
INTERNATIONAL PROGRAMME ON CHEMICAL SAFETY
ENVIRONMENTAL HEALTH CRITERIA 132
TRICHLORFON
This report contains the collective views of an international group of
experts and does not necessarily represent the decisions or the stated
policy of the United Nations Environment Programme, the International
Labour Organisation, or the World Health Organization.
Published under the joint sponsorship of
the United Nations Environment Programme,
the International Labour Organisation,
and the World Health Organization
First draft prepared by Dr J. Sekizawa, Dr M. Takeda
and Dr K. Matsumoto (National Institute of
Hygienic Sciences, Japan) and Dr M. Eto
(Kyushu University, Japan), with the assistance
of Dr J. Miyamoto and Dr M. Matsuo (Sumitomo
Chemical Company)
World Health Orgnization
Geneva, 1992
The International Programme on Chemical Safety (IPCS) is a
joint venture of the United Nations Environment Programme, the
International Labour Organisation, and the World Health
Organization. The main objective of the IPCS is to carry out and
disseminate evaluations of the effects of chemicals on human health
and the quality of the environment. Supporting activities include
the development of epidemiological, experimental laboratory, and
risk-assessment methods that could produce internationally
comparable results, and the development of manpower in the field of
toxicology. Other activities carried out by the IPCS include the
development of know-how for coping with chemical accidents,
coordination of laboratory testing and epidemiological studies, and
promotion of research on the mechanisms of the biological action of
chemicals.
WHO Library Cataloguing in Publication Data
Trichlorfon.
(Environmental health criteria ; 132)
1.Trichlorfon - poisoning 2. Trichlorfon - toxicity
3.Environmental exposure I.Series
ISBN 92 4 157132 2 (NLM Classification: WA 240)
ISSN 0250-863X
The World Health Organization welcomes requests for permission
to reproduce or translate its publications, in part or in full.
Applications and enquiries should be addressed to the Office of
Publications, World Health Organization, Geneva, Switzerland, which
will be glad to provide the latest information on any changes made
to the text, plans for new editions, and reprints and translations
already available.
(c) World Health Organization 1992
Publications of the World Health Organization enjoy copyright
protection in accordance with the provisions of Protocol 2 of the
Universal Copyright Convention. All rights reserved.
The designations employed and the presentation of the material
in this publication do not imply the expression of any opinion
whatsoever on the part of the Secretariat of the World Health
Organization concerning the legal status of any country, territory,
city or area or of its authorities, or concerning the delimitation
of its frontiers or boundaries.
The mention of specific companies or of certain manufacturers'
products does not imply that they are endorsed or recommended by the
World Health Organization in preference to others of a similar
nature that are not mentioned. Errors and omissions excepted, the
names of proprietary products are distinguished by initial capital
letters.
CONTENTS
ENVIRONMENTAL HEALTH CRITERIA FOR TRICHLORFON
INTRODUCTION
1. SUMMARY AND EVALUATION, CONCLUSIONS, AND RECOMMENDATIONS
1.1 Summary and evaluation
1.1.1 Exposure
1.1.2 Uptake, metabolism,
and excretion
1.1.3 Effects on organisms
in the environment
1.1.4 Effects on experimental
animals and in vitro
test systems
1.1.5 Effects on human beings
1.2 Conclusions
1.3 Recommendations
2. IDENTITY, PHYSICAL AND CHEMICAL PROPERTIES,
ANALYTICAL METHODS
2.1 Identity
2.2 Physical and chemical properties
2.3 Conversion factors
2.4 Analytical methods
3. SOURCES OF HUMAN AND
ENVIRONMENTAL EXPOSURE
3.1 Natural occurrence
3.2 Industrial production
3.3 Uses
4. ENVIRONMENTAL TRANSPORT,
DISTRIBUTION, AND TRANSFORMATION
4.1 Transport and distribution
4.1.1 Air
4.1.2 Water
4.1.3 Soil
4.2 Abiotic degradation
4.3 Biodegradation
4.4 Environmental fate
5. ENVIRONMENTAL LEVELS AND
HUMAN EXPOSURE
5.1 Environmental levels
5.1.1 Air
5.1.2 Water
5.1.3 Soil
5.1.4 Residues in plants
and animals
5.2 Residues in food
5.2.1 Crops
5.2.2 Milk
5.2.3 Meat
5.2.4 Poultry and eggs
5.2.5 Fish
5.3 Occupational exposure
6. KINETICS AND METABOLISM
6.1 Absorption and distribution
6.1.1 Animal
6.1.2 Human
6.2 Biotransformation
6.3 Elimination and excretion
6.4 Reaction with body components
6.4.1 In vitro studies
6.4.2 In vivo studies
7. EFFECTS ON ORGANISMS IN THE ENVIRONMENT
7.1 Microorganisms
7.2 Invertebrates
7.3 Aquatic vertebrates
7.4 Terrestrial vertebrates
7.5 Ecosystems
8. EFFECTS ON EXPERIMENTAL ANIMALS
AND IN VITRO TEST SYSTEMS
8.1 Acute toxicity
8.2 Short-term exposure
8.3 Skin and eye irritation;
sensitization
8.3.1 Skin irritation
8.3.2 Skin sensitization
8.3.3 Eye irritation
8.4 Long-term exposure
8.4.1 Oral administration
8.4.1.1 Mouse
8.4.1.2 Rat
8.4.1.3 Dog
8.4.1.4 Monkey
8.4.2 Intraperitoneal administration
8.4.2.1 Mouse
8.4.2.2 Rat
8.4.2.3 Hamster
8.4.3 Dermal administration
8.4.3.1 Mouse
8.5 Mutagenicity
8.5.1 DNA methylation
8.5.2 Mutagenicity
8.6 Carcinogenicity
8.7 Teratogenicity and
reproductive toxicity
8.7.1 Mouse
8.7.2 Rat
8.7.3 Hamster
8.7.4 Rabbit
8.7.5 Congenital tremor
8.8 Neurotoxicity
8.9 Immunological studies
8.10 Toxicity of dichlorvos
8.11 Mechanism of toxicity -
mode of action
9. EFFECTS ON HUMAN BEINGS
9.1 Acute poisoning -
poisoning incidents
9.2 Therapeutic use of
trichlorfon
9.3 Occupational exposures
9.4 Treatment of acute trichlorfon poisoning
10. PREVIOUS EVALUATIONS BY
INTERNATIONAL BODIES
REFERENCES
ANNEX I. Treatment of organophosphate
insecticide poisoning in man
ANNEX II. No-observed-effect levels (NOELs)
in animals treated with
trichlorfon
RESUME ET EVALUATION, CONCLUSIONS,
RECOMMANDATIONS
RESUMEN Y EVALUACION, CONCLUSIONES,
RECOMENDACIONES
WHO TASK GROUP ON ENVIRONMENTAL HEALTH
CRITERIA FOR TRICHLORFON AND FENITROTHION
Members
Dr V. Benes, Department of Toxicology and Reference Laboratory,
Institute of Hygiene and Epidemiology, Prague, Czech and Slovak
Federal Republic
Dr C. Carrington, Division of Toxicological Review and Evaluation,
Food and Drug Administration, Washington DC, USA (Joint Rapporteur)
Dr W. Dedek, Department of Chemical Toxicology Academic of Sciences,
Leipzig, Germany
Dr S. Dobson, Institute of Terrestrial Ecology, Monks Wood
Experimental Station, Huntingdon, United Kingdom
Dr D.J. Ecobichon, Department of Pharmacology and Therapeutics, McGill
University, Montreal, Canada
Dr M. Eto, Department of Agricultural Chemistry, Kyushu University,
Fukuoka-shi, Japan (Vice-Chairman)
Dr Bo Holmstedt, Department of Toxicology, Swedish Medical Research
Council, Karolinska Institute, Stockholm, Sweden
Dr S.K. Kashyap, National Institute of Occupational Health, Ahmedabad,
India
Dr J. Miyamoto, Takarazuka Research Centre, Hyogo, Japan
Dr H. Spencer, United States Environmental Protection Agency,
Washington DC, USA (Chairman)
Dr M. Takeda, National Institute of Hygienic Sciences, Tokyo, Japan
Observers
Dr M. Matsuo, Biochemistry and Toxicology Laboratory, Sumitomo
Chemical Co. Ltd, Osaka-shi, Japan (representing GIFAP)
Secretariat
Dr J. Sekizawa, National Institute of Hygienic Sciences, Tokyo, Japan
(Joint Rapporteur)
Dr K.W. Jager, IPCS, World Health Organization, Geneva, Switzerland
(Secretary)
NOTE TO READERS OF THE CRITERIA DOCUMENTS
Every effort has been made to present information in the criteria
documents as accurately as possible without unduly delaying their
publication. In the interest of all users of the Environmental Health
Criteria documents, readers are kindly requested to communicate any
errors that may have occurred to the Director of the International
Programme on Chemical Safety, World Health Organization, Geneva,
Switzerland, in order that they may be included in corrigenda, which
will appear in subsequent volumes.
* * *
A detailed data profile and a legal file can be obtained from the
International Register of Potentially Toxic Chemicals, Palais des
Nations, 1211 Geneva 10, Switzerland (Telephone no. 7988400 -
7985850).
ENVIRONMENTAL HEALTH CRITERIA FOR TRICHLORFON AND
FENITROTHION
A WHO Task Group on Environmental Health Criteria for Trichlorfon
and Fenitrothion met at the World Health Organization, Geneva, from 10
to 14 December 1990. Dr K.W. Jager, IPCS, welcomed the participants on
behalf of Dr M. Mercier, Director of the IPCS, and the three IPCS
cooperating organizations (UNEP/ILO/WHO). The Group reviewed and
revised the drafts and made evaluations of the risks for human health
and the environment from exposure to trichlorfon and fenitrothion.
The first draft of the EHC on trichlorfon was prepared
collaboratively by Dr M. Eto of the Kyushu University, Dr J. Miyamoto
and Dr M. Matsuo of the Sumitomo Chemical Company, and Dr M. Takeda
and Dr K. Matsumoto of the National Institute of Hygienic Sciences of
Japan. The scientific editing was performed by Dr J. Sekizawa of the
National Institute of Hygienic Sciences of Japan. Dr K.W. Jager of the
International Programme on Chemical Safety, assisted in the
preparation of the second draft, incorporating comments received
following the circulation of the first drafts to the IPCS contact
points for Environmental Health Criteria documents.
Dr K.W. Jager of the IPCS Central Unit was responsible for the
scientific content of the documents, and Mrs M.O. Head of Oxford for
the editing.
The fact that Sumitomo Chemical Company Limited, Japan
(trichlorfon and fenitrothion) and Bayer AG, Germany (trichlorfon)
made available to the IPCS and the Task Group their proprietary
toxicological information on the products under discussion is
gratefully acknowledged. This allowed the Task Group to make its
evaluation on the basis of more complete data.
The efforts of all who helped in the preparation and finalization
of the documents are gratefully acknowledged.
INTRODUCTION
The major transformation product of trichlorfon in mammals,
including human beings, is dichlorvos, the cholinesterase inhibiting
activity of which is at least 100 times that of trichlorfon (Hofer,
1981). Trichlorfon can be said to act in the mammalian body as a "slow
release source" for dichlorvos, which may be of essential importance
for, among others, its schistosomicidal effect (Nordgren, 1981;
Nordgren et al., 1978).
Only information directly related to trichlorfon will be
discussed and evaluated in this publication.
For an evaluation of the health and environmental hazards of
dichlorvos, the reader should refer to EHC No. 79: Dichlorvos (WHO,
1989). A more complete treatise on the effects of organophosphorus
insecticides in general, especially their short- and long-term effects
on the nervous system, and their treatment, can be found in EHC No.
63: Organophosphorus insecticides - A general introduction (WHO,
1986).
A comprehensive review of Russian literature up to 1983, on the
toxicity and hazards of trichlorfon, has been published by the
International Register of Potentially Toxic Chemicals (IRPTC/GKNT,
1983).
1. SUMMARY AND EVALUATION, CONCLUSIONS, AND RECOMMENDATIONS
1.1 Summary and evaluation
1.1.1 Exposure
Trichlorfon is an organophosphorus insecticide that has been in
use since the early 1950s. In agriculture, it is mainly used against
insect pests in field and fruit crops. Trichlorfon is also used to
control forest insects and for the control of parasites in domestic
animals. Under the name of metrifonate, trichlorfon is used for the
treatment of human infestation by Schistosoma haematobium. It is
considered to be a slow release reservoir of dichlorvos. Trichlorfon
is available in the form of an emulsifiable concentrate, powder, dust,
granules, a solution, and ultra-low volume concentrates.
The air concentration of trichlorfon insecticide may be as high
as 0.1 mg/m3, soon after spraying, but levels decrease within days
to below 0.01 mg/m3. Levels of trichlorfon in run-off water from
sprayed areas may be as high as 50 µg/litre, though levels in surface
waters are usually much lower and decrease rapidly.
Trichlorfon degrades rapidly in soil, and levels generally
decrease to negligible amounts within one month of application. It is
relatively stable in water below pH 5.5. At higher pH, trichlorfon is
transformed to dichlorvos. While microorganisms and plants may
metabolize trichlorfon, the most important route of removal is abiotic
hydrolysis.
With a few exceptions, levels of trichlorfon on crops are below
10 mg/kg, the day after application, and fall to below 0.1 mg/kg,
during the two weeks following.
Milk from cows treated with trichlorfon for pest control may
contain residues as high as 1.2 mg/litre, 2 h after application, but
the levels decline to below 0.1 mg/litre, 24 h after treatment.
Significant levels of trichlorfon have not been found in meat from
treated animals. Eggs from treated hens have been found to contain
0.05 mg trichlorfon/kg.
1.1.2 Uptake, metabolism, and excretion
Trichlorfon is readily absorbed via all routes of exposure (oral,
dermal, inhalation) and is rapidly distributed to the tissues of the
body. Peak blood concentrations were detected within 1-2 h, almost
total disappearance from the blood stream occurring in a matter of
1.5-4 h. The biological half-life of trichlorfon in the mammalian
blood was estimated to be in the range of 30 min.
Trichlorfon undergoes transformation to dichlorvos (2,2-dichloro
vinyl dimethyl phosphate), via dehydrochlorination, in water,
biological fluids, and tissues, at pH values higher than 5.5.
Dichlorvos is the physiologically active anticholinesterase. The main
routes of degradation are demethylation, P-C bond cleavage, and ester
hydrolysis via dichlorvos. The major metabolites of trichlorfon found
in vivo are demethyl trichlorfon, demethyl dichlorvos, dimethyl
hydrogen phosphate, methyl hydrogen phosphate, phosphoric acid, and
trichloroethanol. The last metabolite is found in the urine as a
glucuronide conjugate.
Trichlorfon and metabolic products are primarily eliminated via
the urine. Studies conducted with radiolabelled (14C-methyl and
32P-) trichlorfon revealed that the bulk of the chemical was
eliminated in the form of water-soluble material, little being
chloroform-soluble. Some 66-70% of the water-soluble products appeared
in the urine within 12 h while 24% of the 14C-methyl material was
eliminated in the expired air as carbon dioxide (CO2). Low levels of
trichlorfon and metabolites have been detected in bovine milk
following oral and dermal treatment of the animals.
1.1.3 Effects on organisms in the environment
Trichlorfon is moderately toxic for fish (96-h LC50 values
range between 0.45 mg/litre and 51 mg/litre) and moderately to highly
toxic for aquatic arthropods (48-h/96-h LC50 values range between
0.75 µg/litre and 7800 µg/litre). However, reported concentrations of
trichlorfon in surface waters, after application in forests at 6
kg/ha, fall short of these ranges. Thus, in normal usage, trichlorfon
will have little or no effect on populations of aquatic organisms,
since other groups, such as molluscs and microorganisms are less
sensitive than arthropods. LD50 values from laboratory studies
ranging from 40 mg/kg to 180 mg/kg indicate that trichlorfon is
moderately toxic for birds. However, in field studies, no effects on
numbers, breeding pairs, nesting success, or mortality of forest
songbirds were seen following aerial application of trichlorfon. An
observed reduction in singing and increased feeding activity may have
been the result of a reduction in food organisms. There is no
indication that trichlorfon will adversely affect organisms in the
terrestrial environment, other than arthropods. There is no
information on effects on beneficial arthropods.
1.1.4 Effects on experimental animals and in vitro test systems
Trichlorfon is an insecticide that is moderately toxic for
experimental animals. Oral LD50 values for technical trichlorfon in
laboratory animals range from 400 to 800 mg/kg body weight and dermal
LD50 values for the rat are greater than 2000 mg/kg body weight.
Trichlorfon poisoning causes the usual organophosphate
cholinergic signs attributed to accumulation of acetylcholine at nerve
endings.
Technical trichlorfon was shown to be moderately irritating to
the eyes of rats, but was not irritating in skin tests on rabbits.
Skin sensitization potential was demonstrated in guinea-pigs.
Short-term, oral toxicity studies were carried out on rats, dogs,
monkeys, rabbits, and guinea-pigs. In a 16-week study on rats, a
4-year study on dogs, and a 26-week study on monkeys,
no-observed-effect levels (NOELs) of 100 mg/kg diet, 50 mg/kg diet,
and 0.2 mg/kg body weight (based on plasma, erythrocyte, or brain ChE
activity) respectively, were determined. Inhalation exposure of rats,
over 3 weeks, indicated a NOEL of 12.7 mg/m3, based on the
inhibition of plasma, erythrocyte, and brain ChE activity. Long-term
toxicity/carcinogenicity studies were carried out on mice, rats,
monkeys, and hamsters after oral, intraperitoneal, or dermal
administration. An adverse effect on the gonads was seen following
the oral exposure of mice and rats at dose levels of 30 mg/kg body
weight and 400 mg/kg diet, respectively. In a 24-month study on rats
and a 10-year study on monkeys, NOAELs of 50 mg/kg diet and 0.2 mg/kg
body weight, respectively, were determined. Available data do not
provide evidence of carcinogenicity following the long-term exposure
of test animals by several routes of administration.
Under physiological conditions, trichlorfon has been reported to
have a DNA-alkylating property. The trichlorfon mutagenicity results
have been both positive and negative. Dichlorvos may be responsible,
either in part or in full, for the effects observed. Most of the in
vitro mutagenicity studies on both bacterial and mammalian cells were
positive while few of the in vivo studies produced a positive
result.
Studies on mice, rats, and hamsters indicate that trichlorfon
produces a teratogenic response in rats at doses high enough to
produce maternal toxicity. Exposure of rats to 145 mg trichlorfon/kg
diet, during gestation, caused fetal malformations. A gavage dose of
400 mg/kg body weight in hamsters also produced both maternal
toxicity and a teratogenic response. The lowest dose by gavage that
produced teratogenic effects in rats was 80 mg/kg body weight. The
effects appear to be time specific in the gestation period. A NOEL of
8 mg/kg was determined in this gavage study.
NOELs of 8 mg/kg body weight and 200 mg/kg body weight were
demonstrated for rats and hamsters, respectively. Teratogenic
responses involving the central nervous system have also been
reported for the pig and guinea-pig.
However, no teratogenic effects were observed in a 3-generation
reproduction study on rats, in which high dose levels induced adverse
reproductive effects. The NOEL in this study was 300 mg/kg diet.
Very high doses have produced neurotoxic effects in animals.
The active transformation product in mammals is dichlorvos, which
is estimated to be at least 100 times more potent as an
anticholinesterase than trichlorfon.
1.1.5 Effects on human beings
Several cases of acute poisoning from intentional (suicide) or
accidental exposure have occurred. Signs and symptoms of intoxication
were characteristic of AChE inhibition, such as exhaustion, weakness,
confusion, excessive sweating and salivation, abdominal pains,
vomiting, pinpoint pupils, and muscle spasms. In severe cases of
poisoning, unconsciousness and convulsions developed and death usually
resulted from respiratory failure. In cases where victims survived due
to medical intervention, a delayed polyneuropathy, associated with
weakness of the lower limbs, sometimes occurred a few weeks after
exposure. In fatal cases, autopsy findings showed ischaemic changes in
the brain, spinal cord, and vegetative ganglia, damage to the myelin
sheath in the spinal cord and brain peduncles, and structural changes
in the axons of peripheral nerves.
A few cases of occupational poisoning have occurred, mainly
through the neglect of safety precautions. Occupational exposure at
a work-place where air concentrations exceeded 0.5 mg/m3 resulted in
decreased plasma cholinesterase and changes in the EEG pattern.
However, these were completely reversible on cessation of exposure. No
cases of skin sensitization have been reported.
This compound has been extensively used for the treatment of
schistosomiasis in humans. Administration of a single dose (7-12
mg/kg) resulted in cholinesterase inhibition in plasma and
erythrocytes in the range of 40-60%, without cholinergic symptoms.
However, mild symptoms were observed in cases with a repeated dose
regimen. A high dose level (24 mg/kg) caused severe cholinergic
symptoms.
1.2 Conclusions
- Trichlorfon is a moderately toxic organophosphorus ester
insecticide. Over-exposure from handling during manufacture
or use and accidental or intentional ingestion may cause
serious poisoning.
- Trichlorfon exposure of the general population occurs mainly
as a result of agricultural and veterinary practices, and in
the treatment of Schistosoma haematobium.
- The reported trichlorfon intakes are far below the
Acceptable Daily Intake established by FAO/WHO and should
not constitute a health hazard for the general population.
- With good work practices, hygienic measures, and safety
precautions, trichlorfon is unlikely to present a hazard for
those occupationally exposed.
- Despite its high toxicity for non-target arthropods,
trichlorfon has been used with few or no adverse effects on
populations of organisms in the environment.
1.3 Recommendations
- For the health and welfare of workers and the general
population, the handling and application of trichlorfon
should only be entrusted to competently supervised and
well-trained operators, who will follow adequate safety
measures and apply trichlorfon according to good application
practices.
- The manufacture, formulation, agricultural use, and disposal
of trichlorfon should be carefully managed to minimize
contamination of the environment, particularly surface
waters.
- Regularly exposed worker and patient populations should
undergo periodic health evaluations.
- Application rates of trichlorfon should be limited, to avoid
effects on non-target arthropods. The insecticide should
never be sprayed over water bodies or streams.
2. IDENTITY, PHYSICAL AND CHEMICAL PROPERTIES, ANALYTICAL
METHODS
2.1 Identity
Trichlorfon was first prepared by Lorenz in 1952 and then by
Barthel in 1954 by the reaction of dimethyl phosphite with chloral. It
is a racemic mixture of dimethyl 2,2,2-trichloro-1-
hydroxyethylphosphonate. Two molecules of trichlorfon are associated
together (Lorenz et al., 1955).
Chemical structure:
O
"
Cl3CCHP(OCH3)2
'
OH
Chemical formula: C4H8Cl3O4P
Relative molecular mass: 257.44
Common name: trichlorfon (ISO)
Chemical name: dimethyl 2,2,2-trichloro-1-
hydroxyethylphosphonate
Synonyms: chlorofos, DEP, DETF, dipterex,
dimethyl 1-hydroxy- 2,2,2-trichloro
ethanephosphonate, O, O-dimethyl
(2,2,2-trichloro-1-hydroxyethyl)
phosphonate, metrifonate, foschlor,
trichlorofon, trichlorphon
Trade names: Agroforotox, Anthon, L 13/59,
Bilarcil, Cekufon, Danex, Dipterex,
Ditriphon, Dylox, Dyrex, Dyvon,
Masoten, Metrifonate, Neguvon,
Proxol, Tugon, Wotex
CAS registry number: 52-68-6
RTECS registry number: TA 0700000
Impurities: The purity of technical trichlofon
was reported to be more than 98%
(FAO/WHO, 1972). The main
impurities are 2,2-dichlorovinyl
dimethyl phosphate: dichlorvos (0-
0.2%), trichloroacetaldehyde (0-
0.05%), dichloroacetaldehyde (0-
0.03%), methyl hydrogen 2,2,2-
trichloro-1-hydroxyethylphospho
nate; demethyl trichlorfon (0-0.3%),
and water (less than 0.3%). The
technical product also contains
phosphoric acid, 2,2,2-trichloro-1-
hydroxyethylphosphonic acid, and
dimethyl phosphite (FAO/WHO,
1972; Melnikov et al., 1975).
2.2 Physical and chemical properties
Trichlorfon is a colourless crystalline powder that is stable at
room temperature. It is slowly hydrolysed in acid media; the half-life
is 526 days at pH 1-5 and 20 °C (Mühlmann & Schrader, 1957). Cleavage
of one of the methyl ester groups takes place by acid hydrolysis. In
alkaline media, however, trichlorfon is rapidly converted to
dichlorvos and then hydrolytic products (see section 4.2).
Some physical properties are given in Table 1.
2.3 Conversion factors
1 ppm = 11.4 mg/m3
1 mg/m3 = 0.088 ppm, at 25 °C and 760 mmHg.
2.4 Analytical methods
There are several methods for the determination of trichlorfon,
some of which are listed in Table 2. For formulation analysis,
potentiometric titration of liberated chloride with standard silver
nitrate AgNO3 has been recommended (Macdougall, 1964; Bennewitz &
Foth, 1967). The total chlorine content is determined by refluxing
with aqueous NaOH. On the other hand, treatment with ethanol amine at
room temperature gives one molecule of hydrogen chloride from each
molecule of trichlorfon. Polarography is also used (Giang & Caswell,
1957). However, extraction with ethyl acetate and gas-chromatographic
determination are generally applied (Zweig & Sherma, 1972).
Table 1. Physical and chemical properties of trichlorfona
Physical state colourless crystals
Melting point (°C) 83-84
Boiling point (°C) 100 (0.1 mmHg)
Vapour pressure (20 °C) 7.8 x 10-6 mmHg
Volatility (20 °C) 0.022 mg/m3
20
Density 1.73
4
Solubility in g/100 ml (25 °C) water 15.4
benzene 15.2
chloroform 75.0
diethyl ether 17.0
n-hexane 0.08
Partition coefficient log Pow 0.57
(octanol/water)
Corrosiveness corrosive to metals
a From: Giang et al. (1954); FAO/WHO (1972); Dedek (1981); IARC
(1983).
Extraction with acetonitrile, reextraction with ether and
gas-chromatographic determination with flame photometric detection
(FPD) or flame thermionic detection (FTD) are standard procedures for
the determination of residues. Trichlorfon is thermally decomposed
during chromatography to give dimethyl phosphite which is then
determined (Ferreira & Fernandes, 1980). Chloral generated by
decomposition can be determined by electron-capture detector (Zweig &
Sherma, 1972).
Acetylation or trimethylsilylation can stabilize trichlorfon for
gas chromatography without decomposition (Vilceanu et al., 1973;
Bowman & Dame, 1974). Trichlorfon itself has been successfully
determined at a high sensitivity using a column, such as Thermon 3000
on Shimalite TPA. More recently, a GC- FTD method, based on on-column
derivation by acetic anhydride, has been reported by Conrad et al.
(1987). The response is linear over ranges of 0.1-2.0 ng. The method
is applicable for the determination of trichlorfon in technical
products and formulations, as well as for residues in crops and animal
tissue samples.
Table 2. Summary of analytical methods for the determination of trichlorfon
Sample Sample preparation Analytical Detection Recovery (%) Comments Reference
medium conditions limit (added level,
(Detector, column, mg/kg)
column temperature
Formlation refluxing with concentration NaOH potentiometric determination MacDougall
titration with AgNO3 of trichlorfon (1964)
of total Cl- in formulations
standing with ethanolamine for 1 h at potentiometric distinguishable Bennewitz
room temperature titration of liberated between & Foth
HCl with AgNO3 trichlorfon (1967)
and dichlorvos
ethyl acetate ext.a FTD-GC, 25% 0.01 µg ±1-2% determination Zweig &
carbowax 20W, accuracy of trichlorfon Sherma
1.5 m (5 ft) in powder (1972)
195 °C, N2, formulation
120 ml/min
dissolving in CHCl3 and sililating with FID-GC, 3% XE-60, relative 1.3% determination Bowman &
bis-(trimethylsilyl)-trifluoracetamide 1.2 m, 110 °C, He standard of trichlorfon Dame
50 ml/min deviation in soluble (1974)
powder
formulation
acetylating with acetic FTD-GC, 15% pg determination Vilceanu et
anhydridepyridine mixture Apiezon L, 1 m, of trichlorfon al. (1973)
(2:0.5) in CH3CN 160-200 °C, N2, after acetylation
60 ml/min
Residues 0.1 N H2SO4 diethyl ether FTD-GC 16% XF-1150, 0.1 mg/kg 74-86 (0.1) the extracts Zweig &
in food 2 m (6 ft) 102-5 (12.5) should be Sherma
135 °C, He dialysed for (1972)
25 ml/min 24 h before
extraction
Table 2 (continued)
Sample Sample preparation Analytical Detection Recovery (%) Comments Reference
medium conditions limit (added level,
(Detector, column, mg/kg)
column temperature
Crops, CH3CN+H2O/CHCH3 ext.a hexane - FTD-GC, 20% 5 µg/kg 90-100 (0.2) some metabolites Takase et
fish, CH3CN partition CH3CN:+H2O, carbowax 20 M, fat 72 (0.2) are also al. (1972)
chicken evaporation, aq. layer/heptane 1m, 150 °C, N2 determined
washing NaCl, ether ext.a 60 ml/min
Crops, CHCl3 ext.; reext. with activated FPD-GC, 16% 2 µg/kg 100 (0.002-0.25) determination Devine
soil, carbon/acetone (hexane sat.a NaCl ext. XF-1150, 2 m, (water) water, 94-104 of trichlorfon (1973)
animal CHCl3 + NaCl ext.; aq. phase/CHCl3 ext. 125 °C 50 µg/kg (0.05-0.2) in forest
tissues, for animal tissue) (others) soil, 90-99 environmental
water (0.05-50) samples
plants, 82
(0.05-1.0)
animal
Fruit acetone ext.; 2% Na2SO4/hexane then FTD-or FPD-GC, 5% 0.1 mg/kg 90-102% clean-up is Ferreira &
ethyl acetate N2 40 ml/min carbowax-20 M, (1.0) not necessary Fernandes
3 m, 160-180 °C for ethyl (1980)
acetate
extraction
Livestock CH3CN ext.; 5% Na2SO4/CHCl3 ext.a FPD-GC, 3% 2 µg/kg 90 (egg)-102 Salithion Imanaka et
products haxane-CH3CN partition aq. Thermon 3000, (milk) (0.4) (same al. (1981)
CH3CN/CH2Cl2 ext.a 0.3 m, 120-170 °C, retention
N2 60 ml/min time) can be
removed by
washing with
n-hexane
Table 2 (continued)
Sample Sample preparation Analytical Detection Recovery (%) Comments Reference
medium conditions limit (added level,
(Detector, column, mg/kg)
column temperature
Milk CHCl3 ext. TLC, benzenemethyl 5 µg/kg 75-100 semi-quantitative Fechner et
acetate (3:1), (0.02-0.4) determination al. (1971)
enzymic determination separating
after activation dichlorvos
with ammonia
Feed 0.1% HCl ext.a; CHCl3 reext.a FPD-GC DB-1 0.01 mg/kg 88 (50 ppm) feed samples Cox et al.
(FSOT) 0.53 min x (1989)
30 m 120-150 °C
(6 °C/min) He
10 ml/min
Crops acetone or CH3CN ext.; conc.a + NaCl FTD-GC, methyl 0.01 ng 70-99 sep-pak Ishizaka et
reext.a with diethyl ether; Sep-pak C18 silicon or (0.1 mg/kg) cartridge is al. (1986)
(silica gel) benzene/MeOH phenylmethyl very useful
silicon; for simple
(FSOT) 0.53 mm x clean-up
10 m, 160 °C
Serum Serum+0.1 mix well; M HCl (1=1) FTD-GC, CBP-1 2.5 ng/ml extraction is Ameno et
Sep-pak C18 (silica gel) 0.1 M HCl, (FSOT) 0.53 mm x not necessary al. (1989)
10% & 50% aq. MeOH 12 m 120 °C He for wide
30 ml/min range of
calibration
curve (5 approx
500 ng/ml)
a ext. = extraction.
reext. = reextraction.
sat. = saturated.
conc. = concentration.
The simultaneous detection (µg/kg) and identification of
trichlorfon and other organophosphorus pesticides extracted from
foods can be accomplished by using gas chromatography-mass
spectrometry (Stan, 1977; Stan et al., 1977). Although the molecular
ion cannot be measured by the electron impact (EI) ionization mass
spectrum, an intense peak of the protonated molecular ion (M+1)+ is
observed in the chemical ionization (CI) mass spectrum. Thus, the
latter is more sensitive and selective than the former in residue
determination. Field desorption mass spectrum shows the protonated
dimer ion (2M+1)+ of trichlorfon besides the (M+1) ion (Schulten &
Sun, 1981). The occurrence of such ions is helpful in confirming the
identification of trichlorfon.
Thin-layer chromatography (TLC) is particularly useful for
qualitative analysis. Systematic separation schemes for many
organophosphorus pesticides have been proposed (Guth, 1967; Getz &
Wheeler, 1968; Antoine & Mees, 1971; Ambrus et al., 1981). Levels of
0.1 µg trichlorfon can be detected using nitrobenzyl-pyridine reagent
or silver nitrate and UV irradiation on silica gel or polyamide TLC.
A TLC-enzyme inhibition technique, that can be used for the
determination of residues in organophosphorus pesticides was reviewed
by Mendoza (1973). Trichlorfon itself is not a good inhibitor of
cholinesterase (Winterlin et al., 1968), but treatment with ammonia on
the plate, converting it into dichlorvos, is performed to enhance its
sensitivity (Fechner et al., 1971).
Although high performance liquid chromatography (HPLC) has
recently become an important technique in pesticide analysis, few
data are available for trichlorfon (Szalontai, 1976; Daldrup et al.,
1981, 1982).
Colorimetric methods have been applied for determining
trichlorfon, based on the phosphomolybdate reaction (Sissons &
Telling, 1970) and the Fujiwara reaction (Cerna, 1963; Giang et al.,
1954).
A method to preconcentrate water samples for the measurement of
trichlorfon was reported by Dedek et al. (1987).
The use of gas chromatographic detectors in HPLC has recently
received increasing attention because of the growing need for high
sensitivity and selectivity. The on-line combination of HPLC and
these detectors, using a thermo spraying interface (TSP), has been
applied successfully, because the advent of miniaturized HPLC systems
has alleviated many of the difficulties including a loss of
sensitivity associated with direct mobile phase introduction. In most
cases, the techniques of HPLC separation, GC-FTD detection, and GC-MS
confirmation can be successfully used in the analyses with
TSP-HPLC-FTD and TSP-HPLC-MS (Gluckman et al., 1986).
The TSP-HPLC-FTD system has been successfully used to determine
a polar and thermally unstable pesticide like trichlorfon in many
samples, because high sensitivity and less matrix interference are
achieved than with the HPLC-ultraviolet spectrophotometry (UV) system
for pesticide residues. According to Gluckman's report (1986),
trichlorfon can be detected at a level of 40 pg by TSP-HPLC-FTD and
the residues in tomatoes and cabbage can be determined without any
interference.
The characterization of several organophosphorus pesticides has
been achieved using positive and negative ion "filament on"
TSP-HPLC-MS. When ammonia gas is used as a reagent one, the base peak
is [M + NH4]+ in the positive ion mode (PIM) for the
organophosphorus pesticides examined, while the pesticides exhibited
different fragmentation behaviour in the negative ion mode (NIM)
([M]- in the base peak). PIM shows a higher sensitivity for these
compounds than NIM.
Trichlorfon and other organophosphorus pesticides can be detected
at levels of 20-50 ng (minimum detection limit; s/n = 3) in the
reconstructed ion chromatography of PIM-HPLC-MS. Since, a 100-fold to
1000-fold increase in sensitivity will be expected using single ion
monitoring (SIM), the detection limits in PIM-TSP- HPLC-MS are rather
similar to those in GC-NCI-MS and in direct liquid introduction
(DLI)-HPLC-NCI-MS (Barcelo, 1987; Barcelo et al., 1988; Betowski &
Jones, 1988).
The Joint FAO/WHO Codex Alimentarius Commission has given
recommendations for the methods of analysis to be used in the
determination of trichlorfon residues (FAO/WHO, 1989).
Because of the transformation of trichlorfon into dichlorvos, it
is necessary to have a method for the simultaneous quantification of
both of these compounds in biological studies. Such a method has been
worked out by Nordgren (1981), and Nordgren et al. (1978, 1980, 1981),
and has been correlated with the degree of enzyme inhibition. Similar
methods have been used by Yakoub (1990).
3. SOURCES OF HUMAN AND ENVIRONMENTAL EXPOSURE
3.1 Natural occurrence
Trichlorfon is not a natural product. However, it is found as a
metabolite of the insecticide butonate: butyric acid ester of
trichlorfon (Dedek et al., 1979).
3.2 Industrial production
Trichlorfon was introduced as a commercial chemical in 1952. It
is manufactured by reacting dimethyl phosphite with chloral (Barthel
et al., 1954; Lorenz et al., 1955).
There is no record of the world production of trichlorfon. It is
produced in Germany, Japan, and Spain and is believed to be produced
also in Argentina, Brazil, China, Israel, Mexico, the Republic of
Korea, and the USSR. The total production in western Europe was
estimated to be about 2000 tonnes in 1977. The production in Japan has
ranged from 613 to 1095 tonnes per year over the last decade (Japan
Plant Protection Association, 1985, 1986, 1989).
3.3 Uses
Trichlorfon is a broad-spectrum insecticide that is particularly
effective against Diptera. In agriculture, it is used mainly
against insect pests in field and fruit crops. Trichlorfon is also
used to control forest insects, in public health, and for the control
of endo- and ectoparasites in/on domestic animals and fish.
Under the generic name of metrifonate, trichlorfon is used as an
antihelminthic in humans and is one of the treatments of choice for
infestation by Schistosoma haematobium, primarily in Africa
(Snellen, 1981; Davis, 1986; Aden Abdi et al., 1987; Wilkins & Moore,
1987; Aden Abdi & Gustafsson, 1989; Yakoub, 1990; Aden Abdi, 1990).
The usual regimen consists of three doses of 7.5 or 10 mg/kg, given at
intervals of 14-21 days. Because of the lower costs in comparison with
other treatments, metrifonate is particularly attractive for mass
treatments. It has been given to millions of patients with
schistosomiasis with only occasional mild side effects (Nordgren,
1981). In order to obtain better patient compliance, Aden Abdi (1990)
recently proposed a regimen of 3 x 5 mg, administered in one day (see
section 9.2).
Metrifonate is also under consideration as a treatment for
Alzheimer's disease (Hallak & Giacobini, 1989; Becker et al., 1990;
Pomponi et al., 1990).
Table 3 gives an indication of the world-wide consumption of
trichlorfon. Although the quantity is not reported, trichlorfon is
used also in several other countries including Finland, Hungary,
Malaysia, Mongolia, and the USSR. According to a Battelle report
(1987), the total consumption of trichlorfon in 13 countries in 1987
was 851 tonnes as shown in Table 4, which also includes data from
other sources.
The following formulations are used in agriculture: 50%
emulsifiable concentrate, 95, 80, and 50% soluble powders, 50%
wettable powders, 5 and 4% dusts, 5, 2.5, and 1% granules, 75, 50, 40,
and 25% ultra-low volume concentrates. Some formulations mixed with
other organophosphorus insecticides, such as malathion and ESP, or
with carbamate insecticides, such as carbaryl, are also used.
The following formulations are used for animal treatments: 90,
80, and 50% soluble powders, 6% suspension, 11% solution, 50%
injectable solution tablets. A 1% fly bait is also available, and a
0.1% preparation against house-ants. For antihelminthic prep arations,
trichlorfon can be used in combination with atropine, fenbendazole, or
thiabendazole.
Tablets containing 100 mg active ingredient metrifonate are used
in the treatment of schistosomiasis in humans.
Table 3. World usage of trichlorfon
Year Usage (tonne) References
1980 3159 Battelle (1982)
1983 2349 Battelle (1984)
1987 851 Battelle (1987)
Table 4. Usage of trichlorfona
Country Usage Year Main use
(tonne)
France - 1987
Italy 10.2 1987 vines
Turkey 16.4 1987 vegetables
FRGb 19.9 1984 sugar beet
United Kingdomb 0.8 1984 sugar beet
Spain 155.3 1987 vines, vegetables,
olives
Czechoslovakiac - 1983
Swedenc 5.7 1982 agriculture, hygiene
Japan 279.7 1987 potatoes, other
vegetables
Korea, Republic 79.0 forests, apples
India -
Indonesia 6.0 1987 soybeans
Thailandc 19.0 1978
Philippinesb - 1984
USA 454.0d 1978 field crops, alfalfa,
forests, cotton,
vegetables
1.8 1987 alfalfa
Mexico 133.1 1987 maize, cotton,
tobacco, tomatoes,
sugar cane, soybeans
Brazil 145.3 soybeans, cotton,
wheat
Egypt -
South Africa 24.6 maize
Kenyac - 1983
a From: Battelle (1987).
b From: Battelle (1984).
c Information through IRPTC (International Register of
Potentially Toxic Chemicals).
d From: IARC (1983).
4. ENVIRONMENTAL TRANSPORT, DISTRIBUTION, AND TRANSFORMATION
Following aerial application, trichlorfon is distributed to the
air, soil, water, trees, plants, and other media. With rainfall,
trichlorfon penetrates into the lower soil layers and moves into the
aquatic environment.
4.1 Transport and distribution
4.1.1 Air
The air/water partition coefficient of trichlorfon was determined
to be <5.0 x 10-7, indicating that the distributed amount in air is
much smaller than that in water and is, in fact, negligible (Kawamoto
& Urano, 1989).
4.1.2 Water
One and two days after the aerial application of trichlorfon to
a forest at the rate of 1.0 kg/ha, a small amount of the compound was
found in creek water (Pieper & Richmond, 1976). Trichlorfon was also
detected in water samples in sprayed forests in Canada (Sergeant &
Zitko, 1979).
Rainfall caused run-off of trichlorfon from the sprayed steppe
zone into ponds (IRPTC/GKNT, 1983). In an agricultural region of the
USSR, trichlorfon migrated into drainage water and was transferred
across a significant distance, depending on the rainfall (Zakharov,
1980).
4.1.3 Soil
Because of its high water-solubility (15.4 g/100 ml), there was
some downward movement of trichlorfon through soil with water. When
sprayed twice on an apple orchard, the insecticide was detected in
soil layers of 0-10 cm and 10-20 cm depth, 10 days after the last
treatment (Naishtein et al., 1973).
Trichlorfon applied to the soil surface at the rate of 2.4 kg
a.i./ha did not move significantly into the lower layers by leaching
(Baida, 1970); when applied at a high rate of 60 kg/ha, the compound
penetrated into the 60-cm layer of the soil (Naishtein, 1976).
It has been shown, in different soils, that the disappearance of
the insecticide (initial concentration; 10 mg/kg) is very rapid during
the first few days following application and considerably slower
thereafter. The levels of trichlorfon residues in a soil without
plants were 5.2, 2.1, and 0.9 mg/kg on the 5th, 11th, and 21st days
after application, respectively. However, in soils with tomato,
cabbage, and potato plants, trichlorfon levels decreased more rapidly
to 2.1, 1.0, and 0.6 mg/kg, respectively, on the 5th day, 0.7, 0.5,
and 0.3 mg/kg on the 11th day, and 0.6, 0.3, and 0.2 mg/kg on the 21st
day after treatment. Only a small amount of trichlorfon was detected
after 30 days. The rate of disappearance in soils was dependent on
the vegetation (Ivanova & Molozhanova, 1974).
4.2 Abiotic degradation
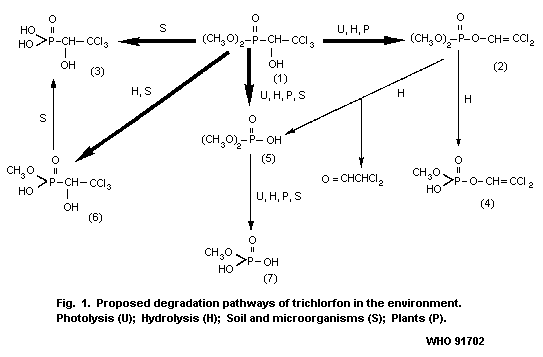
The proposed degradation pathways of trichlorfon in the
environment are shown in Fig. 1.
In alkaline buffers and seawater (pH 8.1), trichlorfon is
rearranged via dehydrochlorination to yield the more potent
cholinesterase inhibitor, dichlorvos; however, in acidic buffers or in
fresh water (pH 5.3), it is stable. At more alkaline pH values, the
anticholinesterase activity disappears slowly (Ecobichon, 1979). At
pH 5.5 and above, degradation to dichlorvos occurs at detectable
rates (Dedek, 1981).
On photolysis in water under ultraviolet radiation (UVR),
trichlorfon was rapidly converted to dichlorvos (2) and two
unidentified products. These two products decomposed further on
prolonged irradiation. Photodegradation appears to be much slower in
the solid state than in aqueous solution (Giovanoli-Jakubczak et al.,
1971).
[32P]-trichlorfon on glass plates was photodecomposed by 7%
after a 5-h exposure to UVR (500 W, 200-600 nm) and by 6% after a
20-h exposure to sunlight. A trace amount of dimethyl hydrogen
phosphate (5) (see Fig. 1) and methyl hydrogen phosphate (7) were
identified as photodegradation-products, whereas dichlorvos (2) was
not detected among the photodegradation-products on the glass plates
in either case (Dedek et al., 1979).
Trichlorfon is fairly stable in acidic solutions, but unstable in
neutral and basic solutions. The half-life of chloroform-extractable
radioactivity in buffer solutions at 40 °C is 46.4 days at pH 2, 16
days at pH 5, 3.75 days at pH 6, 19 h at pH 7, 8.8 h at pH 8, and 75
min at pH 10. Dichlorvos (2), the demethylated derivative of
trichlorfon (6), dimethyl hydrogen phosphate (5) and methyl hydrogen
phosphate (7) were identified as degradation products, but they were
not quantified (see Fig. 1; Dedek et al., 1979).
In other studies, the half-life of trichlorfon at 100 mg/litre in
sterilized water-ethanol (99:1) phosphate buffers at 25 ± 3 °C, was
reported to be more than 1000 weeks at pH 4.5, 3.5 weeks at pH 6.0,
0.4 weeks at pH 7.0, and 0.13 weeks at pH 8.0. It was concluded that
the disappearance was mainly due to the conversion of trichlorfon to
dichlorvos via dehydrochlorination (Chapman & Cole, 1982). In another
study, the half-lives for the formation of dichlorvos from trichlorfon
at pH 7, pH 7.5, and pH 8 were reported by Hofer (1981) to be 27, 9,
and 3 h, respectively.
 Formulated trichlorfon was applied to sterilized and
non-sterilized soils with a 60% moisture content, and incubated at
ambient temper ature under natural sunlight. At concentrations of both
13 and 132 mg/kg, the levels of insecticide decreased to less than
the detection limit within 40 and 50 days, respectively, under both
sterilized and non-sterilized conditions. It appears that the
insecticide is readily subjected to abiotic degradation in soil
(Yurovskaya & Zhulinskaya, 1974).
4.3 Biodegradation
The metabolic fate of [14C]-trichlorfon labelled at the methoxy
group has been studied in culture media of nodule-forming bacteria,
such as Rhizobium leguminosarum and Rhizobium trifolii. After
incubation for 10 days at 30 °C, the unchanged parent compound
(19-25%) together with dimethyl hydrogen phosphate (18- 25%) (5) and
methyl hydrogen phosphate (0.6-1%) (7) was recovered from the media.
In addition, a trace amount of [14C] carbon dioxide was evolved
during the same period (Salama et al., 1975) (see Fig. 1).
In contrast, a demethylated derivative (6) of trichlorfon and
2,2,2- trichlorohydroxyethylphosphonic acid (3) were shown to be the
major metabolites in the culture media of Aspergillus niger,
Penicillium notatum, and Fusarium spp. (Zayed et al., 1965).
4.4 Environmental fate
The metabolism of [14C]-trichlorfon labelled at the methoxy
group has been studied in plants of the broad bean (Vicia faba) and
clover (Trifolium alexandrinum). The roots of the plants were
immersed in a phosphate buffer at pH 6 containing [14C]-trichlorfon
at a concentration of 50 mg/litre, and grown for 5 or 10 days in the
greenhouse. At harvest, the buffer solution as well as the roots of
the two plants contained unchanged parent compound and dimethyl
hydrogen phosphate (5) together with a trace amount of methyl
hydrogen phosphate (7) (Salama et al., 1975) (see Fig. 1).
Following the application of [32P]-trichlorfon to stems or
leaves, the insecticide disappeared from tomato, potato, and cotton
plants with half-lives of 20-57 h in the greenhouse, and from plums,
apples, cherries, peas, and wheat plants with half-lives of 0.5-7.5
days in the field. Characterization of metabolites was not possible
because of their volatility (Dedek et al., 1979).
When formulated trichlorfon (0.2%) was sprayed on cabbage and
onion plants at a rate of 1200 litre/ha, rapid conversion to
dichlorvos occurred. One day after treatment, the treated leaves of
cabbage and onion plants contained the highest residues of dichlorvos
(0.09-0.51 mg/kg) together with the parent compound (0.79-3.1 mg/kg).
Trichlorfon disappeared from the leaves with a half-life of less than
3 days, and dichlorvos decreased to less than the detection limit
within 15 days (Baida, 1975).
5. ENVIRONMENTAL LEVELS AND HUMAN EXPOSURE
5.1 Environmental levels
5.1.1 Air
When 2% trichlorfon was applied at a rate of 30 ml/m2, its
vapour was detected in the air. The initial concentration of 0.1
mg/m3 decreased rapidly to 0.05-0.01 mg/m3 within 2-3 days.
Trichlorfon was not detected after 30 days (Degtyareva et al., 1977).
After handspraying in a vineyard at 2 and 6 kg/ha, air concentrations
of 0.0003 and 0.001 mg/m3, respectively, were measured. Average
daily concentrations of trichlorfon and its maximum single
concentration in the Ukrainian Republic were 0.0003 and 0.0004
mg/m3, respectively (IRPTC/GKNT, 1983).
5.1.2 Water
When an 18% aqueous solution of trichlorfon was sprayed on a
forest at a rate of approximately 1 kg/ha from a helicopter, the
residues were 23.4-51.9 and 8.7-12.1 µg/litre in the creek water on
the first and second day after application, respectively (Pieper &
Richmond, 1976).
According to the monitoring programmes in Canada in 1976 and
1977, water samples in the forests where trichlorfon was sprayed were
contaminated with 0.062-1.0 µg/litre of the insecticide in most of the
year's samples in 1976, but the concentration in samples in 1977 were
considerably lower, with a maximum value of 0.058 µg/litre (Sergeant
& Zitko, 1979).
Trichlorfon was measured in soil water at 0.001 (1970), 0.02
(1971), and 0.001 mg/litre (1972) on average 1.5-2 months after
application (IRPTC/GKNT, 1983).
When trichlorfon was sprayed over a mixed boreal forest in New
Brunswick (Canada) at a rate of 1.14 kg/ha, concentrations in stream
water were approximately 95 µg/litre initially and below detection
limit (0.05 µg/litre) two weeks after treatment (Sundaram & Varty,
1989).
5.1.3 Soil
Trichlorfon was one of the chemicals found at hazardous waste
sites in the USA (Kokoszka & Flood, 1989). Levels were not specified.
After the spraying of a vineyard at 2 or 6 kg/ha, trichlorfon
levels measured in the soil were 0.24 and 0.48 mg/kg, respectively, on
the day of treatment. One day later, the levels in the soil were 0.49
and 1.03 mg/kg, and 15 days later, 0.002 and 0.02 mg/kg. The average
level of trichlorfon in the soil in the Kherson Region was 0.01 mg/kg
(1970), 0.17 mg/kg (1971) and 0.002 mg/kg (1972). After spraying a
forest at 0.8 kg/ha over a 200-ha area (160 kg total), monitoring
soil over the area gave estimates of 29.9 kg trichlorfon remaining in
the soil, 5 days after application and 0.43 and 0.25 kg after 10 and
14 days, respectively. All the trichlorfon measured was in the top 10
cm of soil. No trichlorfon was found 18 days after spraying
(IRPTC/GKNT, 1983).
After aerial spraying of trichlorfon over a mixed boreal forest
in New Brunswick (Canada) at a rate of 1.14 kg/ha, residues in soil
dissipated from 3 mg/kg initially to levels below the detection limit
(0.05 mg/kg) in about two weeks (Sundaram & Varty, 1989).
5.1.4 Residues in plants and animals
An 18% aqueous solution of trichlorfon was applied by helicopter
at 6.1 litre/ha to a forest. The residues on days 1, 2, 8, and 15
after aerial treatment were 11.2-12.6 mg/kg, 3.8-10.4 mg/kg, 0.8-1.6
mg/kg and below detection limit-0.6 mg/kg, respectively, on Douglas
fir, 68.2-81.7 mg/kg, 40.3-59.4 mg/kg, 3.9-4.5 mg/kg and below
detection limit, respectively, on willow, and 43.1-113.0 mg/kg,
4.3-30.0 mg/kg, 5.3-6.3 mg/kg, and below the detection limit-2.1
mg/kg, respectively, on grasses (detection limit: 0.1 mg/kg) (Pieper
& Richmond, 1976).
Trichlorfon was detected in all song birds caught in the area
(60.7 ha), 76 h after spraying at the rate of 1.1 kg/ha. The residues
of trichlorfon were found at 0.01 mg/kg for blue jays and crested
fly-catchers and at 0.013-0.04 mg/kg for baltimore oriols (Kurtz &
Studholme, 1974).
When trichlorfon was sprayed at a rate of 1.14 kg a.i./ha over a
mixed boreal forest in New Brunswick (Canada), the initial residues
in foliage ranged from 10.8 to 17.3 mg/kg fresh weight, but dissipated
rapidly to 2.6 to 6.5 mg/kg in three days (Sundaram & Varty, 1989).
5.2 Residues in food
5.2.1 Crops
The results of supervised trials involving foliar treatment of
various crops with trichlorfon are summarized in Table 5.
Leafy vegetables, such as lettuce, spinach, and Chinese raddish
leaves showed high residues of trichlorfon (several mg/kg or more)
shortly after application. The trichlorfon residues in Chinese raddish
leaves were about ten times higher than those in the roots. Among
fruits, strawberries, raspberries, black currants, and red currants
were found to contain higher trichlorfon residues than other fruits,
such as citrus. The results in Table 5 showed that the residues
decreased rapidly with time after application.
During the period 1987-88, 764 home-grown and imported wheat
samples were analysed for pesticide residues in the United Kingdom.
Trichlorfon was not found at, or above, the reporting limit of 0.1
mg/kg (Osborne et al., 1989).
Following normal field application (in a field trial) of
trichlorfon in Portugal to Portuguese cabbage and broccoli, residue
levels decreased to below the MRL of 0.5 mg/kg in 10 days for the
Portuguese cabbage and two weeks for the broccoli. The difference in
time is mainly ascribed to the much larger surface area exposed in the
case of the broccoli (Magalhaes et al., 1989).
The surveillance of trichlorfon residues on over twenty crops in
Hungary revealed that about 90.1% of the samples contained residues
at levels below 0.02 mg/kg (Anon. 1978a).
Trichlorfon residues on spinach and lettuce after spraying were
somewhat higher in green-house crops than in field crops in West
Germany (Stobwasser & Kirchhoff, 1968).
No trichlorfon residues were found in a survey of pesticide
residues on crops (total samples: 697) collected from a Tokyo market
from April 1984 to March 1989 (detection limit: 0.005 mg/kg) (Nagayama
et al., 1986, 1987, 1988, 1989).
Cucumber vines were sprayed to run-off with 0.05% trichlorfon
aqueous solution, and the fruits sampled over a period of time. The
half-life of trichlorfon was 1.76 days (Hameed et al., 1980).
Grape products were prepared in a laboratory from grapes
harvested 1 day after the last application of trichlorfon. Trichlorfon
residues were detected at levels of 140%, 120%, 159%, and 0.4% of the
originally applied concentration in the grape juice, raisins, wines,
and brandies, respectively, and the dichlorvos concentrations in the
wines were higher than those in the grapes from which the wines were
made (Hiramatsu & Furutani, 1978).
Residues of 0.0002-0.006 mg trichlorfon and dichlorvos/kg were
detected in 8 out of 40 flower honeys in Bulgaria (Tsvetkova et al.,
1981).
Table 5. Residues of trichlorfon in crops
Crop/ Application (spray) Residues (mg/kg) on the day of spraying Reference
Country or at intervals [days] after applicationa
Day
no. kg/ha formulationb
0 1/3 4/6 7/9 10/12 13/15 17/19 21
Cabbage
Finland ns 0.6 0.5 0.4 0.2 Anon. (1978b)
[18-21] [27-29]
Japan 6 2-5 EC-50 0.14-0.05 0.08-0.03 FAO/WHO (1979)
8 2-5 0.23-0.07 0.05-0.03
USSR 4 2.0 EC 0.79-0.27 0.17 0.09 0.07 0.007 Baida (1975)
[30]
Chinese cabbage
Japan 3 2.0 EC-50 0.13-0.04 0.09-0.02 Hiramatsu &
[18-21] Furutani (1977)
5 2.0 0.06 0.05 FAO/WHO (1979)
6 2.0 0.13 0.09
1 0.75 0.19-0.12 0.05 0.03
Lettuce
Finland 1 1.2 WP-80 34-26 5.6 0.75 0.25 0.13 0.01 Abbasov (1972)
USA 1 0.6 WP-50 1.6 0.8 0.3 0.7 0.3 0.2 FAO/WHO (1976)
1 1.2 0.8 0.8 0.6 nd 0.3
1 0.6 1.2 0.3 0.3 nd
5 1.2 0.5
6 1.2 nd
7 1.2 0.2
Table 5 (continued)
Crop/ Application (spray) Residues (mg/kg) on the day of spraying Reference
Country or at intervals [days] after applicationa
Day
no. kg/ha formulationb
0 1/3 4/6 7/9 10/12 13/15 17/19 21
Head lettuce
Finland 1 1.2 WP-50 22.8 1.5 0.31 0.04 0.01 FAO/WHO (1976)
1 1.2 31.2 1.1 0.17 0.06 nd
FRG 1 0.75 EC-50 8.1 1.8 0.54 0.41 0.20 nd nd FAO/WHO (1976)
1 0.75 15.9 4.6 0.21 0.08 0.14 0.05 nd
1 0.45 5.8 1.8 0.22 0.09 nd nd nd
1 0.45 4.8 4.3 0.27 nd nd nd nd
Leaf lettuce
Finland 1 1.2 WP-80 33.5 2.6 0.61 0.03 FAO/WHO (1976)
1 1.2 26.4 1.3 0.23 0.02
USA 2 1.2 WP-50 6.3 1.3 nd nd FAO/WHO (1976)
2 1.2 3.5 1.1 nd nd
2 1.2 3.8-0.8 0.8-0.1 0.4-nd
2 1.2 97.6 9.2 6.0 3.9
Red cabbage
Netherlands 1 1.0 WP-80 0.05-0.03 Anon. (1978c)
Savoy cabbage
Netherlands 1 1.0 WP-50 0.36-0.12 Anon. (1978c)
Parsley
Hungary 1 0.8 WP-50 0.2 0.02 0.01/0.01 FAO/WHO (1976)
Table 5 (continued)
Crop/ Application (spray) Residues (mg/kg) on the day of spraying Reference
Country or at intervals [days] after applicationa
Day
no. kg/ha formulationb
0 1/3 4/6 7/9 10/12 13/15 17/19 21
Brussel
sprouts
Netherlands 3 1.2 WP-80 0.4-0.15 0.4-0.15 0.13-0.08 Anon. (1978c)
4 1.2 0.9-0.08 0.1-0.08 0.05-0.03
Kale
Netherlands 1 1.0 WP-80 0.33-0.12 Anon. (1978c)
Kohlrabi
Netherlands 1 1.0 WP-80 0.02-0.01 Anon. (1978c)
Spinach
Netherlands 1 1.0 WP-80 5.3-3.4 FAO/WHO (1976)
1 1.0 6.2-2.1
Spinach
(greenhouse)
Netherlands 1 1.1 WP-50 33.3-23.3 5.5-2.9 3.4-1.2 1.3-0.4 0.9-0.2 0.4-0.2 0.5-0.1 FAO/WHO (1976)
1 1.1 4.5-1.9 3.1-1.2 0.8-0.4 0.5-0.25 0.3-0.2 0.1-0.06
Spinach
(under frame)
FRG 1 0.75 WP-50 4.4 2.4 1.6 FAO/WHO (1976)
1 0.75 30 21.5 5.2 1.2 2.2 0.25
Spinach
(outdoor)
Canada 2 1.1 WP 0.6 0.2 nd FAO/WHO (1976)
2 1.1 39 3.5 nd
Table 5 (continued)
Crop/ Application (spray) Residues (mg/kg) on the day of spraying Reference
Country or at intervals [days] after applicationa
Day
no. kg/ha formulationb
0 1/3 4/6 7/9 10/12 13/15 17/19 21
FRG 1 0.75 2.0 1.6 0.7 0.35 FAO/WHO (1976)
1 0.75 0.4 0.02
1 ca. 1.5 WP-80 19.8 1.8
1 0.75 WP-50 34 6.0 0.85 0.15 0.15
1 0.75 WP-50 30 1.7 0.3 0.08
1 0.75 30 13.2 5.6 1.0 1.0
Netherlands 1 0.5 WP-50 6.5-5.7 2.7-1.2 2.1-0.08 0.6-0.1 0.3-0.2 FAO/WHO (1976)
1 0.63 2.2-1.8 1.1-0.4 0.6-nd 0.2-0.1 0.2-nd
1 1.9 1.4-0.5 0.3-0.05 0.01-nd
1 0.7 1.4-0.6 0.3-0.2 0.4-nd 0.7-nd
USA 2 1.1 11.2 0.3 0.3 FAO/WHO (1976)
2 1.1 0.9 0.4
2 1.1 1.8 0.2 nd
Lima bean
USA 1 2.22 EC 0.07-0.02FAO/WHO (1976)
[44]
Chinese radish
Japan
(leaf) 5 2.0 EC-50 2.76-0.22 0.26-0.09 0.07-0.02 FAO/WHO (1979)
(root) 5 2.0 0.12-0.07 0.07-0.03 0.05-0.01
(leaf) 8 2.0 1.4-0.9 0.77-0.1 0.09-0.03
Table 5 (continued)
Crop/ Application (spray) Residues (mg/kg) on the day of spraying Reference
Country or at intervals [days] after applicationa
Day
no. kg/ha formulationb
0 1/3 4/6 7/9 10/12 13/15 17/19 21
Chinese radish
Japan
(root) 8 2.0 0.12-0.08 0.04-0.03 0.04-0.01 FAO/WHO (1979)
(leaf) 8 1.5-2.0 not stated 1.4-0.22 0.77-0.10 0.09-0.03
Onion
USSR 4 2.0 EC 2.7-3.1 0.90-1.2 0.02-0.77 nd-0.04 nd [30] Baida (1975)
Potato
Japan 6 2.0 EC-50 0.03-0.02 0.02-nd FAO/WHO (1979)
Egg plant
Japan 5 1.0 EC-50 0.03-0.02 0.01-nd nd FAO/WHO (1979)
8 1.0 0.02 0.007-nd
Green pepper
Hungary 1 1.0 WP-50 0.13 0.06 0.05 0.03 0.02 0.02 Anon. (1978a)
[16]
Tomato
(glasshouse)
Netherlands 1 1.2 WP-50 0.03-nd FAO/WHO (1976)
1 1.2 0.13-nd Abbasov (1972)
Apple
Netherlands 1 1.2 WP 0.9-0.6 Anon. (1978c)
1 1.2 1.9-0.1 Anon. (1978c)
Table 5 (continued)
Crop/ Application (spray) Residues (mg/kg) on the day of spraying Reference
Country or at intervals [days] after applicationa
Day
no. kg/ha formulationb
0 1/3 4/6 7/9 10/12 13/15 17/19 21
Grape
Japan 1-5 1.0 EC-50 1.6-0.8 1.4-0.28 0.9-0.4 0.4-0.26 0.15-0.1 Hiramatsu &
(unsacked) Furutani (1978)
(sacked) 0.1 5 0.07 0.08 0.06 0.06
Lemon
USA 1 4.5 WP-80 nd Iwata at el. (1979)
[52-59]
Orange
USA 1 4.5 WP-80 nd Iwata et el. (1979)
[52-59]
Mandarin
orange
Japan 5 3.0 EC-50 nd FAO/WHO (1979)
[27-29]
Kski
persimmon
Japan 3 3g/tree EC-50 0.57-0.53 0.36 0.3-0.25 FAO/WHO (1979)
Raspberry
Finland 2 0.8g/ not stated 5.6 Anon. (1978b)
linear
0.5-0.6g/ 2.9-0.7 Anon. (1978b)
plant [18-21]
Table 5 (continued)
Crop/ Application (spray) Residues (mg/kg) on the day of spraying Reference
Country or at intervals [days] after applicationa
Day
no. kg/ha formulationb
0 1/3 4/6 7/9 10/12 13/15 17/19 21
Strawberry
Japan 3 2.0 EC-50 1.4 0.31 FAO/WHO (1979)
[18-21]
5 2.0 4.6 0.75
3 3.0 2.12 0.46
[18-21]
5 3.0 3.31 1.06
1 1.6-2.4 0.02
[18-21]
3 2-3 not stated 2.1-1.4 0.46-0.33
[20-21]
5 2-3 not stated 4.6-3.3 1.1-0.74
Water melon
not stated 6 2.0 EC-50 0.008 FAO/WHO (1979)
Black currant
Finland 1.3g/ not stated 2.6 0.2 Anon.(1978b)
plant [27-29]
Red current
Finland 1.6g/ not stated 25 12 Anon.(1978b)
plant [27-29]
Rye-grass
USA 1 1.1 5% bait 37.5 3.1 1.9 0.1 van Middalem et
al. (1972)
Table 5 (continued)
Crop/ Application (spray) Residues (mg/kg) on the day of spraying Reference
Country or at intervals [days] after applicationa
Day
no. kg/ha formulationb
0 1/3 4/6 7/9 10/12 13/15 17/19 21
Cotton
USA 4 3.7-1.4 WP-50 79 4 Isaac at al. (1965)
Sugar beet
Japan 6 1.5 EC-50 0.05-0.02 0.04-0.02 FAO/WHO (1979)
8 1.5 0.04-0.02 0.024-0.020
Finland 2 0.64 not stated 0.05 Anon. (1978b)
[48]
6 1.5 not stated 0.24-0.06 0.07-0.02
8 1.5 not stated 0.26-0.05 0.08-0.02
a nd = not detectable;
ns = not stated.
b WP = Wettable powder;
EC = Emulsifiable concentrate.
5.2.2 Milk
The results of supervised trials in which cows were treated with
trichlorfon via several routes of exposure and the residues in the
milk measured are summerized in Table 6.
Trichlorfon residues in cow's milk were mainly studied in animals
that were administered the pesticide orally. The reports showed
relatively high levels and a gradual disappearance of trichlorfon in
the milk of treated cows. The residue values in FAO/WHO report (1979)
were much higher than those in others (Table 6).
To control botflies, a 11.2% aqueous solution of trichlorfon was
applied dermally to lactating cows. Maximum residues of trichlorfon
and dichlorvos were found in the first milking (0.2 and 0.03 mg/litre,
respectively) and were still detectable in the third milking. Storage
and short-term heating of the milk did not essentially degrade the
insecticide, but, with boiling, an accelerated transformation of
trichlorfon to dichlorvos took place (Fechner et al., 1968).
