuses.
La toxicité du parathion-méthyl est due à sa métabolisation en
paraoxon-méthyl. Cette conversion est très rapide. Aucun signe de
neuropathie retardée induite par les organophosphorés n'a été relevé.
Le parathion-méthyl technique n'a aucun effet irritant sur l'oeil
ni la peau.
Lors d'études de toxicité à court terme utilisant diverses voies
d'administration et portant sur des rats, des chiens et des lapins, on
a observé une inhibition de la cholinestérase du plasma, des
érythrocytes et du cerveau ainsi qu'un certain nombre de signes liés
aux effets cholinergiques. Lors d'une étude d'alimentation de 12
semaines sur des chiens, on a obtenu, pour la dose sans effet nocif
observable, une valeur de 5 mg/kg de nourriture (soit l'équivalent de
0,1 mg/kg de poids corporel par jour). Lors d'une étude de toxicité
par voie percutanée, effectuée pendant trois semaines sur des lapins,
on a obtenu une dose sans effet observable de 10 mg/kg de poids
corporel par jour. Lorsque les animaux étaient exposés par la voie
respiratoire pendant trois semaines, la dose sans effet observable
était de 0,9 mg/m3 d'air. A la dose de 2,6 mg/m3, on n'a observé
qu'une légère inhibition de la cholinestérase plasmatique.
Des études de cancérogénicité et de toxicité à long terme ont été
effectuées sur des souris et des rats. Pour les rats, la dose sans
effet observable basée sur l'inhibition de la cholinestérase était de
0,1 mg/kg de poids corporel par jour. Les résultats de ces études
n'ont fait ressortir aucun signe de cancérogénicité, ni chez les
souris ni chez les rats. Dans une autre étude de deux ans effectuée
sur des rats, on a toutefois relevé les signes d'un effet neurotoxique
périphérique à la dose de 50 mg/kg de nourriture.
Le parathion-méthyl serait capable de provoquer l'alkylation de
l'ADN in vitro. La plupart des études de génotoxicité in vitro
portant sur des cellules bactériennes et mammaliennes ont donné des
résultats positifs, alors que six études in vivo portant sur trois
systèmes d'épreuve différents ont donné des résultats ambigus.
Les études portant sur la reproduction avec administration de
doses toxiques (inhibition de la cholinestérase) n'ont pas produit
d'effets systématiques sur la taille des portées et leur nombre, le
taux de survie des petits ni la lactation. Aucun effet tératogène ou
embryotoxique direct n'a été observé.
1.5 Effets sur l'homme
Plusieurs cas d'intoxication aiguë par le parathion-méthyl ont
été signalés. Les symptômes sont caractéristiques d'une intoxication
générale par les anticholinestérasiques organophosphorés. Il s'agit
d'effets nerveux cholinergiques au niveau périphérique et au niveau
central qui apparaissent dans les minutes qui suivent l'exposition. En
cas d'exposition par voie percutanée, les symptômes peuvent s'aggraver
pendant plus d'une journée et durer plusieurs jours.
Des études sur des volontaires soumis à des expositions répétées
de longue durée ont montré que l'activité cholinestérasique du sang
diminuait sans provoquer de manifestations cliniques.
Aucun cas de neuropathie périphérique retardée induite par les
organophosphorés n'a été signalé. Dans un certain nombre de cas
d'exposition multiple à des pesticides et notamment à du
parathion-méthyl, on a observé des séquelles neurospychiatriques.
Une augmentation du nombre des aberrations chromosomiques a été
signalée dans des cas d'intoxication aiguë.
On ne possède aucune donnée obtenue sur l'homme qui puisse
permettre d'évaluer les effets tératogènes du parathion-méthyl ou ses
effets sur la reproduction.
Les études épidémiologiques disponibles sont consacrées à des
expositions multiples aux pesticides et il n'est pas possible d'en
déduire les effets qu'une exposition de longue durée au
parathion-méthyl pourrait entraîner.
2 Conclusions
Le parathion-méthyl est un insecticide organophosphoré très
toxique. Une exposition excessive due à la manipulation de ce produit
au cours de la production, de l'utilisation ou par suite d'ingestion
accidentelle ou intentionnelle peut entraîner une intoxication grave
voire mortelle. Certaines formulations de parathion-méthyl peuvent,
selon le cas, entraîner une irritation des yeux ou de la peau mais de
toute façon, elles sont toutes facilement absorbées. On peut donc être
dangereusement exposé à cet insecticide sans s'en rendre compte.