Treatment of cows with 0.25 or 0.5% aqueous solution of
trichlorfon resulted in residues in milk of 0.02 and 0.7 mg/litre,
respectively, within the first 72 h following treatment. The levels of
trichlorfon were higher in the morning than in the evening flow
(IRPTC/GKNT, 1983).
The trichlorfon residues in cow's milk were in direct proportion
to the veterinary use of the insecticide on the cows. Heat processing
of milk had little effect on trichlorfon residues. However,
evaporation or spray drying of the milk reduced the residue levels
considerably (Konrad et al., 1975).
5.2.3 Meat
Trichlorfon residues in pigs and sheep treated with the chemical
under supervised trial conditions are shown in Table 7. The results
revealed that trichlorfon residues in pork rapidly disappear; they
were below the detection limit (0.01 mg/kg) 24 h after subcutaneous
application of 25 mg/kg body weight. The residues following spray
treatment of sheep against harmful insects decreased to below the
detection limit (0.01 mg/kg) after 168 h (Dedek & Schwarz, 1970a).
32P-labelled trichlorfon was poured evenly on to 600 cm2
areas of the freshly shorn backs of sheep at a rate of 20 mg/kg. Only
a minimal concentration (0.1 mg/kg) of the insecticide was detected in
the blood of the sheep. However, trichlorfon levels in the blood of
cattle were higher than those in the blood of sheep at a similar dose.
When special solvents were used for the preparation of the trichlor
fon solution and the dose was increased to 50 mg/kg, the level of
residues in the blood of the sheep increased to 1.2 mg/litre (Dedek
& Schwarz, 1970b).
Table 6. Residues of trichlorfon in milk of cows after application
Method of Residues (mg/kg) in milk after application Reference
application
mg/kg 1 h 2 h 3 h 4 h 6 h 8 h 9 h 10 h 12 h 20 h 24 h 32-72 h 96 h 168 h
Dermal 80 0.1-0.25 0.05-0.25 0.05 Mollhoff
(1971)
Dermal 36 0.1-0.2 0.05 Mollhoff
(1971)
Dermal 100 0.1-0.2 0.05-0.1 0.01 Mollhoff
(1971)
Dermal 100 0.05-0.1 0.05 Mollhoff
(1971)
Oral 3 0.033 0.091 0.080 0.052 0.003 0.001 Nakahara
et al.
(1972)
Oral 30 0.56 0.33 0.25 0.16 0.007 0.001 Nakahara
et al.
(1972)
Oral 1a 0.034 0.029 0.021 0.009 0.001 Nakahara
et al.
(1972)
Intramuscularb 25 2.4 1.3 0.7 0.5 0.25 0.1 Dedek &
Schwarz
(1966)
Table 6. (continued)
Method of Residues (mg/kg) in milk after application Reference
application
mg/kg 1 h 2 h 3 h 4 h 6 h 8 h 9 h 10 h 12 h 20 h 24 h 32-72 h 96 h 168 h
Pour-ond 20 1.2 0.5 0.3 0.2 0.1 0.05 FAO/WHOc
(1979)
Pour-one 20 0.2 0.3 0.35 0.35 0.2 0.1 FAO/WHOc
(1979)
Pour-onf 30 0.45 0.05 0.01 Dedek &
Schwarz
(1966)
a 1 mg trichlorfon/kg body weight for 5 days.
b 50% trichlorfon in polyethylene glycol.
c Data cited to fit in this table.
d 2% trichlorfon in mineral oil.
e 2% trichlorfon in vegetable oil.
f 5.7% in aqueous solution.
Six USSR reports were available concerning supervised trials on
pigs (7 mg/kg in meat and 12 mg/kg in fat after unspecified treatment;
Yonova & Zhecheva, 1974), and sheep (Nepoklonov & Bukshtynov, 1971).
According to the English summaries of the reports, the trichlorfon was
rapidly absorbed and distributed among various organs and tissues,
then metabolized and eliminated.
5.2.4 Poultry and eggs
The trichlorfon contents of the organs of hens treated externally
with 1-8% aqueous solutions were 0.03-1.5 mg/kg, 0.01-0.7 mg/kg,
0.04-1.0 mg/kg, 0.02-0.8 mg/kg, 0.01-1.5 mg/kg or 0.02-0.9 mg/kg in
the muscle, liver, lung, heart, kidney and brain, respectively, within
the first 5 days after application. The eggs from hens treated
externally with 6-8% trichlorfon contained trichlorfon levels of
0.01-0.05 mg/kg (IRPTC/GKNT, 1983).
Residues of trichlorfon one day after the spraying of chickens at
the rate of 150 mg/kg body weight were as follows (mg/kg): egg shell,
0.48; egg white, 0.27; egg yolk, not detectable. Trichlorfon was
preserved in chicken carcasses kept for six months at -10 °C, but it
quickly decomposed when the carcasses were boiled (Dmitriyev, 1970).
5.2.5 Fish
Trichlorfon residues in eels were determined 1 and 5 days after
ponds were treated with a 1 mg/litre aqueous solution of the
insecticide. The results showed that the insecticide decomposed in a
short time to form dichlorvos in neutral and weakly alkaline water. In
pond water with a pH of 8-10, less than 10% of the applied
trichlorfon was degraded after 30 min and dichlorvos was detected. The
residues of trichlorfon and dichlorvos in the eels were 0.009-0.032
mg/kg and <0.005-0.02 mg/kg, respectively, on the first day
following treatment, and 0.011 mg/kg and 0.009-0.032 mg/kg,
respectively, on the 4th day after treatment. There was a good
correlation between the residual amounts of trichlorfon in the eels
and the concentrations in the water. Residues of trichlorfon and
dichlorvos, which were detected on the skin of eels in water at pH
7.0, could be removed by rinsing. Insecticide residues were found in
the internal organs of only one out of 7 eels caught in the field
pond. Carps exposed to an aqueous solution of trichlorfon at 0.25
mg/kg were examined on the 2nd, 4th, and 9th days after exposure.
Residues of trichlorfon and dichlorvos could not be detected in the
fish on the second day (Nakahara et al., 1973).
Table 7. Residues of trichlorfon in various meats after experimental application
Animal Method of mg/kg or Residues (mg/kg) after application Reference
application g/m3
1 h 2 h 3 h 4 h 6 h 12 h 24 h 48 h 72 h 120 h 168 h 240 h
Hen spray 150 g/m3 1.26 0.96 ND Dmitriyev
(1970)
Pig subcu- 25 mg/kg 6 5 3-4 2-3 1 0.1 0.01 Dedek &
taneous Schwarz
(1970)
Sheep spray 4 g/m3 2.3 0.8 0.6 trace NDa Nepoklonov&
Bukshtynov
(1971)
a ND = not detectable.
5.3 Occupational exposure
A thousand-fold dilution of 50% trichlorfon emulsifiable
concentrate was applied to apple trees by operators using a speed
sprayer or a power sprayer; the operators wore their usual working
clothes or special protective clothes, plus rubber gloves, full length
rubber boots, and masks with, or without, charcoal filters. The
plasma and red blood cell cholinesterase activity of the operators
following both kinds of spraying did not show any significant changes
compared with pre-exposure values. The calculated cumulative
trichlorfon exposures per person with a speed sprayer and a power
sprayer were 177 ± 54.0 mg and 1179 ± 398 mg, respectively (Kawai et
al., 1982).
Occupational exposures to levels exceeding 0.5 mg/m3 have been
reported (Lu et al., 1984; Hu et al., 1986).
6. KINETICS AND METABOLISM
6.1 Absorption and distribution
6.1.1 Animal
In cattle, percutaneous absorption of 32P-labelled trichlorfon
after pour-on application is extremely affected by the solvent used
(Dedek & Schwarz, 1967). With a 2% aqueous solution, only very little
trichlorfon ended up in the blood (about 0.15 mg/litre). In contrast,
trichlorfon in a 2% mineral oil solution, was absorbed rapidly,
reaching a maximum concentration in the blood of 3.1 mg/litre at 42
min. The percutaneous absorption rate was considerably slower in
sheep, than in cattle (Dedek & Schwarz, 1970). In in vitro
absorption studies on isolated cattle skin, partition of trichlorfon
was dependent on the relative solubilities in the water (blood) and
organic phases (Dedek & Schwarz, 1967).
Trichlorfon administered orally to mammals is rapidly absorbed,
degraded, and eliminated. When 32P-labelled trichlorfon was
administered orally to a cow (25 mg/kg), the radioactivity appeared in
the blood within half an hour and reached a maximum (15.1 mg/litre
trichlorfon equivalent) between 1 and 3 h. It, then decreased rapidly
(less than 1 mg/litre) within 24 h of treatment (Robbins et al.,
1956). In the liver and brain of a mouse treated orally with
32P-trichlorfon (6.2 mg/mouse), chloroform extractable radioactivity
of 188 mg/kg and 28.2 mg/kg trichlorfon equivalents, respectively, was
found, 15 min after treatment (Miyata & Saito, 1973). The
radioactivity decreased rapidly to 6.4 and 1.61 mg/kg trichlorfon
equivalents, respectively, at 4 h. The biological half-life of
trichlorfon in mice was about 80 min, when it was administered orally.
Thirty minutes after radiolabelled trichlorfon was given by
stomach tube to pregnant guinea-pigs on days 35 and 52 of gestation,
the compound had rapidly become distributed to the main organs of the
animals, the highest concentrations being present in the liver,
kidney, and lung. Thirty minutes after dosing, there was a substantial
uptake of trichlorfon into the fetus, and this became more pronounced
at the later stage of gestation (52 days), the concentration in fetal
liver equalling that in the placenta at that time (Berge & Nafstad,
1986).
6.1.2 Human
In the blood of a patient who ingested 10 g of trichlorfon, the
concentration of the insecticide was 270 µg/litre after 24 h,
following which it rapidly decreased and was undetectable after 94 h
(Fournier et al., 1978). In a 70-year-old woman who died from acute
trichlorfon poisoning, caused by the ingestion of a 50% emulsifiable
concentrate, the levels of trichlorfon in the organs (µg/g) were 310
in the blood, 487 in the liver, 465 in the brain, 416 in the kidney,
and 2240 in the urine. In addition, about 7.2 g of trichlorfon was
found in the stomach contents (Yashiki et al., 1982).
A 76-year-old male, who had attempted suicide by ingesting about
50 ml of trichlorfon, died approximately 8 h later. The trichlorfon
concentration was found to be 215 µg/g in a blood sample and 15.0 mg/g
in a gastric lavage liquid sample, both of which were collected about
1 h after intake (Yashiki et al., 1988).
Following the administration of metrifonate to humans at doses of
7.5-10 mg/kg body weight, peak levels of trichlorfon in the plasma (8
µg/ml) were reached in 2 h or less. Detectable levels were still
present in the body after 8 h (Nordgren, 1981).
Four groups of 4 healthy human volunteers each were given
metrifonate at 2.5, 5, 7.5, or 15 mg/kg (single dose). Peak plasma
levels of 5-10, 5-15, 10-25, or 15-100 µmol/litre were observed after
each of the respective doses. There was no evidence of dose- dependent
kinetics (Aden Abdi, 1990).
6.2 Biotransformation
Trichlorfon(1) rearranges readily to form dichlorvos
(2,2-dichlorovinyl dimethyl phosphate)(2) via dehydrochlorination
(Lorenz et al., 1955; Metcalf et al., 1959; Nordgren et al., 1978;
Hofer, 1981; Nordgren, 1981). This transformation occurs under
physiological conditions (Miyamoto, 1959). Dichlorvos has been found
in animal tissues in vivo at less than 5% of the administered dose
following trichlorfon treatment (Metcalf et al., 1959; Nordgren et
al., 1978; Dedek, 1981). However, it could not be detected very often
and only its degradation products, such as demethyl dichlorvos
(2,2-dichlorovinyl methyl hydrogen phosphate)(4) were found, as
evidence of the formation of dichlorvos in vivo (Arthur & Casida,
1957; Bull & Ridgway, 1969; Miyata & Saito, 1973; Otto et al., 1980).
Dichlorvos is also formed from trichlorfon in humans. Following the
administration of metrifonate, dichlorvos was found in erythrocytes
and plasma at levels corresponding to 0.2-1% of the metrifonate
concentrations (Nordgren, 1981; Aden Abdi, 1990).
In in vitro experiments, the conversion of trichlorfon into
dichlorvos was demonstrated by incubating with serum (Dedek & Schwarz,
1966), with the soluble fraction from cow and chicken liver
homogenates (Akhtar, 1982), and with the digestive juice of the
silkworm larvae (Sugiyama & Shigematsu, 1969). Demethylation also
occurred with liver homogenates. The half-life of trichlorfon in the
blood of various mammals in vitro ranged up to 30 min (Dedek &
Schwarz, 1966). Using housefly homogenate, another metabolite was
produced with the same mass spectrum as dichlorvos, but a different
Rf value on TLC; it was proposed that this was dimethyl
2,2-dichloro-1-hydroxyvinylphosphonate (Lange, 1980).
The main metabolites of 32P-trichlorfon found in mammals were
demethyl trichlorfon (Fig. 2)(6), demethyl dichlorvos(4), dimethyl
hydrogen phosphate(5), methyl hydrogen phosphate(7) and phosphoric
acid(8) (Hassan et al., 1965; Bull & Ridgway, 1969; Miyata & Saito,
1973). The percentages of the water-extractable metabolites found in
the whole body in mice, 0.5 and 4 h after oral administration of 6.2
mg 32P-trichlorfon, were respectively, demethyl trichlorfon (4.3,
4.0), demethyl dichlorvos (20.8, 9.9), dimethyl hydrogen phosphate
(34.6, 47.8), methyl hydrogen phosphate (14.3, 17.8), phosphoric acid
(22.4, 20.9), and unknown compounds (3.6, 0.0) (Miyata & Saito, 1973).
The glucuronide of trichloroethanol(9) was isolated from the
urine of a dog in amounts equivalent to 67% of the administered dose
of trichlorfon, indicating the occurrence of hydrolytic P-C bond
cleavage (Arthur & Casida, 1957). Another glucuronide containing
phosphorus and chlorine atoms in 1:2 ratio was found in the urine of
rabbits administered with trichlorfon (Miyamoto, 1961).
Thus, the main degradation routes of trichlorfon are
demethylation, P-C bond cleavage, and ester hydrolysis via dichlorvos.
Proposed metabolic pathways of trichlorfon, together with the
established metabolic pathways of dichlorvos, are illustrated in
Fig. 2.
6.3 Elimination and excretion
Trichlorfon administered to mammals is rapidly eliminated,
primarily via the urine. About 66% of the dose administered orally to
cows was eliminated in the urine within 12 h. Following oral
administration (6.2 mg/animal) of 32P-trichlorfon to mice, 70% of
the total dose was eliminated in the urine and faeces in 12 h (Miyata
& Saito, 1973). More than 80% of the eliminated compound was present
in the urine. The majority of the eliminated radioactive compounds in
both the urine and faeces were degradation products, and only a small
percentage of them were chloroform extractable. The biological
half-life of trichlorfon administered orally to mice was estimated to
be about 80 min (Robbins et al., 1956).
When methyl-14C-trichlorfon was administered intraperitoneally
to rats, 24% of the radioactive carbon was eliminated as carbon
dioxide in the expired air, within 10 h; 32% was present in the urine
as formate and dimethyl hydrogen phosphate, within 24 h (Hassan &
Zayed, 1965).
Residues of trichlorfon were detected in the milk following the
oral treatment of cows. Less than 0.2% of the total dose administered
was recovered in the milk at the end of 144 h (Robbins et al., 1956).
Single doses of 3 or 30 mg/kg body weight resulted in maximum residues
in the milk of 0.09 mg/kg in 3 h and 0.55 mg/kg in 1 h, respectively;
the residues then decreased rapidly to 0.003-0.007 mg/kg at 24 h. A
small amount of dichlorvos (0.04 mg/kg) was detected as a metabolite
Formulated trichlorfon was applied to sterilized and
non-sterilized soils with a 60% moisture content, and incubated at
ambient temper ature under natural sunlight. At concentrations of both
13 and 132 mg/kg, the levels of insecticide decreased to less than
the detection limit within 40 and 50 days, respectively, under both
sterilized and non-sterilized conditions. It appears that the
insecticide is readily subjected to abiotic degradation in soil
(Yurovskaya & Zhulinskaya, 1974).
4.3 Biodegradation
The metabolic fate of [14C]-trichlorfon labelled at the methoxy
group has been studied in culture media of nodule-forming bacteria,
such as Rhizobium leguminosarum and Rhizobium trifolii. After
incubation for 10 days at 30 °C, the unchanged parent compound
(19-25%) together with dimethyl hydrogen phosphate (18- 25%) (5) and
methyl hydrogen phosphate (0.6-1%) (7) was recovered from the media.
In addition, a trace amount of [14C] carbon dioxide was evolved
during the same period (Salama et al., 1975) (see Fig. 1).
In contrast, a demethylated derivative (6) of trichlorfon and
2,2,2- trichlorohydroxyethylphosphonic acid (3) were shown to be the
major metabolites in the culture media of Aspergillus niger,
Penicillium notatum, and Fusarium spp. (Zayed et al., 1965).
4.4 Environmental fate
The metabolism of [14C]-trichlorfon labelled at the methoxy
group has been studied in plants of the broad bean (Vicia faba) and
clover (Trifolium alexandrinum). The roots of the plants were
immersed in a phosphate buffer at pH 6 containing [14C]-trichlorfon
at a concentration of 50 mg/litre, and grown for 5 or 10 days in the
greenhouse. At harvest, the buffer solution as well as the roots of
the two plants contained unchanged parent compound and dimethyl
hydrogen phosphate (5) together with a trace amount of methyl
hydrogen phosphate (7) (Salama et al., 1975) (see Fig. 1).
Following the application of [32P]-trichlorfon to stems or
leaves, the insecticide disappeared from tomato, potato, and cotton
plants with half-lives of 20-57 h in the greenhouse, and from plums,
apples, cherries, peas, and wheat plants with half-lives of 0.5-7.5
days in the field. Characterization of metabolites was not possible
because of their volatility (Dedek et al., 1979).
When formulated trichlorfon (0.2%) was sprayed on cabbage and
onion plants at a rate of 1200 litre/ha, rapid conversion to
dichlorvos occurred. One day after treatment, the treated leaves of
cabbage and onion plants contained the highest residues of dichlorvos
(0.09-0.51 mg/kg) together with the parent compound (0.79-3.1 mg/kg).
Trichlorfon disappeared from the leaves with a half-life of less than
3 days, and dichlorvos decreased to less than the detection limit
within 15 days (Baida, 1975).
5. ENVIRONMENTAL LEVELS AND HUMAN EXPOSURE
5.1 Environmental levels
5.1.1 Air
When 2% trichlorfon was applied at a rate of 30 ml/m2, its
vapour was detected in the air. The initial concentration of 0.1
mg/m3 decreased rapidly to 0.05-0.01 mg/m3 within 2-3 days.
Trichlorfon was not detected after 30 days (Degtyareva et al., 1977).
After handspraying in a vineyard at 2 and 6 kg/ha, air concentrations
of 0.0003 and 0.001 mg/m3, respectively, were measured. Average
daily concentrations of trichlorfon and its maximum single
concentration in the Ukrainian Republic were 0.0003 and 0.0004
mg/m3, respectively (IRPTC/GKNT, 1983).
5.1.2 Water
When an 18% aqueous solution of trichlorfon was sprayed on a
forest at a rate of approximately 1 kg/ha from a helicopter, the
residues were 23.4-51.9 and 8.7-12.1 µg/litre in the creek water on
the first and second day after application, respectively (Pieper &
Richmond, 1976).
According to the monitoring programmes in Canada in 1976 and
1977, water samples in the forests where trichlorfon was sprayed were
contaminated with 0.062-1.0 µg/litre of the insecticide in most of the
year's samples in 1976, but the concentration in samples in 1977 were
considerably lower, with a maximum value of 0.058 µg/litre (Sergeant
& Zitko, 1979).
Trichlorfon was measured in soil water at 0.001 (1970), 0.02
(1971), and 0.001 mg/litre (1972) on average 1.5-2 months after
application (IRPTC/GKNT, 1983).
When trichlorfon was sprayed over a mixed boreal forest in New
Brunswick (Canada) at a rate of 1.14 kg/ha, concentrations in stream
water were approximately 95 µg/litre initially and below detection
limit (0.05 µg/litre) two weeks after treatment (Sundaram & Varty,
1989).
5.1.3 Soil
Trichlorfon was one of the chemicals found at hazardous waste
sites in the USA (Kokoszka & Flood, 1989). Levels were not specified.
After the spraying of a vineyard at 2 or 6 kg/ha, trichlorfon
levels measured in the soil were 0.24 and 0.48 mg/kg, respectively, on
the day of treatment. One day later, the levels in the soil were 0.49
and 1.03 mg/kg, and 15 days later, 0.002 and 0.02 mg/kg. The average
level of trichlorfon in the soil in the Kherson Region was 0.01 mg/kg
(1970), 0.17 mg/kg (1971) and 0.002 mg/kg (1972). After spraying a
forest at 0.8 kg/ha over a 200-ha area (160 kg total), monitoring
soil over the area gave estimates of 29.9 kg trichlorfon remaining in
the soil, 5 days after application and 0.43 and 0.25 kg after 10 and
14 days, respectively. All the trichlorfon measured was in the top 10
cm of soil. No trichlorfon was found 18 days after spraying
(IRPTC/GKNT, 1983).
After aerial spraying of trichlorfon over a mixed boreal forest
in New Brunswick (Canada) at a rate of 1.14 kg/ha, residues in soil
dissipated from 3 mg/kg initially to levels below the detection limit
(0.05 mg/kg) in about two weeks (Sundaram & Varty, 1989).
5.1.4 Residues in plants and animals
An 18% aqueous solution of trichlorfon was applied by helicopter
at 6.1 litre/ha to a forest. The residues on days 1, 2, 8, and 15
after aerial treatment were 11.2-12.6 mg/kg, 3.8-10.4 mg/kg, 0.8-1.6
mg/kg and below detection limit-0.6 mg/kg, respectively, on Douglas
fir, 68.2-81.7 mg/kg, 40.3-59.4 mg/kg, 3.9-4.5 mg/kg and below
detection limit, respectively, on willow, and 43.1-113.0 mg/kg,
4.3-30.0 mg/kg, 5.3-6.3 mg/kg, and below the detection limit-2.1
mg/kg, respectively, on grasses (detection limit: 0.1 mg/kg) (Pieper
& Richmond, 1976).
Trichlorfon was detected in all song birds caught in the area
(60.7 ha), 76 h after spraying at the rate of 1.1 kg/ha. The residues
of trichlorfon were found at 0.01 mg/kg for blue jays and crested
fly-catchers and at 0.013-0.04 mg/kg for baltimore oriols (Kurtz &
Studholme, 1974).
When trichlorfon was sprayed at a rate of 1.14 kg a.i./ha over a
mixed boreal forest in New Brunswick (Canada), the initial residues
in foliage ranged from 10.8 to 17.3 mg/kg fresh weight, but dissipated
rapidly to 2.6 to 6.5 mg/kg in three days (Sundaram & Varty, 1989).
5.2 Residues in food
5.2.1 Crops
The results of supervised trials involving foliar treatment of
various crops with trichlorfon are summarized in Table 5.
Leafy vegetables, such as lettuce, spinach, and Chinese raddish
leaves showed high residues of trichlorfon (several mg/kg or more)
shortly after application. The trichlorfon residues in Chinese raddish
leaves were about ten times higher than those in the roots. Among
fruits, strawberries, raspberries, black currants, and red currants
were found to contain higher trichlorfon residues than other fruits,
such as citrus. The results in Table 5 showed that the residues
decreased rapidly with time after application.
During the period 1987-88, 764 home-grown and imported wheat
samples were analysed for pesticide residues in the United Kingdom.
Trichlorfon was not found at, or above, the reporting limit of 0.1
mg/kg (Osborne et al., 1989).
Following normal field application (in a field trial) of
trichlorfon in Portugal to Portuguese cabbage and broccoli, residue
levels decreased to below the MRL of 0.5 mg/kg in 10 days for the
Portuguese cabbage and two weeks for the broccoli. The difference in
time is mainly ascribed to the much larger surface area exposed in the
case of the broccoli (Magalhaes et al., 1989).
The surveillance of trichlorfon residues on over twenty crops in
Hungary revealed that about 90.1% of the samples contained residues
at levels below 0.02 mg/kg (Anon. 1978a).
Trichlorfon residues on spinach and lettuce after spraying were
somewhat higher in green-house crops than in field crops in West
Germany (Stobwasser & Kirchhoff, 1968).
No trichlorfon residues were found in a survey of pesticide
residues on crops (total samples: 697) collected from a Tokyo market
from April 1984 to March 1989 (detection limit: 0.005 mg/kg) (Nagayama
et al., 1986, 1987, 1988, 1989).
Cucumber vines were sprayed to run-off with 0.05% trichlorfon
aqueous solution, and the fruits sampled over a period of time. The
half-life of trichlorfon was 1.76 days (Hameed et al., 1980).
Grape products were prepared in a laboratory from grapes
harvested 1 day after the last application of trichlorfon. Trichlorfon
residues were detected at levels of 140%, 120%, 159%, and 0.4% of the
originally applied concentration in the grape juice, raisins, wines,
and brandies, respectively, and the dichlorvos concentrations in the
wines were higher than those in the grapes from which the wines were
made (Hiramatsu & Furutani, 1978).
Residues of 0.0002-0.006 mg trichlorfon and dichlorvos/kg were
detected in 8 out of 40 flower honeys in Bulgaria (Tsvetkova et al.,
1981).
Table 5. Residues of trichlorfon in crops
Crop/ Application (spray) Residues (mg/kg) on the day of spraying Reference
Country or at intervals [days] after applicationa
Day
no. kg/ha formulationb
0 1/3 4/6 7/9 10/12 13/15 17/19 21
Cabbage
Finland ns 0.6 0.5 0.4 0.2 Anon. (1978b)
[18-21] [27-29]
Japan 6 2-5 EC-50 0.14-0.05 0.08-0.03 FAO/WHO (1979)
8 2-5 0.23-0.07 0.05-0.03
USSR 4 2.0 EC 0.79-0.27 0.17 0.09 0.07 0.007 Baida (1975)
[30]
Chinese cabbage
Japan 3 2.0 EC-50 0.13-0.04 0.09-0.02 Hiramatsu &
[18-21] Furutani (1977)
5 2.0 0.06 0.05 FAO/WHO (1979)
6 2.0 0.13 0.09
1 0.75 0.19-0.12 0.05 0.03
Lettuce
Finland 1 1.2 WP-80 34-26 5.6 0.75 0.25 0.13 0.01 Abbasov (1972)
USA 1 0.6 WP-50 1.6 0.8 0.3 0.7 0.3 0.2 FAO/WHO (1976)
1 1.2 0.8 0.8 0.6 nd 0.3
1 0.6 1.2 0.3 0.3 nd
5 1.2 0.5
6 1.2 nd
7 1.2 0.2
Table 5 (continued)
Crop/ Application (spray) Residues (mg/kg) on the day of spraying Reference
Country or at intervals [days] after applicationa
Day
no. kg/ha formulationb
0 1/3 4/6 7/9 10/12 13/15 17/19 21
Head lettuce
Finland 1 1.2 WP-50 22.8 1.5 0.31 0.04 0.01 FAO/WHO (1976)
1 1.2 31.2 1.1 0.17 0.06 nd
FRG 1 0.75 EC-50 8.1 1.8 0.54 0.41 0.20 nd nd FAO/WHO (1976)
1 0.75 15.9 4.6 0.21 0.08 0.14 0.05 nd
1 0.45 5.8 1.8 0.22 0.09 nd nd nd
1 0.45 4.8 4.3 0.27 nd nd nd nd
Leaf lettuce
Finland 1 1.2 WP-80 33.5 2.6 0.61 0.03 FAO/WHO (1976)
1 1.2 26.4 1.3 0.23 0.02
USA 2 1.2 WP-50 6.3 1.3 nd nd FAO/WHO (1976)
2 1.2 3.5 1.1 nd nd
2 1.2 3.8-0.8 0.8-0.1 0.4-nd
2 1.2 97.6 9.2 6.0 3.9
Red cabbage
Netherlands 1 1.0 WP-80 0.05-0.03 Anon. (1978c)
Savoy cabbage
Netherlands 1 1.0 WP-50 0.36-0.12 Anon. (1978c)
Parsley
Hungary 1 0.8 WP-50 0.2 0.02 0.01/0.01 FAO/WHO (1976)
Table 5 (continued)
Crop/ Application (spray) Residues (mg/kg) on the day of spraying Reference
Country or at intervals [days] after applicationa
Day
no. kg/ha formulationb
0 1/3 4/6 7/9 10/12 13/15 17/19 21
Brussel
sprouts
Netherlands 3 1.2 WP-80 0.4-0.15 0.4-0.15 0.13-0.08 Anon. (1978c)
4 1.2 0.9-0.08 0.1-0.08 0.05-0.03
Kale
Netherlands 1 1.0 WP-80 0.33-0.12 Anon. (1978c)
Kohlrabi
Netherlands 1 1.0 WP-80 0.02-0.01 Anon. (1978c)
Spinach
Netherlands 1 1.0 WP-80 5.3-3.4 FAO/WHO (1976)
1 1.0 6.2-2.1
Spinach
(greenhouse)
Netherlands 1 1.1 WP-50 33.3-23.3 5.5-2.9 3.4-1.2 1.3-0.4 0.9-0.2 0.4-0.2 0.5-0.1 FAO/WHO (1976)
1 1.1 4.5-1.9 3.1-1.2 0.8-0.4 0.5-0.25 0.3-0.2 0.1-0.06
Spinach
(under frame)
FRG 1 0.75 WP-50 4.4 2.4 1.6 FAO/WHO (1976)
1 0.75 30 21.5 5.2 1.2 2.2 0.25
Spinach
(outdoor)
Canada 2 1.1 WP 0.6 0.2 nd FAO/WHO (1976)
2 1.1 39 3.5 nd
Table 5 (continued)
Crop/ Application (spray) Residues (mg/kg) on the day of spraying Reference
Country or at intervals [days] after applicationa
Day
no. kg/ha formulationb
0 1/3 4/6 7/9 10/12 13/15 17/19 21
FRG 1 0.75 2.0 1.6 0.7 0.35 FAO/WHO (1976)
1 0.75 0.4 0.02
1 ca. 1.5 WP-80 19.8 1.8
1 0.75 WP-50 34 6.0 0.85 0.15 0.15
1 0.75 WP-50 30 1.7 0.3 0.08
1 0.75 30 13.2 5.6 1.0 1.0
Netherlands 1 0.5 WP-50 6.5-5.7 2.7-1.2 2.1-0.08 0.6-0.1 0.3-0.2 FAO/WHO (1976)
1 0.63 2.2-1.8 1.1-0.4 0.6-nd 0.2-0.1 0.2-nd
1 1.9 1.4-0.5 0.3-0.05 0.01-nd
1 0.7 1.4-0.6 0.3-0.2 0.4-nd 0.7-nd
USA 2 1.1 11.2 0.3 0.3 FAO/WHO (1976)
2 1.1 0.9 0.4
2 1.1 1.8 0.2 nd
Lima bean
USA 1 2.22 EC 0.07-0.02FAO/WHO (1976)
[44]
Chinese radish
Japan
(leaf) 5 2.0 EC-50 2.76-0.22 0.26-0.09 0.07-0.02 FAO/WHO (1979)
(root) 5 2.0 0.12-0.07 0.07-0.03 0.05-0.01
(leaf) 8 2.0 1.4-0.9 0.77-0.1 0.09-0.03
Table 5 (continued)
Crop/ Application (spray) Residues (mg/kg) on the day of spraying Reference
Country or at intervals [days] after applicationa
Day
no. kg/ha formulationb
0 1/3 4/6 7/9 10/12 13/15 17/19 21
Chinese radish
Japan
(root) 8 2.0 0.12-0.08 0.04-0.03 0.04-0.01 FAO/WHO (1979)
(leaf) 8 1.5-2.0 not stated 1.4-0.22 0.77-0.10 0.09-0.03
Onion
USSR 4 2.0 EC 2.7-3.1 0.90-1.2 0.02-0.77 nd-0.04 nd [30] Baida (1975)
Potato
Japan 6 2.0 EC-50 0.03-0.02 0.02-nd FAO/WHO (1979)
Egg plant
Japan 5 1.0 EC-50 0.03-0.02 0.01-nd nd FAO/WHO (1979)
8 1.0 0.02 0.007-nd
Green pepper
Hungary 1 1.0 WP-50 0.13 0.06 0.05 0.03 0.02 0.02 Anon. (1978a)
[16]
Tomato
(glasshouse)
Netherlands 1 1.2 WP-50 0.03-nd FAO/WHO (1976)
1 1.2 0.13-nd Abbasov (1972)
Apple
Netherlands 1 1.2 WP 0.9-0.6 Anon. (1978c)
1 1.2 1.9-0.1 Anon. (1978c)
Table 5 (continued)
Crop/ Application (spray) Residues (mg/kg) on the day of spraying Reference
Country or at intervals [days] after applicationa
Day
no. kg/ha formulationb
0 1/3 4/6 7/9 10/12 13/15 17/19 21
Grape
Japan 1-5 1.0 EC-50 1.6-0.8 1.4-0.28 0.9-0.4 0.4-0.26 0.15-0.1 Hiramatsu &
(unsacked) Furutani (1978)
(sacked) 0.1 5 0.07 0.08 0.06 0.06
Lemon
USA 1 4.5 WP-80 nd Iwata at el. (1979)
[52-59]
Orange
USA 1 4.5 WP-80 nd Iwata et el. (1979)
[52-59]
Mandarin
orange
Japan 5 3.0 EC-50 nd FAO/WHO (1979)
[27-29]
Kski
persimmon
Japan 3 3g/tree EC-50 0.57-0.53 0.36 0.3-0.25 FAO/WHO (1979)
Raspberry
Finland 2 0.8g/ not stated 5.6 Anon. (1978b)
linear
0.5-0.6g/ 2.9-0.7 Anon. (1978b)
plant [18-21]
Table 5 (continued)
Crop/ Application (spray) Residues (mg/kg) on the day of spraying Reference
Country or at intervals [days] after applicationa
Day
no. kg/ha formulationb
0 1/3 4/6 7/9 10/12 13/15 17/19 21
Strawberry
Japan 3 2.0 EC-50 1.4 0.31 FAO/WHO (1979)
[18-21]
5 2.0 4.6 0.75
3 3.0 2.12 0.46
[18-21]
5 3.0 3.31 1.06
1 1.6-2.4 0.02
[18-21]
3 2-3 not stated 2.1-1.4 0.46-0.33
[20-21]
5 2-3 not stated 4.6-3.3 1.1-0.74
Water melon
not stated 6 2.0 EC-50 0.008 FAO/WHO (1979)
Black currant
Finland 1.3g/ not stated 2.6 0.2 Anon.(1978b)
plant [27-29]
Red current
Finland 1.6g/ not stated 25 12 Anon.(1978b)
plant [27-29]
Rye-grass
USA 1 1.1 5% bait 37.5 3.1 1.9 0.1 van Middalem et
al. (1972)
Table 5 (continued)
Crop/ Application (spray) Residues (mg/kg) on the day of spraying Reference
Country or at intervals [days] after applicationa
Day
no. kg/ha formulationb
0 1/3 4/6 7/9 10/12 13/15 17/19 21
Cotton
USA 4 3.7-1.4 WP-50 79 4 Isaac at al. (1965)
Sugar beet
Japan 6 1.5 EC-50 0.05-0.02 0.04-0.02 FAO/WHO (1979)
8 1.5 0.04-0.02 0.024-0.020
Finland 2 0.64 not stated 0.05 Anon. (1978b)
[48]
6 1.5 not stated 0.24-0.06 0.07-0.02
8 1.5 not stated 0.26-0.05 0.08-0.02
a nd = not detectable;
ns = not stated.
b WP = Wettable powder;
EC = Emulsifiable concentrate.
5.2.2 Milk
The results of supervised trials in which cows were treated with
trichlorfon via several routes of exposure and the residues in the
milk measured are summerized in Table 6.
Trichlorfon residues in cow's milk were mainly studied in animals
that were administered the pesticide orally. The reports showed
relatively high levels and a gradual disappearance of trichlorfon in
the milk of treated cows. The residue values in FAO/WHO report (1979)
were much higher than those in others (Table 6).
To control botflies, a 11.2% aqueous solution of trichlorfon was
applied dermally to lactating cows. Maximum residues of trichlorfon
and dichlorvos were found in the first milking (0.2 and 0.03 mg/litre,
respectively) and were still detectable in the third milking. Storage
and short-term heating of the milk did not essentially degrade the
insecticide, but, with boiling, an accelerated transformation of
trichlorfon to dichlorvos took place (Fechner et al., 1968).
Treatment of cows with 0.25 or 0.5% aqueous solution of
trichlorfon resulted in residues in milk of 0.02 and 0.7 mg/litre,
respectively, within the first 72 h following treatment. The levels of
trichlorfon were higher in the morning than in the evening flow
(IRPTC/GKNT, 1983).
The trichlorfon residues in cow's milk were in direct proportion
to the veterinary use of the insecticide on the cows. Heat processing
of milk had little effect on trichlorfon residues. However,
evaporation or spray drying of the milk reduced the residue levels
considerably (Konrad et al., 1975).
5.2.3 Meat
Trichlorfon residues in pigs and sheep treated with the chemical
under supervised trial conditions are shown in Table 7. The results
revealed that trichlorfon residues in pork rapidly disappear; they
were below the detection limit (0.01 mg/kg) 24 h after subcutaneous
application of 25 mg/kg body weight. The residues following spray
treatment of sheep against harmful insects decreased to below the
detection limit (0.01 mg/kg) after 168 h (Dedek & Schwarz, 1970a).
32P-labelled trichlorfon was poured evenly on to 600 cm2
areas of the freshly shorn backs of sheep at a rate of 20 mg/kg. Only
a minimal concentration (0.1 mg/kg) of the insecticide was detected in
the blood of the sheep. However, trichlorfon levels in the blood of
cattle were higher than those in the blood of sheep at a similar dose.
When special solvents were used for the preparation of the trichlor
fon solution and the dose was increased to 50 mg/kg, the level of
residues in the blood of the sheep increased to 1.2 mg/litre (Dedek
& Schwarz, 1970b).
Table 6. Residues of trichlorfon in milk of cows after application
Method of Residues (mg/kg) in milk after application Reference
application
mg/kg 1 h 2 h 3 h 4 h 6 h 8 h 9 h 10 h 12 h 20 h 24 h 32-72 h 96 h 168 h
Dermal 80 0.1-0.25 0.05-0.25 0.05 Mollhoff
(1971)
Dermal 36 0.1-0.2 0.05 Mollhoff
(1971)
Dermal 100 0.1-0.2 0.05-0.1 0.01 Mollhoff
(1971)
Dermal 100 0.05-0.1 0.05 Mollhoff
(1971)
Oral 3 0.033 0.091 0.080 0.052 0.003 0.001 Nakahara
et al.
(1972)
Oral 30 0.56 0.33 0.25 0.16 0.007 0.001 Nakahara
et al.
(1972)
Oral 1a 0.034 0.029 0.021 0.009 0.001 Nakahara
et al.
(1972)
Intramuscularb 25 2.4 1.3 0.7 0.5 0.25 0.1 Dedek &
Schwarz
(1966)
Table 6. (continued)
Method of Residues (mg/kg) in milk after application Reference
application
mg/kg 1 h 2 h 3 h 4 h 6 h 8 h 9 h 10 h 12 h 20 h 24 h 32-72 h 96 h 168 h
Pour-ond 20 1.2 0.5 0.3 0.2 0.1 0.05 FAO/WHOc
(1979)
Pour-one 20 0.2 0.3 0.35 0.35 0.2 0.1 FAO/WHOc
(1979)
Pour-onf 30 0.45 0.05 0.01 Dedek &
Schwarz
(1966)
a 1 mg trichlorfon/kg body weight for 5 days.
b 50% trichlorfon in polyethylene glycol.
c Data cited to fit in this table.
d 2% trichlorfon in mineral oil.
e 2% trichlorfon in vegetable oil.
f 5.7% in aqueous solution.
Six USSR reports were available concerning supervised trials on
pigs (7 mg/kg in meat and 12 mg/kg in fat after unspecified treatment;
Yonova & Zhecheva, 1974), and sheep (Nepoklonov & Bukshtynov, 1971).
According to the English summaries of the reports, the trichlorfon was
rapidly absorbed and distributed among various organs and tissues,
then metabolized and eliminated.
5.2.4 Poultry and eggs
The trichlorfon contents of the organs of hens treated externally
with 1-8% aqueous solutions were 0.03-1.5 mg/kg, 0.01-0.7 mg/kg,
0.04-1.0 mg/kg, 0.02-0.8 mg/kg, 0.01-1.5 mg/kg or 0.02-0.9 mg/kg in
the muscle, liver, lung, heart, kidney and brain, respectively, within
the first 5 days after application. The eggs from hens treated
externally with 6-8% trichlorfon contained trichlorfon levels of
0.01-0.05 mg/kg (IRPTC/GKNT, 1983).
Residues of trichlorfon one day after the spraying of chickens at
the rate of 150 mg/kg body weight were as follows (mg/kg): egg shell,
0.48; egg white, 0.27; egg yolk, not detectable. Trichlorfon was
preserved in chicken carcasses kept for six months at -10 °C, but it
quickly decomposed when the carcasses were boiled (Dmitriyev, 1970).
5.2.5 Fish
Trichlorfon residues in eels were determined 1 and 5 days after
ponds were treated with a 1 mg/litre aqueous solution of the
insecticide. The results showed that the insecticide decomposed in a
short time to form dichlorvos in neutral and weakly alkaline water. In
pond water with a pH of 8-10, less than 10% of the applied
trichlorfon was degraded after 30 min and dichlorvos was detected. The
residues of trichlorfon and dichlorvos in the eels were 0.009-0.032
mg/kg and <0.005-0.02 mg/kg, respectively, on the first day
following treatment, and 0.011 mg/kg and 0.009-0.032 mg/kg,
respectively, on the 4th day after treatment. There was a good
correlation between the residual amounts of trichlorfon in the eels
and the concentrations in the water. Residues of trichlorfon and
dichlorvos, which were detected on the skin of eels in water at pH
7.0, could be removed by rinsing. Insecticide residues were found in
the internal organs of only one out of 7 eels caught in the field
pond. Carps exposed to an aqueous solution of trichlorfon at 0.25
mg/kg were examined on the 2nd, 4th, and 9th days after exposure.
Residues of trichlorfon and dichlorvos could not be detected in the
fish on the second day (Nakahara et al., 1973).
Table 7. Residues of trichlorfon in various meats after experimental application
Animal Method of mg/kg or Residues (mg/kg) after application Reference
application g/m3
1 h 2 h 3 h 4 h 6 h 12 h 24 h 48 h 72 h 120 h 168 h 240 h
Hen spray 150 g/m3 1.26 0.96 ND Dmitriyev
(1970)
Pig subcu- 25 mg/kg 6 5 3-4 2-3 1 0.1 0.01 Dedek &
taneous Schwarz
(1970)
Sheep spray 4 g/m3 2.3 0.8 0.6 trace NDa Nepoklonov&
Bukshtynov
(1971)
a ND = not detectable.
5.3 Occupational exposure
A thousand-fold dilution of 50% trichlorfon emulsifiable
concentrate was applied to apple trees by operators using a speed
sprayer or a power sprayer; the operators wore their usual working
clothes or special protective clothes, plus rubber gloves, full length
rubber boots, and masks with, or without, charcoal filters. The
plasma and red blood cell cholinesterase activity of the operators
following both kinds of spraying did not show any significant changes
compared with pre-exposure values. The calculated cumulative
trichlorfon exposures per person with a speed sprayer and a power
sprayer were 177 ± 54.0 mg and 1179 ± 398 mg, respectively (Kawai et
al., 1982).
Occupational exposures to levels exceeding 0.5 mg/m3 have been
reported (Lu et al., 1984; Hu et al., 1986).
6. KINETICS AND METABOLISM
6.1 Absorption and distribution
6.1.1 Animal
In cattle, percutaneous absorption of 32P-labelled trichlorfon
after pour-on application is extremely affected by the solvent used
(Dedek & Schwarz, 1967). With a 2% aqueous solution, only very little
trichlorfon ended up in the blood (about 0.15 mg/litre). In contrast,
trichlorfon in a 2% mineral oil solution, was absorbed rapidly,
reaching a maximum concentration in the blood of 3.1 mg/litre at 42
min. The percutaneous absorption rate was considerably slower in
sheep, than in cattle (Dedek & Schwarz, 1970). In in vitro
absorption studies on isolated cattle skin, partition of trichlorfon
was dependent on the relative solubilities in the water (blood) and
organic phases (Dedek & Schwarz, 1967).
Trichlorfon administered orally to mammals is rapidly absorbed,
degraded, and eliminated. When 32P-labelled trichlorfon was
administered orally to a cow (25 mg/kg), the radioactivity appeared in
the blood within half an hour and reached a maximum (15.1 mg/litre
trichlorfon equivalent) between 1 and 3 h. It, then decreased rapidly
(less than 1 mg/litre) within 24 h of treatment (Robbins et al.,
1956). In the liver and brain of a mouse treated orally with
32P-trichlorfon (6.2 mg/mouse), chloroform extractable radioactivity
of 188 mg/kg and 28.2 mg/kg trichlorfon equivalents, respectively, was
found, 15 min after treatment (Miyata & Saito, 1973). The
radioactivity decreased rapidly to 6.4 and 1.61 mg/kg trichlorfon
equivalents, respectively, at 4 h. The biological half-life of
trichlorfon in mice was about 80 min, when it was administered orally.
Thirty minutes after radiolabelled trichlorfon was given by
stomach tube to pregnant guinea-pigs on days 35 and 52 of gestation,
the compound had rapidly become distributed to the main organs of the
animals, the highest concentrations being present in the liver,
kidney, and lung. Thirty minutes after dosing, there was a substantial
uptake of trichlorfon into the fetus, and this became more pronounced
at the later stage of gestation (52 days), the concentration in fetal
liver equalling that in the placenta at that time (Berge & Nafstad,
1986).
6.1.2 Human
In the blood of a patient who ingested 10 g of trichlorfon, the
concentration of the insecticide was 270 µg/litre after 24 h,
following which it rapidly decreased and was undetectable after 94 h
(Fournier et al., 1978). In a 70-year-old woman who died from acute
trichlorfon poisoning, caused by the ingestion of a 50% emulsifiable
concentrate, the levels of trichlorfon in the organs (µg/g) were 310
in the blood, 487 in the liver, 465 in the brain, 416 in the kidney,
and 2240 in the urine. In addition, about 7.2 g of trichlorfon was
found in the stomach contents (Yashiki et al., 1982).
A 76-year-old male, who had attempted suicide by ingesting about
50 ml of trichlorfon, died approximately 8 h later. The trichlorfon
concentration was found to be 215 µg/g in a blood sample and 15.0 mg/g
in a gastric lavage liquid sample, both of which were collected about
1 h after intake (Yashiki et al., 1988).
Following the administration of metrifonate to humans at doses of
7.5-10 mg/kg body weight, peak levels of trichlorfon in the plasma (8
µg/ml) were reached in 2 h or less. Detectable levels were still
present in the body after 8 h (Nordgren, 1981).
Four groups of 4 healthy human volunteers each were given
metrifonate at 2.5, 5, 7.5, or 15 mg/kg (single dose). Peak plasma
levels of 5-10, 5-15, 10-25, or 15-100 µmol/litre were observed after
each of the respective doses. There was no evidence of dose- dependent
kinetics (Aden Abdi, 1990).
6.2 Biotransformation
Trichlorfon(1) rearranges readily to form dichlorvos
(2,2-dichlorovinyl dimethyl phosphate)(2) via dehydrochlorination
(Lorenz et al., 1955; Metcalf et al., 1959; Nordgren et al., 1978;
Hofer, 1981; Nordgren, 1981). This transformation occurs under
physiological conditions (Miyamoto, 1959). Dichlorvos has been found
in animal tissues in vivo at less than 5% of the administered dose
following trichlorfon treatment (Metcalf et al., 1959; Nordgren et
al., 1978; Dedek, 1981). However, it could not be detected very often
and only its degradation products, such as demethyl dichlorvos
(2,2-dichlorovinyl methyl hydrogen phosphate)(4) were found, as
evidence of the formation of dichlorvos in vivo (Arthur & Casida,
1957; Bull & Ridgway, 1969; Miyata & Saito, 1973; Otto et al., 1980).
Dichlorvos is also formed from trichlorfon in humans. Following the
administration of metrifonate, dichlorvos was found in erythrocytes
and plasma at levels corresponding to 0.2-1% of the metrifonate
concentrations (Nordgren, 1981; Aden Abdi, 1990).
In in vitro experiments, the conversion of trichlorfon into
dichlorvos was demonstrated by incubating with serum (Dedek & Schwarz,
1966), with the soluble fraction from cow and chicken liver
homogenates (Akhtar, 1982), and with the digestive juice of the
silkworm larvae (Sugiyama & Shigematsu, 1969). Demethylation also
occurred with liver homogenates. The half-life of trichlorfon in the
blood of various mammals in vitro ranged up to 30 min (Dedek &
Schwarz, 1966). Using housefly homogenate, another metabolite was
produced with the same mass spectrum as dichlorvos, but a different
Rf value on TLC; it was proposed that this was dimethyl
2,2-dichloro-1-hydroxyvinylphosphonate (Lange, 1980).
The main metabolites of 32P-trichlorfon found in mammals were
demethyl trichlorfon (Fig. 2)(6), demethyl dichlorvos(4), dimethyl
hydrogen phosphate(5), methyl hydrogen phosphate(7) and phosphoric
acid(8) (Hassan et al., 1965; Bull & Ridgway, 1969; Miyata & Saito,
1973). The percentages of the water-extractable metabolites found in
the whole body in mice, 0.5 and 4 h after oral administration of 6.2
mg 32P-trichlorfon, were respectively, demethyl trichlorfon (4.3,
4.0), demethyl dichlorvos (20.8, 9.9), dimethyl hydrogen phosphate
(34.6, 47.8), methyl hydrogen phosphate (14.3, 17.8), phosphoric acid
(22.4, 20.9), and unknown compounds (3.6, 0.0) (Miyata & Saito, 1973).
The glucuronide of trichloroethanol(9) was isolated from the
urine of a dog in amounts equivalent to 67% of the administered dose
of trichlorfon, indicating the occurrence of hydrolytic P-C bond
cleavage (Arthur & Casida, 1957). Another glucuronide containing
phosphorus and chlorine atoms in 1:2 ratio was found in the urine of
rabbits administered with trichlorfon (Miyamoto, 1961).
Thus, the main degradation routes of trichlorfon are
demethylation, P-C bond cleavage, and ester hydrolysis via dichlorvos.
Proposed metabolic pathways of trichlorfon, together with the
established metabolic pathways of dichlorvos, are illustrated in
Fig. 2.
6.3 Elimination and excretion
Trichlorfon administered to mammals is rapidly eliminated,
primarily via the urine. About 66% of the dose administered orally to
cows was eliminated in the urine within 12 h. Following oral
administration (6.2 mg/animal) of 32P-trichlorfon to mice, 70% of
the total dose was eliminated in the urine and faeces in 12 h (Miyata
& Saito, 1973). More than 80% of the eliminated compound was present
in the urine. The majority of the eliminated radioactive compounds in
both the urine and faeces were degradation products, and only a small
percentage of them were chloroform extractable. The biological
half-life of trichlorfon administered orally to mice was estimated to
be about 80 min (Robbins et al., 1956).
When methyl-14C-trichlorfon was administered intraperitoneally
to rats, 24% of the radioactive carbon was eliminated as carbon
dioxide in the expired air, within 10 h; 32% was present in the urine
as formate and dimethyl hydrogen phosphate, within 24 h (Hassan &
Zayed, 1965).
Residues of trichlorfon were detected in the milk following the
oral treatment of cows. Less than 0.2% of the total dose administered
was recovered in the milk at the end of 144 h (Robbins et al., 1956).
Single doses of 3 or 30 mg/kg body weight resulted in maximum residues
in the milk of 0.09 mg/kg in 3 h and 0.55 mg/kg in 1 h, respectively;
the residues then decreased rapidly to 0.003-0.007 mg/kg at 24 h. A
small amount of dichlorvos (0.04 mg/kg) was detected as a metabolite
 in the milk, 1-3 h after the higher dose (Nakahara et al., 1972). When
lactating cows were treated dermally by washing with 11.2%
trichlorfon, 6-8 h before milking, trichlorfon residues (equal to or
more than 0.2 mg/kg) and dichlorvos residues (0.03 mg/kg) were found
in the first milking (Fechner et al., 1968). Trichlorfon was detected
up to the third milking, 32 h after application, but neither
trichlorfon nor dichlorvos could be demonstrated in the fourth
milking.
6.4 Reaction with body components
6.4.1 In vitro studies
Several investigators have reported the considerable inhibitory
activity of trichlorfon on acetylcholinesterase in vitro. The
pI50 for acetylcholinesterase was 5.5 and the bimolecular rate
constant for rat brain acetylcholinesterase was 3.18 × 104/min
(Arthur & Casida, 1957; Buchet & Lauwerys, 1970). The activity is,
however, strongly pH-dependent and is about 30-fold more active at pH
7.4-7.6 than at pH 6.0-6.5. At the lower pH range, trichlorfon is more
stable with practically no inhibiting activity against
cholinesterases. In contrast, dichlorvos is equally active at this pH
range (Metcalf et al., 1959; Miyamoto 1959; Reiner et al., 1975).
However, the rates of non-enzymatic reactivation of the enzymes after
inhibition by trichlorfon and dichlorvos are similar. Thus, it is now
believed that the in vitro inhibitory activity of trichlorfon is due
to its rapid, spontaneous, non-enzymatic conversion into dichlorvos.
Frohlich et al. (1990) determined the competitive and
uncompetitive constants for the action of trichlorfon on bee
p-nitrophenyl acetate hydrolysing esterase to be 4.5 × 10-6
mol/litre and 1.6 × 10-5 mol/litre, respectively.
The inhibition constant for trichlorfon and chicken liver fluora
cetanilidase was determined to be 2.5 × 10-6 mol/litre (Nakamura &
Ueda, 1967).
6.4.2 In vivo studies
In rats given trichlorfon intraperitoneally at 150 mg/kg,
considerable increases in the activities of superoxide dismutase
(× 1.94) and microsomal cytochrome P-450 (× 2.09), and in lipid
peroxidation (× 1.44) in the liver were observed, 2.5 h after
treatment (Matkovics et al., 1980).
In in vivo studies on mice receiving a diet containing 100 mg
trichlorfon/kg, the cholinesterase activities in the brain,
erythrocytes, and plasma were, respectively, 72.1, 115.7, and 92.7% of
control values after one-day (24 h) and 79.9, 83.8, and 81.0%,
respectively, after 20 days (Tsumuki et al., 1970).
A mixture of trichlorfon and phenothiazine, which was
administered to horses with feed as an antihelminthic at a single dose
level of 35.8 mg/kg, reduced the cholinesterase activity in whole
blood and plasma to 32 and 20%, respectively, by 24 h. The activity
increased to about 80% after 4 weeks (Bello et al., 1974).
In humans who received an oral dose of 7.5 mg trichlorfon/kg on
two successive days, the maximum inhibition of erythrocyte and plasma
cholinesterase was 52 and 94%, respectively. The red cell
cholinesterase activity recovered very slowly to reach only 66% of the
pretreatment level, 28 days after treatment, while that of the plasma
cholinesterase recovered rapidly to reach 78% at 22 days (Lebrun &
Cerf, 1960).
When 14CH3-labelled trichlorfon was adminstered intravenously
to rats at 40 mg/kg, the radioactive carbon (14C) was found mainly
in the liver (25 mg/kg) and kidneys (11.6 mg/kg), 3 h after treatment
(Dedek & Lohs, 1970). Similar results were obtained with intra
peritoneal treatment. Only about 1% of the radioactivity found in the
organs was extractable with acetone, indicating that most of the 14C
in the tissues was bound to body components. The bound 14C in the
organs investigated was less than 10% of the total dose. It
disappeared rather rapidly and the bound radioactivity decreased to
about one-tenth by 17 h. In another study, about 10% of the phosphorus
portion, administered orally to mice as 32P-trichlorfon, was found
bound to tissue (Miyata & Saito, 1973).
7. EFFECTS ON ORGANISMS IN THE ENVIRONMENT
Acute toxicity data for trichlorfon on aquatic and terrestrial
non-target organisms are summarized in Tables 8 and 9, respectively.
7.1 Microorganisms
When trichlorfon was applied to a cotton field at 0.5 g/m2, the
total count of soil fungi and the counts of some fungal species, such
as Aspergillus niger, Fusarium oxysporum, and A. fumigatus were
elevated 3 days after treatment. A depressed effect was observed in
Myrothecium verrucaria. After 40 days, trichlorfon did not have any
significant effects on the total count of soil fungi (Abdel-Kader et
al., 1978). Trichlorfon at 8-16 mg/kg in agar affected the mycelial
growth of 4 fungi (A. fumigatus, F. moniliforme, Penicillium
italicum, and Sclerotium cepivorum), even 10 days after application
(El-Hissy & Abdel-Kader, 1980).
In field pond trials, Grygierek & Wasilewska (1981) found that
trichlorfon applied at 1 mg/litre reduced zooplankton numbers within
the first 24 h of application and bottom fauna after 1 week. Rotifers
( Keratella sp.) and cladocerans ( Bosmina) were especially
sensitive to the chemical. The renewal of the affected fauna
communities took about 1 month. When the pond was treated with
trichlorfon at 300-800 µg/litre, the numbers of cladocerans and
copepods decreased, whereas rotifers and phytoplankton increased
markedly in number. However, the total biomass was not affected (Grahl
et al., 1981).
Cain & Cain (1984) incubated the green alga Chlamydomonas
moewusii in a medium containing trichlorfon at concentrations up to
80 µmol/litre (21 mg/litre). At the highest concentration, growth of
the algae was 76% of control levels. Zygospore germination of
Chlamydomonas was unaffected at 80 µmol/litre (21 mg/litre) (108% of
controls).
7.2 Invertebrates
Trichlorfon is highly toxic for aquatic arthropods with LC50
values ranging from 0.75 to 7800 µg/litre in 48-/96-h tests (Table 8).
The toxicity of trichlorfon for amphipods increased 2-fold when
the pH of the media was increased from 7.5 to 8.5; similarly the
toxicity for stonefly naiads increased about 20-fold when the pH was
increased from 6.5 to 8.5 (Woodward & Mauck, 1980). Trichlorfon is
less toxic for mollusca, the 48-h LC50 values ranging up to 25.4
mg/litre. Prolonged exposure (10 days) resulted in a 10-40 times
increase in toxicity (Singh & Agarwal, 1981).
Table 8. Acute toxicity of trichlorfon for non-target aquatic organisms
Species Size Toxicity (mg/litre) Formulationa Systemb Temperature pH Hardnessc Reference
(°C)
Protozoa
Colpidium MAD 10 20 Dive
campylum et al.
(1980)
Fish
Rainbow trout 96-h LC50 4.85 T S 12 Marking
(Salmo & Mauck
gairdneri) (1975)
Cutthroat trout 96-h LC50 1.68 T S 12 7.5 40 Woodward
(Salmo clarki) 96-h LC50 3.25 T S 12 7.5 40 & Mauck
96-h LC50 5.75 T S 7 7.5 40 (1980)
96-h LC50 4.75 T S 12 6.5 40
96-h LC50 0.375 T S 12 8.5 40
96-h LC50 0.620 T S 12 7.8 320
Cherry salmon 2.8 g 12-h LC50 10.2 50%EC SS 17.6 Kimura
(Onchorhyncus 2.8 g 24-h LC50 1.1 50%EC SS 17.6 et el.
masou) 2.8 g 48-h LC50 1.1 50%EC SS 17.6 (1971)
2.8 g 96-h LC50 1.1 50%EC SS 17.6
Carp egg 24-h LC50 15 EC S 25 6.9-7.2 Hashimoto
(Cyprinus 0.60 cm 24-h LC50 11 EC S 25 6.9-7.2 et al.
carpio) 0.73 cm 24-h LC50 8.8 EC S 25 6.9-7.2 (1982)
1.16 cm, 0.013 g 24-h LC50 14 EC S 25 6.9-7.2
1.52 cm, 0.038 g 24-h LC50 15 EC S 25 6.9-7.2
2.51 cm, 0.23 g 24-h LC50 14 EC S 25 6.9-7.2
3.86 cm, 0.87 g 24-h LC50 15 EC S 25 6.9-7.2
5.22 cm, 2.34 g 24-h LC50 15 EC S 25 6.9-7.2
Table 8. (continued)
Species Size Toxicity (mg/litre) Formulationa Systemb Temperature pH Hardnessc Reference
(°C)
Carp 48-h LC50 1200 4%D S 25 Nishiuchi
(Cyprinus 48-h LC50 30 50%EC S 25 (1979a)
carpio) 48-h LC50 50 80%WP S 25
Carp 5.3 cm, 2.2 g 24-h LC50 40 S 15 Nishiuchi
(Cyprinus 5.3 cm, 2.2 g 24-h LC50 40 S 20 (1977)
carpio) 5.3 cm, 2.2 g 24-h LC50 28 S 25
5.3 cm, 2.2 g 24-h LC50 25 S 30
5.3 cm, 2.2 g 24-h LC50 25 S 35
Fathead minnow 96-h LC50 51 Livingston
(Pimephales (1977)
promelas)
American eel black stage 96-h LC50 1.32 S 22 7.2-7.6 40-48 Spehar et
(Anguilla glass stage 96-h LC50 1.31 S 22 7.2.-7.6 40-48 al. (1981)
rostrata) yellow phase 96-h LC50 8.57 S 22 7.2.-7.6 40-48 Hinton &
Eversole
(1980)
Striped bass 6 cm, 2.7 g 24-h LC50 10.4 50%SP S 21 8.2 35 Wellborn
(Roccus 6 cm, 2.7 g 48-h LC50 9.2 50%SP S 21 8.2 35 Jr. (1969)
saxatilis) 6 cm, 2.7 g 96-h LC50 5.2 50%SP S 21 8.2 35
Bluegill 0.87-2.4 g 24-h LC50 41.0a 50%SP S 18 7 51.3 McCann &
(Lepomis Jasper
macrochirus) (1972)
Arthropods
Water flea 3-h LC50 0.18 4%P S 25 Nishiuchi
(Daphnia 3-h LC50 0.030 50%EC S 25 (1979b)
pulex) 3-h LC50 0.019 80%WP S 25
Table 8. (continued)
Species Size Toxicity (mg/litre) Formulationa Systemb Temperature pH Hardnessc Reference
(°C)
Water flea 1-h LC50 0.088 EC S 25 Nishiuchi
(Daphnia 3-h LC50 0.014 EC S 25 (1979c)
carinata) 6-h LC50 0.0064 EC S 25
24-h LC50 0.0012 EC S 25
48-h LC50 0.00075 EC S 25
Amphipod 96-h LC50 0.108 T S 12 7.5 40 Woodward
(Garnmarus 96-h LC50 0.052 T S 12 8.5 40 & Mauck
pseudolimnaeus) (1980)
Mayfly 9.3 mm, 5.6 mg 3-h LC50 1.8 EC S 25 Nishiuchi
(Cloeon 9.3 mm, 5.6 mg 6-h LC50 0.75 EC S 25 & Asano
diptrum) 9.3 mm, 5.6 mg 24-h LC50 0.075 EC S 25 (1979)
9.3 mm, 5.6 mg 48-h LC50 0.056 EC S 25
Dragon fly 2.3 cm, 0.62 g 48-h LC50 0.042 EC 25 Nishiuchi
(Orthetrum (1981)
albistylum
speciosum)
(Sympetrum 2.1 cm, 0.56 g 48-h LC50 0.15 EC 25 Nishiuchi
frequens) (1981)
(Sigara 5.9 mm, 6.1 mg 48-h LC50 0.15 EC 25 Nishiuchi
substriata) (1981)
(Micronecta 3.2 mm, 1.8 mg 48-h LC50 0.075 EC 25 Nishiuchi
sedula) (1981)
Stonefly 96-h LC50 0.100 T S 12 8.5 40 Woodward
(Pteronarcella 96-h LC50 0.0098 T S 12 7.5 40 & Mauck
badia) 96-h LC50 0.0053 T S 12 6.5 40 (1980)
Table 8. (continued)
Species Size Toxicity (mg/litre) Formulationa Systemb Temperature pH Hardnessc Reference
(°C)
(Eretes 1.5 cm, 0.20 g 48-h LC50 0.32 EC 25 Nishiuchi
sticticus) (1981)
Crayfish adults, 15-38 g 96-h LC50 7.8 T S 19 7.8 35 Andreu-Moliner
(Procambarus (1986)
clarkii)
Mollusca
Snail 2.9 cm, 1.6 g 48-h LC50 1.8 EC 22 Nishiuchi
(Semisulcospira & Yoshida
libertina) (1972)
Snail 2.4 cm, 3.3 g 48-h LC50 4.8 EC 22 Nishiuchi
(Cipangopaludina & Yoshida
malleata) (1972)
Red snail 0.72 cm. 1.1 g 48-h LC50 1.8 EC 22 Nishiuchi &
(Indoplanorbis Yoshida
exustus) (1972)
Physa-snail 0.91 cm, 0.11 g 48-h LC50 3.2 EC 22 Nishiuchi
(Physa acuta) & Yoshida
(1972)
10-day LC50 0.05 15-20 7.3 Mandoul et
al. (1967)
Snail 1.2 cm 48-h LC50 2.2 Singh &
(Lymnaea 1.2 cm 72-h LC50 0.65 Agarwal
acuminata) 1.2 cm 96-h LC50 0.3 (1981)
1.2 cm 168-h LC50 0.22
1.2 cm 240-h LC50 0.058
Table 8. (continued)
Species Size Toxicity (mg/litre) Formulationa Systemb Temperature pH Hardnessc Reference
(°C)
(Pila globosa) 3.5 cm 48-h LC50 25.4 Singh &
3.5 cm 72-h LC50 19.0 Agarwal
3.5 cm 96-h LC50 8.0 (1981)
3.5 cm 168-h LC50 2.8
3.5 cm 240-h LC50 2.2
(Gryphaea 10-day LC50 2.45 9-19 SW Mandoul et
angulata) al. (1967)
a T = Technical.
EC = Emulsifiable concentrate.
D = Dust.
WP = Wettable powder.
SP = Soluble powder.
P = Powder.
b S = Static.
SS = Semi-static.
SW = Sea water.
MAD = Minimal active dose.
c Hardness (mg/litre as CaCO3).
Trichlorfon at concentrations of up to 30 mg/litre did not affect
byssal attachment in seed mussels ( Mytilus edulis) (Roberts, 1975).
Trichlorfon showed cholinomimetic properties on the excitor or
inhibitor receptors of acetylcholine in the isolated heart, median
dorsal radula protractor muscle, and rectum of the snail Pila globosa
(Singh & Agarwal, 1979).
When freshwater snail, Lymnaea acuminata, was exposed to 10 or
20 mg trichlorfon/litre for 48 h, the rate of oxygen consumption and
the concentration of glycogen were both reduced, while the levels of
lactic acid and reducing sugars were enhanced. The effects persisted
for 7 days after withdrawal of the trichlorfon. On the basis of these
observations, Mahendru & Agarwal (1981) concluded that trichlorfon may
affect not only cholinesterase activity but also other enzyme systems,
such as the ones involved in carbohydrate metabolism.