Le parathion-méthyl ne subsiste pas dans l'environnement. Il ne
subit pas de bioconcentration et ne se transmet pas le long de la
chaîne alimentaire. Il est rapidement décomposé par un grand nombre de
microorganismes et autres éléments de la faune sauvage. Cet
insecticide peut provoquer des dégâts dans les écosystèmes, mais
seulement en cas d'exposition excessive dues à une utilisation
défectueuse ou à des déversements accidentelles. Toutefois les
insectes utiles et notamment les insectes pollinisateurs peuvent
souffrir des épandages de parathion-méthyl.
C'est principalement par l'intermédiaire des denrées alimentaires
que la population générale peut être exposée à des résidus de
parathion-méthyl. Si l'on respecte les règles de bonne pratique
agricole, il n'y a pas de raison que la dose journalière admissible
fixée par le Comité d'experts FAO/OMS soit dépassée (0-0,02 mg/kg de
poids corporel)). Il peut également y avoir exposition par voie
percutanée lors de contacts accidentels avec des résidus foliaires
dans des champs traités ou des zones voisines contaminées par des
embruns de pesticides.
Moyennant de bonnes méthodes de travail et des précautions
suffisantes en matières d'hygiène et de sécurité, le parathion-méthyl
de devrait pas présenter de danger pour ceux qui lui sont exposés de
par leur profession.
3 Recommandations
* Afin de protéger la santé et le bien-être des travailleurs et de
la population générale il ne faut confier la manipulation et
l'épandage du parathion-méthyl qu'à des personnes bien encadrées
et bien formées qui utiliseront l'insecticide en prenant les
mesures de sécurité nécessaires et se conformeront aux règles de
bonne pratique en la matière.
* La fabrication, la formulation, l'utilisation agricole et
l'élimination du parathion-méthyl doivent être conduites avec
soin afin de réduire au minimum la contamination de
l'environnement.
* Les travailleurs qui sont régulièrement exposés au parathion-
méthyl doivent bénéficier d'un suivi médical approprié.
* Afin de réduire les risques pour l'ensemble de la population, il
est recommandé de ne pas revenir sur une zone traitée avant 48
heures.
* Les autorités nationales devront fixer les délais pour les
épandages avant récolte et les faire respecter.
* En raison de la forte toxicité du parathion-méthyl, cet
insecticide ne doit pas être épandu à très bas volume à l'aide de
dispositifs à main.
* Ne pas pulvériser sur les étendues d'eau. Choisir les horaires
de manière à éviter de détruire les insectes pollinisateurs.
* Les données sur l'état de santé des travailleurs exposés
uniquement au parathion-méthyl (c'est-à-dire employés à la
fabrication et à la formulation de cet insecticide) devront être
publiées afin que l'on puisse mieux en évaluer les risques pour
la santé humaine.
* Des études à caractère plus définitif devront être menées sur les
résidus de parathion-méthyl dans les denrées alimentaires
fraîches.
* Il faudrait procéder à une évaluation plus concluante de la
génotoxicité du parathion-méthyl.
RESUMEN Y EVALUACION, CONCLUSIONES Y RECOMENDACIONES
1 Resumen y evaluación
1.1 Exposición
El metilparatión es un insecticida organofosforado que
sesintetizó por primera vez en la década de 1940. Es
relativamenteinsoluble en agua, poco soluble en éter de petróleo y
aceitesminerales y fácilmente soluble en la mayoría de los
disolventesorgánicos. El metilparatión puro se encuentra en forma de
cristalesblancos; el de calidad técnica tiene un color tostado claro
y olorparecido al del ajo. Es térmicamente inestable y se descompone
conrapidez a un pH superior a 8.
El método más común para la determinación del metilparatión esla
cromatografía de gases con un detector de ionización de llama enálcali
o bien con uno fotométrico de llama. Los límites de detecciónen el
agua oscilan entre 0,01 y 0,1 µg/litro, y en el aire entre 0,1 y 1
ng/m3. También son útiles como métodos de detección lacromatografía
líquida de alta resolución y la cromatografía en capafina.
En la distribución del metilparatión en el aire, el agua, el
sueloy los organismos del medio ambiente influyen varios factores
físicos,químicos y biológicos.