7.3 Aquatic vertebrates
Trichlorfon is moderately toxic for fish, the 96-h LC50 values
ranging from 0.4 to 51 mg/litre (Table 8). The effects of trichlorfon
on the susceptibility of the developmental stages of carp were not
remarkable (Hashimoto et al., 1982). However, with regard to water
quality, the toxicity for fish increased as the water temperature, pH,
and hardness increased. The increase in toxicity was affected least
(3.4 fold) by increasing the temperature from 7 to 12 °C and most
(13-fold) by changing the pH from 6.5 to 8.5 (Woodward & Mauck, 1980).
Cherry salmon fingerlings (Onchorhyncus masou) were exposed to
one-tenth (0.105 mg/litre) and one-third (0.310 mg/litre) of the 96-h
LC50 value (1.1 mg/litre) of trichlorfon for 6 weeks in a
flow-through system. Trichlorfon initially retarded the growth of the
fish at both concentrations, but then the fish grew normally, and the
condition factors (weight-length coefficent) of the test fish did not
differ significantly from those of the controls after 6 weeks of
exposure to trichlorfon. Histopathological examination showed that the
liver cells were swollen with an obscure cell contour at the beginning
of the study, but no compound-induced histopathological changes were
observed after 6 weeks of exposure (Kimura et al., 1971).
When bluegill fingerlings were exposed to formulations of
trichlorfon, haemorrhage along, and fractures of, the caudal vertebrae
(usually extended over three vertebrae) occurred 4-8 h after treatment
at concentrations of 8-52 mg/litre (McCann & Jasper, 1972).
Matton & Laham (1969) treated 1-inch rainbow trout larvae for 16
h with 10-100 mg trichlorfon/litre, or for 40 h with 5 mg/litre.
Histochemical examination revealed that the acetylcholinesterase
activity was inhibited in the septa of the myotomes and at the
myoneural junctions. Furthermore, pathological changes were observed
in the heart, liver, blood cells, pseudogills, and muscular tissues.
7.4 Terrestrial vertebrates
Trichlorfon is relatively toxic for birds with oral LD50 values
of 40-180 mg/kg body weight (Table 9). Peakall & Bart (1983) reported
a 5-day LD50 value of 720 mg/kg for Bobwhite quail.
Residues of trichlorfon and dichlorvos in the bodies of
canopy-feeder birds (Baltimore orioles, crested fly-catcher, and blue
jay) were 0.005-0.04 mg/kg, 3 days following an aerial application of
trichlorfon at 1.12 kg/ha (Kurtz & Studholme, 1974).
Following aerial spraying of trichlorfon at 1.12 kg/ha, the brain
cholinesterase activity was about 20% less than that in the controls
in 2 out of 10 bird-species; in the other 8 species, the activity was
not depressed, even immediately after spraying (Zinkl et al., 1977).
According to a survey based on two, 1-h counts in 20-ha plots, 5
days before and after the spraying of 1120 g trichlorfon/ha, a
virtually identical change in the number of bird species and the total
number of hearings was observed. The application of trichlorfon
resulted in a slight drop in song-bird activity (165 to 151). It did
not produce any effects on cholinesterase activity in fly-catchers and
only 10% inhibition in northern oriols, after 3 days. There was a
considerable decrease in the singing of male birds after spraying; a
marked increase in feeding activity immediately following spraying was
noted. However, examination of the data on individual species does not
show any consistent pattern: the great crested fly-catcher decreased
but other fly-catchers did not, the American redstart decreased, but
another canopy species, the red-eyed vireo, did not. No significant
effects on the numbers of breeding pairs, bird abundance, nesting
success, or mortality were found. Brain ChE levels were reduced by 20%
in 6 out of 103 birds collected during the 5-day period following
spray (Peakall & Bart, 1983).
Japanese quail were given trichlorfon daily for 20 days at an
oral dose of 5 mg/kg body weight. Haematological examinations were
made on the 5th, 10th, 15th, and 20th day of treatment and on the 5th,
10th, 15th, and 30th day after stopping treatment. The number of
erythrocytes dropped on the 5th and 10th days, and the values of the
haematocrit and haemoglobin fell on the 5th day. A significant
increase in erythroblast contents was found on the 5th and 10th day.
No other significant changes of the above parameters were observed up
to the 30th day after treatment. The numbers of leukocytes,
lymphocytes, neutrophils, and monocytes sharply increased from the 5th
to 15th day of treatment, and quickly dropped to normal ranges on the
10th day after stopping treatment (Gromysz-Kalkowska et al., 1985).
Table 9. Acute toxicity of trichlorfon for non-target terrestrial organisms
Species Size Application Toxicity Formulationa Temperature Reference
(°C)
Birds
Red-winged blackbird oral LD50 37-75 mg/kg Schafer Jr et al.
(Agelaius phoeniceus) (1983)
oral LD50 40 mg/kg Schafer (1972)
European starling oral LD50 43 mg/kg Schafer Jr et al.
(Sturnus vulgaris) (1983)
oral LD50 47 mg/kg Schafer (1972)
(Columbia livia) 250-380 g oral LD50 179.9 mg/kg Hattori (1974)
White leghorn hen 1.5-2.0 kg oral LD50 75 mg/kg Kimmerle & Löser
intraperitoneal LD50 75 mg/kg (1974)
Japanese quail oral LD50 50 mg/kg Gromysz-Kalkowska
(Coturnix coturnix (1985)
japonica)
Arthropods
Honey-bee sprayed on glass plate LC50 0.600 W/V% Abdelwahab et
(Apis mellifera, L) al. (1973)
weathered residues (3 h) 24-h 17% mortality 50%SP 26 Johansen (1972)
128.6 mg topical LD50 28.5 µg/g T 16 Ahmad &
Johansen (1973)
Alfalfa leafcutter bee weathered residues (3 h) 24-h 5% mortality 50%SP 31 Johansen (1972)
(Megachile rotundate, F) 22.2 mg topical LD50 250 µg/g T 16 Ahmad &
26.1 mg LD50 515 µg/g T 16 Johansen (1973)
Table 9. (continued)
Species Size Application Toxicity Formulationa Temperature Reference
(°C)
Alkali bee weathered residues (3 h) 24-h 31% mortality 50%SP 31 Johansen (1972)
(Nomia melanderi,
Cockerell)
a SP = Soluble powder.
T = Technical grade.
7.5 Ecosystems
Trichlorfon applied to ponds at the rate of 1 mg/litre water
destroyed the food invertebrates for fish. Large numbers of
zoo-plankton, rotifers, and crustacea, died in the first 24 h after
treatment, whereas benthos died during the first week. The affected
fauna, community recovered slowly. Trichlorfon treatment deprived the
fish of valuable foods, such as crustacean and bottom fauna, for as
long as 1 month after treatment (Grygierek & Wasilewska, 1981).
8. EFFECTS ON EXPERIMENTAL ANIMALS AND IN VITRO TEST SYSTEMS
A more complete treatise on the effects of organophosphorus
insecticides in general, especially their short- and long-term effects
on the nervous system, will be found in the WHO Environmental Health
Criteria 63: Organophosphorus insecticides - A general introduction
(WHO, 1986).
No-observed-effect levels in animals treated with trichlorfon
under various conditions are summarized in Annex II.
8.1 Acute toxicity
LD50 values for trichlorfon in different species and following
different routes of administration are shown in Table 10. Species
differences in LD50 values seem to be rather small.
The acute toxicity of trichlorfon is due to the inhibition of
acetylcholinesterase at the nerve endings by the degradation product
dichlorvos, leading to accumulation of endogenous acetylcholine. The
effects are manifested by muscarinic and central nervous system signs
and symptoms (Taylor, 1980). In the rat, the toxic effects produced by
trichlorfon are characteristics of organophosphorus poisoning, i.e.,
muscular fibrillation, salivation, lacrimation, incontinence,
diarrhoea, respiratory distress, prostration, gasping, tonic and
clonic convulsion, coma, and death. Trichlorfon caused rapid onset of
poisoning, effects occurring within 5 min at approximate LD50 doses
(Edson & Noakes, 1960).
Only a slight increase in mortality was noted in weanling
Holtzman rats compared with that in adults, when treated with
trichlorfon (Brodeur & DuBois, 1963).
Trichlorfon has a low dermal toxicity (LD50: > 2000 mg/kg)
compared with that of dichlorvos (LD50: 75-900 mg/kg). The
difference in lipid solubility between the two compounds may be a
major factor accounting for this difference (Holmstedt et al., 1978).
Since trichlorfon is used as a parasiticide in livestock, studies
have been conducted to assess the effects of the chemical on
cholinesterase activity and clinical conditions. Administration to
horses of a single dose of trichlorfon of 60 or 80 mg/kg body weight,
by stomach tube, resulted in moderately severe and severe colic,
respectively, whereas a single dose of 80 mg/kg mixed into the feed
was associated with only a transient softening of the faeces. Doses of
40 mg/kg or less, by either method of administration, were generally
tolerated without notable adverse effects except for the softening of
the faeces, which tended to be self-limiting. Clinical trials at dose
rates of 35-40 mg/kg in horses, including pregnant and nonpregnant
mares, stallions, suckling and weanling foals, and yearlings, did not
cause any notable adverse effects (Drudge et al., 1976).
Table 10. Acute toxicity of trichlorfon for several animal species
Animal Sex Route Parameter Value Reference
Mouse M oral LD50 800 mg/kg Haley et al. (1975)
F 800 mg/kg
M ip LD50 600 mg/kg Soliman et al. (1984)
Rat M oral LD50 660 mg/kg Benes & Cerna (1970)
M oral LD50 630 mg/kg Gaines (1969)
F 560 mg/kg
M,F dermal LD50 >2000 mg/kg
M dermal LD50 2800 mg/kg Edson & Noakes (1960)
M ip (23-day LD50 190 mg/kg Brodeur & Dubois
old) (1963)
ip (adult) 250 mg/kg
M,F inhalation LC50 533 mg/m3 Kimmerle (1975a)
(4 h)
Guineapig M,F ip LD50 300 mg/kg DuBois & Cotter
(1955)
Rabbit M dermal LD50 5000 mg/kg Deichmann & Lampe
(1955)
Dog M oral LD50 420 mg/kg Deichmann & Lampe
(1955)
Pretreatment of female and castrated male sheep with 1.5 mg
trichlorfon/kg body weight by intravenous injection, which was
insufficient to produce a significant depression of erythrocyte
cholinesterase activity, produced toxic effects that were additive to
those of coumaphos, subsequently administered at 4 mg/kg per day
(Silvestri et al., 1975a,b).
Horses treated with trichlorfon (39.7 mg/kg body weight) combined
with mebendazole (8.8 mg/kg body weight) did not show any, or only a
few, side-effects, except ChE inhibition, whereas horses given higher
dosages (up to 5 times this dose) showed dosage-related increases in
the severity of clinical signs and inhibition of erythrocyte
cholinesterase activity. Depression of the activity was detected
within 1 h of treatment. The maximum depression ranged from 42% (at
the initial dosage) to 75% (at a 5 times higher dosage). Recovery of
base-line activity did not occur in any of the horses within 32 days
after treatment (Gingerich & Mia, 1981).
8.2 Short-term exposure
Wistar albino rats (10 males per group) were fed trichlorfon at
0, 1, 5, 25, or 125 mg/kg diet for 16 weeks. Trichlorfon failed to
cause erythrocyte cholinesterase depression at 125 mg/kg. No effects
were noted on food consumption or growth, or during gross examination
of the tissues (Edson & Noakes, 1960).
Rats (13/sex per group) were fed trichlorfon at dietary levels of
0, 20, 100, or 300 mg/kg diet for 16 weeks. Significant cholinesterase
depression was noted at 300 mg/kg. No effects were observed at the 100
mg/kg level on growth, behaviour, food consumption, or on gross and
microscopic examination of tissues (Doull & Dubois, 1956).
After oral administration of trichlorfon to guinea-pig (100 mg/kg
body weight per day for 60 days) the haemoglobin content was decreased
by 13.5% while the haematocrit value remained unchanged. Trichlorfon
also decreased the serum cholinesterase activity by 50% and increased
the activity of alkaline phosphatase by 36% (Krustev et al., 1976).
Two dogs were administered 45 mg trichlorfon/kg body weight,
orally, for 6 days per week over 3 months. No cumulative effects were
noted. The serum cholinesterase level was 60% of normal at the end of
the study period. No deaths occurred (Deichmann & Lampe, 1955). In
another study, dogs (one male and one female per group, 2 males and 2
females serving as controls) were fed trichlorfon at levels of 0, 50,
200, or 500 mg/kg diet for 12 weeks. Plasma and erythrocyte
cholinesterase activity was depressed at 500 mg/kg diet and unaffected
at 200 mg/kg diet. Recovery of enzyme activity was complete 6 weeks
after the feeding of trichlorfon stopped (Williams et al., 1959).
Rats (10/sex per group) were exposed for 6 h a day over a 3-week
period (total of 15 exposures) to an atmosphere containing trichlorfon
at concentrations of 0, 12.7, 35.4, and 103.5 mg/m3. Exposure to a
concentration of 103.5 mg/m3 slightly affected the health of the
animals (no details available). Body weight gain, parameters of
haematological and clinical chemistry examinations, and urinalyses
were not influenced at any exposure level. Cholinesterase inhibition
of 42, 31, and 22% was found in the plasma, erythrocytes, and the
brain, respectively, in male animals at 103.5 mg/m3; female animals
showed dose-dependent inhibition values of 39, 26, and 26% at 35.4
mg/m3 and 48, 44, and 47% at 103.5 mg/m3 in the plasma,
erythrocyte, and brain, respectively. The only significant alteration
in relative organ weight was found in male animals showing
dose-related increases in relative spleen weights of about 20 and 25%
at the 35.4 and 103.5 mg/m3 exposure levels, respectively. No
abnormal histological findings were observed in any of the tissues
examined microscopically (Kimmerle, 1975b).
In a 13-week study to evaluate target organ toxicity,
dose-response, and maximum tolerated dose for a 2-year study, groups
of 10 male and 10 female Fischer rats and B6C3F1 mice (8-week-old)
were administered trichlorfon in the feed at 0, 62, 185, 555, 1666, or
5000 mg/kg, for 7 days a week. All the rats and mice survived the
13-week treatment, except one male mouse at 5000 mg/kg and one female
mouse at 1666 mg/kg. The body weights of the 5000 mg/kg groups of male
and female rats and mice were significantly lower compared with those
of their respective controls. Plasma and erythrocyte cholinesterase
activity was reduced in a dose-related manner in the rats and mice.
Neurotoxicity tests showed that motor activity and grip strength were
reduced in the 1666 and 5000 mg/kg groups of male and female rats and
mice. No histo pathological changes were observed in the brain, spinal
cord, and sciatic nerve and other organ systems. Absolute and relative
liver, kidney, and spleen weights were increased in the 1666 and 5000
mg/kg groups of male and female rats and mice. The weight increase was
not accompanied by any histopathological findings. Two-year
carcinogenicity studies on trichlorfon administered in the diet to
male and female Fischer rats and B6C3F1 mice are in progress (Chan &
Peters, 1989).
Groups of 7 male and 7 female Wistar rats were fed 1, 5, 10, 30,
50, or 100 mg trichlorfon/kg body weight in their diet for 12 weeks.
A dose of 100 mg/kg, daily, inhibited the cholinesterase activity in
all tissues examined, namely erythrocytes, serum, brain, heart, liver,
and gastrocnemius muscle, but the extent of the inhibition differed
between tissues. No significant histological changes were found in the
parenchymatous organs, such as the brain, heart, lung, pituitary
gland, thyroid gland, liver, intestine, stomach, kidney, adrenal
gland, and testes. Despite the marked inhibition of the tissue
cholinesterase activity in the treated animals, no changes were
observed in nocturnal behaviour, reactivity to external stimuli,
conjunctival and pinna reflexes, and the avoidance reflexes to painful
stimuli (Shimamoto & Hattori, 1965).
Groups of 5 male and 5 female Rhesus monkeys ( Macaca mulatta)
were each administered technical trichlorfon by oral intubation at
dose levels of 0, 0.1, or 0.2 mg/kg body weight per day for 26 weeks,
in order to determine a no-observed-effect level on the cholinesterase
activity in erythrocytes. There were no effects on appearance,
behaviour, nutritional state, feed and water consumption, and body
weight gain. No treatment-related changes were found in haematology,
liver and kidney function, and erythrocyte cholinesterase activity.
The NOEL in this study was 0.2 mg/kg body weight per day (Hoffmann et
al., 1988).
8.3 Skin and eye irritation; sensitization
8.3.1 Skin irritation
A skin irritation test was conducted with technical trichlorfon
applied to the intact and abraded skin of 6 albino rabbits. The
contact time was 24 h and the animals were observed for 7 days.
Technical trichlorfon did not irritate the skin (Thyssen, 1981).
Technical trichlorfon was tested on 6 New Zealand White rabbits
for its dermal primary irritation potential. The test material was
kept in contact with the shaved skin under an occlusion patch for 4 h,
and scoring was performed for 72 h. The results indicated that
technical trichlorfon is not a primary dermal irritant (Bond, 1986).
8.3.2 Skin sensitization
Technical trichlorfon was evaluated in a Magnusson and Kligman
maximization test on guinea-pigs. The concentrations used were:
intra-dermal induction, 1%; topical induction, 25%; first challenge,
25%; and second challenge, 12.5%. The results showed that technical
trichlorfon is a sensitizer for guinea-pigs (Mihail, 1985).
Technical trichlorfon was investigated in the open epicutaneous
test on guinea-pigs for skin-sensitizing potential. Induction (4
weeks, 5 days per week) was carried out using 0, 1, 3, or 10% test
compound. Challenges were done with the same concentrations 4, 6, and
8 weeks after the start of the induction. The 3 and 10% dilutions of
trichlorfon had a skin-sensitizing effect, but not the 1% dilution
(Mihail, 1986a).
8.3.3 Eye irritation
An irritation test was performed using technical trichlorfon on
6 albino rats. The exposure times were 5 minutes and 24 h. Technical
trichlorfon had a moderately irritating effect on the mucosae of the
eye (Thyssen, 1981).
8.4 Long-term exposure
Several reviews on long-term toxicity studies on trichlorfon have
been published (FAO/WHO, 1972, 1976, 1979; Holmstedt et al., 1978;
Machemer, 1981; IARC, 1983).
8.4.1 Oral administration
8.4.1.1 Mouse
A group of 30 male and 28 female AB/Jena strain mice, 8 weeks of
age, received 30 mg trichlorfon/kg body weight, by gavage, twice
weekly for 75 weeks. A group of 30 male and 29 female mice served as
controls. All surviving animals were killed in week 80. Inhibition of
body weight gain and shortening of survival time were noted in the
treated group. There was no statistically significant difference in
the incidence of tumours between treated and control mice (Teichmann
& Hauschild, 1978).
Groups of 60 male and 60 female Charles River CD-1 mice
(6-week-old) were given 100, 300, or 1000 mg trichlorfon/kg diet for
90 weeks, except for males in the 1000 mg/kg group which were treated
for 82 weeks. A group of 60 male and 60 female mice served as
controls. Inhibition of body weight gain was observed in females given
300 or 1000 mg trichlorfon/kg diet. Cholinesterase activity was
depressed in both sexes at 1000 mg/kg diet. The no-observed-effect
level was 100 mg/kg diet. No significant microscopic alterations were
reported. There was no statistically significant difference in the
incidence of tumours between treated and control mice (Machemer,
1981).
Groups of 50 CD-1 mice per sex and dose level were given
technical grade trichlorfon at 0, 300, 900, or 2700 mg/kg diet
(analytical concentrations 0, 275, 891, or 2707 mg/kg diet) for 104
weeks. There were no significant differences between groups in feed
consumption, haematological parameters, or mortality. Increased
incidences of urine stain and ear lesions in males, and vaginal
discharge in females, were regarded as non-specific cholinergic
effects. The body weights of the 2700 mg/kg female mice were
significantly increased throughout the study. There was a
compound-related increase in liver weight in the 900 and 2700 mg/kg
females with no corresponding light-microscopic changes, regarded as
a compound-adaptive mechanism. All trichlorfon-treated groups showed
significant blood cholinesterase depression throughout the study: 20%,
in the 300 mg/kg females, and up to 74% in the 2700 mg/kg group; this
latter dose was compatible with a maximum tolerated dose. No
compound-related tumorigenicity was found in this study (Hayes, 1988).
8.4.1.2 Rat
Groups of 25 male and 25 female, 4-week-old, Sprague-Dawley rats
were fed trichlorfon in the diet at doses of 0, 50, 250, 500, or 1000
mg/kg diet. The treatment continued for 17 months for the males and 24
months for the females. Survival time was shortened in both males and
females given 1000 mg/kg diet. Retarded growth rate was observed in
males given 1000 mg/kg diet: their weight was 15% less than the
controls. Depression of cholinesterase activity was noticed in both
sexes given doses of 500 or 1000 mg/kg. Female rats fed 500 or 1000
mg/kg diet exhibited an absence of primary follicules and primitive
ova and male rats fed 1000 mg/kg diet showed depression of
spermatogenesis. Necrotizing arteritis was observed in both sexes at
doses of 500 or 1000 mg/kg. However, no treatment-related
toxicological changes were reported in rats given 50 or 250 mg/kg
diet. The incidences of mammary tumours in female rats were 14, 8, 20,
21, and 25% in groups given 0, 50, 250, 500, and 1000 mg/kg diet,
respectively. The time to onset of the appearance of tumours was
dose-dependent, being 1.7 years for the controls while at dietary
levels of 50, 250, 500, and 1000 mg/kg, the onset occurred after 1.6,
1.8, 1.5, and 1.1 years, respectively (Doull et al., 1962b).
Another long-term toxicity study was reported in which groups
each of 25 male and 50 female, 4-week-old, Sprague-Dawley rats were
fed trichlorfon at doses of 0, 100, 200, or 400 mg/kg diet for 18
months. Toxicological changes, such as cystic granular alterations of
the ovaries and depression of oocytogenesis were seen only in rats fed
400 mg/kg diet. From these 2 studies, it was concluded that
trichlorfon given orally to rats may enhance some of the normal aging
processes in the reproductive tissues in the females and possibly in
the males (Doull et al., 1965).
Groups each comprising 50 male and 50 female, Long-Evans rats, 4
weeks of age, were fed trichlorfon at doses of 50, 250, 500, or 1000
mg/kg diet. A group of 100 male and 100 female rats served as
controls. There were neither shortening of survival times nor retarded
growth in rats given 1000 mg/kg diet. Depression of cholinesterase
activity in both sexes at 1000 mg/kg diet was the only finding related
to the treatment. There were no morphological changes considered to be
compound-related. All surviving animals were killed after 24 months of
treatment. There was no statistically significant difference in the
incidence of tumours between treated and control rats (Lorke & Löser,
1966; Grundmann & Hobik, 1966).
A group of 30 male and 35 female albino rats, 10 weeks of age,
received 22 mg trichlorfon/kg body weight in saline, by gavage, twice
weekly for 90 weeks. A group of 25 male and 26 female rats served as
controls. All surviving animals were killed in week 118. Survival time
was reduced in the treated group. There were neither biochemical nor
morphological changes attributable to the treatment, and no
statistically significant differences in the incidence of tumours
between treated and control rats (Teichmann et al., 1978).
Groups of 50 male and 50 female Fischer 344 rats, were fed diets
containing mean analytical concentrations of 0, 92.2, 273, or 1518 mg
trichlorfon/kg feed. Dosages were provided as nominal 0, 100, 300,
1750 mg/kg diet. The top dosage was started as 1000 mg/kg for 27
weeks, changed to 1250 mg/kg for 5 weeks, changed to 1500 mg/kg for 8
weeks, and then changed to 1750 mg/kg for the remaining 65 weeks.
Satellite groups of 20 animals per sex served as controls and, at the
highest dose level, the animals were treated for one year and then
sacrificed to provide interim data. Mortality was unaffected by
treatment. Decreased food consumption was slight in females up to 66
weeks and depressed body weight gain occurred in high-dose males.
Increased cholesterol levels were observed in males at doses of 300
mg/kg or more while slight anaemia was present in both sexes at 1750
mg/kg. Cholinesterase activity in plasma, red cells, and the brain was
significantly reduced in both sexes at 1750 mg/kg. Both liver and
kidney weights were increased at 1750 mg/kg in both sexes.
Histological changes were not present in the liver; however, chronic
nephropathy occurred at the highest dose level. Hyperplasia of the
upper small intestine and gastritis occurred at 300 mg/kg and above.
A NOEL equal to 4.45 mg/kg body weight for males and 5.82 mg/kg body
weight for females was established for the study (Hayes, 1989).
Tumorigenicity for this study was not assessed by the Task Group.
8.4.1.3 Dog
Groups each consisting of 2 male and 2 female Beagle dogs were
given trichlorfon in the diet at doses of 0, 50, 250, 500, or 1000
mg/kg diet for 12 months. Depression of cholinesterase activity was
observed at the 500 and 1000 mg/kg diet levels. Spermatogenesis was
inhibited in males given 1000 mg/kg diet (Doull et al., 1962a).
Groups of 4 male and 4 female Beagle dogs were fed dietary
trichlorfon at levels of 0, 50, 200, 800, or 3200 mg/kg diet for 4
years. Cholinesterase activity was depressed in dogs of both sexes at
doses higher than 200 mg/kg diet. Increased mortality, retarded growth
rate, reduced weights of adrenal glands and testes, and impairment of
the kidney function were observed at 800 and 3200 mg/kg diet. In
addition, cholinergic symptoms, such as tremors, cramps, and
salivation were noteworthy at the 3200 mg/kg level. Only one female
dog in the latter group survived for 4 years. Liver injury was
detected biochemically in dogs that died during the study (Löser,
1970).
8.4.1.4 Monkey
Groups of 5 male and 5 female Rhesus monkeys ( Macaca mulatta)
were administered technical trichlorfon by gavage at dose levels of 0,
0.2, 1, or 5 mg/kg body weight per day, 6 days per week for 10 years.
At the 5 mg/kg level, transient pupillary constriction was seen in 2
animals during the first month, and muscular fasciculations in one. At
this dose level, a lowering of the erythrocyte count occurred in both
sexes and, towards the end of the study, the body weights were lower
in this group. Plasma-, erythrocyte-, and brain-cholinesterase
activity was inhibited in the 5 and 1 mg/kg groups, while in the 0.2
mg/kg group inhibition occurred only in erythrocytes in the males.
Liver biopsies done during the first 3 years of the study did not
reveal any evidence of changed liver morphology in any of the groups.
No trichlorfon-related mortality occurred. Complete gross and
microscopic examination of the tissues of the treated groups gave a
non-neoplastic pathology similar to that in the control group. No
significant pathology, including tumorigenicity, related to the
administration of trichlorfon was observed in the treated animals,
compared with the controls (Griffin, 1988).
8.4.2 Intraperitoneal administration
8.4.2.1 Mouse
A group of 30 male and 30 female AB/Jena strain mice, 8 weeks of
age, received 28.2 mg trichlorfon/kg body weight by ip injection,
twice weekly, for 75 weeks. A group of 30 male and 30 female control
mice were given saline intraperitoneally. All surviving animals were
killed in week 80. Inhibition of body weight gain and reduced survival
time were noted in the treated group. There was no statistically
significant difference in the incidences of tumours between treated
and control mice (Teichmann & Hauschild, 1978).
8.4.2.2 Rat
In a study by Teichmann et al. (1978), a group of 30 male and 35
female albino rats, 10 weeks of age, received 12 mg trichlorfon/kg
body weight intraperitoneally, twice weekly, for 90 weeks, while a
group of 25 male and 25 female control rats was given saline. All
surviving animals were killed in week 118. Survival time was reduced
in the treated group. There was no statistically significant
difference in the incidences of tumours between treated and control
rats.
8.4.2.3 Hamster
Syrian golden hamsters, (23 males and 25 females per group) 7-8
weeks of age, were given 20 mg trichlorfon/kg body weight
intraperitoneally, once weekly, for 90 weeks. A group of 22 male and
23 female control hamsters were given saline. All surviving animals
were killed in week 100. Inhibition of body weight gain and reduced
survival time were noted in the treated group. There was no
statistically significant difference in the incidences of tumours
between treated and control hamsters (Teichmann & Schmidt, 1978).
8.4.3 Dermal administration
8.4.3.1 Mouse
A group of 30 male and 30 female AB/Jena strain mice, 8 weeks of
age, received 0.25 ml 1% solution of trichlorfon in acetone, dermally,
twice weekly, for 75 weeks. A group of 30 male and 30 female mice
served as controls. All surviving animals were killed in week 80.
Inhibition of body weight gain and reduced survival time were noted in
the treated group. There was no statistically significant difference
in the incidences of tumours between treated and control mice
(Teichmann & Hauschild, 1978).
8.5 Mutagenicity
8.5.1 DNA methylation
In vitro studies using chemical agents or isolated nucleic
acids with dichlorvos showed that the methylating capability of
dichlorvos was less, by a factor of 10-100, than that of strongly
genotoxic agents (WHO, 1989). In vivo studies using the
determination of 14C-N7-methylguanine in the urine have been used
to calculate the in vivo methylation capability of dichlorvos for
DNA (WHO, 1989) and trichlorfon (Dedek et al., 1976). However, it has
been shown that the excretion of 14CH3-labelled purines in the
urine does not constitute evidence for the methylation of the purines
of DNA, because a natural biosynthetic pathway will give rise to
urinary methylated purines via the C1-pool (Wright et al., 1979;
WHO, 1989). Consequently, only the measurement of 14C methylated
purines in the DNA of organs would provide acceptable results for the
evaluation of the in vivo methylating capability of alkylating
agents (Wright et al., 1979).
Such studies have been performed by Dedek (1981) for trichlorfon
with ip administration of 0.48, 0.40, or 0.065 mmol/kg to male mice
(strain AB Jena/Halle). The extent of methylation in liver DNA was
found to be maximal after 6 h in amounts of 6-8 and 0.8 µmol
N7-MeG/mol guanine for the high and the low dose, respectively, that
is 6-8 methylations in the N7 position per 106 guanine bases.
However, the alkylation at N7 of guanine by small groups, such as
the methyl group, does not show a good correlation with genotoxic
effects.
For obtaining a dose-independent measure of alkylation activity,
the covalent binding index may be used, which is defined as:
micromol bound per mol of nucleotides
CBI =
millimol administered per kg animal
For the strong alkylating agent methyl methanesulfonate (MMS),
the CBI values for guanine N7 have been estimated to be between 135
and 556, depending on the route of administration and the time
intervals. The corresponding CBI values for trichlorfon were 2.3 to
5.1 (Dedek, 1981; Dedek et al., 1984).
Trichlorfon has a DNA-alkylating property and may react with DNA
in vitro to cause depurination and excision (Rosenkranz &
Rosenkranz, 1972).
8.5.2 Mutagenicity
Data on mutagenicity tests in vitro as well as in vivo have
accumulated during the past 20 years. Results indicate that
trichlorfon is partly positive and partly negative, depending on the
purity of the test material, test system, dosage, or source, and
possible effects derived from its degradation products (IARC, 1983).
Results are summarized in Table 11.
Trichlorfon induces gene mutation in S. typhimurium and E.
coli Poole et al., 1977; Carere et al., 1978a; Benigni et al., 1980;
Shirasu et al., 1982; Morya et al., 1983) and in S. cerevisiae
(Riccio et al., 1981; Gilot-Delhalle et al., 1983). It also induces a
mitotic crossing over, gene conversion, and mitotic recombination
(Waters et al., 1980). The positive results in microorganisms indicate
that trichlorfon induces mainly base-pair substitution or mutation in
the absence of metabolic activation. Trichlorfon also induces a
chlorophyl mutation (Panda & Sharma, 1980) and chromosomal damage in
plants (Logvinenko & Morgun, 1978; de Kergommeaux, 1983).
In vitro studies with mammalian cells indicate that trichlorfon
induces unscheduled DNA synthesis (UDS) in human epithelial cells
(Benigni & Dogliotti, 1980; Aquilina et al., 1984) and in human
fibroblasts (Waters et al., 1982). It induces sister chromatid
exchanges (SCE) in Chinese hamster ovary (CHO) cells (Chen et al.,
1981; Waters et al., 1982), and chromosomal aberrations in CHO (Sasaki
et al., 1980; Ishidate et al., 1981) and human lymphocytes (Kurinniy
& Pilinskaya, 1977).
Trichlorfon has also been shown to cause cell transformation in
C3H1OT1/2 CL8 cells (Waters et al., 1981, 1982), and forward mutations
in mouse lymphoma L51784 cells in the absence of metabolic activation
(McGregor et al., 1988).
In vivo studies indicate that trichlorfon induces chromosomal
aberrations in mouse bone marrow cells (Kurinniy, 1975; Kuzmenko et
al., 1980; Ryazanova & Gafurova, 1980; Nehéz et al., 1987). However,
negative results have been reported in a bone marrow micronucleus test
after intraperitoneal injection of trichlorfon in doses up to 400
mg/kg (Paik & Lee, 1977; Jones et al., 1982; Waters et al., 1982). In
the dominant lethal test on mice, trichlorfon was reported to induce
a significant increase in post implantation losses only after
administration of relatively high doses, i.e., 405 mg/kg, as a single
dose, or 54 mg/kg, daily, for 3 weeks (Dedek et al., 1975; Fischer et
al., 1977). The reproducibility of such effects, however, was not
supported by other investigators (Becker & Schöneich, 1980). A
short-term study on mice indicated that trichlorfon did not induce any
chromosomal damage in either bone marrow or germ cells after treatment
with 0.5 mg/litre in the drinking-water, continuously, for 7 weeks
(Degraeve et al., 1984).
Cytogenetic studies on lymphocytes obtained from persons
suffering from acute intoxication or those occupationally exposed to
trichlorfon showed variable increases in the frequency of chromosomal
aberrations (Trinh Van Bao et al., 1979; Kiraly et al., 1974).
In conclusion, trichlorfon is mutagenic for microorganisms or
mammalian cells in in vitro assay systems. However, the data are not
consistent, probably because of the purity of the test material and
possible effects derived from its degradation products, such as
dichlorvos. In vivo studies indicate that trichlorfon induces
chromosomal damage only at relatively high dose levels. In short-term
studies, however, no such effects were found in bone marrow cells or
in germ cells.
Table 11. Summary of mutagenicity studies on trichlorfon
Test system Result Reference
Microorganisms
Rec-assay P. mirabilis + Adler et al. (1976); Braun et al. (1982)
S. typhimurium + Braun et al. (1982)
B. subtilis - Shirasu et al. (1976)
Spot-test E. coli - Nagy et al. (1975)
S. typhimurium - Carere et al. (1978a)
Point mutation E. coli + Hanna & Dyer (1975); Batzinger & Bueding (1977);
Kawachi et al. (1980); Nagy et al. (1975);
Moriya et al. (1983)
S. typhimurium + Poole et al. (1977); Benigni et al. (1980); Carere et al.
(1978a); Kawachi et al. (1980); Shirasu et al. (1982)
A. nidulans - Morpurgo et al. (1977); Moriya et al. (1983)
S. coelicolor + Carere et al. (1978a,b); Benigni et al. (1980)
S. cerevisiae + Riccio et al. (1981)
S. pombe + Gilot-Delhalle et al. (1983)
Mitotic crossing over A. nidulans + Morpurgo et al. (1977); Benigni et al. (1980)
S. cerevisiae + Riccio et al. (1981); Waters et al. (1982)
Gene conversion S. cerevisiae + Riccio et al. (1981); Jones et al. (1982);
Waters et al. (1982)
Mitotic recombination S. cerevisiae + Poole et al. (1977); Riccio et al. (1981);
Simmon (1979); Waters et al. (1982)
Plants
Chlorophyl gene mutation H. vulgare + Panda & Sharma (1980)
Chromosome damage H. vulgare + Panda & Sharma (1980)
Wheat + Logvinenko & Morgun (1978)
Clastogenesis Vicia faba + Amer & Ali (1983); de Kergommeaux et al. (1983)
Mammalian cells in vitro
UDS Human epithelial cells + Benigni & Dogliotti (1980)
Human fibroblasts + Waters et al. (1982)
SCE Chinese hamster cells + Chen et al. (1981, 1982); Waters et al. (1982)
Table 11 (continued).
Test system Result Reference
Mammalian cells in vitro
Chromosome damage Chinese hamster cells + Sasaki et al. (1980); Ishidate et al. (1981)
Human lymphocytes + Kurinniy & Pilinskaya (1977)
Gene mutation Mouse lymphoma + Paik & Lee (1977); Jones et al. (1982);
Waters et al. (1982)
Forward mutation assay Mouse lymphoma cells + McGregor et al. (1988)
Drosophila
Gene mutation D. melanogaster - Benes & Sram (1969); Brzheskii (1973); Lamb (1977)
Valencia (1977); Waters et al. (1980)
Recessive lethal D. melanogaster - Lamb (1977); Waters et al. (1980)
Mammals
Micronucleus test Mouse bone marrow - Paik & Lee (1977); Herbold (1979a);
Waters et al. (1982); Jones et al. (1982);
Chromosome damage Mouse bone marrow - Degraeve et al. (1981, 1985)
+ Kurinniy (1975); Nehez et al. (1982, 1987);
Degraeve et al. (1981)
Hamster bone marrow - Dzwonkowska & Hübner (1986); Volkner (1987)
Dominant lethal Mice - Arnold et al. (1971); Epstein et al. (1972);
Herbold (1979b); Becker & Schoeneich (1980);
Degraeve et al. (1981, 1984a); Moutschen-Dahmen (1981)
+ Schiemann (1975); Dedek et al. (1975)
Chromosome damage Mouse testes - Degraeve et al. (1981, 1984b, 1985)
+ Bulsiewicz et al. (1976);
+ Fischer et al. (1977)
Chromosome damage Human lymphocytes + Trinh Van Bao et al. (1974); Kiraly et al. (1979)
8.6 Carcinogenicity
All long-term studies available for this evaluation have been
described in section 8.4; many of these studies are inadequate as
carcinogenicity studies, or the available reports give insufficient
detail for an evaluation.
A weak, dose-related increase in the incidence of mammary tumours
was reported in female rats fed trichlorfon in the diet (Doull et al.,
1962b). However, reports adequate for evaluation did not show any
evidence for the carcinogenicity of trichlorfon in rats, mice, or
hamsters after oral, intraperitoneal, or skin application.
The carcinogenicity of dichlorvos, the major conversion product
of trichlorfon in the mammalian body, has been discussed in WHO (1989)
and US NTP (1989) (see section 8.10).
8.7 Teratogenicity and reproductive toxicity
8.7.1 Mouse
A single dose of 360 mg trichlorfon/kg, injected
intraperitoneally in 33 AS/Jena mice on the first day of gestation,
caused embryo-toxicity. Post-implantation losses were increased with
60, 120, 240 mg/kg ip on day 9 of gestation (13-15 mice/group), with
120 or 240 mg/kg ip on days 1-7 (25-30 mice/group), and were more
pronounced with 240 mg/kg ip (23 mice) on days 7-14 of gestation.
There were no serious malformations (Scheufler, 1975).
Nehéz et al. (1987) showed that 4 consecutive intraperitoneal
doses of 51.5 mg trichlorfon/kg body weight administered to AB
Jena/Halle mice (12-21 per group) on days 2, 3, 4, and 5, or days 6,
8, 10, and 12 of gestation produced a very weak embryotoxic effect
(slightly elevated post-implantation losses, P <0.05), but no
teratogenic activity was found.
A dose of 500 or 600 mg trichlorfon/kg body weight, given daily
to CD-1 mice by oral gavage on days 10-14 of gestation, produced a
reduction in fetal weight and a slightly increased incidence of cleft
palate. This malformation was found in 4/67 fetuses from dams given
500 mg/kg, in 7/205 fetuses from dams given 600 mg/kg, and in 3/402
fetuses from control dams (Staples & Goulding, 1979).
When trichlorfon was administered by gavage to CD-1 mice (15-24
per group) on days 7-16 of gestation, at a daily dose of 200, 300, or
400 mg/kg, and to CD rats (15-24 per group) at 50, 100, or 200 mg/kg
per day on days 7-19 or 8-20 of gestation, trichlorfon was
teratogenic, fetotoxic, and lethal at the two highest dose levels. At
the lowest dose level, which was not maternally toxic, there was a
significant increase in the number of calcified centres in the
forepaws and hindpaws indicating fetotoxicity and a delay in
maturation (Courtney et al., 1986).
Clemens & Hartnagel (1986) reviewed this study and suggested that
no major or minor birth defects had been demonstrated, and that the
embryotoxicity, fetotoxicity, and rib variations in the mouse study
occurred at maternally toxic dose levels. The fact that a very small
number of fetuses was assessed made conclusions difficult.
Intragastric exposure of male CFW mice (30/group) to 30 mg
trichlorfon/kg for 5-260 days resulted in a significant regression of
the seminiferous epithelium in the testes after 100 days of treatment.
No regeneration of the gonads was observed, 40 days after exposure
ceased (Wenda-Rozewicka, 1983).
8.7.2 Rat
A single dose of 80 mg trichlorfon/kg, given to groups of 11
Wistar rats by oral gavage on day 13 of gestation, produced an
increased incidence of embryonic death and fetal malformations, such
as exencephaly and nonclosing eyelids. When 80 mg trichlorfon/kg was
similarly administered on day 9, these effects were not significant.
A daily dose of 8 mg/kg during gestation did not produce any
teratogenic manifestations (Martson & Voronina, 1976).
When trichlorfon was administered in the diet to groups of 9-26
CD rats on days 6-15 of gestation at a daily intake of 76, 145, 375,
432 or 519 mg/kg body weight, both maternal and fetal body weights
were reduced with ingestion of 432 or 519 mg/kg. There was a
dose-related increase in the incidence of fetal malformations with
ingestion of 145 mg/kg or more. The predominant malformations were
exencephaly, meningocoele, hydrocephaly, syndactily, micrognathia,
cleft palate, and skeletal system alterations. No adverse effects were
found with a dose of 76 mg/kg (Staples et al., 1976).
A daily dose of 480 mg trichlorfon/kg, given to 34 CD rats by
gavage on days 6-15 of gestation, produced a high incidence (86%) of
fetal malformations, such as generalized oedema, herniation of the
brain, hydroencephaly, micrognathia, cleft palate, and skeletal system
alterations (Staples & Goulding, 1979).
Groups of 25 naturally inseminated, female Long Evans rats were
exposed by gavage to technical trichlorfon at 0, 10, 30, or 100 mg/kg
body weight per day from day 6 to day 16 of gestation, and sacrificed
on day 20. None of the doses had a lethal effect. Although at 100
mg/kg diarrhoea was caused in some of the animals, embryonic and fetal
development were not affected at this dose level (Machemer, 1979a).
Groups of 33 naturally inseminated, female Charles River rats
were exposed to technical trichlorfon at 0, 500, 1125, or 2500 mg/kg
(equivalent to 0, 45, 102, or 227 mg/kg body weight per day) from day
6 to day 15 of gestation, and sacrificed on day 20. Trichlorfon was
maternally toxic at dietary levels of 500, 1125, and 2500 mg/kg. There
was no evidence of trichlorfon-related embryotoxicity (increased
resorption), fetotoxicity (decreased fetal weight), or teratogenicity
(malformations) at exposure levels up to and including 2500 mg/kg.
There was an increase in the incidence of delayed ossification and
curved, wavy, and/or bulbous ribs at 2500 mg/kg. On the basis of the
results of this study, 1125 mg/kg, equivalent to 102 mg/kg body weight
per day, is considered the no-effect dose, in terms of reproductive
liability in this study (Kowalski et al., 1987).
A 3-generation (2 litters per generation) rat reproduction study
with levels of 0, 100, 300, 1000, or 3000 mg trichlorfon/kg in the
diet resulted in adverse effects on reproduction at 1000 mg/kg, and
above. At 1000 mg/kg, there was evidence of reduced fertility, smaller
litters, and reduced bodyweight of pups. At 3000 mg/kg, the pregnancy
rate was markedly decreased and the pups were smaller and lighter in
weight with none surviving to the weanling stage. No effects were
noted at 300 mg/kg or below. Microscopic examination of tissues from
the F3b generation did not indicate any adverse effects (Löser, 1969;
Spicer & Urwin, 1971).
8.7.3 Hamster
When 200, 300, or 400 mg trichlorfon/kg body weight was given by
gavage to groups of 10-30 female golden hamsters, each day, on days
7-11 of gestation, the 300 and 400 mg/kg doses produced a reduction in
maternal food consumption. At 400 mg/kg, there were signs of maternal
toxicity and 3 out of 30 animals died; fetal death and malformations
(cleft palate, patagium, and fused ribs) were increased. At 300 mg/kg,
only one fetus (out of 105) had malformations. A dose of 200 mg/kg
body weight did not produce any adverse effects (Staples & Goulding,
1979).
8.7.4 Rabbit
Groups of 15 naturally inseminated, female Himalayan rabbits were
exposed by gavage to technical trichlorfon at 0, 5, 15, or 45 mg/kg
body weight per day from day 6 to day 18 of gestation and sacrificed
on day 29. The average weight gain of the dosed groups was reduced,
but doses of 5 and 15 mg/kg were tolerated well. Because of maternal
toxic effects, two abortions occurred at 45 mg/kg, but the fetuses
delivered in this dose group developed normally. The
no-observed-effect level with respect to embryonic development was 15
mg/kg body weight per day. There were no indications that trichlorfon
produced any teratogenic effects in this study (Machemer, 1979b).
Groups of 20, artificially inseminated, female American Dutch
rabbits were orally exposed to 0, 10, 35, or 110 mg technical
trichlorfon/kg body weight per day from day 6 to day 18 of gestation
and sacrificed on day 28. The highest dose (110 mg/kg) was not well
tolerated and resulted in adverse clinical signs and death,
significantly reduced overall body weight gain and food consumption,
and significantly inhibited cholinesterase. At 35 mg/kg, cholin
esterase was significantly inhibited and one death occurred that was
possibly treatment-related. A dose of 10 mg/kg was devoid of overt
maternal toxicity, while 110 mg/kg was embryotoxic and fetotoxic, but
not teratogenic; 35 mg/kg was a NOEL for developmental toxicity and 10
mg/kg for maternal toxicity (Clemens et al., 1990).
8.7.5 Congenital tremor
Several outbreaks of congenital tremor in piglets have been
described in herds in which the sows had been treated with
antiparasitic trichlorfon preparations between days 45 and 63 of
pregnancy (Kronevi et al., 1975; Dobson, 1977; Bölske et al., 1978;
Hansen et al., 1978; Knox et al., 1978). Clinically, the disease was
characterized by ataxia and tremor and a pronounced hypoplasia of the
cerebellum was found, as well as a reduction in the size of the spinal
cord.
This disease was reproduced experimentally. Four sows were dosed
with Neguvon Vet at 60 mg/kg body weight, mixed in the morning feed on
day 55 and on day 70 of pregnancy. All 40 piglets born alive showed
ataxia and tremor, and, at autopsy, hypoplasia of the cerebellum. In
97 control litters, none of the 892 live-born piglets showed nervous
signs (Knox et al., 1978). Similar experimental results were obtained
following exposure of pigs to multiple oral doses of 50-75 mg
trichlorfon/kg between approximately 45 and 63 days after conception
(Pope et al., 1986; Berge et al., 1987a; Rasmussen et al., 1978). A
similar, but less severe effect resulted from the post-natal exposure
of piglets from week 3 to week 6 to 50 mg/kg body weight per day.
Recovery of animals exposed prenatally was slow; 35 days after birth
they had not reached control values for cerebral and cerebellar
weight. There was still regional loss of Purkinje cells in the
cerebellum (Berge et al., 1987b).
Ten pregnant, white guinea-pigs were given 6 doses of 100 mg
trichlorfon/kg body weight by gavage on days 36, 37, 38, 51, 52, and
53 of pregnancy; 7 additional animals served as controls. The pups
developed trembling and locomotor disturbances. Post-mortem
examination of the pups revealed significantly decreased weights of
the total brain and the cerebellum, compared with controls. There was
also a significant weight reduction, particularly of the medulla
oblongata, but also of the hippocampus, the thalamus, and the
colliculi. Histological examination of the cerebellum revealed
reduction of the external granular layer, and the molecular layer,
together with a regional absence of Purkinje cells. The activities of
the neurotransmitter enzymes cholineacetyltransferase and glutamate
decarboxylase in the cerebellum were reduced compared with the control
values (Berge et al., 1986). Pregnant, albino guinea-pigs were given
radiolabelled trichlorfon on days 37 or 52 of pregnancy and examined
by whole body autoradiography 15, 30, and 45 min after administration.
Trichlorfon had not accumulated in the fetal guinea-pig brain (Berge
& Nafstad, 1986).
8.8 Neurotoxicity
Olajos et al. (1979) reported that the administration of a
divided dose of 300 mg/kg to hens (200 + 100 mg/kg given
subcutaneously, 3 days apart) resulted in levels of neurotoxic
esterase (NTE) inhibition (46-68% at 24 h) approaching the 70% level
that has been correlated with the development of organophosphorus-
ester-induced delayed neurotoxicity (OPIDN), and moderate clinical
signs (ataxia) resembling the early stages of OPIDN. Shiraishi et al.
(1983) produced a peripheral neuropathy in a single monkey (1 out of
1) with a single dose of 250 mg/kg.
Based on a review of the literature on trichlorfon and dichlorvos
(both animal studies and cases of human poisoning), Johnson (1981,
1990), Caroldi & Lotti (1981), and WHO (1989) concluded that only
doses that exceed the lethal dose are likely to result in a level of
NTE inhibition at which OPIDN would be expected. Johnson summarized
reports of neurotoxicity studies on trichlorfon in hens as follows:
The acute maximum tolerated cholinergic dose (200 mg/kg
subcutaneously) produced no marked neuropathy in hens; however,
moderate neuropathy was seen when a further dose of 100 mg/kg was
given subcutaneously, 3 days later. Histological signs of severe
degeneration were seen in the sciatic nerve and spinal cord of the
ataxic bird at autopsy after 3 weeks. Slight changes were reported in
sections of the brain stem of the birds given 200 mg/kg, orally, or
100 mg/kg, subcutaneously (Olajos et al., 1979). But these did not
appear to be entirely typical of organophosphorus neuropathy and no
lesions were reported for the spinal cord or sciatic nerve. Inhibition
of NTE in the spinal cord was only measured in later studies (Hierons
& Johnson, 1978) and lagged markedly behind that in the brain.
The ability of trichlorfon to induce delayed neurotoxicity was
assessed in adult White Leghorn hens, administered single subcutaneous
doses of 100 or 300 mg/kg, and assessed for visible signs of
neurotoxicity 24 h after treatment, prior to killing and collection of
samples of brain and spinal (cervical, thoracic) cord for the
measurement of AChE and NTE activities. In short-term studies, hens
received trichlorfon (100 mg/kg) every 72 h for a total of 6 doses.
Three days after the terminal dose, the hens were killed and the
brains, spinal cords, and distal sciatic nerves were removed for
enzymatic and histological examination. While trichlorfon markedly
inhibited tissue AChE, no reduction in NTE was detected and no overt
signs of neurotoxicity were observed. In the short-term studies,
trichlorfon did not cause any obvious neurotoxicity, an observation
supported by minimal changes in the spinal cord and sciatic nerve
morphology, no impairment of walking ability, and no inhibition of the
brain and spinal cord NTE (Slott & Ecobichon, 1984).
A number of other reports indicate that trichlorfon is either not
neurotoxic, or produces a neurotoxic effect that is distinct from
OPIDN. In addition to the Slott & Ecobichon study, several short-term
dosing regimens that resulted in severe cholinergic toxicity by either
the oral (Olajos et al., 1979) or dermal (Francis et al., 1985) route
did not result in neuropathy. Oral doses of 100 mg trichlorfon/kg were
also reported not to result in significant NTE inhibition (Olajos et
al., 1979; Olajos & Rosenblum, 1981). Finally, rats, which are much
less sensitive to agents that cause delayed neurotoxicity, exhibited
electrophysiological signs of neurotoxicity without accompanying
histological changes when given oral doses of 30 mg/kg for 3 weeks
(Lehotzky, 1982) or ip injections of trichlorfon at 200 mg/kg per day
for 5-15 days (Averbook & Anderson, 1983).
Other adverse neurobehavioural effects that have been observed
may be due to acetylcholinesterase inhibition. Dési (1983) exposed
adult CFY rats (10 males and 10 females per group) at levels of 16 and
32 mg/kg for 3 months. The lower dose resulted in increased high
frequency EEC activity, and enhanced central excitability and the
higher dose in increased low-frequency EEG activity and a significant
overall increase in EEG activity as well as indications of depressed
cortical excitability. In animals treated orally with trichlorfon at
30 mg/kg per day, increased locomotor activity in the open field and
decreased rotorod performance were observed transiently. While the
animals were able to acquire the conditioned escape reflex, latency of
the escape responses in the conditioned escape reflex was markedly
increased (Lehotzky, 1982).
8.9 Immunological studies
Exposure of female mice (BALB/cByJ & C57BL/6J strain; 4 per
group) to 175 mg trichlorfon/100 ml drinking-water for 14 days had no
effect on immune function, measured by response to influenza virus
(Reiss et al., 1987). The dose used was considered to be effective for
prophylactic treatment against helminthic infestation.
8.10 Toxicity of dichlorvos
The major transformation product of trichlorfon in mammals,
including human beings, is dichlorvos, which is at least 100 times
more active as a cholinesterase inhibitor than trichlorfon (Hofer,
1981). An evaluation of the health and environmental hazards of
dichlorvos can be found in WHO (1989). For completeness sake, however,
the summary on effects on experimental animals and in vitro test
systems from this publication is reproduced below.
"Dichlorvos is moderately to highly toxic when administered in
single doses to a variety of animals species by several routes. It
directly inhibits acetylcholinesterase (AChE) activity in the nervous
system and in other tissues. Maximum inhibition generally occurs
within 1 h, and is followed by rapid recovery. The oral LD50 for the
rat is 30-110 mg/kg body weight, depending on the solvent used. The
hazard classification of dichlorvos by WHO is based on an oral LD50
for the rat of 56 mg/kg body weight. The signs of intoxication are
typical of organophosphorus poisoning, i.e., salivation, lachrymation,
diarrhoea, tremors, and terminal convulsions, with death occurring
from respiratory failure. The signs of intoxication are usually
apparent shortly after dosing, and, at lethal doses, death occurs
within 1 h. Survivors recover completely within 24 h.
Potentiation is slight when dichlorvos is given orally in
combination with other organophosphates, but in combination with
malathion it is marked.
In short-term toxicity studies on the mouse, rat, dog, pig, and
monkey, inhibition of plasma, red blood cell, and brain ChE are the
most important signs of toxicity. After oral administration, a dose of
approximately 0.5 mg/kg body weight (range, 0.3-0.7 mg/kg) did not
produce ChE inhibition. In a 2-year study on dogs, ChE inhibition was
noted at 3.2 mg/kg body weight or more.
Flea collar dermatitis has been described in dogs and cats
wearing dichlorvos-impregnated PVC flea collars. This was a primary
irritant contact dermatitis that may have been caused by dichlorvos.
Many short-term, inhalation studies on different animal species
have been carried out. Air concentrations in the range of 0.2-1
mg/m3 do not affect ChE activity significantly. Other effects, such
as growth inhibition and increase in liver weight, have been reported
at dose levels at least 10-20 times higher.
It is possible to produce clinical neuropathy in hens, but the
doses of dichlorvos required are far in excess of the LD50. The
effects are associated with high inhibition of neurotoxic esterase
(NTE) in the brain and spinal cord. In the rat, however, neuropathic
changes in the white matter of the brain have been reported following
repeated daily oral application of an LD50 dose.
Immune suppression has been reported in rabbits. At present, no
evaluation as to the relevance for human beings can be given; more
attention to this aspect is needed.
In a long-term study, rats fed dichlorvos in the diet for 2 years
showed no signs of intoxication. Hepatocellular fatty vacuolization of
the liver and ChE inhibition were significant at the two highest dose
levels (2.5 and 12.5 mg/kg body weight).
In a carefully conducted, long-term, inhalation study on rats
with whole body exposure (23 h/day, for 2 years), results were
comparable with those seen in the oral study. No effects were seen at
0.05 mg/m3, inhibition of ChE activity took place at 0.48 mg/m3 or
more.
In several reproduction studies on rats and domestic animals, no
effects were seen on reproduction, and there was no embryotoxicity at
dose levels that did not cause maternal toxicity. At toxic doses,
dichlorvos may cause reversible disturbances of spermatogenesis in
mice and rats. It was not teratogenic in several studies carried out
on rats and rabbits.
Dichlorvos is an alkylating agent and binds in vitro to
bacterial and mammalian nucleic acids. It is mutagenic in a number of
microbial systems, but there is no evidence of mutagenicity in intact
mammals, where it is rapidly degraded by esterases in blood and other
tissues.
Dichlorvos carcinogenicity has been investigated in mice (oral
studies) and rats (oral and inhalation studies). The dose levels used
in 2-year, oral studies were up to 800 mg/litre drinking-water or 600
mg/kg diet for mice, and up to 280 mg/litre drinking-water or 234
mg/kg diet for rats. In a rat inhalation study, dichlorvos
concentrations in air of up to 4.7 mg/m3 were tested for 2 years. No
statistically significant increase in tumour incidence was found. In
two recent carcinogenicity studies on mice and rats, dichlorvos was
administered by intubation at dose levels between 10 and 40 mg/kg body
weight (mice) and 4 and 8 mg/kg body weight (rat) for up to 2 years.
Only preliminary information has been provided. The evidence for
carcinogenicity in these new studies is difficult to interpret at this
time. Only when complete and final reports become available will it be
possible to draw more definitive conclusions.a
a The US-NTP Peer Review Panel reviewed these studies and came to
the following conclusions: "Under the conditions of these 2-year
gavage studies, there was some evidence of carcinogenic activity
of dichlorvos for male F344/N rats, as shown by increased
incidences of adenomas of the exocrine pancreas and mononuclear
cell leukemia. There was equivocal evidence of carcinogenic
activity of dichlorvos for female F344/N rats, as shown by
increased incidences of adenomas of the exocrine pancreas and
mammary gland fibroadenomas. There was some evidence of
carcinogenic activity of dichlorvos for male B6C3F1 mice and
clear evidence for female B6C3F1 mice, as shown by increased
incidences of forestomach squamous cell papillomas." (US-NTP,
1989).
In a recent evaluation of current data, IARC (in press) concluded
that there is sufficient evidence for the carcinogenicity of
dichlorvos in experimental animals, but that there is inadequate
evidence for the carcinogenicity of dichlorvos in humans.
From acute and short-term studies, it is clear that the
metabolites of dichlorvos are all less toxic than the parent compound.
Only dichloroacetaldehyde was positive in a few mutagenicity tests."
8.11 Mechanism of toxicity - mode of action
While trichlorfon itself is not a potent anticholinesterase
agent, the inhibiting activity of this chemical is attributed to the
transformation product dichlorvos. The slow conversion of trichlorfon
to dichlorvos results in the inhibition of both central and peripheral
cholinergic nerve acetylcholinesterase, with the accumulation of the
neurotransmitter, acetylcholine, at nerve endings, and the generation
of characteristic signs and symptoms of toxicity. A full description
of the mechanism of action of organophosphorus ester insecticides can
be found in Environmental Health Criteria No. 63: Organophosphorus
Insections - A General Introduction (WHO, 1986), and that of
dichlorvos in Environmental Health Criteria No. 79: Dichlorvos (WHO,
1989). The muscarinic, nicotinic, and the central nervous
system-induced signs and symptoms observed in humans have been
described extensively (Ecobichon et al., 1977; Hayes, 1982; Hayes &
Laws, 1991).
Transformation of trichlorfon to dichlorvos in vivo was studied
in relation to its effects on the cholinesterase activity,
acetylcholine content, and acetylcholine-turnover in the mouse brain.
An ip injection of 10 mg dichlorvos/kg caused toxic signs, such as
salivation, diarrhoea and, in some cases, difficulties in breathing,
which were clearly recognized about 15 min after administration and
disappeared almost completely towards the end of 60 min. After an ip
injection of 125 mg trichlorfon/kg, the above mentioned signs were
most intense at around 30 min; and almost complete recovery was
observed towards the end of 2 h. The cholinesterase activity and
acetylcholine levels reached their minimum and maximum, respectively,
at 15 min after the injection of dichlorvos and about 45 min after the
injection of trichlorfon. The delayed decrease in acetylcholine
turnover following to ip injection of trichlorfon was also
demonstrated by measuring the acetylcholine-synthesizing rate in the
brain following intravenous injection of 2H6-choline compared with
dichlorvos pretreatment. The level of dichlorvos in the brain of mice
administered trichlorfon ip reached its maximum a few minutes after
the maximal level of trichlorfon itself; both compounds decreased over
similar curves during a 120-min period (Nordgren et al., 1978).
Thirty minutes after intraventricular injection of trichlorfon
(2.5 mg) in the rat, the activity of the cholinesterase decreased to
20% in the hippocampus; 22% in the medulla; 50% in the cerebellum; 58%
in the striatum, and 72% in the cortex. Levels of acetylcholine
reached a maximum at 45 min in the hippocampus and cortex, and peaked
in the striatum at 60 min. The greatest increases were seen in the
hippocampus and cortex with 60 and 55%, respectively (Hallak &
Giacobini, 1987).
The administration of 80 mg trichlorfon/kg im to male
Sprague-Dawley rats was found to produce an increase in acetylcholine
levels, which peaked at about 170% of control levels, 30 min after
exposure (Hallak & Giacobini, 1989). Acetylcholine levels returned to
normal within 120 min of exposure. A second dose at 120 min resulted
in a second surge in the acetylcholine levels.
9. EFFECTS ON HUMAN BEINGS
Trichlorfon is one of the organophosphorus compounds for which
not only acutely toxic effects have been described, but also delayed
neurotoxicity in humans (WHO, 1986).
In addition to the reports describing cases of human poisoning,
there is wide experience concerning the therapeutic use of trichlorfon
and the side-effects arising from this use.
9.1 Acute poisoning - poisoning incidents
Several hundred cases of acute trichlorfon poisoning, some of
them lethal, have been described in the literature. These were either
accidental, intentional (suicide), or due to gross neglect of
prescriptions or safety precautions. A critical detailed review of
these cases is given by Johnson (1981) and Hayes (1982).
In all cases, the onset of poisoning was rapid, early signs and
symptoms being exhaustion, headache, weakness, confusion, vomiting,
abdominal pain, excessive sweating, and salivation. The pupils are
small. Difficulty in breathing may be experienced, because of either
congestion of the lungs or weakness of the respiratory muscles. In
severe cases of poisoning, muscle spasms, unconsciousness, and
convulsions may develop and death may result from respiratory failure.
In the case of trichlorfon, unconsciousness is disproportionately
common and prolonged and the incidence of mental disturbances is high;
moreover polyneuropathy has been found at a later stage, in
approximately 21% of cases (Hayes, 1982). However, after reviewing the
literature, Johnson (1981; 1990) concluded that only doses of
trichlorfon that exceed the lethal dose, and where the victim survived
because of treatment, are likely to result in a level of NTE
inhibition at which delayed neurotoxicity would be expected.
The onset of polyneuropathy has occurred as early as 3 days after
ingestion and as late as 26 days (Hayes, 1982), the majority of cases
following recovery from the acute effects. The clinical,
electrophysiological, and histopathological features of the neuropathy
were similar to the syndrome resulting from TOCP exposure (Hierons &
Johnson, 1978; Shiraishi et al., 1983; Vasilescu et al., 1984;
Niedziella et al., 1985). Hayes (1982) discusses the possibility that
the polyneuropathy could be caused by other chemical compounds,
present as impurities in the technical product or formulation. Johnson
(1981) mentions higher alkyl analogs as an example. No confirmation
for this hypothesis can be found.
In a case of trichlorfon poisoning, a 21-year-old female
attempted suicide by drinking about 50 ml of a 50% formulation of
trichlorfon. She lost consciousness and recovered after 8 h. Two weeks
after ingestion, the patient developed a tingling sensation in all
extremities followed by weakness in the lower limbs and knees,
characterized as motor dominant polyneuropathy (Shiraishi et al.,
1977).
Progressive neuropathy developed 2-8 weeks after acute poisoning
(i.e., unconsciousness for 16 h) in a 20-year-old man who had taken
orally a handful of granular solid formulation (trichlorfon content:
80%). The clinical symptoms were typical of the delayed neuropathy
that is caused by organophosphates, such as tri- o-cresyl-phosphate,
with normal conduction velocity in surviving motor nerve fibres as an
electrophysiological finding. However, a single dose of the above
granular solid sample did not produce acute delayed neurotoxicity in
hens (Hierons & Johnson, 1978).
A 42-year-old man was in deep coma after ingestion of 100-200 ml
Soldep (25% trichlorfon) and had to be artificially ventilated for 37
days. Plasma cholinesterase activity was significantly decreased.
Three weeks after ingestion, there was severe weakness of the lower
limbs. The EMG (at 40 and 70 days, 4,6,9, and 14 months) indicated
denervation in the lower extremities and peripheral motorneuron
lesions in the upper extremities. Twenty-one months after poisoning,
there was a slow improvement in mobility and the patient was able to
walk by himself for a short distance (Bátora et al., 1988).
Akimov & Kolesnichenko (1985) examined the morphological changes
in the nervous system of 14 patients who had died from acute
chlorophos poisoning. They found congested blood vessels with
perivascular oedema and degeneration of the collagenous- and elastic
fibres of the vascular walls. Diffuse cellular changes, such as
swelling and ischaemic changes, were found in the brain, spinal cord,
and vegetative ganglia. There was a moderate destruction of the myelin
sheaths in the lateral columns of the spinal cord and the brain
peduncles, and there were structural changes in the axons of the
peripheral nerves.
Csik et al. (1986) observed 70 cases of trichlorfon poisoning
(mainly suicide attempts) between 1971 and 1983. Twenty-five of them
were re-examined in 1984. Nine of these (36%) had severe residual
signs of delayed polyneuropathy, mainly of the distal motor type. In
one case, signs of CNS lesions had persisted. Four had had complaints
(paraesthesia, weakness of hands) 2-3 months after poisoning, but were
healthy at the time of re-examination.
9.2 Therapeutic use of trichlorfon
Under the name metrifonate, trichlorfon is used to treat
infection by Schistosomiasis haematobium in humans (WHO, 1985).
Metrifonate has, by now, been given to millions of patients in the
tropics in the treatment of schistosomiasis.
Following a dose of 7-12 mg/kg, severe cholinergic symptoms are
rare in spite of almost complete inhibition of plasma cholinesterase
and 40-60% inhibition of erythrocyte acetylcholin esterase (Nordgren
et al., 1981; Davis, 1986). However, trials in which several doses
were administered on either the same day or one day apart resulted in
abdominal colic, nausea, salivation, dizziness, and headache, which
precluded further treatment (Aden-Abdi et al., 1987), and the
subject-reported incidence of one or more of these effects after a
single dose of 10 mg/kg (46%) was generally higher than in subjects
given a vitamin placebo (34%) (Wilkins & Moore, 1987). There is one
report of a possible human birth defect resulting from metrifonate
treatment (Monson & Alexander, 1984).
In another report on 6000 people, mostly in South Africa and
South America, who had been treated with trichlorfon for a few years
to control various intestinal and body parasites, the dosages varied
from 7.5 up to 70 mg/kg. The dose of 7.5 mg/kg, given 2-4 times at
two-week intervals, caused cholinesterase inhibition, weakness,
nausea, diarrhoea, and abdominal pain. Higher doses (24 mg/kg) caused
more severe symptoms including tachycardia, salivation, colic pain,
vomiting, nausea, fatigue, tremors, and sweating. The effects were not
cumulative and recovery in all cases was rapid. In a few human cases,
an indication was given that spermatogenesis (size and shape of sperm)
and sperm mobility might be affected (Wegner, 1970).
In a large-scale programme in rural villages in Somalia,
metrifonate was given at a current dosage regimen of 3 single doses of
7.5 mg/kg each, on three separate days at intervals of 2 weeks (Aden
Abdi & Gustafsson, 1989). In a large number of cases, the results were
not satisfactory, due to poor patient compliance with the treatment
regimen. In an attempt to simplify this regimen, 5 mg/kg, given thrice
during one day, gave the best results as regards safety and the cure
rates were comparable with those with the standard regimen (Aden Abdi,
1990).
In a clinical trial on 20 patients with Alzheimer disease, Becker
et al. (1990) gave single oral doses of 2.5, 5, 7.5, or 15 mg
metrifonate/kg per week, for 1-3 months. A statistically significant
improvement was obtained with the 5 mg/kg per week dose level. A 60%
depression in red cell ChE and 80% depression in plasma ChE were
accompanied by only minor side effects (nausea, vomiting, and/or
diarrhoea); there were no effects at 2.5 mg/kg, 5 patients were
affected at 5 mg/kg, 9 at 7.5 mg/kg, and 13 at 15 mg/kg per week.
9.3 Occupational exposures
Few cases of occupational poisoning by trichlorfon have been
reported. See section 5.3 for occupational exposures.
Occupational exposure to trichlorfon at a factory where air
concentrations exceeded 0.5 mg/m3, but where skin contamination also
occurred, resulted in decreased plasma cholinesterase levels and
changes in EEG patterns; particularly slow and paroxysmal waves were
observed (Lu et al., 1984; Hu et al., 1986). Both the biochemical and
electrophysiological indices returned to normal after exposure ceased.
Although trichlorfon has been widely used for many years, no
cases of skin sensitization have been reported (Mihail, 1986b).
9.4 Treatment of acute trichlorfon poisoning
The advice on the treatment of organophosphate poisoning from EHC
63: Organophosphorus Insecticides - A general introduction has been
reproduced in Annex I.
10. PREVIOUS EVALUATIONS BY INTERNATIONAL BODIES
Trichlorfon was evaluated by the Joint FAO/WHO Expert Committee
on Pesticide Residues (JMPR) in 1971, 1975, and 1978 (FAO/WHO, 1972,
1976, 1979). In 1978, the JMPR established an Acceptable Daily Intake
(ADI) for man of 0-0.01 mg/kg body weight, based on the fact that the
following levels cause no toxicological effects:
Rat: 50 mg/kg in the diet equivalent to 2.5 mg/kg body weight.
Dog: 50 mg/kg in the diet equivalent to 1.25 mg/kg body weight.
In 1986, the FAO/WHO CODEX Committee advised a range of maximum
residue limits (MRLs) for specified food commodities (FAO/WHO, 1986).
These ranged from 0.05 to 2 mg/kg product.
Trichlorfon was evaluated by an IARC Working Group in 1983. There
were no data on its carcinogenicity in humans and the evidence of
carcinogenicity in experimental animals was inadequate. Trichlorfon
was classified in Group 3, i.e., cannot be classified as to its
carcinogenicity to humans (IARC, 1983, 1987).
WHO classified technical trichlorfon as "slightly hazardous"
(Class III) (WHO, 1990). A data sheet on trichlorfon (No. 27) has been
published by the WHO (WHO/FAO, 1977).
REFERENCES
ABBASOV, T.A. (1972) [Residues of organophosphorus insecticides in
clover and potato plants.] Khim. Sel'sk. Khoz., 10: 597-598 (in
Russian).
ABDEL-KADER, M.I.A., MOUBASHER, A.H., & ABDEL-HAFEZ, S.I. (1978)
Selective effects of five pesticides on soil and cotton-rhizosphere
and -rhizoplane fungus flora. Mycopathologia, 66: 117-123.
ABDELWAHAB, A.M., KHALIL, F.M., & HUSSEIN, M.H. (1973) Toxicity of
insecticides to honey bees in laboratory and field. Bull. Entomo.
Soc. Egypt, Econ. Ser., VII: 83-89.
ADEN ABDI, Y. (1990) A rational approach to the treatment of
Schistosoma haematobium infection with metrifonate - clinical and
pharmacological studies, Stockholm, Karolinska Institute, Department
of Clinical Pharmacology (Thesis).
ADEN ABDI, Y. & GUSTAFSSON, L.L. (1989) Poor patient compliance
reduces the efficacy of metrifonate treatment of Schistosoma
haematobium in Somalia. Eur. J. clin. Pharmacol., 36: 161-164.
ADEN ABDI, Y., GUSTAFSSON, L.L., & ELMI, S.A. (1987) A simplified
dosage schedule of metrifonate in the treatment of Schistosoma
haematobium infection in Somalia. Eur. J. clin. Pharmacol., 32:
437- 441.
ADLER, B., BRAUN, R., SCHÖNEICH, J., & BÖHME, H. (1976)
Repair-defective mutants of Proteus mirabilis as a prescreening
system for the detection of potential carcinogens. Biol. Zentralbl.,
95: 463- 469.
AHMAD, Z. & JOHANSEN, C. (1973) Selective toxicity of carbophenothion
and trichlorfon to the honey bee and the alfalfa leafcutting bee.
Environ. Entomol., 2: 27-30.
AKHTAR, M.H. (1982) Fate of trichlorfon in buffer and soluble fraction
(105 000g) from cow and chicken liver homogenates. J. agric. food
Chem., 30: 551-554.
AKIMOV, G.A. & KOLESNICHENKO, I.P. (1985) [Morphological changes in
the nervous system in acute peroral chlorophos poisoning.] Arkh.
Patol., 47(1): 44-51 (in Russian).
AKSNES, G. & YÜKSEKISIK, N. (1974) Kinetic study of the alkaline
hydrolysis of 2,2,2-trichloro-1-hydroxyethyl, dimethyl phosphonate,
Dipterex, and its ethyl analog. Phosphorus, 4: 33-38.
AMBRUS, A., HARGITAI, E., KAROLY, G., FULOP, A., & LANTOS, J. (1981)
General method for determination of pesticide residues in samples of
plant origin, soil, and water. II. Thin layer chromatographic
determination. J. Assoc. Off. Anal. Chem., 64: 743-748.
AMENO, K., FUKE, C., AMENO, S., KIRIU, T., & IJIRI, I. (1989) A rapid
and sensitive quantitation of dipterex in serum by solid-phase
extraction and gas chromatography with flame thermionic detection.
J. anal. Toxicol., 13: 150-151.
AMER, S.M. & ALI, E.M. (1983) Cytological effects of pesticides. XIV.
Effect of the insecticide Dipterex "trichlorfon" on Vicia faba
plant. Cytologia, 48: 761-770.
ANDREU-MOLINER, E.S., ALMAR, M.M., LEGARRA, I., & NUNEZ, A. (1986)
Toxicity of some rice-field pesticides to the crayfish P. Clarkii
under laboratory and field conditions in Lake Albuferra (Spain). J.
environ. Sci. Health, B 21: 529-537.
ANON (1978a) Information on trichlorfon residues surveillance
available to FAO/WHO from Hungary.
ANON (1978b) Information on trichlorfon residues available to FAO/WHO
from Finland.
ANON (1978c) Information on trichlofon residues available to FAO/WHO
from Netherlands.
ANTOINE, O. & MEES, G. (1971) Test de routine pour la détermination
des insecticides organo-phosphorés par la chromatographie en couche
mince. J. Chromatogr., 58: 247-256.
AQUILINA, G., BENIGNI, R., BIGNAMI, M., CALCAGNILE, A., DOGLIOTTI, E.,
FALCONE, E., & CARERE, A. (1984) Genotoxic activity of dichlorvos,
trichlorfon, and dichloroacetaldehyde. Pestic. Sci., 15: 439-442.
ARNOLD, D., KEPLINGER, M.L., FANCHER, O.E. & CALANDRA, J.C. (1971)
Mutagenic study with Dylox in Albino mice. Industrial Biotest
Laboratories (Unpublished report submitted to WHO by Farbenfabriken
Bayer A.G., Germany).
ARTHUR, B.W. & CASIDA, J.E. (1957) Metabolism and selectivity of
O,O-dimethyl 2,2,2-trichloro-1-hydroxyethylphosphonate and its
acetyl and vinyl derivatives. J. agric. food Chem., 5: 186-192.
AVERBOOK, B.J. & ANDERSON, R.J. (1983) Electrophysiological changes
associated with chronic administration of organophosphates. Arch.
Toxicol., 52: 167-172.
BAIDA, T.A. (1970) [Soil pollution by organophosphorus pesticides.]
Kazakstana, 30: 72 (in Russian).
BAIDA, T.A. (1975) [Dynamics and metabolism of chlorophos in cabbage
and onion plants.] Khim. Sel'sk Khoz., 13: 67-68 (in Russian).
BARCELO, D. (1988) Application of thermospray liquid chromatography/
mass spectrometry for determination of organophosphorus pesticides
and trialkyl and triaryl phosphates. Biomed. environ. mass Spectrom.,
17: 363-369.
BARCELO, D., MARIS, F.A., GEERDINK, R.B., FREI, R.W., DE JONG, G.J.,
& BRINKMAN, U.A.T. (1987) Comparison between positive, negative and
chloride-enhanced negative chemical ionization of organophosphorus
pesticides in on-line liquid chromatography-mass spectrometry. J.
Chromatogr., 394: 65-67.
BARTHEL, W.F., GIANG, P.A., & HALL, S.A. (1954)
Dialkyl-alpha-hydroxyphosphonates derived from chloral. J. Am. Chem.
Soc., 76: 4186-4187.
BATORA, I., KUKUMBERG, P., KUCHAR, M., & KOUTUN, J. (1988) [Delayed
neurotoxic effect of Soldep (trichlorfon) after acute poisoning.]
Prac. Lèk., 40: 172-174 (in Slovak).
BATTELLE (1982) Pesticide programme of research and market planning.
Part II. Insecticides, Geneva, Battelle Institute, 40 pp.
BATTELLE (1984) Trichlorfon. In: World pesticide programme.
Insecticides II. World summary: USA, Canada, Argentina, Brazil,
Colombia, Mexico, Central America, Geneva, Battelle Research
Centres, pp. 16, 26, 30, 34.
BATTELLE (1987) Trichlorfon. In: World pesticide programme.
Insecticides III. Summary: USA, Mexico, Brazil, Geneva, Battelle
Research Centres, Vol. 1, pp. 6-8, 11, 14, 16-17, 27.
BATZINGER, R.P. & BUEDING, E. (1977) Mutagenic activities in vitro
and in vivo of five antischistosomal compounds. J. Pharmacol. exp.
Ther., 200: 1-9.
BECKER, K. & SCHÖNEICH, J. (1980) Dominant lethal test with
trichlorphon in male mice. Mutat. Res., 74: 224, (Meeting abstract).
BECKER, R.E., COLLIVER, J., ELBLE, R., FELDMAN, E., GIACOBINI, E.,
KUMAR, V., MARKWELL, S., MORIEARTY, P., PARKS, R., SHILLCUT, S.D.,
UNNI, L., VICARI, S., WOMACK, C., & ZEC, R.F. (1990) Effects of
metrifonate, a long-acting cholinesterase inhibitor in Alzheimer
Disease: report of an open trial. Drug Dev. Res., 19: 425-434.
BEDFORD, C.T. & ROBINSON, J. (1972) The alkylating properties of
organophosphates. Xenobiotica, 2: 307-337.
BELLO, T.R., AMBORSKI, G.F., & TORBERT, B.J. (1974) Effects of organic
phosphorus anthelmintics on blood cholinesterase values in horses and
ponies. Am. J. vet. Res., 35: 73-78.
BENES, V. & CERNA, V. (1970) Contribution to the toxicological
evaluation of fenitrothion ( O,O-dimethyl- O-/3-methyl-4-
nitrophenyl/thio-phosphate) and its residues. In: Halas et al., ed.
Proceedings of pesticide Symposia Inter-American Conferences on
Toxicology and Occupational Medicine, Miami.
BENES, V. & SRAM, R. (1969) Mutagenic activity of some pesticides in
Drosophila melanogaster. Ind. Med. Surg., 38: 442-444.
BENIGNI, R. & DOGLIOTTI, E. (1980) UDS studies on selected
environmental chemicals. Mutat. Res., 74: 217, 248-249 (Meeting
abstract).
BENIGNI, R., BIGNAMI, M., CARERE, A., DOGLIOTTI, E., NOVELLETO, A., &
PRINCIPE, R. (1980) Comparative mutational studies with dichlorvos,
trichlorfon and dichloroacetaldehyde. Toxicol. Lett., 1(Spec.
Issue): 250 (Meeting abstract).
BENNEWITZ, R. & FOTH, G. (1967) [Chemical behavior and analytical
determination of trichlorfon, dichlorvos and tribufon.] Fresenius Z.
anal. Chem., 229: 110-117 (in German).
BERGE, G.N. & NAFSTAD, I. (1986) Distribution and placental transfer
of trichlorfon in guinea-pigs. Arch. Toxicol., 59: 26-29.
BERGE, G.N., NAFSTAD, I., & FONNUM, F. (1986) Prenatal effects of
trichlorfon on the guinea-pig brain. Arch. Toxicol., 59: 30-35.
BERGE, G.N., FONNUM, F., & BRODAL, P. (1987a) Neurotoxic effects of
prenatal trichlorfon administration in pigs. Acta vet. Scand., 28:
321-332.
BERGE, G.N., FONNUM, F., SOLI, N.E., & SOGNEN, E. (1987b)
Neurotoxicological examination of the piglet brain after prenatal and
postnatal exposure to trichlorfon. Acta vet. Scand., 28: 313-320.
BETOWSKI, L.D. & JONES, T.L. (1988) Analysis of organophosphorus
pesticide samples by high-performance liquid chromatography/mass
spectrometry and high-performance liquid chromatography/mass
spectrometry/mass spectrometry. Environ. Sci. Technol., 22:
1430-1434.
BOLOTNYI, A.V., PISMENNAYA, M.V., & AKORONKO, S.A. (1978)
Transformation of the organophosphorus pesticides anthiophos and
trichlorfon in the environment.] Gig. i Sanit., 1978(5): 28-31 (in
Russian).
BOLSKE, G., KRONEVI, T., & LINDGREN, N.O. (1978) Congenital tremor in
pigs in Sweden. Nord. vet. Med., 30: 534-537.
BOND, G.P. (1986) Primary dermal irritation of technical trichlorfon
(Dylox (R) ) in albino rabbits, Metcalf, USA, Mobay Corporation,
(Proprietary report No. 751, submitted to WHO by Bayer AG, Germany).
BOWMAN, P.B. & DAME, P.W. (1974) Gas-liquid chromatography of
trichlorfon in soluble powder formulations. J. Assoc. Off. Anal.
Chem., 57: 1128-1131.
BRAUN, R., SCHÖNEICH, J., WEISSFLOG, L., & DEDEK, W. (1982) Activity
of organophosphorus insecticides in bacterial tests for mutagenicity
and DNA repair - direct alkylation vs. metabolic activation and
breakdown. 1. Butonate, vinylbutonate, trichlorfon, dichlorvos,
demethyl dichlorvos and demethyl vinylbutonate. Chem.-biol.
Interact., 39: 339-350.
BRODEUR, J. & DUBOIS, K.P. (1963) Comparison of acute toxicity of
anticholinesterase insecticides to weanling and adult male rats.
Proc. Soc. Exp. Biol. Med., 114: 509-511.
BRZHESKII, V.V. (1973) [Possibility of induction of mutations in
Drosophila melanogaster with chlorophos.] Med. Parazitol.
Parazit. Bolezn., 42: 703-706 (in Russian).
BUCHET, J.P. & LAUWERYS, R. (1970) Inhibition of rat heart diolein
hydrolase and brain acetylcholinesterase by organophosphate esters in
vitro. Biochim. biophys. Acta., 218: 369-371.
BULL, D.L. & RIDGWAY, R.L. (1969) Metabolism of trichlorfon in animals
and plants. J. agric. food Chem., 17(4): 837-841.
BULSIEWICZ, H., ROZEWICKA, L., JANUSZEWSKA, H., & BAJKO, J. (1976)
Aberrations of meiotic chromosomes induced in mice with insecticides.
Folia morphol. (Warsaw), 35: 361-368.
CAIN, J.R. & CAIN, R.K. (1984) Effects of five insecticides on
zygospore germination and growth of the green alga Chlamydomonas
moewusii. Bull. environ. Contam. Toxicol., 33(5): 571-574.
CARERE, A., ORTALI, V.A., CARDAMONE, G., & MORPURGO, G. (1978a)
Mutagenicity of dichlorvos and other structurally related pesticides
in Salmonella and Streptomyces. Chem.-biol. Interact., 22:
297-308.
CARERE, A., ORTALI, V.A., CARDAMONE, G., TORRACA, A.M., & RASCHETTI,
R. (1978b) Microbiological mutagenicity studies of pesticides in
vitro. Mutat. Res., 57: 277-286.
CAROLDI, S. & LOTTI, M. (1981) Delayed neurotoxicity caused by a
single massive dose of dichlorvos to adult hens. Toxicol. Lett., 9:
157-159.
CERNA, V. (1963) [Colorimetric determination of dipterex residues in
foods.] Nahrung, 7: 60-66 (in German).
CHAN, P.C. & PETERS, A.C. (1989) Thirteen-week subchronic dosed feed
study of trichlorfon in Fischer rats and B6C3F1 mice. Proc. Am.
Assoc. Cancer Res., 30: 137.
CHAPMAN, R.A. & COLE, C.M. (1982) Observations on the influence of
water and soil pH on the persistence of insecticides. J. environ.
Sci. Health, B17: 487-504.
CHEN, H.H., HSUEH, J.L., SIRIANNI, S.R., & HUANG, C.C. (1981)
Induction of sister chromatid exchanges and cell cycle delay in
cultured mammalian cells treated with eight organophosphorus
pesticides. Mutat. Res., 88: 307-316.
CHEN, H.H., SIRIANNI, S.R., & HUANG, C.C. (1982) Sister chromatid
exchanges in Chinese hamster cells treated with seventeen
organophosphorus compounds in the presence of a metabolic system.
Environ. Mutagen., 4: 621-624.
CLEMENS, G.R. & HARTNAGEL, R.E. (1986) Critique: Assessment of the
teratogenic potential of trichlorfon in mice and rats, Elkhart,
USA, Miles Laboratories, Toxicology Department, (Proprietary report
submitted to WHO by Bayer AG, Germany).
CLEMENS, G.R., TROUP, C.M., & HARTNAGEL, R.E. (1990) Teratology study
in the rabbit with Dylox technical (Trichlorfon), Elkhart, USA,
Miles Laboratories, Toxicology Department, (Proprietary report No. MTD
0134, submitted to WHO by Bayer AG, Germany).
CONRAD, R., DEDEK, W., & ENGEWALD, W. (1987) [Determination of
trichlorfon in biological media and technical products following
derivatization with acetic anhydride by gas chromatography].
Fresenius Z. anal. Chem., 326: 241-246 (in German).
COURTNEY, K.D. & ANDREWS, J.E. (1980) Extra ribs indicate fetotoxicity
and maldevelopment. Teratology, 21(2): 35A (Meeting abstract).
COURTNEY, K.D., ANDREWS, J.E., & SPRINGER, J. (1986) Assessment of
teratogenic potential of trichlorfon in mice and rats. J. environ.
Sci. Health, B 21: 207-227.
COX, C.L., TIMMONS, L.L., & RIDLEN, R.L. (1989) Capillary gas
chromatographic analysis of trichlorfon in dosed feed formulations.
J. anal. Toxicol., 13: 84-88.
CSIK, V., MOTIKA, D., & MAROSI, G.Y. (1986) Delayed neuropathy after
trichlorfon intoxication. J. Neurol. Neurosurg. Psychiatry, 49(2):
222.
DALDRUP, T., SUSANTO, F., & MICHALKE, P. (1981) [Combination of TLC,
GLC (OV 1 and OV 17) and HPLC (RP 18) for A rapid detection of drugs,
intoxicants and related compounds.] Fresenius Z. anal. Chem., 308:
413-427 (in German).
DALDRUP, T., MICHALKE, P., & BOEHME, W. (1982) A screening test for
pharmaceuticals, drug and insecticides with reversed-phase liquid
chromatography-retention data of 560 compounds. Chromatogr. Newsl.,
10: 1-7.
DAVIS, A. (1986) Recent advances in schistomiasis. Q. J. Med., 226:
95-110.
DEDEK, W. (1968) [Degradation and residues of butonate-32p in fruits.]
Z. Naturforsch., B 23: 504-506 (in German).
DEDEK, W. (1981) Guanine N7-alkylation in mice in vivo by
metrifonate-discussion of possible genotoxic risk in mammals. Acta
pharmacol. toxicol., Suppl.49: 40-50.
DEDEK, W. & LOHS, K. (1970) [Alkylating effect of trichlorphon in warm
blooded animals. II. Distribution of 14C in organs and liver
proteins of rats after application of trichlorfon-14C.] Z.
Naturforsch., B 25: 1110-1113 (in German).
DEDEK, W. & SCHWARZ, H. (1966) [Metabolism and application of
trichlorfon in veterinary praxis.] Atompraxis, 12: 603-606 (in
German).
DEDEK, W. & SCHWARZ, H. (1967) [Studies on percutaneous resorption of
32P-labeled systemic organic phosphorus compounds of relatively
small toxicity in cattle.] Z. Naturforsch., B 22: 702-706 (in
German).
DEDEK, W. & SCHWARZ, H. (1970a) [Residues in farm animals following
the application of 32P-labelled organophosphorus insecticides.]
Arch. exp. Veterinaermed, 24: 719-726 (in German).
DEDEK, W. & SCHWARZ, H. (1970b) [Studies of the percutaneous
resorption of 32P-trichlorfon and 32P-dimethoate in sheep.] Z.
Naturforsch., B 25: 1193-1194 (in German).
DEDEK, W., KOCH, H., UHLENHUT, G., & BROSE, F. (1969) [Decomposition
of 3H-trichlorfon to dimethyl dichlorovinyl phosphate.] Z.
Naturforsch., B 24: 663-664 (in German).
DEDEK, W., LOHS, K., & GIBEL,W. (1975a) Studies on the alkylating
properties of trichlorphone and dimethoate in mammals. Environ. Qual.
Saf., 3(Suppl.): 648-651.
DEDEK, W., SCHEUFLER, H., & FISCHER, G.W. (1975b) Mutagenicity of
desmethyltrichlorfon in the dominant lethal test in mice. Arch.
Toxicol., 33: 163-168.
DEDEK, W., LOHS, K., FISCHER, G.W., & SCHMIDT, R. (1976) Alkylation of
guanine in mice in vivo by organophosphorus insecticides. I.
Trichlorfon and butonate. Pestic. Biochem. Physiol., 6: 101-110.
DEDEK, W., GEORGI, W., & GRAHL, R. (1979) Comparative degradation and
metabolism of 32P labeled butonate, trichlorfon and dichlorvos in
crop plants. Biochem. Physiol. Pflanzen, 174: 707-722.
DEDEK, W., GRAHL, R., & SCHMIDT, R. (1984) A comparative study of
guanine N7-alkylation in mice in vivo by the organophosphorus
insecticides trichlorfon, dimethoate, phosmet and bromophos. Acta
pharmacol. toxicol., 55: 104-109.
DEDEK, W., WENZEL, K.D., LUFT, F., OBERLÄNDER, H., & MOTHES, B. (1987)
Preconcentration of hydrophilic pesticides from aqueous solutions and
extraction of residues using the polymeric sorbent Wofatil Y77. I.
Preconcentration of hydrophilic pesticides from water. Fresenius Z.
anal. Chem., 328: 484-486.
DEGRAEVE, N., GILOT-DELHALLE, J., COLIZZI, A., CHOLLET, M.C.,
MOUTSCHEN, J., & MOUTSCHEN-DAHMEN, M. (1981) Evaluation des risques
génétiques d'un insecticide organophosphoré: le trichlorfon
( O,O-diméthyl-(2,2,2 trichloro-1-hydroxy)-éthylphosphonate). Bull.
Soc. R. Sci. Liege, 50(1-2): 85-98.
DEGRAEVE, N., CHOLLET, M.C., & MOUTSCHEN, J. (1984a) Cytogenetic and
genetic effects of subchronic treatments with organophosphorus
insecticides. Arch. Toxicol., 56: 66-67.
DEGRAEVE, N., CHOLLET, M.C., & MOUTSCHEN, J. (1984b) Cytogenetic
effects induced by organophosphorus pesticides in mouse
spermatocytes. Toxicol. Lett., 21: 315-319.
DEGRAEVE, N., CHOLLET, M.C., & MOUTSCHEN, J. (1985) Mutagenic
efficiency of organophosphorus insecticides used in combined
treatments. Environ. Health Perspect., 60: 395-398.
DEGTYAREVA, L.A., POLITOVA, K.S., FOMICHENKO, E.L., & KOCHKINA, N.P.
(1977) [Hygienic evaluation of the air in buildings where
chlorophos-containing insecticides are processed.] Probl. Dezinfekts
Steril., Mater. Simp., 3: 26-27 (in Russian).
DEICHMANN, W.B. & LAMPE, K. (1955) Dipterex, its pharmacological
action. Bull. Univ. Miami Sch. Med., 9: 7-12.
DE KERGOMMEAUX, D.J., GRANT, W.F., & SANDHU, S.S. (1983) Clastogenic
and physiological response of chromosomes to nine pesticides in the
Vicia faba in vivo root tip assay system. Mutat. Res., 124: 69-84.
DÉSI, I. (1983) Neurotoxicological investigations of pesticides in
animal experiments. Neurobehav. Toxicol. Teratol., 5: 503-515.
DEVINE, J.M. (1973) Determination of trichlorfon ( O,O-Dimethyl-
(2,2,2-trichloro-1-hydroxyethyl)phosphonate) in forest environmental
samples. J. agric. food Chem., 21: 1095-1098.
DIVE, D., LECLERC, H., & PERSOONE, G. (1980) Pesticide toxicity on the
ciliate protozoan. Colpidium campylum: possible consequences of the
effect of pesticides in the aquatic environment. Ecotoxicol.
environ. Saf., 4: 129-133.
DMITRIYEV, A.I. (1970) [Trichlorfon action on the quality of chicken
slaughter products.] Veterinariya, 46: 104-106 (in Russian).
DOBSON, K.J. (1977) Trichlorfon toxicity in pigs. Aust. vet. J., 53:
115-117.
DOULL, J. & DUBOIS, K.P. (1956) The effects of diets containing
Dipterex on rats, (Unpublished report by Department of Pharmacology,
University of Chicago).
DOULL, J., ROOT, M., VESSELINOVITCH, D., MESKAUSKAS, J., & FITCH, F.
(1962a) Chronic oral toxicity of Dylox to male and female dogs,
(Unpublished report submitted to WHO by Farbenfabriken Bayer A.G.,
Germany).
DOULL, J., VESSELINOVITCH, D., ROOT, M., COWAN, J., MESKAUSKAS, J., &
FITCH, F. (1962b) Chronic oral toxicity of Dylox to male and female
rats, University of Chicago, Department of Pharmacology,
(Unpublished report).
DOULL, J., VESSELINOVITCH, D., ROOT, M., COWAN, J., MESKAUSKAS, J., &
COWAN, J. (1965) Chronic oral toxicity of Dylox to male and female
rats, University of Chicago, Department of Pharmacology,
(Unpublished report).
DRUDGE, J.H., LYONS, E.T., & TAYLOR, E.L. (1976) Critical tests and
safety studies on trichlorfon as an antiparasitic agent in the horse.
Am. J. vet. Res., 37: 139-144.
DUBOIS, K.P. & COTTER, G.J. (1955) Studies on the toxicity and
mechanism of action of dipterex. Am. Med. Assoc. Arch. ind. Health,
11: 53-60.
DUNACHIE, J.F. & FLETCHER, W.W. (1969) An investigation of the
toxicity of insecticides to birds' eggs using the egg-injection
technique. Ann. appl. Biol., 64: 409-423.
DZWONKOWSKA, A. & HUBNER, H. (1986) Induction of chromosomal
aberrations in the Syrian hamster by insecticides tested in vivo.
Arch. Toxicol., 58: 152-156.
ECOBICHON, D.J. (1979) The chemical stability of trichlorfon in fresh
and salt water as measured by changes in anticholinesterase potency.
Toxicol. Lett., 3: 117-120.
ECOBICHON, D.J., OZERE, R.L., REID, E., & CROCKER, J.F.S. (1977) Acute
fenitrothion poisoning. Can. Med. Assoc. J., 116: 377-379.
EDSON, E.F. & NOAKES, D.N. (1960) The comparative toxicity of six
organophosphorus insecticides. Toxicol. appl. Pharmacol., 2:
523-539.
EL-HISSY, F.T. & ABDEL-KADER, M.I.A. (1980) Effect of five pesticides
on the mycelial growth of some soil and pathogenic fungi. Z. allg.
Mikrobiol., 20: 257-263.
EPSTEIN, S.S., ARNOLD, E., ANDREA, J., BASS, W., & BISHOP, Y. (1972)
Detection of chemical mutagens by the dominant lethal assay in the
mouse. Toxicol. appl. Pharmacol., 23: 288-325.
FAO (1982) Second Government Consultation on International
Harmonization of Pesticide Registration Requirements, Rome, 11-15
October, 1982, Rome, Food and Agriculture Organization of the
United Nations.
FAO/WHO (1972) Trichlorfon. In: 1971 Evaluations of some pesticide
residues in food, Geneva, World Health Organization, pp. 183-230
(WHO Pesticide Residues Series, No. 1).
FAO/WHO (1976) Trichlorfon. In: 1975 Evaluations of some pesticide
residues in food, Geneva, World Health Organization, pp. 379-392
(WHO Pesticide Residues Series, No. 5).
FAO/WHO (1979) Trichlorfon. In: Pesticide residues in food: 1978
Evaluations, Rome, Food and Agriculture Organization of the United
Nations. pp. 235-255 (FAO Plant Production and Protection Paper 15
Sup.).
FAO/WHO (1986) Codex maximum limits for pesticide residues, Rome,
Codex Alimentarius Commission, Food and Agriculture Organization of
the United Nations. (CAC/Vol. XIII-Ed. 2).
FAO/WHO (1989) Guide to Codex recommendations concerning pesticide
residues. Part 8. Recommendations for methods of analysis of
pesticide residues, 4th ed., Rome, Codex Committee on Pesticide
Residues, Food and Agriculture Organization of the United Nations.
(CAC/PR8-1989).
FECHNER, G., FUEHRER, G., & ACKERMANN, H. (1968) [Studies of milk
contamination following trichlorfon treatment of milk cows.] Monatsh.
Veterinärmed., 23: 529-531 (in German).
FECHNER, G., KRETZSCHMANN, F., ACKERMANN, H., & TOPFER, H. (1971)
[Improved method of thin-layer chromatographic and enzymic
determination of trichlorfon and dichlorvos residues in milk.]
Monatsh. Veterinärmed., 26: 860-863 (in German).
FEDORENKO, A.P., ALEEVA, L.V., & TITOK, V.A. (1981) [Organophosphorus
pesticide accumulation in warm-blood animals after chemical treatment
of the forest.] Vestn. Zool., 4: 89-92 (in Russian).
FERREIRA, J.R. & FERNANDES, A.M.S.S. (1980) Gas-liquid chromatographic
determination of organophosphorus insecticide residues in fruits and
vegetables. J. Assoc. Off. Anal. Chem., 63: 517-522.
FILATOV, G.V., SIVOKHIN, P.A., & METELITSA, V.K. (1972) Trichlorfon
excretion in milk. Veterinariya, 5: 102 (in Russian).
FISCHER, G.W., SCHNEIDER, P., & SCHEUFLER, H. (1977) Mutagenicity of
dichloroacetaldehyde and methyl-2,2- dichloro-1,1-dihydroxyethane
phosphonate, possible trichlorfon metabolites. Chem.-biol.
Interact., 19: 205-213.
FOURNIER, E., SONNIER, M., & DALLY, S. (1978) Detection and assay of
organophosphate pesticides in human blood by gas chromatography.
Clin. Toxicol., 12: 457-462.
FRANCIS, B.M., METCALF, R.L., & HANSEN, L.G. (1985) Toxicity of
organophosphorus esters to laying hens after oral and dermal
administration. J. environ. Sci. Health, B 20: 74-95.
FROHLICH, D.R., BOEKER, E.A., & BRINDLEY, W.A. (1990) The kinetics and
inhibition of p-nitrophenylacetate-hydrolising esterases in a
solitary bee, Megachile rotundata (Fab.). Xenobiotica, 20(5):
481-487.
GAINES, T.B. (1969) Acute toxicity of pesticides. Toxicol. appl.
Pharmacol., 14: 515-534.
GARRETT, N.E., STACK, H.F., & WATERS, M.D. (1986) Evaluation of the
genetic activity profiles of 65 pesticides. Mutat. Res., 168:
301-325.
GETZ, M.E. & WHEELER, H.G. (1968) Thin-layer chromatography of
organophosphorus insecticides with several adsorbents and ternary
solvent systems. J. Assoc. Off. Anal. Chem., 51: 1101-1107.
GIANG, P.A. & CASWELL, R.L. (1957) Polarographic determination of
O,O-dimethyl 2,2,2-trichloro-1-hydroxyethylphosphonate (Bayer
L13/59). J. agric. food Chem., 5: 753-754.
GIANG, P.A., BARTHEL, W.F., & HALL, S.A. (1954) Colorimetric
determination of O,O-dialkyl 1-hydroxyphosphonates derived from
chloral. J. agric. food Chem., 2: 1281-1284.
GIBEL, W., VON, LOHS, K.H., WILDNER, G.P., & ZIEBARTH, D. (1971)
[Experimental studies on hepatotoxic and carcinogenic activity of
organophosphorus compounds, Trichlorfon.] Arch. Geschwülstforsch.,
37: 303-312 (in German).
GIBEL, W., VON LOHS, K.H., WILDNER, G.P., ZIEBARTH, D., & STIEGLITS,
R. (1973) [Experimental study on cancerogenic haematotoxic and
hepatotoxic activity of organophosphorus pesticides.] Arch.
Geschwülstforsch, 41: 311-328 (in German).
GILOT-DELHALLE, J., COLIZZI, A., MOUTSCHEN, J., & MOUTSCHEN-DAHMEN, M.
(1983) Mutagenicity of some organophosphorus compounds at the ADE6
locus of Schizosaccharomyces pombe. Mutat. Res., 117: (1-2) 139-148.
GINGERICH, D.A. & MIA, A.S. (1981) Clinical toxicosis and erythrocyte
cholinesterase inhibition of trichlorfon combined with mebendazole in
horses. Am. J. vet. Res., 42: 1645-1650.
GIOVANOLI-JAKUBCZAK, T., FITAK, B., & CHODKOWSKI, J. (1971) Photolysis
of Dipterex and DDVP under the influence of UV irradiation. Rocz.
Chem., 45(4): 689-694.
GLUCKMAN, J.C., BARCELO, D., DE JONG, G.J., FREI, R.W., MARIS, F.A.,
& BRINKMAN, U.A.T. (1986) Improved design and applications of an
on-line thermionic detector for narrow-bore liquid chromatography.
J. Chromatogr., 367: 35-44.
GONCHARUK, E.I., PERELYGIN, V.M., & MELENEVSKAYA, A.V. (1978)
[Analysis of migration of the chemicals from the soil into the
atmosphere.] Gig. i Sanit., 43: 41-45 (in Russian).
GRAHL, K., HORN, H., & HALLEBACH, R. (1981) [Effect of butonate,
trichlorfon and dichlorvos on plankton populations.] Acta hydrochim.
hydrobiol., 9: 147-161 (in German).
GRIFFIN, T.B. (1988) Safety evaluation and tumorigenesis of
trichlorfon in Rhesus monkeys: A ten-year study. Alamogordo, New
Mexico, White Sands Research Center, (Proprietary study report No.
800108 submitted to WHO by Bayer AG, Germany).
GROMYSZ-KALKOWSKA, K., SZUBARTOWSKA, E., & KACZANOWSKA, E. (1985)
Peripheral blood in the Japanese quail ( Coturnix coturnix japonica)
in acute poisoning by different insecticides. Comp. Biochem.
Physiol., C81: 290-312.
GROMYSZ-KALKOWSKA, K., SZUBARTOWSKA, E., SULIKOWSKA, J., & TROCEWICZ,
K. (1985) Peripheral blood changes of the Japanese quail ( Coturnix
coturnix japonica) following repeated small doses of trichlorfon.
Bull. environ. Contam. Toxicol., 35: 757-766.
GRUNDMANN, E. & HOBIK, H.P. (1966) Bay 15922/2 year feeding
experiment in rats/histology (Unpublished report submitted to WHO by
Farbenfabriken Bayer AG, Germany).
GRYGIEREK, E. & WASILEWSKA, B.E. (1981) The effect of Foschlor
(organic phosphorous acid esters) on zooplankton and benthos of fish
ponds. Ekol. Pol., 29: 257-270.
GUTH, J.A. (1967) [Thin-layer chromatographic separation scheme for
insecticidally active phosphoric acid esters.]
Pflanzenschutzberichte, 35: 129-143 (in German).
HAGAN, E.C., JENNER, P.M., & JONES, W.I. (1971) Increased lethal
effects of acutely administered anticholinesterases in female rats
prefed with similar agents. Toxicol. appl. Pharmacol., 18: 235-237.
HALEY, T.J., HARMER, J.H., HARMON, J.R., & DOOLEY, K.L. (1975)
Estimation of the LD1 and extrapolation of the LD0.1 for five
organophosphate pesticides. Arch. Toxicol., 34: 103-109.
HALLAK, M. & GIACOBINI, E. (1987) A comparison of the effects of two
inhibitors on brain cholinesterase. Neuropharmacology, 26: 521-530.
HALLAK, M. & GIACOBINI, E. (1989) Physostigmine, tacrine, and
metrifonate: The effect of multiple doses on acetylcholine metabolism
in rat brain. Neuropharmacology, 28: 199-206.
HAMEED, S.F., SURI, S.M., & KASHYAP, N.P. (1980) Toxicity and
persistence of residues of some organophosphorus insecticides applied
for the control of Dacus cucurbitae Coquillet on the fruits of
cucumber. Indian J. agric. Sci., 50: 73-77.
HANNA, P.J. & DYER, K.F. (1975) Mutagenicity of organophosphorus
compounds in bacteria and Drosophila. Mutat. Res., 28: 405-420.
HANNA, S., BASMY, K., OSAIMA, S., SHOEB, S.M., & AWNY, A.Y. (1966)
Effects of administration of an organophosphorus compound as an
antibilharzial agent with special reference to plasma cholinesterase.
Br. med. J., 1: 1390-1392.
HANSEN, H.J., KRONEVI, T., & BJÖRKLUND, N.E. (1978) On cerebellar
hypoplasia in domestic animals. In: 13th Nord. Vet. Congress Abo,
July 1978.
HASHIMOTO, Y., OKUBO, E., ITO, T., YAMAGUCHI, M., & TANAKA, S. (1982)
Changes in susceptibility of carp to several pesticides with growth.
J. pestic. Sci., 7: 457-461.
HASSAN, A. & ZAYED, S.M.A.D. (1965) Metabolism of organophosphorus
insecticides. III. Fate of the methyl groups of Dipterex in vivo.
Can. J. Biochem., 43: 1271-1275.
HASSAN, A., ZAYED, S.M.A.D., & HASHISH, S. (1965) Metabolism of
organophosphorus insecticides. VI. Mechanism of detoxification of
Dipterex in the rat. Biochem. Pharmacol., 14: 1692-1694.
HATTORI, K. (1974) [Toxicological studies on the effect of chemicals
on birds (part 2). Oral acute toxicity of three organophosphate
insecticides in the common pigeon.] Hokkaidoritsu Eisei Kenkyusho
Ho, 24: 154 (in Japanese).
HAYES, R.H. (1988) Oncogenicity study of technical grade trichlorfon
with mice, Stillwell, Kansas, Mobay Corporation (Proprietary study
report No. 1039 submitted to WHO by Bayer AG, Germany).
HAYES, R.H. (1989) Chronic toxicity/Oncogenicity study of technical
grade trichlorfon (Dylox (R) ) with rats, Stillwell, Kansas,
Mobay Corporation (Proprietary study report No. 1108, submitted to
WHO by Bayer AG, Germany).
HAYES, W.J. Jr (1982) Pesticides studied in man, Baltimore,
Maryland, Williams & Wilkins Co., pp. 351-356.
HAYES, W.J. Jr & LAWS, E.R. Jr (1991) Fenitrothion. In: Handbook of
pesticide toxicology, New York Academic Press, pp. 1020-1023.
HERBOLD, B. (1979a) Micronucleus test on mouse to evaluate L 13/59
(Trichlorfon) for potential mutagenic effects, Wuppertal-Elberfeld,
Bayer AG, Institute of Toxicology, (Proprietary report No. 8505,
submitted to WHO by Bayer AG, Germany).
HERBOLD, B. (1979b) L 13/59 (Trichlorfon) -Dominant lethal study on
male mouse to test for mutagenic effects, Wuppertal-Elberfeld, Bayer
AG, Institute of Toxicology, (Proprietary report No. 8745, submitted
to WHO by Bayer AG, Germany).
HIERONS, R. & JOHNSON, M.K. (1978) Clinical and toxicological
investigations of a case of delayed neuropathy in man after acute
poisoning by an organophosphorus pesticide. Arch. Toxicol., 40:
279-284.
HINTON, M.J. & EVERSOLE, A.G. (1980) Toxicity and tolerance studies
with yellow-phase eels: five chemicals. Prog. Fish Cult., 42:
201-203.
HIRAMATSU, R. & FURUTANI, F. (1977) [Persistence of sprayed
insecticides in head vegetables.] Bull. Yamaguchi-ken agric. Explor.
Stn, 28: 159-163 (in Japanese).
HIRAMATSU, R. & FURUTANI, F. (1978) [Persistence of sprayed malathion
and trichlorfon in grape berries and grape products.] Bull.
Yamaguchi-ken agric. Explor. Stn, 30: 37-44 (in Japanese).
HOFER, W. (1981) Chemistry of metrifonate and dichlorvos. Acta
pharmacol. Scand., 49(suppl.5): 7-14.
HOFFMANN, K., RUF, J., & MAGER, H. (1988) Study of the chronic
toxicity to Rhesus monkeys, oral administration for 26 weeks (oral
intubation tube administration). Wuppertal, Bayer AG, (Proprietary
study report No. 90237, submitted to WHO by Bayer, AG, Germany).
HOLMSTEDT, B., NORDGREN, I., SANDOZ, M., & SUNDWALL, A. (1978)
Metrifonate, summary of toxicological and pharmacological information
available. Arch. Toxicol., 41: 3-29.
HU, X., LU, Y., XUE, S., LING, Y., & GU, X. (1986) Toxicity of
dipterex: A field study. Br. J. ind. Med., 43: 414-419.
IARC (1983) Trichlorfon. In: Miscellaneous pesticides, Lyon,
International Agency for Research on Cancer, pp. 207-231 (IARC
Monographs on the evaluation of the carcinogenic risk of chemicals to
humans, Vol. 30).
IARC (1987) Overall evaluations of carcinogenicity: An updating of
IARC Monographs volumes 1 to 42, Lyon, International Agency for
Research on Cancer, p. 73 (IARC Monographs on the evaluation of the
carcinogenic risk of chemicals to humans, Suppl. 7).
IARC (in press) Re-evaluation of dichlorvos. Lyon, France, IARC
(IARC Monographs on the evaluation of the carcinogenic risk of
chemicals to humans.
IMANAKA, M., MATSUNAGA, K., & ISHIDA, T. (1981) [Gas-liquid
chromatographic determination of trichlorfon (Dipterex) in livestock
products using a flame photometric detector.] J. Food Hyg. Soc.
Jpn., 22: 472-478 (in Japanese).
IRPTC/GKNT (1983) In: Izmerov, N.F., ed. Trichlorfon, Moscow, Centre
of International Projects, GKNT, 24 pp. (Scientific Reviews of Soviet
Literatures on Toxicity and Hazards of Chemicals No. 33).
ISAAC, I.G., SHAFIC, M.T., TANTAWY, G., & EL-KHISHEN, S.A. (1965)
Trichlorfon residues on cotton. Alexandria J. agric. Res., 14: 3-4.
ISHIDATE, M., Jr, SOFUNI, T., & YOSHIKAWA, K. (1981) Chromosomal
aberration tests in vitro as a primary screening tool for
environmental mutagens and/or carcinogens. Gann, 27: 95-108.
ISHIZAKA, T., SEKITA, H., SUZUKI, T., & SAITOU, Y. (1986) Analytical
method for trichlorfon in agricultural foods by gas chromatography.
Shokuhin Eiseigaku Zasshi, 27: 288-295.
IVANOVA, L.N. & MOLOZHANOVA, E.G. (1974) [Kinetics of the
transformation of some pesticides in the soil.] Khim. Sel'sk. Khoz.,
12: 363-365 (in Russian).
IWATA, Y., DUSH, M.E., CARMAN, G.E., & GUNTHER, F.A. (1979) Worker
environment research, residues from carbaryl, chlorobenzilate,
dimethoate and trichlorfon applied to citrus trees. J. agric. food
Chem., 27: 1141-1145.
JAPAN PLANT PROTECTION ASSOCIATION (1985) [Pesticides handbook],
Tokyo, Japan Plant Protection Association (in Japanese).
JAPAN PLANT PROTECTION ASSOCIATION (1986) [Production statistics of
major pesticides - fenitrothion and trichlorfon.] In: [Pesticides
handbook], Tokyo, Japan Plant Protection Association, p. 54 (in
Japanese).
JAPAN PLANT PROTECTION ASSOCIATION (1988) [Production statistics of
major pesticides - fenitrothion and trichlorfon.] In: [Pesticides
handbook], Tokyo, Japan Plant Protection Association, p. 57 (in
Japanese).
JAPAN PLANT PROTECTION ASSOCIATION (1989) [Production statistics of
major pesticides - fenitrothion and trichlorfon.] In: [Pesticides
handbook], Tokyo, Japan Plant Protection Association, p. 60 (in
Japanese).
JOHANSEN, C.A. (1972) Toxicity of field-weathered insecticide residues
to four kinds of bees. Environ. Entomol., 1: 393-394.
JOHNSON, M.K. (1981) Delayed neurotoxicity - do trichlorfon and/or
dichlorvos cause delayed neuropathy in man or in test animals? Acta
pharmacol. toxicol., 49(Suppl.V): 87-98.
JOHNSON, M.K. (1990) Organophosphates and delayed neuropathy - is NTE
alive and well? Toxicol. appl. Pharmacol., 102: 385-399.
JONES, D.C.L., SIMMON, V.F., MORTELMANS, K.E., MITCHELL, A.D., EVANS,
E.L., JOTZ, M.M., RICCIO, E.S., ROBINSON, D.E., & KIRKHART, B.A.
(1982) In vitro mutagenicity studies of environmental chemicals,
Washington, DC, National Technical Information Service.
KAWACHI, T., KOMATSU, T., KADA, T., ISHIDATE, M., Jr, SASAKI, M.,
SUGIYAMA, T., & TAZIMA, Y. (1980) Results of recent studies on the
relevance of various short-term screening tests in Japan. In: The
predictive value of short-term screening tests in carcinogenicity
evaluation, Amsterdam, Elsevier/North-Holland Biomedical Press, pp.
253-267.
KAWAI, M., YOSHIDA, M., IYATOMI, A., KOYAMA, M., & KANEKO, Y. (1982)
[Exposure of spray-operators to trichlorfon.] Nippon koshu Eisei
Zasshi, 29: 165-171 (in Japanese).
KAWAMOTO, K. & URANO, K. (1989) Parameters for predicting fate of
organochlorine pesticides in the environment (I) Octanol-water and
air-water partition coefficients. Chemosphere, 18: 1987-1996.
KIMMERLE, G. & LOSER, E. (1974) Delayed neurotoxicity of
organophosphorus compounds and copper concentration in the serum of
hens. Environ. Qual. Saf., 3: 173-178.
KIMMERLE, G. (1975a) Acute inhalation toxicity study on rats
(Unpublished report No. 5581 from Bayer AG, Germany).
KIMMERLE, G. (1975b) Subacute inhalation toxicity study on rats
(Unpublished report No. 5582 from Bayer AG, Germany).
KIMURA, S., YOKOTE, M., & MATIDA, Y. (1971) Study on the toxicity of
agricultural control chemicals in relation to freshwater fisheries
management No.6 Chronic toxicity of dieldrin, lindane and dipterex to
cherry salmon fingerlings. Bull. Freshwater Fish. Res. Lab., 21:
107-116.
KIRALY, J., SZENTESI, I., RUZICSKA, M., & CZEIZE, A. (1979) Chromosome
studies in workers producing organophosphate insecticides. Arch.
environ. Contam. Toxicol., 8: 309-319.
KLISENKO, M.A. & PISMENNAYA, M.V. (1979) [Photochemical conversion of
organophosphorus pesticides in the air.] Gig. Tr. prof. Zabol.,
56-58 (in Russian).
KNOX, B., ASKAA, J., BASSE, A., BITSCH, V., ESKILDSEN, M., MANDRUP,
M., OTTOSEN, H.E., OVERBY, E., PEDERSEN, K.B., & RASMUSSEN, F. (1978)
Congenital ataxia and tremor with cerebellar hypoplasia in piglets
borne by sows treated with Neguvon(R) vet (Metrifonate, Trichlorfon)
during pregnancy. Nord. vet. Med., 30: 538-545.
KOKOSZKA, L.C. & FLOOD, J.W. (1989) Environmental management handbook.
Appendix 15: Common chemical toxicants found at hazardous waste sites,
New York, Basel, Marcel Dekker.
KONRAD, H., GABRIO, T., & PENNER, G. (1975) [Pesticide contamination
of milk and milk products.] Milchforsch-Milchprax. 17: 67-69 (in
German).
KOWALSKI, R.L., CLEMENS, G.R., BARE, J.J., & HARTNAGEL, R.E. Jr,
(1987) A teratology study with Dylox (R) , technical (Trichlorfon)
in the rat. Elkhart, USA, Miles Laboratories, Toxicology Department,
(Proprietary report No. MTD0017, submitted to WHO by Bayer AG,
Germany).
KRONEVI, T., HANSEN, H.J., & JONSSON, O.J. (1975) Cerebellar
hypoplasia of unknown etiology in pigs. Vet. Rec., May 1975:
403-404.
KRUSTEV, E., KONSTANTINOV, P., & VASILEV, V. (1976) [The effect of
subtoxic doses of dipterex on guinea pigs.] Vet.-Med. Nauki, 13(1):
48-51 (in Russian).
KURINNIY, A.I. (1975) [Comparative investigation of the cytogenetic
effect of certain organophosphorus pesticides.] Genetika, 11: 64-69
(in Russian).
KURINNIY, A.I. & PILINSKAYA, M.A. (1977) [Cytogenetic acticity of the
pesticides imidan, dipterex and gardona in cultured human
lymphocytes.] Genetika, 13: 337-339 (in Russian).
KURTZ, D.A. & STUDHOLME, C.R. (1974) Recovery of trichlorfon
(Dylox(R)) and carbaryl (Sevin(R)) in songbirds following spraying
of forest for gypsy moth. Bull. environ. Contam. Toxicol., 11:
78-84.
KUZMENKO, N.M., DIDENKO, G.G., KURINNIY, A.L., MARTSON, L.V.,
PETROVSKAYA, O.G., PILINSKAYA, M.A., & SHEPELSKAYA, N.R. (1980)
[Long-term health effects of pesticides.] Gig. i Sanit., 45: 88-89
(in Russian).
LAMB, M.J. (1977) Mutagenicity tests with metrifonate in Drosophilla
melanogaster. Mutat. Res., 56: 157-162.
LANGE, C. (1980) [Identification of a metabolite of trichlorfon.] Z.
Chem., 20: 446-447 (in German).
LEBRUN, A. & CERF, C. (1960) Note préliminaire sur la toxicité pour
l'homme d'un insecticide organophosphoré (Dipterex). Bull. World
Health Organ., 22: 579-582.
LEHOTZKY, K. (1982) Effect of pesticides on central and peripheral
nervous system function in rats. Neurobehav. Toxicol. Teratol., 4:
665-669.
LIVINGSTON, R.J. (1977) Review of current literature concerning the
acute and chronic effects of pesticides on aquatic organisms. CRC
Crit. Rev. environ. Control, November 1977: 325-351.
LOGVINENKO, V.F. & MORGUN, V.V. (1978) Study of mutagenic action of
certain pesticides on spring durum wheat. Cytol. Genet. (USSR), 12:
207-212.
LORENZ, W., HENGLEIN, A., & SCHRADER, G. (1955) The new insecticide
O,O-dimethyl 2,2,2-trichloro-1-hydroxy ethylphosphonate. J. Am.
Chem. Soc., 77: 2554-2556.
LORKE, D. & LOSER, E. (1966) Chronic toxicological studies on rats
(Unpublished report submitted to WHO by Farbenfabriken Bayer AG,
Germany).
LOSER, E. (1969) [Propagation trials in rats.] (Unpublished report
submitted to WHO by Farbenfabriken Bayer AG, Germany) (in German).
LOSER, E. (1970) Bay 15992/Chronic toxicological studies on dogs
(Unpublished report submitted by Farbenfabriken Bayer AG, Germany).
LU, Y.P., LU, P., XUE, S., & GU, X. (1984) Investigation on the
chronic effects of dipterex in occupational exposure. Med. Lav.,
75: 376-384.
MCCANN, J.A. & JASPER, R.L. (1972) Vertebral damage to bluegills
exposed to acutely toxic levels of pesticides. Trans. Amer. Fish.
Soc., 101: 317-322.
MCGREGOR, D.B., BROWN, A., CATTANACH, P., EDWARDS, I., MCBRIDE, D.,
RIACH, C., & CASPARY, W.J. (1988) Responses of the L5178Y tk+/tk-
mouse lymphoma cell forward mutation assay: III. 72 coded chemicals.
Environ. mol. Mutagen., 12(1): 85-154.
MACDOUGALL, D. (1964) Dylox. In: Zweig, G. ed. Analytical methods for
pesticides and plant growth regulators, New York, London, Academic
Press, Vol. 2, pp. 199-208.
MACHEMER, L. (1979a) Trichlorfon - Evaluation for embryotoxic and
teratogenic effects on orally dosed rats, Wuppertal-Elberfeld, Bayer
AG, Institute of Toxicology (Proprietary report No. 8400, submitted to
WHO by Bayer AG, Germany).
MACHEMER, L. (1979b) Trichlorfon - Evaluation for embryotoxic and
teratogenic effects on orally dosed rabbits, Wuppertal-Elberfeld,
Bayer AG, Institute of Toxicology (Proprietary report No. 8430,
submitted to WHO by Bayer AG, Germany).
MACHEMER, L. (1981) Chronic toxicity of metrifonate. Acta pharmacol.
toxicol., 49(Suppl.V): 15-28.
MAGALHAES, M.J.A., FERREIRA, J.R., FRUTUOSA, L., & TAINHA, A.A. (1989)
Study of the disappearance of endosulfan, parathion, trichlorfon, and
pirimicarb from broccoli and portuguese cabbage. Pestic. Sci., 27:
23-31.
MAHENDRU, V.K. & AGARWAL, R.A. (1981) Changes in carbohydrate
metabolism in various organs of the snail, Lymnaea acuminata
following exposure to trichlorfon. Acta pharmacol. toxicol., 48:
377-381.
MANDOUL, R., DUBOS, M., DE COURNUAUD, M., & MOULINIER, CL. (1967) In
vitro effects of organophosphorus compounds on the Portugese oyster,
some molluscs, and freshwater microplankton. Bull. Soc. Pathol.
Exot., 60: 568-580.
MARKING, L.L. & MAUCK, W.L. (1975) Toxicity of paired mixtures of
candidate forest insecticides to rainbow trout. Bull. environ.
Contam. Toxicol., 13: 518-523.
MARTSON, L.V. & VORONINA, V.M. (1976) Experimental study of the effect
of a series of phosphoroorganic pesticides (dipterex and imidan) on
embryogenesis. Environ. Health Perspect., 13: 121-125.
MATKOVICS, B., SZABO, L., MINDSZENTY, L., & IVAN, J. (1980) The
effects of organophosphate pesticides on some liver enzymes and lipid
peroxidation. Gen. Pharmacol., 11: 353-356.
MATKOVICS, B., SZABO, L., IVAN, J., & GAAL, I. (1983) Some further
data on the effects of two organophosphate pesticides on the oxidative
metabolism in the liver. Gen. Pharmacol., 124: 689-691.
MATTON, P. & LAHAM, Q.N. (1969) Effect of the organophosphate Dylox on
rainbow trout larvae. J. Fish. Res. Board Can., 26: 2193-2200.
MELNIKOV, N.N., VORONKOVA, V.V., VORONTSOVA, N.A., GUNAR, M.I., SUPIN,
G.S., MOROZOVA, T.N., SHCHERBATYKH, YU. I., & SHVETSOVA-SHILVOSKAYA,
K.D. (1975) [Identification of impurities in technical chlorofos.]
Khim. Sel'sk. Khoz., 13: 142-145 (in Russian).
MENDOZA, C.E. (1973) Analysis of pesticides by the thin-layer
chromatographic-enzyme inhibition technique. Residue Rev., 50:
43-72.
METCALF, R.L., FUKUTO, T.R., & MARCH, R.B. (1959) Toxic action of
Dipterex and DDVP to the house fly. J. econ. Entomol., 52: 44-49.
MIHAIL, F. (1985) L 13/59 (Trichlorfon) - Study for skin-sensitizing
effect on guinea-pigs, Wuppertal-Elberfeld, Bayer AG, Institute of
Toxicology (Proprietary report No. 13323, submitted to WHO by Bayer
AG, Germany).
MIHAIL, F. (1986a) L 13/59 (Trichlorfon) - Study for skin-sensitizing
effect on guinea-pigs in the open epicutaneous test,
Wuppertal-Elberfeld, Bayer AG, Institute of Toxicology (Proprietary
report No. 14348, submitted to WHO by Bayer AG, Germany).
MIHAIL, F. (1986b) L 13/59 (Trichlorfon) - Skin sensitizing effect -
an evaluation, Wuppertal-Elberfeld, Bayer AG, Institute of
Toxicology, (Proprietary statement 11 March 1986, submitted to WHO by
Bayer AG, Germany).
MIYAMOTO, J. (1959) Non-enzymatic conversion of Dipterex into DDVP and
their inhibitory action on enzymes. Botyu-Kagaku, 24: 130-137.
MIYAMOTO, J. (1961) Studies on the mode of action of Dipterex. II. New
glucuronides obtained from the urine of rabbit following
administration of Dipterex. Agric. biol. Chem., 25: 566-572.
MIYATA, T. & SAITO, T. (1973) [Metabolism of NS 2662
( O,O-dimethyl-2,2-dichloro-1-hydroxyethylphosphonate).]
Botyu-Kagaku, 38: 81-86 (in Japanese).
MOLLHOFF, E. (1971) Determination of trichlorfon and fenthion residues
in animals of different species. Pestic. Sci., 2: 179-181.
MONSON, M.H. & ALEXANDER, K. (1984) Metrifonate in pregnancy. Trans.
R. Soc. Trop. Med. Hyg., 78: 565.
MORIYA, M., OHTA, T., WATANABE, K., MIYAZAWA, T., KATA, K., & SHIRASU,
Y. (1983) Further mutagenicity studies on pesticides in bacterial
reversion assay systems. Mutat. Res., 116: 185-216.
MORPURGO, G., AULICINO, F., BIGNAMI, M., CONTI, L., VELCICH, A. &
MONTALENTI, S.G. (1977) Relationship between structure and
mutagenicity dichlorvos and other pesticides. Lincei Rend. Sci. fis.
mat. nat., 62: 692-701.
MOUTSCHEN-DAHMEN, J. (1981) Metrifonate and dichlorvos cytogenetic
investigations. Acta pharmacol. toxicol., 49(Suppl.): 29-39.
MÜHLMANN, R. & SCHRADER, G. (1957) [Hydrolysis of the phosphoric acid
ester insecticide.] Z. Naturforsch., B12: 196-208 (in German).
NAGAYAMA, T., MAKI, T., KAN, K., MAKI, M., & NISHIMA, T. (1986) Survey
of pesticide residues in vegetables and fruits. Ann. Rep. Tokyo
Metrop. Res. Lab. P.H., 37: 173-183 (in Japanese).
NAGAYAMA, T., MAKI, T., IIDA, M., KAN, K., & NISHIMA, T. (1987) Survey
of pesticide residues in vegetables and fruits. Ann. Rep. Tokyo
Metrop. Res. Lab. P.H., 38: 222-228 (in Japanese).
NAGAYAMA, T., MAKI, T., IIDA, M., KAN, K., KAWAI, Y., & NISHIMA, T.
(1988) Survey of pesticide residues in vegetables and fruits. Ann.
Rep. Tokyo Metrop. Res. Lab. P.H., 39: 130-138 (in Japanese).
NAGAYAMA, T., MAKI, T., IIDA, M., KAN, K., KAWAI, Y., & NISHIMA, T.
(1989) Survey of pesticide residues in vegetables and fruits. Ann.
Rep. Tokyo Metrop. Res. Lab. P.H., 40: 155-162 (in Japanese).
NAGY, Z.S., MILE, I., & ANTONI, F. (1975) The mutagenic effect of
pesticides on Escherichia coli WP2 try. Acta Microbiol. Acad. Sci.
Hung., 22: 309-314.
NAISHTEIN, S.Ya. (1976) [Trichlorfon and malathion tolerances in
soil.] Khim. Sel'sk. Khoz., 14: 32-34 (in Russian).
NAISHTEIN, S.Ya., ZHULINSKAYA, V.A., & YUROVSKAYA, E.M. (1973)
[Stability of certain organophosphorous pesticides in soil.] Gig. i
Sanit., 42-45 (in Russian).
NAKAHARA, T., TAKASE, I., KAWAGOE, I., KUBOTA, T., & SAITO, T. (1972)
[Residues of trichlorfon and its metabolites in fresh milk.]
Bochu-kagaku, 37: 149-154 (in Japanese).
NAKAHARA, T., TAKASE, I., & YOSHIMOTO, T. (1973) [Residues of
trichlorfon in eel and carp.] Noyaku Kagaku, 1(2): 75-76 (in
Japanese).
NAKAMURA, T. & UEDA, T. (1967) Studies on the mechanisms of the
monofluoroacetanilides poisoning to mammals; Part VIII.
Organophosphorous compounds as a specific inhibitor to
fluoroacetanilide amidohydrolase. Agric. biol. Chem., 31: 1288-1293.
NEHEZ, M., SELYPES, A., & SCHEUFLER, H. (1982) Mutagenic effect of
trichlorphon and dimethoate on mouse bone marrow cells in vivo. Wiss.
Z. Martin-Luther-Univ. Halle-Wittenberg, 31(2): 57- 62.
NEHEZ, M., HUSZTA, E., MAZZAG, E., SCHEUFLER, H., SCHNEIDER, P., &
FISCHER, G.W. (1987) Cytogenetic, genetic, and embryotoxicity studies
with dimethyl-2,2,2,-trichloro-1-(2,2,2-trichloro-1-hydroxy-
ethoxy)-ethylphosphonate, a hypothetical impurity in technical grade
trichlorfon. Ecotoxicol. environ. Saf., 13: 216-224.
NEPOKLONOV, A.A. & BUKSHTYNOV, V.I. (1971) [Trichlorfon release time
from sheep after aerosol treatment.] Tr. Vses. Nauchn. Issled. Inst.
Sanit., 39: 200-202 (in Russian).
NEPOKLONOV, A.A. & METELITSA, V.K. (1970) [Hypoderminchlorofos used in
the prophylaxis of the hypodermatidae.] Veterinariya, 46: 70-72 (in
Russian).
NIEDZIELLA, S.W., GÖPEL, W., & BANZHAF, E. (1985) [Acute
alkylphosphate poisoning (Trichlorfon) with delayed polyneuropathy.]
Z. gesamte inn. Med., 40(8): 237-239 (in German).
NIKOLAYEV, P.I. (1972) [Distribution of trichlorfon and amidophos in
horses and the secretion of those compounds with mare's milk.] Tr.
Vses. Nauchn. Issled. Vet. Sanit., 44: 91-98 (in Russian).
NISHIUCHI, Y. (1977) [Acute toxicity of pesticide formulations to
Cyprinus carpio at 5 different temperatures.] Suisan Zoshoku, 24:
140-145 (in Japanese).
NISHIUCHI, Y. (1979a) [Acute toxicity of pesticide formulations to
Cyprinus carpio .] Suisan Zoshoku, 27: 36-41 (in Japanese).
NISHIUCHI, Y. (1979b) [Acute toxicity of pesticide formulations to
Daphnia pulex. ] Suisan Zoshoku, 27: 42-47 (in Japanese).
NISHIUCHI, Y. (1979c) [Acute toxicity of pesticide formulations to
Daphnia carinata.] Suisan Zoshoku, 27: 119-124 (in Japanese).
NISHIUCHI, Y. (1981) [Evaluation of the effect of pesticides on some
aquatic organisms. I. Effect of pesticides on some aquatic insects.]
Seitai Kagaku, 4: 31-46 (in Japanese).
NISHIUCHI, Y. & ASANO, K. (1979) [Acute toxicity of pesticide
formulations to Cloeon dipterum.] Suisan Zoshoku, 27: 48-55 (in
Japanese).
NISHIUCHI, Y. & YOSHIDA, K. (1972) Toxicity of pesticides to fresh
water snails. Bull. agric. Chem. Insp. Stn., 12: 86-92.
NORDGREN, I. (1981) Quantitation of metrifonate and dichlorvos in
blood and tissues by gas chromatography - mass spectrophotometry.
Fundam. appl. Toxicol., 1: 230-237.
NORDGREN, I., BERGSTRÖM, M., HOLMSTEDT, B., & SANDOZ, M. (1978)
Transformation and action of Metrifonate. Arch. Toxicol., 41: 31-41.
NORDGREN, I., HOLMSTEDT, B., BENGTSSON, E., & FINKEL, Y. (1980) Plasma
levels of metrifonate and dichlorvos during treatment of
schistosomiasis with bilarcil. Am. J. trop. Med. Hyg., 29(3):
426-430.
NORDGREN, I., BENGTSSON, E., HOLMSTEDT, B., & PETTERSSON, B.M. (1981)
Levels of metrifonate and dichlorvos in plasma and erythrocytes during
treatment of schistosomiasis with bilarcil. Acta pharmacol.
toxicol., 49(Suppl.V): 79-86.
OLAJOS, E.J. & ROSENBLUM, I. (1981) Membrane-bound and soluble
esterase activities in various hen brain regions after diisopropyl
phosphorofluoridate and trichlorfon treatment. Neurotoxicology, 2:
463- 470.
OLAJOS, E.J., ROSENBLUM, I., COULSTON, F., & STROMINGER, N. (1979) The
dose response relationship of trichlorfon neurotoxicity in hens.
Ecotoxicol. environm. Saf., 3: 245-255.
ORDOG, V. (1979) [Effect of organophosphate insecticides on green
algae.] Hydrol. Kozl., 59: 36-42 (in Hungarian).
OSBORNE, B.G., BARRETT, G.M., LAAL-KHOSHAB, A., & WILLIS, K.H. (1989)
The occurrence of pesticide residues in UK home-grown and imported
wheat. Pestic. Sci., 27: 103-109.
OTTO, D., BEHNISCH, I., PFEIFFER, G., DEDEK, W., & GEORGI, W. (1980)
Metabolismus von Trichlorfon in sensiblen und resistenten Fliegen,
Musca domestica L., Arch. Phytopathol. Pflanzenschutz, 16: 397-411.
PAIK, S.G. & LEE, S.Y. (1977) Genetic effects of pesticides in the
mammalian cells. 1. Induction of micronucleus. Tongmul Hakhoe Chi,
20: 19-28.