Los estudios realizados utilizando modelos de ecosistemas y
laelaboración de modelos matemáticos indican que en el medioambiente
el metilparatión se reparte principalmente entre el aire y elsuelo,
con cantidades menores en las plantas y los animales.Prácticamente no
hay desplazamiento a través del suelo, y ni elcompuesto original ni
los productos derivados de su degradaciónllegan normalmente al agua
subterránea. El metilparatión presente enel aire procede sobre todo
del rociado del compuesto, aunque seproduce cierta volatilización con
la evaporación del agua de las hojasy de la superficie del suelo. Los
niveles habituales de metilparatiónen la atmósfera en las zonas
agrícolas oscilan entre una cantidad nodetectable y unos 70 ng/m3.
Se ha observado que las concentracionesen el aire después del rociado
disminuyen con rapidez en tres días,alcanzando los niveles habituales
en unos nueve días. Laconcentración en el agua fluvial (en estudios de
laboratorio)descendió al 80% de la inicial después de una hora, y
transcurridauna semana era del 10%. El metilparatión se mantiene en el
suelomás tiempo que en el aire o el agua, aunque en la retención
influyemucho el tipo de suelo; el arenoso pierde los residuos del
compuestocon mayor rapidez que las margas. Los residuos de la
superficie delas plantas y del interior de las hojas disminuyen
rápidamente, conuna semivida del orden de unas horas; el metilparatión
desaparecetotalmente en unos 6-7 días.
Los animales pueden degradar el metilparatión y eliminar
losproductos de degradación en un período muy breve de tiempo.
Elproceso es más lento en los vertebrados inferiores y en
losinvertebrados que en los mamíferos y las aves. Los factores
debioconcentración son bajos y los niveles acumulados de
metilparatióntransitorios.
La descomposición microbiana es con diferencia el mecanismomás
importante de degradación del metilparatión en el medioambiente. La
desaparición del compuesto en el campo y enecosistemas utilizados como
modelo es más rápida de lo que habíanpermitido suponer los estudios de
laboratodeloorio. Esto se debe a lavariedad de microorganismos que son
capaces de degradarlo endistintos hábitats y circunstancias. La
presencia de sedimentos o desuperficies de plantas, que aumenta la
población microbiana, acelerael ritmo de degradación del
metilparatión.
El metilparatión puede sufrir degradación oxidativa por acciónde
la radiación ultravioleta o la luz solar, convirtiéndose enmetil
paraoxón, que es menos estable; las películas de rociado sedegradan
por acción de la radiación ultravioleta con una semivida aproximada de
40 horas. Sin embargo, se ha estimado que lacontribución de la
fotolisis a la desaparición total en un sistema acuático es sólo de un
4%. También se produce hidrólisis delmetilparatión en condiciones
alcalinas, en las que es más rápida. Lasalinidad elevada favorece
asimismo la hidrólisis del compuesto. Ensedimentos muy reductores se
registraron semividas de unos minutos, aunque el metilparatión es más
estable cuando está adsorbido sobreotros sedimentos.
En las ciudades situadas en el centro de las zonas agrícolas
delos Estados Unidos, las concentraciones de metilparatión en el
airevariaban con las estaciones y alcanzaban el punto más alto en
agostoo septiembre; los niveles máximos registrados durante los
estudios fueron fundamentalmente del orden de 100-800 ng/m3 durante
elperíodo vegetativo. Las concentraciones en el agua natural de
laszonas agrícolas de los Estados Unidos llegaron a 0,46 µg/litro,
conlos niveles más altos en el verano. Son muy pocos los
informespublicados en todo el mundo sobre los residuos de
metilparatión enlos alimentos. En los Estados Unidos, se han
notificado en generalniveles muy bajos de residuos de metilparatión
en los productos alimenticios, con un pequeño número de muestras
aisladas porencima de los límites máximos de residuos (LMR). En todos
losestudios publicados sobre la alimentación sólo se detectaron
niveles ínfimos de metilparatión. En las encuestas sobre la cesta de
lacompra realizadas en los Estados Unidos entre 1966 y 1969,
lascantidades mayores de residuos de metilparatión se encontraron en
lashortalizas de hoja (hasta 2 mg/kg) y en las de raíz (hasta 1
mg/kg). En la preparación, cocción y almacenamiento de los alimentos
se descomponen los residuos de metilparatión, reduciéndose
ulteriormente la exposición humana. Las frutas y hortalizas
sinelaborar pueden contener más residuos después de un uso indebidodel
producto.