PAKHLEVANYAN, A.S., PIRUMOV, S.S., DRAGA, O.N., ZORABYAN, S.L., &
CHARCHOGLYAN, M.S. (1979) [Effect of the treatment of cow with
trichlorfon on some milk composition indexes.] Molochn. Prom-st', 7:
42-44 (in Russian).
PANDA, B.B. & SHARMA, C.B.S.R. (1980) Cytogenetic hazards from
agricultural chemicals. 3. Monitoring the cytogenetic activity of
trichlorfon and dichlorvos in Hordeum vulgare. Mutat. Res., 78:
341-345.
PEAKALL, D.B. & BART, J.R. (1983) Impacts of aerial application of
insecticides on forest birds. CRC crit. Rev. environ. Control, 13:
117-165.
PIEPER, G.R. & RICHMOND, C.E. (1976) Residues of trichlorfon and
lauroyl trichlorfon in Douglas fir, Willow grass, aspen, foliage and
in creek water after aerial application. Bull. environ. Contam.
Toxicol., 15: 250-256.
POMPONI, M., GIACOBINI, E., & BRUFANI, M. (1990) Present state and
future development of Alzheimer diseases. Aging, 2(2): 125-153.
POOLE, D.C., SIMMON, V.F., & NEWELL, G.W. (1977) In vitro mutagenic
activity of fourteen pesticides. Toxicol. appl. Pharmacol., 41:
196 (Meeting abstract).
POPE, A.M., HEAVNER, J.E., GUARNIERI, J.A., & KNOBLOCH, C.P. (1986)
Trichlorfon-induced congenital cerebellar hypoplasia in neonatal pigs.
J. Am. Vet. Med. Assoc., 189: 781-783.
RASMUSSEN, F., BASSE, A., KRAUL, I., & DALGAARD-MIKKELSEN, Sr (1978)
Inhibition of cholinesterase activity in blood from pregnant sows and
hypoplasia of cerebellum in their piglets after oral application of
metrifonate (neguvon(r) vet). In: Nord. Vet. Congress Abo, July
1978.
REINER, E., KRAUTHACKER, B. SIMEON, V., & SKRINJARIC-SPOLIJAR, M.
(1975) Mechanism of inhibition in vitro of mammalian
acetyl-cholinesterase and cholinesterase in solutions of
O,O-dimethyl-2,2,2-trichloro-1-hydroxyethyl phosphonate
(trichlorphon). Biochem. Pharmacol., 24: 717-722.
REISS, C.S., HERRMAN, J.M., & HOPKINS, R.E., II (1987) Effect of
antihelminthic treatment on the immune response of mice. Lab. anim.
Sci., 37: 773-775.
RICCIO, E., SHEPHERD, G., POMEROY, A., MORTELMANS, K., & WATERS, M.D.
(1981) Comparative studies between the S. cerevisiae D3 and D7
assays of eleven pesticides. Environ. Mutagen., 3: 327 (Meeting
abstract).
ROBBINS, W.E., HOPKINS, T.L., & EDDY, G.W. (1956) The metabolism of
32P-labeled Bayer L13/59 in a cow. J. econ. Entomol., 49: 801-806.
ROBERTS, D. (1975) The effect of pesticides on byssus formation in the
common mussel, Mytilus edulis. Environ. Pollut., 8: 241-254.
ROSENKRANZ, H.S. & ROSENKRANZ, S. (1972) Reaction of DNA with
phosphoric acid esters: Gasoline additives and insecticides.
Experientia (Basel), 28: 386-387.
RYAZANOVA, R.A. & GAFUROVA, T.V. (1980) [Mutagenic action of a binary
mixture of the pesticides chlorophos and zineb.] Gig. i Sanit., 1:
80-81 (in Russian).
RYBAK, M. (1973) Effect of phosclor administered in water on the
ontogenetic development of white rats and their offsprings. Rocz.
Panstw. Zakl. Hig., 24: 465-475.
SALAMA, A.M., MOSTAFA, I.Y., & EL-ZAWAHRY, Y.A. (1975) Insecticides
and soil microorganisms: (III) Fate of C-14 labeled Dipterex as
affected by two nodule-forming Rhizobium spp. and roots of their
respective leguminous host plants. Acta biol. Acad. Sci. Hung., 26:
1-7.
SASAKI, M., SUGIMURA, K., YOSHIDA, M., & ABE, S. (1980) Cytogenetic
effects of 60 chemicals on cultured human and Chinese hamster cells.
Senshokutai, 20: 574-584.
SCHAFER, E.W. (1972) The acute oral toxicity of 369 pesticidal,
pharmaceutical and other chemicals to wild birds. Toxicol. appl.
Pharmacol., 21: 315-330.
SCHAFER, E.W., Jr, BOWLES, W.A. J, & HURLBUT, J. (1983) The acute oral
toxicity, repellency, and hazard potential of 998 chemicals to one or
more species of wild and domestic birds. Arch. environ. Contam.
Toxicol., 12: 355-382.
SCHEUFLER, H. (1975) Effect of relatively high doses of dimethoate and
trichlorfon on the embryogenesis of laboratory mice. Biol. Rundsch.,
13: 238-240.
SCHIEMANN, S. (1975) Mutagenic effect of trichlorfon on the mouse,
with special regard to the dominant lethal tests. Wiss. Z.
Martin-Luther-Univ. Halle-Wittenberg Math Naturwiss. R., 24: 85-86.
SCHLINKE, J.C. & PALMER, J.S. (1973) Combined effects of phenothiazine
and organophosphate insecticides in cattle. J. Am. Vet. Med. Assoc.,
163: 756-758.
SCHULTEN, H.-R. & SUN, S.-E. (1981) Field desorption mass spectrometry
of standard organophosphorus pesticides and their identification in
waste water. Int. J. environ. anal. Chem., 10: 247- 264.
SERGEANT, D.B. & ZITKO, V. (1979) The determination of trichlorfon,
dichlorvos, fenitrothion and phosphamidon in water. (Can. Fish Mars.
Tech. Rep, 886, pp. 1-12.
SHIMAMOTO, K. & HATTORI, K. (1965) Effects of chronic feeding of the
organic phosphorus compounds on the tissue cholinesterase in rats.
Acta med. Univ. Kyoto, 39: 128-137.
SHIRAISHI, S., GOTO, I., YAMASHITA, Y., OHNISHI, A., & NAGAO, H.
(1977) [Dipterex polyneuropathy.] Shinkei Naika, 6: 34-38 (in
Japanese).
SHIRAISHI, S., INOUE, N., & MURAI, Y. (1982) Dipterex poisoning. J.
UOEH., 4: 177-183.
SHIRAISHI, S., INOUE, N., MURAI, Y., ONISHI, A., & NODA, S. (1983)
Dipterex (Trichlorfon) poisoning - clinical and pathological studies
in human and monkeys. J. UOEH., 5(Suppl.): 125-132.
SHIRASU, Y., MORIYA, M., KATO, K., FURUHASHI, A., & KADA, T. (1976)
Mutagenicity screening of pesticides in the microbial system. Mutat.
Res., 40: 19-30.
SHIRASU, Y., MORIYA, M., KATO, K., TEZUKA, H., TERAMOTO, S., OHTA, T.,
& INOUE, T. (1982) Mutagenicity screening studies on pesticides. In:
Environmental mutagens and carcinogens, Tokyo, University of Tokyo
Press, pp. 331-335.
SILVESTRI, G.R., HIMES, J.A., & EDDS, G.T. (1975a) Repeated oral
administration of coumaphos in sheep: effects on erythrocyte
acetylcholinesterase and other constituents. Am. J. Vet. Res., 36:
283-287.
SILVESTRI, G.R., HIMES, J.A., & EDDS, G.T. (1975b) Repeated oral
administration of coumaphos in sheep: interactions of coumaphos with
bishydroxycoumarin, trichlorfon, and phenobarbital sodium. Am. J.
Vet. Res., 36: 289-292.
SIMMON, V.F. (1979) In vitro microbiological mutagenicity and
unscheduled DNA synthesis studies of eighteen pesticides, Research
Triangle Park, North Carolina, US Environmental Protection Agency, pp.
79-81, 87, 111 (EPA-600/1-79-041).
SINGH, O. & AGARWAL, R.A. (1979) Effects on certain carbamate and
organophosphorus pesticides on isolated organs of Pila globosa
(Gastropoda). Toxicol. appl. Pharmacol., 50: 485-492.
SINGH, O. & AGARWAL, R.A. (1981) Toxicity of certain pesticides to two
economic species of snails in northern India. J. econ. Entomol., 74:
568-571.
SISSONS, D.J. & TELLING, G.M. (1970) Rapid procedure for the
determination of organophosphorus insecticide residues in vegetables.
II. A screening procedure for water soluble insecticides. J.
Chromatogr., 48: 468-477.
SLOTT, V. & ECOBICHON, D.J. (1984) An acute and subacute neurotoxicity
assessment of trichlorfon. Can. J. Physiol. Pharmacol., 62: 513-518.
SNELLEN, W.M. (1981) Therapeutic properties of metrifonate. Acta
pharmacol. toxicol., 49(Suppl.5): 114-117.
SOLIMAN, M.S., EL-MISSERY, A.G., ABDELMESSIH, M.S., FAYAD, M.E., &
BASSILI, W.R. (1984) The effect of diazinon and neguvon on the liver
of experimentally intoxicated mice. J. Egypt. Soc. Parisitol., 14:
557-562.
SPEHAR, R.L., LEMKE, A.E., PICKERING, Q.H., ROUSH, T.H., RUSSO, R.C.,
& YOUNT, J.D. (1981) Effects of pollution on freshwater fish. J.
Water Pollut. Control. Fed., 53: 1028-1076.
SPICER, E.J.F. & URWIN, C. (1971) Pathology report of Bay 15922
generation experiment in rats (Unpublished report submitted to WHO
by Farbenfabriken Bayer AG, Germany).
STAN, H.J. (1977) [Detection of organophosphorous pesticide residues
in food at the ppb level with open tubular column gas chromatography-
mass spectrometry.] Z. Lebensm. Unters. Forsch., 164: 153-159 (in
German).
STAN, H.J., ABRAHAM, B., JUNG, J., KELLERT, M., & STEINLAND, K. (1977)
[Detection of organophosphorus insecticides by gas chromatography/mass
spectrometry.] Fresenius Z. anal. Chem., 287: 271-285 (in German).
STAPLES, R.E. & GOULDING, E.H. (1979) Dipterex teratogenicity in the
rat, hamster and mouse when given by gavage. Environ. Health
Perspect., 30: 105-113.
STAPLES, R.E., KELLAM, R.G., & HASEMAN, J.K. (1976) Developmental
toxicity in the rat after ingestion or gavage of organophosphate
pesticides (dipterex, imidan) during pregnancy. Environ. Health
Perspect., 13: 133-140.
STOBWASSER, H. & KIRCHHOFF, J. (1968) [Insecticide residues on lettuce
and spinach in outdoor and greenhouse cultivation.] Qual. Plant
Mater. Veg., 15: 273-279 (in German).
SUGIYAMA, H. & SHIGEMATSU, H. (1969) Enzymic dehydrochlorination of
trichlorfon by the digestive juice of the silkworm. Bombyx mori.
Botyu-Kagaku, 34: 79-85.
SUNDARAM, K.M.S. & VARTY, I.W. (1989) Distribution and persistence of
trichlorfon in a forest environment. J. Environ. Sci. Health, B
24(6): 647-659.
SZALONTAI, G. (1976) High-performance liquid chromatography of
organophosphorus insecticides. J. Chromatogr., 124: 9-16.
SZUBARTOWSKA, E. (1983) Peripheral blood picture in rabbits poisoned
by the pesticide trichlorfon. Folia biol., 31: 407-418.
SZUBARTOWSKA, E. & GROMYSZ-KALKOWSKA, K. (1986) Differences between
breeds of rabbits in their susceptibility to the effect of foschlor.
Comp. Biochem. Physiol., 85C: 33-40.
TAKASE, I., NAKAHARA, T., YOSHIMOTO, Y., & NAKAMURA, H. (1972)
[Determination of residues of trichlorfon and its metabolites in
plants, milk, chicken, and fish by gas chromatography.]
Botyu-Kagaku, 37: 142-148 (in Japanese).
TAYLOR, P. (1980) Anticholinesterase agents. In: Gilman, A.G.,
Goodman, L.S. & Gilman, A., ed. The pharmacological basis of
therapeutics, 6th ed., New York, MacMillan Publishing Co., pp.
100-119.
TEICHMANN, B. & HAUSCHILD, F. (1978) [Test of
O,O-dimethyl(1-hydroxy-2,2,2-trichloro-ethyl)phosphonate
(Trichlorfon) for carginogenic activity in mice by oral
(oesophageal-gastric intubation), intraperitoneal and dermal
application.] Arch. Geschwulstforsch, 48: 301-307 (in German).
TEICHMANN, B. & SCHMIDT, A. (1978) [Test of
O,O-dimethyl(1-hydroxy-2,2,2-trichloro-ethyl)phosphonate
(Trichlorfon) for carcinogenic activity in Syrian golden hamsters
( Mesocricetus auratus Waterhouse) by intraperitoneal
administration.] Arch. Geschwulst forsch, 48: 718-721 (in German).
TEICHMANN, B., HAUSCHILD, F., & ECKELMANN, A. (1978) [Test of O,O-
dimethyl (1-hydroxy-2,2,2-trichloroethyl)phosphonate (Trichlorofon)
for carcinogenic activity in rats by oral (oesophageal-gastric
intubation) and intraperitoneal administration.] Arch.
Geschwulstforsch., 48: 112-119, (in German).
TELEUGALIEV, T.M. (1977) [Veterinary hygienic study of fish poisoned
with trichlorfon.] Vestn. S-kh. Nauki Kaz., 20: 75-77 (in Russian).
THYSSEN, J. (1981) L 13/59 (Dipterex active ingredient) tests for
skin and eye irritation, Wuppertal-Elberfeld, Bayer AG, Institute of
Toxicology, (Proprietary report submitted to WHO by Bayer AG,
Germany).
TREFILOV, V.N., FAEDMAN, I.S., & BORISOVA, E.P. (1971) [Contamination
of working clothes and skin of workers engaged in the production of
metaphos and chlorophos.] Gig. Tr. prof. Zabol., 15: 51-53 (in
Russian).
TRINH VAN BAO, SZABO, I., RUZICSKA, P., & CZEIZEL, A. (1974)
Chromosome aberrations in patients suffering acute organic phosphate
insecticide intoxication. Humangenetik, 24: 33-57.
TSUMUKI, H., SAITO, T., MIYATA, T., & IYATOMI, K. (1970) Acute and
subacute toxicity of organophosphorus insecticides to mammals. In:
O'Brien, R.D. & Yamamoto, I., ed. Biochemical toxicology of
insecticides, New York, London, Academic Press, p. 65-73.
TSVETKOVA, T.S., PENEVA, V., & GRIGOROVA, D. (1981) [Residual amount
of pesticides in honey.] Vet. Med. Nauki, 18: 93-98 (in Russian).
UMEHARA, F., FUKUOKA, T., MINAUCHI, Y., OSAME, M., & IGATA, A. (1982)
A case of dipterex poisoning. A histopathological study of peripheral
nerves. Clin. Neurol., 22: 990-996.
US NTP (1989) NTP technical report on the toxicology and
carcinogenesis studies of dichlorvos (CAS No: 62-73-7) in F344/N rats
and B6C3F1 mice (gavage studies), Research Triangle Park, North
Carolina, National Toxicology Program (NTP TR 342).
VALENCIA, R. (1977) Mutagenesis screening of pesticides using
drosophia (Contract No. 68-01-2474), Madison, Wisconsin, WARF
Institute, Inc.
VAN MIDDELEM, C.H., HABECK, D.H., & STRAYER, J.R. (1972) Residues on
ryegrass following preplant or postemergence applications of four
insecticides for mole cricket control. J. econ. Entmol., 65:
495-497.
VASILESCU, C., ALEXIANU, M., & DAN, A. (1984) Delayed neuropathy after
organophosphorus insecticides (Dipterex) poisoning: a clinical,
electrophysiological, and nerve biopsy study. J. Neurol. Neurosurg.
Psychiatry., 47: 543-548.
VILCEANU, R., SCHULZ, P., DRAGHICI, R., & SOIMU, P. (1973) [Gas
chromato graphy of (1-hydroxy-2,2,2-trichlorethyl)-phosphonic acid
esters.] J. Chromatogr., 82: 285-290 (in German).
VÖLKNER, W. (1987) L 13/59 (Trichlorfon) - Sister chromatid exchanges
(SCEs) in Chinese hamster bone marrow cells, Technical University of
Darmstadt, Laboratory for Mutagenicity Testing (Proprietary report
No. R 4079, submitted to WHO by Bayer AG, Germany).
WATERS, M.D., SIMMON, V.F., MITCHELL, A.D., JORGENSON, T.A., &
VALENCIA, R. (1980) An overview of short-term tests for the mutagenic
and carcinogenic potential of pesticides. J. environ. Sci. Health,
B15: 867-906.
WATERS, M.D., NESNOW, S., SIMMON, A.D., MITCHELL, T.A., JORGENSON,
T.A., & VALENCIA, R. (1981) Pesticides: mutagenic and carcinogenic
potential. In: Bandal, S.K., Golberg, L., & Leng, M.L., ed. The
pesticide chemist and modern toxicology, Washington, DC, American
Chemical Society, pp. 80-113 (ACS Symposium Series, No. 160).
WATERS, M.D., SANDHU, S.S., SIMMON, V.F., MORTELMANS, K.E., MITCHELL,
A.D., JORGENSON, T.A., JONES, D.C.L., VALENCIA, R., & GARRET, N.E.
(1982) Study of pesticide genotoxicity. Basic Life Sci., 21:
275-326.
WEGNER, D. (1970) [Bilarcil(R) Bay 2349 - Clinical experience
1960-1969.] (Unpublished report submitted to WHO by Farbenfabriken
Bayer AG, Germany) (in German).
WELLBORN, T.L., Jr (1969) The toxicity of nine therapeutic and
herbicidal compounds to striped bass. Prog. Fish Cult., 31: 27-32.
WENDA-ROZEWICKA, L. (1983) Morphometric studies of male gonads from
mice receiving insecticides (Metox-30, Sadofos-30, and Foschlor-50).
Folia biol., 32: 23-34.
WHO (1985) The control of schistosomiasis: Report of a WHO expert
committee, Geneva, World Health Organization, pp. 113 (Technical
Report Series, No. 728).
WHO (1986) Environmental Health Criteria 63: Organophosphorus
insecticides: A general introduction, Geneva, World Health
Organization, pp. 181.
WHO (1989) Environmental Health Criteria 79: Dichlorvos, Geneva,
World Health Organization, pp. 157.
WHO (1990) The WHO recommended classification of pesticides by hazard
and guidelines to classification 1990-1991, Geneva, World Health
Organization, pp. 39 (Unpublished document WHO/PCS/90.1 Rev.1).
WHO/FAO (1977) Data sheets on pesticides, No. 27: Trichlorfon,
Geneva, World Health Organization, pp. 9 (VBC/D5/77.28).
WILD, D. (1975) Mutagenicity studies on organophosphorus insecticides.
Mutat. Res., 32: 133-150.
WILKINS, H.A. & MOORE, P.J. (1987) Comparative trials of regimes for
the treatment of urinary schistomiasis in the Gambia. J. trop. Med.
Hyg., 90: 83-92.
WILLIAMS, M.W., FUYAT, H.N., & FITZHUGH, O.G. (1959) The subacute
toxicity of four organic phosphates to dogs. Toxicol. appl.
Pharmacol., 1: 1-7.
WINTERLIN, W., WALKER, G., & FRANK, H. (1968) Detection of
cholinesterase-inhibiting pesticides following separation on
thin-layer chromatograms. J. agric. food Chem., 16: 808-812.
WOODWARD, D.F. & MAUCK, W.L. (1980) Toxicity of five forest
insecticides to cutthroat trout Salmo clarki and two species of
aquatic invertebrates. Bull. environ. Contam. Toxicol., 25: 846-853.
WRIGHT, A.S., HUTSON, D.H., & WOODER, A.F. (1979) The chemical and
biochemical reactivity of dichlorvos. Arch. Toxicol., 42: 1-18.
YAKOUB (1990) A rational approach to the treatment of Schistosoma
haematobium infection with metrifonate - clinical and pharmacological
studies, Stockholm, Karolinska Institute, (Dissertation).
YASHIKI, M., KOJIMA, T., & UNE, I. (1982) [Determination of Dipterex
in biological materials by gas chromatography and gas chromatography-
mass spectrometry and the analysis of the distribution of Dipterex in
a case of fatal poisoning.] Nippon Hoigaku Zasshi, 36: 426-430 (in
Japanese).
YASHIKI, M., CHIKASUE, F., MIYAZAKI, T., KOJIMA, T., & OHTANI, M.
(1988) [Gas chromatographic and gas chromatographic-mass spectrometric
determination of trichlorfon in biological fluids collected in a fatal
dipterex poisoning.] Nippon Hoigaku Zasshi, 42: 94-98 (in Japanese,
with English summary).
YONOVA, I. & ZHECHEVA, G. (1974) [Residues of sevin, trichlorfon and
imidan in the meat of pig treated for ectoparasites.] Vet. Med.
Nauki, 11: 28-34 (in Russian).
YÜKSEKISIK, N. (1973) Kinetic study of the rearrangement of O,O-
dialkyl 2,2,2-tricholoro-1-hydroxyethylphosphonates. Commun. Fac.
Sci. Univ. Ankara Ser., B 20: 49-62.
YUROVSKAYA, E.M. & ZHULINSKAYA, V.A. (1974) [Behavior of organo
phosphorous insecticides in the soil.] Khim. Sel'sk. Khoz., 12:
358-361 (in Russian).
ZAKHAROV, B.N. (1980) [Migration of trichlorfon in nature.] Gig. i
Sanit., 45: 62-63 (in Russian).
ZAYED, S.M.A.D., MOSTAFA, I.Y., & HASSAN, A. (1965) Metabolism of
organophosphorus insecticides. (VII) Transformation of 32P-labeled
Dipterex through microorganisms. Arch. Mikrobiol., 51: 118-121.
ZELINSKIY, M.I. (1974) [Toxicity of trichlorfon and its effect on the
quality of meat.] Vestn. S-kh. Nauki Kaz., 17(10): 66-68 (in
Russian).
ZINKL, J.G., HENNY, C.J., & DEWEESE, L.R. (1977) Brain cholinesterase
activities of birds from forests sprayed with trichlorfon (Dylox) and
carbaryl (Sevin-4-oil). Bull. environ. Contam. Toxicol., 17:
379-386.
ZWEIG, G. & SHERMA, J. (1972) Dylox. In: Analytical methods for
pesticides and plant growth regulators, New York, London, Academic
Press, Vol. 6, pp. 387-392.
ANNEX I. TREATMENT OF ORGANOPHOSPHATE
INSECTICIDE POISONING IN MAN
(From EHC 63: Organophosphorus Insecticides - A
General Introduction)
All cases of organophosphorus poisoning should be dealt with as
an emergency and the patient sent to hospital as quickly as possible.
Although symptoms may develop rapidly, delay in onset or a steady
increase in severity may be seen up to 48 h after ingestion of some
formulated organophosphorus insecticides.
Extensive descriptions of treatment of poisoning by
organophosphorus insecticides are given in several major references
(Kagan, 1977; Taylor, 1980; UK DHSS, 1983; Plestina, 1984) and will
also be included in the IPCS Health and Safety Guides to be prepared
for selected organophosphorus insecticides.
The treatment is based on:
(a) minimizing the absorption;
(b) general supportive treatment; and
(c) specific pharmacological treatment.
I.1 Minimizing the absorption
When dermal exposure occurs, decontamination procedures include
removal of contaminated clothes and washing of the skin with alkaline
soap or with a sodium bicarbonate solution. Particular care should be
taken in cleaning the skin area where venepuncture is performed. Blood
might be contaminated with direct-acting organophosphorus esters and,
therefore, inaccurate measures of ChE inhibition might result.
Extensive eye irrigation with water or saline should also be
performed. In the case of ingestion, vomiting might be induced, if the
patient is conscious, by the administration of ipecacuanha syrup
(10-30 ml) followed by 200 ml water. This treatment is, however,
contraindicated in the case of pesticides dissolved in hydrocarbon
solvents. Gastric lavage (with addition of bicarbonate solution or
activated charcoal) can also be performed, particularly in unconscious
patients, taking care to prevent aspiration of fluids into the lungs
(i.e., only after a tracheal tube has been put into place).
The volume of fluid introduced into the stomach should be
recorded and samples of gastric lavage frozen and stored for
subsequent chemical analysis. If the formulation of the pesticide
involved is available, it should also be stored for further analysis
(i.e., detection of toxicologically relevant impurities). A purgative
can be administered to remove the ingested compound.
I.2 General supportive treatment
Artificial respiration (via a tracheal tube) should be started at
the first sign of respiratory failure and maintained for as long as
necessary.
Cautious administration of fluids is advised, as well as general
supportive and symptomatic pharmacological treatment and absolute
rest.
I.3 Specific pharmacological treatment
I.3.1 Atropine
Atropine should be given, beginning with 2 mg iv and given at
15-30-min intervals. The dose and the frequency of atropine treatment
varies from case to case, but should maintain the patient fully
atropinized (dilated pupils, dry mouth, skin flushing, etc.).
Continuous infusion of atropine may be necessary in extreme cases and
total daily doses up to several hundred mg may be necessary during the
first few days of treatment.
I.3.2 Oxime reactivators
Cholinesterase reactivators (e.g., pralidoxime, obidoxime)
specifically restore AChE activity inhibited by organophosphates. This
is not the case with enzymes inhibited by carbamates. The treatment
should begin as soon as possible, because oximes are not effective on
"aged" phosphorylated ChEs. However, if absorption, distribution, and
metabolism are thought to be delayed for any reasons, oximes can be
administered for several days after intoxication. Effective treatment
with oximes reduces the required dose of atropine. Pralidoxime is the
most widely available oxime. A dose of 1 g pralidoxime can be given
either im or iv and repeated 2-3 times per day or, in extreme cases,
more often. If possible, blood samples should be taken for AChE
determinations before and during treatment. Skin should be carefully
cleansed before sampling. Results of the assays should influence the
decision whether to continue oxime therapy after the first 2 days.
There are indications that oxime therapy may possibly have
beneficial effects on CNS-derived symptoms.
I.3.3 Diazepam
Diazepam should be included in the therapy of all but the mildest
cases. Besides relieving anxiety, it appears to counteract some
aspects of CNS-derived symptoms that are not affected by atropine.
Doses of 10 mg sc or iv are appropriate and may be repeated as
required (Vale & Scott, 1974). Other centrally acting drugs and drugs
that may depress respiration are not recommended in the absence of
artificial respiration procedures.
I.3.4 Notes on the recommended treatment
I.3.4.1 Effects of atropine and oxime
The combined effect far exceeds the benefit of either drug
singly.
I.3.4.2 Response to atropine
The response of the eye pupil may be unreliable in cases of
organophosphorus poisoning. A flushed skin and drying of secretions
are the best guide to the effectiveness of atropinization. Although
repeated dosing may well be necessary, excessive doses at any one time
may cause toxic side-effects. Pulse-rate should not exceed 120/min.
I.3.4.3 Persistence of treatment
Some organophosphorus pesticides are very lipophilic and may be
taken into, and then released from, fat depots over a period of many
days. It is therefore quite incorrect to abandon oxime treatment after
1-2 days on the supposition that all inhibited enzyme will be aged.
Ecobichon et al. (1977) noted prompt improvement in both condition and
blood-ChEs in response to pralidoxime given on the 11th-15th days
after major symptoms of poisoning appeared due to extended exposure to
fenitrothion (a dimethyl phosphate with a short half-life for aging of
inhibited AChE).
I.3.4.4 Dosage of atropine and oxime
The recommended doses above pertain to exposures, usually for an
occupational setting, but, in the case of very severe exposure or
massive ingestion (accidental or deliberate), the therapeutic doses
may be extended considerably. Warriner et al. (1977) reported the case
of a patient who drank a large quantity of dicrotophos, in error,
while drunk. Therapeutic dosages were progressively increased up to 6
mg atropine iv every 15 min together with continuous iv infusion of
pralidoxime chloride at 0.5 g/h for 72 h, from days 3 to 6 after
intoxication. After considerable improvement, the patient relapsed and
further aggressive therapy was given at a declining rate from days 10
to 16 (atropine) and to day 23 (oxime), respectively. In total, 92 g
of pralidoxime chloride and 3912 mg of atropine were given and the
patient was discharged on the thirty-third day with no apparent
sequelae.
References to Annex I.
ECOBICHON, D.J., OZERE, R.L., REID, E., & CROCKER, J.F.S (1977)Acute
fenitrothion poisoning. Can. Med. Assoc. J., 116: 377-379.
KAGAN, JU.S. (1977) [ Toxicology of organophosphorus pesticides],
Moscow, Meditsina, pp. 111-121, 219-233, 260-269 (in Russian).
PLESTINA, R. (1984) Prevention, diagnosis, and treatment of
insecticide poisoning, Geneva, World Health Organization
(Unpublished document VBC/84.889).
TAYLOR, P. (1980) Anticholinesterase agents. In: Goodman, L.S. &
Gilman, A., ed. The pharmacological basis of therapeutics, 6th ed.,
New York, Macmillan Publishing Company, pp. 100-119.
UK DHSS (1983) Pesticide poisoning: notes for the guidance of medical
practitioners, London, United Kingdom Department of Health and
Social Security, pp. 41-47.
VALE, J.A. & SCOTT, G.W. (1974) Organophosphorus poisoning. Guy's
Hosp. Rep., 123: 13-25.
WARRINER, R.A., III, NIES, A.S., & HAYES, W.J., Jr (1977) Severe
organophosphate poisoning complicated by alcohol and terpentine
ingestion. Arch. environ. Health, 32: 203-205.
ANNEX II. NO-OBSERVED-EFFECT LEVELS (NOELS) IN ANIMALS TREATED WITH
TRICHLORFON
Annex II No-observed-effect levels (NOELS) in animals treated
with trichlorfon
Animal Exposurea NOEL (paramater) Reference
(strain)
Rat 16 weeks 100 mg/kg diet (ChE) Doull & Dubois (1956)
Dog 12 weeks 200 mg/kg diet Williams et al. (1959)
(plasma, Er-ChE)
Rat 6h/day over 3 weeks 12.7 mg/m3 by inhalation Kimmerle (1975b)
(plasma, Er, brain ChE)
Beagle dog 12 months 250 mg/kg diet (ChE) Doull et al. (1962a)
Beagle dog 4 years 50 mg/kg diet (ChE) Löser (1970)
Rhesus monkey 26 weeks 0.2 mg/kg body weight Hoffmann et al. (1988)
oral intubation (Er-ChE)
CD-1 mouse 90 weeks 100 mg/kg diet (ChE) Machemer (1981)
1000 mg/kg diet
(tumour incidence)
CD-1 mouse 104 weeks 2700 mg/kg diet Hayes (1988)
(tumorigenicity)
Sprague-Dawley 17 months (m) 250 mg/kg diet (ChE, Doull et al. (1962b)
rat 24 months (f) survival, spermatogenesis,
ovarian changes) 50 mg/kg
diet (mammary tumours)
Annex II. (continued)
Animal Exposurea NOEL (paramater) Reference
(strain)
Sprague-Dawley 18 months 200 mg/kg diet Doull et al. (1965)
rat (ovarian changes)
Long-Evans rat 24 months 500 mg/kg diet (ChE) Lorke & Löser (1966);
1000 mg/kg diet Grundmann & Hobik (1966)
(tumorigenicity)
Rat twice/week 22 mg/kg body weight Teichmann et al. (1978)
for 90 weeks by gavage
(incidence of tumours)
Fischer 344 105 weeks 100 mg/kg diet Hayes (1989)
rat (morphological
parameters)
Rhesus monkey 6 days/week 0.2 mg/kg body weight Griffin (1988)
for 10 years oral intubation
(brain-ChE)
AB/Jena mouse twice/week 28.2 mg/body weight Teichmann & Hauschild
for 75 weeks ip injection (1978)
(incidence of tumours)
Rat twice/week 12.0 mg/kg body weight Teichmann et al. (1978)
for 90 weeks ip injection
(incidence of tumours)
Syrian Golden once/week 20 mg/kg body weight Teichmann & Schmidt
hamster for 90 weeks ip injection (1978)
(incidence of tumours)
Annex II. (continued)
Animal Exposurea NOEL (paramater) Reference
(strain)
AB/Jena mouse twice/week 0.25 ml of 1% solution Teichmann & Hauschild
for 75 weeks in acetone dermal (1978)
administration
(incidence of tumours)
a m = male.
f = female.
RESUME ET EVALUATION, CONCLUSIONS ET RECOMMANDATIONS
1. Résumé et évaluation
1.1 Exposition
Le trichlorfon est un insecticide organophosphoré utilisé depuis
le début des années 1950. En agriculture, on l'emploie principalement
contre les ravageurs des cultures de plein champ et des vergers. On
l'utilise également comme insecticide en forêt et pour débarrasser des
animaux domestiques de leurs parasites. Sous le nom de métrifonate, il
est utilisé pour traiter l'infestation humaine à Schistosoma
haematobium. Il agit en libérant lentement du dichlorvos. Le
trichlorfon est commercialisé sous forme de concentré émulsionnable,
de poudre dispersable, de poudre pour poudrage, de granulés, de
solution et de concentré à très bas volume.
Peu après l'épandage, la concentration atmosphérique du
trichlorfon peut atteindre 0,1 mg/m3 mais cette valeur diminue
rapidement pour tomber à moins de 0,01 mg/m3 en quelques jours. Les
eaux de ruissellement provenant des zones traitées peuvent contenir
des concentrations de trichlorfon atteignant 50 µg/litre, mais la
teneur des eaux de surface est généralement beaucoup faible et diminue
rapidement.
Le trichlorfon se décompose rapidement dans le sol et sa
concentration y devient généralement négligeable dans le mois qui suit
l'épandage. Il est relativement stable dans l'eau aux pH inférieurs à
5,5. A pH plus élevé, il se transforme de dichlorvos. Les
microorganismes et les plantes métabolisent probablement le
trichlorfon mais son mode d'élimination principal est l'hydrolyse
abiotique.
A quelques exceptions près, la concentration de trichlorfon sur
les récoltes est inférieure à 10 mg/kg dans le jour qui suit
l'épandage et tombe en-dessous de 0,1 mg/kg une quinzaine jours après.
Le lait des vaches que l'on a traitées au trichlorfon pour les
débarrasser de leur vermine peut contenir des résidus atteignant 1,2
mg/litre deux heures après l'application, mais cette valeur tombe
en-dessous de 0,1 mg/litre dans les 24 heures. On n'a pas constaté la
présence de concentrations importantes de trichlorfon dans la viande
des animaux traités. Dans les oeufs de poules traitées on a mesuré des
concentrations de 0,05 mg/kg.
1.2 Absorption, métabolisme et excrétion
Le trichlorfon est rapidement absorbé par l'ensemble des voies
d'exposition (orale, dermique, respiratoire) et il se répartit
rapidement dans les tissus de l'organisme. Le taux sanguin passe par
un maximum au bout d'une à deux heures, le produit disparaissant
presque totalement du courant sanguin au bout 1,5 à 4 heures. La
demi-vie biologique du trichlorfon dans le sang des mammifères a été
estimée à environ 30 minutes.
Dans l'eau, les liquides biologiques et les tissus, à des valeurs
du pH supérieures à 5,5 le trichlorfon subit une transformation en
dichlorvos (phosphate de 2,2-dichlorovinyle et de diméthyle) par
déshydrochloration. C'est le dichlorvos qui constitue le principe
actif antichlolinestérasique. Les principales voies de dégradation
sont la déméthylation, la coupure de la liaison phosphore-carbone et
l'hydrolyse de l'ester via le dichlorvos. Les principaux métabolites
du trichlorfon que l'on trouve in vivo sont le déméthyltrichlorfon,
le déméthyldichlorvos, l'hydrogénophosphate de diméthyle,
l'hydrogénophosphate de méthyle l'acide phosphorique et de
trichloréthanol. Ce dernier métabolite se retrouve dans l'urine,
conjugué sous forme de glucuronide.
Le trichlorfon et ses métabolites sont principalement éliminés
par voie urinaire. Des études effectuées avec du trichlorfon
radiomarqué (14C-méthyl et 32P) ont montré que la majeure partie
du produit s'éliminait sous la forme de dérivés hydrosolubles et une
faible fraction sous la forme de dérivés solubles dans le chloroforme.
Environ 66 à 70 % des produits hydrosolubles apparaissent dans l'urine
dans les 12 heures, 24 % du produit radiomarqué au niveau du
groupement méthyl étant éliminés dans l'air expiré sous forme de
dioxyde de carbone (CO2). Après avoir traité des vaches soit par
administration orale soit par voie cutanée, on a retrouvé dans leur
lait de faibles quantités de trichlorfon et de ses métabolites.
1.3 Effets sur les êtres vivants dans leur milieu naturel
Le trichlorfon est modérément toxique pour les poissons (les
valeurs de la CL50 à 96 heures vont de 0,45 mg/litre à 51 mg/litre)
et modérément à fortement toxique pour les arthropodes aquatiques
(valeur de la CL50 à 48 et 96 heures comprises en entre 0,75
µg/litre et 7800 µg/litre). Toutefois les concentrations dont il est
fait état dans les eaux superficielles après épandage dans des forêts
à la dose de 6kg/ha, sont inférieures à ces valeurs. Il s'en suit
qu'en utilisation normale, le trichlorfon n'aura guère d'effets sur
les organismes aquatiques car les autres types d'organismes tels que
les mollusques et les microorganismes sont moins sensibles que les
arthropodes. Les valeurs de la DL50 tirées d'études en laboratoire
se situent entre 40 et 180 mg/kg et montrent donc que le trichlorfon
est modérément toxique pour les oiseaux. Toutefois des études menées
sur le terrain après épandage de trichlorfon par voie aérienne sur des
forêts, n'ont révélé aucun effet sur l'effectif, la formation de
couples, le nichage ou la mortalité des oiseaux chanteurs. La
diminution du chant et l'accroissement de l'activité trophique qui ont
été constatés sont peut-être la conséquence d'une réduction du nombre
de proies. Rien n'indique que le trichlorfon puisse avoir un effet
nocif sur la faune terrestre, à part les arthropodes. On ne possède
aucune donnée au sujet des effets de cet insecticide sur les
arthropodes utiles.
1.4 Effets sur les animaux d'expérience et sur les systèmes
d'épreuves in vitro
Le trichlorfon est un insecticide modérément toxique pour les
animaux d'expérience. Chez l'animal de laboratoire, les valeurs de la
DL50 pour le trichlorfon technique s'étagent de 400 à 800 mg/kg de
poids corporel et pour le rat, la DL50 cutanée dépasse 2000 mg/kg de
poids corporel.
L'intoxication par le trichlorfon offre le tableau clinique
habituel de l'atteinte cholinergique due aux organophosphorés et qui
résulte d'une accumulation d'acétylcholine au niveau des terminaisons
nerveuses.
On a montré que le trichlorfon technique était modérément
irritant pour la conjonctive chez le rat, aucun effet de ce genre
n'ayant été noté lors de tests cutanés sur des lapins. Sa capacité de
sensibilisation cutanée a été mise en évidence chez le cobaye.
Des études de toxicité par voie orale de brève durée ont été
effectuées sur des rats, des chiens, des singes, des lapins et des
cobayes. Lors d'une étude de 16 semaines sur des rats, de 4 ans sur
des chiens et de 26 semaines sur des singes, on a fixé respectivement
à 100 mg/kg de nourriture, 5' mg/kg de nourriture et 0,2 mg/kg de
nourriture la dose sans effet observé (sur la base de l'activité
cholinestérasique, plasmatique, érythrocytaire ou cérébrale). En
exposant par voie respiratoire des rats pendant trois semaines, on a
obtenu une dose sans effet observé de 12,7 mg/m3 en se basant sur
l'inhibition de l'activité cholinestérasique du plasma, des
érythrocytes et du cerveau. Des études de toxicité et de
cancérogénicité à long terme ont été menées sur des souris, des rats,
des singes et des hamsters à qui l'on a administré du trichlorfon par
voie orale,intrapéritonéale ou percutanée. Après exposition par voie
orale des souris et des rats à des doses de 30 mg/kg de poids corporel
et de 400 mg/kg de nourriture respectivement, on a observé des
anomalies au niveau des gonades. Lors d'une étude de 24 mois sur des
rats et de 10 ans sur des singes, on a obtenu des doses sans effet
observé respectivement égales à 50 mg/kg de nourriture et 0,2 mg/kg de
poids corporel. Les données sont on dispose ne permettent pas de
conclure que la substance est cancérogène après administration à des
animaux de laboratoire pendant une longue période par diverses voies.
Dans les conditions physiologiques , le trichlorfon est
susceptible d'alkyler l'ADN. Les tests de mutagénicité ont donné des
résultats tantôt positifs tantôt négatifs. Il a peut que les effets
observés soient imputables en tout ou partie au dichlorvos. La plupart
des études de mutagénicité effectuées in vitro sur des bactéries ou
des cellules mammaliennes ont donné des résultats positifs alors que
rares sont les études in vivo qui ont donné de tels résultats.
L'expérimentation sur la souris, le rat et le hamster montre que
le trichlorfon suscite une réaction tératogène chez le rat à des doses
suffisantes pour être toxiques chez la mère. Après avoir administré à
des rattes en cours de gestation une dose de 145 mg de trichlorfon par
kg de nourriture, on a observé des malformations chez les foetus. Une
dose de 400 mg/kg de poids corporel administrée par gavage à des
hamsters a également été toxique pour les mères et a provoqué des
effets tératogènes. La dose la plus faible administrée par gavage et
ayant provoqué des effets tératogènes chez le rat était de 80 mg/kg de
poids corporel. Au cours de la période de gestation, les effets
produits présentent une spécificité chronologique. Cette étude a
permis de fixer à 8 mg/kg la dose sans effet observé.
Des doses sans effet observé respectivement égales à 8 mg/kg et
à 200 mg/kg de poids corporel ont été observées chez le rat et le
hamster. Chez le porc et le cobaye, on a constaté une action
tératogène sur le système nerveux central.
Cependant, aucun effet tératogène n'a été observé lors d'une
étude de reproduction portant sur trois générations de rats, au cours
de laquelle on a relevé des effets nocifs sur la fonction de
reproduction. Dans cette étude, la dose sans effet observé était de
300 mg/kg de nourriture.
A très hautes doses, le trichlorfon produit des effets
neurotoxiques chez l'animal.
Chez les mammifères, le métabolite actif est le dichlorvos, dont
l'activité anticholinestérasique est au moins 100 fois plus forte que
celle du trichlorfon.
1.5 Effets sur l'homme
Plusieurs cas d'intoxication aiguë délibérée (suicide) ou
accidentelle se sont produits. L'intoxication présente une
symptomatologie caractéristique de l'inhibition de la cholinestérase:
épuisement, faiblesse, confusion, sueur et salivation profuses,
douleurs abdominales, vomissements, myosis et spasmes musculaires.
Dans les cas graves, l'intoxication entraîne la perte de conscience et
des convulsions et la mort survient généralement par insuffisance
respiratoire. Chez les victimes qui ont survécu à l'intoxication grâce
à une intervention médicale, on a observé quelquefois, plusieurs
semaines après l'exposition, une polyneuropathie retardée accompagnée
d'une faiblesse des membres inférieurs. Dans les cas mortels,
l'autopsie a révélé des foyers d'ischémie dans le cerveau, la moelle
épinière et les ganglions végétatifs, ainsi que des lésions de la
gaine de myéline dans la moelle épinière et les pédoncules cérébraux,
avec des altérations dans la structure des axones des nerfs
périphériques.
Les quelques cas d'intoxication d'origine professionnelle qui se
sont produits, s'expliquent essentiellement par des négligences au
niveau de la sécurité. L'exposition professionnelle sur les lieux de
travail à des concentrations atmosphériques supérieures à 0,5 mg/m3
a entraîné une réduction de la cholinestérase plasmatique et une
altération du tracé électroencéphalographique. Toutefois ces anomalies
ont complètement régressé à l'arrêt de l'exposition. Aucun cas de
sensibilisation cutanée n'a été signalé.
Ce composé est très largement utilisé pour le traitement de la
schistosomiase chez l'homme. L'administration d'une dose unique (7 à
12 mg/kg) entraîne une inhibition de la cholinestérase plasmatique et
érythrocytaire à hauteur de 40-60 %, sans entraîner de symptômes
cholinergiques. Toutefois, des symptômes légers ont été observés chez
des malades qui avaient pris de ce produit à plusieurs reprises. A
forte dose (24 mg/kg) on a observé de graves symptômes cholinergiques
2. Conclusions
- Le trichlorfon est un insecticide organophosphoré modérément
toxique. Une intoxication grave peut survenir par suite
d'une exposition excessive au produit lors de sa
manipulation, de sa fabrication ou de son utilisation ou par
suite d'une ingestion accidentelle ou délibérée.
- L'exposition au trichlorfon de la population générale
résulte principalement de son utilisation en agriculture et
en médecine vétérinaire et dans le traitement de la
schistosomiase (bilharziose).
- Les quantités de trichlorfon qui sont absorbées sont très
inférieures à la dose journalière adminissible fixée par la
FAO et l'OMS et ne devraient pas constituer une menace pour
la santé publique.
- Si l'on adopte de bonnes méthodes de travail et, que l'on
respecte les précautions d'hygiène et de sécurité, le
trichlorfon n'est vraisemblablement pas dangereux pour les
personnes qui sont exposées de par leur profession.
- Bien que le trichlorfon soit très toxique pour les
arthropodes non visés, son utilisation n'entraîne guère
d'effets nocifs sur la faune et la flore.
3. Recommandations
- Afin de préserver la santé et le bien-être des ouvriers et
de la population en général, il importe de confier la
manipulation et l'épandage du trichlorfon exclusivement à
des personnes correctement encadrées et expérimentées, qui
sauront appliquer les mesures de sécurité indispeurables et
utiliser convenablementle produit.
- Des précautions sont à observer lors de la production, de la
formulation, de l'utilisation en agriculture et du rejet du
trichlorfon afin de contaminer le moins possible
l'environnement et plus spécialement des eaux de surface.
- Les travailleurs qui sont régulièrement exposés au
trichlorfon ainsi que les malades traités avec ce produit
doivent subir des examens médicaux périodiques.
- Les doses d'emploi en agriculture devront rester faibles
afin d'éviter la destruction des arthropodes non visés.
L'insecticide ne devra jamais être épandu sur des étendues
ou des cours d'eau.
RESUMEN Y EVALUACION, CONCLUSIONES Y RECOMENDACIONES
1. Resumen y evaluación
1.1 Exposición
El triclorfón es un insecticida organofosforado que lleva
utilizándose desde principios de los años cincuenta. En agricultura
sirve principalmente para combatir las plagas de insectos en los
cultivos extensivos y en los frutales. Se utiliza asimismo para
combatir los insectos de los bosques y los parásitos de los animales
domésticos. Bajo la denominación de metrifonato, se emplea para tratar
la infestación del hombre por Schistosoma haematobium. Se considera
un reservorio de liberación lenta de diclorvos. El triclorfón se
presenta en forma de solución concentrada emulsionable, polvos para
disolución o aplicación en seco, gránulos, solución y soluciones
concentradas de volumen muy reducido.
La concentración de triclorfón insecticida en el aire puede
alcanzar 0,1 mg/m3 al poco tiempo del rociamiento, pero a los pocos
días los niveles se sitúan alrededor de 0,01 mg/m3. En las aguas de
escorrentía de zonas rociadas, la concentración de triclorfón puede
alcanzar los 50 µg/litro; en cambio en las aguas de superficie suele
ser mucho más baja y disminuye rápidamente.
El triclorfón se degrada rápidamente en el suelo; las
concentraciones suelen disminuir hasta cantidades insignificantes
durante el mes que sigue a la aplicación. Es relativamente estable en
agua si el pH es inferior a 5,5; con un pH más elevado se transforma
en diclorvos. Aunque los microorganismos y las plantas pueden
metabolizar el triclorfón, la vía de eliminación más importante es la
hidrólisis abiótica.
Salvo raras excepciones, las concentraciones de triclorfón en los
cultivos son inferiores a 10 mg/kg al día siguiente de la aplicación,
e inferiores a 0,1 mg/kg durante las dos semanas siguientes.
La leche de vacas tratadas con triclorfón para combatir plagas
puede contener hasta 1,2 mg de residuos/litro a las dos horas de la
aplicación, pero las cifras descienden hasta menos de 0,1 mg/litro a
las 24 horas del tratamiento. No se han encontrado concentraciones
importantes de este compuesto en la carne de animales tratados. En los
huevos de gallinas tratadas se han comprobado valores de 0,05 mg de
triclorfón/kg.
1.2 Ingestión, metabolismo y excreción
El triclorfón se absorbe fácilmente por todas las vías de
exposición (oral, cutánea, respiratoria) y se distribuye rápidamente
a los tejidos del cuerpo. Se detectaron concentraciones máximas en la
sangre al cabo de 1-2 h, y la sustancia desapareció casi por completo
del torrente sanguíneo en cuestión de 1,5-4 h. Se calculó que la
semivida biológica del triclorfón en la sangre de mamíferos es de
alrededor de 30 minutos.
Por deshidrocloración, el triclorfón se transforma en diclorvos
(2,2-diclorovinil dimetil fosfato) en el agua y en los humores y
tejidos de los seres vivos, si el pH es superior a 5,5. El diclorvos
es la anticolinesterasa fisiológicamente activa. Las principales rutas
de degradación son la desmetilación, la escisión del enlace P-C y la
hidrólisis del éster con el diclorvos como producto intermediario. Los
principales metabolitos del triclorfón que se encuentran in vivo son
el demetil triclorfón, el demetil diclorvos, el dimetil
hidrogenofosfato, el metil hidrogenofosfato, el ácido fosfórico y el
tricloroetanol. El último metabolito se encuentra en la orina en forma
de conjugado de glucurónido.
El triclorfón y sus productos metabólicos se eliminan
principalmente con la orina. Los estudios realizados con triclorfón
radiomarcado (14C-metilo y 32P-metilo) revelaron que la mayor
parte de la sustancia se eliminaba en forma hidrosoluble, y una
pequeña parte en forma soluble en cloroformo. Alrededor del 66%-70% de
los productos hidrosolubles aparecían en la orina al cabo de 12 horas,
mientras que el 24% del material marcado con 14C-metilo se eliminaba
en el aire espirado en forma de dióxido de carbono (CO2). Se han
detectado concentraciones bajas de triclorfón y sus metabolitos en la
leche de bóvidos tras el tratamiento de éstos por vía oral y cutánea.
1.3 Efectos en organismos del medio ambiente
El triclorfón es moderadamente tóxico para los peces (los valores
de la CL50 a las 96 h varían entre 0,45 mg/litro y 51 mg/litro) y de
toxicidad moderada a elevada para los artrópodos acuáticos (la CL50
a las 48 h/96 h varía entre 0,75 µg/litro y 7800 µg/litro). En cambio,
los valores observados en aguas de superficie tras aplicar el
compuesto en bosques a razón de 6 kg/ha quedan por debajo de estos
límites. Así pues, si es objeto de un uso normal, el triclorfón tendrá
un efecto muy reducido o nulo en las poblaciones de organismos
acuáticos, puesto que otros grupos como los moluscos y los
microorganismos, son menos sensibles que los artrópodos. Los valores
de la DL50 obtenidos en estudios de laboratorio (de 40 a 180 mg/kg)
indican que este compuesto es moderadamente tóxico para las aves. En
cambio, en estudios de campo no se observó efecto alguno en la
población total, el número de parejas en época de cría, la viabilidad
de los nidos ni la mortalidad de las aves canoras de los bosques
tratados mediante aplicaciones aéreas del insecticida. Se observó
cierta disminución de la actividad canora y mayor actividad de
búsqueda de alimento, tal vez por haberse reducido las poblaciones de
los organismos de que se nutren. Nada indica que el triclorfón
perjudique a los organismos terrestres excepto los artrópodos. No se
dispone de información sobre sus efectos en artrópodos beneficiosos.
1.4 Efectos en animales de laboratorio y en sistemas de
ensayo in vitro
El triclorfón es un insecticida moderadamente tóxico para los
animales de laboratorio. Los valores de la DL50 para el producto
técnico administrado por vía oral a éstos varían entre 400 y 800 mg/kg
de peso corporal; en la rata, los valores de la DL50 cuando se
administra por vía cutánea superan los 2000 mg/kg de peso corporal.
La intoxicación por este compuesto origina los signos
colinérgicos comúnmente relacionados con los organofosfatos y que se
atribuyen a la acumulación de acetilcolina en las terminaciones
nerviosas.
Se ha demostrado que el triclorfón técnico es moderadamente
irritante para los ojos de la rata, aunque no para la piel del conejo.
Se ha observado potencial de sensibilización cutánea en conejillos de
Indias.
Se llevaron a cabo estudios de corto plazo sobre toxicidad por
vía oral en ratas, perros, monos, conejos y conejillos de Indias. En
uno de 16 semanas en ratas, en otro de 4 años en perros y en un
tercero de 26 semanas en monos, se registraron respectivamente las
siguientes concentraciones sin efectos observados (NOEL): 100 mg/kg
de ración alimenticia, 50 mg/kg de ración alimenticia y 0,2 mg/kg de
peso corporal (calculados respecto de la actividad de la colinesterasa
en plasma, eritrocitos o encéfalo). La exposición de ratas por
inhalación durante más de tres semanas indicó una NOEL de 12,7
mg/m3, calculados respecto de la inhibición de la actividad de la
colinesterasa en plasma, eritrocitos y encéfalo. Se llevaron a cabo
estudios de toxicidad/carcinogenicidad a largo plazo en ratones,
ratas, monos y hámsters tras la administración oral, intraperitoneal
o cutánea. Se observaron efectos adversos en las gónadas de ratones y
ratas expuestos por vía oral a 30 mg/kg de peso corporal y 400 mg/kg
de ración alimenticia, respectivamente. En un estudio de 24 meses en
ratas y otro de 10 años en monos, se determinaron valores de NOEL de
50 mg/kg de ración alimentaria y de 0,2 mg/kg de peso corporal,
respectivamente. Los datos disponibles no aportan pruebas de
carcinogenicidad tras la exposición prolongada de animales de
laboratorio por diversas vías de administración.
Se ha comunicado que, en condiciones fisiológicas, el triclorfón
tiene la propiedad de alquilizar al ADN. Los ensayos de mutagenicidad
han dado resultados tanto positivos como negativos. Es posible que el
diclorvos sea la causa, parcial o totalmente, de los efectos
observados. La mayoría de los estudios de mutagenicidad in vitro en
células tanto bacterianas como de mamíferos dieron resultado positivo;
en cambio, pocos de los estudios in vivo dieron ese resultado.
Investigaciones realizadas en el ratón, la rata y el hámster
indican que, en dosis lo bastante elevadas como para producir
toxicidad materna, el triclorfón produce una respuesta teratógena en
ratas. La exposición de ratas gestantes a dosis de 145 mg/kg de ración
alimenticia provocó malformaciones fetales. La administración oral
forzada de 400 mg/kg de peso corporal a hámsters produjo también
toxicidad materna y respuesta teratógena. Por esta vía, la dosis más
baja que produjo efectos teratógenos en la rata fue de 80 mg/kg de
peso corporal. Los efectos son específicos según el momento del
periodo de gestación en que se ingiere el producto. En este estudio de
administración oral forzada se obtuvo una NOEL de 8 mg/kg.
En ratas y hámsters se han encontrado, respectivamente, NOEL de
8 mg/kg de peso corporal y 200 mg/kg de peso corporal. También se han
comunicado respuestas teratógenas que afectaban al sistema nervioso
central en el cerdo y el conejillo de Indias.
En cambio, no se observaron efectos teratógenos en un estudio de
reproducción en tres generaciones de ratas, en el que con dosis
elevadas se indujeron efectos reproductivos adversos. La NOEL en este
estudio fue de 300 mg/kg de ración alimenticia.
Con dosis muy elevadas se han producido efectos neurotóxicos en
animales.
En los mamíferos, el producto activo de la transformación es el
diclorvos, cuya actividad como anticolinesterasa es como mínimo 100
veces mayor que la del triclorfón.
1.5 Efectos en el ser humano
Se han producido varios casos de envenenamiento agudo por
exposición intencional (suicidio) o accidental. Los signos y síntomas
de la intoxicación fueron los característicos de la inhibición de la
acetilcolinesterasa: agotamiento, debilidad, confusión, sudación y
salivación excesivas, dolores abdominales, vómitos, pupilas
puntiformes y espasmos musculares. En casos graves se observaron
pérdida de la consciencia y convulsiones, y la muerte se produjo en
general por fallo respiratorio. En las víctimas que sobrevivieron
gracias a la intervención médica, a veces se observó una
polineuropatía diferida, acompañada de debilidad de los miembros
inferiores, la cual apareció algunas semanas después de la exposición.
En los casos mortales, la autopsia reveló alteraciones isquémicas en
el encéfalo, la médula espinal y los ganglios vegetativos, lesiones de
la vaina mielínica en la médula espinal y los pedúnculos cerebrales,
y cambios estructurales en los axones de los nervios periféricos.
Se han producido algunos casos de envenenamiento ocupacional,
principalmente por no observar las normas de seguridad. La exposición
en un lugar de trabajo con concentraciones en el aire superiores a 0,5
mg/m3 ocasionó la disminución de la colinesterasa plasmática y
cambios del trazado electroencefalográfico. No obstante, estos signos
desaparecieron por completo al cesar la exposición. No se ha
comunicado ningún caso de sensibilización cutánea.
Este compuesto se ha usado extensamente para tratar la
esquistosomiasis del ser humano. La administración de una dosis única
(7-12 mg/kg) inhibió entre el 40 y el 60% de la colinesterasa del
plasma y los eritrocitos, sin que aparecieran síntomas colinérgicos.
En cambio, se observaron síntomas leves en personas tratadas con dosis
repetidas. Las dosis elevadas (24 mg/kg) produjeron síntomas
colinérgicos graves.
2. Conclusiones
- El insecticida triclorfón es un éster organofosforado
moderadamente tóxico. La exposición excesiva que puede
producirse al fabricarlo o utilizarlo y la ingestión
accidental o intencional pueden provocar envenenamientos
graves.
- La exposición de la población general al triclorfón se
produce principalmente como resultado de las prácticas
agrícolas y veterinarias y del tratamiento de la infestación
por Schistosoma haematobium.
- Las ingestas de triclorfón comunicadas se encuentran muy por
debajo de la ingesta diaria admisible establecida por la
FAO/OMS y en principio no constituyen un riesgo para la
salud de la población general.
- Si se siguen prácticas correctas de trabajo, medidas
higiénicas y precauciones de seguridad, es poco probable que
el triclorfón represente un riesgo para las personas
expuestas por su trabajo.
- A pesar de su gran toxicidad para otros artrópodos que no se
pretende destruir, el triclorfón se ha utilizado con escasos
o nulos efectos adversos para las poblaciones de organismos
del medio ambiente.
3. Recomendaciones
- Para proteger la salud de los trabajadores y de la población
general, la manipulación y la aplicación del triclorfón
deben encomendarse solamente a operarios bien supervisados
y adiestrados, que observarán medidas adecuadas de seguridad
y utilizarán el insecticida siguiendo prácticas correctas.
- La fabricación, la formulación, el uso agrícola y la
evacuación del triclorfón deben sujetarse a una gestión
cuidadosa para reducir al mínimo la contaminación del medio,
en particular las aguas de superficie.
- Las poblaciones de trabajadores y de personas regularmente
expuestos deben someterse a exámenes médicos periódicos.
- Las tasas de aplicación del triclorfón deben limitarse a fin
de evitar efectos sobre artrópodos que no se pretende
combatir. Este insecticida nunca debe rociarse sobre masas
ni corrientes de agua.
in the milk, 1-3 h after the higher dose (Nakahara et al., 1972). When
lactating cows were treated dermally by washing with 11.2%
trichlorfon, 6-8 h before milking, trichlorfon residues (equal to or
more than 0.2 mg/kg) and dichlorvos residues (0.03 mg/kg) were found
in the first milking (Fechner et al., 1968). Trichlorfon was detected
up to the third milking, 32 h after application, but neither
trichlorfon nor dichlorvos could be demonstrated in the fourth
milking.
6.4 Reaction with body components
6.4.1 In vitro studies
Several investigators have reported the considerable inhibitory
activity of trichlorfon on acetylcholinesterase in vitro. The
pI50 for acetylcholinesterase was 5.5 and the bimolecular rate
constant for rat brain acetylcholinesterase was 3.18 × 104/min
(Arthur & Casida, 1957; Buchet & Lauwerys, 1970). The activity is,
however, strongly pH-dependent and is about 30-fold more active at pH
7.4-7.6 than at pH 6.0-6.5. At the lower pH range, trichlorfon is more
stable with practically no inhibiting activity against
cholinesterases. In contrast, dichlorvos is equally active at this pH
range (Metcalf et al., 1959; Miyamoto 1959; Reiner et al., 1975).
However, the rates of non-enzymatic reactivation of the enzymes after
inhibition by trichlorfon and dichlorvos are similar. Thus, it is now
believed that the in vitro inhibitory activity of trichlorfon is due
to its rapid, spontaneous, non-enzymatic conversion into dichlorvos.
Frohlich et al. (1990) determined the competitive and
uncompetitive constants for the action of trichlorfon on bee
p-nitrophenyl acetate hydrolysing esterase to be 4.5 × 10-6
mol/litre and 1.6 × 10-5 mol/litre, respectively.
The inhibition constant for trichlorfon and chicken liver fluora
cetanilidase was determined to be 2.5 × 10-6 mol/litre (Nakamura &
Ueda, 1967).
6.4.2 In vivo studies
In rats given trichlorfon intraperitoneally at 150 mg/kg,
considerable increases in the activities of superoxide dismutase
(× 1.94) and microsomal cytochrome P-450 (× 2.09), and in lipid
peroxidation (× 1.44) in the liver were observed, 2.5 h after
treatment (Matkovics et al., 1980).
In in vivo studies on mice receiving a diet containing 100 mg
trichlorfon/kg, the cholinesterase activities in the brain,
erythrocytes, and plasma were, respectively, 72.1, 115.7, and 92.7% of
control values after one-day (24 h) and 79.9, 83.8, and 81.0%,
respectively, after 20 days (Tsumuki et al., 1970).
A mixture of trichlorfon and phenothiazine, which was
administered to horses with feed as an antihelminthic at a single dose
level of 35.8 mg/kg, reduced the cholinesterase activity in whole
blood and plasma to 32 and 20%, respectively, by 24 h. The activity
increased to about 80% after 4 weeks (Bello et al., 1974).
In humans who received an oral dose of 7.5 mg trichlorfon/kg on
two successive days, the maximum inhibition of erythrocyte and plasma
cholinesterase was 52 and 94%, respectively. The red cell
cholinesterase activity recovered very slowly to reach only 66% of the
pretreatment level, 28 days after treatment, while that of the plasma
cholinesterase recovered rapidly to reach 78% at 22 days (Lebrun &
Cerf, 1960).
When 14CH3-labelled trichlorfon was adminstered intravenously
to rats at 40 mg/kg, the radioactive carbon (14C) was found mainly
in the liver (25 mg/kg) and kidneys (11.6 mg/kg), 3 h after treatment
(Dedek & Lohs, 1970). Similar results were obtained with intra
peritoneal treatment. Only about 1% of the radioactivity found in the
organs was extractable with acetone, indicating that most of the 14C
in the tissues was bound to body components. The bound 14C in the
organs investigated was less than 10% of the total dose. It
disappeared rather rapidly and the bound radioactivity decreased to
about one-tenth by 17 h. In another study, about 10% of the phosphorus
portion, administered orally to mice as 32P-trichlorfon, was found
bound to tissue (Miyata & Saito, 1973).
7. EFFECTS ON ORGANISMS IN THE ENVIRONMENT
Acute toxicity data for trichlorfon on aquatic and terrestrial
non-target organisms are summarized in Tables 8 and 9, respectively.
7.1 Microorganisms
When trichlorfon was applied to a cotton field at 0.5 g/m2, the
total count of soil fungi and the counts of some fungal species, such
as Aspergillus niger, Fusarium oxysporum, and A. fumigatus were
elevated 3 days after treatment. A depressed effect was observed in
Myrothecium verrucaria. After 40 days, trichlorfon did not have any
significant effects on the total count of soil fungi (Abdel-Kader et
al., 1978). Trichlorfon at 8-16 mg/kg in agar affected the mycelial
growth of 4 fungi (A. fumigatus, F. moniliforme, Penicillium
italicum, and Sclerotium cepivorum), even 10 days after application
(El-Hissy & Abdel-Kader, 1980).
In field pond trials, Grygierek & Wasilewska (1981) found that
trichlorfon applied at 1 mg/litre reduced zooplankton numbers within
the first 24 h of application and bottom fauna after 1 week. Rotifers
( Keratella sp.) and cladocerans ( Bosmina) were especially
sensitive to the chemical. The renewal of the affected fauna
communities took about 1 month. When the pond was treated with
trichlorfon at 300-800 µg/litre, the numbers of cladocerans and
copepods decreased, whereas rotifers and phytoplankton increased
markedly in number. However, the total biomass was not affected (Grahl
et al., 1981).
Cain & Cain (1984) incubated the green alga Chlamydomonas
moewusii in a medium containing trichlorfon at concentrations up to
80 µmol/litre (21 mg/litre). At the highest concentration, growth of
the algae was 76% of control levels. Zygospore germination of
Chlamydomonas was unaffected at 80 µmol/litre (21 mg/litre) (108% of
controls).
7.2 Invertebrates
Trichlorfon is highly toxic for aquatic arthropods with LC50
values ranging from 0.75 to 7800 µg/litre in 48-/96-h tests (Table 8).
The toxicity of trichlorfon for amphipods increased 2-fold when
the pH of the media was increased from 7.5 to 8.5; similarly the
toxicity for stonefly naiads increased about 20-fold when the pH was
increased from 6.5 to 8.5 (Woodward & Mauck, 1980). Trichlorfon is
less toxic for mollusca, the 48-h LC50 values ranging up to 25.4
mg/litre. Prolonged exposure (10 days) resulted in a 10-40 times
increase in toxicity (Singh & Agarwal, 1981).
Table 8. Acute toxicity of trichlorfon for non-target aquatic organisms
Species Size Toxicity (mg/litre) Formulationa Systemb Temperature pH Hardnessc Reference
(°C)
Protozoa
Colpidium MAD 10 20 Dive
campylum et al.