La producción, formulación, manipulación y uso delmetilparatión
como insecticida pueden ser, en principio, fuente deexposición para
las personas. Las principales vías de exposición delos trabajadores
son el contacto cutáneo y, en menor medida, lainhalación.
En un estudio sobre personas encargadas del rociado en fincas
(trabajadores no protegidos que utilizaban rociadores manuales
devolumen ultrabajo), a partir del p-nitrofenol excretado en la
orina secalculó una ingestión de 0,4-13 mg de metilparatión cada 24
horas. También se puede sufrir exposición si se entra en los
cultivosdemasiado pronto después de tratarlos.
La población general puede estar expuesta a residuos
demetilparatión presentes en el aire, el agua y los alimentos como
consecuencia de prácticas agrícolas y forestales con un uso
indebidodel producto, que provoca la contaminación de los campos,
loscultivos, el agua y el aire debido al rociado parcial fuera del
objetivo.
1.2 Ingestión, metabolismo y excreción
El metilparatión se absorbe fácilmente por todas las vías de
exposición (oral, cutánea, respiratoria) y se distribuye con rapidez
por los tejidos del cuerpo. Se detectaron concentraciones máximas en
diversos órganos 1-2 horas después del tratamiento. Después de la
administración, la transformación del metilparatión en metilparaoxón
se produce en unos minutos. En perros se determinó una semivida
terminal media de 7,2 horas tras la administración intravenosa de
metilparatión. El hígado es el principal órgano de metabolización y
desintoxicación. El metilparatión o el metilparaoxón se destoxifican
en el hígado sobre todo mediante oxidación, hidrólisis y desmetilación
o desarilación con glutatión reducido. Los productos de la reacción
son el O-metil O-p-nitrofenilfosfotioato, o bien los ácidos
dimetilfosfotioico o dimetilfosfórico, y el p-nitrofenol. Por
consiguiente, se puede estimar la exposición midiendo la excreción
urinaria de p-nitrofenol; en voluntarios humanos fue del 60% en
cuatro horas y prácticamente del 100% en 24 horas. El metabolismo del
metilparatión es importante para la toxicidad específica selectiva y
la aparición de resistencia. Le eliminación de esta sustancia y sus
productos derivados tiene lugar primordialmente por la orina. En
estudios realizados en ratones con 32P-metilparatión (marcado
radiactivamente) se observó un 75% de radiactividad en la orina y
hasta un 10% en las heces después de 72 horas.
1.3 Efectos en los seres vivos del medio ambiente
Los microorganismos pueden utilizar el metilparatión como fuente
de carbono, y en el estudio de una comunidad natural se vio que
concentraciones de hasta 5 mg/litro aumentaban la biomasa y la
actividad reproductora. En las bacterias y los actinomicetos se
observó un efecto positivo del metilparatión, mientras que los hongos
y las levaduras tenían menor capacidad para utilizar la sustancia. Con
una concentración aproximada de 5 mg/litro se produjo una inhibición
del 50% del crecimiento de una diatomea. Concentraciones de
metilparatión comprendidas entre 25 y 80 µg/litro redujeron el
crecimiento celular de las algas clorofíceas unicelulares. Las
poblaciones de algas adquirieron tolerancia tras varias semanas de
exposición.
El metilparatión es muy tóxico para los invertebrados acuáticos,
oscilando casi siempre la CL50 entre < 1 µg y alrededor de 40
µg/litro. Hay un pequeño número de especies de artrópodos que son
menos susceptibles. El nivel sin efecto para Daphnia magna es de 1,2
µg/litro. Los moluscos son mucho menos susceptibles, con CL50 entre
12 y 25 mg/litro.
La mayoría de las especies de peces, tanto de agua dulce como de
mar, tienen una CL50 de 6 a 25 mg/litro, pero hay un pequeño número
de especies cuya sensibilidad al metilparatión es considerablemente
mayor o menor. La toxicidad aguda para los anfibios es análoga a la de
los peces.
Se han observado los efectos sobre poblaciones en las comunidades
de invertebrados acuáticos de estanques experimentales tratados con
metilparatión. Las concentraciones necesarias para producir esos
efectos se alcanzarían sólo con un rociado excesivo de las masas de
agua, e incluso en este caso durarían muy poco tiempo. Por
consiguiente, en condiciones normales no es probable que se observen
efectos sobre las poblaciones. Tampoco los es que la acción letal
sobre los invertebrados acuáticos provoque efectos duraderos.