(1980)
Fish
Rainbow trout 96-h LC50 4.85 T S 12 Marking
(Salmo & Mauck
gairdneri) (1975)
Cutthroat trout 96-h LC50 1.68 T S 12 7.5 40 Woodward
(Salmo clarki) 96-h LC50 3.25 T S 12 7.5 40 & Mauck
96-h LC50 5.75 T S 7 7.5 40 (1980)
96-h LC50 4.75 T S 12 6.5 40
96-h LC50 0.375 T S 12 8.5 40
96-h LC50 0.620 T S 12 7.8 320
Cherry salmon 2.8 g 12-h LC50 10.2 50%EC SS 17.6 Kimura
(Onchorhyncus 2.8 g 24-h LC50 1.1 50%EC SS 17.6 et el.
masou) 2.8 g 48-h LC50 1.1 50%EC SS 17.6 (1971)
2.8 g 96-h LC50 1.1 50%EC SS 17.6
Carp egg 24-h LC50 15 EC S 25 6.9-7.2 Hashimoto
(Cyprinus 0.60 cm 24-h LC50 11 EC S 25 6.9-7.2 et al.
carpio) 0.73 cm 24-h LC50 8.8 EC S 25 6.9-7.2 (1982)
1.16 cm, 0.013 g 24-h LC50 14 EC S 25 6.9-7.2
1.52 cm, 0.038 g 24-h LC50 15 EC S 25 6.9-7.2
2.51 cm, 0.23 g 24-h LC50 14 EC S 25 6.9-7.2
3.86 cm, 0.87 g 24-h LC50 15 EC S 25 6.9-7.2
5.22 cm, 2.34 g 24-h LC50 15 EC S 25 6.9-7.2
Table 8. (continued)
Species Size Toxicity (mg/litre) Formulationa Systemb Temperature pH Hardnessc Reference
(°C)
Carp 48-h LC50 1200 4%D S 25 Nishiuchi
(Cyprinus 48-h LC50 30 50%EC S 25 (1979a)
carpio) 48-h LC50 50 80%WP S 25
Carp 5.3 cm, 2.2 g 24-h LC50 40 S 15 Nishiuchi
(Cyprinus 5.3 cm, 2.2 g 24-h LC50 40 S 20 (1977)
carpio) 5.3 cm, 2.2 g 24-h LC50 28 S 25
5.3 cm, 2.2 g 24-h LC50 25 S 30
5.3 cm, 2.2 g 24-h LC50 25 S 35
Fathead minnow 96-h LC50 51 Livingston
(Pimephales (1977)
promelas)
American eel black stage 96-h LC50 1.32 S 22 7.2-7.6 40-48 Spehar et
(Anguilla glass stage 96-h LC50 1.31 S 22 7.2.-7.6 40-48 al. (1981)
rostrata) yellow phase 96-h LC50 8.57 S 22 7.2.-7.6 40-48 Hinton &
Eversole
(1980)
Striped bass 6 cm, 2.7 g 24-h LC50 10.4 50%SP S 21 8.2 35 Wellborn
(Roccus 6 cm, 2.7 g 48-h LC50 9.2 50%SP S 21 8.2 35 Jr. (1969)
saxatilis) 6 cm, 2.7 g 96-h LC50 5.2 50%SP S 21 8.2 35
Bluegill 0.87-2.4 g 24-h LC50 41.0a 50%SP S 18 7 51.3 McCann &
(Lepomis Jasper
macrochirus) (1972)
Arthropods
Water flea 3-h LC50 0.18 4%P S 25 Nishiuchi
(Daphnia 3-h LC50 0.030 50%EC S 25 (1979b)
pulex) 3-h LC50 0.019 80%WP S 25
Table 8. (continued)
Species Size Toxicity (mg/litre) Formulationa Systemb Temperature pH Hardnessc Reference
(°C)
Water flea 1-h LC50 0.088 EC S 25 Nishiuchi
(Daphnia 3-h LC50 0.014 EC S 25 (1979c)
carinata) 6-h LC50 0.0064 EC S 25
24-h LC50 0.0012 EC S 25
48-h LC50 0.00075 EC S 25
Amphipod 96-h LC50 0.108 T S 12 7.5 40 Woodward
(Garnmarus 96-h LC50 0.052 T S 12 8.5 40 & Mauck
pseudolimnaeus) (1980)
Mayfly 9.3 mm, 5.6 mg 3-h LC50 1.8 EC S 25 Nishiuchi
(Cloeon 9.3 mm, 5.6 mg 6-h LC50 0.75 EC S 25 & Asano
diptrum) 9.3 mm, 5.6 mg 24-h LC50 0.075 EC S 25 (1979)
9.3 mm, 5.6 mg 48-h LC50 0.056 EC S 25
Dragon fly 2.3 cm, 0.62 g 48-h LC50 0.042 EC 25 Nishiuchi
(Orthetrum (1981)
albistylum
speciosum)
(Sympetrum 2.1 cm, 0.56 g 48-h LC50 0.15 EC 25 Nishiuchi
frequens) (1981)
(Sigara 5.9 mm, 6.1 mg 48-h LC50 0.15 EC 25 Nishiuchi
substriata) (1981)
(Micronecta 3.2 mm, 1.8 mg 48-h LC50 0.075 EC 25 Nishiuchi
sedula) (1981)
Stonefly 96-h LC50 0.100 T S 12 8.5 40 Woodward
(Pteronarcella 96-h LC50 0.0098 T S 12 7.5 40 & Mauck
badia) 96-h LC50 0.0053 T S 12 6.5 40 (1980)
Table 8. (continued)
Species Size Toxicity (mg/litre) Formulationa Systemb Temperature pH Hardnessc Reference
(°C)
(Eretes 1.5 cm, 0.20 g 48-h LC50 0.32 EC 25 Nishiuchi
sticticus) (1981)
Crayfish adults, 15-38 g 96-h LC50 7.8 T S 19 7.8 35 Andreu-Moliner
(Procambarus (1986)
clarkii)
Mollusca
Snail 2.9 cm, 1.6 g 48-h LC50 1.8 EC 22 Nishiuchi
(Semisulcospira & Yoshida
libertina) (1972)
Snail 2.4 cm, 3.3 g 48-h LC50 4.8 EC 22 Nishiuchi
(Cipangopaludina & Yoshida
malleata) (1972)
Red snail 0.72 cm. 1.1 g 48-h LC50 1.8 EC 22 Nishiuchi &
(Indoplanorbis Yoshida
exustus) (1972)
Physa-snail 0.91 cm, 0.11 g 48-h LC50 3.2 EC 22 Nishiuchi
(Physa acuta) & Yoshida
(1972)
10-day LC50 0.05 15-20 7.3 Mandoul et
al. (1967)
Snail 1.2 cm 48-h LC50 2.2 Singh &
(Lymnaea 1.2 cm 72-h LC50 0.65 Agarwal
acuminata) 1.2 cm 96-h LC50 0.3 (1981)
1.2 cm 168-h LC50 0.22
1.2 cm 240-h LC50 0.058
Table 8. (continued)
Species Size Toxicity (mg/litre) Formulationa Systemb Temperature pH Hardnessc Reference
(°C)
(Pila globosa) 3.5 cm 48-h LC50 25.4 Singh &
3.5 cm 72-h LC50 19.0 Agarwal
3.5 cm 96-h LC50 8.0 (1981)
3.5 cm 168-h LC50 2.8
3.5 cm 240-h LC50 2.2
(Gryphaea 10-day LC50 2.45 9-19 SW Mandoul et
angulata) al. (1967)
a T = Technical.
EC = Emulsifiable concentrate.
D = Dust.
WP = Wettable powder.
SP = Soluble powder.
P = Powder.
b S = Static.
SS = Semi-static.
SW = Sea water.
MAD = Minimal active dose.
c Hardness (mg/litre as CaCO3).
Trichlorfon at concentrations of up to 30 mg/litre did not affect
byssal attachment in seed mussels ( Mytilus edulis) (Roberts, 1975).
Trichlorfon showed cholinomimetic properties on the excitor or
inhibitor receptors of acetylcholine in the isolated heart, median
dorsal radula protractor muscle, and rectum of the snail Pila globosa
(Singh & Agarwal, 1979).
When freshwater snail, Lymnaea acuminata, was exposed to 10 or
20 mg trichlorfon/litre for 48 h, the rate of oxygen consumption and
the concentration of glycogen were both reduced, while the levels of
lactic acid and reducing sugars were enhanced. The effects persisted
for 7 days after withdrawal of the trichlorfon. On the basis of these
observations, Mahendru & Agarwal (1981) concluded that trichlorfon may
affect not only cholinesterase activity but also other enzyme systems,
such as the ones involved in carbohydrate metabolism.
7.3 Aquatic vertebrates
Trichlorfon is moderately toxic for fish, the 96-h LC50 values
ranging from 0.4 to 51 mg/litre (Table 8). The effects of trichlorfon
on the susceptibility of the developmental stages of carp were not
remarkable (Hashimoto et al., 1982). However, with regard to water
quality, the toxicity for fish increased as the water temperature, pH,
and hardness increased. The increase in toxicity was affected least
(3.4 fold) by increasing the temperature from 7 to 12 °C and most
(13-fold) by changing the pH from 6.5 to 8.5 (Woodward & Mauck, 1980).
Cherry salmon fingerlings (Onchorhyncus masou) were exposed to
one-tenth (0.105 mg/litre) and one-third (0.310 mg/litre) of the 96-h
LC50 value (1.1 mg/litre) of trichlorfon for 6 weeks in a
flow-through system. Trichlorfon initially retarded the growth of the
fish at both concentrations, but then the fish grew normally, and the
condition factors (weight-length coefficent) of the test fish did not
differ significantly from those of the controls after 6 weeks of
exposure to trichlorfon. Histopathological examination showed that the
liver cells were swollen with an obscure cell contour at the beginning
of the study, but no compound-induced histopathological changes were
observed after 6 weeks of exposure (Kimura et al., 1971).
When bluegill fingerlings were exposed to formulations of
trichlorfon, haemorrhage along, and fractures of, the caudal vertebrae
(usually extended over three vertebrae) occurred 4-8 h after treatment
at concentrations of 8-52 mg/litre (McCann & Jasper, 1972).
Matton & Laham (1969) treated 1-inch rainbow trout larvae for 16
h with 10-100 mg trichlorfon/litre, or for 40 h with 5 mg/litre.
Histochemical examination revealed that the acetylcholinesterase
activity was inhibited in the septa of the myotomes and at the
myoneural junctions. Furthermore, pathological changes were observed
in the heart, liver, blood cells, pseudogills, and muscular tissues.
7.4 Terrestrial vertebrates
Trichlorfon is relatively toxic for birds with oral LD50 values
of 40-180 mg/kg body weight (Table 9). Peakall & Bart (1983) reported
a 5-day LD50 value of 720 mg/kg for Bobwhite quail.
Residues of trichlorfon and dichlorvos in the bodies of
canopy-feeder birds (Baltimore orioles, crested fly-catcher, and blue
jay) were 0.005-0.04 mg/kg, 3 days following an aerial application of
trichlorfon at 1.12 kg/ha (Kurtz & Studholme, 1974).
Following aerial spraying of trichlorfon at 1.12 kg/ha, the brain
cholinesterase activity was about 20% less than that in the controls
in 2 out of 10 bird-species; in the other 8 species, the activity was
not depressed, even immediately after spraying (Zinkl et al., 1977).
According to a survey based on two, 1-h counts in 20-ha plots, 5
days before and after the spraying of 1120 g trichlorfon/ha, a
virtually identical change in the number of bird species and the total
number of hearings was observed. The application of trichlorfon
resulted in a slight drop in song-bird activity (165 to 151). It did
not produce any effects on cholinesterase activity in fly-catchers and
only 10% inhibition in northern oriols, after 3 days. There was a
considerable decrease in the singing of male birds after spraying; a
marked increase in feeding activity immediately following spraying was
noted. However, examination of the data on individual species does not
show any consistent pattern: the great crested fly-catcher decreased
but other fly-catchers did not, the American redstart decreased, but
another canopy species, the red-eyed vireo, did not. No significant
effects on the numbers of breeding pairs, bird abundance, nesting
success, or mortality were found. Brain ChE levels were reduced by 20%
in 6 out of 103 birds collected during the 5-day period following
spray (Peakall & Bart, 1983).
Japanese quail were given trichlorfon daily for 20 days at an
oral dose of 5 mg/kg body weight. Haematological examinations were
made on the 5th, 10th, 15th, and 20th day of treatment and on the 5th,
10th, 15th, and 30th day after stopping treatment. The number of
erythrocytes dropped on the 5th and 10th days, and the values of the
haematocrit and haemoglobin fell on the 5th day. A significant
increase in erythroblast contents was found on the 5th and 10th day.
No other significant changes of the above parameters were observed up
to the 30th day after treatment. The numbers of leukocytes,
lymphocytes, neutrophils, and monocytes sharply increased from the 5th
to 15th day of treatment, and quickly dropped to normal ranges on the
10th day after stopping treatment (Gromysz-Kalkowska et al., 1985).
Table 9. Acute toxicity of trichlorfon for non-target terrestrial organisms
Species Size Application Toxicity Formulationa Temperature Reference
(°C)
Birds
Red-winged blackbird oral LD50 37-75 mg/kg Schafer Jr et al.
(Agelaius phoeniceus) (1983)
oral LD50 40 mg/kg Schafer (1972)
European starling oral LD50 43 mg/kg Schafer Jr et al.
(Sturnus vulgaris) (1983)
oral LD50 47 mg/kg Schafer (1972)
(Columbia livia) 250-380 g oral LD50 179.9 mg/kg Hattori (1974)
White leghorn hen 1.5-2.0 kg oral LD50 75 mg/kg Kimmerle & Löser
intraperitoneal LD50 75 mg/kg (1974)
Japanese quail oral LD50 50 mg/kg Gromysz-Kalkowska
(Coturnix coturnix (1985)
japonica)
Arthropods
Honey-bee sprayed on glass plate LC50 0.600 W/V% Abdelwahab et
(Apis mellifera, L) al. (1973)
weathered residues (3 h) 24-h 17% mortality 50%SP 26 Johansen (1972)
128.6 mg topical LD50 28.5 µg/g T 16 Ahmad &
Johansen (1973)
Alfalfa leafcutter bee weathered residues (3 h) 24-h 5% mortality 50%SP 31 Johansen (1972)
(Megachile rotundate, F) 22.2 mg topical LD50 250 µg/g T 16 Ahmad &
26.1 mg LD50 515 µg/g T 16 Johansen (1973)
Table 9. (continued)
Species Size Application Toxicity Formulationa Temperature Reference
(°C)
Alkali bee weathered residues (3 h) 24-h 31% mortality 50%SP 31 Johansen (1972)
(Nomia melanderi,
Cockerell)
a SP = Soluble powder.
T = Technical grade.
7.5 Ecosystems
Trichlorfon applied to ponds at the rate of 1 mg/litre water
destroyed the food invertebrates for fish. Large numbers of
zoo-plankton, rotifers, and crustacea, died in the first 24 h after
treatment, whereas benthos died during the first week. The affected
fauna, community recovered slowly. Trichlorfon treatment deprived the
fish of valuable foods, such as crustacean and bottom fauna, for as
long as 1 month after treatment (Grygierek & Wasilewska, 1981).
8. EFFECTS ON EXPERIMENTAL ANIMALS AND IN VITRO TEST SYSTEMS
A more complete treatise on the effects of organophosphorus
insecticides in general, especially their short- and long-term effects
on the nervous system, will be found in the WHO Environmental Health
Criteria 63: Organophosphorus insecticides - A general introduction
(WHO, 1986).
No-observed-effect levels in animals treated with trichlorfon
under various conditions are summarized in Annex II.
8.1 Acute toxicity
LD50 values for trichlorfon in different species and following
different routes of administration are shown in Table 10. Species
differences in LD50 values seem to be rather small.
The acute toxicity of trichlorfon is due to the inhibition of
acetylcholinesterase at the nerve endings by the degradation product
dichlorvos, leading to accumulation of endogenous acetylcholine. The
effects are manifested by muscarinic and central nervous system signs
and symptoms (Taylor, 1980). In the rat, the toxic effects produced by
trichlorfon are characteristics of organophosphorus poisoning, i.e.,
muscular fibrillation, salivation, lacrimation, incontinence,
diarrhoea, respiratory distress, prostration, gasping, tonic and
clonic convulsion, coma, and death. Trichlorfon caused rapid onset of
poisoning, effects occurring within 5 min at approximate LD50 doses
(Edson & Noakes, 1960).
Only a slight increase in mortality was noted in weanling
Holtzman rats compared with that in adults, when treated with
trichlorfon (Brodeur & DuBois, 1963).
Trichlorfon has a low dermal toxicity (LD50: > 2000 mg/kg)
compared with that of dichlorvos (LD50: 75-900 mg/kg). The
difference in lipid solubility between the two compounds may be a
major factor accounting for this difference (Holmstedt et al., 1978).
Since trichlorfon is used as a parasiticide in livestock, studies
have been conducted to assess the effects of the chemical on
cholinesterase activity and clinical conditions. Administration to
horses of a single dose of trichlorfon of 60 or 80 mg/kg body weight,
by stomach tube, resulted in moderately severe and severe colic,
respectively, whereas a single dose of 80 mg/kg mixed into the feed
was associated with only a transient softening of the faeces. Doses of
40 mg/kg or less, by either method of administration, were generally
tolerated without notable adverse effects except for the softening of
the faeces, which tended to be self-limiting. Clinical trials at dose
rates of 35-40 mg/kg in horses, including pregnant and nonpregnant
mares, stallions, suckling and weanling foals, and yearlings, did not
cause any notable adverse effects (Drudge et al., 1976).
Table 10. Acute toxicity of trichlorfon for several animal species
Animal Sex Route Parameter Value Reference
Mouse M oral LD50 800 mg/kg Haley et al. (1975)
F 800 mg/kg
M ip LD50 600 mg/kg Soliman et al. (1984)
Rat M oral LD50 660 mg/kg Benes & Cerna (1970)
M oral LD50 630 mg/kg Gaines (1969)
F 560 mg/kg
M,F dermal LD50 >2000 mg/kg
M dermal LD50 2800 mg/kg Edson & Noakes (1960)
M ip (23-day LD50 190 mg/kg Brodeur & Dubois
old) (1963)
ip (adult) 250 mg/kg
M,F inhalation LC50 533 mg/m3 Kimmerle (1975a)
(4 h)
Guineapig M,F ip LD50 300 mg/kg DuBois & Cotter
(1955)
Rabbit M dermal LD50 5000 mg/kg Deichmann & Lampe
(1955)
Dog M oral LD50 420 mg/kg Deichmann & Lampe
(1955)
Pretreatment of female and castrated male sheep with 1.5 mg
trichlorfon/kg body weight by intravenous injection, which was
insufficient to produce a significant depression of erythrocyte
cholinesterase activity, produced toxic effects that were additive to
those of coumaphos, subsequently administered at 4 mg/kg per day
(Silvestri et al., 1975a,b).
Horses treated with trichlorfon (39.7 mg/kg body weight) combined
with mebendazole (8.8 mg/kg body weight) did not show any, or only a
few, side-effects, except ChE inhibition, whereas horses given higher
dosages (up to 5 times this dose) showed dosage-related increases in
the severity of clinical signs and inhibition of erythrocyte
cholinesterase activity. Depression of the activity was detected
within 1 h of treatment. The maximum depression ranged from 42% (at
the initial dosage) to 75% (at a 5 times higher dosage). Recovery of
base-line activity did not occur in any of the horses within 32 days
after treatment (Gingerich & Mia, 1981).
8.2 Short-term exposure
Wistar albino rats (10 males per group) were fed trichlorfon at
0, 1, 5, 25, or 125 mg/kg diet for 16 weeks. Trichlorfon failed to
cause erythrocyte cholinesterase depression at 125 mg/kg. No effects
were noted on food consumption or growth, or during gross examination
of the tissues (Edson & Noakes, 1960).
Rats (13/sex per group) were fed trichlorfon at dietary levels of
0, 20, 100, or 300 mg/kg diet for 16 weeks. Significant cholinesterase
depression was noted at 300 mg/kg. No effects were observed at the 100
mg/kg level on growth, behaviour, food consumption, or on gross and
microscopic examination of tissues (Doull & Dubois, 1956).
After oral administration of trichlorfon to guinea-pig (100 mg/kg
body weight per day for 60 days) the haemoglobin content was decreased
by 13.5% while the haematocrit value remained unchanged. Trichlorfon
also decreased the serum cholinesterase activity by 50% and increased
the activity of alkaline phosphatase by 36% (Krustev et al., 1976).
Two dogs were administered 45 mg trichlorfon/kg body weight,
orally, for 6 days per week over 3 months. No cumulative effects were
noted. The serum cholinesterase level was 60% of normal at the end of
the study period. No deaths occurred (Deichmann & Lampe, 1955). In
another study, dogs (one male and one female per group, 2 males and 2
females serving as controls) were fed trichlorfon at levels of 0, 50,
200, or 500 mg/kg diet for 12 weeks. Plasma and erythrocyte
cholinesterase activity was depressed at 500 mg/kg diet and unaffected
at 200 mg/kg diet. Recovery of enzyme activity was complete 6 weeks
after the feeding of trichlorfon stopped (Williams et al., 1959).
Rats (10/sex per group) were exposed for 6 h a day over a 3-week
period (total of 15 exposures) to an atmosphere containing trichlorfon
at concentrations of 0, 12.7, 35.4, and 103.5 mg/m3. Exposure to a
concentration of 103.5 mg/m3 slightly affected the health of the
animals (no details available). Body weight gain, parameters of
haematological and clinical chemistry examinations, and urinalyses
were not influenced at any exposure level. Cholinesterase inhibition
of 42, 31, and 22% was found in the plasma, erythrocytes, and the
brain, respectively, in male animals at 103.5 mg/m3; female animals
showed dose-dependent inhibition values of 39, 26, and 26% at 35.4
mg/m3 and 48, 44, and 47% at 103.5 mg/m3 in the plasma,
erythrocyte, and brain, respectively. The only significant alteration
in relative organ weight was found in male animals showing
dose-related increases in relative spleen weights of about 20 and 25%
at the 35.4 and 103.5 mg/m3 exposure levels, respectively. No
abnormal histological findings were observed in any of the tissues
examined microscopically (Kimmerle, 1975b).
In a 13-week study to evaluate target organ toxicity,
dose-response, and maximum tolerated dose for a 2-year study, groups
of 10 male and 10 female Fischer rats and B6C3F1 mice (8-week-old)
were administered trichlorfon in the feed at 0, 62, 185, 555, 1666, or
5000 mg/kg, for 7 days a week. All the rats and mice survived the
13-week treatment, except one male mouse at 5000 mg/kg and one female
mouse at 1666 mg/kg. The body weights of the 5000 mg/kg groups of male
and female rats and mice were significantly lower compared with those
of their respective controls. Plasma and erythrocyte cholinesterase
activity was reduced in a dose-related manner in the rats and mice.
Neurotoxicity tests showed that motor activity and grip strength were
reduced in the 1666 and 5000 mg/kg groups of male and female rats and
mice. No histo pathological changes were observed in the brain, spinal
cord, and sciatic nerve and other organ systems. Absolute and relative
liver, kidney, and spleen weights were increased in the 1666 and 5000
mg/kg groups of male and female rats and mice. The weight increase was
not accompanied by any histopathological findings. Two-year
carcinogenicity studies on trichlorfon administered in the diet to
male and female Fischer rats and B6C3F1 mice are in progress (Chan &
Peters, 1989).
Groups of 7 male and 7 female Wistar rats were fed 1, 5, 10, 30,
50, or 100 mg trichlorfon/kg body weight in their diet for 12 weeks.
A dose of 100 mg/kg, daily, inhibited the cholinesterase activity in
all tissues examined, namely erythrocytes, serum, brain, heart, liver,
and gastrocnemius muscle, but the extent of the inhibition differed
between tissues. No significant histological changes were found in the
parenchymatous organs, such as the brain, heart, lung, pituitary
gland, thyroid gland, liver, intestine, stomach, kidney, adrenal
gland, and testes. Despite the marked inhibition of the tissue
cholinesterase activity in the treated animals, no changes were
observed in nocturnal behaviour, reactivity to external stimuli,
conjunctival and pinna reflexes, and the avoidance reflexes to painful
stimuli (Shimamoto & Hattori, 1965).
Groups of 5 male and 5 female Rhesus monkeys ( Macaca mulatta)
were each administered technical trichlorfon by oral intubation at
dose levels of 0, 0.1, or 0.2 mg/kg body weight per day for 26 weeks,
in order to determine a no-observed-effect level on the cholinesterase
activity in erythrocytes. There were no effects on appearance,
behaviour, nutritional state, feed and water consumption, and body
weight gain. No treatment-related changes were found in haematology,
liver and kidney function, and erythrocyte cholinesterase activity.
The NOEL in this study was 0.2 mg/kg body weight per day (Hoffmann et
al., 1988).
8.3 Skin and eye irritation; sensitization
8.3.1 Skin irritation
A skin irritation test was conducted with technical trichlorfon
applied to the intact and abraded skin of 6 albino rabbits. The
contact time was 24 h and the animals were observed for 7 days.
Technical trichlorfon did not irritate the skin (Thyssen, 1981).
Technical trichlorfon was tested on 6 New Zealand White rabbits
for its dermal primary irritation potential. The test material was
kept in contact with the shaved skin under an occlusion patch for 4 h,
and scoring was performed for 72 h. The results indicated that
technical trichlorfon is not a primary dermal irritant (Bond, 1986).
8.3.2 Skin sensitization
Technical trichlorfon was evaluated in a Magnusson and Kligman
maximization test on guinea-pigs. The concentrations used were:
intra-dermal induction, 1%; topical induction, 25%; first challenge,
25%; and second challenge, 12.5%. The results showed that technical
trichlorfon is a sensitizer for guinea-pigs (Mihail, 1985).
Technical trichlorfon was investigated in the open epicutaneous
test on guinea-pigs for skin-sensitizing potential. Induction (4
weeks, 5 days per week) was carried out using 0, 1, 3, or 10% test
compound. Challenges were done with the same concentrations 4, 6, and
8 weeks after the start of the induction. The 3 and 10% dilutions of
trichlorfon had a skin-sensitizing effect, but not the 1% dilution
(Mihail, 1986a).
8.3.3 Eye irritation
An irritation test was performed using technical trichlorfon on
6 albino rats. The exposure times were 5 minutes and 24 h. Technical
trichlorfon had a moderately irritating effect on the mucosae of the
eye (Thyssen, 1981).
8.4 Long-term exposure
Several reviews on long-term toxicity studies on trichlorfon have
been published (FAO/WHO, 1972, 1976, 1979; Holmstedt et al., 1978;
Machemer, 1981; IARC, 1983).
8.4.1 Oral administration
8.4.1.1 Mouse
A group of 30 male and 28 female AB/Jena strain mice, 8 weeks of
age, received 30 mg trichlorfon/kg body weight, by gavage, twice
weekly for 75 weeks. A group of 30 male and 29 female mice served as
controls. All surviving animals were killed in week 80. Inhibition of
body weight gain and shortening of survival time were noted in the
treated group. There was no statistically significant difference in
the incidence of tumours between treated and control mice (Teichmann
& Hauschild, 1978).
Groups of 60 male and 60 female Charles River CD-1 mice
(6-week-old) were given 100, 300, or 1000 mg trichlorfon/kg diet for
90 weeks, except for males in the 1000 mg/kg group which were treated
for 82 weeks. A group of 60 male and 60 female mice served as
controls. Inhibition of body weight gain was observed in females given
300 or 1000 mg trichlorfon/kg diet. Cholinesterase activity was
depressed in both sexes at 1000 mg/kg diet. The no-observed-effect
level was 100 mg/kg diet. No significant microscopic alterations were
reported. There was no statistically significant difference in the
incidence of tumours between treated and control mice (Machemer,
1981).
Groups of 50 CD-1 mice per sex and dose level were given
technical grade trichlorfon at 0, 300, 900, or 2700 mg/kg diet
(analytical concentrations 0, 275, 891, or 2707 mg/kg diet) for 104
weeks. There were no significant differences between groups in feed
consumption, haematological parameters, or mortality. Increased
incidences of urine stain and ear lesions in males, and vaginal
discharge in females, were regarded as non-specific cholinergic
effects. The body weights of the 2700 mg/kg female mice were
significantly increased throughout the study. There was a
compound-related increase in liver weight in the 900 and 2700 mg/kg
females with no corresponding light-microscopic changes, regarded as
a compound-adaptive mechanism. All trichlorfon-treated groups showed
significant blood cholinesterase depression throughout the study: 20%,
in the 300 mg/kg females, and up to 74% in the 2700 mg/kg group; this
latter dose was compatible with a maximum tolerated dose. No
compound-related tumorigenicity was found in this study (Hayes, 1988).
8.4.1.2 Rat
Groups of 25 male and 25 female, 4-week-old, Sprague-Dawley rats
were fed trichlorfon in the diet at doses of 0, 50, 250, 500, or 1000
mg/kg diet. The treatment continued for 17 months for the males and 24
months for the females. Survival time was shortened in both males and
females given 1000 mg/kg diet. Retarded growth rate was observed in
males given 1000 mg/kg diet: their weight was 15% less than the
controls. Depression of cholinesterase activity was noticed in both
sexes given doses of 500 or 1000 mg/kg. Female rats fed 500 or 1000
mg/kg diet exhibited an absence of primary follicules and primitive
ova and male rats fed 1000 mg/kg diet showed depression of
spermatogenesis. Necrotizing arteritis was observed in both sexes at
doses of 500 or 1000 mg/kg. However, no treatment-related
toxicological changes were reported in rats given 50 or 250 mg/kg
diet. The incidences of mammary tumours in female rats were 14, 8, 20,
21, and 25% in groups given 0, 50, 250, 500, and 1000 mg/kg diet,
respectively. The time to onset of the appearance of tumours was
dose-dependent, being 1.7 years for the controls while at dietary
levels of 50, 250, 500, and 1000 mg/kg, the onset occurred after 1.6,
1.8, 1.5, and 1.1 years, respectively (Doull et al., 1962b).
Another long-term toxicity study was reported in which groups
each of 25 male and 50 female, 4-week-old, Sprague-Dawley rats were
fed trichlorfon at doses of 0, 100, 200, or 400 mg/kg diet for 18
months. Toxicological changes, such as cystic granular alterations of
the ovaries and depression of oocytogenesis were seen only in rats fed
400 mg/kg diet. From these 2 studies, it was concluded that
trichlorfon given orally to rats may enhance some of the normal aging
processes in the reproductive tissues in the females and possibly in
the males (Doull et al., 1965).
Groups each comprising 50 male and 50 female, Long-Evans rats, 4
weeks of age, were fed trichlorfon at doses of 50, 250, 500, or 1000
mg/kg diet. A group of 100 male and 100 female rats served as
controls. There were neither shortening of survival times nor retarded
growth in rats given 1000 mg/kg diet. Depression of cholinesterase
activity in both sexes at 1000 mg/kg diet was the only finding related
to the treatment. There were no morphological changes considered to be
compound-related. All surviving animals were killed after 24 months of
treatment. There was no statistically significant difference in the
incidence of tumours between treated and control rats (Lorke & Löser,
1966; Grundmann & Hobik, 1966).
A group of 30 male and 35 female albino rats, 10 weeks of age,
received 22 mg trichlorfon/kg body weight in saline, by gavage, twice
weekly for 90 weeks. A group of 25 male and 26 female rats served as
controls. All surviving animals were killed in week 118. Survival time
was reduced in the treated group. There were neither biochemical nor
morphological changes attributable to the treatment, and no
statistically significant differences in the incidence of tumours
between treated and control rats (Teichmann et al., 1978).
Groups of 50 male and 50 female Fischer 344 rats, were fed diets
containing mean analytical concentrations of 0, 92.2, 273, or 1518 mg
trichlorfon/kg feed. Dosages were provided as nominal 0, 100, 300,
1750 mg/kg diet. The top dosage was started as 1000 mg/kg for 27
weeks, changed to 1250 mg/kg for 5 weeks, changed to 1500 mg/kg for 8
weeks, and then changed to 1750 mg/kg for the remaining 65 weeks.
Satellite groups of 20 animals per sex served as controls and, at the
highest dose level, the animals were treated for one year and then
sacrificed to provide interim data. Mortality was unaffected by
treatment. Decreased food consumption was slight in females up to 66
weeks and depressed body weight gain occurred in high-dose males.
Increased cholesterol levels were observed in males at doses of 300
mg/kg or more while slight anaemia was present in both sexes at 1750
mg/kg. Cholinesterase activity in plasma, red cells, and the brain was
significantly reduced in both sexes at 1750 mg/kg. Both liver and
kidney weights were increased at 1750 mg/kg in both sexes.
Histological changes were not present in the liver; however, chronic
nephropathy occurred at the highest dose level. Hyperplasia of the
upper small intestine and gastritis occurred at 300 mg/kg and above.
A NOEL equal to 4.45 mg/kg body weight for males and 5.82 mg/kg body
weight for females was established for the study (Hayes, 1989).
Tumorigenicity for this study was not assessed by the Task Group.
8.4.1.3 Dog
Groups each consisting of 2 male and 2 female Beagle dogs were
given trichlorfon in the diet at doses of 0, 50, 250, 500, or 1000
mg/kg diet for 12 months. Depression of cholinesterase activity was
observed at the 500 and 1000 mg/kg diet levels. Spermatogenesis was
inhibited in males given 1000 mg/kg diet (Doull et al., 1962a).
Groups of 4 male and 4 female Beagle dogs were fed dietary
trichlorfon at levels of 0, 50, 200, 800, or 3200 mg/kg diet for 4
years. Cholinesterase activity was depressed in dogs of both sexes at
doses higher than 200 mg/kg diet. Increased mortality, retarded growth
rate, reduced weights of adrenal glands and testes, and impairment of
the kidney function were observed at 800 and 3200 mg/kg diet. In
addition, cholinergic symptoms, such as tremors, cramps, and
salivation were noteworthy at the 3200 mg/kg level. Only one female
dog in the latter group survived for 4 years. Liver injury was
detected biochemically in dogs that died during the study (Löser,
1970).
8.4.1.4 Monkey
Groups of 5 male and 5 female Rhesus monkeys ( Macaca mulatta)
were administered technical trichlorfon by gavage at dose levels of 0,
0.2, 1, or 5 mg/kg body weight per day, 6 days per week for 10 years.
At the 5 mg/kg level, transient pupillary constriction was seen in 2
animals during the first month, and muscular fasciculations in one. At
this dose level, a lowering of the erythrocyte count occurred in both
sexes and, towards the end of the study, the body weights were lower
in this group. Plasma-, erythrocyte-, and brain-cholinesterase
activity was inhibited in the 5 and 1 mg/kg groups, while in the 0.2
mg/kg group inhibition occurred only in erythrocytes in the males.
Liver biopsies done during the first 3 years of the study did not
reveal any evidence of changed liver morphology in any of the groups.
No trichlorfon-related mortality occurred. Complete gross and
microscopic examination of the tissues of the treated groups gave a
non-neoplastic pathology similar to that in the control group. No
significant pathology, including tumorigenicity, related to the
administration of trichlorfon was observed in the treated animals,
compared with the controls (Griffin, 1988).
8.4.2 Intraperitoneal administration
8.4.2.1 Mouse
A group of 30 male and 30 female AB/Jena strain mice, 8 weeks of
age, received 28.2 mg trichlorfon/kg body weight by ip injection,
twice weekly, for 75 weeks. A group of 30 male and 30 female control
mice were given saline intraperitoneally. All surviving animals were
killed in week 80. Inhibition of body weight gain and reduced survival
time were noted in the treated group. There was no statistically
significant difference in the incidences of tumours between treated
and control mice (Teichmann & Hauschild, 1978).
8.4.2.2 Rat
In a study by Teichmann et al. (1978), a group of 30 male and 35
female albino rats, 10 weeks of age, received 12 mg trichlorfon/kg
body weight intraperitoneally, twice weekly, for 90 weeks, while a
group of 25 male and 25 female control rats was given saline. All
surviving animals were killed in week 118. Survival time was reduced
in the treated group. There was no statistically significant
difference in the incidences of tumours between treated and control
rats.
8.4.2.3 Hamster
Syrian golden hamsters, (23 males and 25 females per group) 7-8
weeks of age, were given 20 mg trichlorfon/kg body weight
intraperitoneally, once weekly, for 90 weeks. A group of 22 male and
23 female control hamsters were given saline. All surviving animals
were killed in week 100. Inhibition of body weight gain and reduced
survival time were noted in the treated group. There was no
statistically significant difference in the incidences of tumours
between treated and control hamsters (Teichmann & Schmidt, 1978).
8.4.3 Dermal administration
8.4.3.1 Mouse
A group of 30 male and 30 female AB/Jena strain mice, 8 weeks of
age, received 0.25 ml 1% solution of trichlorfon in acetone, dermally,
twice weekly, for 75 weeks. A group of 30 male and 30 female mice
served as controls. All surviving animals were killed in week 80.
Inhibition of body weight gain and reduced survival time were noted in
the treated group. There was no statistically significant difference
in the incidences of tumours between treated and control mice
(Teichmann & Hauschild, 1978).
8.5 Mutagenicity
8.5.1 DNA methylation
In vitro studies using chemical agents or isolated nucleic
acids with dichlorvos showed that the methylating capability of
dichlorvos was less, by a factor of 10-100, than that of strongly
genotoxic agents (WHO, 1989). In vivo studies using the
determination of 14C-N7-methylguanine in the urine have been used
to calculate the in vivo methylation capability of dichlorvos for
DNA (WHO, 1989) and trichlorfon (Dedek et al., 1976). However, it has
been shown that the excretion of 14CH3-labelled purines in the
urine does not constitute evidence for the methylation of the purines
of DNA, because a natural biosynthetic pathway will give rise to
urinary methylated purines via the C1-pool (Wright et al., 1979;
WHO, 1989). Consequently, only the measurement of 14C methylated
purines in the DNA of organs would provide acceptable results for the
evaluation of the in vivo methylating capability of alkylating
agents (Wright et al., 1979).
Such studies have been performed by Dedek (1981) for trichlorfon
with ip administration of 0.48, 0.40, or 0.065 mmol/kg to male mice
(strain AB Jena/Halle). The extent of methylation in liver DNA was
found to be maximal after 6 h in amounts of 6-8 and 0.8 µmol
N7-MeG/mol guanine for the high and the low dose, respectively, that
is 6-8 methylations in the N7 position per 106 guanine bases.
However, the alkylation at N7 of guanine by small groups, such as
the methyl group, does not show a good correlation with genotoxic
effects.
For obtaining a dose-independent measure of alkylation activity,
the covalent binding index may be used, which is defined as:
micromol bound per mol of nucleotides
CBI =
millimol administered per kg animal
For the strong alkylating agent methyl methanesulfonate (MMS),
the CBI values for guanine N7 have been estimated to be between 135
and 556, depending on the route of administration and the time
intervals. The corresponding CBI values for trichlorfon were 2.3 to
5.1 (Dedek, 1981; Dedek et al., 1984).
Trichlorfon has a DNA-alkylating property and may react with DNA
in vitro to cause depurination and excision (Rosenkranz &
Rosenkranz, 1972).
8.5.2 Mutagenicity
Data on mutagenicity tests in vitro as well as in vivo have
accumulated during the past 20 years. Results indicate that
trichlorfon is partly positive and partly negative, depending on the
purity of the test material, test system, dosage, or source, and
possible effects derived from its degradation products (IARC, 1983).
Results are summarized in Table 11.
Trichlorfon induces gene mutation in S. typhimurium and E.
coli Poole et al., 1977; Carere et al., 1978a; Benigni et al., 1980;
Shirasu et al., 1982; Morya et al., 1983) and in S. cerevisiae
(Riccio et al., 1981; Gilot-Delhalle et al., 1983). It also induces a
mitotic crossing over, gene conversion, and mitotic recombination
(Waters et al., 1980). The positive results in microorganisms indicate
that trichlorfon induces mainly base-pair substitution or mutation in
the absence of metabolic activation. Trichlorfon also induces a
chlorophyl mutation (Panda & Sharma, 1980) and chromosomal damage in
plants (Logvinenko & Morgun, 1978; de Kergommeaux, 1983).
In vitro studies with mammalian cells indicate that trichlorfon
induces unscheduled DNA synthesis (UDS) in human epithelial cells
(Benigni & Dogliotti, 1980; Aquilina et al., 1984) and in human
fibroblasts (Waters et al., 1982). It induces sister chromatid
exchanges (SCE) in Chinese hamster ovary (CHO) cells (Chen et al.,
1981; Waters et al., 1982), and chromosomal aberrations in CHO (Sasaki
et al., 1980; Ishidate et al., 1981) and human lymphocytes (Kurinniy
& Pilinskaya, 1977).
Trichlorfon has also been shown to cause cell transformation in
C3H1OT1/2 CL8 cells (Waters et al., 1981, 1982), and forward mutations
in mouse lymphoma L51784 cells in the absence of metabolic activation
(McGregor et al., 1988).
In vivo studies indicate that trichlorfon induces chromosomal
aberrations in mouse bone marrow cells (Kurinniy, 1975; Kuzmenko et
al., 1980; Ryazanova & Gafurova, 1980; Nehéz et al., 1987). However,
negative results have been reported in a bone marrow micronucleus test
after intraperitoneal injection of trichlorfon in doses up to 400
mg/kg (Paik & Lee, 1977; Jones et al., 1982; Waters et al., 1982). In
the dominant lethal test on mice, trichlorfon was reported to induce
a significant increase in post implantation losses only after
administration of relatively high doses, i.e., 405 mg/kg, as a single
dose, or 54 mg/kg, daily, for 3 weeks (Dedek et al., 1975; Fischer et
al., 1977). The reproducibility of such effects, however, was not
supported by other investigators (Becker & Schöneich, 1980). A
short-term study on mice indicated that trichlorfon did not induce any
chromosomal damage in either bone marrow or germ cells after treatment
with 0.5 mg/litre in the drinking-water, continuously, for 7 weeks
(Degraeve et al., 1984).
Cytogenetic studies on lymphocytes obtained from persons
suffering from acute intoxication or those occupationally exposed to
trichlorfon showed variable increases in the frequency of chromosomal
aberrations (Trinh Van Bao et al., 1979; Kiraly et al., 1974).
In conclusion, trichlorfon is mutagenic for microorganisms or
mammalian cells in in vitro assay systems. However, the data are not
consistent, probably because of the purity of the test material and
possible effects derived from its degradation products, such as
dichlorvos. In vivo studies indicate that trichlorfon induces
chromosomal damage only at relatively high dose levels. In short-term
studies, however, no such effects were found in bone marrow cells or
in germ cells.
Table 11. Summary of mutagenicity studies on trichlorfon
Test system Result Reference
Microorganisms
Rec-assay P. mirabilis + Adler et al. (1976); Braun et al. (1982)
S. typhimurium + Braun et al. (1982)
B. subtilis - Shirasu et al. (1976)
Spot-test E. coli - Nagy et al. (1975)
S. typhimurium - Carere et al. (1978a)
Point mutation E. coli + Hanna & Dyer (1975); Batzinger & Bueding (1977);
Kawachi et al. (1980); Nagy et al. (1975);
Moriya et al. (1983)
S. typhimurium + Poole et al. (1977); Benigni et al. (1980); Carere et al.
(1978a); Kawachi et al. (1980); Shirasu et al. (1982)
A. nidulans - Morpurgo et al. (1977); Moriya et al. (1983)
S. coelicolor + Carere et al. (1978a,b); Benigni et al. (1980)
S. cerevisiae + Riccio et al. (1981)
S. pombe + Gilot-Delhalle et al. (1983)
Mitotic crossing over A. nidulans + Morpurgo et al. (1977); Benigni et al. (1980)
S. cerevisiae + Riccio et al. (1981); Waters et al. (1982)
Gene conversion S. cerevisiae + Riccio et al. (1981); Jones et al. (1982);
Waters et al. (1982)
Mitotic recombination S. cerevisiae + Poole et al. (1977); Riccio et al. (1981);
Simmon (1979); Waters et al. (1982)
Plants
Chlorophyl gene mutation H. vulgare + Panda & Sharma (1980)
Chromosome damage H. vulgare + Panda & Sharma (1980)
Wheat + Logvinenko & Morgun (1978)
Clastogenesis Vicia faba + Amer & Ali (1983); de Kergommeaux et al. (1983)
Mammalian cells in vitro
UDS Human epithelial cells + Benigni & Dogliotti (1980)
Human fibroblasts + Waters et al. (1982)
SCE Chinese hamster cells + Chen et al. (1981, 1982); Waters et al. (1982)
Table 11 (continued).
Test system Result Reference
Mammalian cells in vitro
Chromosome damage Chinese hamster cells + Sasaki et al. (1980); Ishidate et al. (1981)
Human lymphocytes + Kurinniy & Pilinskaya (1977)
Gene mutation Mouse lymphoma + Paik & Lee (1977); Jones et al. (1982);
Waters et al. (1982)
Forward mutation assay Mouse lymphoma cells + McGregor et al. (1988)
Drosophila
Gene mutation D. melanogaster - Benes & Sram (1969); Brzheskii (1973); Lamb (1977)
Valencia (1977); Waters et al. (1980)
Recessive lethal D. melanogaster - Lamb (1977); Waters et al. (1980)
Mammals
Micronucleus test Mouse bone marrow - Paik & Lee (1977); Herbold (1979a);
Waters et al. (1982); Jones et al. (1982);
Chromosome damage Mouse bone marrow - Degraeve et al. (1981, 1985)
+ Kurinniy (1975); Nehez et al. (1982, 1987);
Degraeve et al. (1981)
Hamster bone marrow - Dzwonkowska & Hübner (1986); Volkner (1987)
Dominant lethal Mice - Arnold et al. (1971); Epstein et al. (1972);
Herbold (1979b); Becker & Schoeneich (1980);
Degraeve et al. (1981, 1984a); Moutschen-Dahmen (1981)
+ Schiemann (1975); Dedek et al. (1975)
Chromosome damage Mouse testes - Degraeve et al. (1981, 1984b, 1985)
+ Bulsiewicz et al. (1976);
+ Fischer et al. (1977)
Chromosome damage Human lymphocytes + Trinh Van Bao et al. (1974); Kiraly et al. (1979)
8.6 Carcinogenicity
All long-term studies available for this evaluation have been
described in section 8.4; many of these studies are inadequate as
carcinogenicity studies, or the available reports give insufficient
detail for an evaluation.
A weak, dose-related increase in the incidence of mammary tumours
was reported in female rats fed trichlorfon in the diet (Doull et al.,
1962b). However, reports adequate for evaluation did not show any
evidence for the carcinogenicity of trichlorfon in rats, mice, or
hamsters after oral, intraperitoneal, or skin application.
The carcinogenicity of dichlorvos, the major conversion product
of trichlorfon in the mammalian body, has been discussed in WHO (1989)
and US NTP (1989) (see section 8.10).
8.7 Teratogenicity and reproductive toxicity
8.7.1 Mouse
A single dose of 360 mg trichlorfon/kg, injected
intraperitoneally in 33 AS/Jena mice on the first day of gestation,
caused embryo-toxicity. Post-implantation losses were increased with
60, 120, 240 mg/kg ip on day 9 of gestation (13-15 mice/group), with
120 or 240 mg/kg ip on days 1-7 (25-30 mice/group), and were more
pronounced with 240 mg/kg ip (23 mice) on days 7-14 of gestation.
There were no serious malformations (Scheufler, 1975).
Nehéz et al. (1987) showed that 4 consecutive intraperitoneal
doses of 51.5 mg trichlorfon/kg body weight administered to AB
Jena/Halle mice (12-21 per group) on days 2, 3, 4, and 5, or days 6,
8, 10, and 12 of gestation produced a very weak embryotoxic effect
(slightly elevated post-implantation losses, P <0.05), but no
teratogenic activity was found.
A dose of 500 or 600 mg trichlorfon/kg body weight, given daily
to CD-1 mice by oral gavage on days 10-14 of gestation, produced a
reduction in fetal weight and a slightly increased incidence of cleft
palate. This malformation was found in 4/67 fetuses from dams given
500 mg/kg, in 7/205 fetuses from dams given 600 mg/kg, and in 3/402
fetuses from control dams (Staples & Goulding, 1979).
When trichlorfon was administered by gavage to CD-1 mice (15-24
per group) on days 7-16 of gestation, at a daily dose of 200, 300, or
400 mg/kg, and to CD rats (15-24 per group) at 50, 100, or 200 mg/kg
per day on days 7-19 or 8-20 of gestation, trichlorfon was
teratogenic, fetotoxic, and lethal at the two highest dose levels. At
the lowest dose level, which was not maternally toxic, there was a
significant increase in the number of calcified centres in the
forepaws and hindpaws indicating fetotoxicity and a delay in
maturation (Courtney et al., 1986).
Clemens & Hartnagel (1986) reviewed this study and suggested that
no major or minor birth defects had been demonstrated, and that the
embryotoxicity, fetotoxicity, and rib variations in the mouse study
occurred at maternally toxic dose levels. The fact that a very small
number of fetuses was assessed made conclusions difficult.
Intragastric exposure of male CFW mice (30/group) to 30 mg
trichlorfon/kg for 5-260 days resulted in a significant regression of
the seminiferous epithelium in the testes after 100 days of treatment.
No regeneration of the gonads was observed, 40 days after exposure
ceased (Wenda-Rozewicka, 1983).
8.7.2 Rat
A single dose of 80 mg trichlorfon/kg, given to groups of 11
Wistar rats by oral gavage on day 13 of gestation, produced an
increased incidence of embryonic death and fetal malformations, such
as exencephaly and nonclosing eyelids. When 80 mg trichlorfon/kg was
similarly administered on day 9, these effects were not significant.
A daily dose of 8 mg/kg during gestation did not produce any
teratogenic manifestations (Martson & Voronina, 1976).
When trichlorfon was administered in the diet to groups of 9-26
CD rats on days 6-15 of gestation at a daily intake of 76, 145, 375,
432 or 519 mg/kg body weight, both maternal and fetal body weights
were reduced with ingestion of 432 or 519 mg/kg. There was a
dose-related increase in the incidence of fetal malformations with
ingestion of 145 mg/kg or more. The predominant malformations were
exencephaly, meningocoele, hydrocephaly, syndactily, micrognathia,
cleft palate, and skeletal system alterations. No adverse effects were
found with a dose of 76 mg/kg (Staples et al., 1976).
A daily dose of 480 mg trichlorfon/kg, given to 34 CD rats by
gavage on days 6-15 of gestation, produced a high incidence (86%) of
fetal malformations, such as generalized oedema, herniation of the
brain, hydroencephaly, micrognathia, cleft palate, and skeletal system
alterations (Staples & Goulding, 1979).
Groups of 25 naturally inseminated, female Long Evans rats were
exposed by gavage to technical trichlorfon at 0, 10, 30, or 100 mg/kg
body weight per day from day 6 to day 16 of gestation, and sacrificed
on day 20. None of the doses had a lethal effect. Although at 100
mg/kg diarrhoea was caused in some of the animals, embryonic and fetal
development were not affected at this dose level (Machemer, 1979a).
Groups of 33 naturally inseminated, female Charles River rats
were exposed to technical trichlorfon at 0, 500, 1125, or 2500 mg/kg
(equivalent to 0, 45, 102, or 227 mg/kg body weight per day) from day
6 to day 15 of gestation, and sacrificed on day 20. Trichlorfon was
maternally toxic at dietary levels of 500, 1125, and 2500 mg/kg. There
was no evidence of trichlorfon-related embryotoxicity (increased
resorption), fetotoxicity (decreased fetal weight), or teratogenicity
(malformations) at exposure levels up to and including 2500 mg/kg.
There was an increase in the incidence of delayed ossification and
curved, wavy, and/or bulbous ribs at 2500 mg/kg. On the basis of the
results of this study, 1125 mg/kg, equivalent to 102 mg/kg body weight
per day, is considered the no-effect dose, in terms of reproductive
liability in this study (Kowalski et al., 1987).
A 3-generation (2 litters per generation) rat reproduction study
with levels of 0, 100, 300, 1000, or 3000 mg trichlorfon/kg in the
diet resulted in adverse effects on reproduction at 1000 mg/kg, and
above. At 1000 mg/kg, there was evidence of reduced fertility, smaller
litters, and reduced bodyweight of pups. At 3000 mg/kg, the pregnancy
rate was markedly decreased and the pups were smaller and lighter in
weight with none surviving to the weanling stage. No effects were
noted at 300 mg/kg or below. Microscopic examination of tissues from
the F3b generation did not indicate any adverse effects (Löser, 1969;
Spicer & Urwin, 1971).
8.7.3 Hamster
When 200, 300, or 400 mg trichlorfon/kg body weight was given by
gavage to groups of 10-30 female golden hamsters, each day, on days
7-11 of gestation, the 300 and 400 mg/kg doses produced a reduction in
maternal food consumption. At 400 mg/kg, there were signs of maternal
toxicity and 3 out of 30 animals died; fetal death and malformations
(cleft palate, patagium, and fused ribs) were increased. At 300 mg/kg,
only one fetus (out of 105) had malformations. A dose of 200 mg/kg
body weight did not produce any adverse effects (Staples & Goulding,
1979).
8.7.4 Rabbit
Groups of 15 naturally inseminated, female Himalayan rabbits were
exposed by gavage to technical trichlorfon at 0, 5, 15, or 45 mg/kg
body weight per day from day 6 to day 18 of gestation and sacrificed
on day 29. The average weight gain of the dosed groups was reduced,
but doses of 5 and 15 mg/kg were tolerated well. Because of maternal
toxic effects, two abortions occurred at 45 mg/kg, but the fetuses
delivered in this dose group developed normally. The
no-observed-effect level with respect to embryonic development was 15
mg/kg body weight per day. There were no indications that trichlorfon
produced any teratogenic effects in this study (Machemer, 1979b).
Groups of 20, artificially inseminated, female American Dutch
rabbits were orally exposed to 0, 10, 35, or 110 mg technical
trichlorfon/kg body weight per day from day 6 to day 18 of gestation
and sacrificed on day 28. The highest dose (110 mg/kg) was not well
tolerated and resulted in adverse clinical signs and death,
significantly reduced overall body weight gain and food consumption,
and significantly inhibited cholinesterase. At 35 mg/kg, cholin
esterase was significantly inhibited and one death occurred that was
possibly treatment-related. A dose of 10 mg/kg was devoid of overt
maternal toxicity, while 110 mg/kg was embryotoxic and fetotoxic, but
not teratogenic; 35 mg/kg was a NOEL for developmental toxicity and 10
mg/kg for maternal toxicity (Clemens et al., 1990).
8.7.5 Congenital tremor
Several outbreaks of congenital tremor in piglets have been
described in herds in which the sows had been treated with
antiparasitic trichlorfon preparations between days 45 and 63 of
pregnancy (Kronevi et al., 1975; Dobson, 1977; Bölske et al., 1978;
Hansen et al., 1978; Knox et al., 1978). Clinically, the disease was
characterized by ataxia and tremor and a pronounced hypoplasia of the
cerebellum was found, as well as a reduction in the size of the spinal
cord.
This disease was reproduced experimentally. Four sows were dosed
with Neguvon Vet at 60 mg/kg body weight, mixed in the morning feed on
day 55 and on day 70 of pregnancy. All 40 piglets born alive showed
ataxia and tremor, and, at autopsy, hypoplasia of the cerebellum. In
97 control litters, none of the 892 live-born piglets showed nervous
signs (Knox et al., 1978). Similar experimental results were obtained
following exposure of pigs to multiple oral doses of 50-75 mg
trichlorfon/kg between approximately 45 and 63 days after conception
(Pope et al., 1986; Berge et al., 1987a; Rasmussen et al., 1978). A
similar, but less severe effect resulted from the post-natal exposure
of piglets from week 3 to week 6 to 50 mg/kg body weight per day.
Recovery of animals exposed prenatally was slow; 35 days after birth
they had not reached control values for cerebral and cerebellar
weight. There was still regional loss of Purkinje cells in the
cerebellum (Berge et al., 1987b).
Ten pregnant, white guinea-pigs were given 6 doses of 100 mg
trichlorfon/kg body weight by gavage on days 36, 37, 38, 51, 52, and
53 of pregnancy; 7 additional animals served as controls. The pups
developed trembling and locomotor disturbances. Post-mortem
examination of the pups revealed significantly decreased weights of
the total brain and the cerebellum, compared with controls. There was
also a significant weight reduction, particularly of the medulla
oblongata, but also of the hippocampus, the thalamus, and the
colliculi. Histological examination of the cerebellum revealed
reduction of the external granular layer, and the molecular layer,
together with a regional absence of Purkinje cells. The activities of
the neurotransmitter enzymes cholineacetyltransferase and glutamate
decarboxylase in the cerebellum were reduced compared with the control
values (Berge et al., 1986). Pregnant, albino guinea-pigs were given
radiolabelled trichlorfon on days 37 or 52 of pregnancy and examined
by whole body autoradiography 15, 30, and 45 min after administration.
Trichlorfon had not accumulated in the fetal guinea-pig brain (Berge
& Nafstad, 1986).
8.8 Neurotoxicity
Olajos et al. (1979) reported that the administration of a
divided dose of 300 mg/kg to hens (200 + 100 mg/kg given
subcutaneously, 3 days apart) resulted in levels of neurotoxic
esterase (NTE) inhibition (46-68% at 24 h) approaching the 70% level
that has been correlated with the development of organophosphorus-
ester-induced delayed neurotoxicity (OPIDN), and moderate clinical
signs (ataxia) resembling the early stages of OPIDN. Shiraishi et al.
(1983) produced a peripheral neuropathy in a single monkey (1 out of
1) with a single dose of 250 mg/kg.
Based on a review of the literature on trichlorfon and dichlorvos
(both animal studies and cases of human poisoning), Johnson (1981,
1990), Caroldi & Lotti (1981), and WHO (1989) concluded that only
doses that exceed the lethal dose are likely to result in a level of
NTE inhibition at which OPIDN would be expected. Johnson summarized
reports of neurotoxicity studies on trichlorfon in hens as follows:
The acute maximum tolerated cholinergic dose (200 mg/kg
subcutaneously) produced no marked neuropathy in hens; however,
moderate neuropathy was seen when a further dose of 100 mg/kg was
given subcutaneously, 3 days later. Histological signs of severe
degeneration were seen in the sciatic nerve and spinal cord of the
ataxic bird at autopsy after 3 weeks. Slight changes were reported in
sections of the brain stem of the birds given 200 mg/kg, orally, or
100 mg/kg, subcutaneously (Olajos et al., 1979). But these did not
appear to be entirely typical of organophosphorus neuropathy and no
lesions were reported for the spinal cord or sciatic nerve. Inhibition
of NTE in the spinal cord was only measured in later studies (Hierons
& Johnson, 1978) and lagged markedly behind that in the brain.
The ability of trichlorfon to induce delayed neurotoxicity was
assessed in adult White Leghorn hens, administered single subcutaneous
doses of 100 or 300 mg/kg, and assessed for visible signs of
neurotoxicity 24 h after treatment, prior to killing and collection of
samples of brain and spinal (cervical, thoracic) cord for the
measurement of AChE and NTE activities. In short-term studies, hens
received trichlorfon (100 mg/kg) every 72 h for a total of 6 doses.
Three days after the terminal dose, the hens were killed and the
brains, spinal cords, and distal sciatic nerves were removed for
enzymatic and histological examination. While trichlorfon markedly
inhibited tissue AChE, no reduction in NTE was detected and no overt
signs of neurotoxicity were observed. In the short-term studies,
trichlorfon did not cause any obvious neurotoxicity, an observation
supported by minimal changes in the spinal cord and sciatic nerve
morphology, no impairment of walking ability, and no inhibition of the
brain and spinal cord NTE (Slott & Ecobichon, 1984).
A number of other reports indicate that trichlorfon is either not
neurotoxic, or produces a neurotoxic effect that is distinct from
OPIDN. In addition to the Slott & Ecobichon study, several short-term
dosing regimens that resulted in severe cholinergic toxicity by either
the oral (Olajos et al., 1979) or dermal (Francis et al., 1985) route
did not result in neuropathy. Oral doses of 100 mg trichlorfon/kg were
also reported not to result in significant NTE inhibition (Olajos et
al., 1979; Olajos & Rosenblum, 1981). Finally, rats, which are much
less sensitive to agents that cause delayed neurotoxicity, exhibited
electrophysiological signs of neurotoxicity without accompanying
histological changes when given oral doses of 30 mg/kg for 3 weeks
(Lehotzky, 1982) or ip injections of trichlorfon at 200 mg/kg per day
for 5-15 days (Averbook & Anderson, 1983).
Other adverse neurobehavioural effects that have been observed
may be due to acetylcholinesterase inhibition. Dési (1983) exposed
adult CFY rats (10 males and 10 females per group) at levels of 16 and
32 mg/kg for 3 months. The lower dose resulted in increased high
frequency EEC activity, and enhanced central excitability and the
higher dose in increased low-frequency EEG activity and a significant
overall increase in EEG activity as well as indications of depressed
cortical excitability. In animals treated orally with trichlorfon at
30 mg/kg per day, increased locomotor activity in the open field and
decreased rotorod performance were observed transiently. While the
animals were able to acquire the conditioned escape reflex, latency of
the escape responses in the conditioned escape reflex was markedly
increased (Lehotzky, 1982).
8.9 Immunological studies
Exposure of female mice (BALB/cByJ & C57BL/6J strain; 4 per
group) to 175 mg trichlorfon/100 ml drinking-water for 14 days had no
effect on immune function, measured by response to influenza virus
(Reiss et al., 1987). The dose used was considered to be effective for
prophylactic treatment against helminthic infestation.
8.10 Toxicity of dichlorvos
The major transformation product of trichlorfon in mammals,
including human beings, is dichlorvos, which is at least 100 times
more active as a cholinesterase inhibitor than trichlorfon (Hofer,
1981). An evaluation of the health and environmental hazards of
dichlorvos can be found in WHO (1989). For completeness sake, however,
the summary on effects on experimental animals and in vitro test
systems from this publication is reproduced below.
"Dichlorvos is moderately to highly toxic when administered in
single doses to a variety of animals species by several routes. It
directly inhibits acetylcholinesterase (AChE) activity in the nervous
system and in other tissues. Maximum inhibition generally occurs
within 1 h, and is followed by rapid recovery. The oral LD50 for the
rat is 30-110 mg/kg body weight, depending on the solvent used. The
hazard classification of dichlorvos by WHO is based on an oral LD50
for the rat of 56 mg/kg body weight. The signs of intoxication are
typical of organophosphorus poisoning, i.e., salivation, lachrymation,
diarrhoea, tremors, and terminal convulsions, with death occurring
from respiratory failure. The signs of intoxication are usually
apparent shortly after dosing, and, at lethal doses, death occurs
within 1 h. Survivors recover completely within 24 h.
Potentiation is slight when dichlorvos is given orally in
combination with other organophosphates, but in combination with
malathion it is marked.
In short-term toxicity studies on the mouse, rat, dog, pig, and
monkey, inhibition of plasma, red blood cell, and brain ChE are the
most important signs of toxicity. After oral administration, a dose of
approximately 0.5 mg/kg body weight (range, 0.3-0.7 mg/kg) did not
produce ChE inhibition. In a 2-year study on dogs, ChE inhibition was
noted at 3.2 mg/kg body weight or more.
Flea collar dermatitis has been described in dogs and cats
wearing dichlorvos-impregnated PVC flea collars. This was a primary
irritant contact dermatitis that may have been caused by dichlorvos.
Many short-term, inhalation studies on different animal species
have been carried out. Air concentrations in the range of 0.2-1
mg/m3 do not affect ChE activity significantly. Other effects, such
as growth inhibition and increase in liver weight, have been reported
at dose levels at least 10-20 times higher.
It is possible to produce clinical neuropathy in hens, but the
doses of dichlorvos required are far in excess of the LD50. The
effects are associated with high inhibition of neurotoxic esterase
(NTE) in the brain and spinal cord. In the rat, however, neuropathic
changes in the white matter of the brain have been reported following
repeated daily oral application of an LD50 dose.
Immune suppression has been reported in rabbits. At present, no
evaluation as to the relevance for human beings can be given; more
attention to this aspect is needed.
In a long-term study, rats fed dichlorvos in the diet for 2 years
showed no signs of intoxication. Hepatocellular fatty vacuolization of
the liver and ChE inhibition were significant at the two highest dose
levels (2.5 and 12.5 mg/kg body weight).
In a carefully conducted, long-term, inhalation study on rats
with whole body exposure (23 h/day, for 2 years), results were
comparable with those seen in the oral study. No effects were seen at
0.05 mg/m3, inhibition of ChE activity took place at 0.48 mg/m3 or
more.
In several reproduction studies on rats and domestic animals, no
effects were seen on reproduction, and there was no embryotoxicity at
dose levels that did not cause maternal toxicity. At toxic doses,
dichlorvos may cause reversible disturbances of spermatogenesis in
mice and rats. It was not teratogenic in several studies carried out
on rats and rabbits.
Dichlorvos is an alkylating agent and binds in vitro to
bacterial and mammalian nucleic acids. It is mutagenic in a number of
microbial systems, but there is no evidence of mutagenicity in intact
mammals, where it is rapidly degraded by esterases in blood and other
tissues.
Dichlorvos carcinogenicity has been investigated in mice (oral
studies) and rats (oral and inhalation studies). The dose levels used
in 2-year, oral studies were up to 800 mg/litre drinking-water or 600
mg/kg diet for mice, and up to 280 mg/litre drinking-water or 234
mg/kg diet for rats. In a rat inhalation study, dichlorvos
concentrations in air of up to 4.7 mg/m3 were tested for 2 years. No
statistically significant increase in tumour incidence was found. In
two recent carcinogenicity studies on mice and rats, dichlorvos was
administered by intubation at dose levels between 10 and 40 mg/kg body
weight (mice) and 4 and 8 mg/kg body weight (rat) for up to 2 years.
Only preliminary information has been provided. The evidence for
carcinogenicity in these new studies is difficult to interpret at this
time. Only when complete and final reports become available will it be
possible to draw more definitive conclusions.a
a The US-NTP Peer Review Panel reviewed these studies and came to
the following conclusions: "Under the conditions of these 2-year
gavage studies, there was some evidence of carcinogenic activity
of dichlorvos for male F344/N rats, as shown by increased
incidences of adenomas of the exocrine pancreas and mononuclear
cell leukemia. There was equivocal evidence of carcinogenic
activity of dichlorvos for female F344/N rats, as shown by
increased incidences of adenomas of the exocrine pancreas and
mammary gland fibroadenomas. There was some evidence of
carcinogenic activity of dichlorvos for male B6C3F1 mice and
clear evidence for female B6C3F1 mice, as shown by increased
incidences of forestomach squamous cell papillomas." (US-NTP,
1989).
In a recent evaluation of current data, IARC (in press) concluded
that there is sufficient evidence for the carcinogenicity of
dichlorvos in experimental animals, but that there is inadequate
evidence for the carcinogenicity of dichlorvos in humans.
From acute and short-term studies, it is clear that the
metabolites of dichlorvos are all less toxic than the parent compound.
Only dichloroacetaldehyde was positive in a few mutagenicity tests."
8.11 Mechanism of toxicity - mode of action
While trichlorfon itself is not a potent anticholinesterase
agent, the inhibiting activity of this chemical is attributed to the
transformation product dichlorvos. The slow conversion of trichlorfon
to dichlorvos results in the inhibition of both central and peripheral
cholinergic nerve acetylcholinesterase, with the accumulation of the
neurotransmitter, acetylcholine, at nerve endings, and the generation
of characteristic signs and symptoms of toxicity. A full description
of the mechanism of action of organophosphorus ester insecticides can
be found in Environmental Health Criteria No. 63: Organophosphorus
Insections - A General Introduction (WHO, 1986), and that of
dichlorvos in Environmental Health Criteria No. 79: Dichlorvos (WHO,
1989). The muscarinic, nicotinic, and the central nervous
system-induced signs and symptoms observed in humans have been
described extensively (Ecobichon et al., 1977; Hayes, 1982; Hayes &
Laws, 1991).
Transformation of trichlorfon to dichlorvos in vivo was studied
in relation to its effects on the cholinesterase activity,
acetylcholine content, and acetylcholine-turnover in the mouse brain.
An ip injection of 10 mg dichlorvos/kg caused toxic signs, such as
salivation, diarrhoea and, in some cases, difficulties in breathing,
which were clearly recognized about 15 min after administration and
disappeared almost completely towards the end of 60 min. After an ip
injection of 125 mg trichlorfon/kg, the above mentioned signs were
most intense at around 30 min; and almost complete recovery was
observed towards the end of 2 h. The cholinesterase activity and
acetylcholine levels reached their minimum and maximum, respectively,
at 15 min after the injection of dichlorvos and about 45 min after the
injection of trichlorfon. The delayed decrease in acetylcholine
turnover following to ip injection of trichlorfon was also
demonstrated by measuring the acetylcholine-synthesizing rate in the
brain following intravenous injection of 2H6-choline compared with
dichlorvos pretreatment. The level of dichlorvos in the brain of mice
administered trichlorfon ip reached its maximum a few minutes after
the maximal level of trichlorfon itself; both compounds decreased over
similar curves during a 120-min period (Nordgren et al., 1978).
Thirty minutes after intraventricular injection of trichlorfon
(2.5 mg) in the rat, the activity of the cholinesterase decreased to
20% in the hippocampus; 22% in the medulla; 50% in the cerebellum; 58%
in the striatum, and 72% in the cortex. Levels of acetylcholine
reached a maximum at 45 min in the hippocampus and cortex, and peaked
in the striatum at 60 min. The greatest increases were seen in the
hippocampus and cortex with 60 and 55%, respectively (Hallak &
Giacobini, 1987).
The administration of 80 mg trichlorfon/kg im to male
Sprague-Dawley rats was found to produce an increase in acetylcholine
levels, which peaked at about 170% of control levels, 30 min after
exposure (Hallak & Giacobini, 1989). Acetylcholine levels returned to
normal within 120 min of exposure. A second dose at 120 min resulted
in a second surge in the acetylcholine levels.
9. EFFECTS ON HUMAN BEINGS
Trichlorfon is one of the organophosphorus compounds for which
not only acutely toxic effects have been described, but also delayed
neurotoxicity in humans (WHO, 1986).
In addition to the reports describing cases of human poisoning,
there is wide experience concerning the therapeutic use of trichlorfon
and the side-effects arising from this use.
9.1 Acute poisoning - poisoning incidents
Several hundred cases of acute trichlorfon poisoning, some of
them lethal, have been described in the literature. These were either
accidental, intentional (suicide), or due to gross neglect of
prescriptions or safety precautions. A critical detailed review of
these cases is given by Johnson (1981) and Hayes (1982).
In all cases, the onset of poisoning was rapid, early signs and
symptoms being exhaustion, headache, weakness, confusion, vomiting,
abdominal pain, excessive sweating, and salivation. The pupils are
small. Difficulty in breathing may be experienced, because of either
congestion of the lungs or weakness of the respiratory muscles. In
severe cases of poisoning, muscle spasms, unconsciousness, and
convulsions may develop and death may result from respiratory failure.
In the case of trichlorfon, unconsciousness is disproportionately
common and prolonged and the incidence of mental disturbances is high;
moreover polyneuropathy has been found at a later stage, in
approximately 21% of cases (Hayes, 1982). However, after reviewing the
literature, Johnson (1981; 1990) concluded that only doses of
trichlorfon that exceed the lethal dose, and where the victim survived
because of treatment, are likely to result in a level of NTE
inhibition at which delayed neurotoxicity would be expected.
The onset of polyneuropathy has occurred as early as 3 days after
ingestion and as late as 26 days (Hayes, 1982), the majority of cases
following recovery from the acute effects. The clinical,
electrophysiological, and histopathological features of the neuropathy
were similar to the syndrome resulting from TOCP exposure (Hierons &
Johnson, 1978; Shiraishi et al., 1983; Vasilescu et al., 1984;
Niedziella et al., 1985). Hayes (1982) discusses the possibility that
the polyneuropathy could be caused by other chemical compounds,
present as impurities in the technical product or formulation. Johnson
(1981) mentions higher alkyl analogs as an example. No confirmation
for this hypothesis can be found.
In a case of trichlorfon poisoning, a 21-year-old female
attempted suicide by drinking about 50 ml of a 50% formulation of
trichlorfon. She lost consciousness and recovered after 8 h. Two weeks
after ingestion, the patient developed a tingling sensation in all
extremities followed by weakness in the lower limbs and knees,
characterized as motor dominant polyneuropathy (Shiraishi et al.,
1977).
Progressive neuropathy developed 2-8 weeks after acute poisoning
(i.e., unconsciousness for 16 h) in a 20-year-old man who had taken
orally a handful of granular solid formulation (trichlorfon content:
80%). The clinical symptoms were typical of the delayed neuropathy
that is caused by organophosphates, such as tri- o-cresyl-phosphate,
with normal conduction velocity in surviving motor nerve fibres as an
electrophysiological finding. However, a single dose of the above
granular solid sample did not produce acute delayed neurotoxicity in
hens (Hierons & Johnson, 1978).
A 42-year-old man was in deep coma after ingestion of 100-200 ml
Soldep (25% trichlorfon) and had to be artificially ventilated for 37
days. Plasma cholinesterase activity was significantly decreased.
Three weeks after ingestion, there was severe weakness of the lower
limbs. The EMG (at 40 and 70 days, 4,6,9, and 14 months) indicated
denervation in the lower extremities and peripheral motorneuron
lesions in the upper extremities. Twenty-one months after poisoning,
there was a slow improvement in mobility and the patient was able to
walk by himself for a short distance (Bátora et al., 1988).
Akimov & Kolesnichenko (1985) examined the morphological changes
in the nervous system of 14 patients who had died from acute
chlorophos poisoning. They found congested blood vessels with
perivascular oedema and degeneration of the collagenous- and elastic
fibres of the vascular walls. Diffuse cellular changes, such as
swelling and ischaemic changes, were found in the brain, spinal cord,
and vegetative ganglia. There was a moderate destruction of the myelin
sheaths in the lateral columns of the spinal cord and the brain
peduncles, and there were structural changes in the axons of the
peripheral nerves.
Csik et al. (1986) observed 70 cases of trichlorfon poisoning
(mainly suicide attempts) between 1971 and 1983. Twenty-five of them
were re-examined in 1984. Nine of these (36%) had severe residual
signs of delayed polyneuropathy, mainly of the distal motor type. In
one case, signs of CNS lesions had persisted. Four had had complaints
(paraesthesia, weakness of hands) 2-3 months after poisoning, but were
healthy at the time of re-examination.
9.2 Therapeutic use of trichlorfon
Under the name metrifonate, trichlorfon is used to treat
infection by Schistosomiasis haematobium in humans (WHO, 1985).
Metrifonate has, by now, been given to millions of patients in the
tropics in the treatment of schistosomiasis.
Following a dose of 7-12 mg/kg, severe cholinergic symptoms are
rare in spite of almost complete inhibition of plasma cholinesterase
and 40-60% inhibition of erythrocyte acetylcholin esterase (Nordgren
et al., 1981; Davis, 1986). However, trials in which several doses
were administered on either the same day or one day apart resulted in
abdominal colic, nausea, salivation, dizziness, and headache, which
precluded further treatment (Aden-Abdi et al., 1987), and the
subject-reported incidence of one or more of these effects after a
single dose of 10 mg/kg (46%) was generally higher than in subjects
given a vitamin placebo (34%) (Wilkins & Moore, 1987). There is one
report of a possible human birth defect resulting from metrifonate
treatment (Monson & Alexander, 1984).
In another report on 6000 people, mostly in South Africa and
South America, who had been treated with trichlorfon for a few years
to control various intestinal and body parasites, the dosages varied
from 7.5 up to 70 mg/kg. The dose of 7.5 mg/kg, given 2-4 times at
two-week intervals, caused cholinesterase inhibition, weakness,
nausea, diarrhoea, and abdominal pain. Higher doses (24 mg/kg) caused
more severe symptoms including tachycardia, salivation, colic pain,
vomiting, nausea, fatigue, tremors, and sweating. The effects were not
cumulative and recovery in all cases was rapid. In a few human cases,
an indication was given that spermatogenesis (size and shape of sperm)
and sperm mobility might be affected (Wegner, 1970).
In a large-scale programme in rural villages in Somalia,
metrifonate was given at a current dosage regimen of 3 single doses of
7.5 mg/kg each, on three separate days at intervals of 2 weeks (Aden
Abdi & Gustafsson, 1989). In a large number of cases, the results were
not satisfactory, due to poor patient compliance with the treatment
regimen. In an attempt to simplify this regimen, 5 mg/kg, given thrice
during one day, gave the best results as regards safety and the cure
rates were comparable with those with the standard regimen (Aden Abdi,
1990).
In a clinical trial on 20 patients with Alzheimer disease, Becker
et al. (1990) gave single oral doses of 2.5, 5, 7.5, or 15 mg
metrifonate/kg per week, for 1-3 months. A statistically significant
improvement was obtained with the 5 mg/kg per week dose level. A 60%
depression in red cell ChE and 80% depression in plasma ChE were
accompanied by only minor side effects (nausea, vomiting, and/or
diarrhoea); there were no effects at 2.5 mg/kg, 5 patients were
affected at 5 mg/kg, 9 at 7.5 mg/kg, and 13 at 15 mg/kg per week.
9.3 Occupational exposures
Few cases of occupational poisoning by trichlorfon have been
reported. See section 5.3 for occupational exposures.
Occupational exposure to trichlorfon at a factory where air
concentrations exceeded 0.5 mg/m3, but where skin contamination also
occurred, resulted in decreased plasma cholinesterase levels and
changes in EEG patterns; particularly slow and paroxysmal waves were
observed (Lu et al., 1984; Hu et al., 1986). Both the biochemical and
electrophysiological indices returned to normal after exposure ceased.
Although trichlorfon has been widely used for many years, no
cases of skin sensitization have been reported (Mihail, 1986b).
9.4 Treatment of acute trichlorfon poisoning
The advice on the treatment of organophosphate poisoning from EHC
63: Organophosphorus Insecticides - A general introduction has been
reproduced in Annex I.
10. PREVIOUS EVALUATIONS BY INTERNATIONAL BODIES
Trichlorfon was evaluated by the Joint FAO/WHO Expert Committee
on Pesticide Residues (JMPR) in 1971, 1975, and 1978 (FAO/WHO, 1972,
1976, 1979). In 1978, the JMPR established an Acceptable Daily Intake
(ADI) for man of 0-0.01 mg/kg body weight, based on the fact that the
following levels cause no toxicological effects:
Rat: 50 mg/kg in the diet equivalent to 2.5 mg/kg body weight.
Dog: 50 mg/kg in the diet equivalent to 1.25 mg/kg body weight.
In 1986, the FAO/WHO CODEX Committee advised a range of maximum
residue limits (MRLs) for specified food commodities (FAO/WHO, 1986).
These ranged from 0.05 to 2 mg/kg product.
Trichlorfon was evaluated by an IARC Working Group in 1983. There
were no data on its carcinogenicity in humans and the evidence of
carcinogenicity in experimental animals was inadequate. Trichlorfon
was classified in Group 3, i.e., cannot be classified as to its
carcinogenicity to humans (IARC, 1983, 1987).
WHO classified technical trichlorfon as "slightly hazardous"
(Class III) (WHO, 1990). A data sheet on trichlorfon (No. 27) has been
published by the WHO (WHO/FAO, 1977).
REFERENCES
ABBASOV, T.A. (1972) [Residues of organophosphorus insecticides in
clover and potato plants.] Khim. Sel'sk. Khoz., 10: 597-598 (in
Russian).
ABDEL-KADER, M.I.A., MOUBASHER, A.H., & ABDEL-HAFEZ, S.I. (1978)
Selective effects of five pesticides on soil and cotton-rhizosphere
and -rhizoplane fungus flora. Mycopathologia, 66: 117-123.
ABDELWAHAB, A.M., KHALIL, F.M., & HUSSEIN, M.H. (1973) Toxicity of
insecticides to honey bees in laboratory and field. Bull. Entomo.
Soc. Egypt, Econ. Ser., VII: 83-89.
ADEN ABDI, Y. (1990) A rational approach to the treatment of
Schistosoma haematobium infection with metrifonate - clinical and
pharmacological studies, Stockholm, Karolinska Institute, Department
of Clinical Pharmacology (Thesis).
ADEN ABDI, Y. & GUSTAFSSON, L.L. (1989) Poor patient compliance
reduces the efficacy of metrifonate treatment of Schistosoma
haematobium in Somalia. Eur. J. clin. Pharmacol., 36: 161-164.
ADEN ABDI, Y., GUSTAFSSON, L.L., & ELMI, S.A. (1987) A simplified
dosage schedule of metrifonate in the treatment of Schistosoma
haematobium infection in Somalia. Eur. J. clin. Pharmacol., 32:
437- 441.
ADLER, B., BRAUN, R., SCHÖNEICH, J., & BÖHME, H. (1976)
Repair-defective mutants of Proteus mirabilis as a prescreening
system for the detection of potential carcinogens. Biol. Zentralbl.,
95: 463- 469.
AHMAD, Z. & JOHANSEN, C. (1973) Selective toxicity of carbophenothion
and trichlorfon to the honey bee and the alfalfa leafcutting bee.
Environ. Entomol., 2: 27-30.
AKHTAR, M.H. (1982) Fate of trichlorfon in buffer and soluble fraction
(105 000g) from cow and chicken liver homogenates. J. agric. food
Chem., 30: 551-554.
AKIMOV, G.A. & KOLESNICHENKO, I.P. (1985) [Morphological changes in
the nervous system in acute peroral chlorophos poisoning.] Arkh.
Patol., 47(1): 44-51 (in Russian).
AKSNES, G. & YÜKSEKISIK, N. (1974) Kinetic study of the alkaline
hydrolysis of 2,2,2-trichloro-1-hydroxyethyl, dimethyl phosphonate,
Dipterex, and its ethyl analog. Phosphorus, 4: 33-38.
AMBRUS, A., HARGITAI, E., KAROLY, G., FULOP, A., & LANTOS, J. (1981)
General method for determination of pesticide residues in samples of
plant origin, soil, and water. II. Thin layer chromatographic
determination. J. Assoc. Off. Anal. Chem., 64: 743-748.
AMENO, K., FUKE, C., AMENO, S., KIRIU, T., & IJIRI, I. (1989) A rapid
and sensitive quantitation of dipterex in serum by solid-phase
extraction and gas chromatography with flame thermionic detection.
J. anal. Toxicol., 13: 150-151.
AMER, S.M. & ALI, E.M. (1983) Cytological effects of pesticides. XIV.
Effect of the insecticide Dipterex "trichlorfon" on Vicia faba
plant. Cytologia, 48: 761-770.
ANDREU-MOLINER, E.S., ALMAR, M.M., LEGARRA, I., & NUNEZ, A. (1986)
Toxicity of some rice-field pesticides to the crayfish P. Clarkii
under laboratory and field conditions in Lake Albuferra (Spain). J.
environ. Sci. Health, B 21: 529-537.
ANON (1978a) Information on trichlorfon residues surveillance
available to FAO/WHO from Hungary.
ANON (1978b) Information on trichlorfon residues available to FAO/WHO
from Finland.
ANON (1978c) Information on trichlofon residues available to FAO/WHO
from Netherlands.
ANTOINE, O. & MEES, G. (1971) Test de routine pour la détermination
des insecticides organo-phosphorés par la chromatographie en couche
mince. J. Chromatogr., 58: 247-256.
AQUILINA, G., BENIGNI, R., BIGNAMI, M., CALCAGNILE, A., DOGLIOTTI, E.,
FALCONE, E., & CARERE, A. (1984) Genotoxic activity of dichlorvos,
trichlorfon, and dichloroacetaldehyde. Pestic. Sci., 15: 439-442.
ARNOLD, D., KEPLINGER, M.L., FANCHER, O.E. & CALANDRA, J.C. (1971)
Mutagenic study with Dylox in Albino mice. Industrial Biotest
Laboratories (Unpublished report submitted to WHO by Farbenfabriken
Bayer A.G., Germany).
ARTHUR, B.W. & CASIDA, J.E. (1957) Metabolism and selectivity of
O,O-dimethyl 2,2,2-trichloro-1-hydroxyethylphosphonate and its
acetyl and vinyl derivatives. J. agric. food Chem., 5: 186-192.
AVERBOOK, B.J. & ANDERSON, R.J. (1983) Electrophysiological changes
associated with chronic administration of organophosphates. Arch.
Toxicol., 52: 167-172.
BAIDA, T.A. (1970) [Soil pollution by organophosphorus pesticides.]
Kazakstana, 30: 72 (in Russian).
BAIDA, T.A. (1975) [Dynamics and metabolism of chlorophos in cabbage
and onion plants.] Khim. Sel'sk Khoz., 13: 67-68 (in Russian).
BARCELO, D. (1988) Application of thermospray liquid chromatography/
mass spectrometry for determination of organophosphorus pesticides
and trialkyl and triaryl phosphates. Biomed. environ. mass Spectrom.,
17: 363-369.
BARCELO, D., MARIS, F.A., GEERDINK, R.B., FREI, R.W., DE JONG, G.J.,
& BRINKMAN, U.A.T. (1987) Comparison between positive, negative and
chloride-enhanced negative chemical ionization of organophosphorus
pesticides in on-line liquid chromatography-mass spectrometry. J.
Chromatogr., 394: 65-67.
BARTHEL, W.F., GIANG, P.A., & HALL, S.A. (1954)
Dialkyl-alpha-hydroxyphosphonates derived from chloral. J. Am. Chem.
Soc., 76: 4186-4187.
BATORA, I., KUKUMBERG, P., KUCHAR, M., & KOUTUN, J. (1988) [Delayed
neurotoxic effect of Soldep (trichlorfon) after acute poisoning.]
Prac. Lèk., 40: 172-174 (in Slovak).
BATTELLE (1982) Pesticide programme of research and market planning.
Part II. Insecticides, Geneva, Battelle Institute, 40 pp.
BATTELLE (1984) Trichlorfon. In: World pesticide programme.
Insecticides II. World summary: USA, Canada, Argentina, Brazil,
Colombia, Mexico, Central America, Geneva, Battelle Research
Centres, pp. 16, 26, 30, 34.
BATTELLE (1987) Trichlorfon. In: World pesticide programme.
Insecticides III. Summary: USA, Mexico, Brazil, Geneva, Battelle
Research Centres, Vol. 1, pp. 6-8, 11, 14, 16-17, 27.
BATZINGER, R.P. & BUEDING, E. (1977) Mutagenic activities in vitro
and in vivo of five antischistosomal compounds. J. Pharmacol. exp.
Ther., 200: 1-9.
BECKER, K. & SCHÖNEICH, J. (1980) Dominant lethal test with
trichlorphon in male mice. Mutat. Res., 74: 224, (Meeting abstract).
BECKER, R.E., COLLIVER, J., ELBLE, R., FELDMAN, E., GIACOBINI, E.,
KUMAR, V., MARKWELL, S., MORIEARTY, P., PARKS, R., SHILLCUT, S.D.,
UNNI, L., VICARI, S., WOMACK, C., & ZEC, R.F. (1990) Effects of
metrifonate, a long-acting cholinesterase inhibitor in Alzheimer
Disease: report of an open trial. Drug Dev. Res., 19: 425-434.
BEDFORD, C.T. & ROBINSON, J. (1972) The alkylating properties of
organophosphates. Xenobiotica, 2: 307-337.
BELLO, T.R., AMBORSKI, G.F., & TORBERT, B.J. (1974) Effects of organic
phosphorus anthelmintics on blood cholinesterase values in horses and
ponies. Am. J. vet. Res., 35: 73-78.
BENES, V. & CERNA, V. (1970) Contribution to the toxicological
evaluation of fenitrothion ( O,O-dimethyl- O-/3-methyl-4-
nitrophenyl/thio-phosphate) and its residues. In: Halas et al., ed.
Proceedings of pesticide Symposia Inter-American Conferences on
Toxicology and Occupational Medicine, Miami.
BENES, V. & SRAM, R. (1969) Mutagenic activity of some pesticides in
Drosophila melanogaster. Ind. Med. Surg., 38: 442-444.
BENIGNI, R. & DOGLIOTTI, E. (1980) UDS studies on selected
environmental chemicals. Mutat. Res., 74: 217, 248-249 (Meeting
abstract).
BENIGNI, R., BIGNAMI, M., CARERE, A., DOGLIOTTI, E., NOVELLETO, A., &
PRINCIPE, R. (1980) Comparative mutational studies with dichlorvos,
trichlorfon and dichloroacetaldehyde. Toxicol. Lett., 1(Spec.
Issue): 250 (Meeting abstract).
BENNEWITZ, R. & FOTH, G. (1967) [Chemical behavior and analytical
determination of trichlorfon, dichlorvos and tribufon.] Fresenius Z.
anal. Chem., 229: 110-117 (in German).
BERGE, G.N. & NAFSTAD, I. (1986) Distribution and placental transfer
of trichlorfon in guinea-pigs. Arch. Toxicol., 59: 26-29.
BERGE, G.N., NAFSTAD, I., & FONNUM, F. (1986) Prenatal effects of
trichlorfon on the guinea-pig brain. Arch. Toxicol., 59: 30-35.
BERGE, G.N., FONNUM, F., & BRODAL, P. (1987a) Neurotoxic effects of
prenatal trichlorfon administration in pigs. Acta vet. Scand., 28:
321-332.
BERGE, G.N., FONNUM, F., SOLI, N.E., & SOGNEN, E. (1987b)
Neurotoxicological examination of the piglet brain after prenatal and
postnatal exposure to trichlorfon. Acta vet. Scand., 28: 313-320.
BETOWSKI, L.D. & JONES, T.L. (1988) Analysis of organophosphorus
pesticide samples by high-performance liquid chromatography/mass
spectrometry and high-performance liquid chromatography/mass
spectrometry/mass spectrometry. Environ. Sci. Technol., 22:
1430-1434.
BOLOTNYI, A.V., PISMENNAYA, M.V., & AKORONKO, S.A. (1978)
Transformation of the organophosphorus pesticides anthiophos and
trichlorfon in the environment.] Gig. i Sanit., 1978(5): 28-31 (in
Russian).
BOLSKE, G., KRONEVI, T., & LINDGREN, N.O. (1978) Congenital tremor in
pigs in Sweden. Nord. vet. Med., 30: 534-537.
BOND, G.P. (1986) Primary dermal irritation of technical trichlorfon
(Dylox (R) ) in albino rabbits, Metcalf, USA, Mobay Corporation,
(Proprietary report No. 751, submitted to WHO by Bayer AG, Germany).
BOWMAN, P.B. & DAME, P.W. (1974) Gas-liquid chromatography of
trichlorfon in soluble powder formulations. J. Assoc. Off. Anal.
Chem., 57: 1128-1131.
BRAUN, R., SCHÖNEICH, J., WEISSFLOG, L., & DEDEK, W. (1982) Activity
of organophosphorus insecticides in bacterial tests for mutagenicity
and DNA repair - direct alkylation vs. metabolic activation and
breakdown. 1. Butonate, vinylbutonate, trichlorfon, dichlorvos,
demethyl dichlorvos and demethyl vinylbutonate. Chem.-biol.
Interact., 39: 339-350.
BRODEUR, J. & DUBOIS, K.P. (1963) Comparison of acute toxicity of
anticholinesterase insecticides to weanling and adult male rats.
Proc. Soc. Exp. Biol. Med., 114: 509-511.
BRZHESKII, V.V. (1973) [Possibility of induction of mutations in
Drosophila melanogaster with chlorophos.] Med. Parazitol.
Parazit. Bolezn., 42: 703-706 (in Russian).
BUCHET, J.P. & LAUWERYS, R. (1970) Inhibition of rat heart diolein
hydrolase and brain acetylcholinesterase by organophosphate esters in
vitro. Biochim. biophys. Acta., 218: 369-371.
BULL, D.L. & RIDGWAY, R.L. (1969) Metabolism of trichlorfon in animals
and plants. J. agric. food Chem., 17(4): 837-841.
BULSIEWICZ, H., ROZEWICKA, L., JANUSZEWSKA, H., & BAJKO, J. (1976)
Aberrations of meiotic chromosomes induced in mice with insecticides.
Folia morphol. (Warsaw), 35: 361-368.
CAIN, J.R. & CAIN, R.K. (1984) Effects of five insecticides on
zygospore germination and growth of the green alga Chlamydomonas
moewusii. Bull. environ. Contam. Toxicol., 33(5): 571-574.
CARERE, A., ORTALI, V.A., CARDAMONE, G., & MORPURGO, G. (1978a)
Mutagenicity of dichlorvos and other structurally related pesticides
in Salmonella and Streptomyces. Chem.-biol. Interact., 22:
297-308.
CARERE, A., ORTALI, V.A., CARDAMONE, G., TORRACA, A.M., & RASCHETTI,
R. (1978b) Microbiological mutagenicity studies of pesticides in
vitro. Mutat. Res., 57: 277-286.
CAROLDI, S. & LOTTI, M. (1981) Delayed neurotoxicity caused by a
single massive dose of dichlorvos to adult hens. Toxicol. Lett., 9:
157-159.
CERNA, V. (1963) [Colorimetric determination of dipterex residues in
foods.] Nahrung, 7: 60-66 (in German).
CHAN, P.C. & PETERS, A.C. (1989) Thirteen-week subchronic dosed feed
study of trichlorfon in Fischer rats and B6C3F1 mice. Proc. Am.
Assoc. Cancer Res., 30: 137.
CHAPMAN, R.A. & COLE, C.M. (1982) Observations on the influence of
water and soil pH on the persistence of insecticides. J. environ.
Sci. Health, B17: 487-504.
CHEN, H.H., HSUEH, J.L., SIRIANNI, S.R., & HUANG, C.C. (1981)
Induction of sister chromatid exchanges and cell cycle delay in
cultured mammalian cells treated with eight organophosphorus
pesticides. Mutat. Res., 88: 307-316.
CHEN, H.H., SIRIANNI, S.R., & HUANG, C.C. (1982) Sister chromatid
exchanges in Chinese hamster cells treated with seventeen
organophosphorus compounds in the presence of a metabolic system.
Environ. Mutagen., 4: 621-624.
CLEMENS, G.R. & HARTNAGEL, R.E. (1986) Critique: Assessment of the
teratogenic potential of trichlorfon in mice and rats, Elkhart,
USA, Miles Laboratories, Toxicology Department, (Proprietary report
submitted to WHO by Bayer AG, Germany).
CLEMENS, G.R., TROUP, C.M., & HARTNAGEL, R.E. (1990) Teratology study
in the rabbit with Dylox technical (Trichlorfon), Elkhart, USA,
Miles Laboratories, Toxicology Department, (Proprietary report No. MTD
0134, submitted to WHO by Bayer AG, Germany).
CONRAD, R., DEDEK, W., & ENGEWALD, W. (1987) [Determination of
trichlorfon in biological media and technical products following
derivatization with acetic anhydride by gas chromatography].
Fresenius Z. anal. Chem., 326: 241-246 (in German).
COURTNEY, K.D. & ANDREWS, J.E. (1980) Extra ribs indicate fetotoxicity
and maldevelopment. Teratology, 21(2): 35A (Meeting abstract).
COURTNEY, K.D., ANDREWS, J.E., & SPRINGER, J. (1986) Assessment of
teratogenic potential of trichlorfon in mice and rats. J. environ.
Sci. Health, B 21: 207-227.
COX, C.L., TIMMONS, L.L., & RIDLEN, R.L. (1989) Capillary gas
chromatographic analysis of trichlorfon in dosed feed formulations.
J. anal. Toxicol., 13: 84-88.
CSIK, V., MOTIKA, D., & MAROSI, G.Y. (1986) Delayed neuropathy after
trichlorfon intoxication. J. Neurol. Neurosurg. Psychiatry, 49(2):
222.
DALDRUP, T., SUSANTO, F., & MICHALKE, P. (1981) [Combination of TLC,
GLC (OV 1 and OV 17) and HPLC (RP 18) for A rapid detection of drugs,
intoxicants and related compounds.] Fresenius Z. anal. Chem., 308:
413-427 (in German).
DALDRUP, T., MICHALKE, P., & BOEHME, W. (1982) A screening test for
pharmaceuticals, drug and insecticides with reversed-phase liquid
chromatography-retention data of 560 compounds. Chromatogr. Newsl.,
10: 1-7.
DAVIS, A. (1986) Recent advances in schistomiasis. Q. J. Med., 226:
95-110.
DEDEK, W. (1968) [Degradation and residues of butonate-32p in fruits.]
Z. Naturforsch., B 23: 504-506 (in German).
DEDEK, W. (1981) Guanine N7-alkylation in mice in vivo by
metrifonate-discussion of possible genotoxic risk in mammals. Acta
pharmacol. toxicol., Suppl.49: 40-50.
DEDEK, W. & LOHS, K. (1970) [Alkylating effect of trichlorphon in warm
blooded animals. II. Distribution of 14C in organs and liver
proteins of rats after application of trichlorfon-14C.] Z.
Naturforsch., B 25: 1110-1113 (in German).
DEDEK, W. & SCHWARZ, H. (1966) [Metabolism and application of
trichlorfon in veterinary praxis.] Atompraxis, 12: 603-606 (in
German).
DEDEK, W. & SCHWARZ, H. (1967) [Studies on percutaneous resorption of
32P-labeled systemic organic phosphorus compounds of relatively
small toxicity in cattle.] Z. Naturforsch., B 22: 702-706 (in
German).
DEDEK, W. & SCHWARZ, H. (1970a) [Residues in farm animals following
the application of 32P-labelled organophosphorus insecticides.]
Arch. exp. Veterinaermed, 24: 719-726 (in German).
DEDEK, W. & SCHWARZ, H. (1970b) [Studies of the percutaneous
resorption of 32P-trichlorfon and 32P-dimethoate in sheep.] Z.
Naturforsch., B 25: 1193-1194 (in German).
DEDEK, W., KOCH, H., UHLENHUT, G., & BROSE, F. (1969) [Decomposition
of 3H-trichlorfon to dimethyl dichlorovinyl phosphate.] Z.
Naturforsch., B 24: 663-664 (in German).
DEDEK, W., LOHS, K., & GIBEL,W. (1975a) Studies on the alkylating
properties of trichlorphone and dimethoate in mammals. Environ. Qual.
Saf., 3(Suppl.): 648-651.
DEDEK, W., SCHEUFLER, H., & FISCHER, G.W. (1975b) Mutagenicity of
desmethyltrichlorfon in the dominant lethal test in mice. Arch.
Toxicol., 33: 163-168.
DEDEK, W., LOHS, K., FISCHER, G.W., & SCHMIDT, R. (1976) Alkylation of
guanine in mice in vivo by organophosphorus insecticides. I.
Trichlorfon and butonate. Pestic. Biochem. Physiol., 6: 101-110.
DEDEK, W., GEORGI, W., & GRAHL, R. (1979) Comparative degradation and
metabolism of 32P labeled butonate, trichlorfon and dichlorvos in
crop plants. Biochem. Physiol. Pflanzen, 174: 707-722.
DEDEK, W., GRAHL, R., & SCHMIDT, R. (1984) A comparative study of
guanine N7-alkylation in mice in vivo by the organophosphorus
insecticides trichlorfon, dimethoate, phosmet and bromophos. Acta
pharmacol. toxicol., 55: 104-109.
DEDEK, W., WENZEL, K.D., LUFT, F., OBERLÄNDER, H., & MOTHES, B. (1987)
Preconcentration of hydrophilic pesticides from aqueous solutions and
extraction of residues using the polymeric sorbent Wofatil Y77. I.
Preconcentration of hydrophilic pesticides from water. Fresenius Z.
anal. Chem., 328: 484-486.
DEGRAEVE, N., GILOT-DELHALLE, J., COLIZZI, A., CHOLLET, M.C.,
MOUTSCHEN, J., & MOUTSCHEN-DAHMEN, M. (1981) Evaluation des risques
génétiques d'un insecticide organophosphoré: le trichlorfon
( O,O-diméthyl-(2,2,2 trichloro-1-hydroxy)-éthylphosphonate). Bull.
Soc. R. Sci. Liege, 50(1-2): 85-98.
DEGRAEVE, N., CHOLLET, M.C., & MOUTSCHEN, J. (1984a) Cytogenetic and
genetic effects of subchronic treatments with organophosphorus
insecticides. Arch. Toxicol., 56: 66-67.
DEGRAEVE, N., CHOLLET, M.C., & MOUTSCHEN, J. (1984b) Cytogenetic
effects induced by organophosphorus pesticides in mouse
spermatocytes. Toxicol. Lett., 21: 315-319.
DEGRAEVE, N., CHOLLET, M.C., & MOUTSCHEN, J. (1985) Mutagenic
efficiency of organophosphorus insecticides used in combined
treatments. Environ. Health Perspect., 60: 395-398.
DEGTYAREVA, L.A., POLITOVA, K.S., FOMICHENKO, E.L., & KOCHKINA, N.P.
(1977) [Hygienic evaluation of the air in buildings where
chlorophos-containing insecticides are processed.] Probl. Dezinfekts
Steril., Mater. Simp., 3: 26-27 (in Russian).
DEICHMANN, W.B. & LAMPE, K. (1955) Dipterex, its pharmacological
action. Bull. Univ. Miami Sch. Med., 9: 7-12.
DE KERGOMMEAUX, D.J., GRANT, W.F., & SANDHU, S.S. (1983) Clastogenic
and physiological response of chromosomes to nine pesticides in the
Vicia faba in vivo root tip assay system. Mutat. Res., 124: 69-84.
DÉSI, I. (1983) Neurotoxicological investigations of pesticides in
animal experiments. Neurobehav. Toxicol. Teratol., 5: 503-515.
DEVINE, J.M. (1973) Determination of trichlorfon ( O,O-Dimethyl-
(2,2,2-trichloro-1-hydroxyethyl)phosphonate) in forest environmental
samples. J. agric. food Chem., 21: 1095-1098.
DIVE, D., LECLERC, H., & PERSOONE, G. (1980) Pesticide toxicity on the
ciliate protozoan. Colpidium campylum: possible consequences of the
effect of pesticides in the aquatic environment. Ecotoxicol.
environ. Saf., 4: 129-133.
DMITRIYEV, A.I. (1970) [Trichlorfon action on the quality of chicken
slaughter products.] Veterinariya, 46: 104-106 (in Russian).
DOBSON, K.J. (1977) Trichlorfon toxicity in pigs. Aust. vet. J., 53:
115-117.
DOULL, J. & DUBOIS, K.P. (1956) The effects of diets containing
Dipterex on rats, (Unpublished report by Department of Pharmacology,
University of Chicago).
DOULL, J., ROOT, M., VESSELINOVITCH, D., MESKAUSKAS, J., & FITCH, F.
(1962a) Chronic oral toxicity of Dylox to male and female dogs,
(Unpublished report submitted to WHO by Farbenfabriken Bayer A.G.,
Germany).
DOULL, J., VESSELINOVITCH, D., ROOT, M., COWAN, J., MESKAUSKAS, J., &
FITCH, F. (1962b) Chronic oral toxicity of Dylox to male and female
rats, University of Chicago, Department of Pharmacology,
(Unpublished report).
DOULL, J., VESSELINOVITCH, D., ROOT, M., COWAN, J., MESKAUSKAS, J., &
COWAN, J. (1965) Chronic oral toxicity of Dylox to male and female
rats, University of Chicago, Department of Pharmacology,
(Unpublished report).
DRUDGE, J.H., LYONS, E.T., & TAYLOR, E.L. (1976) Critical tests and
safety studies on trichlorfon as an antiparasitic agent in the horse.
Am. J. vet. Res., 37: 139-144.
DUBOIS, K.P. & COTTER, G.J. (1955) Studies on the toxicity and
mechanism of action of dipterex. Am. Med. Assoc. Arch. ind. Health,
11: 53-60.
DUNACHIE, J.F. & FLETCHER, W.W. (1969) An investigation of the
toxicity of insecticides to birds' eggs using the egg-injection
technique. Ann. appl. Biol., 64: 409-423.
DZWONKOWSKA, A. & HUBNER, H. (1986) Induction of chromosomal
aberrations in the Syrian hamster by insecticides tested in vivo.
Arch. Toxicol., 58: 152-156.
ECOBICHON, D.J. (1979) The chemical stability of trichlorfon in fresh
and salt water as measured by changes in anticholinesterase potency.
Toxicol. Lett., 3: 117-120.
ECOBICHON, D.J., OZERE, R.L., REID, E., & CROCKER, J.F.S. (1977) Acute
fenitrothion poisoning. Can. Med. Assoc. J., 116: 377-379.
EDSON, E.F. & NOAKES, D.N. (1960) The comparative toxicity of six
organophosphorus insecticides. Toxicol. appl. Pharmacol., 2:
523-539.
EL-HISSY, F.T. & ABDEL-KADER, M.I.A. (1980) Effect of five pesticides
on the mycelial growth of some soil and pathogenic fungi. Z. allg.
Mikrobiol., 20: 257-263.
EPSTEIN, S.S., ARNOLD, E., ANDREA, J., BASS, W., & BISHOP, Y. (1972)
Detection of chemical mutagens by the dominant lethal assay in the
mouse. Toxicol. appl. Pharmacol., 23: 288-325.
FAO (1982) Second Government Consultation on International
Harmonization of Pesticide Registration Requirements, Rome, 11-15
October, 1982, Rome, Food and Agriculture Organization of the
United Nations.
FAO/WHO (1972) Trichlorfon. In: 1971 Evaluations of some pesticide
residues in food, Geneva, World Health Organization, pp. 183-230
(WHO Pesticide Residues Series, No. 1).
FAO/WHO (1976) Trichlorfon. In: 1975 Evaluations of some pesticide
residues in food, Geneva, World Health Organization, pp. 379-392
(WHO Pesticide Residues Series, No. 5).
FAO/WHO (1979) Trichlorfon. In: Pesticide residues in food: 1978
Evaluations, Rome, Food and Agriculture Organization of the United
Nations. pp. 235-255 (FAO Plant Production and Protection Paper 15
Sup.).
FAO/WHO (1986) Codex maximum limits for pesticide residues, Rome,
Codex Alimentarius Commission, Food and Agriculture Organization of
the United Nations. (CAC/Vol. XIII-Ed. 2).
FAO/WHO (1989) Guide to Codex recommendations concerning pesticide
residues. Part 8. Recommendations for methods of analysis of
pesticide residues, 4th ed., Rome, Codex Committee on Pesticide
Residues, Food and Agriculture Organization of the United Nations.
(CAC/PR8-1989).
FECHNER, G., FUEHRER, G., & ACKERMANN, H. (1968) [Studies of milk
contamination following trichlorfon treatment of milk cows.] Monatsh.
Veterinärmed., 23: 529-531 (in German).
FECHNER, G., KRETZSCHMANN, F., ACKERMANN, H., & TOPFER, H. (1971)
[Improved method of thin-layer chromatographic and enzymic
determination of trichlorfon and dichlorvos residues in milk.]
Monatsh. Veterinärmed., 26: 860-863 (in German).
FEDORENKO, A.P., ALEEVA, L.V., & TITOK, V.A. (1981) [Organophosphorus
pesticide accumulation in warm-blood animals after chemical treatment
of the forest.] Vestn. Zool., 4: 89-92 (in Russian).
FERREIRA, J.R. & FERNANDES, A.M.S.S. (1980) Gas-liquid chromatographic
determination of organophosphorus insecticide residues in fruits and
vegetables. J. Assoc. Off. Anal. Chem., 63: 517-522.
FILATOV, G.V., SIVOKHIN, P.A., & METELITSA, V.K. (1972) Trichlorfon
excretion in milk. Veterinariya, 5: 102 (in Russian).
FISCHER, G.W., SCHNEIDER, P., & SCHEUFLER, H. (1977) Mutagenicity of
dichloroacetaldehyde and methyl-2,2- dichloro-1,1-dihydroxyethane
phosphonate, possible trichlorfon metabolites. Chem.-biol.
Interact., 19: 205-213.
FOURNIER, E., SONNIER, M., & DALLY, S. (1978) Detection and assay of
organophosphate pesticides in human blood by gas chromatography.
Clin. Toxicol., 12: 457-462.
FRANCIS, B.M., METCALF, R.L., & HANSEN, L.G. (1985) Toxicity of
organophosphorus esters to laying hens after oral and dermal
administration. J. environ. Sci. Health, B 20: 74-95.
FROHLICH, D.R., BOEKER, E.A., & BRINDLEY, W.A. (1990) The kinetics and
inhibition of p-nitrophenylacetate-hydrolising esterases in a
solitary bee, Megachile rotundata (Fab.). Xenobiotica, 20(5):
481-487.
GAINES, T.B. (1969) Acute toxicity of pesticides. Toxicol. appl.
Pharmacol., 14: 515-534.
GARRETT, N.E., STACK, H.F., & WATERS, M.D. (1986) Evaluation of the
genetic activity profiles of 65 pesticides. Mutat. Res., 168:
301-325.
GETZ, M.E. & WHEELER, H.G. (1968) Thin-layer chromatography of
organophosphorus insecticides with several adsorbents and ternary
solvent systems. J. Assoc. Off. Anal. Chem., 51: 1101-1107.
GIANG, P.A. & CASWELL, R.L. (1957) Polarographic determination of
O,O-dimethyl 2,2,2-trichloro-1-hydroxyethylphosphonate (Bayer
L13/59). J. agric. food Chem., 5: 753-754.
GIANG, P.A., BARTHEL, W.F., & HALL, S.A. (1954) Colorimetric
determination of O,O-dialkyl 1-hydroxyphosphonates derived from
chloral. J. agric. food Chem., 2: 1281-1284.
GIBEL, W., VON, LOHS, K.H., WILDNER, G.P., & ZIEBARTH, D. (1971)
[Experimental studies on hepatotoxic and carcinogenic activity of
organophosphorus compounds, Trichlorfon.] Arch. Geschwülstforsch.,
37: 303-312 (in German).
GIBEL, W., VON LOHS, K.H., WILDNER, G.P., ZIEBARTH, D., & STIEGLITS,
R. (1973) [Experimental study on cancerogenic haematotoxic and
hepatotoxic activity of organophosphorus pesticides.] Arch.
Geschwülstforsch, 41: 311-328 (in German).
GILOT-DELHALLE, J., COLIZZI, A., MOUTSCHEN, J., & MOUTSCHEN-DAHMEN, M.
(1983) Mutagenicity of some organophosphorus compounds at the ADE6
locus of Schizosaccharomyces pombe. Mutat. Res., 117: (1-2) 139-148.
GINGERICH, D.A. & MIA, A.S. (1981) Clinical toxicosis and erythrocyte
cholinesterase inhibition of trichlorfon combined with mebendazole in
horses. Am. J. vet. Res., 42: 1645-1650.
GIOVANOLI-JAKUBCZAK, T., FITAK, B., & CHODKOWSKI, J. (1971) Photolysis
of Dipterex and DDVP under the influence of UV irradiation. Rocz.
Chem., 45(4): 689-694.
GLUCKMAN, J.C., BARCELO, D., DE JONG, G.J., FREI, R.W., MARIS, F.A.,
& BRINKMAN, U.A.T. (1986) Improved design and applications of an
on-line thermionic detector for narrow-bore liquid chromatography.
J. Chromatogr., 367: 35-44.
GONCHARUK, E.I., PERELYGIN, V.M., & MELENEVSKAYA, A.V. (1978)
[Analysis of migration of the chemicals from the soil into the
atmosphere.] Gig. i Sanit., 43: 41-45 (in Russian).
GRAHL, K., HORN, H., & HALLEBACH, R. (1981) [Effect of butonate,
trichlorfon and dichlorvos on plankton populations.] Acta hydrochim.
hydrobiol., 9: 147-161 (in German).
GRIFFIN, T.B. (1988) Safety evaluation and tumorigenesis of
trichlorfon in Rhesus monkeys: A ten-year study. Alamogordo, New
Mexico, White Sands Research Center, (Proprietary study report No.
800108 submitted to WHO by Bayer AG, Germany).
GROMYSZ-KALKOWSKA, K., SZUBARTOWSKA, E., & KACZANOWSKA, E. (1985)
Peripheral blood in the Japanese quail ( Coturnix coturnix japonica)
in acute poisoning by different insecticides. Comp. Biochem.
Physiol., C81: 290-312.
GROMYSZ-KALKOWSKA, K., SZUBARTOWSKA, E., SULIKOWSKA, J., & TROCEWICZ,
K. (1985) Peripheral blood changes of the Japanese quail ( Coturnix
coturnix japonica) following repeated small doses of trichlorfon.
Bull. environ. Contam. Toxicol., 35: 757-766.
GRUNDMANN, E. & HOBIK, H.P. (1966) Bay 15922/2 year feeding
experiment in rats/histology (Unpublished report submitted to WHO by
Farbenfabriken Bayer AG, Germany).
GRYGIEREK, E. & WASILEWSKA, B.E. (1981) The effect of Foschlor
(organic phosphorous acid esters) on zooplankton and benthos of fish
ponds. Ekol. Pol., 29: 257-270.
GUTH, J.A. (1967) [Thin-layer chromatographic separation scheme for
insecticidally active phosphoric acid esters.]
Pflanzenschutzberichte, 35: 129-143 (in German).
HAGAN, E.C., JENNER, P.M., & JONES, W.I. (1971) Increased lethal
effects of acutely administered anticholinesterases in female rats
prefed with similar agents. Toxicol. appl. Pharmacol., 18: 235-237.
HALEY, T.J., HARMER, J.H., HARMON, J.R., & DOOLEY, K.L. (1975)
Estimation of the LD1 and extrapolation of the LD0.1 for five
organophosphate pesticides. Arch. Toxicol., 34: 103-109.
HALLAK, M. & GIACOBINI, E. (1987) A comparison of the effects of two
inhibitors on brain cholinesterase. Neuropharmacology, 26: 521-530.
HALLAK, M. & GIACOBINI, E. (1989) Physostigmine, tacrine, and
metrifonate: The effect of multiple doses on acetylcholine metabolism
in rat brain. Neuropharmacology, 28: 199-206.
HAMEED, S.F., SURI, S.M., & KASHYAP, N.P. (1980) Toxicity and
persistence of residues of some organophosphorus insecticides applied
for the control of Dacus cucurbitae Coquillet on the fruits of
cucumber. Indian J. agric. Sci., 50: 73-77.
HANNA, P.J. & DYER, K.F. (1975) Mutagenicity of organophosphorus
compounds in bacteria and Drosophila. Mutat. Res., 28: 405-420.
HANNA, S., BASMY, K., OSAIMA, S., SHOEB, S.M., & AWNY, A.Y. (1966)
Effects of administration of an organophosphorus compound as an
antibilharzial agent with special reference to plasma cholinesterase.
Br. med. J., 1: 1390-1392.
HANSEN, H.J., KRONEVI, T., & BJÖRKLUND, N.E. (1978) On cerebellar
hypoplasia in domestic animals. In: 13th Nord. Vet. Congress Abo,
July 1978.
HASHIMOTO, Y., OKUBO, E., ITO, T., YAMAGUCHI, M., & TANAKA, S. (1982)
Changes in susceptibility of carp to several pesticides with growth.
J. pestic. Sci., 7: 457-461.
HASSAN, A. & ZAYED, S.M.A.D. (1965) Metabolism of organophosphorus
insecticides. III. Fate of the methyl groups of Dipterex in vivo.
Can. J. Biochem., 43: 1271-1275.
HASSAN, A., ZAYED, S.M.A.D., & HASHISH, S. (1965) Metabolism of
organophosphorus insecticides. VI. Mechanism of detoxification of
Dipterex in the rat. Biochem. Pharmacol., 14: 1692-1694.
HATTORI, K. (1974) [Toxicological studies on the effect of chemicals
on birds (part 2). Oral acute toxicity of three organophosphate
insecticides in the common pigeon.] Hokkaidoritsu Eisei Kenkyusho
Ho, 24: 154 (in Japanese).
HAYES, R.H. (1988) Oncogenicity study of technical grade trichlorfon
with mice, Stillwell, Kansas, Mobay Corporation (Proprietary study
report No. 1039 submitted to WHO by Bayer AG, Germany).
HAYES, R.H. (1989) Chronic toxicity/Oncogenicity study of technical
grade trichlorfon (Dylox (R) ) with rats, Stillwell, Kansas,
Mobay Corporation (Proprietary study report No. 1108, submitted to
WHO by Bayer AG, Germany).
HAYES, W.J. Jr (1982) Pesticides studied in man, Baltimore,
Maryland, Williams & Wilkins Co., pp. 351-356.
HAYES, W.J. Jr & LAWS, E.R. Jr (1991) Fenitrothion. In: Handbook of
pesticide toxicology, New York Academic Press, pp. 1020-1023.
HERBOLD, B. (1979a) Micronucleus test on mouse to evaluate L 13/59
(Trichlorfon) for potential mutagenic effects, Wuppertal-Elberfeld,
Bayer AG, Institute of Toxicology, (Proprietary report No. 8505,
submitted to WHO by Bayer AG, Germany).
HERBOLD, B. (1979b) L 13/59 (Trichlorfon) -Dominant lethal study on
male mouse to test for mutagenic effects, Wuppertal-Elberfeld, Bayer
AG, Institute of Toxicology, (Proprietary report No. 8745, submitted
to WHO by Bayer AG, Germany).
HIERONS, R. & JOHNSON, M.K. (1978) Clinical and toxicological
investigations of a case of delayed neuropathy in man after acute
poisoning by an organophosphorus pesticide. Arch. Toxicol., 40:
279-284.
HINTON, M.J. & EVERSOLE, A.G. (1980) Toxicity and tolerance studies
with yellow-phase eels: five chemicals. Prog. Fish Cult., 42:
201-203.
HIRAMATSU, R. & FURUTANI, F. (1977) [Persistence of sprayed
insecticides in head vegetables.] Bull. Yamaguchi-ken agric. Explor.
Stn, 28: 159-163 (in Japanese).
HIRAMATSU, R. & FURUTANI, F. (1978) [Persistence of sprayed malathion
and trichlorfon in grape berries and grape products.] Bull.
Yamaguchi-ken agric. Explor. Stn, 30: 37-44 (in Japanese).
HOFER, W. (1981) Chemistry of metrifonate and dichlorvos. Acta
pharmacol. Scand., 49(suppl.5): 7-14.
HOFFMANN, K., RUF, J., & MAGER, H. (1988) Study of the chronic
toxicity to Rhesus monkeys, oral administration for 26 weeks (oral
intubation tube administration). Wuppertal, Bayer AG, (Proprietary
study report No. 90237, submitted to WHO by Bayer, AG, Germany).
HOLMSTEDT, B., NORDGREN, I., SANDOZ, M., & SUNDWALL, A. (1978)
Metrifonate, summary of toxicological and pharmacological information
available. Arch. Toxicol., 41: 3-29.
HU, X., LU, Y., XUE, S., LING, Y., & GU, X. (1986) Toxicity of
dipterex: A field study. Br. J. ind. Med., 43: 414-419.
IARC (1983) Trichlorfon. In: Miscellaneous pesticides, Lyon,
International Agency for Research on Cancer, pp. 207-231 (IARC
Monographs on the evaluation of the carcinogenic risk of chemicals to
humans, Vol. 30).
IARC (1987) Overall evaluations of carcinogenicity: An updating of
IARC Monographs volumes 1 to 42, Lyon, International Agency for
Research on Cancer, p. 73 (IARC Monographs on the evaluation of the
carcinogenic risk of chemicals to humans, Suppl. 7).
IARC (in press) Re-evaluation of dichlorvos. Lyon, France, IARC
(IARC Monographs on the evaluation of the carcinogenic risk of
chemicals to humans.
IMANAKA, M., MATSUNAGA, K., & ISHIDA, T. (1981) [Gas-liquid
chromatographic determination of trichlorfon (Dipterex) in livestock
products using a flame photometric detector.] J. Food Hyg. Soc.
Jpn., 22: 472-478 (in Japanese).
IRPTC/GKNT (1983) In: Izmerov, N.F., ed. Trichlorfon, Moscow, Centre
of International Projects, GKNT, 24 pp. (Scientific Reviews of Soviet
Literatures on Toxicity and Hazards of Chemicals No. 33).
ISAAC, I.G., SHAFIC, M.T., TANTAWY, G., & EL-KHISHEN, S.A. (1965)
Trichlorfon residues on cotton. Alexandria J. agric. Res., 14: 3-4.
ISHIDATE, M., Jr, SOFUNI, T., & YOSHIKAWA, K. (1981) Chromosomal
aberration tests in vitro as a primary screening tool for
environmental mutagens and/or carcinogens. Gann, 27: 95-108.
ISHIZAKA, T., SEKITA, H., SUZUKI, T., & SAITOU, Y. (1986) Analytical
method for trichlorfon in agricultural foods by gas chromatography.
Shokuhin Eiseigaku Zasshi, 27: 288-295.
IVANOVA, L.N. & MOLOZHANOVA, E.G. (1974) [Kinetics of the
transformation of some pesticides in the soil.] Khim. Sel'sk. Khoz.,
12: 363-365 (in Russian).
IWATA, Y., DUSH, M.E., CARMAN, G.E., & GUNTHER, F.A. (1979) Worker
environment research, residues from carbaryl, chlorobenzilate,
dimethoate and trichlorfon applied to citrus trees. J. agric. food
Chem., 27: 1141-1145.
JAPAN PLANT PROTECTION ASSOCIATION (1985) [Pesticides handbook],
Tokyo, Japan Plant Protection Association (in Japanese).
JAPAN PLANT PROTECTION ASSOCIATION (1986) [Production statistics of
major pesticides - fenitrothion and trichlorfon.] In: [Pesticides
handbook], Tokyo, Japan Plant Protection Association, p. 54 (in
Japanese).
JAPAN PLANT PROTECTION ASSOCIATION (1988) [Production statistics of
major pesticides - fenitrothion and trichlorfon.] In: [Pesticides
handbook], Tokyo, Japan Plant Protection Association, p. 57 (in
Japanese).
JAPAN PLANT PROTECTION ASSOCIATION (1989) [Production statistics of
major pesticides - fenitrothion and trichlorfon.] In: [Pesticides
handbook], Tokyo, Japan Plant Protection Association, p. 60 (in
Japanese).
JOHANSEN, C.A. (1972) Toxicity of field-weathered insecticide residues
to four kinds of bees. Environ. Entomol., 1: 393-394.
JOHNSON, M.K. (1981) Delayed neurotoxicity - do trichlorfon and/or
dichlorvos cause delayed neuropathy in man or in test animals? Acta
pharmacol. toxicol., 49(Suppl.V): 87-98.
JOHNSON, M.K. (1990) Organophosphates and delayed neuropathy - is NTE
alive and well? Toxicol. appl. Pharmacol., 102: 385-399.
JONES, D.C.L., SIMMON, V.F., MORTELMANS, K.E., MITCHELL, A.D., EVANS,
E.L., JOTZ, M.M., RICCIO, E.S., ROBINSON, D.E., & KIRKHART, B.A.
(1982) In vitro mutagenicity studies of environmental chemicals,
Washington, DC, National Technical Information Service.
KAWACHI, T., KOMATSU, T., KADA, T., ISHIDATE, M., Jr, SASAKI, M.,
SUGIYAMA, T., & TAZIMA, Y. (1980) Results of recent studies on the
relevance of various short-term screening tests in Japan. In: The
predictive value of short-term screening tests in carcinogenicity
evaluation, Amsterdam, Elsevier/North-Holland Biomedical Press, pp.
253-267.
KAWAI, M., YOSHIDA, M., IYATOMI, A., KOYAMA, M., & KANEKO, Y. (1982)
[Exposure of spray-operators to trichlorfon.] Nippon koshu Eisei
Zasshi, 29: 165-171 (in Japanese).
KAWAMOTO, K. & URANO, K. (1989) Parameters for predicting fate of
organochlorine pesticides in the environment (I) Octanol-water and
air-water partition coefficients. Chemosphere, 18: 1987-1996.
KIMMERLE, G. & LOSER, E. (1974) Delayed neurotoxicity of
organophosphorus compounds and copper concentration in the serum of
hens. Environ. Qual. Saf., 3: 173-178.
KIMMERLE, G. (1975a) Acute inhalation toxicity study on rats
(Unpublished report No. 5581 from Bayer AG, Germany).
KIMMERLE, G. (1975b) Subacute inhalation toxicity study on rats
(Unpublished report No. 5582 from Bayer AG, Germany).
KIMURA, S., YOKOTE, M., & MATIDA, Y. (1971) Study on the toxicity of
agricultural control chemicals in relation to freshwater fisheries
management No.6 Chronic toxicity of dieldrin, lindane and dipterex to
cherry salmon fingerlings. Bull. Freshwater Fish. Res. Lab., 21:
107-116.
KIRALY, J., SZENTESI, I., RUZICSKA, M., & CZEIZE, A. (1979) Chromosome
studies in workers producing organophosphate insecticides. Arch.
environ. Contam. Toxicol., 8: 309-319.
KLISENKO, M.A. & PISMENNAYA, M.V. (1979) [Photochemical conversion of
organophosphorus pesticides in the air.] Gig. Tr. prof. Zabol.,
56-58 (in Russian).
KNOX, B., ASKAA, J., BASSE, A., BITSCH, V., ESKILDSEN, M., MANDRUP,
M., OTTOSEN, H.E., OVERBY, E., PEDERSEN, K.B., & RASMUSSEN, F. (1978)
Congenital ataxia and tremor with cerebellar hypoplasia in piglets
borne by sows treated with Neguvon(R) vet (Metrifonate, Trichlorfon)
during pregnancy. Nord. vet. Med., 30: 538-545.
KOKOSZKA, L.C. & FLOOD, J.W. (1989) Environmental management handbook.
Appendix 15: Common chemical toxicants found at hazardous waste sites,
New York, Basel, Marcel Dekker.
KONRAD, H., GABRIO, T., & PENNER, G. (1975) [Pesticide contamination
of milk and milk products.] Milchforsch-Milchprax. 17: 67-69 (in
German).
KOWALSKI, R.L., CLEMENS, G.R., BARE, J.J., & HARTNAGEL, R.E. Jr,
(1987) A teratology study with Dylox (R) , technical (Trichlorfon)
in the rat. Elkhart, USA, Miles Laboratories, Toxicology Department,
(Proprietary report No. MTD0017, submitted to WHO by Bayer AG,
Germany).
KRONEVI, T., HANSEN, H.J., & JONSSON, O.J. (1975) Cerebellar
hypoplasia of unknown etiology in pigs. Vet. Rec., May 1975:
403-404.
KRUSTEV, E., KONSTANTINOV, P., & VASILEV, V. (1976) [The effect of
subtoxic doses of dipterex on guinea pigs.] Vet.-Med. Nauki, 13(1):
48-51 (in Russian).
KURINNIY, A.I. (1975) [Comparative investigation of the cytogenetic
effect of certain organophosphorus pesticides.] Genetika, 11: 64-69
(in Russian).
KURINNIY, A.I. & PILINSKAYA, M.A. (1977) [Cytogenetic acticity of the
pesticides imidan, dipterex and gardona in cultured human
lymphocytes.] Genetika, 13: 337-339 (in Russian).
KURTZ, D.A. & STUDHOLME, C.R. (1974) Recovery of trichlorfon
(Dylox(R)) and carbaryl (Sevin(R)) in songbirds following spraying
of forest for gypsy moth. Bull. environ. Contam. Toxicol., 11:
78-84.
KUZMENKO, N.M., DIDENKO, G.G., KURINNIY, A.L., MARTSON, L.V.,
PETROVSKAYA, O.G., PILINSKAYA, M.A., & SHEPELSKAYA, N.R. (1980)
[Long-term health effects of pesticides.] Gig. i Sanit., 45: 88-89
(in Russian).
LAMB, M.J. (1977) Mutagenicity tests with metrifonate in Drosophilla
melanogaster. Mutat. Res., 56: 157-162.
LANGE, C. (1980) [Identification of a metabolite of trichlorfon.] Z.
Chem., 20: 446-447 (in German).
LEBRUN, A. & CERF, C. (1960) Note préliminaire sur la toxicité pour
l'homme d'un insecticide organophosphoré (Dipterex). Bull. World
Health Organ., 22: 579-582.
LEHOTZKY, K. (1982) Effect of pesticides on central and peripheral
nervous system function in rats. Neurobehav. Toxicol. Teratol., 4:
665-669.
LIVINGSTON, R.J. (1977) Review of current literature concerning the
acute and chronic effects of pesticides on aquatic organisms. CRC
Crit. Rev. environ. Control, November 1977: 325-351.
LOGVINENKO, V.F. & MORGUN, V.V. (1978) Study of mutagenic action of
certain pesticides on spring durum wheat. Cytol. Genet. (USSR), 12:
207-212.
LORENZ, W., HENGLEIN, A., & SCHRADER, G. (1955) The new insecticide
O,O-dimethyl 2,2,2-trichloro-1-hydroxy ethylphosphonate. J. Am.
Chem. Soc., 77: 2554-2556.
LORKE, D. & LOSER, E. (1966) Chronic toxicological studies on rats
(Unpublished report submitted to WHO by Farbenfabriken Bayer AG,
Germany).
LOSER, E. (1969) [Propagation trials in rats.] (Unpublished report
submitted to WHO by Farbenfabriken Bayer AG, Germany) (in German).
LOSER, E. (1970) Bay 15992/Chronic toxicological studies on dogs
(Unpublished report submitted by Farbenfabriken Bayer AG, Germany).
LU, Y.P., LU, P., XUE, S., & GU, X. (1984) Investigation on the
chronic effects of dipterex in occupational exposure. Med. Lav.,
75: 376-384.
MCCANN, J.A. & JASPER, R.L. (1972) Vertebral damage to bluegills
exposed to acutely toxic levels of pesticides. Trans. Amer. Fish.
Soc., 101: 317-322.
MCGREGOR, D.B., BROWN, A., CATTANACH, P., EDWARDS, I., MCBRIDE, D.,
RIACH, C., & CASPARY, W.J. (1988) Responses of the L5178Y tk+/tk-
mouse lymphoma cell forward mutation assay: III. 72 coded chemicals.
Environ. mol. Mutagen., 12(1): 85-154.
MACDOUGALL, D. (1964) Dylox. In: Zweig, G. ed. Analytical methods for
pesticides and plant growth regulators, New York, London, Academic
Press, Vol. 2, pp. 199-208.
MACHEMER, L. (1979a) Trichlorfon - Evaluation for embryotoxic and
teratogenic effects on orally dosed rats, Wuppertal-Elberfeld, Bayer
AG, Institute of Toxicology (Proprietary report No. 8400, submitted to
WHO by Bayer AG, Germany).
MACHEMER, L. (1979b) Trichlorfon - Evaluation for embryotoxic and
teratogenic effects on orally dosed rabbits, Wuppertal-Elberfeld,
Bayer AG, Institute of Toxicology (Proprietary report No. 8430,
submitted to WHO by Bayer AG, Germany).
MACHEMER, L. (1981) Chronic toxicity of metrifonate. Acta pharmacol.
toxicol., 49(Suppl.V): 15-28.
MAGALHAES, M.J.A., FERREIRA, J.R., FRUTUOSA, L., & TAINHA, A.A. (1989)
Study of the disappearance of endosulfan, parathion, trichlorfon, and
pirimicarb from broccoli and portuguese cabbage. Pestic. Sci., 27:
23-31.
MAHENDRU, V.K. & AGARWAL, R.A. (1981) Changes in carbohydrate
metabolism in various organs of the snail, Lymnaea acuminata
following exposure to trichlorfon. Acta pharmacol. toxicol., 48:
377-381.
MANDOUL, R., DUBOS, M., DE COURNUAUD, M., & MOULINIER, CL. (1967) In
vitro effects of organophosphorus compounds on the Portugese oyster,
some molluscs, and freshwater microplankton. Bull. Soc. Pathol.
Exot., 60: 568-580.
MARKING, L.L. & MAUCK, W.L. (1975) Toxicity of paired mixtures of
candidate forest insecticides to rainbow trout. Bull. environ.
Contam. Toxicol., 13: 518-523.
MARTSON, L.V. & VORONINA, V.M. (1976) Experimental study of the effect
of a series of phosphoroorganic pesticides (dipterex and imidan) on
embryogenesis. Environ. Health Perspect., 13: 121-125.
MATKOVICS, B., SZABO, L., MINDSZENTY, L., & IVAN, J. (1980) The
effects of organophosphate pesticides on some liver enzymes and lipid
peroxidation. Gen. Pharmacol., 11: 353-356.
MATKOVICS, B., SZABO, L., IVAN, J., & GAAL, I. (1983) Some further
data on the effects of two organophosphate pesticides on the oxidative
metabolism in the liver. Gen. Pharmacol., 124: 689-691.
MATTON, P. & LAHAM, Q.N. (1969) Effect of the organophosphate Dylox on
rainbow trout larvae. J. Fish. Res. Board Can., 26: 2193-2200.
MELNIKOV, N.N., VORONKOVA, V.V., VORONTSOVA, N.A., GUNAR, M.I., SUPIN,
G.S., MOROZOVA, T.N., SHCHERBATYKH, YU. I., & SHVETSOVA-SHILVOSKAYA,
K.D. (1975) [Identification of impurities in technical chlorofos.]
Khim. Sel'sk. Khoz., 13: 142-145 (in Russian).
MENDOZA, C.E. (1973) Analysis of pesticides by the thin-layer
chromatographic-enzyme inhibition technique. Residue Rev., 50:
43-72.
METCALF, R.L., FUKUTO, T.R., & MARCH, R.B. (1959) Toxic action of
Dipterex and DDVP to the house fly. J. econ. Entomol., 52: 44-49.
MIHAIL, F. (1985) L 13/59 (Trichlorfon) - Study for skin-sensitizing
effect on guinea-pigs, Wuppertal-Elberfeld, Bayer AG, Institute of
Toxicology (Proprietary report No. 13323, submitted to WHO by Bayer
AG, Germany).
MIHAIL, F. (1986a) L 13/59 (Trichlorfon) - Study for skin-sensitizing
effect on guinea-pigs in the open epicutaneous test,
Wuppertal-Elberfeld, Bayer AG, Institute of Toxicology (Proprietary
report No. 14348, submitted to WHO by Bayer AG, Germany).
MIHAIL, F. (1986b) L 13/59 (Trichlorfon) - Skin sensitizing effect -
an evaluation, Wuppertal-Elberfeld, Bayer AG, Institute of
Toxicology, (Proprietary statement 11 March 1986, submitted to WHO by
Bayer AG, Germany).
MIYAMOTO, J. (1959) Non-enzymatic conversion of Dipterex into DDVP and
their inhibitory action on enzymes. Botyu-Kagaku, 24: 130-137.
MIYAMOTO, J. (1961) Studies on the mode of action of Dipterex. II. New
glucuronides obtained from the urine of rabbit following
administration of Dipterex. Agric. biol. Chem., 25: 566-572.
MIYATA, T. & SAITO, T. (1973) [Metabolism of NS 2662
( O,O-dimethyl-2,2-dichloro-1-hydroxyethylphosphonate).]
Botyu-Kagaku, 38: 81-86 (in Japanese).
MOLLHOFF, E. (1971) Determination of trichlorfon and fenthion residues
in animals of different species. Pestic. Sci., 2: 179-181.
MONSON, M.H. & ALEXANDER, K. (1984) Metrifonate in pregnancy. Trans.
R. Soc. Trop. Med. Hyg., 78: 565.
MORIYA, M., OHTA, T., WATANABE, K., MIYAZAWA, T., KATA, K., & SHIRASU,
Y. (1983) Further mutagenicity studies on pesticides in bacterial
reversion assay systems. Mutat. Res., 116: 185-216.
MORPURGO, G., AULICINO, F., BIGNAMI, M., CONTI, L., VELCICH, A. &
MONTALENTI, S.G. (1977) Relationship between structure and
mutagenicity dichlorvos and other pesticides. Lincei Rend. Sci. fis.
mat. nat., 62: 692-701.
MOUTSCHEN-DAHMEN, J. (1981) Metrifonate and dichlorvos cytogenetic
investigations. Acta pharmacol. toxicol., 49(Suppl.): 29-39.
MÜHLMANN, R. & SCHRADER, G. (1957) [Hydrolysis of the phosphoric acid
ester insecticide.] Z. Naturforsch., B12: 196-208 (in German).