Hay que tener cuidado para evitar un rociado excesivo de
estanques, ríos y lagos al utilizar el metilparatión. Nunca se debe
efectuar la operación con viento.
El metilparatión es un insecticida no selectivo que mata especies
beneficiosas tan fácilmente como las plagas. Se ha notificado la
muerte de abejas después de su aplicación. Sus efectos sobre esta
especie fueron más graves que los de otros insecticidas. Las abejas
africanizadas son más tolerantes al metilparatión que las razas
europeas.
El metilparatión fue moderadamente tóxico para las aves en
estudios de laboratorio, con una DL50 oral aguda comprendida entre
3 y 8 mg/kg de peso corporal. La CL50 en la dieta osciló entre 70 y
680 mg/kg de alimentos. No hay indicios de que las aves puedan verse
afectadas negativamente con la utilización recomendada en el campo.
Hay que tener el máximo cuidado al programar el rociado con
metilparatión, a fin de evitar los efectos adversos sobre las abejas.
1.4 Efectos en los animales de experimentación y en sistemas de
prueba in vitro
Los valores de la DL50 del metilparatión por vía oral en
roedores oscilan entre 3 y 35 mg/kg de peso corporal, y los valores
por vía cutánea entre 44 y 67 mg/kg de peso corporal.
El envenenamiento por metilparatión provoca los signos
colinérgicos habituales de los organofosfatos, atribuidos a la
acumulación de acetilcolina en la terminaciones nerviosas. El
metilparatión adquiere la toxicidad al metabolizarse a metilparaoxón,
en un proceso que es muy rápido. No se han observado indicios de
neuropatía retardada inducida por compuestos organofosforados.
Se ha comprobado que el metilparatión de calidad técnica no tiene
potencial de irritación primaria de los ojos o la piel.
En estudios de toxicidad de corta duración, utilizando diversas
vías de administración en ratas, perros y conejos, se observó
inhibición de la colinesterasa del plasma, los eritrocitos y el
cerebro, así como signos colinérgicos conexos. En un estudio de
alimentación durante 12 semanas con perros, el nivel sin efectos
adversos observados (NOAEL) fue de 5 mg/kg de la dieta (equivalente a
0,1 mg/kg de peso corporal al día). En un estudio de toxicidad cutánea
de tres semanas en conejos, el nivel sin efectos observados (NOEL) fue
de 10 mg/kg de peso corporal al día. La exposición por inhalación
durante tres semanas dio como resultado un NOEL de 0,9 mg/m3 de
aire. Con 2,6 mg/m3 solamente se observó una ligera inhibición de la
colinesterasa del plasma.
Se realizaron estudios de toxicidad/teratogenicidad de larga
duración con ratones y ratas. El NOEL para las ratas fue de 0,1 mg/kg
de peso corporal al día, basado en la inhibición de la colinesterasa.
No hay pruebas de carcinogenicidad en ratones y ratas tras una
exposición de larga duración. Sin embargo, en otro estudio de dos años
con ratas se detectó un efecto neurotóxico periférico con una dosis de
50 mg/kg de la dieta.
Se ha informado que el metilparatión tiene propiedades
alquilizantes del ADN in vitro. Los resultados de la mayoría de los
estudios de genotoxicidad in vitro con células tanto bacterianas
como de mamífero fueron positivos, mientras que en seis estudios in
vivo, utilizando tres sistemas de prueba distintos, los resultados
fueron equívocos.
En estudios de reproducción con niveles de dosificación tóxicos
(inhibición de la colinesterasa), no se observaron efectos constantes
sobre el tamaño de la camada, el número de partos, la tasa de
supervivencia de las crías y el rendimiento de la lactación. No se
detectó ningún efecto teratogénico o embriotóxico primario.
1.5 Efectos en la especie humana
Se han registrado varios casos de intoxicación aguda por
metilparatión. Los signos y síntomas son los característicos de la
intoxicación sistémica por compuestos organofosforados inhibidores de
la colinesterasa. Cabe mencionar entre ellos las manifestaciones del
sistema nervioso colinérgico periférico y central, que aparecen apenas
unos minutos después de la exposición. En el caso de la exposición
cutánea, la gravedad de los síntomas puede ir en aumento durante más
de un día y pueden durar varios días.