NAGAYAMA, T., MAKI, T., KAN, K., MAKI, M., & NISHIMA, T. (1986) Survey
of pesticide residues in vegetables and fruits. Ann. Rep. Tokyo
Metrop. Res. Lab. P.H., 37: 173-183 (in Japanese).
NAGAYAMA, T., MAKI, T., IIDA, M., KAN, K., & NISHIMA, T. (1987) Survey
of pesticide residues in vegetables and fruits. Ann. Rep. Tokyo
Metrop. Res. Lab. P.H., 38: 222-228 (in Japanese).
NAGAYAMA, T., MAKI, T., IIDA, M., KAN, K., KAWAI, Y., & NISHIMA, T.
(1988) Survey of pesticide residues in vegetables and fruits. Ann.
Rep. Tokyo Metrop. Res. Lab. P.H., 39: 130-138 (in Japanese).
NAGAYAMA, T., MAKI, T., IIDA, M., KAN, K., KAWAI, Y., & NISHIMA, T.
(1989) Survey of pesticide residues in vegetables and fruits. Ann.
Rep. Tokyo Metrop. Res. Lab. P.H., 40: 155-162 (in Japanese).
NAGY, Z.S., MILE, I., & ANTONI, F. (1975) The mutagenic effect of
pesticides on Escherichia coli WP2 try. Acta Microbiol. Acad. Sci.
Hung., 22: 309-314.
NAISHTEIN, S.Ya. (1976) [Trichlorfon and malathion tolerances in
soil.] Khim. Sel'sk. Khoz., 14: 32-34 (in Russian).
NAISHTEIN, S.Ya., ZHULINSKAYA, V.A., & YUROVSKAYA, E.M. (1973)
[Stability of certain organophosphorous pesticides in soil.] Gig. i
Sanit., 42-45 (in Russian).
NAKAHARA, T., TAKASE, I., KAWAGOE, I., KUBOTA, T., & SAITO, T. (1972)
[Residues of trichlorfon and its metabolites in fresh milk.]
Bochu-kagaku, 37: 149-154 (in Japanese).
NAKAHARA, T., TAKASE, I., & YOSHIMOTO, T. (1973) [Residues of
trichlorfon in eel and carp.] Noyaku Kagaku, 1(2): 75-76 (in
Japanese).
NAKAMURA, T. & UEDA, T. (1967) Studies on the mechanisms of the
monofluoroacetanilides poisoning to mammals; Part VIII.
Organophosphorous compounds as a specific inhibitor to
fluoroacetanilide amidohydrolase. Agric. biol. Chem., 31: 1288-1293.
NEHEZ, M., SELYPES, A., & SCHEUFLER, H. (1982) Mutagenic effect of
trichlorphon and dimethoate on mouse bone marrow cells in vivo. Wiss.
Z. Martin-Luther-Univ. Halle-Wittenberg, 31(2): 57- 62.
NEHEZ, M., HUSZTA, E., MAZZAG, E., SCHEUFLER, H., SCHNEIDER, P., &
FISCHER, G.W. (1987) Cytogenetic, genetic, and embryotoxicity studies
with dimethyl-2,2,2,-trichloro-1-(2,2,2-trichloro-1-hydroxy-
ethoxy)-ethylphosphonate, a hypothetical impurity in technical grade
trichlorfon. Ecotoxicol. environ. Saf., 13: 216-224.
NEPOKLONOV, A.A. & BUKSHTYNOV, V.I. (1971) [Trichlorfon release time
from sheep after aerosol treatment.] Tr. Vses. Nauchn. Issled. Inst.
Sanit., 39: 200-202 (in Russian).
NEPOKLONOV, A.A. & METELITSA, V.K. (1970) [Hypoderminchlorofos used in
the prophylaxis of the hypodermatidae.] Veterinariya, 46: 70-72 (in
Russian).
NIEDZIELLA, S.W., GÖPEL, W., & BANZHAF, E. (1985) [Acute
alkylphosphate poisoning (Trichlorfon) with delayed polyneuropathy.]
Z. gesamte inn. Med., 40(8): 237-239 (in German).
NIKOLAYEV, P.I. (1972) [Distribution of trichlorfon and amidophos in
horses and the secretion of those compounds with mare's milk.] Tr.
Vses. Nauchn. Issled. Vet. Sanit., 44: 91-98 (in Russian).
NISHIUCHI, Y. (1977) [Acute toxicity of pesticide formulations to
Cyprinus carpio at 5 different temperatures.] Suisan Zoshoku, 24:
140-145 (in Japanese).
NISHIUCHI, Y. (1979a) [Acute toxicity of pesticide formulations to
Cyprinus carpio .] Suisan Zoshoku, 27: 36-41 (in Japanese).
NISHIUCHI, Y. (1979b) [Acute toxicity of pesticide formulations to
Daphnia pulex. ] Suisan Zoshoku, 27: 42-47 (in Japanese).
NISHIUCHI, Y. (1979c) [Acute toxicity of pesticide formulations to
Daphnia carinata.] Suisan Zoshoku, 27: 119-124 (in Japanese).
NISHIUCHI, Y. (1981) [Evaluation of the effect of pesticides on some
aquatic organisms. I. Effect of pesticides on some aquatic insects.]
Seitai Kagaku, 4: 31-46 (in Japanese).
NISHIUCHI, Y. & ASANO, K. (1979) [Acute toxicity of pesticide
formulations to Cloeon dipterum.] Suisan Zoshoku, 27: 48-55 (in
Japanese).
NISHIUCHI, Y. & YOSHIDA, K. (1972) Toxicity of pesticides to fresh
water snails. Bull. agric. Chem. Insp. Stn., 12: 86-92.
NORDGREN, I. (1981) Quantitation of metrifonate and dichlorvos in
blood and tissues by gas chromatography - mass spectrophotometry.
Fundam. appl. Toxicol., 1: 230-237.
NORDGREN, I., BERGSTRÖM, M., HOLMSTEDT, B., & SANDOZ, M. (1978)
Transformation and action of Metrifonate. Arch. Toxicol., 41: 31-41.
NORDGREN, I., HOLMSTEDT, B., BENGTSSON, E., & FINKEL, Y. (1980) Plasma
levels of metrifonate and dichlorvos during treatment of
schistosomiasis with bilarcil. Am. J. trop. Med. Hyg., 29(3):
426-430.
NORDGREN, I., BENGTSSON, E., HOLMSTEDT, B., & PETTERSSON, B.M. (1981)
Levels of metrifonate and dichlorvos in plasma and erythrocytes during
treatment of schistosomiasis with bilarcil. Acta pharmacol.
toxicol., 49(Suppl.V): 79-86.
OLAJOS, E.J. & ROSENBLUM, I. (1981) Membrane-bound and soluble
esterase activities in various hen brain regions after diisopropyl
phosphorofluoridate and trichlorfon treatment. Neurotoxicology, 2:
463- 470.
OLAJOS, E.J., ROSENBLUM, I., COULSTON, F., & STROMINGER, N. (1979) The
dose response relationship of trichlorfon neurotoxicity in hens.
Ecotoxicol. environm. Saf., 3: 245-255.
ORDOG, V. (1979) [Effect of organophosphate insecticides on green
algae.] Hydrol. Kozl., 59: 36-42 (in Hungarian).
OSBORNE, B.G., BARRETT, G.M., LAAL-KHOSHAB, A., & WILLIS, K.H. (1989)
The occurrence of pesticide residues in UK home-grown and imported
wheat. Pestic. Sci., 27: 103-109.
OTTO, D., BEHNISCH, I., PFEIFFER, G., DEDEK, W., & GEORGI, W. (1980)
Metabolismus von Trichlorfon in sensiblen und resistenten Fliegen,
Musca domestica L., Arch. Phytopathol. Pflanzenschutz, 16: 397-411.
PAIK, S.G. & LEE, S.Y. (1977) Genetic effects of pesticides in the
mammalian cells. 1. Induction of micronucleus. Tongmul Hakhoe Chi,
20: 19-28.
PAKHLEVANYAN, A.S., PIRUMOV, S.S., DRAGA, O.N., ZORABYAN, S.L., &
CHARCHOGLYAN, M.S. (1979) [Effect of the treatment of cow with
trichlorfon on some milk composition indexes.] Molochn. Prom-st', 7:
42-44 (in Russian).
PANDA, B.B. & SHARMA, C.B.S.R. (1980) Cytogenetic hazards from
agricultural chemicals. 3. Monitoring the cytogenetic activity of
trichlorfon and dichlorvos in Hordeum vulgare. Mutat. Res., 78:
341-345.
PEAKALL, D.B. & BART, J.R. (1983) Impacts of aerial application of
insecticides on forest birds. CRC crit. Rev. environ. Control, 13:
117-165.
PIEPER, G.R. & RICHMOND, C.E. (1976) Residues of trichlorfon and
lauroyl trichlorfon in Douglas fir, Willow grass, aspen, foliage and
in creek water after aerial application. Bull. environ. Contam.
Toxicol., 15: 250-256.
POMPONI, M., GIACOBINI, E., & BRUFANI, M. (1990) Present state and
future development of Alzheimer diseases. Aging, 2(2): 125-153.
POOLE, D.C., SIMMON, V.F., & NEWELL, G.W. (1977) In vitro mutagenic
activity of fourteen pesticides. Toxicol. appl. Pharmacol., 41:
196 (Meeting abstract).
POPE, A.M., HEAVNER, J.E., GUARNIERI, J.A., & KNOBLOCH, C.P. (1986)
Trichlorfon-induced congenital cerebellar hypoplasia in neonatal pigs.
J. Am. Vet. Med. Assoc., 189: 781-783.
RASMUSSEN, F., BASSE, A., KRAUL, I., & DALGAARD-MIKKELSEN, Sr (1978)
Inhibition of cholinesterase activity in blood from pregnant sows and
hypoplasia of cerebellum in their piglets after oral application of
metrifonate (neguvon(r) vet). In: Nord. Vet. Congress Abo, July
1978.
REINER, E., KRAUTHACKER, B. SIMEON, V., & SKRINJARIC-SPOLIJAR, M.
(1975) Mechanism of inhibition in vitro of mammalian
acetyl-cholinesterase and cholinesterase in solutions of
O,O-dimethyl-2,2,2-trichloro-1-hydroxyethyl phosphonate
(trichlorphon). Biochem. Pharmacol., 24: 717-722.
REISS, C.S., HERRMAN, J.M., & HOPKINS, R.E., II (1987) Effect of
antihelminthic treatment on the immune response of mice. Lab. anim.
Sci., 37: 773-775.
RICCIO, E., SHEPHERD, G., POMEROY, A., MORTELMANS, K., & WATERS, M.D.
(1981) Comparative studies between the S. cerevisiae D3 and D7
assays of eleven pesticides. Environ. Mutagen., 3: 327 (Meeting
abstract).
ROBBINS, W.E., HOPKINS, T.L., & EDDY, G.W. (1956) The metabolism of
32P-labeled Bayer L13/59 in a cow. J. econ. Entomol., 49: 801-806.
ROBERTS, D. (1975) The effect of pesticides on byssus formation in the
common mussel, Mytilus edulis. Environ. Pollut., 8: 241-254.
ROSENKRANZ, H.S. & ROSENKRANZ, S. (1972) Reaction of DNA with
phosphoric acid esters: Gasoline additives and insecticides.
Experientia (Basel), 28: 386-387.
RYAZANOVA, R.A. & GAFUROVA, T.V. (1980) [Mutagenic action of a binary
mixture of the pesticides chlorophos and zineb.] Gig. i Sanit., 1:
80-81 (in Russian).
RYBAK, M. (1973) Effect of phosclor administered in water on the
ontogenetic development of white rats and their offsprings. Rocz.
Panstw. Zakl. Hig., 24: 465-475.
SALAMA, A.M., MOSTAFA, I.Y., & EL-ZAWAHRY, Y.A. (1975) Insecticides
and soil microorganisms: (III) Fate of C-14 labeled Dipterex as
affected by two nodule-forming Rhizobium spp. and roots of their
respective leguminous host plants. Acta biol. Acad. Sci. Hung., 26:
1-7.
SASAKI, M., SUGIMURA, K., YOSHIDA, M., & ABE, S. (1980) Cytogenetic
effects of 60 chemicals on cultured human and Chinese hamster cells.
Senshokutai, 20: 574-584.
SCHAFER, E.W. (1972) The acute oral toxicity of 369 pesticidal,
pharmaceutical and other chemicals to wild birds. Toxicol. appl.
Pharmacol., 21: 315-330.
SCHAFER, E.W., Jr, BOWLES, W.A. J, & HURLBUT, J. (1983) The acute oral
toxicity, repellency, and hazard potential of 998 chemicals to one or
more species of wild and domestic birds. Arch. environ. Contam.
Toxicol., 12: 355-382.
SCHEUFLER, H. (1975) Effect of relatively high doses of dimethoate and
trichlorfon on the embryogenesis of laboratory mice. Biol. Rundsch.,
13: 238-240.
SCHIEMANN, S. (1975) Mutagenic effect of trichlorfon on the mouse,
with special regard to the dominant lethal tests. Wiss. Z.
Martin-Luther-Univ. Halle-Wittenberg Math Naturwiss. R., 24: 85-86.
SCHLINKE, J.C. & PALMER, J.S. (1973) Combined effects of phenothiazine
and organophosphate insecticides in cattle. J. Am. Vet. Med. Assoc.,
163: 756-758.
SCHULTEN, H.-R. & SUN, S.-E. (1981) Field desorption mass spectrometry
of standard organophosphorus pesticides and their identification in
waste water. Int. J. environ. anal. Chem., 10: 247- 264.
SERGEANT, D.B. & ZITKO, V. (1979) The determination of trichlorfon,
dichlorvos, fenitrothion and phosphamidon in water. (Can. Fish Mars.
Tech. Rep, 886, pp. 1-12.
SHIMAMOTO, K. & HATTORI, K. (1965) Effects of chronic feeding of the
organic phosphorus compounds on the tissue cholinesterase in rats.
Acta med. Univ. Kyoto, 39: 128-137.
SHIRAISHI, S., GOTO, I., YAMASHITA, Y., OHNISHI, A., & NAGAO, H.
(1977) [Dipterex polyneuropathy.] Shinkei Naika, 6: 34-38 (in
Japanese).
SHIRAISHI, S., INOUE, N., & MURAI, Y. (1982) Dipterex poisoning. J.
UOEH., 4: 177-183.
SHIRAISHI, S., INOUE, N., MURAI, Y., ONISHI, A., & NODA, S. (1983)
Dipterex (Trichlorfon) poisoning - clinical and pathological studies
in human and monkeys. J. UOEH., 5(Suppl.): 125-132.
SHIRASU, Y., MORIYA, M., KATO, K., FURUHASHI, A., & KADA, T. (1976)
Mutagenicity screening of pesticides in the microbial system. Mutat.
Res., 40: 19-30.
SHIRASU, Y., MORIYA, M., KATO, K., TEZUKA, H., TERAMOTO, S., OHTA, T.,
& INOUE, T. (1982) Mutagenicity screening studies on pesticides. In:
Environmental mutagens and carcinogens, Tokyo, University of Tokyo
Press, pp. 331-335.
SILVESTRI, G.R., HIMES, J.A., & EDDS, G.T. (1975a) Repeated oral
administration of coumaphos in sheep: effects on erythrocyte
acetylcholinesterase and other constituents. Am. J. Vet. Res., 36:
283-287.
SILVESTRI, G.R., HIMES, J.A., & EDDS, G.T. (1975b) Repeated oral
administration of coumaphos in sheep: interactions of coumaphos with
bishydroxycoumarin, trichlorfon, and phenobarbital sodium. Am. J.
Vet. Res., 36: 289-292.
SIMMON, V.F. (1979) In vitro microbiological mutagenicity and
unscheduled DNA synthesis studies of eighteen pesticides, Research
Triangle Park, North Carolina, US Environmental Protection Agency, pp.
79-81, 87, 111 (EPA-600/1-79-041).
SINGH, O. & AGARWAL, R.A. (1979) Effects on certain carbamate and
organophosphorus pesticides on isolated organs of Pila globosa
(Gastropoda). Toxicol. appl. Pharmacol., 50: 485-492.
SINGH, O. & AGARWAL, R.A. (1981) Toxicity of certain pesticides to two
economic species of snails in northern India. J. econ. Entomol., 74:
568-571.
SISSONS, D.J. & TELLING, G.M. (1970) Rapid procedure for the
determination of organophosphorus insecticide residues in vegetables.
II. A screening procedure for water soluble insecticides. J.
Chromatogr., 48: 468-477.
SLOTT, V. & ECOBICHON, D.J. (1984) An acute and subacute neurotoxicity
assessment of trichlorfon. Can. J. Physiol. Pharmacol., 62: 513-518.
SNELLEN, W.M. (1981) Therapeutic properties of metrifonate. Acta
pharmacol. toxicol., 49(Suppl.5): 114-117.
SOLIMAN, M.S., EL-MISSERY, A.G., ABDELMESSIH, M.S., FAYAD, M.E., &
BASSILI, W.R. (1984) The effect of diazinon and neguvon on the liver
of experimentally intoxicated mice. J. Egypt. Soc. Parisitol., 14:
557-562.
SPEHAR, R.L., LEMKE, A.E., PICKERING, Q.H., ROUSH, T.H., RUSSO, R.C.,
& YOUNT, J.D. (1981) Effects of pollution on freshwater fish. J.
Water Pollut. Control. Fed., 53: 1028-1076.
SPICER, E.J.F. & URWIN, C. (1971) Pathology report of Bay 15922
generation experiment in rats (Unpublished report submitted to WHO
by Farbenfabriken Bayer AG, Germany).
STAN, H.J. (1977) [Detection of organophosphorous pesticide residues
in food at the ppb level with open tubular column gas chromatography-
mass spectrometry.] Z. Lebensm. Unters. Forsch., 164: 153-159 (in
German).
STAN, H.J., ABRAHAM, B., JUNG, J., KELLERT, M., & STEINLAND, K. (1977)
[Detection of organophosphorus insecticides by gas chromatography/mass
spectrometry.] Fresenius Z. anal. Chem., 287: 271-285 (in German).
STAPLES, R.E. & GOULDING, E.H. (1979) Dipterex teratogenicity in the
rat, hamster and mouse when given by gavage. Environ. Health
Perspect., 30: 105-113.
STAPLES, R.E., KELLAM, R.G., & HASEMAN, J.K. (1976) Developmental
toxicity in the rat after ingestion or gavage of organophosphate
pesticides (dipterex, imidan) during pregnancy. Environ. Health
Perspect., 13: 133-140.
STOBWASSER, H. & KIRCHHOFF, J. (1968) [Insecticide residues on lettuce
and spinach in outdoor and greenhouse cultivation.] Qual. Plant
Mater. Veg., 15: 273-279 (in German).
SUGIYAMA, H. & SHIGEMATSU, H. (1969) Enzymic dehydrochlorination of
trichlorfon by the digestive juice of the silkworm. Bombyx mori.
Botyu-Kagaku, 34: 79-85.
SUNDARAM, K.M.S. & VARTY, I.W. (1989) Distribution and persistence of
trichlorfon in a forest environment. J. Environ. Sci. Health, B
24(6): 647-659.
SZALONTAI, G. (1976) High-performance liquid chromatography of
organophosphorus insecticides. J. Chromatogr., 124: 9-16.
SZUBARTOWSKA, E. (1983) Peripheral blood picture in rabbits poisoned
by the pesticide trichlorfon. Folia biol., 31: 407-418.
SZUBARTOWSKA, E. & GROMYSZ-KALKOWSKA, K. (1986) Differences between
breeds of rabbits in their susceptibility to the effect of foschlor.
Comp. Biochem. Physiol., 85C: 33-40.
TAKASE, I., NAKAHARA, T., YOSHIMOTO, Y., & NAKAMURA, H. (1972)
[Determination of residues of trichlorfon and its metabolites in
plants, milk, chicken, and fish by gas chromatography.]
Botyu-Kagaku, 37: 142-148 (in Japanese).
TAYLOR, P. (1980) Anticholinesterase agents. In: Gilman, A.G.,
Goodman, L.S. & Gilman, A., ed. The pharmacological basis of
therapeutics, 6th ed., New York, MacMillan Publishing Co., pp.
100-119.
TEICHMANN, B. & HAUSCHILD, F. (1978) [Test of
O,O-dimethyl(1-hydroxy-2,2,2-trichloro-ethyl)phosphonate
(Trichlorfon) for carginogenic activity in mice by oral
(oesophageal-gastric intubation), intraperitoneal and dermal
application.] Arch. Geschwulstforsch, 48: 301-307 (in German).
TEICHMANN, B. & SCHMIDT, A. (1978) [Test of
O,O-dimethyl(1-hydroxy-2,2,2-trichloro-ethyl)phosphonate
(Trichlorfon) for carcinogenic activity in Syrian golden hamsters
( Mesocricetus auratus Waterhouse) by intraperitoneal
administration.] Arch. Geschwulst forsch, 48: 718-721 (in German).
TEICHMANN, B., HAUSCHILD, F., & ECKELMANN, A. (1978) [Test of O,O-
dimethyl (1-hydroxy-2,2,2-trichloroethyl)phosphonate (Trichlorofon)
for carcinogenic activity in rats by oral (oesophageal-gastric
intubation) and intraperitoneal administration.] Arch.
Geschwulstforsch., 48: 112-119, (in German).
TELEUGALIEV, T.M. (1977) [Veterinary hygienic study of fish poisoned
with trichlorfon.] Vestn. S-kh. Nauki Kaz., 20: 75-77 (in Russian).
THYSSEN, J. (1981) L 13/59 (Dipterex active ingredient) tests for
skin and eye irritation, Wuppertal-Elberfeld, Bayer AG, Institute of
Toxicology, (Proprietary report submitted to WHO by Bayer AG,
Germany).
TREFILOV, V.N., FAEDMAN, I.S., & BORISOVA, E.P. (1971) [Contamination
of working clothes and skin of workers engaged in the production of
metaphos and chlorophos.] Gig. Tr. prof. Zabol., 15: 51-53 (in
Russian).
TRINH VAN BAO, SZABO, I., RUZICSKA, P., & CZEIZEL, A. (1974)
Chromosome aberrations in patients suffering acute organic phosphate
insecticide intoxication. Humangenetik, 24: 33-57.
TSUMUKI, H., SAITO, T., MIYATA, T., & IYATOMI, K. (1970) Acute and
subacute toxicity of organophosphorus insecticides to mammals. In:
O'Brien, R.D. & Yamamoto, I., ed. Biochemical toxicology of
insecticides, New York, London, Academic Press, p. 65-73.
TSVETKOVA, T.S., PENEVA, V., & GRIGOROVA, D. (1981) [Residual amount
of pesticides in honey.] Vet. Med. Nauki, 18: 93-98 (in Russian).
UMEHARA, F., FUKUOKA, T., MINAUCHI, Y., OSAME, M., & IGATA, A. (1982)
A case of dipterex poisoning. A histopathological study of peripheral
nerves. Clin. Neurol., 22: 990-996.
US NTP (1989) NTP technical report on the toxicology and
carcinogenesis studies of dichlorvos (CAS No: 62-73-7) in F344/N rats
and B6C3F1 mice (gavage studies), Research Triangle Park, North
Carolina, National Toxicology Program (NTP TR 342).
VALENCIA, R. (1977) Mutagenesis screening of pesticides using
drosophia (Contract No. 68-01-2474), Madison, Wisconsin, WARF
Institute, Inc.
VAN MIDDELEM, C.H., HABECK, D.H., & STRAYER, J.R. (1972) Residues on
ryegrass following preplant or postemergence applications of four
insecticides for mole cricket control. J. econ. Entmol., 65:
495-497.
VASILESCU, C., ALEXIANU, M., & DAN, A. (1984) Delayed neuropathy after
organophosphorus insecticides (Dipterex) poisoning: a clinical,
electrophysiological, and nerve biopsy study. J. Neurol. Neurosurg.
Psychiatry., 47: 543-548.
VILCEANU, R., SCHULZ, P., DRAGHICI, R., & SOIMU, P. (1973) [Gas
chromato graphy of (1-hydroxy-2,2,2-trichlorethyl)-phosphonic acid
esters.] J. Chromatogr., 82: 285-290 (in German).
VÖLKNER, W. (1987) L 13/59 (Trichlorfon) - Sister chromatid exchanges
(SCEs) in Chinese hamster bone marrow cells, Technical University of
Darmstadt, Laboratory for Mutagenicity Testing (Proprietary report
No. R 4079, submitted to WHO by Bayer AG, Germany).
WATERS, M.D., SIMMON, V.F., MITCHELL, A.D., JORGENSON, T.A., &
VALENCIA, R. (1980) An overview of short-term tests for the mutagenic
and carcinogenic potential of pesticides. J. environ. Sci. Health,
B15: 867-906.
WATERS, M.D., NESNOW, S., SIMMON, A.D., MITCHELL, T.A., JORGENSON,
T.A., & VALENCIA, R. (1981) Pesticides: mutagenic and carcinogenic
potential. In: Bandal, S.K., Golberg, L., & Leng, M.L., ed. The
pesticide chemist and modern toxicology, Washington, DC, American
Chemical Society, pp. 80-113 (ACS Symposium Series, No. 160).
WATERS, M.D., SANDHU, S.S., SIMMON, V.F., MORTELMANS, K.E., MITCHELL,
A.D., JORGENSON, T.A., JONES, D.C.L., VALENCIA, R., & GARRET, N.E.
(1982) Study of pesticide genotoxicity. Basic Life Sci., 21:
275-326.
WEGNER, D. (1970) [Bilarcil(R) Bay 2349 - Clinical experience
1960-1969.] (Unpublished report submitted to WHO by Farbenfabriken
Bayer AG, Germany) (in German).
WELLBORN, T.L., Jr (1969) The toxicity of nine therapeutic and
herbicidal compounds to striped bass. Prog. Fish Cult., 31: 27-32.
WENDA-ROZEWICKA, L. (1983) Morphometric studies of male gonads from
mice receiving insecticides (Metox-30, Sadofos-30, and Foschlor-50).
Folia biol., 32: 23-34.
WHO (1985) The control of schistosomiasis: Report of a WHO expert
committee, Geneva, World Health Organization, pp. 113 (Technical
Report Series, No. 728).
WHO (1986) Environmental Health Criteria 63: Organophosphorus
insecticides: A general introduction, Geneva, World Health
Organization, pp. 181.
WHO (1989) Environmental Health Criteria 79: Dichlorvos, Geneva,
World Health Organization, pp. 157.
WHO (1990) The WHO recommended classification of pesticides by hazard
and guidelines to classification 1990-1991, Geneva, World Health
Organization, pp. 39 (Unpublished document WHO/PCS/90.1 Rev.1).
WHO/FAO (1977) Data sheets on pesticides, No. 27: Trichlorfon,
Geneva, World Health Organization, pp. 9 (VBC/D5/77.28).
WILD, D. (1975) Mutagenicity studies on organophosphorus insecticides.
Mutat. Res., 32: 133-150.
WILKINS, H.A. & MOORE, P.J. (1987) Comparative trials of regimes for
the treatment of urinary schistomiasis in the Gambia. J. trop. Med.
Hyg., 90: 83-92.
WILLIAMS, M.W., FUYAT, H.N., & FITZHUGH, O.G. (1959) The subacute
toxicity of four organic phosphates to dogs. Toxicol. appl.
Pharmacol., 1: 1-7.
WINTERLIN, W., WALKER, G., & FRANK, H. (1968) Detection of
cholinesterase-inhibiting pesticides following separation on
thin-layer chromatograms. J. agric. food Chem., 16: 808-812.
WOODWARD, D.F. & MAUCK, W.L. (1980) Toxicity of five forest
insecticides to cutthroat trout Salmo clarki and two species of
aquatic invertebrates. Bull. environ. Contam. Toxicol., 25: 846-853.
WRIGHT, A.S., HUTSON, D.H., & WOODER, A.F. (1979) The chemical and
biochemical reactivity of dichlorvos. Arch. Toxicol., 42: 1-18.
YAKOUB (1990) A rational approach to the treatment of Schistosoma
haematobium infection with metrifonate - clinical and pharmacological
studies, Stockholm, Karolinska Institute, (Dissertation).
YASHIKI, M., KOJIMA, T., & UNE, I. (1982) [Determination of Dipterex
in biological materials by gas chromatography and gas chromatography-
mass spectrometry and the analysis of the distribution of Dipterex in
a case of fatal poisoning.] Nippon Hoigaku Zasshi, 36: 426-430 (in
Japanese).
YASHIKI, M., CHIKASUE, F., MIYAZAKI, T., KOJIMA, T., & OHTANI, M.
(1988) [Gas chromatographic and gas chromatographic-mass spectrometric
determination of trichlorfon in biological fluids collected in a fatal
dipterex poisoning.] Nippon Hoigaku Zasshi, 42: 94-98 (in Japanese,
with English summary).
YONOVA, I. & ZHECHEVA, G. (1974) [Residues of sevin, trichlorfon and
imidan in the meat of pig treated for ectoparasites.] Vet. Med.
Nauki, 11: 28-34 (in Russian).
YÜKSEKISIK, N. (1973) Kinetic study of the rearrangement of O,O-
dialkyl 2,2,2-tricholoro-1-hydroxyethylphosphonates. Commun. Fac.
Sci. Univ. Ankara Ser., B 20: 49-62.
YUROVSKAYA, E.M. & ZHULINSKAYA, V.A. (1974) [Behavior of organo
phosphorous insecticides in the soil.] Khim. Sel'sk. Khoz., 12:
358-361 (in Russian).
ZAKHAROV, B.N. (1980) [Migration of trichlorfon in nature.] Gig. i
Sanit., 45: 62-63 (in Russian).
ZAYED, S.M.A.D., MOSTAFA, I.Y., & HASSAN, A. (1965) Metabolism of
organophosphorus insecticides. (VII) Transformation of 32P-labeled
Dipterex through microorganisms. Arch. Mikrobiol., 51: 118-121.
ZELINSKIY, M.I. (1974) [Toxicity of trichlorfon and its effect on the
quality of meat.] Vestn. S-kh. Nauki Kaz., 17(10): 66-68 (in
Russian).
ZINKL, J.G., HENNY, C.J., & DEWEESE, L.R. (1977) Brain cholinesterase
activities of birds from forests sprayed with trichlorfon (Dylox) and
carbaryl (Sevin-4-oil). Bull. environ. Contam. Toxicol., 17:
379-386.
ZWEIG, G. & SHERMA, J. (1972) Dylox. In: Analytical methods for
pesticides and plant growth regulators, New York, London, Academic
Press, Vol. 6, pp. 387-392.
ANNEX I. TREATMENT OF ORGANOPHOSPHATE
INSECTICIDE POISONING IN MAN
(From EHC 63: Organophosphorus Insecticides - A
General Introduction)
All cases of organophosphorus poisoning should be dealt with as
an emergency and the patient sent to hospital as quickly as possible.
Although symptoms may develop rapidly, delay in onset or a steady
increase in severity may be seen up to 48 h after ingestion of some
formulated organophosphorus insecticides.
Extensive descriptions of treatment of poisoning by
organophosphorus insecticides are given in several major references
(Kagan, 1977; Taylor, 1980; UK DHSS, 1983; Plestina, 1984) and will
also be included in the IPCS Health and Safety Guides to be prepared
for selected organophosphorus insecticides.
The treatment is based on:
(a) minimizing the absorption;
(b) general supportive treatment; and
(c) specific pharmacological treatment.
I.1 Minimizing the absorption
When dermal exposure occurs, decontamination procedures include
removal of contaminated clothes and washing of the skin with alkaline
soap or with a sodium bicarbonate solution. Particular care should be
taken in cleaning the skin area where venepuncture is performed. Blood
might be contaminated with direct-acting organophosphorus esters and,
therefore, inaccurate measures of ChE inhibition might result.
Extensive eye irrigation with water or saline should also be
performed. In the case of ingestion, vomiting might be induced, if the
patient is conscious, by the administration of ipecacuanha syrup
(10-30 ml) followed by 200 ml water. This treatment is, however,
contraindicated in the case of pesticides dissolved in hydrocarbon
solvents. Gastric lavage (with addition of bicarbonate solution or
activated charcoal) can also be performed, particularly in unconscious
patients, taking care to prevent aspiration of fluids into the lungs
(i.e., only after a tracheal tube has been put into place).
The volume of fluid introduced into the stomach should be
recorded and samples of gastric lavage frozen and stored for
subsequent chemical analysis. If the formulation of the pesticide
involved is available, it should also be stored for further analysis
(i.e., detection of toxicologically relevant impurities). A purgative
can be administered to remove the ingested compound.
I.2 General supportive treatment
Artificial respiration (via a tracheal tube) should be started at
the first sign of respiratory failure and maintained for as long as
necessary.
Cautious administration of fluids is advised, as well as general
supportive and symptomatic pharmacological treatment and absolute
rest.
I.3 Specific pharmacological treatment
I.3.1 Atropine
Atropine should be given, beginning with 2 mg iv and given at
15-30-min intervals. The dose and the frequency of atropine treatment
varies from case to case, but should maintain the patient fully
atropinized (dilated pupils, dry mouth, skin flushing, etc.).
Continuous infusion of atropine may be necessary in extreme cases and
total daily doses up to several hundred mg may be necessary during the
first few days of treatment.
I.3.2 Oxime reactivators
Cholinesterase reactivators (e.g., pralidoxime, obidoxime)
specifically restore AChE activity inhibited by organophosphates. This
is not the case with enzymes inhibited by carbamates. The treatment
should begin as soon as possible, because oximes are not effective on
"aged" phosphorylated ChEs. However, if absorption, distribution, and
metabolism are thought to be delayed for any reasons, oximes can be
administered for several days after intoxication. Effective treatment
with oximes reduces the required dose of atropine. Pralidoxime is the
most widely available oxime. A dose of 1 g pralidoxime can be given
either im or iv and repeated 2-3 times per day or, in extreme cases,
more often. If possible, blood samples should be taken for AChE
determinations before and during treatment. Skin should be carefully
cleansed before sampling. Results of the assays should influence the
decision whether to continue oxime therapy after the first 2 days.
There are indications that oxime therapy may possibly have
beneficial effects on CNS-derived symptoms.
I.3.3 Diazepam
Diazepam should be included in the therapy of all but the mildest
cases. Besides relieving anxiety, it appears to counteract some
aspects of CNS-derived symptoms that are not affected by atropine.
Doses of 10 mg sc or iv are appropriate and may be repeated as
required (Vale & Scott, 1974). Other centrally acting drugs and drugs
that may depress respiration are not recommended in the absence of
artificial respiration procedures.
I.3.4 Notes on the recommended treatment
I.3.4.1 Effects of atropine and oxime
The combined effect far exceeds the benefit of either drug
singly.
I.3.4.2 Response to atropine
The response of the eye pupil may be unreliable in cases of
organophosphorus poisoning. A flushed skin and drying of secretions
are the best guide to the effectiveness of atropinization. Although
repeated dosing may well be necessary, excessive doses at any one time
may cause toxic side-effects. Pulse-rate should not exceed 120/min.
I.3.4.3 Persistence of treatment
Some organophosphorus pesticides are very lipophilic and may be
taken into, and then released from, fat depots over a period of many
days. It is therefore quite incorrect to abandon oxime treatment after
1-2 days on the supposition that all inhibited enzyme will be aged.
Ecobichon et al. (1977) noted prompt improvement in both condition and
blood-ChEs in response to pralidoxime given on the 11th-15th days
after major symptoms of poisoning appeared due to extended exposure to
fenitrothion (a dimethyl phosphate with a short half-life for aging of
inhibited AChE).
I.3.4.4 Dosage of atropine and oxime
The recommended doses above pertain to exposures, usually for an
occupational setting, but, in the case of very severe exposure or
massive ingestion (accidental or deliberate), the therapeutic doses
may be extended considerably. Warriner et al. (1977) reported the case
of a patient who drank a large quantity of dicrotophos, in error,
while drunk. Therapeutic dosages were progressively increased up to 6
mg atropine iv every 15 min together with continuous iv infusion of
pralidoxime chloride at 0.5 g/h for 72 h, from days 3 to 6 after
intoxication. After considerable improvement, the patient relapsed and
further aggressive therapy was given at a declining rate from days 10
to 16 (atropine) and to day 23 (oxime), respectively. In total, 92 g
of pralidoxime chloride and 3912 mg of atropine were given and the
patient was discharged on the thirty-third day with no apparent
sequelae.
References to Annex I.
ECOBICHON, D.J., OZERE, R.L., REID, E., & CROCKER, J.F.S (1977)Acute
fenitrothion poisoning. Can. Med. Assoc. J., 116: 377-379.
KAGAN, JU.S. (1977) [ Toxicology of organophosphorus pesticides],
Moscow, Meditsina, pp. 111-121, 219-233, 260-269 (in Russian).
PLESTINA, R. (1984) Prevention, diagnosis, and treatment of
insecticide poisoning, Geneva, World Health Organization
(Unpublished document VBC/84.889).
TAYLOR, P. (1980) Anticholinesterase agents. In: Goodman, L.S. &
Gilman, A., ed. The pharmacological basis of therapeutics, 6th ed.,
New York, Macmillan Publishing Company, pp. 100-119.
UK DHSS (1983) Pesticide poisoning: notes for the guidance of medical
practitioners, London, United Kingdom Department of Health and
Social Security, pp. 41-47.
VALE, J.A. & SCOTT, G.W. (1974) Organophosphorus poisoning. Guy's
Hosp. Rep., 123: 13-25.
WARRINER, R.A., III, NIES, A.S., & HAYES, W.J., Jr (1977) Severe
organophosphate poisoning complicated by alcohol and terpentine
ingestion. Arch. environ. Health, 32: 203-205.
ANNEX II. NO-OBSERVED-EFFECT LEVELS (NOELS) IN ANIMALS TREATED WITH
TRICHLORFON
Annex II No-observed-effect levels (NOELS) in animals treated
with trichlorfon
Animal Exposurea NOEL (paramater) Reference
(strain)
Rat 16 weeks 100 mg/kg diet (ChE) Doull & Dubois (1956)
Dog 12 weeks 200 mg/kg diet Williams et al. (1959)
(plasma, Er-ChE)
Rat 6h/day over 3 weeks 12.7 mg/m3 by inhalation Kimmerle (1975b)
(plasma, Er, brain ChE)
Beagle dog 12 months 250 mg/kg diet (ChE) Doull et al. (1962a)
Beagle dog 4 years 50 mg/kg diet (ChE) Löser (1970)
Rhesus monkey 26 weeks 0.2 mg/kg body weight Hoffmann et al. (1988)
oral intubation (Er-ChE)
CD-1 mouse 90 weeks 100 mg/kg diet (ChE) Machemer (1981)
1000 mg/kg diet
(tumour incidence)
CD-1 mouse 104 weeks 2700 mg/kg diet Hayes (1988)
(tumorigenicity)
Sprague-Dawley 17 months (m) 250 mg/kg diet (ChE, Doull et al. (1962b)
rat 24 months (f) survival, spermatogenesis,
ovarian changes) 50 mg/kg
diet (mammary tumours)
Annex II. (continued)
Animal Exposurea NOEL (paramater) Reference
(strain)
Sprague-Dawley 18 months 200 mg/kg diet Doull et al. (1965)
rat (ovarian changes)
Long-Evans rat 24 months 500 mg/kg diet (ChE) Lorke & Löser (1966);
1000 mg/kg diet Grundmann & Hobik (1966)
(tumorigenicity)
Rat twice/week 22 mg/kg body weight Teichmann et al. (1978)
for 90 weeks by gavage
(incidence of tumours)
Fischer 344 105 weeks 100 mg/kg diet Hayes (1989)
rat (morphological
parameters)
Rhesus monkey 6 days/week 0.2 mg/kg body weight Griffin (1988)
for 10 years oral intubation
(brain-ChE)
AB/Jena mouse twice/week 28.2 mg/body weight Teichmann & Hauschild
for 75 weeks ip injection (1978)
(incidence of tumours)
Rat twice/week 12.0 mg/kg body weight Teichmann et al. (1978)
for 90 weeks ip injection
(incidence of tumours)
Syrian Golden once/week 20 mg/kg body weight Teichmann & Schmidt
hamster for 90 weeks ip injection (1978)
(incidence of tumours)
Annex II. (continued)
Animal Exposurea NOEL (paramater) Reference
(strain)
AB/Jena mouse twice/week 0.25 ml of 1% solution Teichmann & Hauschild
for 75 weeks in acetone dermal (1978)
administration
(incidence of tumours)
a m = male.
f = female.
RESUME ET EVALUATION, CONCLUSIONS ET RECOMMANDATIONS
1. Résumé et évaluation
1.1 Exposition
Le trichlorfon est un insecticide organophosphoré utilisé depuis
le début des années 1950. En agriculture, on l'emploie principalement
contre les ravageurs des cultures de plein champ et des vergers. On
l'utilise également comme insecticide en forêt et pour débarrasser des
animaux domestiques de leurs parasites. Sous le nom de métrifonate, il
est utilisé pour traiter l'infestation humaine à Schistosoma
haematobium. Il agit en libérant lentement du dichlorvos. Le
trichlorfon est commercialisé sous forme de concentré émulsionnable,
de poudre dispersable, de poudre pour poudrage, de granulés, de
solution et de concentré à très bas volume.
Peu après l'épandage, la concentration atmosphérique du
trichlorfon peut atteindre 0,1 mg/m3 mais cette valeur diminue
rapidement pour tomber à moins de 0,01 mg/m3 en quelques jours. Les
eaux de ruissellement provenant des zones traitées peuvent contenir
des concentrations de trichlorfon atteignant 50 µg/litre, mais la
teneur des eaux de surface est généralement beaucoup faible et diminue
rapidement.
Le trichlorfon se décompose rapidement dans le sol et sa
concentration y devient généralement négligeable dans le mois qui suit
l'épandage. Il est relativement stable dans l'eau aux pH inférieurs à
5,5. A pH plus élevé, il se transforme de dichlorvos. Les
microorganismes et les plantes métabolisent probablement le
trichlorfon mais son mode d'élimination principal est l'hydrolyse
abiotique.
A quelques exceptions près, la concentration de trichlorfon sur
les récoltes est inférieure à 10 mg/kg dans le jour qui suit
l'épandage et tombe en-dessous de 0,1 mg/kg une quinzaine jours après.
Le lait des vaches que l'on a traitées au trichlorfon pour les
débarrasser de leur vermine peut contenir des résidus atteignant 1,2
mg/litre deux heures après l'application, mais cette valeur tombe
en-dessous de 0,1 mg/litre dans les 24 heures. On n'a pas constaté la
présence de concentrations importantes de trichlorfon dans la viande
des animaux traités. Dans les oeufs de poules traitées on a mesuré des
concentrations de 0,05 mg/kg.
1.2 Absorption, métabolisme et excrétion
Le trichlorfon est rapidement absorbé par l'ensemble des voies
d'exposition (orale, dermique, respiratoire) et il se répartit
rapidement dans les tissus de l'organisme. Le taux sanguin passe par
un maximum au bout d'une à deux heures, le produit disparaissant
presque totalement du courant sanguin au bout 1,5 à 4 heures. La
demi-vie biologique du trichlorfon dans le sang des mammifères a été
estimée à environ 30 minutes.
Dans l'eau, les liquides biologiques et les tissus, à des valeurs
du pH supérieures à 5,5 le trichlorfon subit une transformation en
dichlorvos (phosphate de 2,2-dichlorovinyle et de diméthyle) par
déshydrochloration. C'est le dichlorvos qui constitue le principe
actif antichlolinestérasique. Les principales voies de dégradation
sont la déméthylation, la coupure de la liaison phosphore-carbone et
l'hydrolyse de l'ester via le dichlorvos. Les principaux métabolites
du trichlorfon que l'on trouve in vivo sont le déméthyltrichlorfon,
le déméthyldichlorvos, l'hydrogénophosphate de diméthyle,
l'hydrogénophosphate de méthyle l'acide phosphorique et de
trichloréthanol. Ce dernier métabolite se retrouve dans l'urine,
conjugué sous forme de glucuronide.
Le trichlorfon et ses métabolites sont principalement éliminés
par voie urinaire. Des études effectuées avec du trichlorfon
radiomarqué (14C-méthyl et 32P) ont montré que la majeure partie
du produit s'éliminait sous la forme de dérivés hydrosolubles et une
faible fraction sous la forme de dérivés solubles dans le chloroforme.
Environ 66 à 70 % des produits hydrosolubles apparaissent dans l'urine
dans les 12 heures, 24 % du produit radiomarqué au niveau du
groupement méthyl étant éliminés dans l'air expiré sous forme de
dioxyde de carbone (CO2). Après avoir traité des vaches soit par
administration orale soit par voie cutanée, on a retrouvé dans leur
lait de faibles quantités de trichlorfon et de ses métabolites.
1.3 Effets sur les êtres vivants dans leur milieu naturel
Le trichlorfon est modérément toxique pour les poissons (les
valeurs de la CL50 à 96 heures vont de 0,45 mg/litre à 51 mg/litre)
et modérément à fortement toxique pour les arthropodes aquatiques
(valeur de la CL50 à 48 et 96 heures comprises en entre 0,75
µg/litre et 7800 µg/litre). Toutefois les concentrations dont il est
fait état dans les eaux superficielles après épandage dans des forêts
à la dose de 6kg/ha, sont inférieures à ces valeurs. Il s'en suit
qu'en utilisation normale, le trichlorfon n'aura guère d'effets sur
les organismes aquatiques car les autres types d'organismes tels que
les mollusques et les microorganismes sont moins sensibles que les
arthropodes. Les valeurs de la DL50 tirées d'études en laboratoire
se situent entre 40 et 180 mg/kg et montrent donc que le trichlorfon
est modérément toxique pour les oiseaux. Toutefois des études menées
sur le terrain après épandage de trichlorfon par voie aérienne sur des
forêts, n'ont révélé aucun effet sur l'effectif, la formation de
couples, le nichage ou la mortalité des oiseaux chanteurs. La
diminution du chant et l'accroissement de l'activité trophique qui ont
été constatés sont peut-être la conséquence d'une réduction du nombre
de proies. Rien n'indique que le trichlorfon puisse avoir un effet
nocif sur la faune terrestre, à part les arthropodes. On ne possède
aucune donnée au sujet des effets de cet insecticide sur les
arthropodes utiles.
1.4 Effets sur les animaux d'expérience et sur les systèmes
d'épreuves in vitro
Le trichlorfon est un insecticide modérément toxique pour les
animaux d'expérience. Chez l'animal de laboratoire, les valeurs de la
DL50 pour le trichlorfon technique s'étagent de 400 à 800 mg/kg de
poids corporel et pour le rat, la DL50 cutanée dépasse 2000 mg/kg de
poids corporel.
L'intoxication par le trichlorfon offre le tableau clinique
habituel de l'atteinte cholinergique due aux organophosphorés et qui
résulte d'une accumulation d'acétylcholine au niveau des terminaisons
nerveuses.
On a montré que le trichlorfon technique était modérément
irritant pour la conjonctive chez le rat, aucun effet de ce genre
n'ayant été noté lors de tests cutanés sur des lapins. Sa capacité de
sensibilisation cutanée a été mise en évidence chez le cobaye.
Des études de toxicité par voie orale de brève durée ont été
effectuées sur des rats, des chiens, des singes, des lapins et des
cobayes. Lors d'une étude de 16 semaines sur des rats, de 4 ans sur
des chiens et de 26 semaines sur des singes, on a fixé respectivement
à 100 mg/kg de nourriture, 5' mg/kg de nourriture et 0,2 mg/kg de
nourriture la dose sans effet observé (sur la base de l'activité
cholinestérasique, plasmatique, érythrocytaire ou cérébrale). En
exposant par voie respiratoire des rats pendant trois semaines, on a
obtenu une dose sans effet observé de 12,7 mg/m3 en se basant sur
l'inhibition de l'activité cholinestérasique du plasma, des
érythrocytes et du cerveau. Des études de toxicité et de
cancérogénicité à long terme ont été menées sur des souris, des rats,
des singes et des hamsters à qui l'on a administré du trichlorfon par
voie orale,intrapéritonéale ou percutanée. Après exposition par voie
orale des souris et des rats à des doses de 30 mg/kg de poids corporel
et de 400 mg/kg de nourriture respectivement, on a observé des
anomalies au niveau des gonades. Lors d'une étude de 24 mois sur des
rats et de 10 ans sur des singes, on a obtenu des doses sans effet
observé respectivement égales à 50 mg/kg de nourriture et 0,2 mg/kg de
poids corporel. Les données sont on dispose ne permettent pas de
conclure que la substance est cancérogène après administration à des
animaux de laboratoire pendant une longue période par diverses voies.
Dans les conditions physiologiques , le trichlorfon est
susceptible d'alkyler l'ADN. Les tests de mutagénicité ont donné des
résultats tantôt positifs tantôt négatifs. Il a peut que les effets
observés soient imputables en tout ou partie au dichlorvos. La plupart
des études de mutagénicité effectuées in vitro sur des bactéries ou
des cellules mammaliennes ont donné des résultats positifs alors que
rares sont les études in vivo qui ont donné de tels résultats.
L'expérimentation sur la souris, le rat et le hamster montre que
le trichlorfon suscite une réaction tératogène chez le rat à des doses
suffisantes pour être toxiques chez la mère. Après avoir administré à
des rattes en cours de gestation une dose de 145 mg de trichlorfon par
kg de nourriture, on a observé des malformations chez les foetus. Une
dose de 400 mg/kg de poids corporel administrée par gavage à des
hamsters a également été toxique pour les mères et a provoqué des
effets tératogènes. La dose la plus faible administrée par gavage et
ayant provoqué des effets tératogènes chez le rat était de 80 mg/kg de
poids corporel. Au cours de la période de gestation, les effets
produits présentent une spécificité chronologique. Cette étude a
permis de fixer à 8 mg/kg la dose sans effet observé.
Des doses sans effet observé respectivement égales à 8 mg/kg et
à 200 mg/kg de poids corporel ont été observées chez le rat et le
hamster. Chez le porc et le cobaye, on a constaté une action
tératogène sur le système nerveux central.
Cependant, aucun effet tératogène n'a été observé lors d'une
étude de reproduction portant sur trois générations de rats, au cours
de laquelle on a relevé des effets nocifs sur la fonction de
reproduction. Dans cette étude, la dose sans effet observé était de
300 mg/kg de nourriture.
A très hautes doses, le trichlorfon produit des effets
neurotoxiques chez l'animal.
Chez les mammifères, le métabolite actif est le dichlorvos, dont
l'activité anticholinestérasique est au moins 100 fois plus forte que
celle du trichlorfon.
1.5 Effets sur l'homme
Plusieurs cas d'intoxication aiguë délibérée (suicide) ou
accidentelle se sont produits. L'intoxication présente une
symptomatologie caractéristique de l'inhibition de la cholinestérase:
épuisement, faiblesse, confusion, sueur et salivation profuses,
douleurs abdominales, vomissements, myosis et spasmes musculaires.
Dans les cas graves, l'intoxication entraîne la perte de conscience et
des convulsions et la mort survient généralement par insuffisance
respiratoire. Chez les victimes qui ont survécu à l'intoxication grâce
à une intervention médicale, on a observé quelquefois, plusieurs
semaines après l'exposition, une polyneuropathie retardée accompagnée
d'une faiblesse des membres inférieurs. Dans les cas mortels,
l'autopsie a révélé des foyers d'ischémie dans le cerveau, la moelle
épinière et les ganglions végétatifs, ainsi que des lésions de la
gaine de myéline dans la moelle épinière et les pédoncules cérébraux,
avec des altérations dans la structure des axones des nerfs
périphériques.
Les quelques cas d'intoxication d'origine professionnelle qui se
sont produits, s'expliquent essentiellement par des négligences au
niveau de la sécurité. L'exposition professionnelle sur les lieux de
travail à des concentrations atmosphériques supérieures à 0,5 mg/m3
a entraîné une réduction de la cholinestérase plasmatique et une
altération du tracé électroencéphalographique. Toutefois ces anomalies
ont complètement régressé à l'arrêt de l'exposition. Aucun cas de
sensibilisation cutanée n'a été signalé.
Ce composé est très largement utilisé pour le traitement de la
schistosomiase chez l'homme. L'administration d'une dose unique (7 à
12 mg/kg) entraîne une inhibition de la cholinestérase plasmatique et
érythrocytaire à hauteur de 40-60 %, sans entraîner de symptômes
cholinergiques. Toutefois, des symptômes légers ont été observés chez
des malades qui avaient pris de ce produit à plusieurs reprises. A
forte dose (24 mg/kg) on a observé de graves symptômes cholinergiques
2. Conclusions
- Le trichlorfon est un insecticide organophosphoré modérément
toxique. Une intoxication grave peut survenir par suite
d'une exposition excessive au produit lors de sa
manipulation, de sa fabrication ou de son utilisation ou par
suite d'une ingestion accidentelle ou délibérée.
- L'exposition au trichlorfon de la population générale
résulte principalement de son utilisation en agriculture et
en médecine vétérinaire et dans le traitement de la
schistosomiase (bilharziose).
- Les quantités de trichlorfon qui sont absorbées sont très
inférieures à la dose journalière adminissible fixée par la
FAO et l'OMS et ne devraient pas constituer une menace pour
la santé publique.
- Si l'on adopte de bonnes méthodes de travail et, que l'on
respecte les précautions d'hygiène et de sécurité, le
trichlorfon n'est vraisemblablement pas dangereux pour les
personnes qui sont exposées de par leur profession.
- Bien que le trichlorfon soit très toxique pour les
arthropodes non visés, son utilisation n'entraîne guère
d'effets nocifs sur la faune et la flore.
3. Recommandations
- Afin de préserver la santé et le bien-être des ouvriers et
de la population en général, il importe de confier la
manipulation et l'épandage du trichlorfon exclusivement à
des personnes correctement encadrées et expérimentées, qui
sauront appliquer les mesures de sécurité indispeurables et
utiliser convenablementle produit.
- Des précautions sont à observer lors de la production, de la
formulation, de l'utilisation en agriculture et du rejet du
trichlorfon afin de contaminer le moins possible
l'environnement et plus spécialement des eaux de surface.
- Les travailleurs qui sont régulièrement exposés au
trichlorfon ainsi que les malades traités avec ce produit
doivent subir des examens médicaux périodiques.
- Les doses d'emploi en agriculture devront rester faibles
afin d'éviter la destruction des arthropodes non visés.
L'insecticide ne devra jamais être épandu sur des étendues
ou des cours d'eau.
RESUMEN Y EVALUACION, CONCLUSIONES Y RECOMENDACIONES
1. Resumen y evaluación
1.1 Exposición
El triclorfón es un insecticida organofosforado que lleva
utilizándose desde principios de los años cincuenta. En agricultura
sirve principalmente para combatir las plagas de insectos en los
cultivos extensivos y en los frutales. Se utiliza asimismo para
combatir los insectos de los bosques y los parásitos de los animales
domésticos. Bajo la denominación de metrifonato, se emplea para tratar
la infestación del hombre por Schistosoma haematobium. Se considera
un reservorio de liberación lenta de diclorvos. El triclorfón se
presenta en forma de solución concentrada emulsionable, polvos para
disolución o aplicación en seco, gránulos, solución y soluciones
concentradas de volumen muy reducido.
La concentración de triclorfón insecticida en el aire puede
alcanzar 0,1 mg/m3 al poco tiempo del rociamiento, pero a los pocos
días los niveles se sitúan alrededor de 0,01 mg/m3. En las aguas de
escorrentía de zonas rociadas, la concentración de triclorfón puede
alcanzar los 50 µg/litro; en cambio en las aguas de superficie suele
ser mucho más baja y disminuye rápidamente.
El triclorfón se degrada rápidamente en el suelo; las
concentraciones suelen disminuir hasta cantidades insignificantes
durante el mes que sigue a la aplicación. Es relativamente estable en
agua si el pH es inferior a 5,5; con un pH más elevado se transforma
en diclorvos. Aunque los microorganismos y las plantas pueden
metabolizar el triclorfón, la vía de eliminación más importante es la
hidrólisis abiótica.
Salvo raras excepciones, las concentraciones de triclorfón en los
cultivos son inferiores a 10 mg/kg al día siguiente de la aplicación,
e inferiores a 0,1 mg/kg durante las dos semanas siguientes.
La leche de vacas tratadas con triclorfón para combatir plagas
puede contener hasta 1,2 mg de residuos/litro a las dos horas de la
aplicación, pero las cifras descienden hasta menos de 0,1 mg/litro a
las 24 horas del tratamiento. No se han encontrado concentraciones
importantes de este compuesto en la carne de animales tratados. En los
huevos de gallinas tratadas se han comprobado valores de 0,05 mg de
triclorfón/kg.
1.2 Ingestión, metabolismo y excreción
El triclorfón se absorbe fácilmente por todas las vías de
exposición (oral, cutánea, respiratoria) y se distribuye rápidamente
a los tejidos del cuerpo. Se detectaron concentraciones máximas en la
sangre al cabo de 1-2 h, y la sustancia desapareció casi por completo
del torrente sanguíneo en cuestión de 1,5-4 h. Se calculó que la
semivida biológica del triclorfón en la sangre de mamíferos es de
alrededor de 30 minutos.
Por deshidrocloración, el triclorfón se transforma en diclorvos
(2,2-diclorovinil dimetil fosfato) en el agua y en los humores y
tejidos de los seres vivos, si el pH es superior a 5,5. El diclorvos
es la anticolinesterasa fisiológicamente activa. Las principales rutas
de degradación son la desmetilación, la escisión del enlace P-C y la
hidrólisis del éster con el diclorvos como producto intermediario. Los
principales metabolitos del triclorfón que se encuentran in vivo son
el demetil triclorfón, el demetil diclorvos, el dimetil
hidrogenofosfato, el metil hidrogenofosfato, el ácido fosfórico y el
tricloroetanol. El último metabolito se encuentra en la orina en forma
de conjugado de glucurónido.
El triclorfón y sus productos metabólicos se eliminan
principalmente con la orina. Los estudios realizados con triclorfón
radiomarcado (14C-metilo y 32P-metilo) revelaron que la mayor
parte de la sustancia se eliminaba en forma hidrosoluble, y una
pequeña parte en forma soluble en cloroformo. Alrededor del 66%-70% de
los productos hidrosolubles aparecían en la orina al cabo de 12 horas,
mientras que el 24% del material marcado con 14C-metilo se eliminaba
en el aire espirado en forma de dióxido de carbono (CO2). Se han
detectado concentraciones bajas de triclorfón y sus metabolitos en la
leche de bóvidos tras el tratamiento de éstos por vía oral y cutánea.
1.3 Efectos en organismos del medio ambiente
El triclorfón es moderadamente tóxico para los peces (los valores
de la CL50 a las 96 h varían entre 0,45 mg/litro y 51 mg/litro) y de
toxicidad moderada a elevada para los artrópodos acuáticos (la CL50
a las 48 h/96 h varía entre 0,75 µg/litro y 7800 µg/litro). En cambio,
los valores observados en aguas de superficie tras aplicar el
compuesto en bosques a razón de 6 kg/ha quedan por debajo de estos
límites. Así pues, si es objeto de un uso normal, el triclorfón tendrá
un efecto muy reducido o nulo en las poblaciones de organismos
acuáticos, puesto que otros grupos como los moluscos y los
microorganismos, son menos sensibles que los artrópodos. Los valores
de la DL50 obtenidos en estudios de laboratorio (de 40 a 180 mg/kg)
indican que este compuesto es moderadamente tóxico para las aves. En
cambio, en estudios de campo no se observó efecto alguno en la
población total, el número de parejas en época de cría, la viabilidad
de los nidos ni la mortalidad de las aves canoras de los bosques
tratados mediante aplicaciones aéreas del insecticida. Se observó
cierta disminución de la actividad canora y mayor actividad de
búsqueda de alimento, tal vez por haberse reducido las poblaciones de
los organismos de que se nutren. Nada indica que el triclorfón
perjudique a los organismos terrestres excepto los artrópodos. No se
dispone de información sobre sus efectos en artrópodos beneficiosos.
1.4 Efectos en animales de laboratorio y en sistemas de
ensayo in vitro
El triclorfón es un insecticida moderadamente tóxico para los
animales de laboratorio. Los valores de la DL50 para el producto
técnico administrado por vía oral a éstos varían entre 400 y 800 mg/kg
de peso corporal; en la rata, los valores de la DL50 cuando se
administra por vía cutánea superan los 2000 mg/kg de peso corporal.
La intoxicación por este compuesto origina los signos
colinérgicos comúnmente relacionados con los organofosfatos y que se
atribuyen a la acumulación de acetilcolina en las terminaciones
nerviosas.
Se ha demostrado que el triclorfón técnico es moderadamente
irritante para los ojos de la rata, aunque no para la piel del conejo.
Se ha observado potencial de sensibilización cutánea en conejillos de
Indias.
Se llevaron a cabo estudios de corto plazo sobre toxicidad por
vía oral en ratas, perros, monos, conejos y conejillos de Indias. En
uno de 16 semanas en ratas, en otro de 4 años en perros y en un
tercero de 26 semanas en monos, se registraron respectivamente las
siguientes concentraciones sin efectos observados (NOEL): 100 mg/kg
de ración alimenticia, 50 mg/kg de ración alimenticia y 0,2 mg/kg de
peso corporal (calculados respecto de la actividad de la colinesterasa
en plasma, eritrocitos o encéfalo). La exposición de ratas por
inhalación durante más de tres semanas indicó una NOEL de 12,7
mg/m3, calculados respecto de la inhibición de la actividad de la
colinesterasa en plasma, eritrocitos y encéfalo. Se llevaron a cabo
estudios de toxicidad/carcinogenicidad a largo plazo en ratones,
ratas, monos y hámsters tras la administración oral, intraperitoneal
o cutánea. Se observaron efectos adversos en las gónadas de ratones y
ratas expuestos por vía oral a 30 mg/kg de peso corporal y 400 mg/kg
de ración alimenticia, respectivamente. En un estudio de 24 meses en
ratas y otro de 10 años en monos, se determinaron valores de NOEL de
50 mg/kg de ración alimentaria y de 0,2 mg/kg de peso corporal,
respectivamente. Los datos disponibles no aportan pruebas de
carcinogenicidad tras la exposición prolongada de animales de
laboratorio por diversas vías de administración.
Se ha comunicado que, en condiciones fisiológicas, el triclorfón
tiene la propiedad de alquilizar al ADN. Los ensayos de mutagenicidad
han dado resultados tanto positivos como negativos. Es posible que el
diclorvos sea la causa, parcial o totalmente, de los efectos
observados. La mayoría de los estudios de mutagenicidad in vitro en
células tanto bacterianas como de mamíferos dieron resultado positivo;
en cambio, pocos de los estudios in vivo dieron ese resultado.
Investigaciones realizadas en el ratón, la rata y el hámster
indican que, en dosis lo bastante elevadas como para producir
toxicidad materna, el triclorfón produce una respuesta teratógena en
ratas. La exposición de ratas gestantes a dosis de 145 mg/kg de ración
alimenticia provocó malformaciones fetales. La administración oral
forzada de 400 mg/kg de peso corporal a hámsters produjo también
toxicidad materna y respuesta teratógena. Por esta vía, la dosis más
baja que produjo efectos teratógenos en la rata fue de 80 mg/kg de
peso corporal. Los efectos son específicos según el momento del
periodo de gestación en que se ingiere el producto. En este estudio de
administración oral forzada se obtuvo una NOEL de 8 mg/kg.
En ratas y hámsters se han encontrado, respectivamente, NOEL de
8 mg/kg de peso corporal y 200 mg/kg de peso corporal. También se han
comunicado respuestas teratógenas que afectaban al sistema nervioso
central en el cerdo y el conejillo de Indias.
En cambio, no se observaron efectos teratógenos en un estudio de
reproducción en tres generaciones de ratas, en el que con dosis
elevadas se indujeron efectos reproductivos adversos. La NOEL en este
estudio fue de 300 mg/kg de ración alimenticia.
Con dosis muy elevadas se han producido efectos neurotóxicos en
animales.
En los mamíferos, el producto activo de la transformación es el
diclorvos, cuya actividad como anticolinesterasa es como mínimo 100
veces mayor que la del triclorfón.
1.5 Efectos en el ser humano
Se han producido varios casos de envenenamiento agudo por
exposición intencional (suicidio) o accidental. Los signos y síntomas
de la intoxicación fueron los característicos de la inhibición de la
acetilcolinesterasa: agotamiento, debilidad, confusión, sudación y
salivación excesivas, dolores abdominales, vómitos, pupilas
puntiformes y espasmos musculares. En casos graves se observaron
pérdida de la consciencia y convulsiones, y la muerte se produjo en
general por fallo respiratorio. En las víctimas que sobrevivieron
gracias a la intervención médica, a veces se observó una
polineuropatía diferida, acompañada de debilidad de los miembros
inferiores, la cual apareció algunas semanas después de la exposición.
En los casos mortales, la autopsia reveló alteraciones isquémicas en
el encéfalo, la médula espinal y los ganglios vegetativos, lesiones de
la vaina mielínica en la médula espinal y los pedúnculos cerebrales,
y cambios estructurales en los axones de los nervios periféricos.
Se han producido algunos casos de envenenamiento ocupacional,
principalmente por no observar las normas de seguridad. La exposición
en un lugar de trabajo con concentraciones en el aire superiores a 0,5
mg/m3 ocasionó la disminución de la colinesterasa plasmática y
cambios del trazado electroencefalográfico. No obstante, estos signos
desaparecieron por completo al cesar la exposición. No se ha
comunicado ningún caso de sensibilización cutánea.
Este compuesto se ha usado extensamente para tratar la
esquistosomiasis del ser humano. La administración de una dosis única
(7-12 mg/kg) inhibió entre el 40 y el 60% de la colinesterasa del
plasma y los eritrocitos, sin que aparecieran síntomas colinérgicos.
En cambio, se observaron síntomas leves en personas tratadas con dosis
repetidas. Las dosis elevadas (24 mg/kg) produjeron síntomas
colinérgicos graves.
2. Conclusiones
- El insecticida triclorfón es un éster organofosforado
moderadamente tóxico. La exposición excesiva que puede
producirse al fabricarlo o utilizarlo y la ingestión
accidental o intencional pueden provocar envenenamientos
graves.
- La exposición de la población general al triclorfón se
produce principalmente como resultado de las prácticas
agrícolas y veterinarias y del tratamiento de la infestación
por Schistosoma haematobium.
- Las ingestas de triclorfón comunicadas se encuentran muy por
debajo de la ingesta diaria admisible establecida por la
FAO/OMS y en principio no constituyen un riesgo para la
salud de la población general.
- Si se siguen prácticas correctas de trabajo, medidas
higiénicas y precauciones de seguridad, es poco probable que
el triclorfón represente un riesgo para las personas
expuestas por su trabajo.
- A pesar de su gran toxicidad para otros artrópodos que no se
pretende destruir, el triclorfón se ha utilizado con escasos
o nulos efectos adversos para las poblaciones de organismos
del medio ambiente.
3. Recomendaciones
- Para proteger la salud de los trabajadores y de la población
general, la manipulación y la aplicación del triclorfón
deben encomendarse solamente a operarios bien supervisados
y adiestrados, que observarán medidas adecuadas de seguridad
y utilizarán el insecticida siguiendo prácticas correctas.
- La fabricación, la formulación, el uso agrícola y la
evacuación del triclorfón deben sujetarse a una gestión
cuidadosa para reducir al mínimo la contaminación del medio,
en particular las aguas de superficie.
- Las poblaciones de trabajadores y de personas regularmente
expuestos deben someterse a exámenes médicos periódicos.
- Las tasas de aplicación del triclorfón deben limitarse a fin
de evitar efectos sobre artrópodos que no se pretende
combatir. Este insecticida nunca debe rociarse sobre masas
ni corrientes de agua.