Los estudios con voluntarios sometidos a exposiciones repetidas
de larga duración parecen indicar que hay una disminución de la
actividad de la colinesterasa de la sangre, sin manifestaciones
clínicas.
No se ha informado de ningún caso de neuropatía periférica
retardada inducida por compuestos organofosforados. Se han descrito
secuelas neuropsiquiátricas en casos de exposición múltiple a
plaguicidas, entre ellos el metilparatión.
En casos de intoxicaciones agudas, se ha detectado un aumento de
las aberraciones cromosómicas.
No se dispone de datos relativos al metilparatión en la especie
humana que permitan evaluar los efectos teratogénicos y sobre la
reproducción.
Los estudios epidemiológicos disponibles se refieren a una
exposición múltiple a plaguicidas, y no es posible evaluar los efectos
de una exposición de larga duración al metilparatión.
2 Conclusiones
El metilparatión es un éster organofosfórico muy tóxico,
utilizado como insecticida. Una exposición excesiva al manejarlo
durante su fabricación y uso o por ingestión accidental o intencionada
puede ocasionar una intoxicación grave o letal. Las formulaciones de
metilparatión unas veces son irritantes y otras no para los ojos o la
piel, pero se absorben fácilmente. Por consiguiente, pueden producirse
exposiciones peligrosas sin advertirlo.
El metilparatión no se mantiene mucho tiempo en el medio
ambiente, no se produce bioconcentración y no se desplaza a través de
la cadena alimentaria. Lo degradan con rapidez numerosos
microorganismos y otros tipos de seres vivos presentes en el medio
ambiente. Este insecticida puede ocasionar daños a ecosistemas
solamente en casos de una exposición muy intensa causada por el uso
indebido o escapes accidentales; sin embargo, el rociado con
metilparatión representa un riesgo para los insectos polinizadores y
otros que son beneficiosos.
La exposición de la población general a los residuos del
metilparatión tiene lugar fundamentalmente por medio de los alimentos.
Si se siguen buenas prácticas agrícolas, no se supera la ingesta
diaria admisible (0-0,02 mg/kg de peso corporal) establecida por la
FAO/OMS. Puede haber exposición cutánea accidental por contacto con
residuos foliares en campos rociados o en zonas adyacentes a los
lugares que se están rociando, como consecuencia de pérdidas del
producto que no llegan a su objetivo.
Con buenas prácticas de trabajo, medidas higiénicas y
precauciones de seguridad, no es probable que el metilparatión
represente un riesgo para las personas con exposición profesional.
3 Recomendaciones
* Para salvaguardar la salud y el bienestar de los trabajadores y
de la población general, el manejo y la aplicación del
metilparatión sólo se debería encomendar, bajo una atenta
supervisión, a personas bien capacitadas que se ajusten a las
medidas de seguridad adecuadas y utilicen el producto de acuerdo
con las buenas prácticas de aplicación.
* Se debe prestar particular atención a la fabricación, la
formulación, el uso agrícola y la eliminación del metilparatión,
a fin de reducir al mínimo la contaminación del medio ambiente.
* Los trabajadores regularmente expuestos deberían ser objeto de
vigilancia y exámenes médicos adecuados.
* A fin de reducir al mínimo el riesgo para todas las personas, se
recomienda esperar 48 horas después del rociado antes de entrar
de nuevo en cualquier zona tratada.
* Las autoridades nacionales deberían establecer intervalos sin
tratamiento antes de la recolección y obligar a respetarlos.
* A la vista de la elevada toxicidad del metilparatión, se debe
excluir este producto de la aplicación mediante rociado de
volumen ultrabajo aplicado manualmente.
* No se han de rociar masas de agua. Hay que elegir los momentos de
la aplicación de manera que se evite la muerte de insectos
polinizadores.
* Se debe hacer pública la información relativa al estado de salud
de los trabajadores expuestos exclusivamente al metilparatión (es
decir, en la fabricación, la formulación), con objeto de evaluar
mejor los riesgos de este producto químico para la salud humana.
* Deberían llevarse a cabo estudios más definitivos sobre los
residuos de metilparatión en los alimentos frescos.
* Debería realizarse una evaluación genotóxica más definitiva del
metilparatión.